

Abstract
Gastric adenocarcinoma (GC) is a devastating disease and is the 3rd leading cause of cancer deaths worldwide. This heterogenous disease has several different classification systems that consider histological appearance and genomic alterations. Understanding the etiology of GC, including infection, hereditary conditions, and environmental factors is of particular importance and is discussed in this review. To improve survival in GC, we also must improve our therapeutic strategies. Here, we discuss new targets that warrant further exploration.
Introduction
Gastric adenocarcinoma (GC) is currently the 5th most common type of cancer and 3rd leading cause of cancer related deaths worldwide. This disease is very heterogenous, resulting in variable effectiveness with current treatments. GC is very common in East Asian populations with men having a two times greater likelihood of developing gastric cancer [1, 2]. Regional differences in etiology and disease progression are important in disease prognosis and therapeutic effectiveness.
Current standard of care for GC can be broken up into two categories; surgical candidates, and non-surgical/metastatic patients. Individuals who are candidates for surgical resection may receive neo-adjuvant chemotherapy or chemoradiotherapy followed by surgery, then adjuvant chemotherapy or chemoradiotherapy [3]. Commonly utilized chemotherapy regimens include perioperative fluorouracil, leucovorin, oxaliplatin, and docetaxel (FLOT) and the 3 drug combination, epirubicin, cisplatin, and fluorouracil (ECF). In the phase 2/3 FLOT4-AIO trial, patients in the FLOT arm saw substantial increase in overall survival (median overall survival (OS) 50 vs 35 months) and 9% increase in 5-year survival rate (45% vs 36%) compared with ECF [4]. Depending on the etiology of disease, other therapeutic strategies might be implemented as standard of care treatment, such anti-HER2, immune checkpoint therapy, or anti-VEGF therapy. Patients with surgically unresectable GC generally receive systemic chemotherapy with the goal to reduce symptoms, prolong survival, and control disease. Chemotherapy choice is dependent on patient comorbidities and current health. Generally, a fluoropyrimidine and platinum double is chosen, with an additional chemotherapy added to patients who will be able to tolerate high toxicity [5]. Targeted therapy is also used in advanced disease such as anti-HER2, anti-PD-L1, or anti-VEGF depending on patient tumor characteristics. The major problem with treatment selection and efficacy in this patient population is intra-tumoral heterogeneity, which facilitates treatment resistance.
Classification of Gastric Adenocarcinoma
Histological
Gastric adenocarcinoma is classified using two different schema based on histological and molecular subtypes (Figure 1). The Lauren classification of GC divides this cancer into three different subtypes; intestinal, diffuse, or mixed/intermediate [6]. This is based on the histological appearance of the tumor and is associated with specific disease characteristics. Intestinal subtype is the most common subtype and is often found in the gastric antrum of elderly male patients. Histologically, this subtype appears with tubular or glandular formations with intestinal metaplasia and is associated with lymphatic and vascular invasion. Diffuse subtype is characterized by tumor cells that lack adhesion and infiltrate as single or small group of cells. This subgroup includes signet ring cell carcinoma where intracellular mucin pushes the nucleus to the side of the cell. This subtype is most often found in younger female patients and is more common in low-risk areas (Western countries). It is commonly found in the body of the stomach with a short course and worse prognosis compared to the intestinal subtype. Diffuse GC is more commonly associated with genetic/hereditary causes (CDH1, APC, etc) while intestinal subtype is associated with environmental causes (Helicobacter pylori, diet, reflux, etc.) [6, 7]. Mixed or intermediate classification has characteristics of both intestinal and diffuse and can account for 20% of gastric cancers. Unlike the other two subtypes, mixed gastric adenocarcinoma has not been extensively studied but has been associated with aggressive disease and worst overall survival [8].
Figure 1.
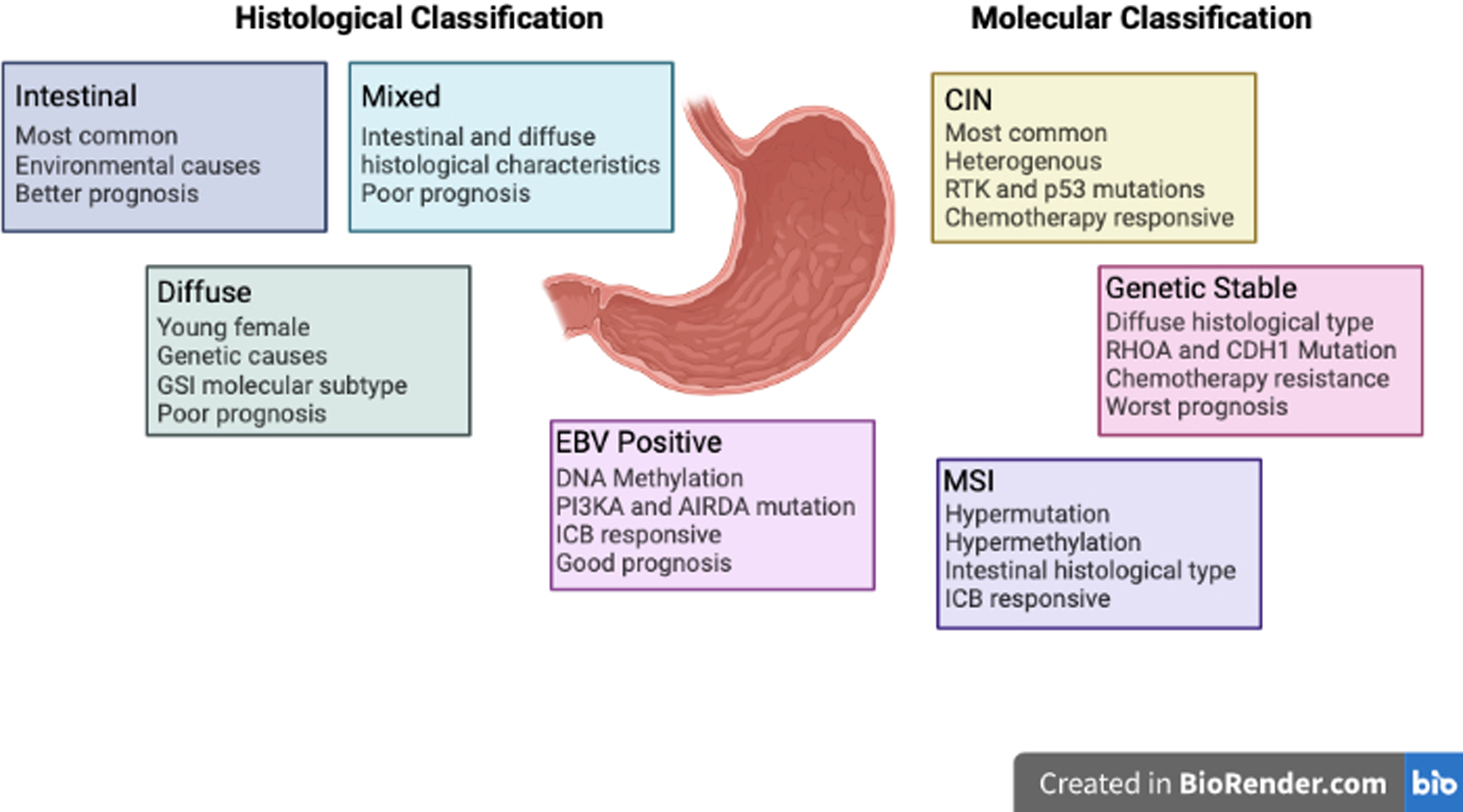
Histological and molecular subtypes of gastric cancer. (Created with BioRender.com)
The World Health Organization (WHO) has also classified GC based on histological appearance into four subtypes. This includes tubular, papillary, poorly cohesive, and mucinous subtypes. These classifications are based on histological appearance and each subtype has specific characteristics like the Lauren’s classification. The tubular, papillary, and mucinous subtype is most like the intestinal subtype of Lauren’s classification. The poorly cohesive subtype (including signet ring carcinoma) is most like diffuse subtype [9, 10]. Other groups have also attempted to classify this cancer into different subtypes. Carnerio et al divided this disease into 4 different groups based on immunophenotype [11]. Goseki et al classified gastric cancer based on mucin production and level of tubular differentiation [12]. Ming et al and Grundman et al also created their own classification system for gastric cancer based on morphological differences [13, 14]. The existence of multiple classifications for gastric cancer based on histology speaks to the vast heterogeneity found in this patient population.
Molecular
The Cancer Genome Atlas (TCGA) has classified GC molecularly using genomic and RNA sequencing and protein arrays on 295 gastric adenocarcinoma tumors. TCGA has categorized this disease into four distinct molecular subtypes; EBV positive, microsatellite instability, genomic stable, and chromosomal instability [15].
Chromosomal instable subtype (CIN) is the most common, accounting for 49% of gastric cancer cases. CIN is associated with mutations in p53, receptor tyrosine kinases, and amplification mutations for VEGFA [15]. This group is the most heterogenous of the four TCGA subtypes, which lack a universal marker for identification. This subtype prognosis is worse than the EBV subtype but better than the genomic stable group. This patient group benefits the most with adjuvant chemotherapy treatment with significant increases in recurrence free survival (RFS) [16].
EBV subtype is associated with Epstein Barr Virus infection and found in 9% of gastric cancers. It is often found in the fundus of the stomach in younger males. This subtype has a high incidence of DNA hypermethylation. It is also associated with a PIK3CA and ARID1A mutation. TCGA analysis also found upregulation of immune checkpoint molecules such as PD-L1/2 and responsiveness to immunotherapies [15]. Kim et al found that the overall response rate in EBV positive gastric cancer was 100% when treated with pembrolizumab (anti-PD1) [17]. Similarly, EBV positive patients treated with immunotherapies had favorable outcomes compared to EBV negative GC cohort [18]. Out of the four TCGA subtypes, this one has the best prognosis, which is thought to be due to a robust immune response related to the viral infection [16].
Genomic stable subtype (GS) is often found within the group of diffuse histological subtype and accounts for 20% of gastric cancer cases. This subtype often has a mutation in the Ras homolog gene family member A (RHOA) and CDH1 (E-Cadherin) [15]. RHOA is a GTPasae-activating protein that is responsible for regulating actomyosin dynamics, cell adhesion, proliferation, and survival [19]. E-cadherin is a cell adhesion molecule and important in regulating cell signaling and cell adhesion and polarity [20]. In a study assessing the OS of all four subtypes, GS was found to have the worst overall prognosis. The GS subtype had little to no response to adjuvant chemotherapy and was most likely to have chemoresistance to 5-fluoruracil [16].
The microsatellite instability subtype (MSI) accounts for 22% of gastric cancers and often found in the body and pyloric area of the stomach. MSI is associated with hypermutation, MLH1 silencing and hypermethylation of CpG islands. This correlates to the Lauren intestinal subtype. The mutations in this subtype often affect PIK3CA, ERRB3, ERBB2, and EGFR but lack the common BRAF V600E mutation often seen in other cancers with MSI [15]. The overall prognosis of this subtype is in the middle of the four categories and is thought to be due to the increased immune response due to neoantigen production [16]. MSI tumors have a very high expression of immune checkpoint molecules like CTLA4, PD-1, PD-L1, and LAG-3 which suggests that immune checkpoint therapy would be very effective in this subtype of gastric cancer [21, 22]. In a study analyzing the effect of microsatellite status on immunotherapy effectiveness, it found that pembrolizumab + chemotherapy improved OS and PFS compared to the placebo/chemotherapy only group [23]. The CHECKMATE 032 study tested the combination of nivolumab (anti-PDL1) with ipilimumab (anti-CTLA4). When breaking the patients up based on microsatellite status, the disease control rate was 71% for MSI with an ORR of 28% and a longer median OS of 14.75 months compared to microsatellite stable group [24].
