

Abstract
Brain metastasis is a significant cause of morbidity and mortality in multiple cancer types and represents an unmet clinical need. The mechanisms that mediate metastatic cancer growth in the brain parenchyma are largely unknown. Melanoma, which has the highest rate of brain metastasis among common cancer types, is an ideal model to study how cancer cells adapt to the brain parenchyma. Our unbiased proteomics analysis of melanoma short-term cultures revealed that proteins implicated in neurodegenerative pathologies are differentially expressed in melanoma cells explanted from brain metastases compared to those derived from extracranial metastases. We showed that melanoma cells require amyloid beta (Aβ) for growth and survival in the brain parenchyma. Melanoma-secreted Aβ activates surrounding astrocytes to a pro-metastatic, anti-inflammatory phenotype and prevents phagocytosis of melanoma by microglia. Finally, we demonstrate that pharmacological inhibition of Aβ decreases brain metastatic burden.
Introduction
Brain metastasis is the most common form of adult intracranial malignancy (1) and results in severe morbidity and mortality. 40–75% of Stage IV melanoma patients develop brain metastasis (2,3), reflecting melanoma’s striking ability to colonize the brain. Although recent clinical trials have shown brain metastases to be similarly responsive to approved targeted and immune therapies as extracranial metastases (4–6), responses are less durable and the majority of patients eventually succumb to disease (7). Thus, there is an urgent need for new clinical options for patients with brain metastasis. In recent years, research has started to elucidate the molecular mechanisms contributing to the multi-step process of brain metastasis. Most findings have focused on cancer extravasation across the blood-brain barrier (BBB), which cannot be leveraged therapeutically given that the vast majority of patients who eventually develop brain metastasis already have extravasated cancer cells in the brain parenchyma at the time of cancer diagnosis. The main bottleneck in the brain metastatic process has been shown to be the successful expansion of a single cell in the brain parenchyma to a macro-metastasis (8). Recent studies have begun to demonstrate the role of the brain microenvironment in this process. In particular, reactive astrocytes have been shown to interact with cancer cells in the brain (9,10) and exhibit both pro- and anti-brain metastatic activity (11,12). Astrocytes have several roles in normal brain physiology, including neurotransmitter uptake (13), metabolic support (14), and response to injury (15). Furthermore, astrocytes have been heavily implicated as both neurotoxic and neuroprotective in a variety of neurodegenerative pathologies, including Alzheimer’s disease (16–18). These findings suggest the intriguing possibility of a functional connection between neurodegenerative pathologies and brain metastasis, which has not yet been explored.
Here, we demonstrate that melanoma cells require amyloid beta (Aβ), a polypeptide heavily implicated in Alzheimer’s disease, for survival and growth in the brain parenchyma. Melanoma cells cleave Amyloid Precursor Protein (APP) to produce and secrete Aβ, and Aβ secreted from cancer cells triggers local astrocytes to adopt a pro-metastatic, anti-inflammatory phenotype. Melanoma-secreted Aβ promotes anti-inflammatory microglial polarization and protects melanoma cells from phagocytosis by microglia. Targeting Aβ production by pharmacologic inhibition of β-secretase activity suppresses metastatic growth of human cancer cells in the brain parenchyma of mice.
Results
Proteomics analysis links melanoma brain metastasis and neurodegeneration
To study mechanisms of brain metastasis, we leveraged pairs of brain metastasis-derived (BM) and non-brain metastasis-derived (NBM) melanoma short term cultures (STCs) obtained from the same patient (Fig. 1A) as a novel model of brain metastasis. Comparing patient-matched BM and NBM STC pairs reduces the confounding inter-patient heterogeneity characteristic of melanoma. Upon intracardiac injection in immunocompromised mice, BM STCs (12–273 BM, 10–230 BM) exhibited increased ability to metastasize to the brain compared to their paired NBM STCs (12–273 NBM, 10–230 NBM) (Fig. 1B–D; Supplementary Fig. S1A–D). These findings were confirmed in additional patient-matched STCs obtained from collaborators at an independent institution (WM-4071 BM vs. NBM, WM-4257 BM vs. NBM) (Supplementary Fig. S1F–G, I–J). Generally, BM STCs also demonstrated increased ability to metastasize to extracranial organs (Supplementary Fig. S1E and H). In some pairs, however, we observed a disproportionate increase in brain metastasis as compared to extracranial metastases as indicated by increased brain to body luminescence ratio (Fig. 1B and C; Supplementary Fig. S1A and B). Therefore, molecular differences between paired BM and NBM STCs likely reflect a combination of generally pro-metastatic features and adaptations that specifically favor brain metastasis.
Figure 1.
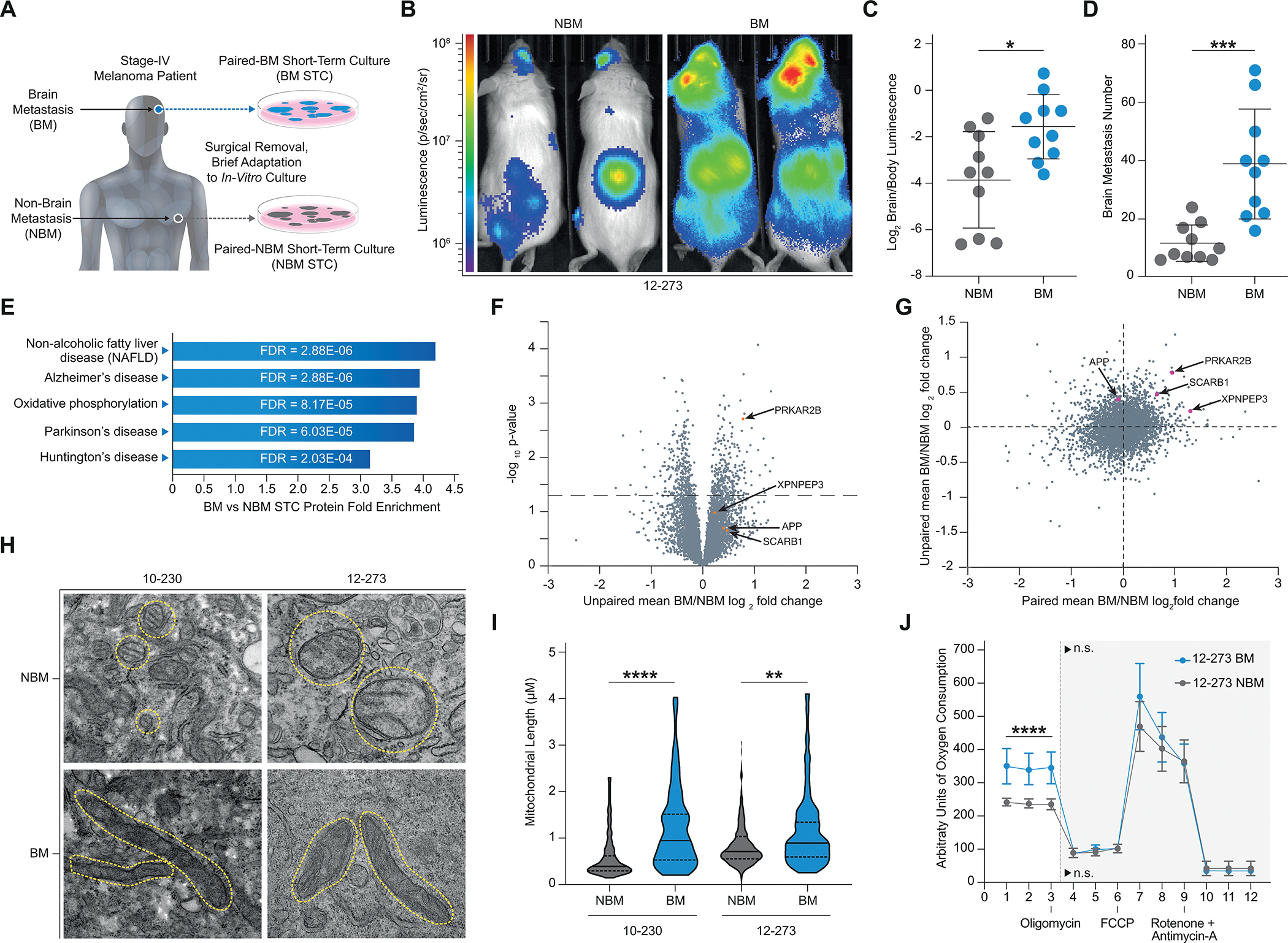
Proteomics Links Melanoma Brain Metastasis and Neurodegeneration. A, Diagram of the generation of patient-matched STC pairs. B-D, Intracardiac injection of 12–273 paired STCs in mice (10 NSG mice per group, representative experiment shown). B, Representative IVIS images of 12–273 STC pair at 29 days post intracardiac injection. C, Quantified brain/body luminescence ratio on day 29. 12–273 NBM vs. BM (* p<0.05). D, Number of brain metastases quantified by microscopy of serial FFPE sections with H&E staining. 12–273 NBM vs. BM (***, p<0.0005). E-G, Mass spectrometry analysis of whole cell lysates from a cohort of 12 BM and 12 NBM STCs, including 3 patient-matched pairs. E, Top enriched pathways (p<0.001) in BM vs NBM STCs identified by KEGG Pathway analysis of global protein levels. F, Volcano plot of mean Log2 BM/NBM fold change of global protein levels and -Log10 p-values. Orange – candidates selected for in-vivo mini-screen. G, Comparison of mean paired STC BM/NBM Log2 fold change to unpaired STC BM/NBM mean Log2 fold change. Pink – candidates selected for in-vivo mini-screen. H, Representative electron microscopy images of paired STCs. Yellow circles outline mitochondria. I, Quantification of average mitochondrial length in paired STCs. 10–230 NBM vs. BM (**** p<.00005), 12–273 NBM vs. BM (** p<.005). J, Seahorse MitoStress analysis of oxygen consumption rate in 12–273 NBM vs. BM. Basal oxygen consumption rate. 12–273 NBM vs BM (**** p<.00005).
Using a cohort of 12 BM and 12 NBM derived STCs, which included 3 isogenic patient-matched pairs (Supplementary Table S1 for clinicopathological information), we performed an unbiased mass spectrometry analysis of whole cell protein lysates to identify novel candidate pathways and proteins that may mediate melanoma brain metastasis. KEGG pathway analysis of differentially expressed proteins revealed enrichment in proteins related to neurodegenerative pathologies in the BM vs. NBM STCs (Fig. 1E–G, Supplementary Table S2). Of the top five enriched categories in BM vs NBM STCs (Fig. 1E), three were neurodegenerative diseases (Alzheimer’s, Parkinson’s and Huntington’s diseases) and one (oxidative phosphorylation) has been previously implicated in neurodegenerative pathologies. Proteomics results were validated by western blot analyses (Supplementary Fig. S1K and L). Metabolic analyses and transmission electron microscopy demonstrated that BM STCs have increased mitochondrial length and electron density (Fig. 1H and I), elevated rate of mitochondrial oxygen consumption (Fig. 1J; Supplementary Fig. S1M–O), and decreased rate of glycolysis (Supplementary Fig. S1P–R) than their respective paired NBM STCs. Data mining of published transcriptomic datasets from independent cohorts (19,20) confirmed a previously unreported enrichment of gene expression associated with neurodegenerative pathways in BM vs. NBM samples (Fig. 2A and B; Supplementary Fig. S2A; Supplementary Table S3). Altogether, these results provide further evidence of a recently reported connection between melanoma brain metastasis and oxidative phosphorylation (20,21), demonstrate the capability of our proteomic results to reflect phenotypic differences related to brain metastatic ability, and reveal a novel association between brain metastasis and neurodegenerative disorders.
Figure 2.
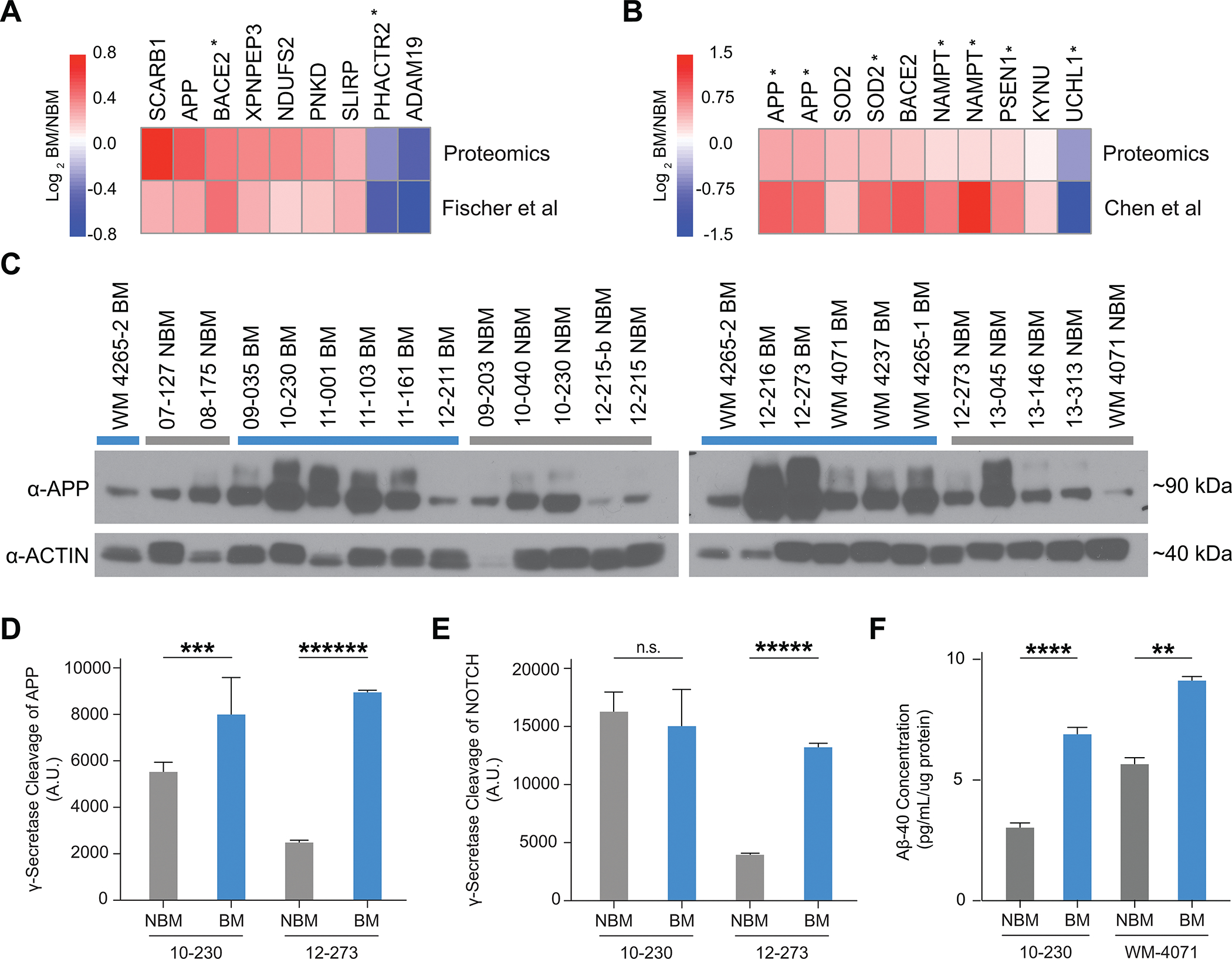
Production of Aβ by Melanoma Cells is Associated with Brain Metastasis. A,B, Heatmap comparing unpaired Log 2 BM/NBM protein levels from proteomics dataset (Figure 1E–G) to Log 2 BM/NBM gene expression from previously published Fischer et al (20)(A) (EGA Accession #EGAS00001003672) and Chen et al (19) (B) (GEO Accession # GSE50496), respectively. * = p-value < 0.05 for BM vs NBM gene expression in Fischer et al (A) or Chen et al (B). C, Western blot analysis of APP (22C11) in full cohort of STCs (12 BM and 12 NBM). WM4265–2 included twice (once in each blot) for reference of signal across blots. D-E, Quantification of gamma-secretase cleavage of APP (D) and NOTCH (E) (n=4 biological replicates per group, representative experiment shown). D, APP cleavage. 10–230 NBM vs. BM (*** p<0.0005). E, NOTCH cleavage. 12–273 NBM vs BM (***** p<0.000005). F, Quantification of Aβ-40 secretion by ELISA (n=2–3 biological replicates per group). 10–230 NBM vs BM (**** p<0.00005), WM-4071 NBM vs BM (** p<0.005).
