

Conspectus
Reagent instability reduces the efficiency of chemical processes, and while much effort is devoted to reaction optimization, less attention is paid to the mechanistic causes of reagent decomposition. Indeed, the response is often to simply use an excess of the reagent. Two reaction classes with ubiquitous examples of this are the Suzuki–Miyaura cross-coupling of boronic acids/esters and the transfer of CF3 or CF2 from the Ruppert–Prakash reagent, TMSCF3. This Account describes some of the overarching features of our mechanistic investigations into their decomposition. In the first section we summarize how specific examples of (hetero)arylboronic acids can decompose via aqueous protodeboronation processes: Ar–B(OH)2 + H2O → ArH + B(OH)3. Key to the analysis was the development of a kinetic model in which pH controls boron speciation and heterocycle protonation states. This method revealed six different protodeboronation pathways, including self-catalysis when the pH is close to the pKa of the boronic acid, and protodeboronation via a transient aryl anionoid pathway for highly electron-deficient arenes. The degree of “protection” of boronic acids by diol-esterification is shown to be very dependent on the diol identity, with six-membered ring esters resulting in faster protodeboronation than the parent boronic acid. In the second section of the Account we describe 19F NMR spectroscopic analysis of the kinetics of the reaction of TMSCF3 with ketones, fluoroarenes, and alkenes. Processes initiated by substoichiometric “TBAT” ([Ph3SiF2][Bu4N]) involve anionic chain reactions in which low concentrations of [CF3]− are rapidly and reversibly liberated from a siliconate reservoir, [TMS(CF3)2][Bu4N]. Increased TMSCF3 concentrations reduce the [CF3]− concentration and thus inhibit the rates of CF3 transfer. Computation and kinetics reveal that the TMSCF3 intermolecularly abstracts fluoride from [CF3]− to generate the CF2, in what would otherwise be an endergonic α-fluoride elimination. Starting from [CF3]− and CF2, a cascade involving perfluoroalkene homologation results in the generation of a hindered perfluorocarbanion, [C11F23]−, and inhibition. The generation of CF2 from TMSCF3 is much more efficiently mediated by NaI, and in contrast to TBAT, the process undergoes autoacceleration. The process involves NaI-mediated α-fluoride elimination from [CF3][Na] to generate CF2 and a [NaI·NaF] chain carrier. Chain-branching, by [(CF2)3I][Na] generated in situ (CF2 + TFE + NaI), causes autoacceleration. Alkenes that efficiently capture CF2 attenuate the chain-branching, suppress autoacceleration, and lead to less rapid difluorocyclopropanation. The Account also highlights how a collaborative approach to experiment and computation enables mechanistic insight for control of processes.
Key References
Cox P. A.; Leach A. G.; Campbell A. D.; Lloyd-Jones G. C.. Protodeboronation of Heteroaromatic, Vinyl, and Cyclopropyl Boronic Acids: pH-Rate Profiles, Autocatalysis, and Disproportionation J. Am. Chem. Soc. 2016, 138, 9145–9157.1 Development of a speciation-kinetics model to account for empirical pH–log kobs profiles in protodeboronation of heteroaromatic boronic acids.
Hayes H. L. D.; Wei R.; Assante M.; Geogheghan K. J.; Jin N.; Tomasi S.; Noonan G.; Leach A. G.; Lloyd-Jones G. C.. Protodeboronation of (Hetero)Arylboronic Esters: Direct versus Prehydrolytic Pathways and Self-/Auto-Catalysis. J. Am. Chem. Soc. 2021, 143, 14814–14826.2 A rationalization of the protolytic instability of boronic esters, under aqueous basic conditions, using stopped-flow NMR and computation.
Johnston C. P.; West T. H.; Dooley R. E.; Reid M.; Jones A. B.; King E. J.; Leach A. G.; Lloyd-Jones G. C.. Anion-Initiated Trifluoromethylation by TMSCF3: Deconvolution of the Siliconate-Carbanion Dichotomy by Stopped-Flow NMR/IR. J. Am. Chem. Soc. 2018, 140, 11112–11124.3 Detailed kinetic analysis of the anion-initiated reaction of TMSCF3with ketones, demonstrating the intermediacy of a CF3anionoid, and the introduction of a novel variable-ratio stopped flow NMR system.
García-Domínguez A.; West T. H.; Primozic J. J.; Grant K. M.; Johnston C. P.; Cumming G. G.; Leach A. G.; Lloyd-Jones G. C.. Difluorocarbene Generation from TMSCF3: Kinetics and Mechanism of NaI-Mediated and Si-Induced Anionic Chain Reactions. J. Am. Chem. Soc. 2020, 142, 14649–14663.4 The use of partitioning analysis and computation to rationalize the behavior of the Ruppert–Prakash reagent under anionic initiation.
1. Introduction
Many organoboron and organosilicon species benefit from low toxicity, low cost, and ease of preparation, leading to numerous uses, including industrial processes.5 However, in some cases these reagents become unstable under the conditions of their application, leading to loss of yield or function. This Account discusses the elucidation of some of the key mechanistic features that lead to this instability in arylboronic acids/esters6 and in the remarkably versatile fluorochemical TMSCF3.7−10 Throughout the Account we try to highlight how strategic combinations of NMR spectroscopy,11,12 kinetics, byproduct analysis, pH–rate profiles, isotopes, and computation have allowed us to dissect competing reaction pathways involving organoboron and organosilicon “ate” complexes, and to explain several counterintuitive prior observations.
2. (Hetero)aryl Boronates
The Suzuki–Miyaura (SM) cross-coupling of arylboronic acids13 revolutionalized biaryl synthesis and remains highly valued in industry. A base is usually required to induce transfer of the aryl group from boron to the metal catalyst.14,15 Competing base-mediated processes (Scheme 1), including oxidation and protodeboronation, are detrimental to the efficiency.16 Although the oxidative processes can be minimized by careful choice of reaction conditions, the protodeboronation is mostly dependent on the identity of the boronic acid. The development of protected, or “masked”,17 reagents has been one of several effective strategies for mitigating the “protodeboronation problem”.18−20
Scheme 1. Organoboron Reagents and Suzuki–Miyaura Coupling.

In 2006, we began a collaborative project on ligand descriptors21 and used SM coupling to generate parametrization data. We encountered extensive side reactions with boronic acids and instead used ArBF3K reagents22 under Molander’s conditions.23 This gave substantially cleaner couplings and naturally led to a curiosity as to why this is the case.24 Further investigation identified that many of the beneficial effects arose from the controlled hydrolytic release of arylboronic acids in situ,24a a process modulated by the glass surface of the reaction vessel, the stirring rate, and the pH.24b With a new-found interest in “slow-release”,17 we collaborated with Burke, Houk, and Cheong on the base-mediated hydrolysis of BMIDA boronates.25In situ NMR, kinetics, heavy atom kinetic isotope effects (KIEs), and DFT calculations revealed two pathways, controlled by pH. One involves attack at C=O by hydroxide ion, the other B–NMe bond cleavage by neutral water.25
2.1. Protodeboronation pH–Rate Profiles
The above investigations made us appreciate that the stability of boronic acids under the conventional aqueous–organic basic conditions of SM coupling was not fully understood.13,16 Indeed, although mechanistic work by Kuivila26 in the 1960s on the protodeboronation of simple aryl boronic acids had been expanded on by Fröhn,27 Cammidge,28 Buchwald,20 and Perrin,29 reactivity trends were not readily compared, and there was scant detail on heteroaromatic boronic acids, which are systems perceived to be the most sensitive.13,16−18
A key first step was our identification that a medium of 50% aq. dioxane at 70 °C allowed the kinetics of a very wide array of boronic acids to be monitored in the presence of exogenous acids, bases, buffers, and metal salts, at concentrations amenable to NMR analysis.11,12 At the heart of the analysis was the nonlinear regression of pH–log kobs profiles using a model comprising weighted combinations of six pathways (Figure 1A), where kobs is the overall empirical pseudo-first-order rate constant.
Figure 1.

(A) Overarching kinetic model (pathways i–v) for pH-mediated speciation and protodeboronation. (B) Example pH–rate profiles for the protodeboronation of selected (hetero)arylboronic acids (50 mM, 50% vol. aq. dioxane, 70 °C). (C) Modified Swain–Lupton analysis of the protodeboronation of arylboronic acids. (D) Competing direct and indirect protodeboronation of boronate esters. Data from refs (1), (2), and (30).
We then analyzed the kinetics of protodeboronation of 52 different boronic acids, R–B(OH)2, where R is aryl, heteroaryl, (cyclo)alkyl, and vinyl.1,30 For 20 of these we explored the full pH scale (Figure 1B). While the impact of pH on the protodeboronation rate is, a priori, difficult to predict, the empirical data provide insight into the pH-controlled speciation of the boronic acid, catalysis by [H3O]+ and [OH]− ions, and the identification of auto/self-catalysis, vide infra. This allowed categorization of the boronic acids according to specific features of the R-group, such as electron-demand, basicity, number and position of heteroatoms, and ability to coordinate metal ions.1
2.2. Nonbasic (Hetero)aromatics
Fitting the kinetic model to the pH–log kobs profiles for simple aromatic and nonbasic heterocyclic systems required three general pathways. These proceed via a deprotonated boronate (i, Figure 1A), as suggested by Perrin;29 the boronate (ii); and the boronic acid (iii), the latter two pathways having been identified by Kuivila26 for substituted phenylboronic acids. However, for technical reasons, Kuivila’s studies were conducted at low boron concentrations and were limited to pH ≤ 6.7;26 in other words, under conditions very different from those commonly employed in Suzuki–Miyaura cross-couplings.13−20
Exploration of the basic pH region of the pH–log kobs profiles, Figure 1B, revealed some unexpected features.1,30 For example, 2,6-difluorophenylboronic acid (1) shows a simple rise in rate to reach a plateau at a pH above the pKa, consistent with water-mediated unimolecular decomposition of the boronate (see solid line for pathway ii in Figure 1A). In contrast, the 3-thienyl (2), p-anisyl (3), and derivatives reach a rate maximum when the boronic/boronate speciation is equal (pH = pKa).30 The extent of this deviation in behavior is dependent on the initial concentration of boronic acid.