The Asian Cancer Research Group has also identified a molecular classification system for gastric cancer. This classification system is based around microsatellite instability and is broken up into 4 different groups, microsatellite instability, microsatellite stable and epithelial to mesenchymal transition, microsatellite stable and p53 intact, and microsatellite stable and p53 mutated [25].
Etiology of Gastric Adenocarcinoma
EBV
Epstein Barr virus (EBV) is an extremely common virus found the blood of in 90% of adults. This virus is known to cause Burkitt’s lymphoma nasopharyngeal, T-cell lymphomas, and gastric cancer [26]. EBV positive gastric cancer has specific clinical and pathological characteristics with a good overall prognosis. Individuals with the viral induced GC make up their own molecular subtype defined by TCGA, as mentioned above [15, 16]. This virus induces GC through translocation of the viral genome to the nucleus, resulting in expression of several virulence factors needed to induce genome wide hypermethylation. During this latency period of infection, it disrupts gene expression, generates a suitable tumor microenvironment, and infects other gastric epithelium[27]. EBV can also infect cells through a latent viral cycle resulting in expression of latent proteins EBERs, BARF-0, EBNA-1, and LMP2A. These proteins are responsible for disrupting the miR-200 family, leading to reduced expression of E-cadherin [28]. Each latent protein also has specific roles that have been implicated in GC disease progression [29–32]
Helicobacter pylori
Helicobacter pylori is a gram negative spiral shaped bacteria known to cause gastric inflammation, uclers, and GC. This bacterium infects the gastric mucosa resulting in disruption of normal epithelium and induction of chronic inflammation resulting in carcinogenesis. Of individuals infected with H. pylori, only a small percentage developing gastric ulcers and only 2–3% will progress to GC [33]. The propensity for this pathogen to induce GC is dependent on several aspects including patient genetic and environmental influences and virulence of the strain of H. pylori. H. pylori induced GC is associated with the Lauren intestinal subtype and often found in the non-cardia portion of the stomach [7, 33]. This bacterium has several different mechanisms that are responsible for carcinogenesis including induction of chronic inflammation and virulence factors that disrupt cell homeostasis. Pre-clinical studies on H. pylori induced GC have identified that this bacterium can induce autophagy, NF-κB signaling, disrupt cell adhesions, inhibit anti-tumor immunity, and promote epithelial to mesenchymal transition (EMT) [34–38]. Primary and secondary prevention strategies in Eastern Asian countries against H. pylori infection, including awareness of bacterial infection and symptoms and subsequent treatment with antibiotics, have shown to be very effective in reducing incidence of gastric cancer [39, 40].
Hereditary
Around 10% of gastric cancer have familial association but only 1–3% are connected to inherited genetic disorders (hereditary diffuse gastric carcinoma (HDGC), familial adenomatous polyposis, hereditary nonpolyposis colorectal carcinoma (or Lynch syndrome), Peutz-Jeghers syndrome, etc.) [41]. Hereditary causes of GC can be broken up into two different categories, polyp forming and non-polyp forming diseases, and are discussed below.
Non-polyp forming hereditary disease is the most common cause of hereditary gastric cancer. The most common non-polyp forming hereditary disease associated with gastric cancer is hereditary diffuse gastric carcinoma. This disease is an autosomal dominant predisposition to having either diffuse gastric cancer or breast cancer. Individuals with this disease have a 70% chance of having diffuse GC in their lifetime for males and 56% for females [42]. Molecularly, this disease is driven by a heterozygous mutation to the CDH1 gene. CDH1 codes for the calcium dependent cell adhesion protein and tumor suppressor, E-cadherin [43]. E-cadherin plays an important role in regulating the signaling of several pathways responsible for tumorigenesis. This includes the Wnt signaling pathway through binding and sequestering of B-catenin, preventing signaling through that pathway [20]. When E-cadherin is mutated, it was found that Wnt signaling regulates both proliferation and chemotherapy resistance in GC [44, 45]. E-cadherin also plays an important role in maintaining cell adhesion and polarity and when mutated, there is a loss of contact inhibition and cell migration [46, 47]. Hereditary nonpolyposis colon cancer syndrome is another autosomal dominant disorder associated with an increased risk of GC. This disease is characterized by a mutation in the DNA repair system (MLH1, MSH2, MSH6, PMS2, etc.) resulting is microsatellite instability, or repeats of DNA sequences throughout the genome [48]. The lifetime risk for GC is regionally different for this disorder, with higher risk in Eastern Asia (30–40%) and lower risk in Western countries [49, 50]. Other non-polyp forming hereditary diseases that are associated with gastric cancer include hereditary breast cancer (BRCA1/2 mutations) and Li Fraumeni syndrome (p53 mutation). Both of these hereditary syndromes have a slight increased risk of gastric cancer [51–53].
Polyp forming diseases associated with an increased risk of gastric cancer include syndromes like familial adenomatous polyposis (FAP), gastric adenocarcinoma and proximal polyposis syndrome (GAPPS), Peutz-Jegher Syndrome, and others. These diseases are associated with polyp formation in the gastro-intestinal system due to hereditary mutations that result in an increased propensity to have cancer in several locations, including the stomach. Familial adenomatous polyposis (FAP) is an autosomal dominant disorder with a heterozygous mutation in the tumor suppressor gene APC. Gastric polyp incidence in this disease is very high with the risk of gastric adenocarcinoma regionally determined. Eastern Asian has a risk around 5–10% while in western countries it is around 0.6−.4.2% [54–56]. Gastric adenocarcinoma and proximal polyposis syndrome (GAPPS) is a relatively new syndrome and is generally a diagnosis of exclusion after ruling out other polyp forming diseases. These individuals have an increased risk of gastric cancer with multiple polyps with areas of dysplasia followed by development of carcinomas [57]. This is a rare disorder that is associated with APC dysregulation due to a mutation in the promoter IB which results in reduced binding of the Yin Yang 1 transcription factor. Promoter IB for APC is reportedly the more transcriptionally active promoter for APC and so when this promoter is lost through mutation there is a marked reduction in APC production [58–60]. The alternative promoter for APC, promoter IA, is often methylated in gastric epithelial cells which is thought to contribute to the localization of this phenomena to the stomach [61]. Generally, the polyps start to form in adolescence in the fundus and body of the stomach. These polyps progress with high grade dysplasia to carcinomas often associated with other somatic mutations in genes such as APC, TP53, GNAs, and FBXW7 [57]. Peutz-Jeghers syndrome is another autosomal dominant hereditary syndrome associated with an increased risk of gastrointestinal cancers, including GC. This syndrome is characterized by a germline mutation in the tumor suppressor gene STK11 resulting in gastrointestinal hamartomatous polyps and melanocytic macules. The driver mutation for this disease is generally considered the “initiator” followed by other somatic mutations required for the development of cancer [62].
Environmental factors
Environmental factors play a very important role in GC pathogenesis. The Lauren intestinal subtype is associated with environmental factors [7]. As the stomach is a part of the digestive system, diet plays a very important role in carcinogenesis. An EPIC study found that a diet’s inflammatory index is directly correlative to a patient’s risk of having GC [63]. A diet high in red meat, processed meat, or smoked/cured meats has also been associated with an increased risk of GC due to the formation of carcinogens during cooking or digestion [64, 65]. Both a high salt diet and smoking are also risk factors for GC [66]. Foods high in antioxidants (fruits and vegetables), have been shown to reduce the risk of GC through combating the generation of the carcinogens mentioned above [67, 68]
Targetable pathways in gastric adenocarcinoma
Advances in our understanding of the pathways involved in gastric cancer has led to several emerging treatment targets for pre-clinical development and ongoing clinical trials (Table 1).
Table 1.
Completed clinical trials for the targetable pathways in gastric cancer.
| Pathway | Name | Treatment | Conclusions | Reference |
|---|---|---|---|---|
| Angiogenesis | REGARD | Ramucirumab | OS 5.2 mo vs. 3.8 mo | Fuchs et al. 2014 |
| RAINBOW | Ramucirumab + Pacitaxel | OS 9.6 mo vs. 7.4 mo | Wilke et al. 2014 | |
| Apatinib | Apatinib | PFS 2.6 mo vs 1.8 mo OS 6.5 mo vs. 4.7 mo |
Li et al. 2016 | |
| INTEGRATE | Regorafenib | PFS 2.6 mo vs. 0.9 mo | Pavlakis et al. 2016 | |
| AVAGAST | Bevacizumab + Capecitabine/Cisplatin | PFS 6.7 mo vs. 5.3 mo OS 12.1 vs 10.1 mo |
Ohtsu et al. 2011 | |
| AVATAR | Bevacizumab + Capecitabine/Cisplatin | PFS 6.3 mo vs. 6.0 mo OS 10.5 mo vs 11.6 mo |
Shen et al. 2013 | |
| Ma et al | Bevacizumab + DOF | OS 17.6 mo vs. 16.4 mo DFS 15.2 mo vs. 12.3 TRR 65% vs 42.5% |
Ma et al. 2015 | |
| Moy et al | Regorafenib + mFOLFOX6 | 6 mo PFS 56% | Moy et al. 2020 | |
| HER2 | TRIO-013/LOGiC | Lapatinib + Capecitabine/Oxaliplatin | PFS 6.0 mo vs. 5.4 mo OS 12.2 mo vs. 10.5 mo Response rate 53% vs 39% |
Hecht et al. 2016 |
| Iqbal et al | Lapatinib | OS 4.8 mo Well tolerated with minimal adverse effects |
Iqbal et al. 2011 | |
| TyTAN | Lapatinib + Pacitaxel | OS 11 mo vs. 8.9 mo PFS 5.4 mo vs 4.4 mo |
Satoh et al. 2014 | |
| ToGA | Trastuzumab + Capecitabine/Cisplatin or Fluorouracil/Cisplatin | OS 13.8 mo vs. 11.1 mo | Bang et al. 2010 | |
| T-ACT | Trastuzumab + Pacitaxel | PFS 3.2 mo vs. 3.7 mo OS 10 mo vs. 10 mo ORR 32% vs. 33% |
Makiyama et al. 2020 | |
| GATSBY | Trastuzumab + Pacitaxel/Docetaxel | OS 7.9 mo vs. 8.6 mo | Thuss-Patience et al. 2017 | |
| JACOB | Pertuzumab + Trastuzumab + Capecitabine/Cisplatin or 5-fluorouracil | OS 17.5 mo vs. 14.2 mo | Tabernero et al. 2018 | |
| Shitara et al | Trastuzumab + Chemotherapy | OS 24.7 mo vs. 13.9 mo in HER2+ patients | Shitara et al. 2013 | |
| Shitara et al | Trastuzumab + Irinotectan/Pacitaxel | OS 12.5 mo vs. 8.4 mo | Shitara et al. 2020 | |
| Janjigian et al | Trastuzumab + Pembrolizumab + Capecitabine/Cisplatin or Oxaliplatin | 6 mo PFS 70% Safe combination with minimal adverse effects |
Janjigian et al. 2020 | |
| Bang et al | Margetuximab | Tumor reduction in 78% Well tolerated with only Grade 1/2 toxicities | Bang et al. 2017 | |
| CP-MGAH22-05 | Margetuximab + Pembrolizumab | Well tolerated | Catenacci et al 2020 | |
| RTK | FOLFETUX | Cetuximab + FOLFIRI | ORR 44.1% Well tolerated |