Increased Aβ production by melanoma cells is associated with brain metastasis
An additional review of the literature on the top differentially expressed proteins in our proteomics profiling revealed multiple factors involved in APP cleavage. APP can be processed to produce Aβ, a polypeptide heavily implicated in Alzheimer’s disease, through sequential cleavage by β- and γ-secretases (22). Both BACE2, a β-secretase enzyme, and PSEN1, the catalytic subunit of the γ-secretase complex, were found enriched in BM vs NBM STCs (Fig. 2A and B). Furthermore, both UCHL1, a ubiquitin ligase that targets β-secretase for degradation and decreases Aβ production, and ADAM19, an α-secretase enzyme that cleaves APP within the Aβ sequence and precludes Aβ production, were found decreased in BM vs NBM STCs (Fig. 2A and B). Taken together, these findings suggested the possibility of a connection between increased Aβ production by melanoma cells and brain metastasis.
While APP has been shown to be expressed in a wide variety of normal tissues and tumor types, Aβ synthesis by cancer cells has not been documented or explored. Therefore, we sought to investigate whether melanoma cells can produce Aβ and, if so, whether Aβ is altered in BM vs. NBM STCs. We began by investigating APP expression, a requirement for Aβ production, in melanoma STCs. By proteomics analysis, APP was increased in unpaired BM vs. NBM STCs (Fig. 1F) but was not consistently increased in paired BM vs. NBM STCs (Fig. 1G). By western blot analysis, APP was generally increased in both paired and unpaired BM STCs relative to NBM STCs by western blot (Fig. 2C, Supplementary Fig. S2B).
Using probe specific γ-secretase assays (23), we investigated whether melanoma cells can cleave APP with γ- secretase, which is required for production of Aβ. We found that melanoma STCs can cleave APP with gamma secretase and observed consistently increased cleavage of APP in BM vs. paired NBM STCs (Fig. 2D). Notably, increased cleavage of NOTCH, a canonical γ- secretase substrate, was not consistently observed in paired BM vs. NBM STCs (Fig. 2E), which may reflect a specific association between γ- secretase cleavage of APP and brain metastasis.To directly investigate whether melanoma cells produce and secrete Aβ, we analyzed melanoma-conditioned media by ELISA and observed increased Aβ levels in BM vs NBM melanoma-conditioned media. This demonstrated that melanoma cells produce and secrete Aβ, and that Aβ secretion is increased in BM relative to paired NBM STCs (Fig. 2F). Given that Aβ can have profound effects on the brain microenvironment and is implicated in the development of Alzheimer’s disease (22), we hypothesized that melanoma cells may require Aβ for survival or growth in the brain parenchyma.
APP is specifically required for melanoma brain metastasis
To investigate whether proteins related to neurodegeneration are functional mediators of melanoma brain metastasis, we performed an in vivo brain metastasis mini-screen. In addition to Aβ, we selected three proteins (PRKAR2B, SCARB1, XPNPEP3) that were found to be consistently increased in both paired and unpaired BM STCs relative to NBM STCs (Fig. 1F and G) and are implicated in mitochondrial metabolism and/or neurodegeneration (24–26). We silenced PRKAR2B, SCARB1, and XPNPEP3 directly with lentiviral shRNA in the 12–273 BM STC (Supplementary Fig. S3A). Because Aβ is a post-translational product of APP cleavage, we targeted Aβ indirectly through silencing of APP (Fig. 3A). Silencing of APP, PRKAR2B, SCARB1, or XPNPEP3 did not affect the proliferative capacity of melanoma cells in vitro (Fig. 3A, Supplementary Fig. S3B–D). We measured the effects of candidate silencing on metastatic capability upon intracardiac injection in mice. Silencing of APP and PRKAR2B resulted in a significantly decreased brain to body luminescence signal (Fig. 3B), indicating that these proteins have differential effects on metastatic potential to the brain as compared to other organs. However, since mice injected with PRKAR2B-depleted cells did not have a statistically significant decrease in brain luminescence (Supplementary Fig. S3E) and had increased metastasis to extracranial organs (Supplementary Fig. S3F and S3G), we decided to focus on APP. Silencing of APP resulted in reduced colonization of the brain and a decreased brain/body luminescence signal (Fig. 3C and D). Ex vivo MRI of mouse brains revealed that loss of APP decreases overall brain tumor burden (Fig. 3E and F; Supplementary Video S1), average brain metastasis size (Supplementary Fig. S3H), and number of brain metastases (Supplementary Fig. S3I). Metastatic burden to specific organs was further quantified by NuMA immunohistochemistry, which specifically labels the nuclei of human cells within mouse organs. Loss of APP resulted in a dramatic reduction of brain metastatic burden (Fig. 3G and H; Supplementary Fig. S3J and S3K), but had no significant effect on metastasis to either the liver (Fig. 3I, Supplementary Fig. S3L and S3M) or the kidneys (Supplementary Fig. S3N). To further confirm the specific role of APP in brain metastasis, we knocked-out APP via CRISPR/Cas9 genome editing with 2 independent small guide RNA (sg-RNA) in 12–273 BM and an additional melanoma cell line (131/4–5B1. hereafter 5B1; ref. 23), which also displays brain tropism upon intracardiac injection in immunodeficient mice. As observed with sh-RNA, APP depletion with both sg-RNAs (Supplementary Fig. S3O) did not affect in vitro cell proliferation in either cell line (Supplementary Fig. S3P and S3Q), but significantly reduced brain metastasis in vivo (Fig. 3J and K; Supplementary Fig. S3R and S3S) without affecting extracranial metastasis (Fig. 3L). Altogether, these data demonstrate that APP is required for melanoma to colonize the brain but not to metastasize to other organs.
Figure 3.
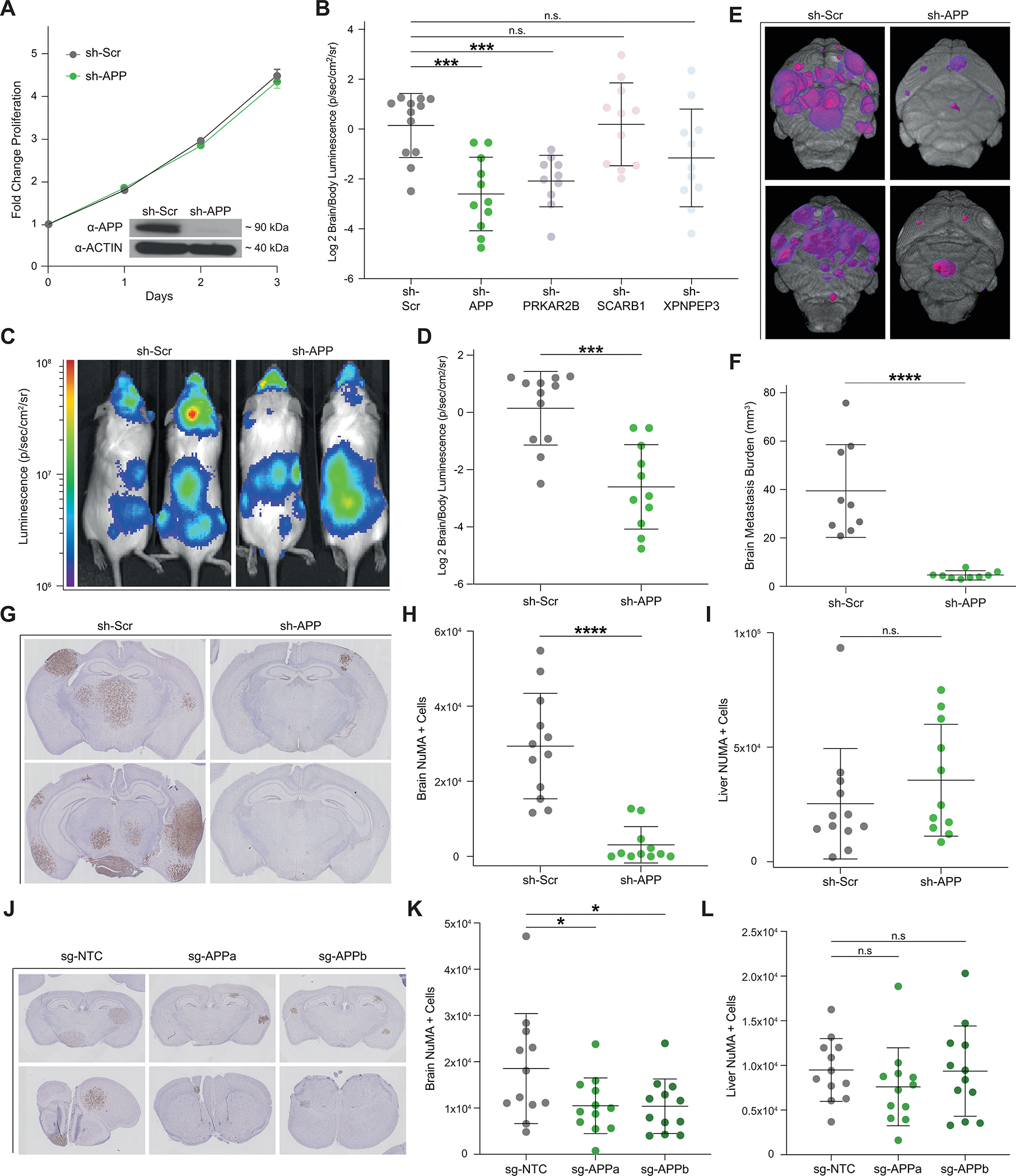
APP is Specifically Required for Melanoma Brain Metastasis. A, In vitro proliferation curve and western blot analysis of 12–273BM cells transduced with sh-APP vs. sh-Scr. B, Quantified Log2 Brain/Body luminescence 35 days post intracardiac injection in NSG mice of 12–273 BM with shRNA-mediated silencing of selected candidates or scrambled hairpin control (sh-Scr) (10–12 NSG mice per group). sh-Scr vs. sh-APP (*** p<0.0005), sh-Scr vs. sh-PRKAR2B (*** p<0.0005). C, Representative IVIS images at day 35. D, Quantified Log2 Brain/Body luminescence at day 35 of mice injected with 12–273 BM cells transduced with sh-Scr vs. sh-APP lentivirus (*** p<0.0005). E-F, Ex-vivo brain MRI of mice injected with 12–273 BM sh-Scr vs. sh-APP (9 mice per group). E, Representative images. Pink-purple – brain metastasis. F, Quantification of brain metastatic burden by MRI. sh-Scr vs. sh-APP (**** p<0.00005). (G-I), Staining of metastatic cells by anti-NuMA immunohistochemistry on FFPE sections of mice injected with sh-Scr vs. sh-APP (10–12 NSG mice per group). G, Representative brain images. H-I, Quantification of NuMA+ metastatic cells in mouse brains (H), sh-Scr vs. sh-APP (**** p<0.00005), and livers (I). J-L, Staining of metastatic cells by anti-NuMA immunohistochemistry on FFPE brain sections of mice injected with 12–273 BM cells with CRISPR/Cas9 mediated knockdown using a non-targeting control single guide RNA (sg-NTC) or one of two single guide RNAs targeting APP (sg-APPa, sg-APPb) (11–12 mice per group). J, Representative brain images. K-L, Quantification of NuMA+ metastatic cells in mouse brains (K), sg-NTC vs. sg-APPa (* p<0.05) and sg-NTC vs. sg-APPb (* p<0.05), and livers (L).
Aβ is the form of APP required for melanoma brain metastasis
We hypothesized that Aβ is required for melanoma brain metastasis and, to test that, we indirectly targeted Aβ through APP silencing. Silencing of APP blocks production of Aβ but also removes full length APP and blocks production of other cleavage products. Therefore, the loss of brain metastasis observed upon silencing of APP does not definitively establish which form of APP is required for melanoma brain metastasis. While our observation of increased cleavage of APP into Aβ in BM vs NBM STCs (Fig. 2C and 2E) suggests Aβ is the form of APP required for melanoma brain metastasis, additional experimentation is required to unequivocally attribute the role to Aβ.
To investigate this, we asked whether Aβ is sufficient to rescue the inhibition of metastasis observed upon APP silencing. We cloned SPA4CT-T43P (27), a heavily truncated (>75% of APP amino acid sequence removed), mutant form of APP that retains the ability to produce Aβ but cannot produce other major cleavage products, such as sAPP-α and sAPP-β (Fig. 4A). The substitution of proline for threonine at position 43 partially inhibits gamma secretase cleavage (28) and prevents excessive Aβ generation (Supplementary Fig. S4A and S4B). We knocked out APP in 12–273 BM STC using CRISPR/Cas9 and reintroduced either APP-770, the major full-length APP isoform expressed in melanoma cells (Supplementary Fig. S4C), or SPA4CT-T43P (Fig. 4B). We confirmed that knockout of APP reduces Aβ secretion and that both wild type APP (APP-770) and SPA4CT-T43P are able to rescue Aβ secretion to near physiologic levels (Fig. 4C). In vivo, the loss of brain metastasis observed upon APP knockout was rescued by introduction of either APP-770 or SPA4CT-T43P (Fig. 4D and E), indicating that Aβ is the form of APP required for melanoma brain metastasis.
Figure 4.
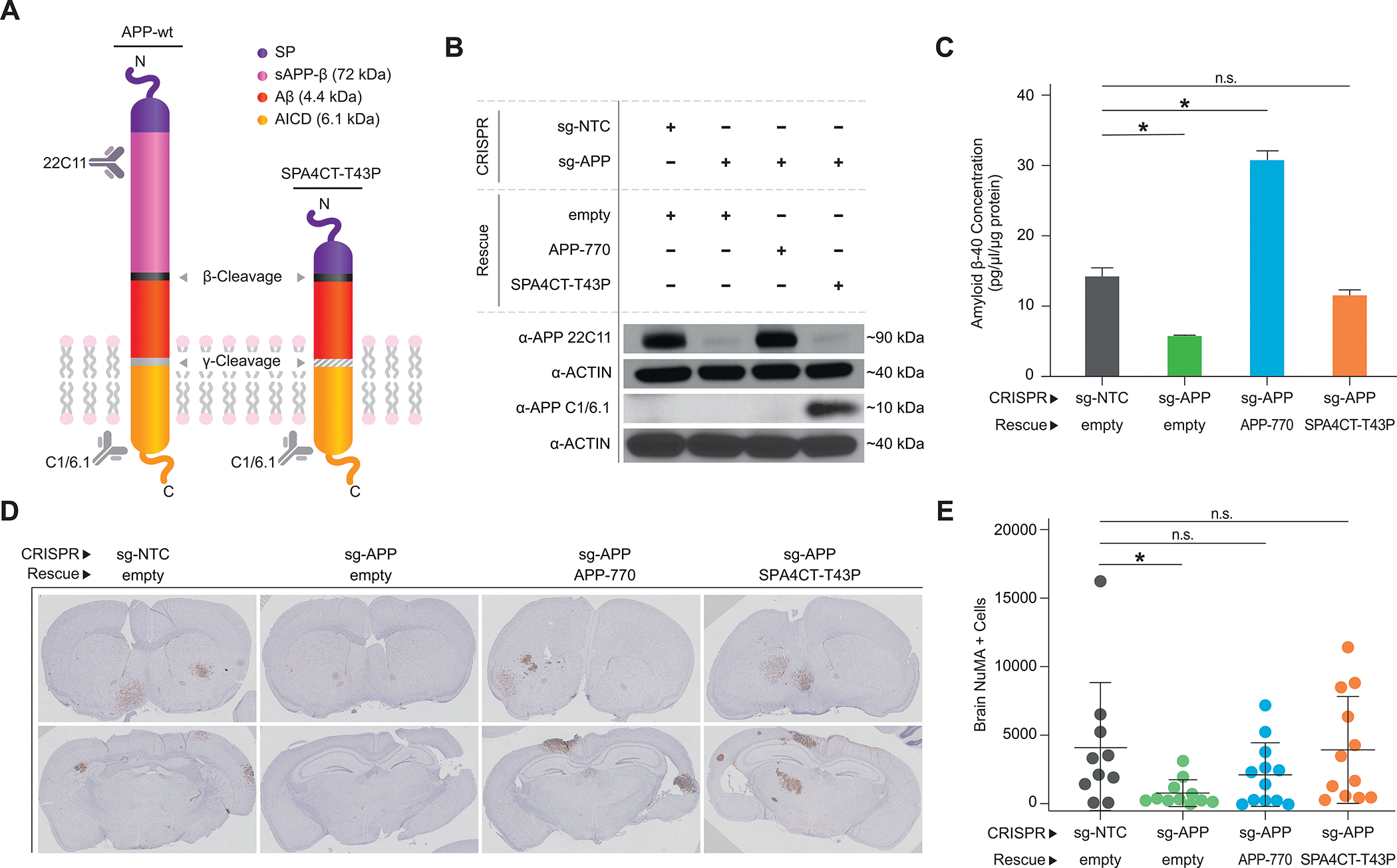
Aβ is the form of APP required for Melanoma Brain Metastasis. A, Diagram of wildtype APP and SPA4CT-T43P. B, Western blot analysis using anti-APP 22C11 and APP C1/6.1 in 12–273 BM cells transduced with the indicated constructs. C, Quantification of Aβ secretion by ELISA in 12–273 BM infected cells. sg-NTC empty vs. sg-APP empty (* p<0.05), sg-NTC empty vs. sg-APP APP-770 (* p<0.05). D-E, Intracardiac injection in mice of 12–273 BM sg-NTC or sg-APP transduced with indicated constructs (10–12 NSG mice per group). D, Representative images of FFPE brain slides with metastatic cells stained by anti-NuMA immunohistochemistry. D, Quantification of NuMA+ metastatic cells in mouse brains. sg-NTC vs. sg-APP (* p<0.05).