Both features were indicative that the protodeboronation of [ArB(OH)3]− is catalyzed by ArB(OH)2 (Figure 1A, path iicat). However, an initially confusing aspect was that overall kinetics were still first-order. This was resolved by showing, experimentally and computationally,1 that the process is similarly catalyzed by endogenous B(OH)3, i.e., kcat ≈ k([B(OH)3] + [ArB(OH)2]). In other words, as the protodeboronation proceeds, one catalyst is replaced by the other and pseudo-first-order kinetics are observed. Thus, conducting Suzuki–Miyaura cross-couplings at pH values close to the pKa of the boronic acid can result in exacerbated protodeboronation.1 This is especially the case at high initial concentrations and another illustration of the benefits of slow-release methods which maintain a low steady-state concentration of the unstable boronate.17
2.3. Electron-Deficient Aromatics
The rapid base-mediated decomposition of 2,6-dihalogenated arylboronic acids was reported by Perrin.29 Their reactivity contrasted the acceleration by electron-donating para- and meta-substituents reported by Kuivila.26 Reassessment of the effect of aryl substituents, with a much-expanded set of 30 substrates, proved very revealing.30 Using Swain–Lupton parameters to weight field (F) and resonance (R), together with an empirical correction for ortho fluorine (σo-F = 1.24) gave a very asymmetric “V-shaped” plot (Figure 1C).30 The correlation is indicative of a change in mechanism from simple aryl rings (regime I) to very electron-deficient ones (regime II), with a significant accumulation of negative charge at the transition state in the latter.
As noted above, the pH–log kobs profile for the 2,6-difluorophenyl system (1) is indicative of exclusive reaction via pathway ii, where the boronate has a half-life of about 5 s. The analogous pentafluorophenyl boronate ([C6F5B(OH)3]−) was found to have a half-life of 2.6 ms. Analysis of 2H, 11B, and 13C KIEs suggested rate-limiting B–C cleavage in regime II, with aryl protonation occurring after this step. Detailed computational dissection of the water networks associated with boronate fragmentation rationalized the experimental KIEs, activation entropy (ΔS‡ = +6.2 cal/molK), and substituent effects (regime II, ρ = +3.4, Figure 1C). Reinvestigation of regime I suggested concerted protonation–deboronation,30 rather than the stepwise SEAr mechanism proposed by Kuivila.26
2.4. Basic Heterocycles
Nonlinear regression of pH–log kobs profiles for systems containing basic nitrogen-sites required additional pathways involving zwitterionic and cationic speciation (iv and v, Figure 1A).1 The studies showed the protodeboronation rates to be highly dependent on the relative positions of the boron and heteroatom substituents, sometimes in surprising ways (4 to 8, Figure 1B). For example, 5-pyrazolylboronic acid (6) exhibits a relatively simple profile (pathways ii and iv), whereas the regioisomer (7) has a much more nuanced one (pathways i, ii, iv, and v). DFT identified several key interactions that assist boronate departure in the protodeboronation transition states. For example, hydrogen bonding assists B(OH)3 departure for 2-pyridyl boronic acid (8), Figure 2, leading to the highest reactivity at neutral pH, where the species is zwitterionic (5H,OH, via pathway iv). This interaction is absent in the 3-pyridyl isomer (8), leading to much greater stability. In the 5-thiazolyl system (4), the σ*C–S orbitals assist B(OH)3 departure, and the addition of N-coordinating metal salts, e.g. ZnCl2, enhances this, leading to rate acceleration. The opposite effect is observed with 2-pyridyl boronic acid (6) where metal salts block the H-bonding.1
Figure 2.
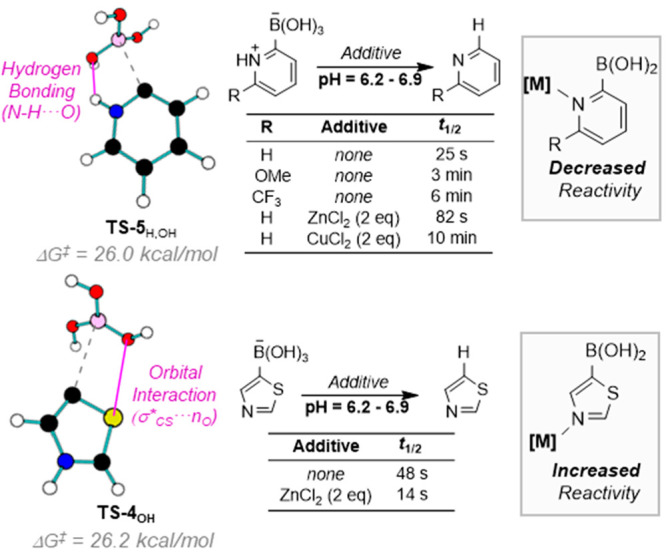
Key interactions in the protodeboronaton of 2-pyridinium (5H,OH) and 5-thiazolyl (4OH) boronates and the effects of metal-coordination. Data from ref (1).
2.5. Boronic Esters
Use of a boronic ester rather than the acid can provide increased shelf life, ease of manipulation/purification, and stability toward protodeboronation under basic cross-coupling conditions.13,14,16 Prime examples of this are the ubiquitous pinacol boronic esters, e.g., 9 (Figure 3). However, in a recent in situ19F NMR investigation we showed that this stabilization is not general, with some classes of ester undergoing substantially accelerated protodeboronation.
Figure 3.
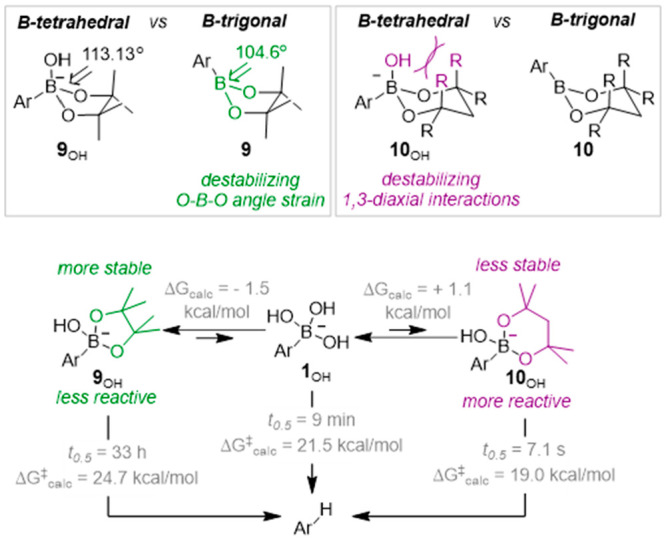
Contrasting effects of ring size on stability of esters (9/10) and their hydroxyboronate anions (9OH/10OH). Ar = 2,6-difluorophenyl. Data from ref (2).
DFT calculations showed that the acceleration arises when there is significant steric strain in the tetrahedral boronate that is generated on addition of the hydroxide ion to the trigonal boron center of the ester.2 This is typically found in esters generated from highly alkylated 1,3-propanediols, in other words, those that lead to 1,3-diaxial ring strain in the cyclic boronate (e.g., 10OH, Figure 3). These can undergo base-mediated protodeboronation 2 orders of magnitude more quickly than the corresponding boronic acid. Conversely, considerably less strain is present in the tetrahedral boronates generated from five-membered ring esters, e.g., 9OH,31,32 resulting in enhanced stability and genuine “protection”.2 The range of stability of the esters can be compared with boronic acids in Figure 4, where they are arranged in order of half-lives of the hydroxyboronate anions at 70 °C.
Figure 4.

Comparison of the protodeboronation rates of hydroxyboronate anions generated from the corresponding boronic acid or ester at pH 13, in 50% aq. dioxane at 70 °C; some structural features are highlighted in color to aid comparison. Vertical axes indicate approximate half-lives (log10-scale). Half-lives for the ester are for direct protodeboronation (kBE) only, see Figure 1D, and have been extrapolated by reference to their rate-ratio with the corresponding boronic acid at 21 °C. The half-lives at 21 °C can generally be estimated using t0.5 ≈ 10(2.15+1.13log{t}) where {t} is the half-life at 70 °C, in seconds. Data from refs (1), (2), and (30).
