

Abstract
Aneuploidy is a hallmark of cancer with tissue-specific prevalence patterns that suggest it plays a driving role in cancer initiation and progression. However, the contribution of aneuploidy to tumorigenesis depends on both cellular and genomic contexts. Whole-genome duplication (WGD) is a common macro-evolutionary event that occurs in >30% of human tumors early in tumorigenesis. Although tumors that have undergone WGD are reported to be more permissive to aneuploidy, it remains unknown whether WGD also affects aneuploidy prevalence patterns. Here we analyzed clinical tumor samples from 5,586 WGD− tumors and 3,435 WGD+ tumors across 22 tumor types and found distinct patterns of aneuploidy in WGD− and WGD+ tumors. WGD+ tumors were characterized by more promiscuous aneuploidy patterns, in line with increased aneuploidy tolerance. Moreover, the genetic interactions between chromosome arms differed between WGD− and WGD+ tumors, giving rise to distinct co-occurrence and mutual exclusivity aneuploidy patterns. The proportion of whole-chromosome aneuploidy vs. arm-level aneuploidy was significantly higher in WGD+ tumors, indicating distinct dominant mechanisms for aneuploidy formation. Human cancer cell lines successfully reproduced these WGD/aneuploidy interactions, confirming the relevance of studying this phenomenon in culture. Lastly, induction of WGD and assessment of aneuploidy in isogenic WGD−/WGD+ human colon cancer cell lines under standard or selective conditions validated key findings from the clinical tumor analysis, supporting a causal link between WGD and altered aneuploidy landscapes. We conclude that WGD shapes the aneuploidy landscape of human tumors and propose that this interaction contributes to tumor evolution.
Keywords: Cancer genomics, Aneuploidy, Whole-genome duplication (WGD)
Introduction
Whole-genome duplication (WGD) is a macro-evolutionary genetic alteration that affects about a third of human tumors (1,2). WGD promotes tumorigenesis by propagating further genomic instability, thereby creating a diverse substrate for tumor evolution (3,4); attenuating selection against mutations in essential genes (5); and increasing the tolerance for chromosome mis-segregation (6,7). WGD has been associated with several molecular and clinical tumor features, including TP53 mutations, higher mutational burden, increased proliferation signatures, and worse overall survival (2,8). While promoting tumorigenesis, WGD has also been associated with altered cellular vulnerabilities in cancer cells, such as increased sensitivity to the inhibition of the mitotic kinesin KIF18A (8–10).
Aneuploidy, defined as copy number alterations of whole chromosomes or chromosome arms, is a hallmark of cancer, and its contribution to cancer development and progression greatly depends on the cellular and genomic context (11). Aneuploidy patterns are shaped by tumor type (1,12,13), the tumors’ active oncogenic pathways (14), and even the specific driver mutations (15). Moreover, aneuploidies genetically interact, such that pairs of aneuploidies can sometimes co-occur or be mutually exclusive with one another (16,17). Tumors that have undergone WGD (hereinafter referred to as WGD+ tumors) present an elevated degree of aneuploidy, and of chromosome losses in particular, in comparison to tumors that have not undergone WGD (hereinafter referred to as WGD− tumors) (2,4,9,12,18). However, whether and how WGD contributes to shaping the aneuploidy patterns of human cancers remains unknown.
In yeast, polyploidy has been associated with genomic instability and with unique cellular vulnerabilities (19), as well as with a greater ability to explore genotypic and phenotypic solutions to stressful culture conditions in in vitro evolution experiments (20). Moreover, specific aneuploidies were shown to confer fitness advantage in a ploidy-dependent manner (21), and the adaptive value of specific chromosome-chromosome genetic interactions depended on the ploidy of the organism (17). Induction of certain chromosome losses in mouse embryonic fibroblasts (MEFs) could drive tumorigenesis in vivo on a tetraploid, but not diploid, genomic background (22). These studies raise the possibility that WGD may also alter the adaptive value of specific aneuploidies and their genetic interactions with other aneuploidies in human tumors, potentially resulting in distinct aneuploidy patterns between WGD− and WGD+ tumors.
Here, we compared the aneuploidy landscapes between WGD− and WGD+ tumors across all The Cancer Genome Atlas (TCGA) tumor types. We identified distinct aneuploidy prevalence patterns and chromosome-arm genetic interactions in WGD− vs. WGD+ tumors, then validated these results in human cancer cell lines. Furthermore, we studied the relationship between WGD and aneuploidy using genetically-matched systems of human colon cancer cell lines before and after WGD (7,23,24). Our findings suggest that WGD is a major determinant of aneuploidy evolution in human cancer.
Materials and Methods
Data collection and preparation
The WGD status and chromosome arm aneuploidy landscapes were determined for 33 TCGA tumor types, as reported by Taylor et al. (12). Samples of each of the 33 tumor types were separated into WGD− and WGD+ groups based on the absence or presence of genome doubling events. Only the 22 tumor types with a minimum of 20 samples in each group were retained for further analyses.
The WGD status and chromosome arm aneuploidy landscapes were determined for the CCLE cell lines, as reported in Cohen-Sharir et al. (9). Cell lines were classified as WGD− if their ploidy was <2.5, and were classified as WGD+ if their ploidy was >3. Only the 14 lineages with a minimum of 5 samples in each group were retained for further analyses. One stomach cancer cell line, KE97, was omitted from the analyses due to its later removal from the CCLE data set due to apparent misclassification (https://web.expasy.org/cellosaurus/CVCL_3386).
For each group, WGD− and WGD+, the number of chromosome-arm gains and chromosome-arm losses was computed per sample, and the degree of aneuploidy was determined as the number of whole-chromosome and arm-level events per sample (relative to the sample’s ploidy, rounded to the nearest integer). For the calculation of the fraction of whole-chromosome aneuploidies vs. chromosome-arm aneuploidies, the acrocentric human chromosomes (13, 14, 15, 21, 22) were excluded. All analyses were performed for the TCGA and CCLE data sets separately.
To assess whether chromosome-arm copy number calls were affected by discontinuous copy number changes (such as chromothripsis), 40 tumor samples per tumor type (20 in each WGD group) were randomly chosen, and visualized using IGV (25). For COAD, MSI and MSS were considered separately, so that 20 samples of each MS status were analyzed within each WGD group. Arm-levels CN calls were manually evaluated for copy number continuity. Continuous events were defined as events that encompass at least 80% of the chromosome-arm, with 3 or fewer disruptions of the copy number state within the aberration. The IGV heat map parameters used for visualization: max/min values = +/− 0.3.
Aneuploidy prevalence analysis
Prevalent chromosome-arm aneuploidies in the WGD− and WGD+ groups of each tumor type were determined by GISTIC 2.0 (26) (RRID:SCR_000151). GISTIC 2.0.23 was applied with the hg19 build of the human genome. Segmentation files were obtained separately for each TCGA tumor type from the GDAC Firehose portal (https://gdac.broadinstitute.org/). A single segmentation file for CCLE, containing all cell-line types, was downloaded from the Broad Institute CCLE portal (https://data.broadinstitute.org/ccle_legacy_data/dna_copy_number/). Each data set was split based on the WGD status of the samples, as described above. GISTIC was run with the ‘broad’ setting (using the default settings for all parameters, except for –brlen that was set to 0.9) in order to obtain results for each chromosome arm (as opposed to focal regions). The sex chromosomes and the short arms of acrocentric chromosomes were removed. For each WGD group of each tumor type, prevalent aneuploidies were determined as those with a prevalence threshold of >=10% of the samples and a significance threshold of q<0.05. To identify significant differences in these prevalent aneuploidies between tumors and cell lines, their prevalence was compared between the TCGA and CCLE datasets using a two-tailed Fisher’s Exact test. For this comparison, CCLE lineages that correspond to two TCGA cohorts were considered against the average prevalence of the two TCGA tumor types (Large_intestine against COAD+READ; Central_Nervous_System against GBM+LGG; Lung against LUAD+LUSC). Events were determined to differentially recur in tumors and in cell lines if the difference in their prevalence was > 0.2 and q-value<0.05.
The positive and negative genetic interactions of all possible chromosome-arm pairs were determined, considering gains and losses of each chromosome-arm separately. First, within the WGD− and WGD+ groups of each tumor type, the chromosome-arm pairs were split into two categories – inter-chromosomal pairs and intra-chromosomal pairs – based on whether the two arms involved belonged to different chromosomes or to the same one. Chromosome-arm events whose co-prevalence was significantly higher than expected by chance (empirical p-value<0.05; empirical q-value<0.25) were defined as co-occurring. Chromosome-arm pairs whose co-prevalence was significantly lower than expected by chance (empirical p-value<0.05; empirical q-value<0.25) were defined as mutually exclusive. A genetic interaction score (also referred to as an enrichment score) was defined as the log10(p-value) of the interaction, with co-occurring events receiving positive scores and mutually exclusive events receiving negative scores.
Permutation analysis
For each tumor type, the tumor samples were split into the WGD− and WGD+ groups, as described above. For each group, a matrix was created for recording the aneuploidy profiles, where rows were sample identifiers, columns were chromosome arms, and the values in each cell indicated gain (1), loss (−1) or neutral (0). To calibrate the significance of genetic interactions between WGD− and WGD+ data sets, therefore controlling for a potentially inflated rate of significant interactions in the WGD+ group, a permutation analysis was performed based on a simulated annealing approach described in Zack et al. (1). Specifically, permuted matrices were generated while maintaining the prevalence of each aneuploidy event. We repeatedly drew two samples and an aneuploidy event and swapped the values of the aneuploidy event between the samples. First, 100K such swaps were performed. Then, for additional 1M iterations, an E score was calculated as following:
Where is the # of gains in sample after iteration is the # of losses after iteration in sample , and T is the “temperature” factor that increases in each iteration (); and the swap was conducted with a probability of . This process was repeated 10,000 times, resulting in 10,000 permuted matrices for each WGD group in each tumor type. To enable parallelization and reproducible results, a sequence of seeds was used for the swaps: for the first 100K iterations, the sequences of seeds were unique per permuted matrix, whereas for the following 1M swaps a constant sequence of seeds was used.
Next, for each permuted matrix, the significance of co-occurring and mutually exclusive events was calculated using a hypergeometric test, thereby generating a background distribution of 10,000 (permuted) p-values per genetic interaction. Finally, for each genetic interaction, the observed p-value was compared to the background p-value distribution, and the empirical p-value for that interaction was calculated based on its relative ranking.