Pinto et al. 2007 |
| Han et al | Cetuximab + FOLFOX6 | 50% response rate OS 9.9 mo |
Han et al. 2009 | |
| Moehler et al | Cetuximab + Irinotecan and 5-fluouracil | ORR 46% Disease control 79% PFS 9 mo OS 16.5 mo |
Moehler et al. 2011 | |
| Kim et al | Cetuximab + Oxaliplatin/Capecitabine | ORR 52.3% PFS 6.5 mo OS 11.8 mo Well tolerated |
Kim et al. 2011 | |
| EXPAND | Cetuximab + Cisplatin/Capecitabine | PFS 4.4 mo vs. 5.6 mo | Lordick et al. 2013 | |
| Rojo et al | Gefitinib | Reduced pEGFR levels in tumors | Rojo et al. 2006 | |
| MMP-9 | Shah et al | Adencalazimab + mFOLFOX6 | ORR 47.5% | Shah et al. 2018 |
| DNA Damage | Bang et al | Olaparib + Paclitaxel | PFS 3.91 mo vs. 3.55 mo OS 13.1 mo vs. 8.3 mo |
Bang et al 2015 |
| GOLD | Olaparib + Paclitaxel | OS 8.8 mo vs. 6.9 mo | Bang et al. 2017 | |
| Friedlander et al | Tislelizumab + Pamiparib | 49 patients achieved objective response | Friedlander et al. 2019 | |
| Immunotherapy | ATTRACTION-2 | Nivolumab | OS 5.26 mo vs. 4.14 mo 12 mo ORR 26.2% vs. 10.9% |
Kang et al. 2017 |
| JAVELIN Gastric 300 | Avelumab + Pacitaxel/Irinotecan | OS 4.6 vs. 5.0 mo PFS 1.4 vs. 2.7 mo ORR 2.2% vs 4.3% |
Bang et al. 2018 | |
| ATTRACTION-4 | Nivolumab + SOX/CapeOX | ORR 57.1% (SOX) 76.5% (CapeOX) PFS 9.7 mo (SOX) 10.6 mo (CapeOX) |
Boku et al. 2019 | |
| KEYNOTE-059 | Pembrolizumab | PD-L1+ ObRR 15.5% Response duration 16.3 mo PD-L1- ObRR 6.4% Response duration 6.9 mo |
Fuchs et al. 2018 | |
| KEYNOTE-061 | Pembrolizumab + Pacitaxel | OS 9.1 vs. 8.3 mo PFS 1.5 mo vs 4.1 mo |
Shitara et al. 2018 | |
| KEYNOTE-062 | Pembrolizumab + Cisplatin/Fluoruracil/Capecitabine | OS 17.4 mo vs. 10.8 mo for CPS>10 for pembro alone OS 12.5 mo vs. 11.1 mo for CPS>10 for pembro + chemo |
Shitara et al. 2020 | |
| KEYNOTE-063 | Pembrolizumab + Pacitaxel | OS 8 mo vs. 8 mo PFS 2 mo vs. 4 mo ObRR 13% vs 19% |
Chung et al. 2022 | |
| JAVELIN Gastric 100 | Avelumab + Chemotherapy | Whole population OS 10.4 mo vs. 10.9 mo PD-L1+ OS 14.9 mo vs 11.6 mo |
Moehler et al. 2021 | |
| CheckMate 649 | Nivolumab + Ipilimumab + Chemotherapy | OS 13.1 mo (Nivo + Chemo) vs. 11.1 mo (Chemo alone) | Janjigian et al. 2021 | |
| KEYNOTE-158 | Pembrolizumab | ObRR 34.3% PFS 4.1 mo OS 23.5 mo |
Marabelle et al. 2020 | |
| Claudin 18.2 | FAST | IMAB362 + EOX | PFS 7.9 mo vs 5.7 mo OS 12.5 mo vs 8.7 mo |
Al-Batran et al. 2016, Sahin et al. 2021 |
OS= Overall Survival, PFS= Progression free survival, ORR= Overall response rate, ObRR= Objective response rate, DFS= Disease free survival, TRR= Total response rate
Autophagy
Autophagy typically refers to macroautophagy, or the process of degrading damaged proteins and organelles. Autophagy can provide a cell with resources for energy and survival in stressful conditions, such as hypoxia in the tumor setting; however, excessive autophagy can be detrimental to cell survival. Thus, the role of autophagy in cancer progression, including gastric and gastro-esophageal cancer, appears to be context dependent and remains controversial.
Markers of autophagy have been identified in gastric cancer tissues [69, 70]. Microtubule associated light chain 3 (LC3) is a commonly used marker of autophagy [71] and is increased in the cytoplasm of malignant GC cells compared to gastric epithelial cells [69]. Protein p62, also known as sequestrome 1 (SQSTM1), is a protein adaptor that can deliver polyubiquitinated cargo to autophagic complexes and may also be used as a measure of autophagic flux [72]. While the cytoplasm and nuclei within GC tissue samples stain positive for p62 via immunohistologic staining, non-malignant gastric tissue is negative for p62 [70]. Autophagy inhibition by Bafilomycin A1 in the SGC-7901 gastric cancer cell line increased sensitivity to 5-fluorouracil treatment, and apoptosis increased in these cells when dual-treatment was given [73]. Similarly, inhibition of autophagy via a small interfering RNA (siRNA) of class 3 PI3K in combination with 5-fluorouracil significantly increased apoptosis of SGC7901 cells compared to either treatment alone, suggesting that autophagic inhibition may enhance sensitivity to chemotherapy in gastric cancer [74]. Interestingly, in patients treated with epirubicin, cisplatin, and 5-fluorouracil adjuvant chemotherapy, 77.78% of GC tissue and 83.70% of adjacent non-cancerous tissue specimens were positive for autophagy-related gene-5 (ATG-5). ATG-5 levels were correlated with multidrug resistance-associated protein-1 (MRP-1) in these tissues. Those patients expressing the highest ATG-5 and MRP-1 had the worst prognosis. Thus, chemotherapy resistance may be due to increased autophagy [75].
Chloroquine and hydroxychloroquine have been explored for their ability to inhibit autophagy. Chloroquine was shown to interfere with the fusion of the autophagosome and lysosome, which differs from other autophagy inhibitors such as Bafilomycin A1, which alters the pH of the lysosome [76]. Chloroquine improved response to cisplatin in a SGC7901 murine xenograft model of GC [77]. Since the safety and efficacy of hydroxychloroquine as an inhibitor of autophagy has been studied in pancreatic cancer [78] and multiple other malignancies, hydroxychloroquine could be implemented as an autophagy inhibitor in the setting of GC. Adding chloroquine/hydroxychloroquine to other treatments may be of benefit in GC. Hydroxychloroquine combined with a novel vascular endothelial factor receptor 2 (VEFGR2) monoclonal antibody, which inhibits tumor angiogenesis, improved tumor control more than either treatment alone in a BGC823 murine xenograft model of GC. This combined therapy may be particularly beneficial since anti-angiogenic drugs could induce autophagy [79]. Wang, et al. demonstrated that autophagy inhibition with Bafilomycin A1, chloroquine, or 3-methyladenine, resulted in increased expression of PD-L1 on gastric cancer cell lines AGS and NCI-n87 [80], suggesting that dual therapy with immune checkpoint blockade is a potential therapeutic strategy.
Although autophagy has been linked to potential chemoresistance in GC, additional studies have shown an anti-tumor effect of autophagy in gastric cancer. In a recent study by Li, et al., elevated staining for DAPK3 was correlated with elevated markers of autophagy. These markers were elevated primarily in early stages of disease and correlated with improved prognosis. This study demonstrates a potential tumor limiting effect of DAPK3-driven autophagy in GC [81]. Further, expression of the tumor suppressor gene klotho is inhibited by methylation in GC; however, restoration of this gene expression increases apoptotic and autophagic cell death, which may inhibit tumor growth [82]. Given the conflicting findings regarding the role of autophagy in GC progression and therapeutic resistance, further preclinical studies are warranted.
Angiogenesis
Angiogenesis is the process of making and remodeling blood vessels that is often hijacked by tumors, leading to aberrant vessel formation with non-functional vasculature found throughout the tumor. This biological process is regulated by several different cellular pathways but most notably through the vascular endothelial factor (VEGF) family, angiopoietin/Tie, hypoxia, and others that tumors manipulate to their advantage [83, 84]. VEGFA has been identified as a biomarker for GC, with patients who have high serum levels associated with advanced disease and poor outcomes[85]. Pre-clinical models of GC express angiogenic factors at a much higher level than healthy gastric epithelium. These molecules are responsible for both angiogenesis and autocrine signaling to support tumor cell growth. Inhibition of these molecules in vitro, results in a marked reduction in tumor cell proliferation and migration [86, 87]. These findings highlight the importance of these molecules in not only vessel homeostasis but also tumorigenesis. When inhibiting angiogenesis in in vivo GC models, there was reduced tumor size and volume [88]. With abundant pre-clinical data supporting the transition to patients, several different clinical trials have been implemented to test the efficacy of anti-angiogenic therapy for GC. The REGARD trial ran from 2009–2012 with previously treated GC patients given either ramucirumab (VEGFR-2 antibody) or placebo. Patients in the ramucirumab arm faired far better than the placebo arm with an OS of 5.2 months vs 3.8 months and progression free survival (PFS) of 2.1 months vs. 1.3 months [89]. The RAINBOW trial followed to test ramucirumab with paclitaxel. In the combination treatment arm, patient OS benefit was 2.2 months and PFS benefit was 1.5 months [90]. These two studies lead to the FDA approval of ramucirumab for second line treatment of GC. A clinical trial is currently active testing the efficacy of treating patients with advanced carcinomas, including gastric adenocarcinoma, with ramucirumab and pembrolizumab. This is testing the safety of this drug combination but also looking at the effect it has on disease progression (NCT02443324). Apatinib, a small molecule inhibitor of VEGFR2, was tested in advanced gastric adenocarcinoma and found to increase PFS and OS [91]. Regorafenib, a multi kinase inhibitor (including VEGFR-1/2, tie-2, stromal and oncogenic kinases) was also tested in advanced gastric adenocarcinoma and found to prolong PFS [92]. This trial lead to the creation of the INTEGRATEIIa trial that is currently underway. The primary outcome of this trial is to evaluate the effect of regorafenib on overall survival in advanced gastric adenocarcinoma (NCT02773524). Another trial looked at the effect of regorafenib with FOLFOX chemotherapy regimen (5-Fluorouracil, leucovorin, and oxaliplatin). They found that while the drug was safe to give with the FOLFOX regimen, there was no benefit to the progression free survival in this cohort [93]. There are several clinical trials underway testing the efficacy of this drug in combination with other chemotherapies for gastric cancer (NCT03722108, NCT02406170, NCT03627728) Several other trials using anti-angiogenic therapy were less successful in GC. The AVAGAST and AVATAR trials showed no additional benefit to overall survival with bevacizumab (anti-VEGF antibody) added to standard of care [94, 95]. Ma et al tested bevacizumab with DOF (docetaxel/oxaliplatin/5-FU) and found that patients had prolonged PFS and increased incidence of complete surgical resection but again no overall survival benefit. This is possibly due to the heterogenic nature of GC and regional differences [96]. Bevacizumab is still involved in ongoing clinical trials in combination with other therapeutics to evaluate its therapeutic potential for GC (NCT03990103, NCT01359397) More study in the regional difference of gastric cancer and how that affects anti-angiogenic therapeutic potential is required to advance this treatment strategy. Common issues with anti-angiogenic therapy are tumor escape methods, identifying the most effective treatment timing, adverse reactions, and complications with surgical treatment. Further trials are needed to help overcome these obstacles as well as help identify patients who are most likely to benefit from this treatment.