However, the situation is not as simple as the generalizations in Figure 3 suggest. A key issue is that the aqueous organic medium that induces direct protodeboronation (kBE, Figure 1D) also mediates ester hydrolysis,2,33 resulting in a competing indirect “prehydrolytic” route (khyd, kBA) via the trihydroxyboronate.2 This “leakage” has the effect of reducing the effective stabilization by the five-membered ring esters. For example, at high pH the pinacol ester-ate complex 9OH undergoes about 70% indirect protodeboronation, even though the trihydroxyboronate (1OH) does not significantly accumulate (≤1%, see inset in Figure 1D).2 Computed barriers for boronic ester protodeboronation indicated concerted fragmentation–protonation and direct fragmentation mechanisms, analogous to I and II, Figure 1C.2 Although the lowest-energy pathways correlated well with observed rates, with the more reactive examples, typically electron-deficient aromatics, proceeding via pathway II, the absolute barriers were anomalously low. This triggered our development of an improved computational protocol for the systematic placement of solvent molecules for specific solvation of the boronates.2
3. The Ruppert–Prakash Reagent, TMSCF3
TMSCF3 was introduced in 1984 by Ruppert as a de novo CF3-source,7 and its use in organic synthesis was pioneered soon after by Prakash.8 It is now a core reagent in the synthesis of fluorochemicals, available at scale, easy to handle, relatively cheap, and the starting material for many other CF3-transfer reagents.9,10 Recent advances by Prakash and Hu have greatly expanded the application of TMSCF3 as a versatile CF2-source,34−36e.g., for generation of difluorocyclopropa(e)nes,34,37 TFE,35 perfluoroalkylmetallics,36 and other difluoromethylenes.38 In all applications, the reagent is used in excess, typically 2–5 equiv.
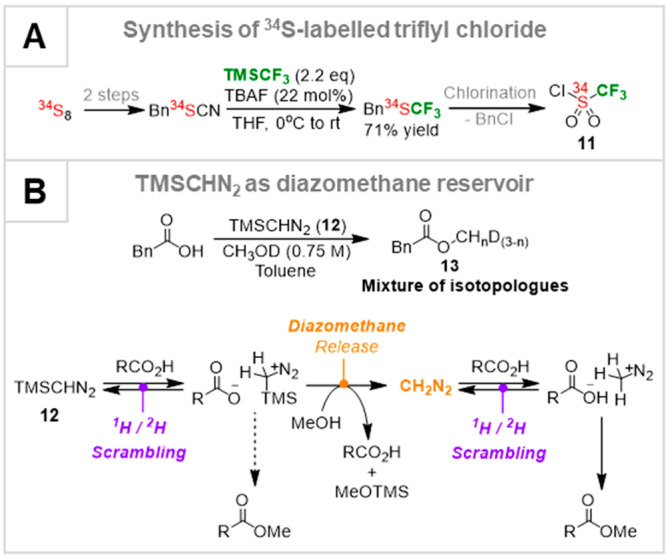
In 2008 we needed to prepare 34S-triflyl chloride (11), for a mechanistic study of the anionic thia-Fries rearrangement.39 After considerable exploration of other methods, we developed a route from 34S8, in a sequence involving delivery of CF3 from TMSCF3, Scheme 2A.39,40 At about the same time we required various 2H-labeled methyl esters for a study of homoallylcyclopropanation.41 Given the accepted mechanism for Aoyama–Shiori methylesterification with TMS-diazomethane (12, Scheme 2B),42 replacing MeOH by MeOD should have given monodeutero esters. Instead, we obtained all four isotopologues, RCO2CHnD(3–n), (13). After detailed investigation, we elucidated that CH2N2 is generated transiently in situ,43 which is another example of a benefit of “slow-release”. These investigations led us to develop an interest in the role of anions as initiators for nucleophilic transfer of organic fragments from organosilanes to electrophiles, including from Ar-TMS to Au,44 and eventually to detailed studies of TMSCF3.3,4,45
Scheme 2. (A) TMSCF3 in the Synthesis of 34S-11; (B) Mechanism of Methyl-Esterification by TMS-diazomethane (12).
3.1. Siliconates and the Trifluoromethanide Anion
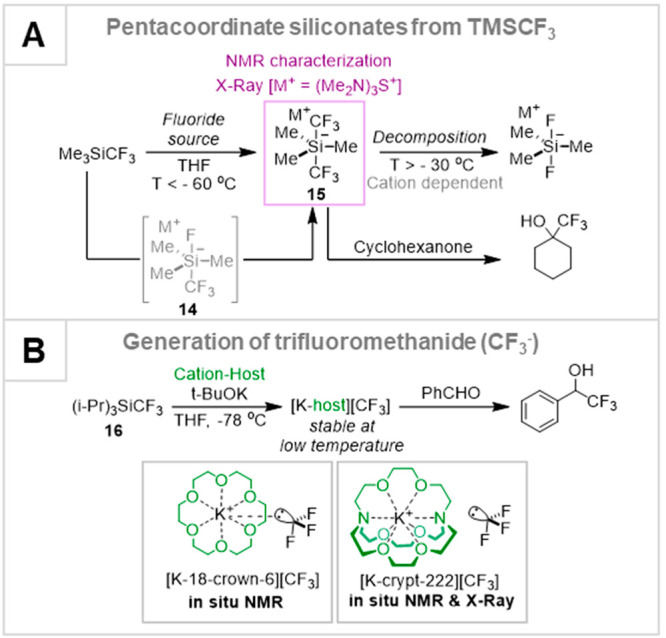
The stoichiometric reaction of TMSCF3 with nucleophilic anions had already been studied experimentally, and in considerable detail.46−50 The two principal findings are summarized in Scheme 3. In 1999, Naumann46 and Kolomeitsev and Röschenthaler47 independently showed that addition of a silaphilic anion to TMSCF3 generates pentacoordinate siliconates 14 and 15 that rapidly decomposed above −30 °C. Most, but not all, interpretations of anion-mediated reactions of TMSCF3 invoke direct transfer of CF3 from a siliconate (e.g., 14 or 15) to the electrophile, Scheme 3A.10 About 15 years later, Prakash48 and Grushin49 independently showed that trifluoromethanide ([CF3]−) could be generated at low temperatures from bulky silane 16, by using t-BuOK with a crown ether48 or a cryptand.49 The free carbanion ([CF3]−) was even characterized by cryogenic single-crystal X-ray diffraction.49c In all cases, addition of electrophiles such as ketones and aldehydes to the reaction mixtures at low temperature generated the corresponding CF3-addition products.46−48,49a
Scheme 3. (A) Anion Addition to TMSCF3 to Generate Thermally Labile Siliconates 14 and 15; (B) Generation of Trifluoromethanide ([CF3]−[ML]+) from Bulky Silane 16.
These prior analyses provided us with a framework to interpret the kinetics and mechanism of CF3 transfer, and later also CF2, from TMSCF3 after initiation with substoichiometric anion at ambient temperature. We focused on the addition of CF3 to p-F-acetophenone (17),3 Kondo silylation of 1,3-difluorobenzene (18),4,51 and the difluorocyclopropanation of p-F-α-methylstryene (19)4 (Figure 5). Intriguingly, all three processes can be conducted using the same anhydrous fluoride-based initiator (“TBAT”; 20)52 in THF at ambient temperature. This feature allowed us to interrogate how the substrate, the only variable, affects the behavior of the system. After careful adjustment of concentrations, and use of high-purity TMSCF3,3 all three reactions (Figure 5i–iii) were amenable to detailed in situ analysis by 19F NMR spectroscopy.3,4,12,45
Figure 5.
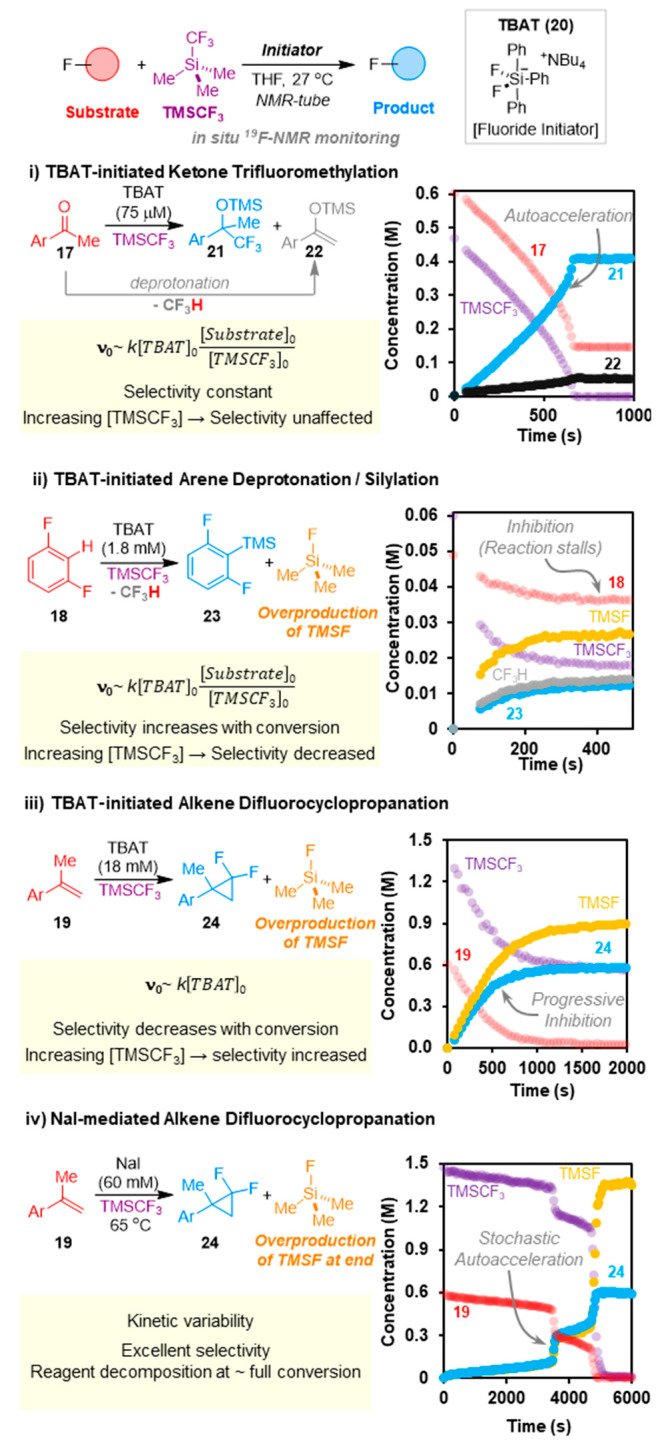
Examples of in situ19F NMR reaction profiles for the reactions of TMSCF3 with ketone (17), arene (18), and alkene (19), together with factors affecting selectivity and the initial rate (v0) of TMSCF3-consumption. Data from refs (3), (4), and (45).