Multiple chromosome-arm co-occurrence analysis
Inter-chromosomal genetic interactions that involve between 2 to 5 chromosome-arms were determined and compared between WGD− and WGD+ tumors, for each tumor type separately. The genetic interaction between each combination of chromosome-arms was determined using SuperExactTest R package (27), which was recently developed to assess statistical significance of intersections among three or more sets. To limit the number of tested interactions, only prevalent events were considered in this analysis (GISTIC 2.0 prevalence >=0.1 and q-value<0.05 in at least one of the WGD groups). Genetic interactions were determined to be different between WGD− and WGD+ tumors if they met the following criteria: (a) prevalence>0.02 and # of samples>10 in one of the WGD groups; (b) q<0.05 for one WGD status and q>0.95 for the other WGD status; (c) observed over expected occurrence fold-change>=1.1 for one WGD status and <=0.9 for the other WGD status. As a significant interaction between >2 chromosome-arms may merely reflect a significant interaction between a subset of the chromosome-arms included in it, a power set analysis was performed, and only interactions that were more significant than all possible subset interactions were retained.
Cell lines and culture conditions
DLD1 cells (ATCC CCL-221; RRID:CVCL_0248) were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were maintained in RPMI 1640 medium with ATCC modification (Thermo Fisher Scientific-Gibco, CA, USA), supplemented with 10% fetal bovine serum (FBS; Corning 35-015-CV) and 1% antibiotic-antimycotic (Thermo Fisher Scientific), in a humidified incubator at 37°C and 5% CO2. RPE1 cells (ATCC CRL-4000; RRID:CVCL_4388) were maintained in a 1:1 mixture of DMEM/F-12 with HEPES (Thermo Fisher Scientific-Gibco, CA, USA), supplemented with 10% fetal bovine serum (FBS; Corning #35-015-CV) and 1% antibiotic-antimycotic (Thermo Fisher Scientific), in a humidified incubator at 37°C and 5% CO2.
Tetraploid DLD1 and RPE1 clones were generated by treating diploid cells with 1.5 μg/mL dihydrocytochalasin B (DCB; CAS #39156-67-7; Sigma Aldrich, St. Louis, MO) for 20 hrs to induce cytokinesis failure. After treatment, the cells were washed four times with cell culture medium and allowed to grow an additional 1–2 days before the isolation of single cells by limiting dilution in 96-well-plates. Only wells containing a single cell were expanded into clonal cell lines and used for further experimentation after confirming ploidy by chromosome counting. For 12-day evolution experiments, DLD1 cells were treated with 1.5 μg/mL DCB for 20 hrs, followed by four washes in cell culture media. Following washout (day 0), cells were either used for chromosome spread harvesting or kept in culture with normal supplemented cell culture medium and passaged on days 2, 6 and 10 into new tissue culture flasks. On day 10, a tissue culture flask was prepared for chromosomes spread harvesting on day 12. Because <100% of the cells went through mitosis during the 20 hr treatment (28), both WGD− and WGD+ cells could be analyzed both at day 0 and at day 12.
HCT116 H2B-GFP cells (ATCC CL-247; RRID:CVCL_0291) were cultured in DMEM (Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific-GIBCO #10270), 50 IU/ml penicillin, and 50μg/mL streptomycin (Thermo Fisher Scientific), in a humidified incubator at 37°C and 5% CO2. Tetraploid HCT116 clones were previously generated, as described in Kuznetsova et al.(7), by treating diploid cells with 0.75 μg/mL dihydrocytochalasin D (DCD; CAS #22144-77-0; Sigma Aldrich) for 18 hrs to induce cytokinesis failure. After treatment, the cells were washed, placed into a drug-free medium and subcloned by limiting dilution in 96-well plates. Only wells containing a single cell were expanded into clonal cell lines and used for further experimentation after confirming ploidy by chromosome counting.
Cell lines were authenticated by validating their characteristic karyotypes, and monitored for possible mycoplasma infection by DNA staining or by Mycoplasmacheck Test (Eurofins BioPharma; https://eurofinsgenomics.eu/en/genotyping-gene-expression/applied-genomics-services/mycoplasmacheck/). After thawing, cells were kept in culture for <30 passages prior to their profiling and/or experimentation.
Chromosome spreads and chromosome counting
DLD1 cells were seeded in T-25 flasks, allowed to adhere for at least 12 hours, then treated with 50 ng/mL colcemid (Invitrogen – Karyomax, Waltham, MA) for 5 hrs. HCT116 cells were treated with 50 ng/mL colchicine (SERVA, Heidelberg, Germany) for 4.5 hrs. Cells were trypsinized, and centrifuged at 1,000 rpm for 5 min. Pre-warmed (37°C) hypotonic solution (0.075 M KCl) was added dropwise to the disrupted cell pellet and the cell suspension was incubated for 15-18 min at 37°C. Freshly prepared fixative (3:1 methanol:glacial acetic acid) was added. Chromosome spreads were dropped on wet glass slides, and either stained with 300 nM DAPI (Thermo Fisher Scientific – Invitrogen), or processed for mFISH (see next section). DAPI stained slides were mounted with antifade solution (90% glycerol, 0.5% N-propyl gallate) and sealed under 22x50 mm coverslips (Corning Incorporated, Corning, NY) with nail polish. Images of DAPI-stained chromosome spreads were acquired with a Nikon Eclipse Ti inverted microscope equipped with a ProScan automated stage (Prior Scientific, Cambridge, UK), CoolSNAP HQ2 CCD camera (Photometrics, AZ, USA), and Lumen200PRO light source (Prior Scientific, Cambridge, UK) using either a 60X/1.4 NA or 100X/1.4 NA Plan-Apochromatic phase contrast objective. After image acquisition, chromosomes in individual spreads were counted using NIS elements (RRID:SCR_014329; Nikon instruments Inc., NY, USA) or FIJI(29).
Multicolor fluorescence in situ hybridization (mFISH)
For HCT116 cells, mFISH was performed with a DNA probe mixture (24XCyte Human Multicolor FISH Probe Kit, MetaSystems) as previously described (7). Briefly, fixed cells were dropped on a slide, pepsinized, incubated in a coplin jar for 2 min at 37°C and rinsed twice for 7 min each time in 1xPBS. Subsequently, the slides were dehydrated in EtOH series, 3 min each, and baked at 61.4°C for 1 h. Denaturation at 72°C for 1min 30 sec in 70% Formamide/2xSSC, and dehydration in EtOH series was followed by in situ hybridization. Here, 4 μl of denatured, pre-annealed probe was applied on a slide, covered with a coverslip, sealed with rubber cement and incubated in a dark chamber overnight at 37°C. Subsequently, rubber cement was removed, coverslip was removed by soaking the slide in 4XSSCT and the slides were washed 3x5min in 0.1xSSC at 62°C. For biotin detection, additional steps with streptavidin-Alexa Fluor 488 (Molecular Probes) were performed. Finally, the slides were washed twice each time 7 min in 4xSSCT at 42°C, mounted in Vectashield Antifade solution (4’,6 diamidino-2-phenylindole, Vector laboratories H-1200, Axxora/Alexis, Lörrach, Germany) with DAPI and cover with 24x60 mm coverslip sealed with a nail polish. The spreads were manually analyzed on the Zeiss Observer.Z1 microscope, Plan Apochromat 63x magnification oil objective in DAPI, CFP, GFP, Cy3, Texas Red and Cy5 channels.
For DLD1 cells, mFISH was performed with 24XCyte human probe cocktail (MetaSystems) according to manufacturer’s protocol. Briefly, chromosome spreads were incubated in 2X saline-sodium citrate (SSC) buffer at 70°C for 30 min, cooled to room temp for 20 mins, and washed for 1min each in 0.1X SSC and 0.07M NaOH at room temperature. Next, slides were washed at 4°C in 0.1X SSC for 1 min, followed by 2X SSC for 1 min, then 70% ethanol, 90% ethanol, and 100% ethanol for 1 min each. Probes were added to the slide, sealed under a coverslip with rubber cement, and hybridized overnight at 37°C. The coverslip was then removed, and slides were washed for 2 min in 0.4X SSC at 70°C and for 30 sec in 2XSCC, followed by a quick rinse in DIH2O and mounting in DAPI/Antifade counterstain buffer (MetaSystems) for imaging. Images were acquired with a Zeiss Axioplan 2 motorized microscope (Carl Zeiss, Inc., Oberkochen, Germany) equipped with an Ikaros4/Isis4 system (MetaSystems). Analysis was performed using Isis 4 software.
Single cell DNA sequencing (scDNAseq) data analysis
Single cell DNA sequencing data of HCT116 and HPT cells were obtained and processed as described in Cohen-Sharir et al. (9) For each single cell, the number of whole-chromosome, chromosome-arm and sub-chromosomal (structural) copy number changes were extracted from the AneuFinder plots (30). Single cell DNA sequencing data of colon cancer patient-derived organoids were obtained from Bolhaqueiro et al. and Bollen et al. (31,32). For each single cell, the number of whole-chromosome, chromosome-arm and sub-chromosomal (structural) copy number changes were extracted from the copy number plots. Single cell DNA sequencing data of breast cancer patient-derived xenografts were obtained from Laks et al. and Salehi et al. (33,34) For each single cell, the number of whole-chromosome, chromosome-arm and sub-chromosomal (structural) copy number changes were extracted from the copy number plots provided in https://www.cellmine.org/dashboards. Due to the high resolution and high number of single cells included in the PDX analyses, only CNAs that encompass >5Mb were considered in the analysis, and only up to 20 cells were analyzed per WGD group.
Soft agar assay
Cells were grown in T-25 flasks. On the day of experimental set up, melted 2% agar (Fisher Scientific, NJ, USA) was mixed 1:1 with 2X DMEM supplemented with 20% FBS (Thermo Fisher Scientific – Gibco) and 2% antibiotic-antimycotic (Thermo Fisher Scientific – Gibco). 1.5 mL of this 1% agar/1X DMEM solution was transferred into 35 mm tissue culture dishes and allowed to cool to room temperature. Cells were trypsinized, harvested, counted, and resuspended at a concentration of 5 million cells/mL. 1.4% agarose was melted, cooled to 42°C in a water bath, and 750 μL of the agar was mixed with 650 μL of the 2X DMEM and 100 μL of cell suspension (500,000 cells) to be seeded in each dish. The mixture was added on top of the 1% agar layer and allowed to cool for 30 min before cell culture media was added to the dishes. The dishes were kept in a humidified tissue culture incubator at 37°C and 5% CO2 for three weeks and cell culture media was replenished every three days for the course of the experiment. After three weeks, macroscopic colonies were imaged with the Nikon Eclipse Ti inverted microscope described above using a 20X/0.3 NA A Plan corrected phase contrast objective lens. These large colonies were imaged only at a single focal plane (in which the outermost edges of the colony were most in focus) and images were captured using the Large Image tiling function in NIS Elements software. Individual macroscopic colonies were picked from soft agar using a sterile 200 μL pipette tip and placed in sterile 1.5 mL Eppendorf tubes with 1 mL trypsin. Tubes were placed at 37°C in an incubator for 20 min with occasional pipetting to disrupt the colony. The tubes were then centrifuged for 5 min at 1000 rpm, supernatant was carefully aspirated, cells were resuspended in 1 mL cell culture media, and transferred to a 35 mm tissue culture dish containing another 1 mL of media. Cells were allowed to adhere overnight before preparation for chromosome spreads, following the protocol described above.