HER2
Overexpression of human epidermal growth factor 2 (HER2), also known as Cerb2 and ERRB2, is a receptor tyrosine kinase (RTK) that has been identified as a driver of tumor growth in breast cancer and successfully targeted by antibody therapy, primarily with trastuzumab. HER2 overexpression has more recently been identified in 12–32% of gastric and gastro-esophageal cancer [97], and HER2 overexpression is more common in patients with intestinal-type gastric adenocarcinoma compared to patients with diffuse-type [98].
Given the amplification of HER2 in gastric cancer, many studies have focused on targeting this molecule as a therapy. In HER2 overexpressing gastric cancer cell lines, lapatinib, a small molecule inhibitor of EGFR and HER2, decreased phosphorylation of AKT and ERK in pre-clinical analyses of in vitro and in vivo effects of lapatinib in combination with trastuzumab [99]. In a Phase 2 clinical trial of lapatinib, there was an 11% response rate in chemonaive patients with metastatic gastric cancer [100]. However, in the Phase 3 trial, TRIO-013/LOGiC, lapatinib did not have improved survival when added to a treatment regimen of capecitabine and oxaliplatin [101]. The median overall survival and progression free survival were not improved in lapatinib plus paclitaxel vs paclitaxel alone as a second-line therapy for patients with HER2-positive advanced GC in the Phase 3 TyTAN trial, but there was an improvement in overall response rate [102].
Trastuzumab has also been explored for its ability to improve outcomes in patients with gastric cancer. In a Phase 3 trial of patients with HER2-positive gastric or gastroesophageal junction cancer (Trastuzumab for Gastric Cancer or ToGA), treatment with trastuzumab plus chemotherapy resulted in improved overall survival, tumor response rate, time to progression, and duration of response compared to chemotherapy alone. Further, patients with high HER2 expression had improved response to trastuzumab plus chemotherapy compared to those with lower HER2 expression [103]. Despite promising results in the ToGA trials, additional trials of trastuzumab therapy in GC have not seen as much success. In a Phase 2 clinical trial including patients who received first-line trastuzumab with chemotherapy (T-ACT, WJOG7112G), treatment with trastuzumab plus paclitaxel did not improve overall survival or overall response rate compared to paclitaxel alone. It was noted that HER2-positivity was only maintained in 31% of patients following first-line therapy [104]. Further, treatment with trastuzumab emtansine alone did not improve overall survival as a second-line therapy for patients with HER2-positive gastric cancer in a Phase 2/3 clinical trial [105]. In the JACOB Phase 3 clinical trial, pertuzumab, another anti-HER2 monoclonal antibody, was added to trastuzumab plus chemotherapy as a first-line treatment for gastric or gastroesophageal junction cancer. Two-thirds of the patients had a HER2 immunohistochemistry of 3+ in this study. The primary endpoint of improved overall survival was not reached; however, there was a significant improvement in progression free survival for the pertuzumab plus trastuzumab group (8.5 months) compared to the trastuzumab alone group (7.0 months) [106]. Given that several clinical trials have failed to meet their endpoints for HER2-targeted therapy, additional studies are warranted to explore the role of HER2 in GC progression
Despite some of the negative clinical trials targeting HER2 in GC, there have also been some promising results. In a retrospective analysis of 364 patients with advanced GC, the overall survival rate for HER2-positive patients treated with trastuzumab was significantly longer (24.7 months) than HER2-negative patients (13.9 months) and HER2-positive patients not treated with trastuzumab (13.5 months) [107]. A Phase 2 clinical trial of patients with advanced GC having received at least two previous systemic therapies showed that use of trastuzumab deruxtecan, a monoclonal HER-2 antibody conjugated to a topoisomerase I inhibitor, improved overall survival to 12.5 months compared to treatment with chemotherapy (8.4 months). The objective response rate was greater in patients with a HER2 score of 3+ via immunohistochemistry [108]. Treatment with trastuzumab was shown to increase PD-L1 expression in HER2-overexpressing cells [109]; thus, dual treatment with trastuzumab and PD-1/PD-L1-targeting antibodies may be warranted [110]. In a Phase 2 clinical trial (NCT02954536), patients with HER2-positive metastatic esophagogastric cancer received first-line trastuzumab, pembrolizumab, capecitabine, and oxaliplatin irrespective of PD-L1 expression status. The 6-month progression free survival was 70% [110]. Margetuximab (MGAH22), a monoclonal antibody that targets HER2 and is derived from the same parent compound as trastuzumab, has improved antitumor activity in vivo due to increased binding to its activating receptor CD16A and decreased binding to its inhibitory receptor CD32A [111]. In a Phase 1 clinical trial, MGAH22 had improved ex vivo antibody dependent cytotoxicity compared to trastuzumab. In this trial (NCT01148849), which included patients with HER2-positive gastric cancer, margetuximab demonstrated clinical activity in patients previously treated with HER2 antibodies [112]. Currently, in the ongoing MAHOGANY Phase 2/3 clinical (NCT04082364), patients with HER2-positive gastric and gastro-esophageal cancer will be treated with checkpoint inhibitors in addition to margetuximab [113]. This clinical trial is in response to the Phase 1b-2 trial CP-MGAH22–05, which demonstrated safety and efficacy of such a treatment strategy [114]. Given the conflicting results of HER2-targeted therapies in GC, a more targeted approach to HER2 therapy may be needed for gastric cancer.
Other receptor tyrosine kinases
Other RTKs have been explored in GC using a variety of different methods. Genomic analysis of gastric tumor samples identified amplification of RTKs including fibroblast growth factor receptor 2 (FGFR2), epidermal growth factor receptor (EGFR), and c-MET (MET) in addition to HER2 [115]. Amplification of these RTKs was associated with a poor prognosis compared to those without RTK mutations [116]. Using a phosphor-RTK array system, EGFR, HER2, FGFR21, FGFR2a, insulin R and EphA4 were found to be upregulated and activated RTKs in GC tissues [117]. Similarly, in a tissue microarray of 950 GC tumor samples, overexpression of MET, FGFR2, EGFR, or HER2 was found in 63.1% of samples. Of those, the rate of amplification was 31.3% for FGFR2, 24.9% for MET, and 23.5% for EGFR. Nearly a quarter of the samples expressed more than one RTK, although expression of each RTK was exclusive from the others [115].
Given that EGFR protein expression is significantly correlated with worse prognosis, including lymphatic invasion, lymph node metastasis, advanced disease, and higher TNM stage, EGFR has been targeted in gastric cancer [118]. In addition to lapatinib discussed in the previous section, more specific EGFR antibodies have been explored. In the FOLFETUX trial, the overall response rate was 44.1% when cetuximab, a monoclonal antibody to EGFR, was added to 5-flurouracil, folinic acid, and irinotecan, irrespective of EGFR expression level [119]. In a Phase 2 clinical trial, adding cetuximab to mFOLFOX6 (5-fluoruracil, leucovorin, and oxaliplatin) therapy showed a response rate of 47.5% and disease control rate was 87.5% in an intent-to-treat analysis; however, the primary end point of overall response rate, was not met. Nonetheless, this trial identified tumor EGFR expression and low serum EGF and TNFα as potential biomarkers to optimize this therapeutic strategy [120]. In a Phase 2 trial of cetuximab plus irinotecan/folinic acid/5-fluorouracil, the progression free survival and overall survival were 9.0 months and 16.5 months, respectively. PTEN expression was associated with a longer progression free survival and may also be a biomarker for cetuximab therapy [121]. A Phase 2 clinical trial of cetuximab plus capecitabine and oxaliplatin showed an overall response rate of 52.3%. The progression free survival and overall survival were 6.6 months and 11.7 months, respectively [122]. However, in the Phase 3 EXPAND clinical trial adding cetuximab to capecitabine and cisplatin, there was no additional benefit to chemotherapy alone [123]. Treatment with gefitinib, an orally active EGFR inhibitor, demonstrated penetration of the gastric tumor through decreased activation of EGFR following treatment; however, the clinical benefit of this therapy was limited as downstream signaling molecules, such as MAPK and Akt, were not affected [124]. Although the EGFR and HER2 pathways has been the primary focus of RTK targeted therapies in GC, other RTKs have been shown to be overexpressed and could be explored in future studies.
MMP-9
Matrix metalloproteinase 9 (MMP-9) is known for its ability to degrade basement membranes, which can promote invasion and metastasis of tumor cells [125]. Expression of MMP-9 was found in 86.7% of GC tissue samples but only 10% of healthy gastric mucosa samples. Importantly, expression of MMP-9 correlates with depth of invasion of the tumor cells [126, 127]. In an analysis of 100 gastric carcinomas, 69% of tumors were positive for MMP9 expression. MMP-9 expression is correlated with STAT3, or signal transducers and activators of transcription 3, in immunohistochemical analysis of GC tissues. NF-κB is an upstream regulator of STAT3, and NF-κB inhibition downregulates markers of EMT, including MMP-9. Thus, this signaling pathway may be of potential interest for therapeutic intervention. However, given that more tumor cells express MMP-9 than STAT3 and NF-κB, additional pathways may regulate MMP-9, suggesting a need for additional strategies to target MMP-9 [128].
The anti-MMP-9 antibody andecalaximab (andeca) has been explored for its effectiveness in treating GC. In an orthotopic murine model of GC, andeca therapy reduced tumor growth. Combined andeca and anti-PD-L1 treatment increased infiltration of CD3+, CD4+, and CD8+ T cells and a decreased CD25+FoxP3+ T regulatory cells (Tregs). The effects may be due to decreased MMP-9 mediated lysis of IL-7 [129] since IL-7 production is higher in GC tissues compared to normal adjacent tissue [130] and enhances survival of naïve and memory T cells but does not have such an effect on Tregs [131]. In a Phase 1 clinical trial (NCT01803282), overall response rate for patients treated with andeca and modified FOLFIRINOX6 (mFOLFOX6) was 47.5% in HER2-negative advanced gastric/gastro-esophageal junction cancer. Biomarker analysis in these patients showed that all circulating MMP-9 was bound to andeca after monotherapy at 600mg and 1800mg doses. Further, patients with the highest levels of circulating C1M, a collagen fragment, had the greatest decrease in C1M following treatment with andeca [132]. Interestingly, HER2 can upregulate expression of MMP-9. Inhibition of both MMP9 and HER2 in MKN-45 and SGC-7901 gastric cancer cell lines decreases invasion, suggesting a role for dual inhibition of both MMP9 and HER2 [125]. Future studies could identify additional combination treatments that could improve survival in gastric cancer patients.
Stem Cells
Targeting cancer stem cells is another potential therapeutic strategy that could be employed in GC. CD44 has been used as a stem cell marker in several cancer types and has been used to identify gastric cancer stem cells [133–136]. In early GC (Stage I), immunohistochemical staining of 3+ for CD44v9 was associated with worse overall survival; however, there was no difference for Stage II and III disease [137]. In patients with early GC, high CD44v9 staining can predict recurrence after submucosal dissection [138]. Further, gastric cancer stem cells expressing high levels of aldehyde dehydrogenase (ALDH) are resistant to cisplatin and 5-fluorouracil in culture [139].