3.2. Fluoride-Initiated CF3 Transfer
Differences in behavior between the three reaction classes, in terms of both initial rates (ν0) and selectivities, are evident in Figure 5i–iii. The addition of CF3 to ketone 17 proceeds with autoacceleration when [17]0 > [TMSCF3]0, as in the example shown in Figure 4i, where the product (21) curve has a rising gradient. Conversely, when [17]0 < [TMSCF3]0, the reactions become progressively slower. The only major side reaction involves the O-silylation (22) of enolizable ketones. This cogenerates CF3H and proceeds throughout the reaction in a constant proportion relative to the addition.3
The Kondo silylation51 of arene 18 displays kinetics analogous to the ketone, but in the example shown in Figure 4ii, [18]0 < [TMSCF3]0 and the reaction becomes progressively slower, eventually stalling.45 Moreover, a major side reaction, not involving arene 18, converts TMSCF3 into TMSF and a range of perfluoroalkenes, vide infra. The kinetics of the difluorocyclopropanation of 19 (Figure 5iii) are very distinct from the other two cases, with the initial rate of TMSF generation independent of both [19]0 and [TMSCF3]0.4 The major side reaction is the overproduction of TMSF. This is also found for the NaI-mediated process (Figure 5iv)—but only in the final phases of reaction.4
In all three of the TBAT-initiated reactions (Figure 5i-iii), the in situ19F NMR signal of the TMSCF3 at ambient temperature exhibits dynamic line-broadening.3,4,45 At lower temperatures, the siliconate 15 is detected, and variable temperature line-shape analysis (Figure 5A) allowed extraction of the rate of CF3-decomplexation (kex; ΔH‡ = 20 kcal/mol; ΔS‡ = 23 cal/mol K). Although DFT calculations indicate the equilibrium very strongly favors 15 over free [CF3]−, the rapidly reversible decomplexation (kex; ΔG‡ = 13.1 kcal/mol at 27 °C) leads to fast exchange of CF3-groups between TMSCF3 and 15 and dynamic line-broadening in both. In terms of the productive reactions (Figure 5i,ii), siliconate 15 is either a passive anionic reservoir (scenario I, Figure 6A) or directly transfers the CF3 to the substrate (scenario II).10 Analysis of the kinetics allowed dissection of this dichotomy: only in scenario I can the TMSCF3 inhibit the rate of CF3-transfer to the substrate. It does this by sequestering (Kc) the [CF3]−, thus attenuating the rate of the anionic chain reaction. DFT-calculations strongly supported these findings by showing that direct anionic CF3-transfer from the siliconate 15 (scenario II) to any electrophile or acid involves a prohibitive (>50 kcal/mol) umbrella-like CF3-inversion. Instead, transfers must proceed via predissociation of the [CF3]−.3 Bulkier R3SiCF3 reagents have a lower affinity (Kc) for [CF3]− and lead to more efficient CF3-transfer (R = Et) or to a change in rate-limiting step (R = iPr).3
Figure 6.

(A) Overarching mechanism highlighting the kinetics and mechanism of CF3 and CF2 transfer from TMSCF3, after initiation by substoichiometric TBAT (20). The ticks and crosses indicate which mechanistic scenarios (I, II, III, IV) are consistent with the experimentally determined kinetics. S = substrate, e.g.17 or 18; R-SiMe3 = product, e.g.21 or 23. (B) Pathways for TMSCF3 decomposition leading to anion-sequestration and inhibition. Data from ref (4).
3.3. Fluoride-Initiated CF2 Transfer
A variety of tests, including heavy-atom KIEs,53 relative reactivity of E/Z-alkenes, and linear free-energy relationships, confirmed that the difluorocyclopropanation reactions proceed via concerted capture of singlet difluorocarbene, CF2 (19 → 24, Figure 5iii,iv).4 The consensus in the literature was that the CF2 is generated via spontaneous α-fluoride elimination from [CF3]− (scenario III, Figure 6A).54 However, the kinetics under fluoride initiation were not at all consistent with this, because TMSCF3 should inhibit the reaction (Kc), via generation of siliconate 15 (Figure 6A, scenario III). We thus computationally explored the direct extrusion of CF2 from siliconate 15; however, all attempts to locate a transition state for this diverted to an intermolecular fluoride transfer from C to Si, scenario IV. Detailed stopped-flow 19F NMR spectroscopic analyses of the difluorocyclopropanation of 19 between 2 and 18 °C gave activation parameters and kinetics fully consistent with scenario IV,4 with fluoride-abstraction (kF) occurring approximately once in every 105 reassociations (Kc) of [CF3]− with TMSCF3.
3.4. Perfluoroalkenes and Inhibition of the Chain Reaction
During in situ19F NMR spectroscopic analysis of the Kondo silylation of 18 and the difluorocyclopropanation of 19 (Figure 5), numerous low-intensity complex multiplets appear in the 19F NMR spectra.4,45
This phenomenon was kinetically linked with progressive inhibition. Both reactions proceed by anionic chain reactions (Figure 6A, scenarios I and IV), and thus, inhibition involves diversion of the active chain-carrier(s) into inert, i.e., nonsilaphilic, anions. The major component of these was identified as the known perfluoroalkyl anion [C11F23]− (25),50 albeit with a structure revised on the basis of 19F–19F NOESY and nJFF values.45
Computational investigation of the thermodynamics of sequential CF3-addition, 1,2-fluorine shifts, and fluoride-elimination55 allowed us to understand why a CnF2n/[CnF2n+1]− cascade leads to, and ceases at, C11, i.e., 25 (Figure 6B). Each alkene homologation step is favorable (ΔGav = −29.2 kcal/mol) until [CF3]− adds to C10F20 (26) to generate anion 25. At this point, fluoride-elimination becomes disfavored (ΔG = +10.0 kcal/mol) because of the steric strain in the resulting alkene, C11F22 (27).4 In other words, anion 25 acts as a thermodynamic “sink”, trapping [CF3]− and F– and terminating the desired anionic chain reactions.4,45
3.5. Productive Fractionation, f
TMSF evolution acts as reporter for the net loss of CF2 from TMSCF3. This can be used to quantify the extent of side-reactions versus product in the form of a productive fractionation parameter: f = d[Product]/d[TMSF].4,45 Graphical analysis of f, Figure 7A, allows assessment of how the changes in concentration of the various reaction components, within or between reactions, affects the efficiency. These analyses proved fruitful: by deliberately keeping a low concentration of the “problematic” component, the productive fractionations can be enhanced. For example, the Kondo silylation could be improved to near-quantitative conversion of 18 to 23 by slow addition45,56 of TMSCF3, Figure 7B.
Figure 7.
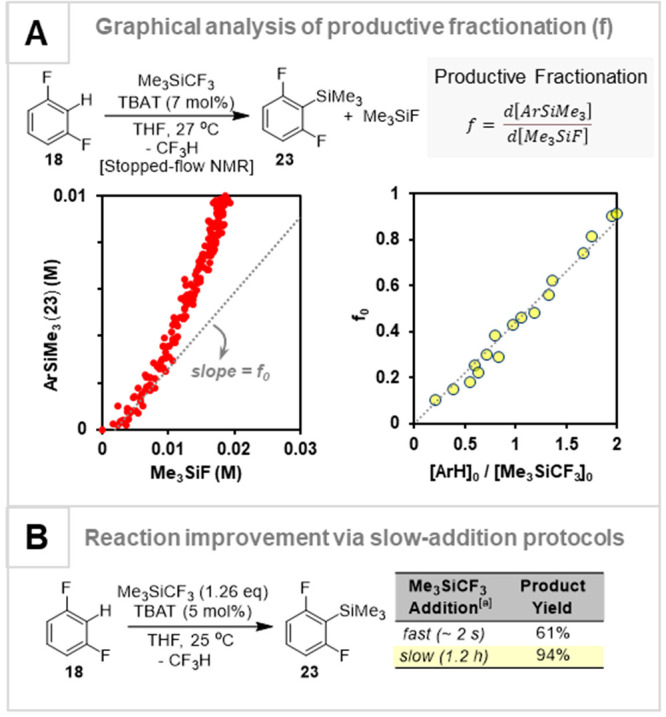
(A) Analysis of the productive fractionation, f, of TMSCF3 into desired (Ar-TMS, 23) and undesired (TMSF) products during silylation of 18. The fractionation increases as the reactions proceed and as the initial ratio [18]0/[TMSCF3]0 is raised. (B) Insight from the changes in f, informing the slow addition of TMSCF3. Data from ref (45).