Statistical analysis
Kullback-Leibler (KL) divergence and L2 norm values were determined for each WGD group of each tumor type in order to obtain a measure of the deviation of the aneuploidy landscapes from a uniform distribution. KL divergence was computed using the ‘KL.plugin’ function of the ‘entropy’ package in R, with the event distribution and a uniform distribution as inputs. Similarly, the L2 norm was calculated using the ‘l2norm’ function of the ‘wavethresh’ package in R, with the event distribution and a uniform distribution as inputs.
The significance of the differences in the number of aneuploidies and in the fraction of whole-chromosome aneuploidies between WGD− and WGD+ samples within each tumor type, were determined using a two-sided unpaired Student’s t-test. The significance of the differences in the relative prevalence of the top-3 aneuploidies, the L2 norm values, and the KL-divergence values, between WGD− and WGD+ groups across cancer types, were determined using a two-sided paired Student’s t-test.
Prevalent GISTIC 2.0 events were determined using the p-values and q-values provided by the GISTIC 2.0.23 ‘broad’ analysis output, with the following thresholds used for determining prevalence: q-value<0.05 and prevalence >=0.1. Co-occurrence and mutual-exclusivity of chromosome-arm aneuploidy pairs were determined using the empirical p-values from the permutation analysis (described above), with the following thresholds: p<0.05 and q<0.25.
The number of aneuploidy events of the HCT116 and DLD1 cell lines and their various derivatives, were determined as the sum of whole-chromosome and chromosome-arm aneuploidies in each sample, excluding chromosome Y from the analysis. Karyotypic heterogeneity was determined by counting the fraction of cells with at least one non-clonal whole-chromosome aneuploidy. The significance of the difference between the number of aneuploidy events was determined using a one-sided one-way ANOVA. The significance of the differences in the fraction of whole-chromosome aneuploidies, in the fraction of cells with non-clonal aneuploidies, and in the fraction of gains/losses, were determined using a one-sided Fisher’s Exact test. The significance of the differences in the prevalence of chromosome-arm aneuploidies between tumors (TCGA) and cell lines (CCLE) was determined using a two-sided Fisher’s Exact test. The significance of the difference in chromosome count distributions between day 0 and day 12 of each WGD group, was calculated using a two-sided Mann-Whitney test. The significance of the increased fraction of whole-chromosome aneuploidies out of all aneuploidies in WGD+ vs. WGD− tumors was determined using McNemar’s test.
Data visualization
Boxplots were plotted using R Base graphics, with the following parameters: bar, median; box, 25th and 75th percentile; whiskers, 1.5 X interquartile range. Heatmaps and histograms were plotted using the ‘ggplot2’ R package, or the ‘matplotlib’ and ‘seaborn’ packages in Python (RRID:SCR_001658). Venn diagrams were plotted using ‘matplotlib_venn2’ in Python (RRID:SCR_001658). Whenever data are presented separately for each tumor type, plots were generated only for the 11 tumor types with the highest number of samples, with each WGD group consisting of at least 100 tumor samples.
Ethics
This study was conducted in accordance to the U.S. Common Rule. All tumor data analyses were based on de-identified human tumor data that is publically-available from TCGA.
Data Availability Statement
All datasets are available within the article and its Supplementary Tables, or from the Corresponding Author upon request. The code used to analyze the data is available at https://github.com/BenDavidLab/WGD_Aneuploidy.
Results
The prevalence and general features of aneuploidy differ between WGD− and WGD+ tumors
To compare the aneuploidy landscape of WGD− vs. WGD+ tumors, we compiled genomic data from TCGA. We determined WGD status and chromosome-arm copy number alterations per sample, as previously described (9,12), and confirmed that the aneuploidy calls were not affected by the accumulation of multiple discontinuous copy number changes within chromosome arms (Supplementary Fig. 1a–c) Aneuploidy was defined as deviation from a diploid karyotype for WGD− tumors and as deviation from the sample’s basal ploidy (rounded to the nearest integer), and the number of whole-chromosome and chromosome-arm gains and losses were computed for each tumor sample. We focused our downstream analyses on the 22 tumor types with >20 samples in each of the WGD groups (Supplementary Table 1).
Across tumor types, WGD+ tumors were significantly more aneuploid than WGD− tumors (Fig. 1a and Supplementary Fig. 2a). In line with the role proposed for WGD in “buffering” the cellular consequences of chromosome-arm losses (35), and as previously reported (1,2,12), the median ploidy of WGD− tumors was indeed 2 (median=2.0; mean=1.99), whereas that of tumors that have undergone one round of WGD was significantly lower than 4 (median=3.26; mean=3.26) and that of tumors that have undergone two rounds of WGD was very far from 8 (median=4.41; mean=4.54) (Fig. 1b and Supplementary Fig. 2b). The increased aneuploidy levels of WGD+ tumors might stem from an increased prevalence of the most common aneuploidies, or from an elevated tolerance to aneuploidy in general. To distinguish between these two options, we compared the relative prevalence of each aneuploidy between WGD− and WGD+ tumors. For both gains and losses, although the absolute prevalence of most common aneuploidies was higher in the WGD+ tumors in all tumor types, their relative prevalence (defined as the cumulative fraction of the most common events) was significantly lower than in WGD− tumors (Fig. 1c). Moreover, in line with WGD+ tumors being more permissive to aneuploidy in general, the distribution of aneuploidy across the genome was more uniform in WGD+ than in WGD− tumors (Fig. 1d,e and Supplementary Fig. 2c,d), as measured by quantifying the deviation of the aneuploidy distributions from uniform distributions (KL divergence; see Methods). Notably, these results do not merely reflect the ongoing CIN in the WGD+ cells, as the observed aneuploidies are clonal (or at least exist in a dominant clone). Together, these data suggest that aneuploidy is not only more pervasive in WGD+ than in WGD− tumors, but also that the aneuploidy landscapes of WGD+ tumors are more promiscuous.
Figure 1: Distinct prevalence and features of aneuploidy in WGD− and WGD+ tumors.
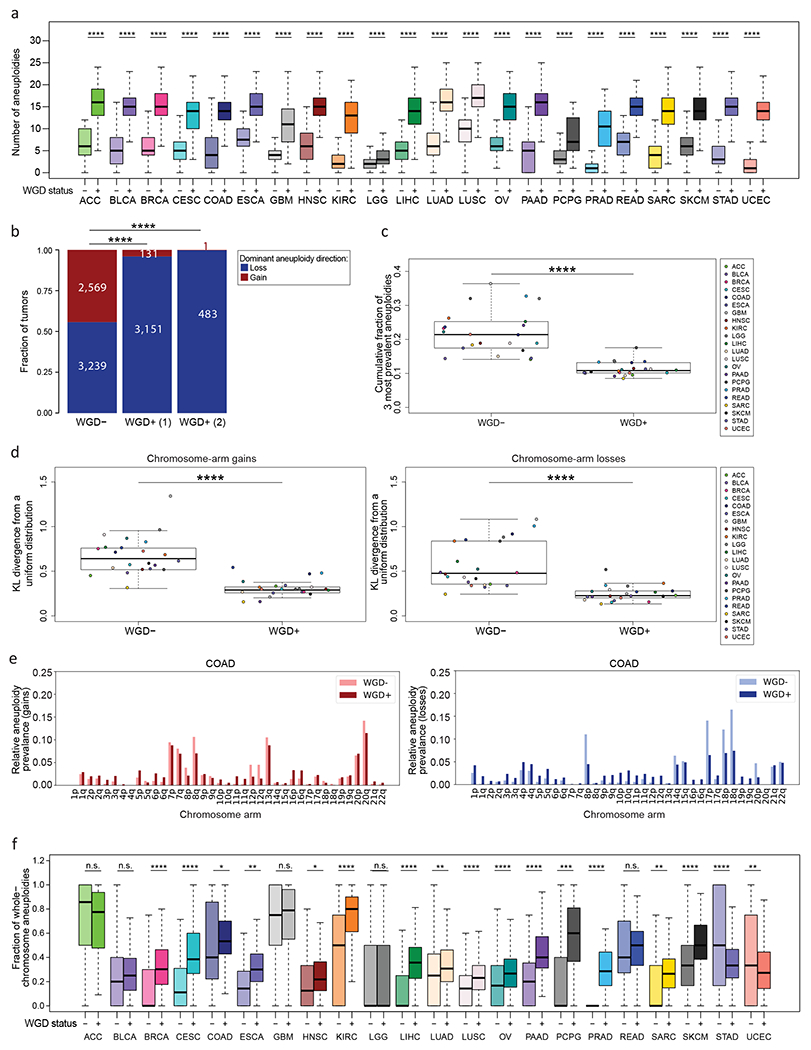
(a) Comparison of the number of aneuploidies between WGD− and WGD+ tumors across 22 tumor types. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001; adjusted two-tailed Student’s t-test. (b) Comparison of the dominant whole-chromosome aneuploidy direction (i.e., the fraction of WGD−, WGD+ (1) and WGD+ (2) tumors with ploidy below or above the basal ploidies of 2n, 4n or 8n, respectively) between WGD− and WGD+ tumors, across all cancer types combined. p=2<2-16; two-tailed Fisher’s Exact test. (c) Comparison of the cumulative fraction of the top three aneuploidies (out of all aneuploidies) between the WGD− and WGD+ tumors, across the 22 tumor types. ****, p=3e-9; two-tailed Student’s t-test. (d) Comparison of the deviation of chromosome-arm gains (left) and losses (right) from a uniform distribution, between WGD− and WGD+ tumors, across the 22 tumor types. ****, p=8e-08 and p=4e-07, for gains and losses, respectively; two-tailed paired Student’s paired t-test. (e) The relative prevalence of chromosome-arm gains (left) and losses (right) in WGD− and WGD+ colon adenocarcinoma (COAD) tumors. (f) Comparison of the fraction of whole-chromosome aneuploidies (out of all whole-chromosome and arm-level aneuploidies) between WGD− and WGD+ tumors within 22 tumor types. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001; adjusted two-tailed Student’s t-test. ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, Cervical squamous cell carcinoma and endocervical adenocarcinoma; COAD, colon adenocarcinoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, Head and Neck squamous cell carcinoma; KIRC, Kidney renal clear cell carcinoma; LGG, Brain lower grad glioma; LIHC, Liver hepatocellular carcinoma; LUAD, Lung adenocarcinoma; LUSC, Lung squamous cell carcinoma; OV, Ovarian serous cystadenocarcinoma; PAAD, Pancreatic adenocarcinoma; PCPG, Pheochromocytoma and Paraganglioma; PRAD, Prostate adenocarcinoma; READ, Rectum adenocarcinoma; SARC, sarcoma; SKCM, Skin Cutaneous Melanoma; STAD, Stomach adenocarcinoma; UCEC., Uterine Corpus Endometrial Carcinoma
Interestingly, in 15 of the 22 tumor types, whole-chromosome aneuploidies contributed significantly more than chromosome-arm aneuploidies to the overall aneuploidy landscape in the WGD+ tumors compared to WGD− tumors (Fig. 1f). This finding is consistent with a high rate of chromosome mis-segregation induced by WGD (3,4,6,7,36), and also with an increased tolerance of WGD+ tumors to whole-chromosome aneuploidies, in comparison to WGD− tumors (6).