There are several methods that may be used to target gastric cancer stem cells. Treatment with all-trans retinoic acid (ATRA) increased expression of cell cycle regulators and decreased expression of cell cycle promoting genes as well as markers of stemness, such as CD44, ALDH1A1, KLF4, and SOX2 in MKN45 diffuse type gastric cancer cells. In murine xenograft models with GC06 and GC10 cell lines, tumor growth was inhibited by ATRA and ex vivo tumor sphere formation, an assay which indicates the stemness of tumor cells, was decreased. However, tumors relapsed two weeks after treatment withdrawal, suggesting that CD44+ALDH+ stem cells are decreased but not entirely eliminated [133]. MicroRNA-34a, which is a negative regulator of CD44, has been encapsulated in a poly(L-lysine-graft-imidazole) (PLI) nanovesicles for systemic delivery. The PLI/miR-34a nanovesicles accumulated in the tumor and decreased CD44 expression in an orthotopic xenograft model of GC, and tumor growth was suppressed in a subcutaneous xenograft model of MKN-74 GC cells [140]. In a Phase 3 trial (NCT01830621) in advanced colon and rectal tumors, napabucasin, a stemness inhibitor that targets STAT3, improved overall survival in patients with pSTAT3 positive tumors [141]. Such a treatment strategy could be employed in GC. Cancer stem cells have enhanced antioxidant programs that can inhibit success of therapies generating reactive oxygen species as their mechanism of action. One mechanism protecting cells against reactive species is glutathione (GSH) production, which can be inhibited by sulfasalazine. Thus, sulfasalazine has been explored as a treatment in patients with advanced GC in the EPOC1205 dose escalation trial. Administration of sulfasalazine to patients with CD44v-positive cells in their gastric tumor biopsy resulted in half of the patients having a decrease in CD44v-positivity by more than 10%. More than half of the patients had a decrease in intratumoral GSH levels. Shitara, et al. also noted that patients with HER2-positive tumors who had already received a HER2-targeted therapy had a higher proportion of CD44v-positive cancer cells than those with HER2-negative tumors [142]. The gene Bmi-1 for gastric cancer may promote stem cell properties in gastric cancer stem cells [143] and is a poor prognostic indicator [144]. Thus, BMI-1 may be another attractive target for gastric cancer. Using an adenoviral vector to deliver short hairpin (sh)RNA (Ad-Bmi-1i) to silence BMI1 expression in gastric cancer stem cells, growth of subcutaneous xenografts of SGC-7901 (low BMI1 expression) and HGC-27 (high BMI1 expression) was inhibited by Ad-Bmi-1i. Treatment with Ad-Bmi-1i also decreases the proportion of CD133+ and CD44+ cancer stem cells [143]. Overexpression of the helix-loop-helix transcription factor, Atonal homolog 1 (ATOH1) in gastric cancer stem cells reduces tumor growth of gastric cancer stem cells and NCI-N87 cells [145]. Previously, ATOH1 was identified as a tumor suppressor in human colon and rectal cancer [146], so this response may be due to ATOH1-induced differentiation of gastric cancer stem cells [145]. Overall, there are many therapeutic strategies that could be employed to combat gastric cancer stem cells and warrant further exploration in the pre-clinical and clinical settings.
DNA Damage Repair
Targeting DNA damage response pathways has been of interest in many types of cancer. In a retrospective study of 110 patient samples treated with first-line chemotherapy, H2AXhigh/pATMhigh expression was correlated with both progression-free survival and overall survival in patients with advanced GC. H2AX is a histone family member that indicates double stranded breaks [147]; thus, targeting DNA damage response in GC is an important treatment strategy to explore.
When DNA is damaged, poly(ADP-ribose) polymerase (PARP)1, and to a lesser extent PARP2, bind to DNA to aid resealing of single-strand breaks [148]. Olaparib is a PARP inhibitor that enhances DNA binding to PARP and stabilizes PARP-DNA complexes [149]. Gastric cancer cell lines are responsive to olaparib, and low levels of ataxia telangiectasia mutated (ATM) and inactivated p53 promote responsiveness [150, 151], likely due to defective DNA damage response [152]. Olaparib induces apoptosis or aberrant mitoses, indicated by multinucleated cells [151]. In a Phase 2 clinical trial, patients with recurrent or metastatic GC received treatment with olaparib/paclitaxel or placebo/paclitaxel. There was an improvement in overall survival for patients receiving olaparib/paclitaxel (13.1 months) compared to placebo/paclitaxel (8.3 months), and stratifying patients by low ATM status improved the overall survival benefit [149]. However, the Phase 3 GOLD trial of patients who progressed following first-line chemotherapy and were subsequently treated with olaparib/paclitaxel did not meet its primary endpoint of improved overall survival [153]. In a retrospective analysis of tumors from this study, ATM-negative status indicated by immunohistochemistry was an indicator of better prognosis but was independent of treatment response. Thus, a biomarker to indicate olaparib plus paclitaxel therapy is still needed [154]. Another solution may be to combine PARP inhibitor therapies with additional treatments. For example, a Phase 1a/b trial in Australia of pamiparib, an oral PARP1/2 inhibitor, and tislelizumab, a humanized anti-PD-1 monoclonal antibody showed some promise in patients with advanced solid tumors. At median follow-up, 20% of patients had an objective response based on the Response Evaluation Criteria in Solid Tumors (RECIST) [155]. Thus, additional combination therapies should be explored.
Breast and ovarian cancer susceptibility gene 1 (BRCA1) is also involved in the DNA damage response [152]. BRCA1 positivity, determined by an IHC score greater than 1 point, has been identified in about 34% of gastric cancers. GC patients with positive BRCA1 expression have improved overall survival but disease-free survival is not significantly different. This may be due to the finding that poorly differentiated tumors had lower expression of BRCA1. In the group negative for BRCA1 expression, there was improved response to platinum drugs, which function by inducing DNA damage [156]. In a retrospective case series, 10 patients were identified with germline BRCA mutations. Despite 6 of these patients having metastatic disease, the median overall survival was 47.5 months. Patients with operable and metastatic disease had a median overall survival of 55.5 months and 32 months, respectively. Since all these patients were treated with DNA damaging therapies, germline BRCA mutations in GC warrant further investigation [157]. However, the role of BRCA1 and BRCA2 in GC may not be straight forward. In another study of the genetic mutations present in GC, overall survival is significantly longer in patients with BRCA2 mutations [158]. Yet, in a more recent study, BRCA1 and BRCA2 mutations were found not to correlate with survival following platinum drug treatment [159]. Given the conflicting evidence regarding BRCA in GC, additional studies are needed to understand how this pathway could potentially be targeted.
Immunotherapy
Immunotherapies have been successful in several cancer types and have thus been of interest in GC; thus, immune checkpoint blockade, specifically the PD-1/PD-L1 signaling pathway has been targeted.
In the Phase 3 ATTRACTION-2 Trial (NCT02267343) in unresectable advanced or recurrent gastric or gastro-esophageal cancers refractory to more than two chemotherapy regimens, patients receiving anti-PD-1 antibody nivolumab had significantly longer overall survival compared to patients receiving placebo. The median overall survival was 5.26 months for the nivolumab group and 4.14 months for the placebo group [160]. The Phase 3 JAVELIN Gastric 300 trial failed to meet its primary endpoint of improved overall survival and secondary endpoints of improved progression free survival or overall response rate when avelumab, an anti-PD-L1 antibody, was compared to paclitaxel and irinotecan therapy in advanced GC and gastroesophageal cancer [161]. In the ATTRACTION-4 Phase 2 clinical trial (NCT02746796), the safety and efficacy of first line nivolumab plus S-1/capecitabine plus oxaliplatin in advanced gastric cancer patients was measured. The objective response rate was 57.1% in the nivolumab plus S-1/oxaliplatin group and 76.5% in the nivolumab plus capecitabine/oxaliplatin group [162].
The Phase 2 Clinical Trial KEYNOTE-059 (NCT02335411) demonstrated efficacy in for anti-PD-1 antibody pembrolizumab in patients with advanced gastric or gastroesophageal cancer. An objective response based on RECIST criteria was observed in 11.6% of patients and 42.6% of patients with postbaseline radiologic imaging had a decrease in measurable tumor size. Patients with PD-L1 positive tumors had a response rate of 15.5% and response duration of 16.3 months while patients with PD-L1 negative tumors had a response rate of 6.4% and a response duration of 6.9 months [163]. In the Phase 3 KEYNOTE-061 trial, treatment with pembrolizumab did not improve overall survival or progression free survival compared to paclitaxel as a second line therapy for patients with a PD-L1 combined positive score of at least 1. The duration of response was 18.0 months in the pembrolizumab group compared to 5.2 months in the paclitaxel group [164]. A two-year follow-up study, however, showed that the 24-month survival rate in the pembrolizumab group was 19.9% compared to 8.5% in the paclitaxel group. The difference in 24-month survival increased as the PD-L1 combined positive score increased. Thus, the combined positive score may be useful in identifying patients who will benefit from pembrolizumab [165]. To determine any possible benefit from combining pembrolizumab with chemotherapy, the Phase 3 KEYNOTE-062 trial treated treatment naïve advanced gastric or gastro-esophageal cancer patients with pembrolizumab, pembrolizumab plus chemotherapy (cisplatin and fluorouracil or capecitabine), or chemotherapy plus placebo. There was no overall survival or progression free survival patients in the pembrolizumab or pembrolizumab plus chemotherapy groups [166]. The Phase 3 KEYNOTE-063 trial, which was terminated prior to completion, studied the overall survival benefit of pembrolizumab versus paclitaxel treatment in Asian patients with advanced PD-L1 positive GC that had progressed after platinum/fluoropyrimidine therapy [167]. The JAVELIN Gastric 100 trial similarly failed to show a benefit of immune checkpoint blockade therapy following chemotherapy induction. Patients with treatment naïve, unresectable, HER2-negative gastric or gastroesophageal cancer were treated with first line oxaliplatin and a fluoropyridine then received avelumab, an anti-PD-L1 antibody, or continued chemotherapy treatment [168]. The Phase 3 CheckMate 649 (NCT02872116) showed improved overall survival as a first line treatment in patients with CPS 5+ PD-L1 following treatment with nivolumab with or without chemotherapy (leucovorin, fluorouracil, and oxaliplatin) compared to chemotherapy alone [169].
Mismatch repair deficiency has been proposed as a potential biomarker for successful use of immune checkpoint blockade therapies. Interestingly, for tumors with mismatch repair deficiency (including lack of MLH1, MSH2, PMS2, or MSH6), there is an increased incidence of PD-L1 expression [170]. Whole exome sequencing of carcinomas showed 1782 somatic mutations per tumor and 73 mutations per tumor in mismatch repair-deficient and mismatch-repair proficient cancer, respectively. Patients with mismatch-repair deficiency have a higher percentage of potential mutation associated neoantigens, which predicts longer progression free survival [171]. Further, pembrolizumab treatment promotes expansion of peripheral T cells specific for mutation-associated neoantigens [172]. Since mismatch repair deficiency can lead to MSI, the Phase 2 KEYNOTE-158 clinical trial (NCT02628067) explored the impact of pembrolizumab treatment in patients with MMR-deficient/MSI-high tumor status. In this trial, which included GC (10.3%), the overall response rate was 34.3%. The overall response rate for GC patients was 45.8% and the progression free survival was 11.0 months [173].
Overall, these findings suggest a need for further studies of microsatellite instability and mismatch repair deficiency and immunotherapy in gastric cancer. Additional biomarkers should also continue to be explored in future clinical trials.
Claudin 18.2
Claudin 18.2 (CLDN18.2) is a splice variant of the cell surface marker Claudin 18, which functions in the formation of tight junctions. CLDN18.2 was detected in 77% of primary GC and is expressed to a lesser extent in the gastric mucosa, specifically in the epithelial cells at the base of gastric glands. In a small European cohort of tumor samples, CLDN18.2 was expressed in 87.5% of paired primary tumor and lymph node metastases [174]. In a retrospective analysis of a cohort of Japanese patients, 87% of primary tumors and 80% of lymph node metastases expressed CLDN18.2. Staining of 2+ in 40% of tumor cells was observed in 52% of primary tumors and 45% of lymph node metastases. Importantly, CLDN18.2 was more common in Grade 3 and nodal-negative disease than Grade 1/2 and nodal-positive disease. Comparing primary tumors and lymph node metastases from the same patients, CLDN18.2 positivity occurred in both 66% of the time [175].