Melanoma-secreted Aβ is required for growth and survival in the brain parenchyma
To investigate how Aβ functions in melanoma brain metastasis, we began by identifying which step of the brain metastatic process requires Aβ. First, we established the timeline of brain metastasis in sh-Scr 12–273 BM STC by ex vivo immunofluorescence analysis of brain slices (Fig. 5A; Supplementary Videos S2–S6). At day 1 post intracardiac injection, melanoma cells are arrested in the brain microvasculature (Fig. 5Aa; Supplementary Video S2). By day 3, melanoma cells have extravasated into the parenchyma and remain adhered to the surface of blood vessels with a rounded morphology (Fig. 5Ab, Supplementary Video S3). From days 3 to 7, cells begin to divide and spread along the vasculature in an elongated morphology (Fig. 5Ac; Supplementary Video S4), a process known as vascular co-option (8). By day 7, approximately two thirds of the melanoma cells that had initially reached the brain at day 1 have died (Fig. 5B). This occurs when melanoma cells either fail to extravasate (Supplementary Fig. 5A) or undergo apoptosis upon entering the brain parenchyma (Supplementary Fig. 5B). From days 7–14, surviving melanoma cells begin to proliferate in a rounded morphology independent of the vasculature and form micro-metastases (Fig. 5Ad; Supplementary Video S5). After day 14, cells rapidly divide, forming small macro-metastases visible to the naked eye by day 21 (Fig. 5Ae; Supplementary Video S6).
Figure 5.
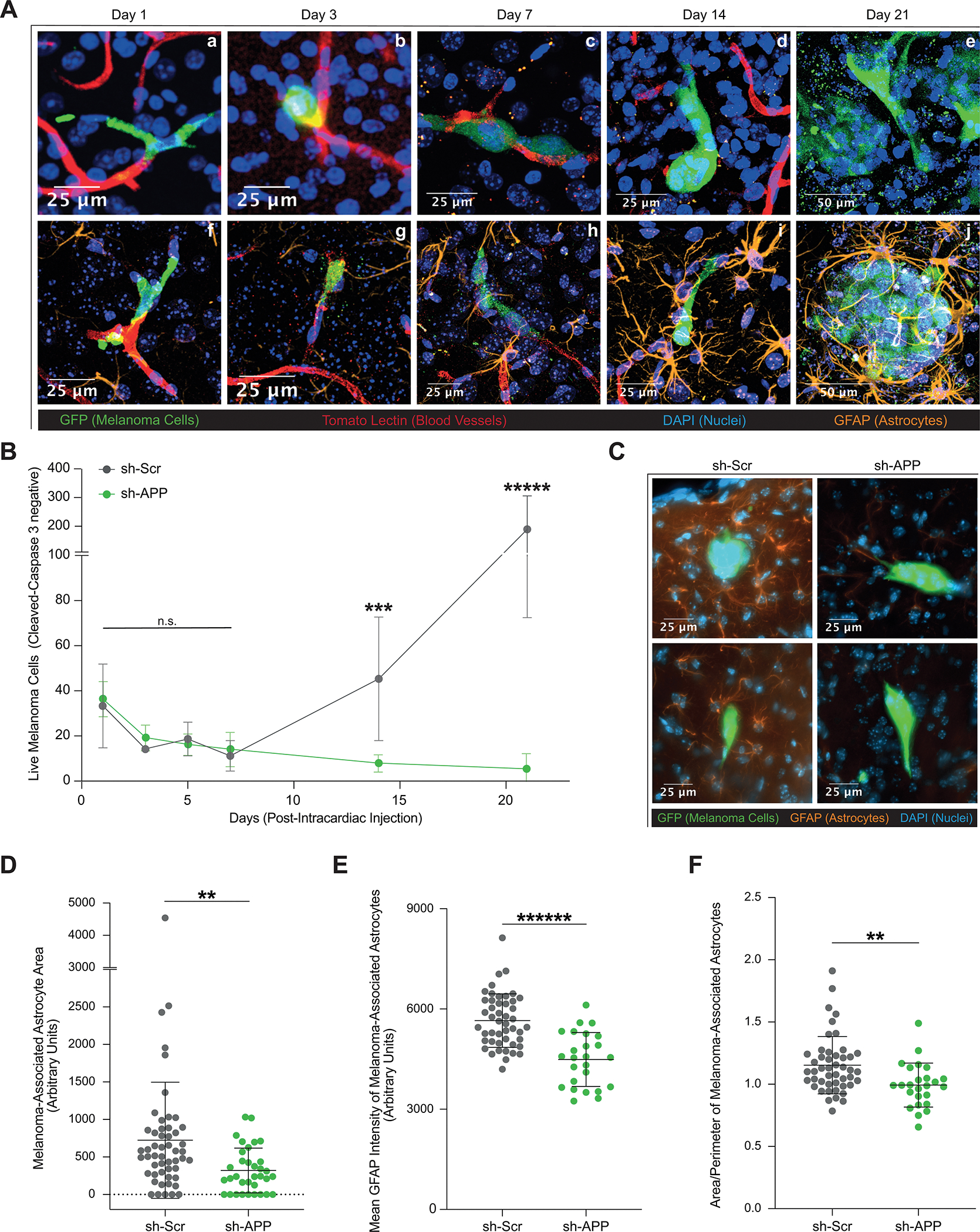
Melanoma-secreted Aβ is Required for Growth and Survival in the Brain Parenchyma. A, Representative images of brain slice immunofluorescence of 12–273 BM cells post intracardiac injection in NSG mice at days 1(Aa,Af), 3 (Ab,Ag), 7(Ac,Ah), 14 (Ad,Ai), and 21 (Ae,Aj). Fluorescent markers: green = anti-GFP (melanoma cells), red = tomato lectin (blood vessels), blue = DAPI (nuclei), orange = anti-GFAP (astrocytes). B, Quantification of live 12–273 BM cells in the brain parenchyma over time after intracardiac injection (6 mice per group per experiment, 4 50uM-thick brain slices per mouse). Day 14 sh-Scr vs. sh-APP (*** p< 0.0005), day 21 sh-Scr vs. sh-APP (***** p<0.000005). C, Representative images of brain slice immunofluorescence at day 10 post intracardiac injection. D-F, Two-dimensional quantification of parameters of astrocyte reactivity in sh-Scr vs. sh-APP melanoma-associated astrocytes (2 mice per group, 3 50uM-thick brain slices per mouse). D, Area. sh-Scr vs. sh-APP (** p<0.005). E, Mean GFAP Intensity. sh-Scr vs. sh-APP (****** p<0.0000005). F, Area/Perimeter – a proxy measurement for hypertrophy. sh-Scr vs. sh-APP (** p<0.005).
When comparing melanoma cells with and without Aβ, we did not observe differences in the number of live cells from day 1 through day 7 post intracardiac injection (Fig. 5B), indicating that Aβ is not required for vascular arrest, extravasation, or immediate survival and proliferation of melanoma cells in the brain parenchyma as they grow along the vasculature. Melanoma cells lacking Aβ, however, were unable to successfully proliferate to form micro- and macro-metastases by days 14 and 21 respectively (Fig. 5B), and instead underwent apoptosis. Therefore, Aβ is required for melanoma cells to progress from vascular co-option to successful metastatic colonization of the brain parenchyma.
Melanoma-secreted Aβ stimulates local reactive astrocytosis
Given that reactive astrocytes are important regulators of brain metastasis (10) and that Aβ can influence astrocyte physiology (29,30), we hypothesize that Aβ secreted by melanoma cells triggers a reactive phenotype in surrounding astrocytes that supports melanoma growth in the brain. GFAP staining revealed an increase in the presence of reactive astrocytes surrounding melanoma cells over time (Fig. 5Af–Aj), with significant physical contacts between melanoma cells and astrocytes developing from days 7 (Fig. 5Ah) to 14 (Fig. 5Ai). By day 21, reactive astrocytes form a glial scar-like structure that envelops the growing brain metastases (Fig. 5Aj; Supplementary Video S7). Notably, the time period during which melanoma cells lacking Aβ (sh-APP) fail to survive aligns with the time in which astrocytes begin to extensively interact with melanoma cells. We analyzed astrocytes surrounding live melanoma cell clusters with (sh-Scr) and without Aβ (sh-APP) in the brain at day 10 post-injection by GFAP staining (Supplementary Fig. S5D–F). Melanoma cells lacking Aβ display significantly decreased local reactive astrocytosis than control cells (Fig. 5C), as evidenced by diminished melanoma-associated astrocyte area (Fig. 5D), perimeter (Supplementary Fig. S5C), mean GFAP intensity (Fig. 5E), and area-to-perimeter ratio (Fig. 5F) – a proxy measurement of astrocyte hypertrophy. These data suggest that melanoma-secreted Aβ stimulates local reactive astrocytosis.
Melanoma-secreted Aβ suppresses inflammatory gene expression in astrocytes
We further interrogated how melanoma-derived Aβ affects the phenotype of astrocytes by exposing primary rat astrocytes to melanoma-conditioned media. Primary rat astrocytes were isolated by immunopanning and maintained in vitro in serum-free media, as previously reported (31). We exposed astrocytes to 12–273 BM melanoma conditioned media (CM) lacking Aβ, either by genetic silencing of APP in melanoma cells (sh-APP IgG) or by immunodepletion of Aβ from control melanoma CM (sh-Scr Anti-Aβ), and compared them to astrocytes exposed to control melanoma CM (sh-Scr IgG) (Fig. 6A). When comparing global transcriptomic changes in astrocytes exposed to melanoma CM with and without Aβ, we found a high degree of correlation (r=0.87, p<1×10−15) between changes induced by APP silencing in melanoma cells and those induced by Aβ immunodepletion from CM of control melanoma cells (Fig. 6B). Furthermore, unsupervised principal component analysis showed clustering of melanoma CM-exposed astrocytes by group on the main axis of variation (PC1, 83.7% of variance), with both Aβ-immunodepletion and genetic silencing of APP inducing variation in the same direction (Fig. 6C). Taken together, these data demonstrate that melanoma-secreted Aβ accounts for the majority of APP-mediated transcriptomic changes that melanoma cells induce in astrocytes and establishes secreted Aβ as a direct mediator of crosstalk between melanoma cells and astrocytes.
Figure 6.
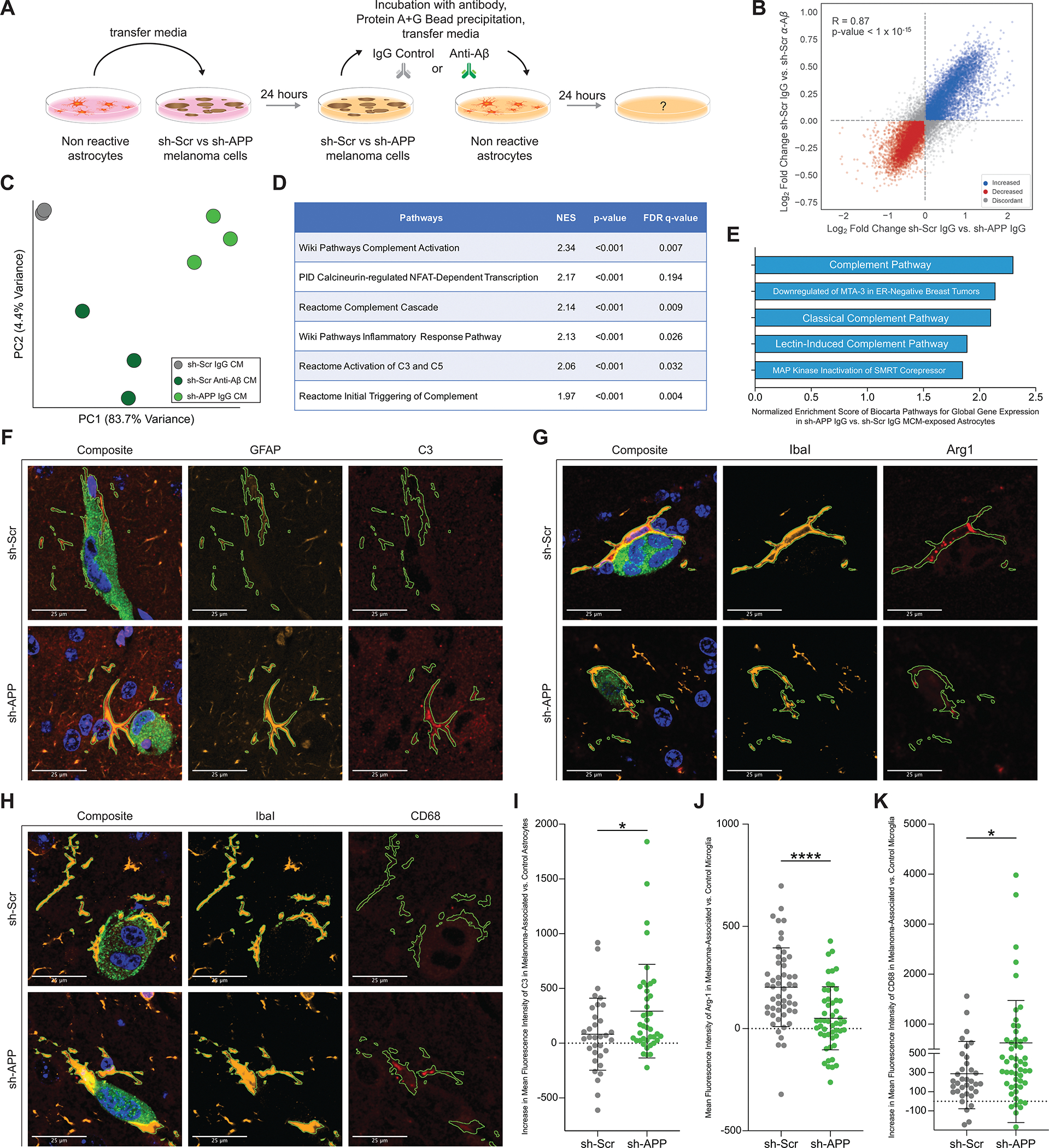
Melanoma-secreted Aβ suppresses inflammatory signaling in astrocytes and protects melanoma from microglial phagocytosis. A, Diagram of experimental method for exposing astrocytes to melanoma-conditioned media with and without Aβ (2–3 biological replicates per group, representative experiment shown B-E). B, Gene expression changes induced in astrocytes upon removal of Aβ from melanoma conditioned media (CM) by genetic silencing of APP in 12–273 BM melanoma cells (x axis – 12–273 BM sh-Scr IgG CM vs. sh-APP IgG CM) compared to changes induced by direct immunodepletion of Aβ from conditioned media (y axis – 12–273 BM sh-Scr IgG CM vs. sh-Scr Anti-Aβ CM). Correlation coefficient r=0.89 (p<1×10–15). C, Unsupervised principal component analysis of gene expression in astrocytes exposed to control melanoma-conditioned media. D, Selected pathways from Gene Set Enrichment Analysis found to be significantly enriched in 12–273 BM sh-APP IgG CM vs. sh-Scr IgG CM exposed astrocytes (NES=Normalized Enrichment Score, FDR=False Discovery Rate). E, Top five Biocarta pathways with the highest Normalized Enrichment Score in 12–273 BM sh-APP IgG CM vs. sh-Scr IgG CM exposed astrocytes. F-H, Representative images of brain slice immunofluorescence at day 10 post intracardiac injection (12–273 BM, 3 mice per group). Fluorescent Markers: green = anti-GFP (melanoma cells), blue = DAPI (nuclei), orange = anti-GFAP (F, astrocytes) or anti-Iba1 (G-H, microglia), red = Complement-3 (F) or Arginase-1 (G) or CD68 (H). I, 3D-quantification of mean fluorescence intensity of Complement-3 in melanoma-associated vs. control astrocytes. 12–273 BM sh-Scr vs. sh-APP (* p<0.05). J, 3D-quantification of mean fluorescence intensity of Arginase-1 in melanoma-associated vs. control microglia. 12–273 BM sh-Scr vs. sh-APP (* p<0.00005). K, 3D-quantification of mean fluorescence intensity of CD68 in melanoma-associated vs. control microglia. 12–273 BM sh-Scr vs. sh-APP (* p<0.05).