WGD is associated with distinct aneuploidy patterns and chromosome-arm genetic interactions
Next, we compared aneuploidy prevalence patterns between WGD− and WGD+ tumors. As expected, the relative tendency of individual chromosomes and chromosome arms to be gained or lost was similar between the WGD groups (Fig. 2a). However, many more aneuploidies were found to be common in the WGD+ group than in the WGD− group, based on a GISTIC 2.0 analysis (26) (Methods; Fig. 2a and Supplementary Table 2). Overall, we identified 406 prevalent chromosome-arm aneuploidies in WGD+ tumors across the 22 tumor types, compared to 305 prevalent events in WGD− tumors (Supplementary Table 2). 248 (81%) of the prevalent events in WGD− tumors were also prevalent in WGD+ tumors (Supplementary Fig. 3a), suggesting overall similar karyotypic patterns between the groups.
Figure 2: WGD is associated with significant changes in aneuploidy prevalence patterns.
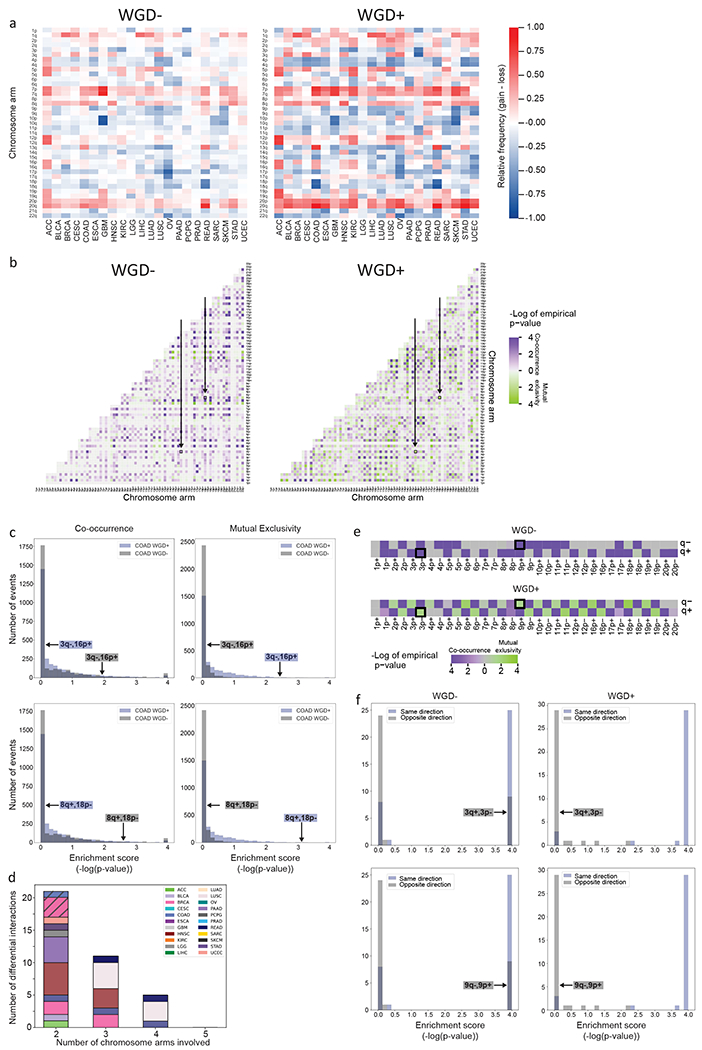
(a) Heatmaps of the relative prevalence of all chromosome-arm gains and losses in WGD− tumors (left) and WGD+ tumors (right), across 22 tumor types. The prevalence of the events was calculated by GISTIC 2.0. (b) Heatmaps of the significance (−log(empirical p-value)) of positive genetic interactions (co-occurrence, purple) and negative genetic interactions (mutual exclusivity, green) between chromosome arms of different chromosomes in WGD− tumors (left) and WGD+ tumors (right) of colon adenocarcinomas (COAD). Note the interactions between loss of chromosome 3q (3q−) and gain of chromosome 16p (16p+), and between gain of chromosome 8q (8q+) and loss of chromosome 18p (18p−), which are significantly co-occurring in WGD− tumors and significantly mutually exclusive in WGD+ tumors (empirical p<0.05; q<0.25) and are highlighted on the heatmaps. (c) Histograms of the distribution of enrichment scores (defined as −log(empirical p-value)) for co-occurrence (left) and mutual exclusivity (right) of inter-chromosomal genetic interactions in WGD− vs. WGD+ COAD tumors. The discordant genetic interactions (3q−,16p+; 8q+,18p−) are highlighted on the histograms. (d) Bar plot presenting the number of differential interactions between WGD− and WGD+ tumors, for n=2, n=3, n=4 and n=5 chromosome arms (using the SuperExactTest algorithm). Samples are colored by tumor type. For n=2, interactions that were also identified as differential in the permutation analysis are denoted by diagonal lines. For n=3, n=4 and n=5, shown are genetic interactions found to be more significant than any of the subset interactions that comprise them. (e) Heatmaps of the significance (−log(empirical p-value)) of positive genetic interactions (co-occurrence, purple) and negative genetic interactions (mutual exclusivity, green) between gains and losses of chromosome arms within the same chromosome in WGD− tumors (top) and WGD+ tumors (bottom) of COAD. Note the interactions between loss of chromosome 3p (3p−) and gain of chromosome 3q (3q+), and between gain of chromosome 9p (9p+) and loss of chromosome 9q (9q−), which are significantly co-occurring in WGD− tumors and significantly mutually exclusive in WGD+ tumors (empirical p<0.05; q<0.25) and are highlighted on the heatmaps. (f) Histograms of the distribution of enrichment scores (defined as −log(empirical p-value)) for co-occurrence of same-direction and opposite-direction intra-chromosomal genetic interactions in WGD− (left) and WGD+ (right) COAD tumors. The discordant genetic interactions, which are co-occurring in one WGD group and mutually exclusive in the other (3p−,3q+; 9p+,9q−), are highlighted on the histograms. Tumor type abbreviations as in Fig. 1.
The complex karyotypes of tumors are shaped by selective pressures that can drive the gain or loss of individual chromosomes, as well as selective pressures that promote or suppress genetic interactions between chromosomes (16,17). We therefore sought to compare the chromosome-arm genetic interactions – namely, the co-occurrence and mutual-exclusivity aneuploidy patterns – between WGD− and WGD+ tumors. It should be noted that given the overall higher degree of aneuploidy in the WGD+, it is expected to observe more chromosome-arm genetic interactions in the WGD+ group, potentially confounding the comparison between the groups. Indeed, the association between copy number alterations (CNAs) in cancer genomes was previously shown to be confounded by the overall genomic disruption of the samples (1). To overcome this challenge, we applied a simulated annealing approach (1,37). For each tumor type and WGD group (i.e., WGD+ and WGD−), we created 10,000 permuted matrices, maintaining both the chromosome-arm aneuploidy prevalence across tumor samples and the overall aneuploidy levels within tumor samples. We then used a hypergeometric test to determine the co-occurrence of all possible pairs of chromosome-arm aneuploidies in each tumor type, and compared the observed co-occurrence to that calculated on the permuted matrices, thus obtaining an empirical p-value for each genetic interaction (Methods).
Across cancer types, we found distinct chromosome-arm genetic interactions between WGD− and WGD+ tumors (Fig. 2b, Supplementary Fig. 3b and Supplementary Table 3). Excluding interactions between chromosome-arms of the same chromosome, a median of 78% of the significant co-occurring aneuploidy pairs in WGD− tumors were not significant in WGD+, and 80% of the significant co-occurring aneuploidy pairs in WGD+ were not significant in WGD− (Supplementary Table 3 and Supplementary Fig. 3c). Surprisingly, we identified 8 events that, within a given tumor type, were significantly co-occurring in WGD− samples, but were significantly mutually exclusive in the WGD+ samples: for example, loss of chromosome-arm 3q (Chr3q) and gain of chromosome-arm 16p (Chr16p) frequently co-occurred in WGD− colon cancer samples, but were mutually exclusive in WGD+ colon cancers samples (Fig. 2b,c). Other such events are gain of Chr8q and loss of Chr18p in colon cancer, losses of Chr5q and Chr9p in breast cancer, and losses of Chr4p and Chr14q in lung adenocarcinoma (Fig. 2c and Supplementary Fig. 3b,d). We also found two events that were significantly co-occurring in WGD+ tumors, but were significantly mutually exclusive in the WGD− tumors: loss of Chr10q and loss of Chr18p in low-grade glioma, and loss of Chr1p and gain of Chr5p in glioblastoma (Supplementary Fig. 3b,d). We conclude that opposite genetic interactions between specific chromosome-arm aneuploidies can exist in WGD− and WGD+ tumors. To the best of our knowledge, this is the first demonstration that the fitness of specific karyotypes is altered in human cancer following WGD, consistent with previous reports in yeast (17,21). Notably, 9 out of these 10 genetic interactions involve a chromosome-arm alteration that is significantly associated with increased aggressiveness or worse outcome in patients (18,38), highlighting the potential clinical relevance of this finding.
We next sought to expand the interaction analysis beyond pairs of chromosome arms. To this end, we applied SuperExactTest, an algorithm for analyzing multi-set intersections (27), to compare the co-occurrence patterns of prevalent chromosome-arm aneuploidies between WGD− and WGD+ tumors within each tumor type (Methods). We set stringent prevalence and significance criteria to determine which genetic interactions are significantly different between the WGD groups (Methods). We first tested pairwise interactions (n=2) and identified 21 interactions that are different between WGD− and WGD+ tumors (Fig. 2d, Supplementary Fig. 4a–d and Supplementary Table 4). Reassuringly, 4 of the 5 interactions identified by the permutation-based approach were also detected by this analysis, with the 5th interaction nearly meeting the stringent thresholds applied (Fig. 2d and Supplementary Table 4). Moreover, all of the 17 additional interactions that were identified by SuperExactTest nearly reached significance in the permutation analysis, suggesting high degree of agreement between the two approaches. We therefore extended the analysis to test the interactions between 3, 4 or 5 chromosome-arm aneuploidies. As a significant interaction between a group of >2 chromosome-arms may merely reflect a significant interaction between a subset of the chromosome-arms included in it, we performed a power set analysis, to identify interactions that were more significant than all possible subset interactions (Methods). We found 11, 5 and 0 differential genetic interactions with n=3, n=4 and n=5 chromosome-arms, respectively (Fig. 2d, Supplementary Fig. 4a–d and Supplementary Table 4). We note that all n>2 differential interactions were co-occurring in the WGD− group and mutually exclusive in the WGD+ group, probably because the lower overall prevalence of aneuploidy in WGD− tumors reduces the power to detect significant mutually exclusive interactions within this group.