In the Phase 2 FAST trial (NCT01630083), patients with 2+ CLDN18.2 expression in 40% of tumor cells were treated with the anti-CLDN18.2 chimeric antibody IMAB362 and chemotherapy or chemotherapy alone. Treatment with IMAB362 (now known as zolbetuximab) in addition to chemotherapy (epirubicin/oxaliplatin/capecitabine) improved progression free survival and overall survival compared to chemotherapy alone. Patients with 2+ CLDN18.2 expression in more than 70% of tumor cells saw a great effect in PFS and OS [176, 177]. Additional analysis of this study showed that patients treated with IMAB362 and chemotherapy had improved health related quality of life compared to chemotherapy alone [178]. Initial studies targeting CLDN18.2 have been promising, and additional studies to identify the safety and efficacy of combination therapies with zolbetuximab are underway [177].
Conclusion
Gastric cancer remains a deadly disease but an enhanced understanding of the molecular pathogenesis of this unique cancer has resulted in the emergence of promising new targets. Additional preclinical and clinic research is critically needed to continue to improve patient outcomes for gastric cancer.
Acknowledgements
Research reported in this publication was supported by the National Institutes of Health under award number P20GM121322 (BAB) and T32GM133369 (AI). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Bibliography
- 1.Balakrishnan M, et al. , Changing Trends in Stomach Cancer Throughout the World. Curr Gastroenterol Rep, 2017. 19(8): p. 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray F, et al. , Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin, 2018. 68(6): p. 394–424. [DOI] [PubMed] [Google Scholar]
- 3.Joshi SS and Badgwell BD, Current treatment and recent progress in gastric cancer. CA Cancer J Clin, 2021. 71(3): p. 264–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Batran S-E, et al. , Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): a randomised, phase 2/3 trial. The Lancet, 2019. 393(10184): p. 1948–1957. [DOI] [PubMed] [Google Scholar]
- 5.Al-Batran SE, et al. , The feasibility of triple-drug chemotherapy combination in older adult patients with oesophagogastric cancer: a randomised trial of the Arbeitsgemeinschaft Internistische Onkologie (FLOT65+). Eur J Cancer, 2013. 49(4): p. 835–42. [DOI] [PubMed] [Google Scholar]
- 6.Lauren P, The Two Histological Main Types of Gastric Carcinoma: Diffuse and So-Called Intestinal-Type Carcinoma. An Attempt at a Histo-Clinical Classification. Acta Pathol Microbiol Scand, 1965. 64: p. 31–49. [DOI] [PubMed] [Google Scholar]
- 7.Chen YC, et al. , Clinicopathological Variation of Lauren Classification in Gastric Cancer. Pathol Oncol Res, 2016. 22(1): p. 197–202. [DOI] [PubMed] [Google Scholar]
- 8.Pyo JH, et al. , Early gastric cancer with a mixed-type Lauren classification is more aggressive and exhibits greater lymph node metastasis. J Gastroenterol, 2017. 52(5): p. 594–601. [DOI] [PubMed] [Google Scholar]
- 9.Flejou JF, [WHO Classification of digestive tumors: the fourth edition]. Ann Pathol, 2011. 31(5 Suppl): p. S27–31. [DOI] [PubMed] [Google Scholar]
- 10.Berlth F, et al. , Pathohistological classification systems in gastric cancer: diagnostic relevance and prognostic value. World J Gastroenterol, 2014. 20(19): p. 5679–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carneiro F, Seixas M, and Sobrinho-Simoes M, New elements for an updated classification of the carcinomas of the stomach. Pathol Res Pract, 1995. 191(6): p. 571–84. [DOI] [PubMed] [Google Scholar]
- 12.Goseki N, Takizawa T, and Koike M, Differences in the mode of the extension of gastric cancer classified by histological type: new histological classification of gastric carcinoma. Gut, 1992. 33(5): p. 606–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ming S-C, Gastric carcinoma:A pathobiological classification. Cancer, 1977. 39(6): p. 2475–2485. [DOI] [PubMed] [Google Scholar]
- 14.Grundmann E and Schlake W, Histological classification of gastric cancer from initial to advanced stages. Pathol Res Pract, 1982. 173(3): p. 260–74. [DOI] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research, N., Comprehensive molecular characterization of gastric adenocarcinoma. Nature, 2014. 513(7517): p. 202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sohn BH, et al. , Clinical Significance of Four Molecular Subtypes of Gastric Cancer Identified by The Cancer Genome Atlas Project. Clin Cancer Res, 2017. 23(15): p. 4441–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim ST, et al. , Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med, 2018. 24(9): p. 1449–1458. [DOI] [PubMed] [Google Scholar]
- 18.Xie T, et al. , Positive Status of Epstein-Barr Virus as a Biomarker for Gastric Cancer Immunotherapy: A Prospective Observational Study. J Immunother, 2020. 43(4): p. 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nam S, Kim JH, and Lee DH, RHOA in Gastric Cancer: Functional Roles and Therapeutic Potential. Front Genet, 2019. 10: p. 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gall TM and Frampton AE, Gene of the month: E-cadherin (CDH1). J Clin Pathol, 2013. 66(11): p. 928–32. [DOI] [PubMed] [Google Scholar]
- 21.Llosa NJ, et al. , The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov, 2015. 5(1): p. 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma C, et al. , Programmed Death-Ligand 1 Expression Is Common in Gastric Cancer Associated With Epstein-Barr Virus or Microsatellite Instability. Am J Surg Pathol, 2016. 40(11): p. 1496–1506. [DOI] [PubMed] [Google Scholar]
- 23.Chao J, et al. , Pembrolizumab (pembro) in microsatellite instability-high (MSI-H) advanced gastric/gastroesophageal junction (G/GEJ) cancer by line of therapy. Journal of Clinical Oncology, 2020. 38(4_suppl): p. 430–430. [Google Scholar]
- 24.Ott PA, et al. , Nivolumab (NIVO) in patients (pts) with advanced (adv) chemotherapy-refractory (CT-Rx) esophagogastric (EG) cancer according to microsatellite instability (MSI) status: checkmate 032. Annals of Oncology, 2017. 28: p. v229–v230. [Google Scholar]
- 25.Cristescu R, et al. , Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med, 2015. 21(5): p. 449–56. [DOI] [PubMed] [Google Scholar]
- 26.Lieberman PM, Virology. Epstein-Barr virus turns 50. Science, 2014. 343(6177): p. 1323–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lieberman PM, Keeping it quiet: chromatin control of gammaherpesvirus latency. Nat Rev Microbiol, 2013. 11(12): p. 863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shinozaki A, et al. , Downregulation of microRNA-200 in EBV-associated gastric carcinoma. Cancer Res, 2010. 70(11): p. 4719–27. [DOI] [PubMed] [Google Scholar]
- 29.Mohidin TB and Ng CC, BARF1 gene silencing triggers caspase-dependent mitochondrial apoptosis in Epstein-Barr virus-positive malignant cells. J Biosci, 2015. 40(1): p. 41–51. [DOI] [PubMed] [Google Scholar]
- 30.Pal AD, et al. , Epstein-Barr virus latent membrane protein-2A alters mitochondrial dynamics promoting cellular migration mediated by Notch signaling pathway. Carcinogenesis, 2014. 35(7): p. 1592–601. [DOI] [PubMed] [Google Scholar]
- 31.Kim SM, et al. , EBV-encoded EBNA1 regulates cell viability by modulating miR34a-NOX2-ROS signaling in gastric cancer cells. Biochem Biophys Res Commun, 2017. 494(3–4): p. 550–555. [DOI] [PubMed] [Google Scholar]
- 32.Banerjee AS, Pal AD, and Banerjee S, Epstein-Barr virus-encoded small non-coding RNAs induce cancer cell chemoresistance and migration. Virology, 2013. 443(2): p. 294–305. [DOI] [PubMed] [Google Scholar]
- 33.Herrera V and Parsonnet J, Helicobacter pylori and gastric adenocarcinoma. Clin Microbiol Infect, 2009. 15(11): p. 971–6. [DOI] [PubMed] [Google Scholar]
- 34.Halder P, et al. , The secreted antigen, HP0175, of Helicobacter pylori links the unfolded protein response (UPR) to autophagy in gastric epithelial cells. Cell Microbiol, 2015. 17(5): p. 714–29. [DOI] [PubMed] [Google Scholar]
- 35.Perri F, et al. , Aberrant DNA methylation in non-neoplastic gastric mucosa of H. Pylori infected patients and effect of eradication. Am J Gastroenterol, 2007. 102(7): p. 1361–71. [DOI] [PubMed] [Google Scholar]
- 36.Soutto M, et al. , Trefoil factor 1 expression suppresses Helicobacter pylori-induced inflammation in gastric carcinogenesis. Cancer, 2015. 121(24): p. 4348–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oster P, et al. , Helicobacter pylori infection has a detrimental impact on the efficacy of cancer immunotherapies. Gut, 2022. 71(3): p. 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baud J, et al. , Helicobacter pylori initiates a mesenchymal transition through ZEB1 in gastric epithelial cells. PLoS One, 2013. 8(4): p. e60315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mera R, et al. , Long term follow up of patients treated for Helicobacter pylori infection. Gut, 2005. 54(11): p. 1536–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Massarrat S, et al. , Precancerous conditions after H. pylori eradication: a randomized double blind study in first degree relatives of gastric cancer patients. Arch Iran Med, 2012. 15(11): p. 664–9. [PubMed] [Google Scholar]
- 41.Oliveira C, et al. , Genetic screening for familial gastric cancer. Hered Cancer Clin Pract, 2004. 2(2): p. 51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guilford P, Humar B, and Blair V, Hereditary diffuse gastric cancer: translation of CDH1 germline mutations into clinical practice. Gastric Cancer, 2010. 13(1): p. 1–10. [DOI] [PubMed] [Google Scholar]
- 43.Fitzgerald RC, et al. , Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet, 2010. 47(7): p. 436–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Selga E, et al. , Role of caveolin 1, E-cadherin, Enolase 2 and PKCalpha on resistance to methotrexate in human HT29 colon cancer cells. BMC Med Genomics, 2008. 1: p. 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao H, et al. , Relationships of MMP-9, E-cadherin, and VEGF expression with clinicopathological features and response to chemosensitivity in gastric cancer. Tumour Biol, 2017. 39(5): p. 1010428317698368. [DOI] [PubMed] [Google Scholar]
- 46.Aban CE, et al. , Downregulation of E-cadherin in pluripotent stem cells triggers partial EMT. Sci Rep, 2021. 11(1): p. 2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park JW, et al. , Establishment and characterization of metastatic gastric cancer cell lines from murine gastric adenocarcinoma lacking Smad4, p53, and E-cadherin. Mol Carcinog, 2015. 54(11): p. 1521–7. [DOI] [PubMed] [Google Scholar]
- 48.Vasen HF, Clinical description of the Lynch syndrome [hereditary nonpolyposis colorectal cancer (HNPCC)]. Fam Cancer, 2005. 4(3): p. 219–25. [DOI] [PubMed] [Google Scholar]
- 49.Vasen HF, et al. , MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol, 2001. 19(20): p. 4074–80. [DOI] [PubMed] [Google Scholar]
- 50.Park YJ, Shin KH, and Park JG, Risk of gastric cancer in hereditary nonpolyposis colorectal cancer in Korea. Clin Cancer Res, 2000. 6(8): p. 2994–8. [PubMed] [Google Scholar]
- 51.Ford D, Risks of cancer in BRCA1-mutation carriers. The Lancet, 1994. 343(8899): p. 692–695. [DOI] [PubMed] [Google Scholar]