To further characterize how melanoma-derived Aβ influences astrocytes, we performed gene set enrichment analysis of differentially expressed transcripts in astrocytes exposed to melanoma CM with (sh-Scr IgG) and without Aβ (sh-APP IgG). Intriguingly, results showed a significantly increased enrichment score in multiple pathways related to inflammatory signaling in astrocytes exposed to CM without Aβ as compared to those exposed to CM with Aβ (Fig. 6D). In particular, gene sets related to the complement cascade, a component of the innate immune system (32), were among the top enriched pathways across multiple analyses (Fig. 6D) and comprised three out of the five top enriched pathways from Biocarta analysis (Fig. 6E). Increased expression of complement cascade components is a key feature that distinguishes neurotoxic, inflammatory A1-type astrocytes from other reactive sub-states that may be neuro-supportive or have other pathophysiological functions (33). To validate these findings in vivo, we performed brain slice immunofluorescence followed by confocal microscopy and three-dimensional digital image analysis. We quantified expression of C3, a component of the complement pathway that is specifically upregulated in A1-astrocytes (33), in melanoma-associated astrocytes. In concordance with the in vitro findings, astrocytes associated with melanoma cells without Aβ (sh-APP) have increased expression of C3 as compared to astrocytes associated with melanoma cells with Aβ (sh-Scr) (Fig. 6F and I), indicating that melanoma-secreted Aβ suppresses inflammatory A1-like polarization of astrocytes in the brain metastasis microenvironment.
Aβ suppresses microglia activation and phagocytic clearance of melanoma cells
Astrocyte-derived C3 has been shown to stimulate microglial neuroinflammation and phagocytosis in multiple neuropathological contexts (34–36). Therefore, we hypothesized that in the absence of melanoma-secreted Aβ, melanoma-associated microglia would be activated to a proinflammatory state and mediate clearance of melanoma cells from the brain parenchyma through phagocytosis. We examined the effect of melanoma-secreted Aβ on melanoma-associated microglia (defined by expression of Iba1 and TMEM119, Supplementary Fig. S6A–F), by performing brain slice immunofluorescence followed by confocal microscopy and three-dimensional digital image analysis (Supplementary Fig. S6G–P) to quantify expression of Arginase-1, a well-defined anti-inflammatory microglial marker (37), and CD68, a marker of active microglial phagocytosis (38), in melanoma-associated microglia. When compared with microglia associated with melanoma cells with Aβ (sh-Scr), microglia associated with melanoma cells lacking Aβ (sh-APP) had significantly lower expression of Arginase-1 (Fig. 6G and J) and higher expression of CD68 (Fig. 6H and K). This demonstrates that a consequence of melanoma-secreted Aβ is anti-inflammatory polarization of microglia and that Aβ promotes brain metastasis by protecting melanoma cells from microglial phagocytosis (Supplementary Video S8).
Aβ is a Promising Therapeutic Target for Treatment of Brain Metastasis
To assess if targeting Aβ could be a promising therapeutic strategy for treatment of melanoma brain metastasis, we investigated whether Aβ is required for growth and survival of established brain metastases. Using a doxycycline inducible sh-RNA system (Supplementary Fig. S7A), we depleted APP in growing melanoma brain macro-metastases (Fig. 7A). Loss of ability to produce Aβ in pre-existing brain metastases resulted in decreased brain metastatic burden (Fig. 7B–D, Supplementary Fig. S7B). Many therapeutic approaches efficiently targeting Aβ have been developed and tested for the treatment of Alzheimer’s disease, including anti-Aβ antibodies (39) and β-secretase inhibitors (BACE-i) (40). We tested LY2886721, a BACE-i that blocks Aβ production (41) by inhibiting the rate limiting step in its generation (Fig. 7E). Treatment of mice with LY2886721 at a dose of 75 mg/kg/day in food resulted in a 75% reduction in plasma Aβ levels (Supplementary Fig. S7C). Pharmacological inhibition of Aβ production decreased brain metastatic burden in both a patient-derived short-term culture (12–273BM; Fig. 7F–H, Supplementary Fig. S7D) and an established melanoma cell line (5B1; Fig. 7I–K, Supplementary Fig. S7E). In addition, we conducted a more therapeutically relevant experiment by initiating BACE-i treatment once brain metastases had been established (Supplementary Fig. S7F). All mice were injected with the same number of cancer cells, and diet-based treatment with BACE-i was initiated at day 11 post-injection. Mice fed BACE-i containing chow from day 11 had significantly decreased brain metastasis compared to those fed control chow (Supplementary Fig. S7F–J). In summary, our data support Aβ-targeting as a promising therapeutic approach against melanoma brain metastasis.
Figure 7.
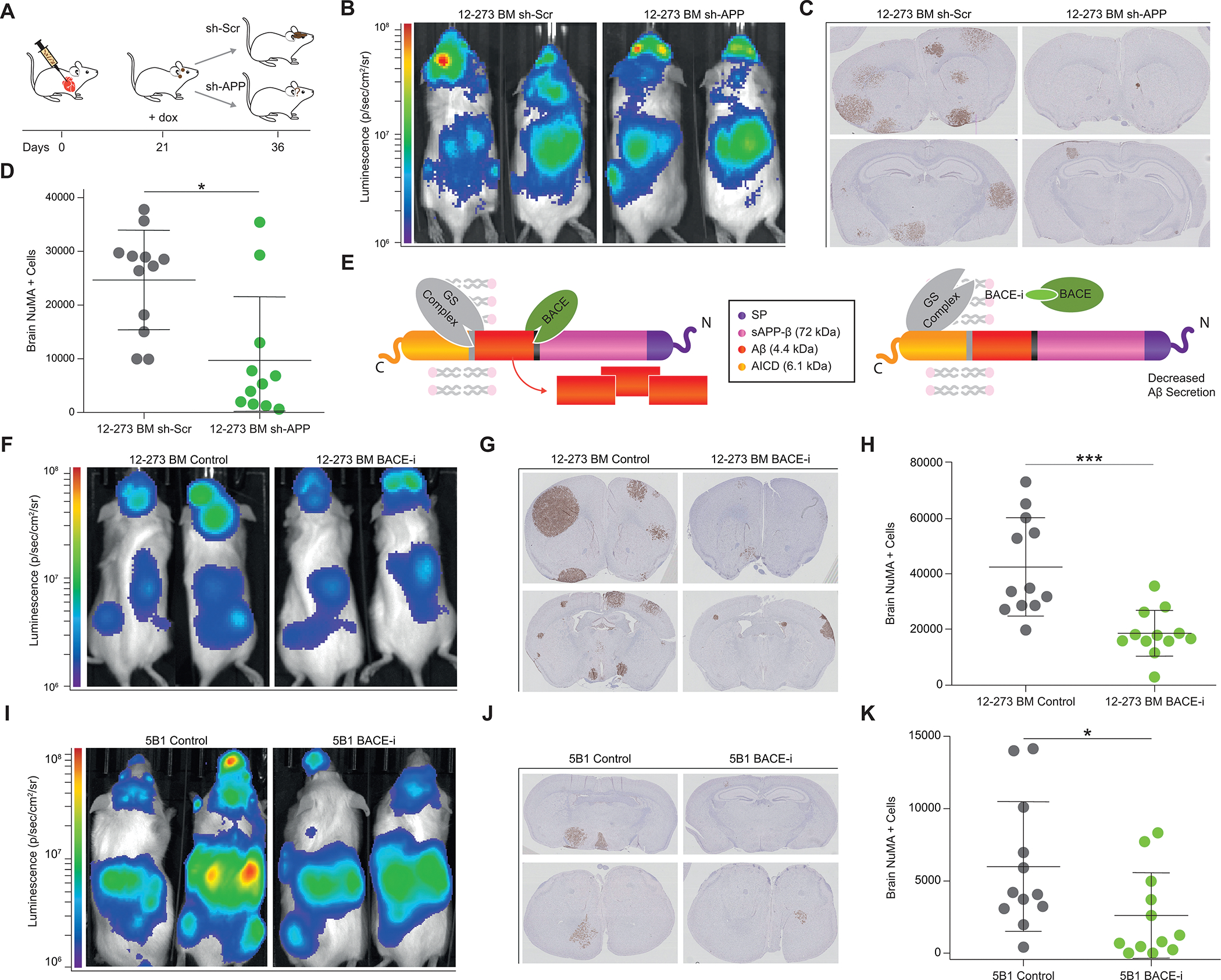
Aβ is a Promising Therapeutic Target for Treatment of Brain Metastasis. A, Diagram of therapeutic simulation experiment inducing silencing of APP in established brain metastases. B-D, Induction of silencing of APP in established brain metastases (11–12 NSG mice per group). B, Representative IVIS images at day 37 post intracardiac injection. C, Representative images of FFPE brain slides with labeling of metastatic cells by anti-NuMA immunohistochemistry. D, Quantification of NuMA+ metastatic cells in mouse brains. sh-Scr vs. sh-APP (* p<0.05). E, Diagram of APP cleavage and beta-secretase inhibition of Aβ production. F, Representative IVIS images at Day 28 post intracardiac injection with 12–273 BM STC (12 NSG mice per group). G, Representative images of FFPE brain slides with 12–273 BM metastatic cells stained by anti-NuMA immunohistochemistry. H, Quantification of NuMA+ metastatic cells in mouse brains. 12–273 BM Control vs BACE-i (*** p<0.0005). I, Representative IVIS images at Day 28 post intracardiac injection with 5B1 melanoma cell line (12 NSG mice per group). J, Representative images of FFPE brain slides with 5B1 metastatic cells stained by anti-NuMA immunohistochemistry. K, Quantification of NuMA+ metastatic cells in mouse brains. 5B1 Control vs BACE-i (* p<0.05).
Discussion
Identifying and characterizing mechanisms that mediate survival of cancer cells in the brain parenchyma has been historically challenging. The majority of studies aiming to identify mechanisms of brain metastasis have utilized cell lines originally derived from extracranial metastases and examined transcriptomic changes after serial transplantation in mice to increase brain tropism (12,42–44). While these models have been an invaluable tool, the process by which they are generated varies greatly from the brain metastatic process occurring in patients (45). In contrast, STC pairs exhibiting differential metastatic behavior and brain tropism (Fig. 1 A–D, Supplementary Fig. S1A–S1J) are derived directly from naturally arising metastases in patients (46). Recently, some studies have directly profiled gene expression of surgically resected patient tumors (20,47). While this method has several advantages, such as strong clinical relevance and ability to capture effects mediated by the brain microenvironment, the limited amount of material available per sample can restrict the type of analyses that are technically feasible. Furthermore, direct use of patient biopsies makes it difficult to establish whether genes identified as upregulated in CNS metastases represent contamination from brain tissue or neural mimicry by cancer cells. Proteomic screening of short-term cultures – which no longer contain normal brain tissue – circumvents this issue and allowed us to identify an association between brain metastasis and proteins implicated in neurodegenerative disorders such as Alzheimer’s disease.
Our results, which revealed an unexpected molecular association between brain metastasis and neurodegenerative disorders, raise the interesting hypothesis of a clinical association between brain metastasis and neurodegeneration. To our knowledge, there is no published epidemiological data that connects Alzheimer’s disease and melanoma brain metastasis. Intriguingly, there is epidemiological data that indirectly associates Parkinson’s disease and melanoma brain metastasis. Parkinson’s and melanoma are comorbid, with both Parkinson’s disease increasing the risk of developing melanoma (48) and melanoma increasing the risk of developing Parkinson’s disease or Parkinsonian symptoms (49). An epidemiologic study of Parkinson’s disease and melanoma demonstrated that 50% of melanoma patients with parkinsonism have a melanoma whose primary site is located on the head or neck as compared to only ~6% of melanoma patients without parkinsonism (50). Given that head and neck melanomas are significantly more likely to metastasize to the brain, this finding indirectly associates development of Parkinson’s disease and brain metastasis. Future interrogation of large clinical datasets is needed to determine whether any direct clinical association exists between brain metastasis and neurodegenerative disorders.
Several studies have demonstrated that reactive astrocytes support the formation of brain metastasis (10,11), provided early antagonistic interactions can be overcome (12). Here, we show that Aβ secreted by melanoma cells stimulates pro-metastatic activation of nearby astrocytes, characterized by suppression of inflammatory gene expression. Previous research has demonstrated that cancer cells in the brain induce a subpopulation of pro-metastatic Stat3+ reactive astrocytes in their microenvironment by an unknown mechanism (51). In addition, Stat3+ reactive astrocytes contribute to the pathology of acute brain injury and several neurodegenerative diseases (17,52), including Alzheimer’s disease. Further investigation is warranted to address whether melanoma-secreted Aβ specifically induces Stat3 activation in astrocytes.
Neurotoxic A1 astrocytes have been observed in a variety of neurological pathologies (16,53), including Alzheimer’s disease, but have yet to be investigated in brain metastasis. Here, we establish that melanoma-secreted Aβ suppresses proinflammatory complement pathway gene expression in astrocytes, which has been previously validated as a hallmark of neurotoxic A1 astrocytes (33). Further studies are necessary to characterize similarities and differences between A1 astrocytes in other neurological disorders and the “A1-like” complement-expressing astrocytes that are suppressed by melanoma-secreted Aβ in the brain metastasis microenvironment.
Astrocytes have been shown to regulate microglial activity in response to inflammatory insults (54). A similar role for astrocytes has been theorized in the brain metastatic microenvironment but remains largely unexplored (55). Our studies demonstrate that melanoma-secreted Aβ suppresses inflammatory signaling in astrocytes, promotes anti-inflammatory microglial polarization, and suppresses phagocytosis of melanoma cells by microglia. Given that astrocyte C3 has been established to promote microglial inflammatory secretion and phagocytosis in multiple other disease contexts (34–36), it is logical to infer that melanoma-secreted Aβ is regulating microglia activity indirectly through astrocytes. However, it is also possible that Aβ impacts microglial response to melanoma cells directly. A recent study showed that acute exposure to Aβ initially stimulated microglial phagocytosis of microparticles, but re-exposure of the same microglia to Aβ days later instead inhibited phagocytosis (56). Given the high Aβ concentrations used and lack of continuous exposure of microglia to Aβ, it is difficult to extrapolate that study’s findings to the context of the brain metastasis microenvironment. Additional studies are needed to clarify whether melanoma-secreted Aβ directly affects microglial function and if so, how melanoma-secreted Aβ impacts astrocyte-microglia crosstalk.