We next considered the intra-chromosome interactions, i.e., the interactions between the p arm and the q arm within chromosomes. We found that opposite-direction chromosome-arm aneuploidies within the same chromosome (that is, loss of the p arm and gain of the q arm, or vice versa) are much more common in WGD− tumors. Whereas such opposite-direction alterations rarely occurred (and were therefore mutually exclusive) in WGD+ tumors, they often co-occurred in WGD− tumors (Fig. 2e,f, Supplementary Fig. 3e and Supplementary Table 5). For example, Chr3p loss and Chr3q gain are mutually exclusive events in WGD+, but are co-occurring events in WGD− colon cancer tumors; and so are Chr9p gain and Chr9q loss (Fig. 2e,f). This is in line with our observation that whole-chromosome aneuploidy (relative to chromosome-arm aneuploidy) accounts for a larger fraction of the aneuploidy landscape in WGD+ than in WGD− tumors (Fig. 1f): when a chromosome-arm is altered in a WGD+ tumor, the reciprocal arm is likely to be altered in the same direction; in contrast, when a chromosome arm is altered in a WGD− tumor, the reciprocal arm may be altered either in the same direction or in the opposite direction (Fig. 2e, Supplementary Fig. 3e and Supplementary Table 3). Together, these findings suggest that the contribution of distinct mechanisms of aneuploidy formation to tumor evolution is WGD-dependent.
Human cancer cell lines successfully recapitulate the association of WGD with aneuploidy features
Cancer cell lines are extensively used for the research of WGD and aneuploidy (39). We therefore wanted to examine whether the effects of WGD on the aneuploidy landscapes are evident in cell lines as well. To compare the aneuploidy landscapes between WGD− and WGD+ cancer cell lines, we compiled data from the Cancer Cell Line Encyclopedia (CCLE) (40) and determined their WGD status and chromosome-arm alterations, as we previously described (9). We focused our analyses on the 14 cancer types with >5 samples in each of the WGD groups (Supplementary Table 6). In agreement with our findings from the TCGA clinical samples, WGD+ cell lines had significantly more aneuploidy events than WGD− cell lines across all lineages (Fig. 3a), and the relative fraction of whole-chromosome aneuploidies tended to be higher in the WGD+ tumors (p=0.003 McNemar’s test; a significant difference between the groups was observed within four of the tumor types; Fig. 3b). The same cancer-type specific common aneuploidies observed in TCGA (Fig. 2a) were observed in the cell lines (Fig. 3c and Supplementary Table 7). Of the chromosome-arm aneuploidies found to be prevalent within each tumor type, 88.6% and 97.1% were concordant between cell lines and tumors, in WGD− and WGD+ samples, respectively (Supplementary Fig. 5). Moreover, although the absolute prevalence of the most common aneuploidies was higher in WGD+ than in WGD− cell lines (Fig. 3c), their relative prevalence was lower (Fig. 3d), consistent with the findings from the primary tumors. We conclude that the associations between WGD and aneuploidy features are conserved between human tumors and human cancer cell lines. (We note that the cancer cell line analysis lacked the statistical power to examine the association between WGD and chromosome-arm genetic interactions.)
Figure 3: The association between WGD and aneuploidy is conserved in human cancer cell lines.
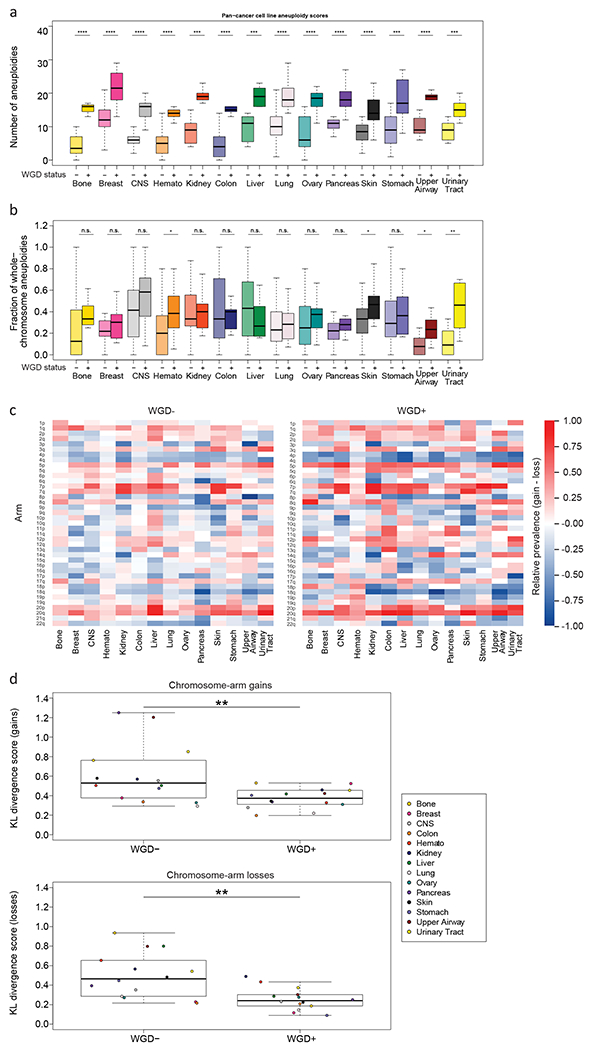
(a) Comparison of the number of aneuploidies between WGD− and WGD+ cell lines within 14 cancer types. ***, p<0.001; ****, p<0.0001; adjusted two-tailed Student’s t-test. (b) Comparison of the fraction of whole-chromosome aneuploidies (out of all whole-chromosome and arm-level aneuploidies) between WGD− and WGD+ cell lines within 14 cancer types. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001; adjusted two-tailed Student’s t-test. (c) Heatmaps of the relative prevalence of all chromosome-arm gains and losses in WGD− cell lines (left) and WGD+ cell lines (right), across 14 cancer types. The prevalence of the events was calculated by GISTIC 2.0. (d) Comparison of the KL-divergence values of chromosome-arm gains (top) and losses (bottom) between WGD− and WGD+ cell lines, across the 14 cancer types. **, p=0.008 and p=0.001, for gains and losses, respectively; two-tailed paired Student’s paired t-test. CNS, central nervous system; Hemato, hematologic malignancies.
Isogenic human cell lines reveal a causal relationship between WGD and aneuploidy landscapes
To study the consequence of WGD in human cells, we previously developed genetically-matched systems of human colon cancer cell lines before and after WGD (7,23,24). These cell lines are aneuploid, chromosomally unstable, and exhibit increased tumorigenic behavior and drug resistance (7,23,24). We therefore turned to these systems to further characterize the relationship between WGD and aneuploidy in human cancer.
We first compared the near-diploid WGD− HCT116 cells to their WGD+ derivatives HPTs (HCT116 Post Tetraploid cells) (7). We used multicolor fluorescence in situ hybridization (mFISH) to karyotype single cells from the parental HCT116 population, from two independent WGD− single cell-derived populations, and from two independent WGD+ HPT single cell-derived populations. In comparison to the HCT116 parental cells and to their WGD− derivatives, HPT cells were more aneuploid in general (compared to their basal ploidy; Fig. 4a), displaying a higher relative fraction of chromosome losses (Fig. 4b) and whole-chromosome aneuploidies (compared to both arm-level and structural aneuploidies; Fig. 4c). The HPT cells remained chromosomally unstable, and did not converge on an optimal karyotype (Fig. 4d), in line with WGD+ cells being more tolerant to a variety of aneuploidies. We confirmed the mFISH findings using data from single-cell DNA sequencing (scDNAseq) of the HCT116 and HPT cells (9) (Supplementary Fig. 6a–d). These results are consistent with our previous observation that HPT cells became more aneuploid than HCT116 cells following CIN induction, and that their karyotypes did not converge on a specific karyotype after drug washout (9). Importantly, we included in the mFISH analysis a population of HCT116 cells with a stable aneuploidy, which have not undergone WGD (41,42). When the clonal aneuploid chromosome itself was excluded, these WGD− aneuploid HCT116 cells showed a similar degree of aneuploidy, losses/gains ratio, whole-chromosome/chromosome-arm ratio, and overall chromosomal stability, as the parental near-diploid HCT116 cells (Fig. 4a–d). These results strengthen the notion that it is WGD per se that underlies the observed changes in the aneuploidy landscape.
Figure 4: Isogenic WGD− and WGD+ HCT116 cell lines demonstrate a causal effect of WGD on aneuploidy landscapes.