3.6. NaI-Mediated CF2 Transfer
The most effective synthetic method for alkene difluorocyclopropanation with TMSCF3 employs NaI, a process pioneered by Prakash and Hu.34 The conditions afford substantially enhanced substrate scope, including alkynes, and are also effective for in situ generation of tetrafluoroethylene.35,36 Grygorenko56 has shown that slow addition of TMSCF3 and NaI allows efficient difluorocyclopropanation of alkenes which are considered “unactivated” toward CF2 cycloaddition (Scheme 4), considerably expanding the scope of application.37
Scheme 4. Counterintuitive Results with Alkenes That Are Deactivated Toward CF2-Cycloaddition.
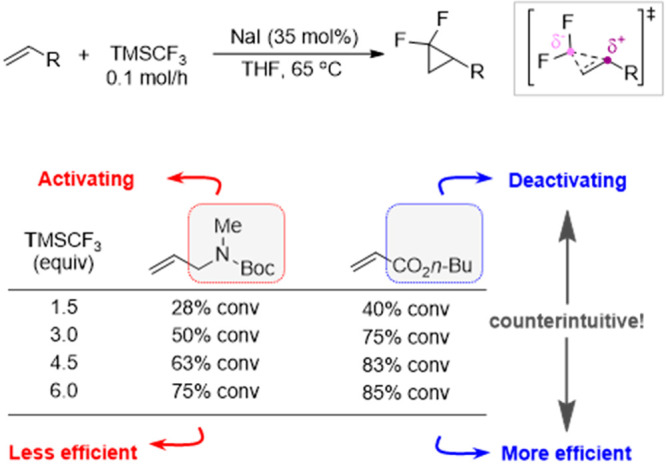
Data from ref (56).
Mechanistically, the NaI-mediated reactions of TMSCF3 proved highly vexing, with many initially counterintuitive features. The reactions are characterized by transient and apparently stochastic autoaccelerations;4 indeed, this exothermic process can proceed violently upon scale-up.37a,56 During this quasi-stochastic phase (see Figure 5iv), the productive fractionations are excellent (f0 > 0.99) with very little overproduction of TMSF, irrespective of the concentrations of any of the reaction components. However, at some point, and in an unpredictable manner, the rate of TMSF generation surges and f drops precipitously. Alkene 19 undergoes quantitative difluorocyclopropanation, and the majority of the excess TMSCF3 is converted into a broad range of fluorocarbons, vide infra.
This distinctive reactivity requires the presence of both the sodium and the iodide, and despite much effort, the primary initiation of these processes remains unclear.61 The NaI concentration does not affect the initiation rate, and DFT calculations indicate prohibitively high energies for all direct reaction pathways with TMSCF3. Overall we concluded that initiation must be “effected by traces of unidentified silaphilic species generated in situ from the NaI, by oxidation, reaction with decomposition products of the TMSCF3, or coreaction with the Lewis basic THF solvent, or by species already present in the NaI from the supplier.”4 What was clear is that suitably reactive carbonyls, e.g., p-F-benzaldehyde (28), undergo addition of both [CF3]− and, in trace quantities, [CF2I]−.57 However, there were none of the characteristic signals3,4,45−47 for siliconate equilibria (Kc) evident in the in situ19F NMR analyses, even at low temperatures.
The timing and magnitude of the transient autoaccelerations varied greatly from run to run making standard time-based kinetic analyses very unreproducible. We thus tackled the problem by competition experiments, using fractional conversion of substrates as a time-independent parameter to characterize the various processes involved. For example, coreaction of alkene 19 with aldehyde 28 provided 24 and 29 as an indirect measurement of the CF2 and [CF3]− present in the reaction, in the form of a first-order partitioning factor (kCF2/kCF3). The linear relationship between kCF2/kCF3 and [NaI]0, Figure 8A, indicated that CF2-generation from [CF3] – involves NaI, and DFT studies suggested an assisted α-fluoride elimination and stabilization58 of the nascent NaF (see “primary chain” in Figure 8B).
Figure 8.

(A) Partitioning analysis by competition of alkene 19 with aldehyde 28 during NaI-mediated difluorocyclopropanation. (B) Chain reaction identified for CF2-generation from TMSCF3. (C) NaI-mediated decomposition of TMSCF3 in the absence of alkene 19, and identification of species arising from chain-branching autoacceleration. Data from ref (4).
Structures that see association of ions in solution, and that can undergo dynamic exchange of partners, present another challenge for computation. We informally described our initial attempts to understand the counterion effects on the speciation in Figure 8A,B as “molecular paintball”: exotically colored spheres representing the cations were placed around the relevant anions, relying on intuition. The protocol that was first applied for solvation of the boronate species2 (vide supra) is now being extended to placement of counterions, this being driven by a need to be able to approach such situations more logically in the future.
Identifying the origins of the autoacceleration was challenging, not least because the onset and duration is very nonpredictable.4,58 The surge in consumption of the TMSCF3 when the alkene is depleted suggested that CF2-accumulation triggers the autoacceleration. Consistent with this, reactions conducted without alkene 19 present enter autoacceleration after a short but variable induction period.4 Detailed 19F NMR spectroscopic analysis of the temporal evolution at high NaI concentrations proved informative (Figure 8C). At first, TFE is generated, followed by an intermediate tentatively identified as TMSCF2CF2CF2I (30)4 and then perfluorocyclopropane (31), with a clear correlation: the temporal concentration of [30] mirrors the rate of consumption of the TMSCF3. Taken together, the observations suggested that transient carbanionoid [Na][(CF2)3I] (32) induces “chain-branching” (Figure 8B), a classic origin of rapidly accelerating reactions.
The requirement for TFE and CF2 accumulation to indirectly induce chain-branching explains several initially confusing or counterintuitive results. For example, alkenes that are activated toward CF2 dampen the autoacceleration, and thus, reactions employing less activated alkenes and alkynes can lead to faster overall difluorocyclopropanation, Scheme 4.4,56 Also, in contrast to fluoride initiation, most alkynes undergo selective reaction under the NaI conditions, without competing double-addition of CF2. This is because the TFE that accumulates in the autoacceleration phase has a low barrier to CF2 cycloaddition (ΔG338‡ = 12 kcal mol–1), allowing bypass of excess CF2 into c-C3F6 (31) and other perfluorocarbons, Figure 8B, rather than consuming the desired difluorocyclopropene product. The mechanistic features also provide an explanation for the greatly improved efficiency under Grygorenko’s conditions.56 Slow addition of the TMSCF3/NaI gives time for the endogenous TFE to dissipate or decay, delaying or attenuating intense autoacceleration and maintaining a high productive fractionation, f.
4. Conclusion
This Account has summarized some of our mechanistic work on popular classes of organoboron1,2,24,25,30 and organosilicon3,4,45 reagents. A recurring theme to the investigations has been the use of kinetics, NMR, isotope-effects, and partitioning analysis, i.e., measuring the selectivity of a process as a function of conversion or reactant concentration. Partitioning analysis has the benefit of removing the time-dependency component for reactions that cannot easily be controlled or that proceed with unpredictable rates.
All of the studies benefitted from a deeply collaborative combination of experiment and computation. Crucially, this was initiated at the very beginning of each investigation and in two separate research groups. This facilitated the development, testing, and revision or elimination of a large variety of hypotheses. Indeed, were it not for this two-centered collaborative arrangement that enforced discussion, reflection, more-rigorous logic, and the tensioning of experiment and theory, we would not have elucidated many of the features outlined above. The collaborative process also highlighted gaps in both experimental and computational methodologies that we then sought to fill.2,3,11
Both areas of investigation were initiated after making unexpected observations in unrelated projects. Indeed, the work has been almost entirely curiosity-driven without predefined goals. It has nonetheless yielded insights that are of considerable practical utility, an outcome inconceivable to some research funding administrators. For example, investigation of the mechanism of hydrolysis of trifluoroborate salts led us to develop nonetching conditions for their synthesis,24c a process that has now been optimized and applied industrially at >10 kg scale.59 Insights into the hydrolytic processes involved in C–O and B–N cleavage in MIDA boronates25 aided Burke in the design of a new class of hydrolysis-resistant TIDA boronates,60 widely expanding the scope of application, and also led us to develop new parameters for nucleofugality at boron.61 By study of over 70 different boronic acids and esters we have shown that their protodeboronation can be defined by six different pathways, modulated by pH speciation; concentration (self/autocatalysis); and their (hetero)aromatic, alkyl, or vinyl structure.1,2,30 The protodeboronation half-lives at high pH span nearly 10 orders of magnitude, Figure 4. Analogously, the anionic chain reactions that lead to decomposition of TMSCF3 have been identified and kinetically delineated.3,4,45 This allows the mechanism-informed design of conditions for maximizing the productive fractionation of the TMSCF3 into the desired CF3- or CF2-derived product and for the safer scale-up of these processes.37,56
Acknowledgments
We are very grateful to members of the Leach and Lloyd-Jones research groups, past and present, and to our collaborators Andrew Campbell and Gary Noonan at AstraZeneca. The authors acknowledge the assistance given by Research IT and the use of the Computational Shared Facility at The University of Manchester. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission.