The role of Aβ, both in Alzheimer’s disease and in normal physiology, remains controversial. Most studies investigating the function of Aβ have been performed in the context of Alzheimer’s pathology using transgenic mice that mimic some pathological components of the human disease (57). It is well established that insoluble aggregates of Aβ contribute to pathological astrocyte- and microglia-mediated neuroinflammation in Alzheimer’s disease (58). Intriguingly, we demonstrate an anti-inflammatory function of soluble Aβ in the context of brain metastasis. The majority of Aβ produced by melanoma cells is Aβ-40 (Supplementary Fig. S4D), the less aggregative form, and none of our models of brain metastasis gives rise to Aβ plaques (Supplementary Fig. S4E). Instead, we hypothesize that melanoma-derived Aβ acts in the form of soluble monomers or oligomers to suppress astrocyte-driven inflammation. Indeed, both monomers and oligomers of Aβ have been shown to affect phenotypic changes in astrocytes (29,30). Whether soluble Aβ also acts as an anti-inflammatory mediator in the brain during normal physiology and other pathophysiologic contexts is an important question that requires further investigation. If present, a similar function of soluble Aβ could profoundly impact our understanding of Alzheimer’s development and shed light on the reported lack of clinical efficacy of anti-Aβ agents in the treatment of advanced Alzheimer’s disease (39).
Our studies show Aβ is a highly promising therapeutic target for melanoma brain metastasis. The brain is often a site of treatment resistance or relapse in patients (4). Although checkpoint blockade immunotherapy, the standard-of-care for metastatic melanoma, can be efficacious in brain metastasis, many patients are non-responsive or develop resistance (5,6). Targeting Aβ, which suppresses neuroinflammation, in combination with immune checkpoint inhibitors could result in a more robust anti-tumor immune response and improve patient outcomes. It is important to note that our in vivo experiments were performed utilizing human melanoma xenografts in immunocompromised mice lacking an adaptive immune system, which is necessary to prevent xenograft rejection secondary to antigenic variation between species. Therefore, testing using syngeneic mouse models of melanoma brain metastasis in immunocompetent mice is needed to both validate our findings in the context of a normal immune system and demonstrate safety and efficacy of targeting Aβ in combination with immunotherapy. We attempted to establish an immune competent model of parenchymal brain metastasis and tested several existing mouse melanoma cell lines. Unfortunately, we did not observe metastasis to brain parenchyma with high penetrance in any of our attempts. Additional work to establish a robust immune-competent in vivo model of melanoma brain metastasis is urgently needed for preclinical evaluation of promising novel therapies that may be combined with immune checkpoint inhibitors, the current first-line therapy.
Several therapeutic agents targeting Aβ have been developed and tested in clinical trials for treatment of Alzheimer’s disease that could potentially be repurposed for treatment of melanoma brain metastasis. One possibility includes BACE small molecule inhibitors, exemplified by LY2886721, which provided proof-of-principle efficacy in our preclinical models. Another possible approach is the use of anti-Aβ antibodies, which have been extensively tested in clinical trials for Alzheimer’s disease. Anti-Aβ antibodies successfully sequestered Aβ in Alzheimer’s patients (59) but lacked clear clinical efficacy (60). In Phase III clinical trials, an anti-Aβ antibody was well tolerated at high doses for extended periods of time without an increase in rates of adverse effects over placebo (60). Given that dose limiting toxicities and adverse effects are major drivers of failure in cancer clinical trials, targeting Aβ in melanoma brain metastasis with anti-Aβ antibodies represents a particularly promising treatment strategy.
In summary, our studies reveal an unexpected anti-inflammatory role for tumor-secreted Aβ in promoting survival of melanoma cells in the brain microenvironment and provide proof-of-principle of Aβ targeting as a novel therapeutic approach in the treatment of melanoma brain metastasis.
Materials & Methods
Cell culture
Melanoma short-term cultures and cell lines.
Low passage melanoma short-term cultures (STCs), derived in the Osman laboratory as described (43). were grown in DMEM with 10% fetal bovine serum (FBS), 1 mM Sodium Pyruvate, 4 mM L-Glutamine, 25 mM D-Glucose, 1% Nonessential Amino Acids (NEAA), 100 units/mL penicillin, and 100 ug/mL streptomycin. Additional STCs (WM-4071 pair, WM-4257 pair, WM-4265 1 and 2, WM-4237–1; (61) ), a kind gift of the Herlyn lab at the Wistar Institute, were grown in Tu2% media (80% MCDB153, 20% Leibovitz’s L-15, 2% FBS, 5 μg/mL Insulin (Bovine), 1.68 mM calcium chloride, 100 units/mL penicillin, 100 μg/mL streptomycin). STCs were kept below lifetime passage number of 40 for all experiments. For details of STCs utilized, see Supplementary Table S1. HEK293T cells (for lentivirus production) and 131/4–5B1 (hereafter 5B1; (42) ) melanoma cells were grown in DMEM with 10% fetal bovine serum (FBS), 1 mM Sodium Pyruvate, 4 mM L-Glutamine, 25 mM D-Glucose, 100 units/mL penicillin, and 100 μg/mL streptomycin. Cell lines were maintained in a 5% CO2 incubator at 37 °C and were routinely tested for Mycoplasma contamination.
Astrocytes isolation and culture.
Astrocytes were purified by immunopanning and cultured in serum-free conditions as previously described (62). Briefly, cortices from 5–6 postnatal day 4–6 Sprague Dawley rat pups (Charles River) were dissected out and meninges and choroid plexus removed. The cortices were minced with a scalpel and digested in Papain for 40 min at 34°C under constant CO2/O2 gas equilibration. The digested brain pieces were washed with CO2/O2-equilibrated Ovomucoid inhibitor solution, triturated, and spun down through a cushion gradient containing low and high Ovomucoid inhibitor layers. The resulting cell pellet was passed through a 20 μm nylon mesh to create a single cell suspension. The cells were then incubated in a 34°C water bath for 30–45 mins to allow cell-specific antigens to return to the cell surface. Negative selection was performed using Goat anti-mouse IgG + IgM (H + L) (Jackson ImmunoResearch Labs #115–005-044, RRID:AB_2338451), Griffonia (Bandeiraea) simplicifolia lectin 1 (BSL-1) (Vector Laboratories #L-1100, RRID:AB_2336491), Rat anti-mouse CD45 (BD Biosciences #550539, RRID:AB_2174426), and O4 hybridoma supernatant mouse IgM (63,64) followed by positive selection for astrocytes using mouse anti-human integrin β5 (ITGB5) (Thermo Fischer #14–0497-82, RRID:AB_467288). Purified astrocytes were detached from the panning plate with trypsin at 37°C for 3–4 min, neutralized by 30% fetal calf serum, counted, pelleted, and resuspended in 0.02% BSA in DPBS. All isolation and immunopanning steps occurred at room temperature, except for the heated digestion, incubation, and trypsinization steps. Cells were plated at 50,000 cells per well in 6 well plates containing 2 mL/well of serum-free Astrocyte Growth Medium (50% Neurobasal Medium, 50% Dulbecco’s Modified Eagle Medium (DMEM), 100 U/mL Penicillin, 100 μg/mL Streptomycin, 1 mM Sodium pyruvate, 292 μg/mL L-Glutamine, 5 μg/mL N-Acetyl-L-cysteine (NAC), 100 μg/mL BSA, 100 μg/mL Transferrin, 16 μg/mL Putrescine dihydrochloride, 60 ng/mL (0.2 μM) Progesterone, and 40 ng/mL Sodium selenite. Immediately before plating, the astrocyte trophic factor Heparin-binding EGF-like growth factor (HBEGF) was added (5 ng/mL) and media equilibrated to 37°C in a 10% CO2 incubator). Cells were incubated at 37°C in 10% CO2. Media was changed (50% well volume) once per week.
Exposure of Astrocytes to Melanoma Conditioned Media.
Astrocytes were plated at 5×104 astrocytes/well in 6-well plates. 12–273 BM melanoma cells were plated at 2.5×105 cells/well in 6-well plates. Astrocyte-conditioned media (ACM) was obtained by culture of astrocytes in astrocyte media for 1 week. 1.5 mL of ACM were then transferred and incubated with 12–273 BM cells in 6-well plates for 24 hours. Media was then removed and incubated with either 4.5 μg of mouse IgG or 2.25 μg of anti-amyloid beta antibody N25 (Marc Mercken/Janssen Research & Development Cat# JRF/AbN/25, RRID:AB_2571524) and 2.25 μg of anti-APP 6E10 antibody (Biolegend Cat# 803001, RRID:AB_25643453) for 1 hr at 4°C and 1 hr at room temperature with rotation mixing. 75 μl of protein A/G beads (Pierce 88803) were washed twice with PBS, added to 1.5 mL of conditioned media, and incubated for 1 hour at room temperature with rotation mixing. Beads were precipitated using a magnetic tube holder and media was concentrated to 10X at 4°C using Amicon Ultra 3-kDa Concentrators (Millipore Z740169). 100 μl of concentrated conditioned media was transferred per well to astrocytes in 2 mL of serum-free Astrocyte Growth Media in a 6-well plate. Astrocytes were incubated for 24 hours.
Astrocyte RNA-Seq and Analysis.
After 24-hour incubation in melanoma-conditioned media, RNA was harvested from astrocytes using the RNeasy Mini Kit (Qiagen 74104). RNA-Seq library preps were made using the Illumina TruSeq® Stranded mRNA LT kit (Cat #RS- RS-122–2101 or RS-122–2102), on a Beckman Biomek FX instrument, using 100 ng of total RNA as input, amplified by 12 cycles of PCR, and run on an Illumina 4000. FastQC v0.11.7 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, RRID:SCR_014583) was used to check fastq files for poor sequencing quality; all samples had high quality. Illumina adapter sequences and poor-quality bases were then trimmed using trimmomatic v0.36 (65) (RRID:SCR_011848). Trimmed sequences were mapped to rn6 using STAR v2.6.0a (66) (RRID:SCR_004463), indexed using samtools v1.9 (67) (RRID:SCR_002105), then quantified for UCSC genes using HTSeq-count v0.11.1 (68) (RRID:SCR_011867). Comparative analysis between conditions was performed using DESeq2 v1.24.0 (69) (RRID:SCR_000154) with default parameters.
Gene Set Enrichment Pathway Analysis.
Pathway analysis was performed using Gene Set Enrichment Analysis (70,71) with a ranked list of global gene expression changes as determined by Log2 Fold Change of sh-APP IgG CM vs. sh-Scr IgG CM. Analysis was performed using the following Molecular Signatures Database (MSigDB) pathways: Biocarta, Pathway Interaction Database (PID), Reactome, and WikiPathways canonical pathways.
Proteomics Analysis of Short-Term Cultures (STCs)
Protein isolation.
STCs at ~80% confluence at ~24 hours post-media change were scraped from 10 or 15cm plates on ice, washed once with cold PBS, and lysed in cold RIPA buffer supplemented with protease inhibitor, for 15 minutes with vortexing every 5 minutes. Protein concentration was determined using Micro BCA Protein Assay Kit (Thermo Scientific 23235).
150 μg of each protein lysate were proteolytically digested and subjected to quantitative mass spectrometry on an Orbitrap Fusion Lumos mass spectrometer using isobaric tandem mass tags (TMT) similar to previous studies (72,73).
Sample preparation for mass spectrometry analysis.
150 μg of each protein lysate were prepared using the filter-aided sample preparation (FASP) method (74). Briefly, each sample was reduced with DTT (final concentration of 20 mM) at 57°C for 1 hour and loaded onto a MicroCon 30-kDa centrifugal filter unit (Millipore) pre-equilibrated with 200 μl of FASP buffer [8 M urea and 0.1 M tris-HCl (pH 7.8)]. Following three washes with FASP buffer, lysates were alkylated on a filter with 50 mM iodoacetamide for 45 min in the dark. Filter-bound lysates were washed three times each with FASP buffer followed by 100 mM ammonium bicarbonate (pH 7.8). The samples were digested overnight at room temperature with trypsin (Promega) at a 1:100 ratio of enzyme to protein. Peptides were eluted twice with 100 μl of 0.5 M NaCl. The tryptic peptides were subsequently desalted using an UltraMicro Spin Column, C18 (Harvard Apparatus) and concentrated in a SpeedVac concentrator.
TMT labeling.
The dried peptide mixture was re-suspended in 100 μl of 100 mM Triethylammonium bicarbonate (TEAB) (pH 8.5). Each sample was labeled with TMT reagent according to the manufacturer’s protocol. In brief, each TMT reagent vial (0.8 mg) was dissolved in 41 μL of anhydrous ethanol and was added to each sample. The reaction was allowed to proceed for 60 min at room temperature and then quenched using 8 μL of 5% (w/v) hydroxylamine. The samples were combined at a 1:1 ratio and the pooled sample was subsequently desalted using strong-cation exchange and strong-anion exchange solid-phase extraction columns (Strata, Phenomenex) as described (73).
Global Proteome Analysis.
A 500 μg aliquot of the pooled sample was fractionated using basic pH reverse-phase HPLC (as described) (66). Briefly, the sample was loaded onto a 4.6 mm × 250 mm Xbridge C18 column (Waters, 3.5 μm bead size) using an Agilent 1260 Infinity Bio-inert HPLC and separated over a 90 min linear gradient from 10 to 50% solvent B at a flow rate of 0.5 mL/min (Buffer A = 10 mM ammonium formate, pH 10.0; Buffer B = 90% ACN, 10 mM ammonium formate, pH 10.0). A total of 120 fractions were collected and non-concatenated fractions combined into 40 final fractions. The final fractions were concentrated in the SpeedVac and stored at −80°C until further analysis.
LC-MS/MS analysis.
An aliquot of each final fraction was loaded onto a trap column (Acclaim® PepMap 100 pre-column, 75 μm × 2 cm, C18, 3 μm, 100 Å, Thermo Scientific) connected to an analytical column (EASY-Spray column, 50 m × 75 μm ID, PepMap RSLC C18, 2 μm, 100 Å, Thermo Scientific) using the autosampler of an Easy nLC 1000 (Thermo Scientific) with solvent A consisting of 2% acetonitrile in 0.5% acetic acid and solvent B consisting of 80% acetonitrile in 0.5% acetic acid. The peptide mixture was gradient eluted into the Orbitrap Lumos Fusion mass spectrometer (Thermo Scientific) using the following gradient: 5%–5% solvent B in 100 min, 23% – 34% solvent B in 20 min, 34% – 56% solvent B in 10 min, followed by 56%- 100% solvent B in 20 min. Full scans were acquired with a resolution of 60,000 (@ m/z 200), a target value of 4e5 and a maximum ion time of 50 ms. After each full scan the most intense ions above 5e4 were selected for fragmentation with HCD using the “Top Speed” algorithm. The MS/MS were acquired in the Orbitrap with a resolution of 60,000 (@ m/z 200), isolation window of 1.5 m/z, target value of 1e5, maximum ion time of 60 ms, normalized collision energy (NCE) of 35, and dynamic exclusion of 30 s.
Data analysis.
The MS/MS spectra were searched against the UniProt human reference proteome with the Andromeda (75) search engine integrated into the MaxQuant (76) environment (version 1.5.2.8) using the following settings: oxidized methionine (M), TMT-labeled N-term and lysine, acetylation (protein N-term) and deamidation (asparagine and glutamine) were selected as variable modifications, and carbamidomethyl (C) as fixed modifications; precursor mass tolerance was set to 10 ppm; fragment mass tolerance was set to 0.01 Th. The identifications were filtered using a false-discovery rate (FDR) of 0.01 using a target-decoy approach at the protein and peptide level. Only unique peptides were used for quantification and only proteins with at least two unique peptides were reported. Data analysis was performed using Perseus (RRID:SCR_015753). Protein levels were median centered and log2 normalized. To identify proteins differentially expressed proteins between the BM and NBM cohorts, a Welch’s t-test was performed between unpaired 14 BM and 11 NBM unpaired samples and a paired t-test was performed on the 3 sample pairs. Top differentially expressed genes (defined here as p-value < 0.05) were assessed at the pathway level using DAVID GO-term enrichment (RRID:SCR_001881) with an FDR < 0.001 (77,78).
Analysis of Independent Transcriptomic Datasets
Chen et al 2014 (19).