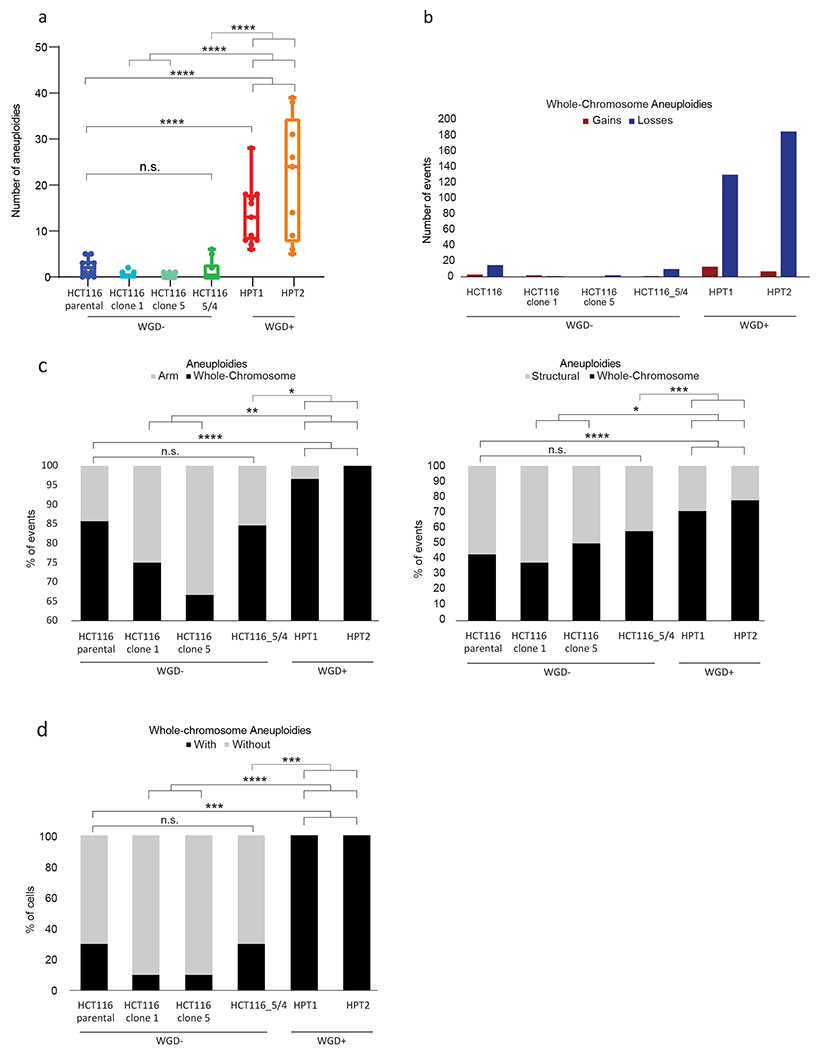
(a) mFISH-based comparison of the number of aneuploidies (relative to basal ploidy) between the near-diploid parental HCT116 cells (HCT116-WT), two WGD− HCT116 clones (#1 and #5), HCT116 cells with two extra copies of chromosome 5 introduced through microcell-mediated chromosome transfer (HCT116–5/4) (42), and two WGD+ HCT116 clones (HPT1 and HPT2) (7). n.s., p>0.05, ***, p<0.001; ****, p<0.0001; two-tailed Student’s t-test. (b) The number of whole-chromosome gains and losses observed by mFISH, in HCT116 and its derived clones (>10 single cells per clone). (c) The relative fraction of whole-chromosome aneuploidies relative to arm-level aneuploidies (left) or structural aneuploidies (right; including arm-level aneuploidies, translocations and smaller structural alterations), in HCT116 and its derived clones. n.s., p>0.05, **, p<0.01, ***, p<0.001; ****, p<0.0001; one-tailed Fisher’s Exact test. (d) The fraction of cells with non-clonal whole-chromosome aneuploidies, a measure of karyotypic heterogeneity, in HCT116 and its derived clones. ***, p<0.001; one-tailed Fisher’s Exact test.
Next, we inhibited cytokinesis to induce WGD in another near-diploid WGD− human colon cancer cell line, DLD1, and isolated 11 WGD+ DLD1 clones and two WGD− DLD1 clones ((24) and the current study; Methods). In comparison to their parental cells and to the WGD− clones, WGD+ clones were generally more aneuploid (Fig. 5a), their aneuploidy landscape was dominated by chromosome losses (Fig. 5b) and whole-chromosome aberrations (Fig. 5c), and were more karyotypically heterogeneous (Fig. 5d), in complete agreement with our findings with the HCT116/HPT cells. Moreover, by inhibiting cytokinesis and comparing the karyotypic evolution of WGD− and WGD+ cells within the same culture (Fig. 5e), we found that aneuploidy and karyotypic heterogeneity accumulated quickly in the WGD+ cells, but not in the WGD− cells (Fig. 5f). 12 days of in vitro culture (as in (28)) were sufficient to reproduce the karyotypic features that we found to be associated with WGD+ tumors: increased levels of karyotypic heterogeneity, chromosome losses, and whole-chromosome aneuploidies, relative to WGD− cells (Fig. 5g,h). Interestingly, despite the selection pressure associated with single-cell cloning, no specific aneuploidy was associated with the derivation of the WGD+ clones (Supplementary Fig. 7a). To further study karyotypic evolution under selection, we transferred the day-12 WGD+ cells to soft-agar and karyotyped four of the macroscopic colonies emerging from them (Supplementary Fig. 7b). Karyotypic heterogeneity was maintained, and no common aneuploidies were detected across these colonies (Supplementary Fig. 7c). Therefore, there was no evidence for rapid selection of a specific karyotype in either of these challenges, confirming that WGD+ cancer cells are permissive to a wide variety of aneuploidies and can find multiple evolutionary solutions to induced stress.
Figure 5: Isogenic WGD− and WGD+ DLD1 cell lines demonstrate a causal effect of WGD on aneuploidy landscapes.
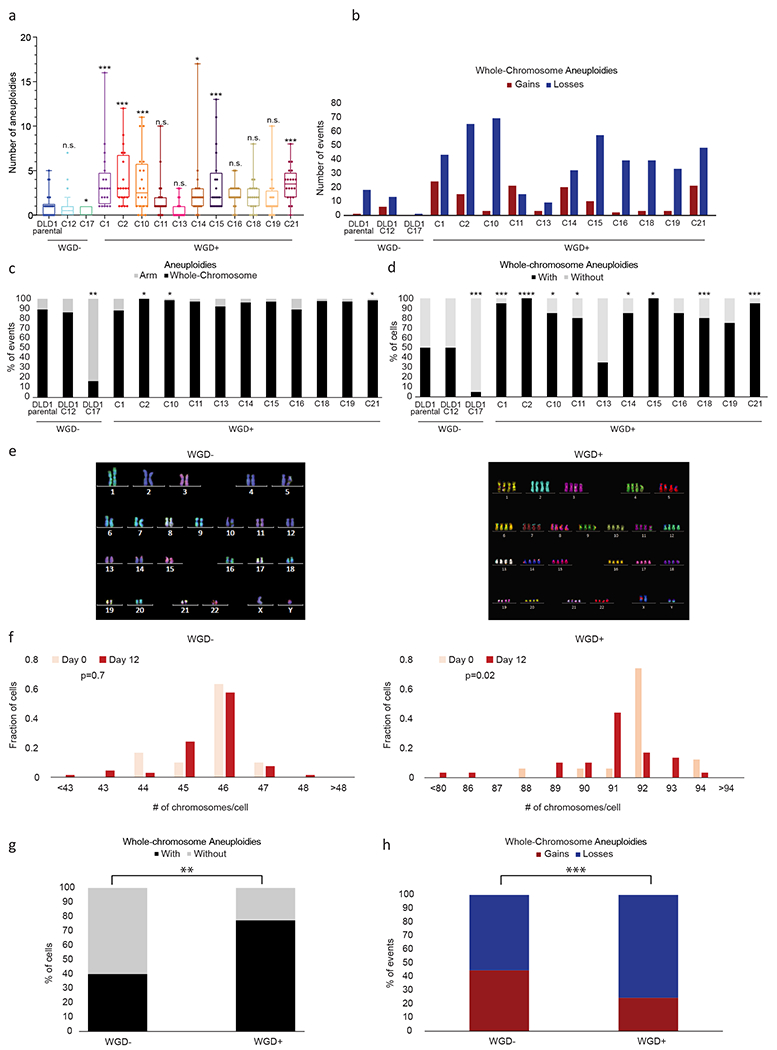
(a) mFISH-based comparison of the number of aneuploidies (relative to basal ploidy) between near-diploid parental DLD1 cells (DLD1-parental), two DLD1-derived WGD− single-cell clones, and 11 DLD1-derived WGD+ single-cell clones. n.s., p>0.05, *, p<0.05, ***, p<0.001; two-tailed Student’s t-test. All comparisons are with the DLD1-parental line. (b) The number of whole-chromosome gains and losses observed by mFISH, in DLD1 and its derived clones (>20 cells per clone). (c) The relative fraction of whole-chromosome aneuploidies relative to arm-level aneuploidies in DLD1 and its derived clones. *, p<0.05; one-tailed Fisher’s Exact test. All comparisons are with the DLD1-parental line. (d) The fraction of cells with non-clonal whole-chromosome aneuploidies, a measure of karyotypic heterogeneity, in DLD1 and its derived clones. *, p<0.05, ***, p<0.001, ****, p<0.0001; one-tailed Fisher’s Exact test. All comparisons are with the DLD1-parental line. (e) Representative karyograms of WGD− and WGD+ DLD1 cells taken from the same cell population right after cytokinesis inhibition. (f) Histograms of the chromosome count of WGD− and WGD+ cells at day 0 (right after cytokinesis inhibition) and at day 12 (twelve days after cytokinesis inhibition). p=0.7 and p=0.02 for the difference in the means of the distributions between day 0 and day 12, in the WGD− and WGD+ populations, respectively; two-tailed Mann-Whitney test. For WGD− day 0: means=45.67, S.D.=0.88; for WGD− day 12: means=45.61, S.D.=1.01; for WGD+ day 0: means=91.82, S.D.=1.33; for WGD+ day 12: means=90.81; S.D.=2.10. (g) Comparison of the fraction of cells with whole-chromosome aneuploidies, a measure of karyotypic heterogeneity, in WGD− and WGD+ DLD1-derived cells at day 12 following cytokinesis inhibition. **, p<0.01; one-tailed Fisher’s Exact test. (h) The fraction of whole-chromosome gains and losses in WGD− and WGD+ DLD1-derived cells at day 12 following cytokinesis inhibition. ***, p<0.001; one-tailed Fisher’s Exact test.
Lastly, we induced WGD in non-transformed RPE1 cells, and compared the near-diploid WGD− RPE1 cells, their single-cell derived WGD− clone, and their WGD+ derivative RPTs (RPE1 Post Tetraploid cells). The parental RPE1 cells and their WGD− single cell-derived clone were highly chromosomally stable (Supplementary Fig. 8a), in contrast to the WGD+ clones, whose aneuploidy landscape was dominated by chromosome losses (Supplementary Fig. 8b) and whole-chromosome aberrations (Supplementary Fig. 8c), and whose karyotypes were much more heterogeneous (Supplementary Fig. 8d). We conclude that the effect of WGD on aneuploidy formation in isogenic cell lines is reproducible across multiple cell line systems.
Human patient-derived cancer models also capture the association of WGD with aneuploidy features
To examine whether the association between WGD and aneuploidy holds true in physiologically-relevant cancer models, we used scDNAseq to analyze the aneuploidy patterns of WGD− vs. WGD+ cancer cells in two patient-derived models: patient-derived colon cancer organoids (PDOs) (31,32) and patient-derived breast cancer xenografts (PDXs) (33,34). An analysis of aneuploidy patterns in these models yielded highly-similar results to those obtained with the human cell lines: WGD+ cells exhibited a higher degree of aneuploidy and karyotypic heterogeneity, increased tendency towards chromosome losses, and increased fraction of whole-chromosome aneuploidies, compared to WGD− cells from the same tumors (Fig. 6). The effect of WGD on aneuploidy formation is therefore reproducible also in patient-derived cancer models. We note that despite a clear overall trend, there were individual tumors in which these associations did not hold true (Fig. 6), suggesting that WGD is a key – but not sole – factor determining aneuploidy patterns in tumors.
Figure 6: Association between WGD and aneuploidy landscapes in patient-derived organoids and xenografts.