Biographies
Andrés García-Domínguez was born in Spain and obtained his Ph.D. under the supervision of Cristina Nevado (Zurich) developing novel transition-metal-catalyzed carbofunctionalizations of multiple bonds. In 2018 he secured an SNSF postdoctoral fellowship to work with Guy Lloyd-Jones at Edinburgh in the kinetic study of reactions involving silicon-based reagents. His interests are in the design and understanding of the reactivity of main-group organometallic reagents.
Andrew G. Leach is a Cestrian and obtained his Ph.D. under the supervision of Steven Ley FRS (Cambridge). After a Fulbright scholarship in the group of Kendal Houk (UCLA), where he investigated pericyclic mechanisms and enzyme catalysis, he joined AstraZeneca in 2003 as a computational chemist. He is a coinventor of capivasertib, currently in Phase III clinical trials. In 2012, he joined Liverpool John Moores University, and in 2019, he moved to the University of Manchester. He applies computational and informatic approaches to reaction mechanisms and develops tools for molecular design.
Guy C. Lloyd-Jones was born in the U.K. and obtained his D.Phil. under the supervision of John Brown FRS (Oxford). After a Royal Society scholarship in the group of Andreas Pfaltz (Basel), he began his independent career at Bristol in 1996 and moved to the Forbes Chair of Organic Chemistry at Edinburgh in 2013. His research interests are in the kinetics and mechanisms of reactions and process and the development of physical-organic methods to enable this.
A.G.D. thanks the SNSF for a postdoctoral fellowship (P2ZHP2_181497). The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement nos. 340163 and 838616.
The authors declare no competing financial interest.
References
- Cox P. A.; Leach A. G.; Campbell A. D.; Lloyd-Jones G. C. Protodeboronation of Heteroaromatic, Vinyl, and Cyclopropyl Boronic Acids: pH-Rate Profiles, Autocatalysis, and Disproportionation. J. Am. Chem. Soc. 2016, 138, 9145–9157. 10.1021/jacs.6b03283. [DOI] [PubMed] [Google Scholar]
- Hayes H. L. D.; Wei R.; Assante M.; Geogheghan K. J.; Jin N.; Tomasi S.; Noonan G.; Leach A. G.; Lloyd-Jones G. C. Protodeboronation of (Hetero)Arylboronic Esters: Direct versus Prehydrolytic Pathways and Self-/Auto-Catalysis. J. Am. Chem. Soc. 2021, 143, 14814–14826. 10.1021/jacs.1c06863. [DOI] [PubMed] [Google Scholar]
- Johnston C. P.; West T. H.; Dooley R. E.; Reid M.; Jones A. B.; King E. J.; Leach A. G.; Lloyd-Jones G. C. Anion-Initiated Trifluoromethylation by TMSCF3: Deconvolution of the Siliconate-Carbanion Dichotomy by Stopped-Flow NMR/IR. J. Am. Chem. Soc. 2018, 140, 11112–11124. 10.1021/jacs.8b06777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Domínguez A.; West T. H.; Primozic J. J.; Grant K. M.; Johnston C. P.; Cumming G. G.; Leach A. G.; Lloyd-Jones G. C. Difluorocarbene Generation from TMSCF3: Kinetics and Mechanism of NaI-Mediated and Si-Induced Anionic Chain Reactions. J. Am. Chem. Soc. 2020, 142, 14649–14663. 10.1021/jacs.0c06751. [DOI] [PubMed] [Google Scholar]
- See for example; Crawley M. L.; Trost B. M.. Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective; J. Wiley and Sons: NJ, 2012; pp 1–356. 10.1002/9781118309872. [DOI] [Google Scholar]
- de Jesus Hiller N.; do Amaral e Silva N. A.; Tavares T. A.; Faria R. X.; Eberlin M. N.; de Luna Martins D. Arylboronic Acids and their Myriad of Applications Beyond Organic Synthesis. Eur. J. Org. Chem. 2020, 2020, 4841–4877. 10.1002/ejoc.202000396. [DOI] [Google Scholar]
- Ruppert I.; Schlich K.; Volbach W. Die Ersten CF3-Substituierten Organyl(Chlor)Silane. Tetrahedron Lett. 1984, 25, 2195–2198. 10.1016/S0040-4039(01)80208-2. [DOI] [Google Scholar]
- Prakash G. K. S.; Krishnamurti R.; Olah G. A. Synthetic methods and reactions. 141. Fluoride-induced trifluoromethylation of carbonyl compounds with trifluoromethyltrimethylsilane (TMS-CF3). A trifluoromethide equivalent. J. Am. Chem. Soc. 1989, 111, 393–395. 10.1021/ja00183a073. [DOI] [Google Scholar]
- Beier P.; Zibinsky M.; Prakash S. G. K. Nucleophilic Additions of Perfluoroalkyl Groups. Organic Reactions 2016, 91, 1–492. 10.1002/0471264180.or091.01. [DOI] [Google Scholar]
- Liu X.; Xu C.; Wang M.; Liu Q. Trifluoromethyltrimethylsilane: Nucleophilic Trifluoromethylation and Beyond. Chem. Rev. 2015, 115, 683–730. and references therein 10.1021/cr400473a. [DOI] [PubMed] [Google Scholar]
- Wei R.; Hall A. M. R.; Behrens R.; Pritchard M. S.; King E. J.; Lloyd-Jones G. C. Stopped-Flow 19F NMR Spectroscopic Analysis of a Protodeboronation Proceeding at the Sub-Second Time-Scale. Eur. J. Org. Chem. 2021, 2021 (17), 2331–2342. 10.1002/ejoc.202100290. [DOI] [Google Scholar]
- Ben-Tal Y.; Boaler P. J.; Dale H. J. A; Dooley R. E.; Fohn N. A.; Gao Y.; García-Domínguez A.; Grant K. M.; Hall A. M. R.; Hayes H. D. L.; Kucharski M. M.; Wei R.; Lloyd-Jones G. C. Mechanistic Analysis by NMR Spectroscopy: a Users Guide. Prog. Nucl. Magn. Reson. Spectrosc. 2022, 129, 28–106. 10.1016/j.pnmrs.2022.01.001. [DOI] [PubMed] [Google Scholar]
- Pagett A. B.; Lloyd-Jones G. C. Suzuki-Miyaura Cross-Coupling. Org. React. 2019, 100, 547–620. 10.1002/0471264180.or100.09. [DOI] [Google Scholar]
- See for example; Thomas A. A.; Zahrt A. F.; Delaney C. P.; Denmark S. E. Elucidating the Role of the Boronic Esters in the Suzuki-Miyaura Reaction: Structural, Kinetic, and Computational Investigations. J. Am. Chem. Soc. 2018, 140, 4401–4416. and references therein 10.1021/jacs.8b00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennox A. J. J.; Lloyd-Jones G. C. Transmetallation in Suzuki-Miyaura Coupling: The Fork in the Trail. Angew. Chem., Int. Ed. 2013, 52, 7362–7370. and references therein 10.1002/anie.201301737. [DOI] [PubMed] [Google Scholar]
- Lennox A. J.; Lloyd-Jones G. C. Selection of boron reagents for Suzuki-Miyaura coupling. Chem. Soc. Rev. 2014, 43, 412–43. 10.1039/C3CS60197H. [DOI] [PubMed] [Google Scholar]
- a Lennox A. J. J.; Lloyd-Jones G. C. The Slow-Release Strategy in Suzuki-Miyaura Coupling. Isr. J. Chem. 2010, 50, 664–674. 10.1002/ijch.201000074. [DOI] [Google Scholar]; b Noguchi H.; Hojo K.; Suginome M. Boron-Masking Strategy for the Selective Synthesis of Oligoarenes via Iterative Suzuki–Miyaura Coupling. J. Am. Chem. Soc. 2007, 129, 758–759. 10.1021/ja067975p. [DOI] [PubMed] [Google Scholar]; c Knapp D. M.; Gillis E. P.; Burke M. D. A General Solution for Unstable Boronic Acids: Slow-Release Cross-Coupling from Air-Stable MIDA Boronates. J. Am. Chem. Soc. 2009, 131, 6961–6963. 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassel V. M.; Hanneman C. M.; Delaney C. P.; Denmark S. E. Heteroaryl-Heteroaryl, Suzuki-Miyaura, Anhydrous Cross-Coupling Reactions Enabled by Trimethylborate. J. Am. Chem. Soc. 2021, 143, 13845–13853. 10.1021/jacs.1c06419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Francis H.; Carrow B. P. An “On-Cycle” Precatalyst Enables Room-Temperature Polyfluoroarylation Using Sensitive Boronic Acids. ACS Catal. 2018, 8, 2989–2994. 10.1021/acscatal.8b00341. [DOI] [Google Scholar]