- 52.Breast Cancer Linkage C, Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst, 1999. 91(15): p. 1310–6. [DOI] [PubMed] [Google Scholar]
- 53.Masciari S, et al. , Gastric cancer in individuals with Li-Fraumeni syndrome. Genet Med, 2011. 13(7): p. 651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mankaney G, et al. , Gastric cancer in FAP: a concerning rise in incidence. Fam Cancer, 2017. 16(3): p. 371–376. [DOI] [PubMed] [Google Scholar]
- 55.Jagelman D, Upper Gastrointestinal Cancer in Familial Adenomatous Polyposis. The Lancet, 1988. 331(8595): p. 1149–1151. [DOI] [PubMed] [Google Scholar]
- 56.Offerhaus GJ, et al. , The risk of upper gastrointestinal cancer in familial adenomatous polyposis. Gastroenterology, 1992. 102(6): p. 1980–2. [DOI] [PubMed] [Google Scholar]
- 57.Rudloff U, Gastric adenocarcinoma and proximal polyposis of the stomach: diagnosis and clinical perspectives. Clin Exp Gastroenterol, 2018. 11: p. 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, et al. , Point Mutations in Exon 1B of APC Reveal Gastric Adenocarcinoma and Proximal Polyposis of the Stomach as a Familial Adenomatous Polyposis Variant. Am J Hum Genet, 2016. 98(5): p. 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Repak R, et al. , The first European family with gastric adenocarcinoma and proximal polyposis of the stomach: case report and review of the literature. Gastrointest Endosc, 2016. 84(4): p. 718–25. [DOI] [PubMed] [Google Scholar]
- 60.Tsuchiya T, et al. , Distinct methylation patterns of two APC gene promoters in normal and cancerous gastric epithelia. Oncogene, 2000. 19(32): p. 3642–6. [DOI] [PubMed] [Google Scholar]
- 61.Hosoya K, et al. , Adenomatous polyposis coli 1A is likely to be methylated as a passenger in human gastric carcinogenesis. Cancer Lett, 2009. 285(2): p. 182–9. [DOI] [PubMed] [Google Scholar]
- 62.Jenne DE, et al. , Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet, 1998. 18(1): p. 38–43. [DOI] [PubMed] [Google Scholar]
- 63.Agudo A, et al. , Inflammatory potential of the diet and risk of gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Am J Clin Nutr, 2018. 107(4): p. 607–616. [DOI] [PubMed] [Google Scholar]
- 64.Skog KI, Johansson MA, and Jagerstad MI, Carcinogenic heterocyclic amines in model systems and cooked foods: a review on formation, occurrence and intake. Food Chem Toxicol, 1998. 36(9–10): p. 879–96. [DOI] [PubMed] [Google Scholar]
- 65.Ferro A, et al. , Meat intake and risk of gastric cancer in the Stomach cancer Pooling (StoP) project. Int J Cancer, 2020. 147(1): p. 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joossens JV, et al. , Dietary salt, nitrate and stomach cancer mortality in 24 countries. European Cancer Prevention (ECP) and the INTERSALT Cooperative Research Group. Int J Epidemiol, 1996. 25(3): p. 494–504. [DOI] [PubMed] [Google Scholar]
- 67.Zamora-Ros R, et al. , Dietary flavonoid and lignan intake and gastric adenocarcinoma risk in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Am J Clin Nutr, 2012. 96(6): p. 1398–408. [DOI] [PubMed] [Google Scholar]
- 68.Tannenbaum SR, Wishnok JS, and Leaf CD, Inhibition of nitrosamine formation by ascorbic acid. Am J Clin Nutr, 1991. 53(1 Suppl): p. 247S–250S. [DOI] [PubMed] [Google Scholar]
- 69.Yoshioka A, et al. , LC3, an autophagosome marker, is highly expressed in gastrointestinal cancers. Int J Oncol, 2008. 33(3): p. 461–8. [PubMed] [Google Scholar]
- 70.Mohamed A, et al. , P62/Ubiquitin IHC Expression Correlated with Clinicopathologic Parameters and Outcome in Gastrointestinal Carcinomas. Front Oncol, 2015. 5: p. 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoshii SR and Mizushima N, Monitoring and Measuring Autophagy. Int J Mol Sci, 2017. 18(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu WJ, et al. , p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett, 2016. 21: p. 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li LQ, et al. , Inhibition of autophagy by bafilomycin A1 promotes chemosensitivity of gastric cancer cells. Tumour Biol, 2016. 37(1): p. 653–9. [DOI] [PubMed] [Google Scholar]
- 74.Zhu BS, et al. , Effects of 5-fluorouracil and class III phosphoinositide 3-kinase small interfering RNA combination therapy on SGC7901 human gastric cancer cells. Mol Med Rep, 2015. 11(3): p. 1891–8. [DOI] [PubMed] [Google Scholar]
- 75.Ge J, et al. , Upregulation of autophagy-related gene-5 (ATG-5) is associated with chemoresistance in human gastric cancer. PLoS One, 2014. 9(10): p. e110293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mauthe M, et al. , Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy, 2018. 14(8): p. 1435–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang HQ, et al. , Antitumor activity of chloroquine in combination with Cisplatin in human gastric cancer xenografts. Asian Pac J Cancer Prev, 2015. 16(9): p. 3907–12. [DOI] [PubMed] [Google Scholar]
- 78.Boone BA, et al. , Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann Surg Oncol, 2015. 22(13): p. 4402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang W, et al. , Hydroxychloroquine enhances the antitumor effects of BC001 in gastric cancer. Int J Oncol, 2019. 55(2): p. 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang X, et al. , Autophagy inhibition enhances PD-L1 expression in gastric cancer. J Exp Clin Cancer Res, 2019. 38(1): p. 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li GM, et al. , DAPK3 inhibits gastric cancer progression via activation of ULK1-dependent autophagy. Cell Death Differ, 2021. 28(3): p. 952–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xie B, et al. , Restoration of klotho gene expression induces apoptosis and autophagy in gastric cancer cells: tumor suppressive role of klotho in gastric cancer. Cancer Cell Int, 2013. 13(1): p. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nishida N, et al. , Angiogenesis in cancer. Vasc Health Risk Manag, 2006. 2(3): p. 213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nienhuser H and Schmidt T, Angiogenesis and Anti-Angiogenic Therapy in Gastric Cancer. Int J Mol Sci, 2017. 19(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Karayiannakis AJ, et al. , `Circulating VEGF levels in the serum of gastric cancer patients: correlation with pathological variables, patient survival, and tumor surgery. Ann Surg, 2002. 236(1): p. 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lv Y, et al. , Inhibitory effects of bevacizumab monoclonal antibodies in combination with chemotherapy in different time sequences on a human gastric carcinoma cell line. Ir J Med Sci, 2017. 186(2): p. 275–280. [DOI] [PubMed] [Google Scholar]
- 87.Lin Y, et al. , Autocrine VEGF signaling promotes cell proliferation through a PLC-dependent pathway and modulates Apatinib treatment efficacy in gastric cancer. Oncotarget, 2017. 8(7): p. 11990–12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sun P, et al. , Lentivirus-mediated siRNA targeting VEGF inhibits gastric cancer growth in vivo. Oncol Rep, 2012. 28(5): p. 1687–92. [DOI] [PubMed] [Google Scholar]
- 89.Fuchs CS, et al. , Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. The Lancet, 2014. 383(9911): p. 31–39. [DOI] [PubMed] [Google Scholar]
- 90.Wilke H, et al. , Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. The Lancet Oncology, 2014. 15(11): p. 1224–1235. [DOI] [PubMed] [Google Scholar]
- 91.Li J, et al. , Randomized, Double-Blind, Placebo-Controlled Phase III Trial of Apatinib in Patients With Chemotherapy-Refractory Advanced or Metastatic Adenocarcinoma of the Stomach or Gastroesophageal Junction. J Clin Oncol, 2016. 34(13): p. 1448–54. [DOI] [PubMed] [Google Scholar]
- 92.Pavlakis N, et al. , Regorafenib for the Treatment of Advanced Gastric Cancer (INTEGRATE): A Multinational Placebo-Controlled Phase II Trial. J Clin Oncol, 2016. 34(23): p. 2728–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moy RH, et al. , Regorafenib in Combination with First-Line Chemotherapy for Metastatic Esophagogastric Cancer. Oncologist, 2020. 25(1): p. e68–e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ohtsu A, et al. , Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol, 2011. 29(30): p. 3968–76. [DOI] [PubMed] [Google Scholar]
- 95.Shen L, et al. , Bevacizumab plus capecitabine and cisplatin in Chinese patients with inoperable locally advanced or metastatic gastric or gastroesophageal junction cancer: randomized, double-blind, phase III study (AVATAR study). Gastric Cancer, 2015. 18(1): p. 168–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ma J, et al. , Neoadjuvant Therapy of DOF Regimen Plus Bevacizumab Can Increase Surgical Resection Ratein Locally Advanced Gastric Cancer: A Randomized, Controlled Study. Medicine (Baltimore), 2015. 94(42): p. e1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gravalos C and Jimeno A, HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann Oncol, 2008. 19(9): p. 1523–9. [DOI] [PubMed] [Google Scholar]
- 98.Van Cutsem E, et al. , HER2 screening data from ToGA: targeting HER2 in gastric and gastroesophageal junction cancer. Gastric Cancer, 2015. 18(3): p. 476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wainberg ZA, et al. , Lapatinib, a dual EGFR and HER2 kinase inhibitor, selectively inhibits HER2-amplified human gastric cancer cells and is synergistic with trastuzumab in vitro and in vivo. Clin Cancer Res, 2010. 16(5): p. 1509–19. [DOI] [PubMed] [Google Scholar]
- 100.Iqbal S, et al. , Southwest Oncology Group study S0413: a phase II trial of lapatinib (GW572016) as first-line therapy in patients with advanced or metastatic gastric cancer. Ann Oncol, 2011. 22(12): p. 2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hecht JR, et al. , Lapatinib in Combination With Capecitabine Plus Oxaliplatin in Human Epidermal Growth Factor Receptor 2-Positive Advanced or Metastatic Gastric, Esophageal, or Gastroesophageal Adenocarcinoma: TRIO-013/LOGiC--A Randomized Phase III Trial. J Clin Oncol, 2016. 34(5): p. 443–51. [DOI] [PubMed] [Google Scholar]
- 102.Satoh T, et al. , Lapatinib plus paclitaxel versus paclitaxel alone in the second-line treatment of HER2-amplified advanced gastric cancer in Asian populations: TyTAN--a randomized, phase III study. J Clin Oncol, 2014. 32(19): p. 2039–49. [DOI] [PubMed] [Google Scholar]
- 103.Bang YJ, et al. , Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet, 2010. 376(9742): p. 687–97. [DOI] [PubMed] [Google Scholar]
- 104.Makiyama A, et al. , Randomized, Phase II Study of Trastuzumab Beyond Progression in Patients With HER2-Positive Advanced Gastric or Gastroesophageal Junction Cancer: WJOG7112G (T-ACT Study). J Clin Oncol, 2020. 38(17): p. 1919–1927. [DOI] [PubMed] [Google Scholar]
- 105.Thuss-Patience PC, et al. , Trastuzumab emtansine versus taxane use for previously treated HER2-positive locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma (GATSBY): an international randomised, open-label, adaptive, phase 2/3 study. Lancet Oncol, 2017. 18(5): p. 640–653. [DOI] [PubMed] [Google Scholar]
- 106.Tabernero J, et al. , Pertuzumab plus trastuzumab and chemotherapy for HER2-positive metastatic gastric or gastro-oesophageal junction cancer (JACOB): final analysis of a double-blind, randomised, placebo-controlled phase 3 study. Lancet Oncol, 2018. 19(10): p. 1372–1384. [DOI] [PubMed] [Google Scholar]