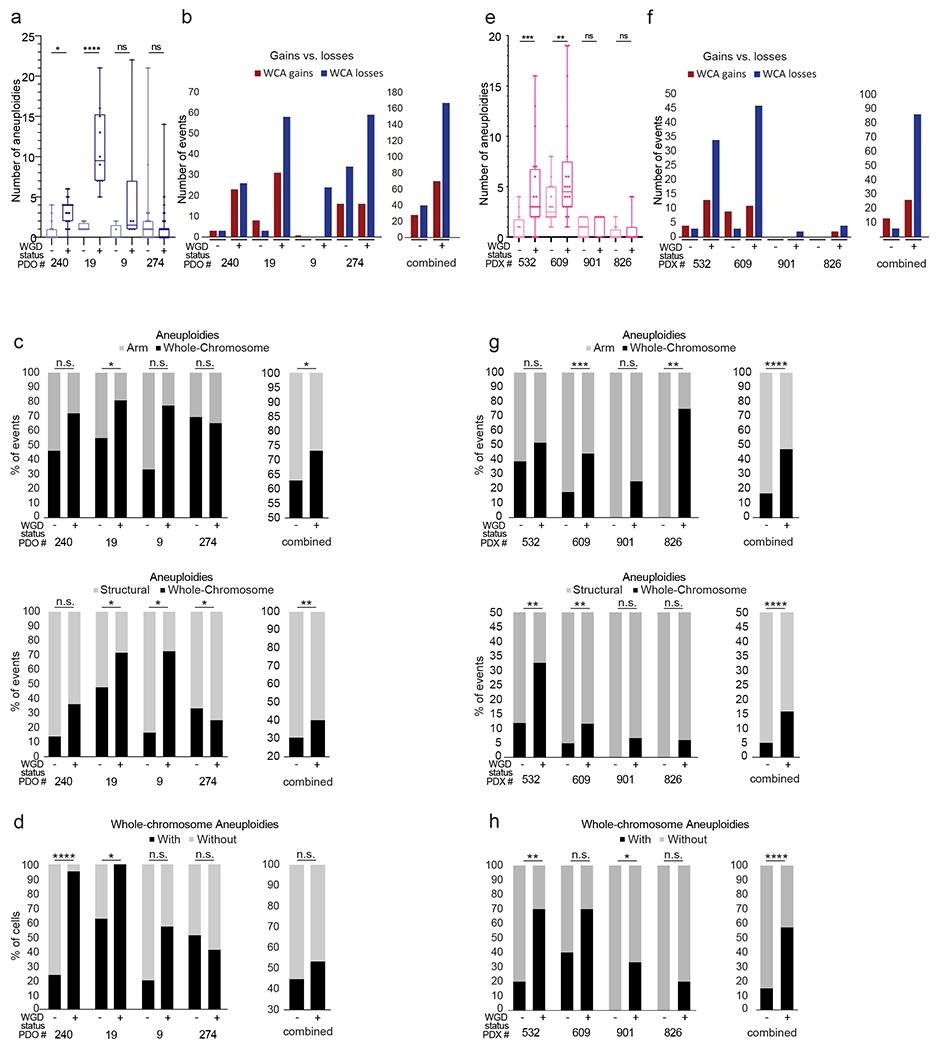
(a) mFISH-based comparison of the number of aneuploidies (relative to basal ploidy) between WGD− and WGD+ cells from 4 colon cancer patient-derived organoids (PDOs) (31,32). n.s., p>0.05, *, p<0.05, ****, p<0.0001; two-tailed Student’s t-test. (b) Left: the number of whole-chromosome gains and losses observed by mFISH in WGD− and WGD+ cells from 4 colon cancer PDOs. Right: a combined quantification of the 4 models. (c) Left: the relative fraction of whole-chromosome aneuploidies relative to arm-level aneuploidies (top) or structural aneuploidies (bottom; including arm-level aneuploidies, translocations and smaller structural alterations) in WGD− and WGD+ cells from 4 colon cancer PDOs. Right: a combined quantification of the 4 models. n.s., p>0.05, *, p<0.05, **, p<0.01; one-tailed Fisher’s Exact test. (d) Left: the fraction of cells with non-clonal whole-chromosome aneuploidies, a measure of karyotypic heterogeneity, in WGD− and WGD+ cells from 4 colon cancer PDOs. Right: a combined quantification of the 4 models. n.s., p>0.05, *, p<0.05, ****, p<0.0001; one-tailed Fisher’s Exact test. (e) mFISH-based comparison of the number of aneuploidies (relative to basal ploidy) between WGD− and WGD+ cells from 4 breast cancer patient-derived xenografts (PDXs) (33,34) . n.s., p>0.05, **, p<0.01, ***, p<0.001; two-tailed Student’s t-test. (f) Left: the number of whole-chromosome gains and losses observed by mFISH in WGD− and WGD+ cells from 4 breast cancer PDXs. Right: a combined quantification of the 4 models. (g) Left: the relative fraction of whole-chromosome aneuploidies relative to arm-level aneuploidies (top) or structural aneuploidies (bottom; including arm-level aneuploidies, translocations and smaller structural alterations) in WGD− and WGD+ cells from 4 breast cancer PDXs. Right: a combined quantification of the 4 models. n.s., p>0.05, **, p<0.01, ***, p<0.001; ****, p<0.0001; one-tailed Fisher’s Exact test. (h) Left: the fraction of cells with non-clonal whole-chromosome aneuploidies, a measure of karyotypic heterogeneity, in WGD− and WGD+ cells from 4 breast cancer PDXs. Right: a combined quantification of the 4 models. n.s., p>0.05, *, p<0.05, **, p<0.01, ****, p<0.0001; one-tailed Fisher’s Exact test.
Discussion
In recent years, it has become clear that the evolution of tumor aneuploidy is context-dependent, and is affected by the cell lineage, tumor developmental stage, the tumor environment, and the immune system (11,43). The genomic background from which aneuploidy arises plays an important role in determining the adaptive value of emerging chromosomal alterations (14,15,22). Here, we systematically compared the aneuploidy landscapes of WGD− and WGD+ tumors across all major tumor types, and found them to be distinct in important ways. The elevated degree of aneuploidy and increased tolerance to chromosome loss in WGD+ tumors were already reported prior to our study (1,2,8,12). However, our analysis yielded several novel insights. First, we show that WGD+ tumors are indeed more tolerant to a wider range of aneuploidies, as evident by their more diverse aneuploidy patterns. Second, whole-chromosome aneuploidies constituted a larger fraction of the karyotypic alterations in WGD+ tumors, suggesting that the increased chromosome mis-segregation and/or aneuploidy tolerance resulting from WGD (3,4,6,7,36,44) shape the aneuploidy landscapes of the tumors. Third, whereas tumor type-specific prevalent aneuploidies are largely shared by WGD− and WGD+ tumors, the genetic interactions between chromosome arms are WGD−dependent; in extreme cases, events that co-occur in WGD− tumors are mutually exclusive in WGD+ tumors, and vice versa. Fourth, opposite-direction alterations of same-chromosome arms are more common in WGD− tumors, again suggesting WGD−dependent mechanisms of aneuploidy formation and selection. Fifth, we show that the abovementioned associations are conserved in human cancer cell lines. Lastly, we use isogenic human cancer cell lines to demonstrate that WGD induction recapitulates the main changes observed in clinical samples, thereby demonstrating that the observed associations are causative rather than merely correlative. These isogenic cell line models are therefore appropriate for studying the interplay between WGD and aneuploidy in human cancer.
Ploidy-specific fitness advantage conferred by an aneuploid chromosome was previously reported in yeast (21). Recently, the aneuploidy landscape induced by CIN in yeast was shown to be more diverse in diploid yeast than in haploid yeast (17), consistent with our findings from human tumors. Furthermore, significantly different aneuploidy prevalence patterns were observed between haploid and diploid yeast, indicating ploidy-dependent genetic interactions between specific chromosomes (17). We now show that the fitness effects of specific chromosome-arm aneuploidy interactions can be ploidy-dependent in human cancer as well. Interestingly, a recent thermodynamic analysis of copy number states in human cancers revealed that WGD+ tumors are characterized by a distinct energy landscape, which is predicted to select for specific amplifications and deletions that favor the fitness of WGD+ tumors (45). This theoretical thermodynamic analysis matches our empirical findings from tumors and cancer cell lines. However, in organisms from yeast to human, further research is required to elucidate the mechanisms underlying specific ploidy-dependent chromosome interactions.
The finding that WGD+ tumors are skewed towards whole-chromosome aneuploidies suggests that chromosome mis-segregation plays a greater role in shaping the aneuploidy landscape of WGD+ tumors compared to WGD− tumors. The elevated prevalence of mis-segregation events observed in WGD+ cells (3,4,19,46) is clearly consistent with this finding. However, increased tolerance of WGD+ cells for whole-chromosome aneuploidies likely plays a role as well. It has been observed in both yeast and mammalian cells that tetraploid cells are more tolerant than diploid cells to numerical aneuploidy (7,17), and that the smaller the extra-chromosome the weaker the selection against it (47,48), lending support to the notion that WGD “buffers” the cellular consequences of aneuploidy. In line with these findings, we recently reported that following transient induction of CIN in HCT116 and HPT cells, the population of WGD− cells quickly selected for the parental near-diploid karyotype, whereas the population of WGD+ cells remained karyotypically heterogeneous, indicating a stronger selection against aneuploid karyotypes in WGD− vs. WGD+ populations (9). The relative contribution of mechanistic vs. selective pressures to the effect of WGD on aneuploidy evolution remains an open question for future research.
The genetically-matched cell line models before and after WGD induction enabled us to move from correlation to causation, demonstrating that WGD induction indeed alters some features of tumor aneuploidy. We note that some of the features that we found to be associated with WGD may still be influenced by other genomic properties that are tightly associated with WGD, such as p53 inactivation and ongoing CIN (2,5,8). Nevertheless, similar results were obtained with the APC-WT/TP53-WT colon cancer cell line HCT116 and with the APC-mutant/TP53-mutant colon cancer cell line DLD1 (40), as well as the non-transformed cell line RPE1, supporting the notion that the observed differences are indeed due to WGD. The same results were also found in colon cancer organoids and in breast cancer PDXs, which are more clinically-relevant than 2D cell lines.
We note that the effect of WGD on aneuploidy evolution may depend on the way by which the genome has doubled. Three distinct causes of tetraploidization have been proposed to occur in cancer (49): cell fusion, cytokinesis failure, and endoreduplication. These mechanisms may affect aneuploidy formation in different ways; for example, cytokinesis failure is more likely to result in additional mitotic aberrations such as whole-chromosome mis-segregation, whereas endoreduplication following persistent DNA damage would likely result in additional structural aneuploidies. As the cell line models used in this study are based on cytokinesis failure, how the way by which the genome is doubled affects aneuploidy evolution remains to be elucidated.