- Kinzel T.; Zhang Y.; Buchwald S. L. A New Palladium Precatalyst Allows for the Fast Suzuki-Miyaura Coupling. J. Am. Chem. Soc. 2010, 132, 14073–14075. 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jover J.; Fey N.; Harvey J. N.; Lloyd-Jones G. C.; Orpen A. G.; Owen-Smith G. J. J.; Murray P.; Hose D. R. J.; Osborne R.; Purdie M. Expansion of the Ligand Knowledge Base for Chelating P,P-Donor Ligands (LKB-PP). Organometallics 2012, 31, 5302–5306. 10.1021/om300312t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darses S.; Genet J.-P. Potassium Organotrifluoroborates: New Perspectives in Organic Synthesis. Chem. Rev. 2008, 108, 288–325. 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]
- For an overview seeMolander G. A. Organotrifluoroborates: Another Branch of the Mighty Oak. J. Org. Chem. 2015, 80, 7837–7848. 10.1021/acs.joc.5b00981. [DOI] [PubMed] [Google Scholar]
- a Butters M.; Harvey J. N.; Jover J.; Lennox A. J. J.; Lloyd-Jones G. C.; Murray P. M. Angew. Chem., Int. Ed. 2010, 49, 5156–5160. 10.1002/anie.201001522. [DOI] [PubMed] [Google Scholar]; b Lennox A. J. J.; Lloyd-Jones G. C. Organotrifluoroborate Hydrolysis: Boronic Acid Release Mechanism and an Acid-Base Paradox in Cross-Coupling. J. Am. Chem. Soc. 2012, 134, 7431–7441. 10.1021/ja300236k. [DOI] [PubMed] [Google Scholar]; c Lennox A. J. J.; Lloyd-Jones G. C. Preparation of Organotrifluoroborate Salts: Precipitation-driven Equilibrium under Non-Etching Conditions. Angew. Chem., Int. Ed. 2012, 51, 9385–9388. 10.1002/anie.201203930. [DOI] [PubMed] [Google Scholar]
- Gonzalez J. A.; Ogba O. M.; Morehouse G. F.; Rosson N.; Houk K. N.; Leach A. G.; Cheong P. H. Y.; Burke M. D.; Lloyd-Jones G. C. MIDA Boronates Are Hydrolysed Fast and Slow by Two Different Mechanisms. Nat. Chem. 2016, 8, 1067–1075. 10.1038/nchem.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kuivila H. G.; Nahabedian K. V. Electrophilic Displacement Reactions, X.General Acid Catalysis in the Protodeboronation of Areneboronic Acids. J. Am. Chem. Soc. 1961, 83, 2159–2163. 10.1021/ja01470a028. [DOI] [Google Scholar]; b Kuivila H. G.; Reuwer J. F.; Mangravite J. A. Electrophilic Displacement Reactions XV. Kinetics and Mechanism of the Base-Catalysed Protodeboronation of Areneboronic Acids. Can. J. Chem. 1963, 41, 3081–3090. and references therein 10.1139/v63-451. [DOI] [Google Scholar]
- Frohn H.-J.; Adonin N. Y.; Bardin V. V.; Starichenko V. F. Base-catalysed Hydrodeboration of Polyfluorophenyl(dihydroxy)boranes. Z. Anorg. Allg. Chem. 2002, 628, 2834–2838. . [DOI] [Google Scholar]
- Cammidge A. N.; Crépy K. V. L. Application of the Suzuki Reaction as the Key Step in the Synthesis of a Novel Atropisomeric Biphenyl Derivative for Use as a Liquid Crystal Dopant. J. Org. Chem. 2003, 68, 6832–6835. 10.1021/jo034652s. [DOI] [PubMed] [Google Scholar]
- Lozada J.; Liu Z.; Perrin D. M. Base-promoted protodeboronation of 2,6-disubstituted arylboronic acids. J. Org. Chem. 2014, 79, 5365–5368. 10.1021/jo500734z. [DOI] [PubMed] [Google Scholar]
- Cox P. A.; Reid M.; Leach A. G.; Campbell A. D.; King E. J.; Lloyd-Jones G. C. Base-Catalyzed Aryl-B(OH)2 Protodeboronation Revisited: From Concerted Proton Transfer to Liberation of a Transient Aryl Anion. J. Am. Chem. Soc. 2017, 139, 13156–13165. 10.1021/jacs.7b07444. [DOI] [PubMed] [Google Scholar]
- This is the opposite trend to the neutral boron esters.Bowie R. A.; Musgrave O. C. Organoboron Compounds. Part V. The Hydrolysis of Cyclic Phenylboronates. J. Chem. Soc. 1963, 3945–3949. 10.1039/jr9630003945. [DOI] [Google Scholar]
- Fasano V.; McFord A. W.; Butts C. P.; Collins B. S. L.; Fey N.; Alder R. W.; Aggarwal V. K. How Big is the Pinacol Boronic Ester as a Substituent?. Angew. Chem., Int. Ed. 2020, 59, 22403–22407. 10.1002/anie.202007776. [DOI] [PubMed] [Google Scholar]
- Pizer R. Boron acid complexation reactions with polyols and α-hydroxy carboxylic acids: Equilibria, reaction mechanisms, saccharide recognition. Inorg. Chim. Acta 2017, 467, 194–197. 10.1016/j.ica.2017.08.003. [DOI] [Google Scholar]
- Wang F.; Luo T.; Hu J.; Wang Y.; Krishnan H. S.; Jog P. V.; Ganesh S. K.; Prakash G. K. S.; Olah G. A. Synthesis of gem- Difluorinated Cyclopropanes and Cyclopropenes; Trifluoromethyl- trimethylsilane as a Difluorocarbene Source. Angew. Chem., Int. Ed. 2011, 50, 7153–7157. 10.1002/anie.201101691. [DOI] [PubMed] [Google Scholar]
- Li L.; Ni C.; Xie Q.; Hu M.; Wang F.; Hu J. TMSCF3 as a Convenient Source of CF2 = CF2 for Pentafluoroethylation, (Aryloxy)Tetrafluoroethylation, and Tetra-fluoroethylation. Angew. Chem., Int. Ed. 2017, 56, 9971–9975. 10.1002/anie.201705734. [DOI] [PubMed] [Google Scholar]
- Xie Q.; Li L.; Zhu Z.; Zhang R.; Ni C.; Hu J. From C1 to C2: TMSCF3 as a Precursor for Pentafluoroethylation. Angew. Chem., Int. Ed. 2018, 57, 13211–13215. 10.1002/anie.201807873. [DOI] [PubMed] [Google Scholar]
- a Recent examples (2020 to date) of difluorocyclopropanation using TMSCF3:Hryshchuk O. V.; Varenyk A. O.; Yurov Y.; Kuchkovska Y.; Tymtsunik A. V.; Grygorenko O. O. Gem-Difluorocyclopropanation of Alkenyl Trifluoroborates with the CF3SiMe3-NaI System. Eur. J. Org. Chem. 2020, 2020, 2217–2224. 10.1002/ejoc.202000346. [DOI] [Google Scholar]; b Herasymchuk M.; Melnykov K. P.; Yarmoliuk D. V.; Serhiichuk D.; Rotar V.; Pukhovoi T.; Kuchkovska Y. O.; Holovach S.; Volochnyuk D. M.; Ryabukhin S. V.; Grygorenko O. O. Last of the gem -Difluorocycloalkanes 2: Synthesis of Fluorinated Cycloheptane Building Blocks. Eur. J. Org. Chem. 2021, 2021, 6561–6569. 10.1002/ejoc.202001530. [DOI] [Google Scholar]
- a Recent examples (2020 to date) of the use of TMSCF3 to transfer CF2 to species other than alkenes and alkynes:Xie Q.; Zhu Z.; Li L.; Ni C.; Hu J. Controllable Double CF2-Insertion into sp2 C-Cu Bond Using TMSCF3: A Facile Access to Tetrafluoroethylene-Bridged Structures. Chem. Sci. 2020, 11, 276–280. 10.1039/C9SC05018C. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang Q.; Ni C.; Hu M.; Xie Q.; Liu Q.; Pan S.; Hu J. From C1 to C3: Copper-Catalyzed Gem-Bis(Trifluoromethyl)Olefination of α-Diazo Esters with TMSCF3. Angew. Chem., Int. Ed. 2020, 59, 8507. 10.1002/anie.202002409. [DOI] [PubMed] [Google Scholar]; c Cai Y.; Zhu W.; Zhao S.; Dong C.; Xu Z.; Zhao Y. Difluorocarbene-Mediated Cascade Cyclization: The Multifunctional Role of Ruppert-Prakash Reagent. Org. Lett. 2021, 23, 3546–3551. 10.1021/acs.orglett.1c00962. [DOI] [PubMed] [Google Scholar]
- Dyke A. M.; Gill D. M.; Harvey J. N.; Hester A. J.; Lloyd-Jones G. C.; Muñoz M. P.; Shepperson I. R. Decoupling Deprotonation from Metallation: thia-Fries rearrangement. Angew. Chem., Int. Ed. 2008, 47, 5067–5070. 10.1002/anie.200800750. [DOI] [PubMed] [Google Scholar]
- Billard T.; Large S.; Langlois B. Preparation of trifluoromethyl sulfides or selenides from trifluoromethyl trimethylsilane and thiocyanates or selenocyanates. Tetrahedron Lett. 1997, 38, 65–68. 10.1016/S0040-4039(96)02216-2. [DOI] [Google Scholar]