EdgeR-limma-voom packages applied to the non-normalized expression value matrix accessed via GEO (Accession GSE50493). Data are comprised of microarray-based gene expression of 72 samples from 52 brain and extracranial melanoma metastases. Linear model defines contrast comparing brain to extracranial metastases for the entire cohort. Moderated t-statistics calculated with empirical Bayes’ shrinkage of the sample variances. All genes resulting from differential expression analysis below adjusted p value of 0.05 were analyzed using the DAVID functional annotation tool (http://david.abcc.ncifcrf.gov) to yield enriched biological modules.
Fischer et al 2019 (20).
HTSeq-count (v0.9.1) tool was applied with its default settings to aligned RNA-seq BAM files acquired from EGA (Accession EGAD00001005046). Differential expression analysis conducted by application of EdgeR-limma-voom packages to generate a linear model comparing brain to extracranial metastases for the cohort of 88 resected melanoma brain metastases and 42 extracranial metastases.
Animal Studies
All experiments were conducted following protocols approved by the NYU Institutional Animal Care Use Committee (IACUC) (protocol numbers s16–00051 and s18–00249). NOD/SCID/IL2rg−/− male mice (Jackson Labs, Cat# 05557) at 6–8 weeks were used for all in-vivo studies with the following exceptions: Athymic nude male mice (Jackson labs, Cat# 002019) at 6–8 weeks were used for CRISPR/Cas9 studies in 5B1 and for WM-4257 STC pair studies.
Long-term brain metastasis assays.
For all experiments utilizing Short Term Cultures, 1 × 105 cells suspended in 100 μl of PBS were injected with ultrasound guidance (Visualsonics Vevo 770 Ultrasound Imaging System) into the left ventricle of mice anesthetized with isoflurane. For 5B1, 2 × 105 cells were injected by the same method as described above. Mice were monitored weekly for metastatic progression by in-vivo bio-luminescent imaging (BLI). Upon substantial weight loss and/or signs of distress (neurological signs, abnormal locomotion) in any experimental mice, experimental endpoint was established. At experimental endpoint, all mice in all experimental groups were euthanized by perfusion.
For all long-term brain metastasis assays, either 10 or 12 mice per group underwent intracardiac injection with cancer cells. Experimental group sizes vary from 8 to 10 (for group size 10) or 10 to 12 (for group size 12) mice due to infrequent instances of unsuccessful intracardiac injection.
In-vivo BLI.
10 minutes prior to imaging, luciferin substrate (150 mg/kg) was administered to mice by intraperitoneal injection. Mice were anesthetized with isoflurane and imaged by IVIS Illumina instrument (PerkinElmer) for an automatically-determined duration (1–120 sec). Signal was quantified by measurement of total luminescent flux (p/sec/cm2/sr) in drawn brain and body regions of interest.
Perfusion.
Mice were anesthetized with a ketamine (100 mg/kg) and xylazine (10 mg/kg) cocktail by intraperitoneal injection. The heart was exposed by gross dissection and an incision was made in the right atrium. Subsequently, 10 mL of PBS followed by 10 mL of 4% PFA was injected into the left ventricle.
Short-term brain metastasis assays and brain slice immunofluorescence.
1×105 (for live melanoma cell quantification) or 5 × 105 (for astrocyte and microglial scoring) 12–273 BM cells were introduced by intracardiac injection. Mice were euthanized by perfusion at specified time points post intracardiac injection. Where relevant, 100 μg of Dylight 647 labeled Lycopersicon Esculentum (Tomato Lectin) were injected into the left ventricle 3 min prior to perfusion. For these assays, 3–6 mice with successful intracardiac injection were used per group per time point.
Drug treatment experiments.
Protocol for long-term brain metastasis assay (as described above) was performed. LY288671 (75 mg/kg/day) was administered to mice in food pellets starting one week prior to intracardiac injection of cancer cells and continuing through the experimental endpoint. For treatment of established brain metastasis (Supplementary Fig. S7F–J), LY288671 (75 mg/kg/day) was administered to mice in food pellets starting 11 days after intracardiac injection of cancer cells and continuing through the experimental endpoint.
Continuous shRNA-mediated gene silencing.
Protocol for long-term brain metastasis assay (as described above) was performed. Doxycycline hyclate (200 mg/kg/day) was administered to mice in food pellets starting two days prior to intracardiac injection of cancer cells and continuing for the experimental duration.
Induction of gene silencing in growing metastases.
Protocol for long-term brain metastasis assay (as described above) was performed. Mice were administered a normal diet at the beginning of the experiment. 21 days after intracardiac injection of cancer cells, gene silencing was induced by administration of Doxycycline hyclate (200 mg/kg/day) in food pellets.
Mouse tissue processing, histology and NuMA Immunohistochemistry
Organs harvested from PFA-perfused mice were fixed in 10% formalin for 48 hours. Prior to embedding, brains were sectioned grossly into thirds coronally and livers were sectioned by lobe. The two kidneys from each mouse were arranged in parallel so that both could be sectioned at a single level. Organs were embedded in paraffin and cut into 5 μM thick sections.
Histological Analysis Comparing Short-Term Culture Pairs.
The embedded brain thirds were sectioned coronally through the entire length of tissue at an interval of 50 μM. All sections were stained with H&E. Number of unique metastases present per brain was assessed by a pathologist by identifying and tracking metastases through serial sections, ensuring each metastasis was counted only once. Slides were blinded to pathologist (R. R.) prior to analysis.
Sectioning and NuMA Immunohistochemistry.
For kidney, sections were obtained from one representative level through both kidneys. In total, this resulted in two transverse kidney sections (one from each kidney per mouse) for metastatic quantification. One section per level was stained with H&E. For liver, sections were obtained from one (Fig. 3I, Supplementary Fig. S3L–M) or two evenly spaced (Supplementary Fig. S1E and H, Supplementary Fig. S3L) levels. In total, this resulted in one or two evenly spaced serial axial liver sections per liver lobe per mouse for metastatic quantification. One section per level was stained with H&E. For brain, sections were obtained from 1 (12–273 BM in-vivo miniscreen – Fig. 3H, Supplementary Fig. S3J and K), 2 (all other 12–273 BM experiments – Fig. 3J and K, Fig. 4D and E, Fig. 7C and D, Fig. 7G and H; WM-4071 – Supplementary Fig. S1G; WM-4257 – Supplementary Fig. S1I and J; 5B1 BACE-i – Fig. 7J and K) or 4 (5B1 APP CRISPR – Supplementary Fig. S3R and S) evenly spaced levels of the embedded coronal thirds. In total, this resulted in 6, 12, or 24 serial coronal brain section levels per mouse for metastatic quantification. One section per level was stained with H&E. Chromogenic Immunohistochemistry was performed on a Ventana Medical Systems Discovery XT instrument with online deparaffinization and using Ventana’s reagents and detection kits unless otherwise noted. Unconjugated, polyclonal rabbit anti-human Nuclear Mitotic Apparatus Protein (NuMA; Abcam Cat# 97585 Lot# B115626 RRID: AB_1855299) was used for labeling. Sections were deparaffinized in xylene and rehydrated in graded ethanol followed by rinsing in deionized water. Epitope retrieval was performed in a 1200-Watt microwave oven at 100% power in 10 mM sodium citrate buffer, pH 6.0 for 10 minutes. NuMA antibody was diluted 1:7000 in Tris-BSA (25 mM Tris, 15 mM NaCL, 1% BSA, pH 7.2) and incubated for 12 hours. Primary antibody was detected with goat anti-rabbit HRP conjugated multimer, and the complexes were visualized with 3,3’-diaminobenzidene and enhanced with copper sulfate. Slides were washed in distilled water, counterstained with hematoxylin, dehydrated and mounted with permanent media. Appropriate positive and negative controls were run in parallel to study sections.
NuMA+ Cell Quantification.
Scanned images of slides were analyzed using Viziopharm software. Briefly, for brain, regions on interest were drawn to include all brain parenchymal tissue and exclude leptomeningeal areas. For kidney and liver, regions of interest were drawn to include all organ parenchyma. A cell identification and binary categorization algorithm was used to quantify the total number of NuMA-positive cells per slide. Settings for successful discrimination between NuMA-positive and NuMA-negative cells were established for each organ through testing on positive and negative control areas in multiple slides. The total number of metastatic cells from all quantified sections per organ per mouse (see Sectioning and NuMA Immunohistochemistry section above for details on number of sections for metastatic quantification) is reported in the data figures. In Supplementary Fig. S3J–M, the total number and average size of brain and liver metastases from all quantified sections per organ per mouse is reported.
Congo Red Staining
5 um sections were deparaffinized and stained with Congo Red solution (American, MasterTech, Item # KTCRE) for 1 hour. Slides were then immersed briefly in 1% sodium hydroxide, rinsed for 5 minutes in running water and counterstained with Modified Mayer’s Hematoxylin before dehydrating and coverslipping.
Brain Slice Immunofluorescence
Brains from perfused mice were fixed overnight in 4% PFA. Brains were sectioned using a vibratome (Leica) into 30 μM (all Fig. 6 and Supplementary Fig. S6 experiments) or 50 μM (All Fig. 5 and Supplementary Fig. S5 experiments)-thick slices. Slices were taken from 3–4 levels evenly spaced through the cortex. Slices were incubated in blocking buffer (10% Normal Goat Serum, 2% BSA, 0.25% Triton) in PBS for 2 hours at room temperature. Primary antibodies were incubated for 24 to 72 hours at 4°C in blocking buffer and were washed 4 times for 5 min in 0.25% Triton in PBS. Slices were incubated in secondary antibody in blocking solution for 2 hours and were washed 4 times for 5 min in 0.25% Triton in PBS. Nuclei were stained with DAPI at 1:1000 for 4 min in PBS. Brain slices were mounted to glass slides and coverslipped in Dako Fluorescence Mounting Medium (Agilent S3023). Confocal images were captured in z-stack using a Zeiss-770 microscope at 60x in oil. Non-confocal images were captured using a Zeiss-880 microscope at 20x or 40x. The same voltages were used for image capture across all images and groups within each experiment. Images were processed using ImageJ (RRID:SCR_003070). Within each experiment, brightness and contrast values were kept the same across all images and groups.
The following primary antibodies were used:
Mouse anti-GFP AlexaFluor 488 Conjugated Ab (Santa Cruz sc-9996 AF488, RRID:AB_627695) 1:200 – 24 hours, Rabbit anti-Cleaved Caspase-3 AlexaFluor 555 Conjugate (Cell Signaling 9604S, RRID:AB_2797708) 1:100 – 24 hours, Rabbit Anti-GFP (Abcam ab6556, RRID:AB_305564) 1:500 – 48 hours, Chicken Anti-GFAP (Aves Labs, RRID:AB_2734372) 1:1000 – 24 hours, Mouse Anti-CD68 (Invitrogen 14–0681-82, RRID:AB_2572857) 1:50 – 72 hours, Mouse Anti-Arginase I (Santa Cruz sc271430, RRID:AB_10648473) 1:50 – 72 hours, Rabbit Anti-Iba1 (Wako Chemicals 019–19741, RRID:AB_839504) 1:500 – 48 hours, Rat Anti-C3 [11H9] (Abcam ab11862, RRID:AB_2066623) 1:50 – 72 hours, Rabbit Anti-TMEM119 (Abcam ab209064, RRID:AB_2800343) 1:100 – 72 hours, Mouse Anti-Iba1 (Millipore #MABN92, RRID:AB_10917271) 1:200 – 72 hours
The following secondary antibodies were used:
Goat anti-Chicken IgG H+L A488 1:500 (Invitrogen A11039, RRID:AB_142924), Goat anti-Chicken IgG H+L A568 1:500 (Invitrogen A11041, RRID:AB_2534098 ), Goat anti-Rabbit IgG H+L A488 1:500 (Invitrogen A32731, RRID:AB_2633280), Goat anti-Rabbit IgG H+L A568 1:500 (Invitrogen A11011, RRID:AB_143157), Goat anti-Rabbit IgG H+L A647 1:500 (Invitrogen A21245, RRID:AB_141775), Goat anti-Mouse IgG H+L A568 1:500 (Invitrogen A11004, RRID:AB_2534072), Goat anti-Mouse IgG H+L A647 1:500 (Invitrogen A21235, RRID:AB_2535804), Goat anti-Rat IgG H+L A647 1:500 (Invitrogen A21247, RRID:AB_141778)
Two-dimensional astrocyte image analysis.
Images were analyzed using ImageJ (RRID:SCR_003070). Images were processed by the following steps: Gaussian blur of the GFAP channel with a radius of 1, subtraction of background from GFAP channel with a rolling ball radius of 50, conversion to a binary image with thresholding on the GFAP fluorescence channel using the intermodes formula, and generation of discrete astrocyte ROIs using object counter with a minimum size filter of 100 μm2. Discrete astrocytes were classified as melanoma-associated or control based on presence of physical contact with GFP-positive melanoma cells. Melanoma-associated astrocyte parameters were quantified on a per-image basis – in images where more than one discrete melanoma-associated astrocyte was detected, the sum of the area and perimeter of all melanoma-associated astrocytes in the image was reported, whereas the average of the GFAP intensity and Area/Perimeter of all melanoma-associated astrocytes was reported. For melanoma cells with no melanoma-associated astrocytes detected, Area and Perimeter values were reported as zero. Melanoma cells with no melanoma-associated astrocytes were excluded from the GFAP intensity and Area/Perimeter analyses.
Three-dimensional astrocyte and microglia image analysis.
Images were analyzed using ImageJ (RRID:SCR_003070). Images were processed by the following steps: 3D-Gaussian blur of the GFAP or Iba1 and the C3, Arg-1, or CD68 channels with radii x=2, y=2, z=2; subtraction of background from GFAP or Iba1 and the C3, Arg-1, CD68 channels with a rolling ball radius of 100 applied to all images in z-stack, conversion to binary z-stack image with thresholding of GFAP or Iba1 fluorescence using the IsoData formula, generation of three-dimensional ROIs of discrete astrocytes or microglia from the binary image using 3D-object counter with a minimum volume threshold of 20,000 voxels. Discrete astrocytes or microglia were classified as melanoma-associated or control based on presence of any physical contact at any level in the z-stack with GFP+ melanoma cells. Using 3D-manager, the mean intensity of C3, Arg-1, or CD68 were quantified. The mean signal intensity of all discrete melanoma-associated astrocytes or microglia for each z-stack was determined. A mean signal intensity of control melanoma-associated astrocytes was determined for each z-stack by averaging values from 4 to 6 representative control astrocytes or microglia. To control for fluorescence and staining variability between images, the mean signal intensity of melanoma-associated astrocytes or microglia were normalized to control astrocytes or microglia by subtracting mean control astrocyte or microglia intensity from each z-stack from the corresponding mean melanoma-associated astrocyte or microglia intensity from the same z-stack and was reported as a value in Fig. 6H–J. For melanoma cells with less than 4 to 6 but greater than one control astrocytes or microglia detected, 1–3 control astrocytes or microglia were used to calculate a control average mean intensity. Z-stack images with no control astrocytes or microglia were excluded from analysis. Z-stack images with no melanoma-associated astrocytes or microglia were also excluded from analysis
Ex Vivo Magnetic Resonance Imaging
After perfusion, the skulls of mice were removed and fixed in formalin for 72 hours. MRI experiments were performed on a Biospec 7030 micro-MRI system (Bruker) composed of an Avance-3 HD console and a zero-boil off 7-Tesla (7 T) (300 MHz) 300-mm horizontal bore magnet equipped with an actively shielded gradient coil insert (Bruker BGA-12S-HP; ID 114-mm, 660-mT/m gradient strength, 130-μs rise time). All scans were performed with a Bruker transmit-receive whole mouse body radiofrequency coil (Bruker 1P T20071V3: OD=59mm, ID=38mm, L=40mm) tuned to 300.16 MHz, the 1H proton Larmor frequency at 7-T commercial. This rf probe enabled the acquisition of 3D datasets with sub-millimetric isotropic resolution (<150 μm) during overnight scans spanning from 8 to 12 hours. As previously described (79), tumor burden was detected using multiple sequences. Hyper-intense signal detected by a T2-weighted. Rapid Imaging with Refocused Echoes (RARE) sequence recognizes edema surrounding tumors. The 3D RARE sequence was performed with the following acquisition parameters: (120 μm)3 isotropic resolution, acquisition time 5h 27 min, repetition time TR = 500 ms, echo spacing ES=12.7 min, Turbo factor TFx=12, effective echo time TEeff=76.2 ms, bandwidth BW= 75 KHz, Matrix size = 2843, field of view FOV= (4.0 mm)3, number of averages Nav=6. Pigmented metastases were detected with signal brightening when using a T1-weighted 3D Gradient echo sequence with the parameters as follow: (120 μm)3 isotropic resolution, acquisition time 2hrs 41 min, repetition time TR = 20 ms, echo time TE=4.0 ms, flip angle FA= 18o, bandwidth BW= 75 KHz, Matrix size = 2843, field of view FOV= (34.0 mm)3, number of averages Nav=6. Unpigmented and/or hemorrhagic metastases were detected with a hypo-intense signal when acquired under a T2*-weighted multi-gradient echo (MGE) sequence (3D MGE, [120 μm]3 isotropic resolution, acquisition time 3h 35 min, repetition time TR = 40 ms, echo time TE=3.6 ms, echo spacing ES=3.2 ms, 4 echoes, flip angle FA= 20o, bandwidth BW= 100 KHz, Matrix size = 2843, field of view FOV= (34.0 mm)3, number of averages Nav=4). All 3 sequences were used to quantify tumor burden. Identified tumor areas from analysis were cross referenced with histological sections to ensure accuracy.