Notably, we did not observe selection for a specific aneuploidy profile in any of our in vitro approaches, which included single-cell cloning and growth in soft-agar. While these findings are in line with WGD being more permissive to aneuploidy in general, this does not mean that specific karyotypes would not get selected for under different selection pressures (50–52). Further studies are necessary in order to systematically map the interactions between WGD and specific aneuploidies across a variety of selection pressures.
Finally, it is tempting to speculate that by comparing within-chromosome aneuploidy patterns between WGD− and WGD+ tumors, the “driver” chromosome-arm could be identified. For example, in WGD+ colon cancer, loss of chromosome 8p arises together with gain of chromosome 8q in ~20% of the tumors (Fig. 2e). At the same time, a gain of the entire chromosome 8 is much more prevalent than a loss of that chromosome in this tumor type (Fig. 2e), suggesting that it is the gain of Chr8q that is likely the driving force underlying the prevalence of this chromosome gain. This reasoning remains to be functionally tested in future studies.
In summary, our study shows that WGD shapes the aneuploidy landscapes of human tumors both quantitatively and qualitatively. Therefore, when examining the tumorigenic role of a specific aneuploidy or of a specific interaction between a pair of cancer aneuploidies, the WGD status of the tumor must be considered. More broadly, WGD should be accounted for when studying and reporting aneuploidy prevalence patterns of human cancer.
Supplementary Material
Statement of significance.
These findings suggest that the interactions between whole-genome duplication and aneuploidy are important for tumor evolution, highlighting the need to consider genome status in the analysis and modeling of cancer aneuploidy.
Acknowledgements
The authors would like to acknowledge Jonathan M. Herrera, Eli Reuveni and Gali Yanovich-Arad for technical assistance with computational analyses; Ran Elkon and Ron Shamir for helpful advice and for sharing computing resources; A. Chan Yong Jie and Peter Walian for assistance with data transfer; Caroline Joseph and Venkat Rangachari for technical assistance with image file formatting; Sharon Tsach for assistance with figure preparation; and Peter Duesberg for allowing us to collect mFISH data in his lab. This work was funded by the European Research Council Starting Grant (U.B.-D.; grant #945674), the Israel Cancer Research Fund Gesher Award (U.B.-D.), the Azrieli Faculty Fellowship (U.B.-D), the Israel Science Foundation (U.B.-D.; grant #1339/18; H.L.), the Virginia Tech College of Science Dean’s Discovery Fund (D.C.), the Virginia Tech ICTAS Center for Engineered Health seed funds (D.C.), the U.S. National Institutes of Health (grant #R01GM140042; D.C.), and Deutsche Forschungsgemeinschaft (STO918-7; Z.S.). Work in the G.R. lab is supported by the Singapore National Research Foundation grant NRFI05-2019-0008. U.B.-D. is an EMBO Young Investigator.
Authors’ Disclosures
G.R. is an employee of F. Hoffman-La Roche AG. U.B.-D. receives grant funding from Novocure Inc. The other authors declare no competing interests.
References
- 1.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bielski CM, Zehir A, Penson AV, Donoghue MTA, Chatila W, Armenia J, et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet. 2018;50:1189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fujiwara T, Bandi M, Nitta M, Ivanova EV., Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–7. [DOI] [PubMed] [Google Scholar]
- 4.Watkins TBK, Lim EL, Petkovic M, Elizalde S, Birkbak NJ, Wilson GA, et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature. 2020;587:126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.López S, Lim EL, Horswell S, Haase K, Huebner A, Dietzen M, et al. Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat Genet. 2020;52:283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Grönroos E, Endesfelder D, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014;4:175–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuznetsova AY, Seget K, Moeller GK, de Pagter MS, de Roos JA, Durrbaum M, et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle. 2015;14:2810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quinton RJ, DiDomizio A, Vittoria MA, Kotýnková K, Ticas CJ, Patel S, et al. Whole-genome doubling confers unique genetic vulnerabilities on tumour cells. Nature. 2021;590:492–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen-Sharir Y, McFarland JM, Abdusamad M, Marquis C, Bernhard SV, Kazachkova M, et al. Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature. 2021;590:486–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marquis C, Fonseca CL, Wood L, Malaby HLH, Clayton JE, Stumpff J. Chromosomally unstable tumor cells specifically require KIF18A for proliferation. bioRxiv. 2020;2020.06.18.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ben-David U, Amon A. Context is everything: aneuploidy in cancer. Nat Rev Genet. 2019;21:44–62. [DOI] [PubMed] [Google Scholar]
- 12.Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell. 2018;33:676–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017;49:1567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gatza ML, Silva GO, Parker JS, Fan C, Perou CM. An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer. Nat Genet. 2014;46:1051–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben-David U, Ha G, Khadka P, Jin X, Wong B, Franke L, et al. The landscape of chromosomal aberrations in breast cancer mouse models reveals driver-specific routes to tumorigenesis. Nat Commun. 2016;7:12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ozery-Flato M, Linhart C, Trakhtenbrot L, Izraeli S, Shamir R. Large-scale analysis of chromosomal aberrations in cancer karyotypes reveals two distinct paths to aneuploidy. Genome Biol. 2011;12:R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ravichandran MC, Fink S, Clarke MN, Hofer FC, Campbell CS. Genetic interactions between specific chromosome copy number alterations dictate complex aneuploidy patterns. Genes Dev. 2018;32:1485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shukla A, Nguyen THM, Moka SB, Ellis JJ, Grady JP, Oey H, et al. Chromosome arm aneuploidies shape tumour evolution and drug response. Nat Commun. 2020; 1:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Storchová Z, Breneman A, Cande J, Dunn J, Burbank K, O’Toole E, et al. Genome-wide genetic analysis of polyploidy in yeast. Nature. 2006;443:541–7. [DOI] [PubMed] [Google Scholar]
- 20.Scott AL, Richmond PA, Dowell RD, Selmecki AM. The Influence of Polyploidy on the Evolution of Yeast Grown in a Sub-Optimal Carbon Source. Mol Biol Evol. 2017;34:2690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selmecki AM, Maruvka YE, Richmond PA, Guillet M, Shoresh N, Sorenson AL, et al. Polyploidy can drive rapid adaptation in yeast. Nature. 2015;519:349–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas R, Marks DH, Chin Y, Benezra R. Whole chromosome loss and associated breakage-fusion-bridge cycles transform mouse tetraploid cells. EMBO J. 2018;37:201–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan Z, Chan YJA, Chua YJK, Rutledge SD, Pavelka N, Cimini D, et al. Environmental stresses induce karyotypic instability in colorectal cancer cells. Mol Biol Cell. 2019;30:42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bloomfield M, Chen J, Cimini D. Spindle architectural features must be considered along with cell size to explain the timing of mitotic checkpoing silencing. Fron Physiol. 2021;11:596263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang M, Zhao Y, Zhang B. Efficient Test and Visualization of Multi-Set Intersections. Sci Rep. 2015;5:16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baudoin NC, Nicholson JM, Soto K, Martin O, Chen J, Cimini D. Asymmetric clustering of centrosomes defines the early evolution of tetraploid cells. Elife. 2020;9:e54565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bakker B, Taudt A, Belderbos ME, Porubsky D, Spierings DCJ, de Jong T V., et al. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol. 2016;17:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolhaqueiro ACF, Ponsioen B, Bakker B, Klaasen SJ, Kucukkose E, van Jaarsveld RH, et al. Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat Genet. 2019;51:824–34. [DOI] [PubMed] [Google Scholar]
- 32.Bollen Y, Stelloo E, van Leenen P, van den Bos M, Ponsioen B, Lu B, et al. Reconstructing single-cell karyotype alterations in colorectal cancer identifies punctuated and gradual diversification patterns. Nat Genet. 2021;53:1187–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laks E, McPherson A, Zhan H, Lai D, Steif A, Brimhall J, et al. Clonal Decomposition and DNA Replication States Defined by Scaled Single-Cell Genome Sequencing. Cell. 2019;179:1207–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salehi S, Kabeer F, Ceglia N, Andronescu M, Williams MJ, Campbell KR, et al. Clonal fitness inferred from time-series modelling of single-cell cancer genomes. Nature. 2021;595:585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157–62. [DOI] [PubMed] [Google Scholar]
- 36.Wangsa D, Quintanilla I, Torabi K, Vila-Casadesús M, Ercilla A, Klus G, et al. Near-tetraploid cancer cells show chromosome instability triggered by replication stress and exhibit enhanced invasiveness. FASEB J. 2018;32:3502–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirkpatrick S, Gelatt CD, Vecchi MP. Optimization by simulated annealing. Science. 1983;220:671–80. [DOI] [PubMed] [Google Scholar]
- 38.Vasudevan A, Baruah PS, Smith JC, Wang Z, Sayles NM, Andrews P, et al. Single-Chromosomal Gains Can Function as Metastasis Suppressors and Promoters in Colon Cancer. Dev Cell. 2020;52:413–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ben-David U, Beroukhim R, Golub TR. Genomic evolution of cancer models: perils and opportunities. Nat Rev Cancer. 2019;19:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV., Lo CC, McDonald ER, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Passerini V, Ozeri-Galai E, De Pagter MS, Donnelly N, Schmalbrock S, Kloosterman WP, et al. The presence of extra chromosomes leads to genomic instability. Nat Commun. 2016;7:10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol. 2012;8:608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baudoin NC, Bloomfield M. Karyotype Aberrations in Action: The Evolution of Cancer Genomes and the Tumor Microenvironment. Genes (Basel). 2021;12:558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Remacle F, Graeber TG, Levine RD. Thermodynamic energetics underlying genomic instability and whole-genome doubling in cancer. Proc Natl Acad Sci U S A. 2020;117:18880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, et al. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 2007;317:916–24. [DOI] [PubMed] [Google Scholar]
- 48.Peter J, De Chiara M, Friedrich A, Yue JX, Pflieger D, Bergstrom A, et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature. 2018; 556:339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davoli T, Lange T de. The Causes and Consequences of Polyploidy in Normal Development and Cancer. Ann Rev Cell Dev Biol. 2011;27:585–610. [DOI] [PubMed] [Google Scholar]
- 50.Rutledge SD, Douglas TA, Nicholson JM, Vila-Casadesus M, Kantzler CL, Wangsa D, et al. Selective advantage of trisomic human cells cultured in non-standard conditions. Sci Rep. 2016;6:22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ippolito MR, Martis V, Martin S, Tijhuis AE, Hong C, Wardenaar R, et al. Gene copy-number changes and chromosomal instability induced by aneuploidy confer resistance to chemotherapy. Dev Cell. 2021;56:2440–54. [DOI] [PubMed] [Google Scholar]
- 52.Lukow DA, Sausville EL, Suri P, Chunduri NK, Leu J, Kendall J, et al. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dec Cell. 2021;56:2427–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets are available within the article and its Supplementary Tables, or from the Corresponding Author upon request. The code used to analyze the data is available at https://github.com/BenDavidLab/WGD_Aneuploidy.