- Slaughter J. L.; Lloyd-Jones G. C. C-O cleavage via InIII alkoxide intermediates: In situ 13C NMR analysis of the mechanism of an enantioselective In-mediated cyclopropanation reaction. Tetrahedron 2021, 78, 131786. 10.1016/j.tet.2020.131786. [DOI] [Google Scholar]
- Hashimoto N.; Aoyama T.; Shioiri T. New Methods and Reagents in Organic Synthesis. 14. A Simple Efficient Preparation of Methyl Esters with Trimethylsilyldiazomethane (TMSCHN2) and Its Application to Gas Chromatographic Analysis of Fatty Acids. Chem. Pharm. Bull. 1981, 29, 1475–1478. 10.1248/cpb.29.1475. [DOI] [Google Scholar]
- Kühnel E.; Laffan D. D. P.; Lloyd-Jones G. C.; Martínez del Campo T.; Shepperson I. R.; Slaughter J. L. Mechanism of Methyl-Esterification of Carboxylic Acids by Trimethylsilyl-diazomethane. Angew. Chem., Int. Ed. 2007, 46, 7075–7078. 10.1002/anie.200702131. [DOI] [PubMed] [Google Scholar]
- Ball L. T.; Corrie T J. A.; Cresswell A. J.; Lloyd-Jones G. C. Kinetic Analysis of Domino Catalysis: a Case Study on Gold-Catalyzed Arylation. ACS Catal. 2020, 10, 10420–10426. and references therein 10.1021/acscatal.0c03178. [DOI] [Google Scholar]
- García-Domínguez A.; Helou de Oliveira P. H. H.; Thomas G. T.; Sugranyes A. R.; Lloyd-Jones G. C. Mechanism of Anion-Catalyzed C-H Silylation Using TMSCF3: Kinetically-Controlled CF3-Anionoid Partitioning As a Key Parameter. ACS Catal. 2021, 11, 3017–3025. 10.1021/acscatal.1c00033. [DOI] [Google Scholar]
- Maggiarosa N.; Tyrra W.; Naumann D.; Kirij N. V.; Yagupolskii Y. L. [Me3Si(CF3)F]− and [Me3Si(CF3)2]−: Reactive Intermediates in Fluoride-Initiated Trifluoromethylation with Me3SiCF3 - An NMR Study. Angew. Chem., Int. Ed. 1999, 38, 2252–2253. . [DOI] [PubMed] [Google Scholar]
- Kolomeitsev A.; Bissky G.; Lork E.; Movchun V.; Rusanov E.; Kirsch P.; Röschenthaler G.-V. Different fluoride anion sources and (trifluoromethyl)trimethylsilane: molecular structure of tris(dimethylamino)sulfonium bis(trifluoromethyl)trimethylsiliconate, the first isolated pentacoordinate silicon species with five Si-C bonds. Chem. Commun. 1999, 1017–1018. 10.1039/a901953g. [DOI] [Google Scholar]
- Prakash G. K. S.; Wang F.; Zhang Z.; Haiges R.; Rahm M.; Christe K. O.; Mathew T.; Olah G. A. Long-Lived Trifluoromethanide Anion: A Key Intermediate in Nucleophilic Trifluoromethylations. Angew. Chem., Int. Ed. 2014, 53, 11575–11578. 10.1002/anie.201406505. [DOI] [PubMed] [Google Scholar]
- a Lishchynskyi A.; Miloserdov F. M.; Martin E.; Benet-Buchholz J.; Escudero-Adán E. C.; Konovalov A. I.; Grushin V. V. The Trifluoromethyl Anion. Angew. Chem., Int. Ed. 2015, 54, 15289–15293. 10.1002/anie.201507356. [DOI] [PubMed] [Google Scholar]; b Miloserdov F. M.; Konovalov A. I.; Martin E.; Benet-Buchholz J.; Escudero-Adán E. C.; Lishchynskyi A.; Grushin V. V. The Trifluoromethyl Anion: Evidence for [K(crypt-222)]+CF3–. Helv. Chim. Acta 2017, 100, e1700032 10.1002/hlca.201700032. [DOI] [PubMed] [Google Scholar]; c Harlow R. L.; Benet-Buchholz J.; Miloserdov F. M.; Konovalov A. I.; Marshall W. J.; Escudero-Adán E. C.; Martin E.; Lishchynskyi A.; Grushin V. V. On the Structure of [K(crypt-222)]+CF3–. Helv. Chim. Acta 2018, 101, e1800015 10.1002/hlca.201800015. [DOI] [Google Scholar]
- Tyrra W.; Kremlev M. M.; Naumann D.; Scherer H.; Schmidt H.; Hoge B.; Pantenburg I.; Yagupolskii Y. L. How Trimethyl(trifluoromethyl)silane Reacts with Itself in the Presence of Naked Fluoride—A One-Pot Synthesis of Bis([15]crown-5)cesium 1,1,1,3,5,5,5-Heptafluoro-2,4-bis(trifluoromethyl)pentenide. Chem.—Eur. J. 2005, 11, 6514–6518. 10.1002/chem.200500799. [DOI] [PubMed] [Google Scholar]
- a Nozawa-Kumada K.; Inagi M.; Kondo Y. Highly Chemoselective DMPU-Mediated Trialkylsilylation of Terminal Alkynes Using Trifluoromethyltrialkylsilane. Asian J. Org. Chem. 2017, 6, 63–66. 10.1002/ajoc.201600472. [DOI] [Google Scholar]; b Nozawa-Kumada K.; Osawa S.; Sasaki M.; Chataigner I.; Shigeno M.; Kondo Y. Deprotonative Silylation of Aromatic C-H Bonds Mediated by a Combination of Trifluoromethyltrialkylsilane and Fluoride. J. Org. Chem. 2017, 82, 9487–9496. 10.1021/acs.joc.7b01525. [DOI] [PubMed] [Google Scholar]; c Sasaki M.; Kondo Y. Deprotonative C-H Silylation of Functionalized Arenes and Heteroarenes Using Trifluoromethyltrialkylsilane with Fluoride. Org. Lett. 2015, 17, 848–851. 10.1021/ol503671b. [DOI] [PubMed] [Google Scholar]
- Pilcher A. S.; DeShong P. Utilization of Tetrabutylammonium Triphenyldifluorosilicate as a Fluoride Source for Silicon-Carbon Bond Cleavage. J. Org. Chem. 1996, 61, 6901–6905. 10.1021/jo960922d. [DOI] [PubMed] [Google Scholar]
- Dale H. J. A.; Leach A. G.; Lloyd-Jones G. C. Heavy-Atom Kinetic Isotope Effects: Primary Interest or Zero Point?. J. Am. Chem. Soc. 2021, 143, 21079–21099. 10.1021/jacs.1c07351. [DOI] [PubMed] [Google Scholar]
- Luo G.; Luo Y.; Qu J. Direct nucleophilic trifluoromethylation using fluoroform: a theoretical mechanistic investigation and insight into the effect of alkali metal cations. New J. Chem. 2013, 37, 3274–3280. 10.1039/c3nj00686g. [DOI] [Google Scholar]
- Farnham W. B. Fluorinated Carbanions. Chem. Rev. 1996, 96, 1633–1640. 10.1021/cr9411435. [DOI] [PubMed] [Google Scholar]
- Nosik P. S.; Ryabukhin S. V.; Grygorenko O. O.; Volochnyuk D. M. Transition Metal-Free Gem-Difluorocyclopropanation of Alkenes with CF3SiMe3-NaI System: A Recipe for Electron-Deficient Substrates. Adv. Synth. Catal. 2018, 360, 4104–4114. 10.1002/adsc.201801006. [DOI] [Google Scholar]
- Dilman A. D.; Levin V. V. Difluorocarbene as a Building Block for Consecutive Bond-Forming Reactions. Acc. Chem. Res. 2018, 51, 1272–1280. and references therein 10.1021/acs.accounts.8b00079. [DOI] [PubMed] [Google Scholar]
- Wann D. A.; Rankin D. W. H.; McCaffrey P. D.; Martin J. M. L.; Mawhorter R. J. Equilibrium Gas-Phase Structures of Sodium Fluoride, Bromide, and Iodide Monomers and Dimers. J. Phys. Chem. A 2014, 118, 1927–1935. 10.1021/jp411202t. [DOI] [PubMed] [Google Scholar]
- Pawar L.; Jayaramaiah R.; Krishnan B.; Arunachalampillai A.; Chen Y.; Parsons A. T.; Robinson J. A.; Tedrow J. S. Process development and manufacture of potassium 2-fluoro-6-hydroxyphenyltrifluoroborate. Tetrahedron 2019, 75, 4266–4270. 10.1016/j.tet.2019.05.063. [DOI] [Google Scholar]
- Blair D. J.; Chitti S.; Trobe M.; Kostyra D. M.; Haley H. M. S.; Hansen R. L.; Ballmer S. G.; Woods T. J.; Wang W.; Mubayi V.; Schmidt M. J.; Pipal R. W.; Morehouse G. F.; Palazzolo Ray A. E.; Gray D. L.; Gill A. L.; Burke M. D. Automated iterative Csp3–C bond formation. Nature 2022, 604, 92. 10.1038/s41586-022-04491-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor N. P.; Gonzalez J. A.; Nichol G. S.; García-Domínguez A.; Leach A. G.; Lloyd-Jones G. C. A Lewis Base Nucleofugality Parameter, NFB, and its Application in an Analysis of MIDA-boronate Hydrolysis Kinetics. J. Org. Chem. 2022, 87, 721–729. 10.1021/acs.joc.1c02729. [DOI] [PubMed] [Google Scholar]