Transmission Electron Microscopy
Cultured cells were fixed in 0.1 M sodium cacodylate buffer (pH 7.2) containing 2.5% glutaraldehyde and 2% paraformaldehyde for 2 hours and post-fixed with 1% osmium tetroxide for one hour, then block stained in 1% aqueous uranyl acetate, dehydrated using a gradient of ethanol and embedded in EMbed 812 (Electron Microscopy Sciences, Hatfield, PA). Ultrathin sections (60 nm) were cut, mounted on copper grids and stained with uranyl acetate and lead citrate. Stained grids were examined under Philips CM-12 electron microscope and photographed with a Gatan (4k x 2.7k) digital camera. Images were analyzed in ImageJ (RRID:SCR_003070) by measuring the largest visible mitochondrial dimension (length or width) for each mitochondrion present in randomly selected images.
Seahorse Metabolic Analysis
4.5 × 104 12–273 NBM, 3.5 × 104 12–273 BM, 8 × 104 10–230 NBM, 1×105 10–230 BM, 3 × 104 WM-4071 NBM, or 3 × 104 WM-4071 BM cells were plated on a XF24 Cell Culture Microplate coated with Cell-Tak (Corning). Simultaneously, cells were also plated from same master mix in 12-well plates for later use for normalization. Seahorse MitoStress test protocol was followed using a Seahorse XF24 instrument (Agilent). Concentrations of inhibitors injected are as follows: 12–273 pair – 1 μM oligomycin, 1.5 μM FCCP, 0.5 μM antimycin A and rotenone; 10–230 pair 0.5 μM oligomycin, 1.5 μM FCCP, 0.5 μM antimycin A and rotenone; WM-4071 pair - 0.75 μM oligomycin, 2 μM FCCP, 0.5 μM antimycin A and rotenone. After MitoStress test, protein was harvested from the normalization 12-well plates using RIPA buffer lysis and quantified by BCA assay. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) values for every STC were normalized to average protein concentration of the corresponding normalization wells.
Plasmid Generation
CMV-Luciferase-EF1α-copGFP (GFP-luc) Lentivector Plasmid was purchased from BD Biosciences (BLIV511PA-1)
sh-RNA plasmids.
Tet-pLKO-puro was purchased from Addgene (RRID:Addgene_21915). sh-RNAs were cloned as previously described (https://mcmanuslab.ucsf.edu/protocol/cloning-small-hairpins-lentiviral-vectors) into Tet-pLKO-puro using AgeI and EcoRI restriction sites. pLKO tet-on scrambled (sh-Scr) was purchased from Addgene (RRID:Addgene_47541). See Supplementary Table S4 for sh-RNA sequences.
CRISPR plasmids.
pLentiCas9-Blast was purchased from Addgene (RRID:Addgene_52962). pLentiGuide-Puro was purchased from Addgene (RRID:Addgene_52963). For 5B1 cells and 12–273 BM CRISPR APP-knockout mutant rescue experiment (Fig. 4A–E), sg-RNA sequences were designed using the GPP sgRNA Designer (Broad Institute) and cloned into pLentiGuide-Puro using the BsmBI restriction site. For 12–273 BM CRISPR APP-knockout experiment (Fig. 3J–L), sg-RNA sequences were designed using the GPP sgRNA Designer (Broad Institute) and cloned into pLentiCRISPRv2 from Addgene (RRID:Addgene_52961) using the BsmBI restriction site. See Supplementary Table S4 for sg-RNA sequences.
APP Expression plasmids.
pLVX-IRES-tdTomato (pLVX) was purchased from Clontech (631238, RRID:Addgene_64831). APP-770 was purchased from Genecopoeia (EX-Z2553-M02) and subcloned into pLVX using SpeI and NotI restriction sites.
SPA4CT plasmids.
A cloning strategy was designed and implemented to generate the SPA4CT sequence (25). pLVX APP-770 was digested with EcoRI and NotI to generate a 366 base pair terminal APP insert. Complementary oligos (purchased from Integrated DNA Technologies) were annealed to form an insert with XhoI and EcoRI overhangs. pLVX was digested with XhoI and NotI and religated together with the two inserts. For specific sequences, see Supplementary Table S4. SPA4CT-T43P was generated using the Q5 Site-Directed Mutagenesis Kit (NEB E0554S) using pLVX SPA4CT as a template. For specific sequences of primers and inserts, please see Supplementary Table S4.
Lentiviral Production and Infection
HEK293T cells at 80% confluency were co-transfected with 12 μg of lentiviral expression constructs, 8 μg of psPAX2 (RRID:Addgene_12260) and 4 μg pMD2.G (RRID:Addgene_12259) vectors using Lipofectamine 2000 (Invitrogen) following manufacturer’s recommendations. At 48 hr post transfection, supernatants were collected, filtered (0.45 μm) and stored at −80°C. Melanoma cells were infected with lentiviral supernatant supplemented with polybrene at a final concentration of 4 μg/mL. 24 hours after infection, the following selection methods were used for infected cells:
Cell sorting by GFP fluorescence for GFP-luc plasmid.Culture in puromycin (2 μg/mL) for sh-RNA and sg-RNA plasmids.
Culture in blasticidin (10 μg/mL) for Cas9 plasmid.
Successful pLVX infection was verified by RFP expression on microscopy; cells were not sorted based on RFP expression.
In vitro Proliferation Assay
2 days prior to assay, genetic silencing was induced by addition of doxycycline (1 μg/mL) to media. 1 × 104 12–273 BM or 1 × 104 WM-4265 BM cells were plated in four 24-well plates. After allowing cells to adhere overnight, a baseline plate was obtained by removal of media and fixation in 0.1% glutaraldehyde for 15 minutes. Remaining plates were fixed every 24 hours thereafter. Wells were stained with 0.5% crystal violet in PBS for one hour and washed extensively with water. Crystal violet retained by cells was then eluted by incubation in 250 μL 15% acetic acid for 1 hour with shaking. 100 μL from each well was transferred to a 96 well plate and absorbance at 590 nm was measured. Absorbances were normalized to the absorbances from the baseline plate to obtain a fold-change value.
Western Blot Analysis
Cells were harvested in cold RIPA lysis buffer supplemented with protease inhibitor and protein was quantified by BCA assay. Cell lysates (15–20 μg of protein) were resolved in 4%–4% Bis-Tris gels (Invitrogen) and transferred to PVDF membranes using wet transfer. Membranes were blocked in either 5% non-fat milk or 5% BSA in Tris-buffered saline 1% Tween 20 (TBST) for 1 hour. Membranes were incubated overnight with primary antibodies diluted in 5% milk or BSA TBST. Membranes were washed 3 times for 10 minutes in TBST and incubated in secondary antibody for 1 hour in 5% milk or BSA TBST. Membranes were washed 3 times for 10 minutes in TBST and incubated with ECL substrate for 3 min and exposed to film for imaging.
The following primary antibodies were used:
Anti-APP 22C11 (Thermo Fisher Scientific 14–9749-82, RRID:AB_2572978 RRID:AB_2572978) 1:1000 in milk, Anti-APP C6/1.1 (Gift from Mathews Lab, NYU School of Medicine) 1:5000 in BSA, anti-PRKAR2B (Thermo Fisher Scientific
PA5–28266, RRID:AB_2545742) 1:1000 in milk, anti-XPNPEP3 (Atlas Antibodies HPA000527, RRID:AB_1079266) 1:500 in milk, anti-SCARB1 (Abcam ab52629, RRID:AB_882458) 1:1000 in milk, anti-SELENBP1 (Abcam ab90135, RRID:AB_042880) 1:1000 milk, anti-EARS2 (Santa Cruz sc-271728, RRID:AB_10708847) 1:500 in milk, anti-FARP1 (Novus #H00010160-M01, RRID:AB_1214432) 1:1000 in milk, anti-MBTD1 (Thermo Fisher 730065, RRID:AB_2532866) 1:1000 in milk, anti-beta-actin-peroxidase (Sigma Aldrich A3854, RRID:AB_262011) 1:100000 in milk, Anti-tubulin (Sigma T9026, RRID:AB_477593) 1:10000 in milk.
The following secondary antibodies were used:
Goat anti-rabbit IgG-Peroxidase (Sigma-Aldrich A0545, RRID:AB_257896) 1:5000, Goat anti-mouse IgG kappa-light chain (Santa Cruz sc-516102, RRID:AB_2687626) 1:1000.
Membrane purification and γ-secretase activity assay
Cell membrane preparation and γ-secretase assays were described previously (80–83). Briefly, cells were harvested from T75 flask 90% confluency and collected by centrifuge (800 g, 10 min). Cell pellets were resuspended with hypotonic buffer (5mM Tris, pH 7.4), incubated in ice for 30 min and homogenized with Glass Teflon Homogenizer. Samples were centrifuged at 1000 g, 30 min and the supernatants that contain the total membrane fraction were collected. Pellets were resuspended with hypotonic buffer, homogenized and centrifuged again. Combined supernatants were centrifugated at 100,000 g for 60 min. Resulted pellets referred to as membrane fractions were resuspended and washed with MES buffer (50 mM MES, pH 6.0, 150 mM KCl, 5 mM CaCl2, 5 mM MgCl2, and protease inhibitors) and spun down (100,000g for 60 min). Membrane fractions were dissolved in MES buffer and protein concentration was determined by the DC assay kit (Biorad). For γ-secretase assays, Sb4 substrate (1 μM) or NTM2 substrate (0.4 μM) and membrane fractions (50 μg/mL) were incubated in PIPES Buffer (50 mM PIPES, pH 7.0, 150 mM KCl, 5 mM CaCl2, 5 mM MgCl2) and 0.25% CHAPSO detergent at 37°C for 3 hours. γ-Secretase products were detected by AlphaLISA methods using G2–10 (Millipore MABN11, RRID:AB_10561738) or SM320 antibodies for Aβ40 or NICD, respectively.
Amyloid Beta ELISA
Media was conditioned with melanoma cells for 24–72 hours and concentrated 10x using Amicon Ultra 3-kDa Concentrators (Millipore Z740169). Aβ-40 ELISA was performed using the Human Aβ-40 ELISA Kit (Invitrogen) and Aβ-40 (aa1–40) Quantikine ELISA Kit (R&D). Aβ-42 ELISA was performed using Amyloid Beta 42 Human ELISA Kit (Invitrogen). Secretion values were normalized to protein content of wells as measured by RIPA harvest and BCA protein quantification.
APP Isoform Semi Quantitative qPCR
RNA was isolated from 10–230 NBM, 10–230 BM, 12–273 NBM, and 12–273 BM cells at 80% confluence in 6 well plates using the RNeasy Mini Kit (Qiagen 74104). 600 ng of RNA was subjected to DNase I treatment and reverse transcription. PCR was performed with primers as previously described (84) to generate bands of different sizes corresponding to the following isoforms: APP-770 – 461bp, APP-751 – 417bp, APP-695 235bp
Statistical Analysis
Statistical analyses were performed with Prism 8 (GraphPad Software, RRID:SCR_002798). Unless otherwise stated, the Student’s two-sided t-test was used for experiments. P-values <0.05 were considered to be statistically significant. Unless otherwise stated, values are averages and error bars are +/− standard deviation.
Data Availability
The proteomics data of BM and NBM melanoma STCs (Fig. 1) has been deposited to the UCSD Mass Spectrometry Interactive Virtual Environment (MassIVE) and are accessible through MassIVE ID number MSV000088814. The RNA-sequencing data of rat astrocytes exposed to melanoma-conditioned media (Fig. 6) has been deposited in NCBI’s Gene Expression Omnibus (85) and are accessible through GEO Series accession number GSE196332.
Supplementary Material
Statement of Significance.
Our results reveal a novel mechanistic connection between brain metastasis and Alzheimer’s disease – two previously unrelated pathologies, establish Aβ as a promising therapeutic target for brain metastasis, and demonstrate suppression of neuroinflammation as a critical feature of metastatic adaptation to the brain parenchyma.
Acknowledgements
E.H. is supported by NIH 5R01CA243446, P01CA206980, an American Cancer Society-Melanoma Research Alliance Team Science Award and a NIH Melanoma SPORE (NCI P50 CA225450; PI: I.O.). K.K. is supported by F30CA221068, and previously by the NIH/NCI 5 T32 CA009161–37 (Training Program in Molecular Oncology and Immunology, PI: D.E. Levy), NIGMS 5 T32 GM007308–41 (Medical Scientist Training Program, PI: M. Philips), and a Vilcek Foundation Scholarship. A.F. is supported by a Fundacion Ramon Areces fellowship. YM.L. is supported by RF1AG057593. S.A.L. is funded by the Cure Alzheimer’s Fund, Anonymous Donors, the Blas Frangione Foundation, the MD Anderson Neurodegenerative Consortium, and NYU Grossman School of Medicine.
We thank Drs. Clemens Krepler and Meenhard Herlyn for patient-derived short-term cultures. We thank Dr. Rana Moubarak and members of the Hernando lab for critical reading of the manuscript. We thank the NYU Center for Biospecimen Research and Development (CBRD), the Experimental Pathology Core Facility (Director, Dr. Cindy Loomis; [RRID:SCR017928]), the Immunohistochemistry Core Laboratory (Dr. Luis Chiriboga), the Microscopy core (Dr. Alice Xiang), the Genomics Technology Center (Dr. Adriana Heguy), and the Flow Cytometry Core (Dr. Peter Lopez), supported in part by the Laura and Isaac Perlmutter Cancer Center Support Grant NIH/NCI P30CA016087, and National Institutes of Health S10 Grants NIH/ORIP S10OD01058 and S10OD018338. Authors also acknowledge the MSK Cancer Center Support Grant/Core Grant (Grant P30 CA008748).
Footnotes
Conflicts of Interest: R.D. is a full-time employee at Eli Lilly. All other authors have no financial interests. E.H., R.J.S. and K.K. are inventors on a pending International Patent Application No. PCT/US2019/033377 filed on May 21, 2019. SAL is a Founder of AstronauTx Ltd, a company making therapies to target astrocytes in neurodegenerative disease.
References
- 1.Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol 2004;22(14):2865–72 doi 10.1200/JCO.2004.12.149. [DOI] [PubMed] [Google Scholar]
- 2.Patel JK, Didolkar MS, Pickren JW, Moore RH. Metastatic pattern of malignant melanoma. A study of 216 autopsy cases. Am J Surg 1978;135(6):807–10 doi 10.1016/0002-9610(78)90171-x. [DOI] [PubMed] [Google Scholar]
- 3.de la Monte SM, Moore GW, Hutchins GM. Patterned distribution of metastases from malignant melanoma in humans. Cancer Res 1983;43(7):3427–33. [PubMed] [Google Scholar]
- 4.Davies MA, Saiag P, Robert C, Grob JJ, Flaherty KT, Arance A, et al. Dabrafenib plus trametinib in patients with BRAF(V600)-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol 2017;18(7):863–73 doi 10.1016/S1470-2045(17)30429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N Engl J Med 2018;379(8):722–30 doi 10.1056/NEJMoa1805453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Long GV, Atkinson V, Lo S, Sandhu S, Guminski AD, Brown MP, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicentre randomised phase 2 study. Lancet Oncol 2018;19(5):672–81 doi 10.1016/S1470-2045(18)30139-6. [DOI] [PubMed] [Google Scholar]
- 7.Zhang D, Wang Z, Shang D, Yu J, Yuan S. Incidence and prognosis of brain metastases in cutaneous melanoma patients: a population-based study. Melanoma Res 2019;29(1):77–84 doi 10.1097/CMR.0000000000000538. [DOI] [PubMed] [Google Scholar]
- 8.Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 2010;16(1):116–22 doi 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- 9.Lin Q, Balasubramanian K, Fan D, Kim SJ, Guo L, Wang H, et al. Reactive astrocytes protect melanoma cells from chemotherapy by sequestering intracellular calcium through gap junction communication channels. Neoplasia 2010;12(9):748–54 doi 10.1593/neo.10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartz H, Blacher E, Amer M, Livneh N, Abramovitz L, Klein A, et al. Incipient Melanoma Brain Metastases Instigate Astrogliosis and Neuroinflammation. Cancer Res 2016;76(15):4359–71 doi 10.1158/0008-5472.CAN-16-0485. [DOI] [PubMed] [Google Scholar]
- 11.Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016;533(7604):493–8 doi 10.1038/nature18268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014;156(5):1002–16 doi 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levi G, Wilkin GP, Ciotti MT, Johnstone S. Enrichment of differentiated, stellate astrocytes in cerebellar interneuron cultures as studied by GFAP immunofluorescence and autoradiographic uptake patterns with [3H]D-aspartate and [3H]GABA. Brain Res 1983;312(2):227–41 doi 10.1016/0165-3806(83)90139-6. [DOI] [PubMed] [Google Scholar]
- 14.Pellerin L, Magistretti PJ. Excitatory amino acids stimulate aerobic glycolysis in astrocytes via an activation of the Na+/K+ ATPase. Dev Neurosci 1996;18(5–6):336–42 doi 10.1159/000111426. [DOI] [PubMed] [Google Scholar]
- 15.Han RT, Kim RD, Molofsky AV, Liddelow SA. Astrocyte-immune cell interactions in physiology and pathology. Immunity 2021;54(2):211–24 doi 10.1016/j.immuni.2021.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017;541(7638):481–7 doi 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben Haim L, Ceyzeriat K, Carrillo-de Sauvage MA, Aubry F, Auregan G, Guillermier M, et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J Neurosci 2015;35(6):2817–29 doi 10.1523/JNEUROSCI.3516-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 2006;129(Pt 10):2761–72 doi 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- 19.Chen G, Chakravarti N, Aardalen K, Lazar AJ, Tetzlaff MT, Wubbenhorst B, et al. Molecular profiling of patient-matched brain and extracranial melanoma metastases implicates the PI3K pathway as a therapeutic target. Clin Cancer Res 2014;20(21):5537–46 doi 10.1158/1078-0432.CCR-13-3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer GM, Jalali A, Kircher DA, Lee WC, McQuade JL, Haydu LE, et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discov 2019;9(5):628–45 doi 10.1158/2159-8290.CD-18-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sundstrom T, Prestegarden L, Azuaje F, Aasen SN, Rosland GV, Varughese JK, et al. Inhibition of mitochondrial respiration prevents BRAF-mutant melanoma brain metastasis. Acta Neuropathol Commun 2019;7(1):55 doi 10.1186/s40478-019-0712-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med 2010;12(1):1–12 doi 10.1007/s12017-009-8104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shelton CC, Tian Y, Frattini MG, Li YM. An exo-cell assay for examining real-time gamma-secretase activity and inhibition. Mol Neurodegener 2009;4:22 doi 10.1186/1750-1326-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thanopoulou K, Fragkouli A, Stylianopoulou F, Georgopoulos S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc Natl Acad Sci U S A 2010;107(48):20816–21 doi 10.1073/pnas.1005888107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brandon EP, Logue SF, Adams MR, Qi M, Sullivan SP, Matsumoto AM, et al. Defective motor behavior and neural gene expression in RIIbeta-protein kinase A mutant mice. J Neurosci 1998;18(10):3639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stames EM, O’Toole JF. Mitochondrial aminopeptidase deletion increases chronological lifespan and oxidative stress resistance while decreasing respiratory metabolism in S. cerevisiae. PLoS One 2013;8(10):e77234 doi 10.1371/journal.pone.0077234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lichtenthaler SF, Multhaup G, Masters CL, Beyreuther K. A novel substrate for analyzing Alzheimer’s disease gamma-secretase. FEBS Lett 1999;453(3):288–92 doi 10.1016/s0014-5793(99)00730-9. [DOI] [PubMed] [Google Scholar]
- 28.Lichtenthaler SF, Ida N, Multhaup G, Masters CL, Beyreuther K. Mutations in the transmembrane domain of APP altering gamma-secretase specificity. Biochemistry 1997;36(49):15396–403 doi 10.1021/bi971071m. [DOI] [PubMed] [Google Scholar]
- 29.Allaman I, Gavillet M, Belanger M, Laroche T, Viertl D, Lashuel HA, et al. Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci 2010;30(9):3326–38 doi 10.1523/JNEUROSCI.5098-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye B, Shen H, Zhang J, Zhu YG, Ransom BR, Chen XC, et al. Dual pathways mediate beta-amyloid stimulated glutathione release from astrocytes. Glia 2015;63(12):2208–19 doi 10.1002/glia.22886. [DOI] [PubMed] [Google Scholar]
- 31.Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung WS, Zhou L, et al. Development of a method for the purification and culture of rodent astrocytes. Neuron 2011;71(5):799–811 doi 10.1016/j.neuron.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement System Part II: Role in Immunity. Front Immunol 2015;6:257 doi 10.3389/fimmu.2015.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci 2012;32(18):6391–410 doi 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J Neurosci 2016;36(2):577–89 doi 10.1523/JNEUROSCI.2117-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen T, Lennon VA, Liu YU, Bosco DB, Li Y, Yi MH, et al. Astrocyte-microglia interaction drives evolving neuromyelitis optica lesion. J Clin Invest 2020;130(8):4025–38 doi 10.1172/JCI134816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei Y, Chen T, Bosco DB, Xie M, Zheng J, Dheer A, et al. The complement C3-C3aR pathway mediates microglia-astrocyte interaction following status epilepticus. Glia 2021;69(5):1155–69 doi 10.1002/glia.23955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 2014;11:98 doi 10.1186/1742-2094-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabinowitz SS, Gordon S. Macrosialin, a macrophage-restricted membrane sialoprotein differentially glycosylated in response to inflammatory stimuli. J Exp Med 1991;174(4):827–36 doi 10.1084/jem.174.4.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Dyck CH. Anti-Amyloid-beta Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol Psychiatry 2018;83(4):311–9 doi 10.1016/j.biopsych.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coimbra JRM, Marques DFF, Baptista SJ, Pereira CMF, Moreira PI, Dinis TCP, et al. Highlights in BACE1 Inhibitors for Alzheimer’s Disease Treatment. Front Chem 2018;6:178 doi 10.3389/fchem.2018.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.May PC, Willis BA, Lowe SL, Dean RA, Monk SA, Cocke PJ, et al. The potent BACE1 inhibitor LY2886721 elicits robust central Abeta pharmacodynamic responses in mice, dogs, and humans. J Neurosci 2015;35(3):1199–210 doi 10.1523/JNEUROSCI.4129-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cruz-Munoz W, Man S, Xu P, Kerbel RS. Development of a preclinical model of spontaneous human melanoma central nervous system metastasis. Cancer Res 2008;68(12):4500–5 doi 10.1158/0008-5472.CAN-08-0041. [DOI] [PubMed] [Google Scholar]
- 43.Cruz-Munoz W, Jaramillo ML, Man S, Xu P, Banville M, Collins C, et al. Roles for endothelin receptor B and BCL2A1 in spontaneous CNS metastasis of melanoma. Cancer Res 2012;72(19):4909–19 doi 10.1158/0008-5472.CAN-12-2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jilaveanu LB, Parisi F, Barr ML, Zito CR, Cruz-Munoz W, Kerbel RS, et al. PLEKHA5 as a Biomarker and Potential Mediator of Melanoma Brain Metastasis. Clin Cancer Res 2015;21(9):2138–47 doi 10.1158/1078-0432.CCR-14-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov 2015;5(11):1164–77 doi 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Miera EV, Friedman EB, Greenwald HS, Perle MA, Osman I. Development of five new melanoma low passage cell lines representing the clinical and genetic profile of their tumors of origin. Pigment Cell Melanoma Res 2012;25(3):395–7 doi 10.1111/j.1755-148X.2012.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JY, Park K, Lee E, Ahn T, Jung HH, Lim SH, et al. Gene Expression Profiling of Breast Cancer Brain Metastasis. Sci Rep 2016;6:28623 doi 10.1038/srep28623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertoni JM, Arlette JP, Fernandez HH, Fitzer-Attas C, Frei K, Hassan MN, et al. Increased melanoma risk in Parkinson disease: a prospective clinicopathological study. Arch Neurol 2010;67(3):347–52 doi 10.1001/archneurol.2010.1. [DOI] [PubMed] [Google Scholar]
- 49.Olsen JH, Friis S, Frederiksen K. Malignant melanoma and other types of cancer preceding Parkinson disease. Epidemiology 2006;17(5):582–7 doi 10.1097/01.ede.0000229445.90471.5e. [DOI] [PubMed] [Google Scholar]
- 50.Walter U, Heilmann E, Voss J, Riedel K, Zhivov A, Schad SG, et al. Frequency and profile of Parkinson’s disease prodromi in patients with malignant melanoma. J Neurol Neurosurg Psychiatry 2016;87(3):302–10 doi 10.1136/jnnp-2014-310239. [DOI] [PubMed] [Google Scholar]
- 51.Priego N, Zhu L, Monteiro C, Mulders M, Wasilewski D, Bindeman W, et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat Med 2018;24(7):1024–35 doi 10.1038/s41591-018-0044-4. [DOI] [PubMed] [Google Scholar]
- 52.O’Callaghan JP, Kelly KA, VanGilder RL, Sofroniew MV, Miller DB. Early activation of STAT3 regulates reactive astrogliosis induced by diverse forms of neurotoxicity. PLoS One 2014;9(7):e102003 doi 10.1371/journal.pone.0102003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gibson EM, Nagaraja S, Ocampo A, Tam LT, Wood LS, Pallegar PN, et al. Methotrexate Chemotherapy Induces Persistent Tri-glial Dysregulation that Underlies Chemotherapy-Related Cognitive Impairment. Cell 2019;176(1–2):43–55 e13 doi 10.1016/j.cell.2018.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kano SI, Choi EY, Dohi E, Agarwal S, Chang DJ, Wilson AM, et al. Glutathione S-transferases promote proinflammatory astrocyte-microglia communication during brain inflammation. Sci Signal 2019;12(569) doi 10.1126/scisignal.aar2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Priego N, Valiente M. The Potential of Astrocytes as Immune Modulators in Brain Tumors. Front Immunol 2019;10:1314 doi 10.3389/fimmu.2019.01314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baik SH, Kang S, Lee W, Choi H, Chung S, Kim JI, et al. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab 2019;30(3):493–507 e6 doi 10.1016/j.cmet.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 57.Esquerda-Canals G, Montoliu-Gaya L, Guell-Bosch J, Villegas S. Mouse Models of Alzheimer’s Disease. J Alzheimers Dis 2017;57(4):1171–83 doi 10.3233/JAD-170045. [DOI] [PubMed] [Google Scholar]
- 58.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci 2008;28(33):8354–60 doi 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Farlow M, Arnold SE, van Dyck CH, Aisen PS, Snider BJ, Porsteinsson AP, et al. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement 2012;8(4):261–71 doi 10.1016/j.jalz.2011.09.224. [DOI] [PubMed] [Google Scholar]
- 60.Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N Engl J Med 2018;378(4):321–30 doi 10.1056/NEJMoa1705971. [DOI] [PubMed] [Google Scholar]
- 61.Krepler C, Sproesser K, Brafford P, Beqiri M, Garman B, Xiao M, et al. A Comprehensive Patient-Derived Xenograft Collection Representing the Heterogeneity of Melanoma. Cell Rep 2017;21(7):1953–67 doi 10.1016/j.celrep.2017.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Foo LC. Purification of rat and mouse astrocytes by immunopanning. Cold Spring Harb Protoc 2013;2013(5):421–32 doi 10.1101/pdb.prot074211. [DOI] [PubMed] [Google Scholar]
- 63.Winzeler A, Wang JT. Culturing hybridoma cell lines for monoclonal antibody production. Cold Spring Harb Protoc 2013;2013(7):640–2 doi 10.1101/pdb.prot074914. [DOI] [PubMed] [Google Scholar]
- 64.Bansal R, Warrington AE, Gard AL, Ranscht B, Pfeiffer SE. Multiple and novel specificities of monoclonal antibodies O1, O4, and R-mAb used in the analysis of oligodendrocyte development. J Neurosci Res 1989;24(4):548–57 doi 10.1002/jnr.490240413. [DOI] [PubMed] [Google Scholar]
- 65.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30(15):2114–20 doi 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25(16):2078–9 doi 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31(2):166–9 doi 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34(3):267–73 doi 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 72.Stafford JM, Lee CH, Voigt P, Descostes N, Saldana-Meyer R, Yu JR, et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 2018;4(10):eaau5935 doi 10.1126/sciadv.aau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Becker SH, Jastrab JB, Dhabaria A, Chaton CT, Rush JS, Korotkov KV, et al. The Mycobacterium tuberculosis Pup-proteasome system regulates nitrate metabolism through an essential protein quality control pathway. Proc Natl Acad Sci U S A 2019;116(8):3202–10 doi 10.1073/pnas.1819468116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 2009;6(5):359–62 doi 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 75.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 2011;10(4):1794–805 doi 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 76.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26(12):1367–72 doi 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 77.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4(1):44–57 doi 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 78.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009;37(1):1–13 doi 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morsi A, Gaziel-Sovran A, Cruz-Munoz W, Kerbel RS, Golfinos JG, Hernando E, et al. Development and characterization of a clinically relevant mouse model of melanoma brain metastasis. Pigment Cell Melanoma Res 2013;26(5):743–5 doi 10.1111/pcmr.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chau DM, Crump CJ, Villa JC, Scheinberg DA, Li YM. Familial Alzheimer disease presenilin-1 mutations alter the active site conformation of gamma-secretase. J Biol Chem 2012;287(21):17288–96 doi 10.1074/jbc.M111.300483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, et al. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci U S A 2000;97(11):6138–43 doi 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tian Y, Bassit B, Chau D, Li YM. An APP inhibitory domain containing the Flemish mutation residue modulates gamma-secretase activity for Abeta production. Nat Struct Mol Biol 2010;17(2):151–8 doi 10.1038/nsmb.1743. [DOI] [PubMed] [Google Scholar]
- 83.Wong E, Liao GP, Chang JC, Xu P, Li YM, Greengard P. GSAP modulates gamma-secretase specificity by inducing conformational change in PS1. Proc Natl Acad Sci U S A 2019;116(13):6385–90 doi 10.1073/pnas.1820160116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Quast T, Wehner S, Kirfel G, Jaeger K, De Luca M, Herzog V. sAPP as a regulator of dendrite motility and melanin release in epidermal melanocytes and melanoma cells. FASEB J 2003;17(12):1739–41 doi 10.1096/fj.02-1059fje. [DOI] [PubMed] [Google Scholar]
- 85.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002;30(1):207–10 doi 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proteomics data of BM and NBM melanoma STCs (Fig. 1) has been deposited to the UCSD Mass Spectrometry Interactive Virtual Environment (MassIVE) and are accessible through MassIVE ID number MSV000088814. The RNA-sequencing data of rat astrocytes exposed to melanoma-conditioned media (Fig. 6) has been deposited in NCBI’s Gene Expression Omnibus (85) and are accessible through GEO Series accession number GSE196332.