- 107.Shitara K, et al. , Prognosis of patients with advanced gastric cancer by HER2 status and trastuzumab treatment. Gastric Cancer, 2013. 16(2): p. 261–7. [DOI] [PubMed] [Google Scholar]
- 108.Shitara K, et al. , Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N Engl J Med, 2020. 382(25): p. 2419–2430. [DOI] [PubMed] [Google Scholar]
- 109.Chaganty BKR, et al. , Trastuzumab upregulates PD-L1 as a potential mechanism of trastuzumab resistance through engagement of immune effector cells and stimulation of IFNγ secretion. Cancer Lett, 2018. 430: p. 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Janjigian YY, et al. , First-line pembrolizumab and trastuzumab in HER2-positive oesophageal, gastric, or gastro-oesophageal junction cancer: an open-label, single-arm, phase 2 trial. Lancet Oncol, 2020. 21(6): p. 821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nordstrom JL, et al. , Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Res, 2011. 13(6): p. R123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bang YJ, et al. , First-in-human phase 1 study of margetuximab (MGAH22), an Fc-modified chimeric monoclonal antibody, in patients with HER2-positive advanced solid tumors. Ann Oncol, 2017. 28(4): p. 855–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Catenacci DV, et al. , MAHOGANY: margetuximab combination in HER2+ unresectable/metastatic gastric/gastroesophageal junction adenocarcinoma. Future Oncol, 2021. 17(10): p. 1155–1164. [DOI] [PubMed] [Google Scholar]
- 114.Catenacci DVT, et al. , Margetuximab plus pembrolizumab in patients with previously treated, HER2-positive gastro-oesophageal adenocarcinoma (CP-MGAH22–05): a single-arm, phase 1b-2 trial. Lancet Oncol, 2020. 21(8): p. 1066–1076. [DOI] [PubMed] [Google Scholar]
- 115.Nagatsuma AK, et al. , Expression profiles of HER2, EGFR, MET and FGFR2 in a large cohort of patients with gastric adenocarcinoma. Gastric Cancer, 2015. 18(2): p. 227–38. [DOI] [PubMed] [Google Scholar]
- 116.Deng N, et al. , A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut, 2012. 61(5): p. 673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gong J, et al. , Use of protein array to investigate receptor tyrosine kinases activated in gastric cancer. Int J Oncol, 2010. 36(1): p. 101–6. [PubMed] [Google Scholar]
- 118.Kim MA, et al. , EGFR in gastric carcinomas: prognostic significance of protein overexpression and high gene copy number. Histopathology, 2008. 52(6): p. 738–46. [DOI] [PubMed] [Google Scholar]
- 119.Pinto C, et al. , Phase II study of cetuximab in combination with FOLFIRI in patients with untreated advanced gastric or gastroesophageal junction adenocarcinoma (FOLCETUX study). Ann Oncol, 2007. 18(3): p. 510–7. [DOI] [PubMed] [Google Scholar]
- 120.Han SW, et al. , Phase II study and biomarker analysis of cetuximab combined with modified FOLFOX6 in advanced gastric cancer. Br J Cancer, 2009. 100(2): p. 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Moehler M, et al. , Cetuximab with irinotecan, folinic acid and 5-fluorouracil as first-line treatment in advanced gastroesophageal cancer: a prospective multi-center biomarker-oriented phase II study. Ann Oncol, 2011. 22(6): p. 1358–1366. [DOI] [PubMed] [Google Scholar]
- 122.Kim C, et al. , A prospective phase II study of cetuximab in combination with XELOX (capecitabine and oxaliplatin) in patients with metastatic and/or recurrent advanced gastric cancer. Invest New Drugs, 2011. 29(2): p. 366–73. [DOI] [PubMed] [Google Scholar]
- 123.Lordick F, et al. , Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open-label phase 3 trial. Lancet Oncol, 2013. 14(6): p. 490–9. [DOI] [PubMed] [Google Scholar]
- 124.Rojo F, et al. , Pharmacodynamic studies of gefitinib in tumor biopsy specimens from patients with advanced gastric carcinoma. J Clin Oncol, 2006. 24(26): p. 4309–16. [DOI] [PubMed] [Google Scholar]
- 125.Shan YQ, et al. , MMP-9 is increased in the pathogenesis of gastric cancer by the mediation of HER2. Cancer Gene Ther, 2015. 22(3): p. 101–7. [DOI] [PubMed] [Google Scholar]
- 126.Chen SZ, et al. , Expression levels of matrix metalloproteinase-9 in human gastric carcinoma. Oncol Lett, 2015. 9(2): p. 915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.R P, N P, and J T, Expression of Matrix Metalloproteinase-9 in Gastric Cancer. Cureus, 2021. 13(9): p. e18195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yoon J, et al. , A synergistic interaction between transcription factors nuclear factor-κB and signal transducers and activators of transcription 3 promotes gastric cancer cell migration and invasion. BMC Gastroenterol, 2013. 13: p. 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Greenstein AE, et al. , Effect of andecaliximab (anti-MMP9) on proteolysis of IL-7 in vitro, TCR diversity in mice, and serum IL-7 in gastric cancer patients in combination with chemotherapy. Journal of Clinical Oncology, 2018. 36(5_suppl): p. 101–101.29220288 [Google Scholar]
- 130.Bednarz-Misa I, Diakowska D, and Krzystek-Korpacka M, Local and Systemic IL-7 Concentration in Gastrointestinal-Tract Cancers. Medicina (Kaunas), 2019. 55(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dwyer CJ, et al. , Fueling Cancer Immunotherapy With Common Gamma Chain Cytokines. Front Immunol, 2019. 10: p. 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shah MA, et al. , Andecaliximab/GS-5745 Alone and Combined with mFOLFOX6 in Advanced Gastric and Gastroesophageal Junction Adenocarcinoma: Results from a Phase I Study. Clin Cancer Res, 2018. 24(16): p. 3829–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nguyen PH, et al. , All-trans retinoic acid targets gastric cancer stem cells and inhibits patient-derived gastric carcinoma tumor growth. Oncogene, 2016. 35(43): p. 5619–5628. [DOI] [PubMed] [Google Scholar]
- 134.Bessède E, et al. , Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene, 2014. 33(32): p. 4123–31. [DOI] [PubMed] [Google Scholar]
- 135.Lau WM, et al. , CD44v8-10 is a cancer-specific marker for gastric cancer stem cells. Cancer Res, 2014. 74(9): p. 2630–41. [DOI] [PubMed] [Google Scholar]
- 136.Takaishi S, et al. , Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells, 2009. 27(5): p. 1006–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Go SI, et al. , CD44 Variant 9 Serves as a Poor Prognostic Marker in Early Gastric Cancer, But Not in Advanced Gastric Cancer. Cancer Res Treat, 2016. 48(1): p. 142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hirata K, et al. , CD44 variant 9 expression in primary early gastric cancer as a predictive marker for recurrence. Br J Cancer, 2013. 109(2): p. 379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nishikawa S, et al. , Aldehyde dehydrogenase high gastric cancer stem cells are resistant to chemotherapy. Int J Oncol, 2013. 42(4): p. 1437–42. [DOI] [PubMed] [Google Scholar]
- 140.Jang E, et al. , Nanovesicle-mediated systemic delivery of microRNA-34a for CD44 overexpressing gastric cancer stem cell therapy. Biomaterials, 2016. 105: p. 12–24. [DOI] [PubMed] [Google Scholar]
- 141.Jonker DJ, et al. , Napabucasin versus placebo in refractory advanced colorectal cancer: a randomised phase 3 trial. Lancet Gastroenterol Hepatol, 2018. 3(4): p. 263–270. [DOI] [PubMed] [Google Scholar]
- 142.Shitara K, et al. , Dose-escalation study for the targeting of CD44v. Gastric Cancer, 2017. 20(2): p. 341–349. [DOI] [PubMed] [Google Scholar]
- 143.Zhang X, et al. , Antitumor activity and inhibitory effects on cancer stem cell-like properties of Adeno-associated virus (AAV) -mediated Bmi-1 interference driven by Bmi-1 promoter for gastric cancer. Oncotarget, 2016. 7(16): p. 22733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang XW, et al. , BMI1 and Mel-18 oppositely regulate carcinogenesis and progression of gastric cancer. Mol Cancer, 2010. 9: p. 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Han ME, et al. , ATOH1 Can Regulate the Tumorigenicity of Gastric Cancer Cells by Inducing the Differentiation of Cancer Stem Cells. PLoS One, 2015. 10(5): p. e0126085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bossuyt W, et al. , Atonal homolog 1 is a tumor suppressor gene. PLoS Biol, 2009. 7(2): p. e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ronchetti L, et al. , DNA damage repair and survival outcomes in advanced gastric cancer patients treated with first-line chemotherapy. Int J Cancer, 2017. 140(11): p. 2587–2595. [DOI] [PubMed] [Google Scholar]
- 148.Murai J, et al. , Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res, 2012. 72(21): p. 5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bang YJ, et al. , Randomized, Double-Blind Phase II Trial With Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. J Clin Oncol, 2015. 33(33): p. 3858–65. [DOI] [PubMed] [Google Scholar]
- 150.Hodgson D, et al. , Abstract 2398: Activity of the PARP inhibitor olaparib in ATM-deficient gastric cancer: from preclinical models to the clinic. Cancer Research, 2014. 74(19 Supplement): p. 2398–2398. [Google Scholar]
- 151.Kubota E, et al. , Low ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell Cycle, 2014. 13(13): p. 2129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rosen EM, BRCA1 in the DNA damage response and at telomeres. Front Genet, 2013. 4: p. 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bang YJ, et al. , Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol, 2017. 18(12): p. 1637–1651. [DOI] [PubMed] [Google Scholar]
- 154.Liu YZ, et al. , Olaparib plus paclitaxel sensitivity in biomarker subgroups of gastric cancer. Annals of Oncology, 2018. 29: p. viii25–viii26. [Google Scholar]
- 155.Friedlander M, et al. , Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. The Lancet Oncology, 2019. 20(9): p. 1306–1315. [DOI] [PubMed] [Google Scholar]
- 156.Chen W, et al. , Prognostic significance of BRCA1 expression in gastric cancer. Med Oncol, 2013. 30(1): p. 423. [DOI] [PubMed] [Google Scholar]
- 157.Halpern N, et al. , Clinical Characteristics and Prognosis of Gastric Cancer Patients with. Onco Targets Ther, 2020. 13: p. 11637–11644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chen K, et al. , Mutational landscape of gastric adenocarcinoma in Chinese: implications for prognosis and therapy. Proc Natl Acad Sci U S A, 2015. 112(4): p. 1107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kim HS, et al. , Clinical significance of BRCA1 and BRCA2 mRNA and protein expression in patients with sporadic gastric cancer. Oncol Lett, 2019. 17(5): p. 4383–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kang YK, et al. , Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538–12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet, 2017. 390(10111): p. 2461–2471. [DOI] [PubMed] [Google Scholar]
- 161.Bang YJ, et al. , Phase III, randomised trial of avelumab versus physician’s choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: primary analysis of JAVELIN Gastric 300. Ann Oncol, 2018. 29(10): p. 2052–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Boku N, et al. , Safety and efficacy of nivolumab in combination with S-1/capecitabine plus oxaliplatin in patients with previously untreated, unresectable, advanced, or recurrent gastric/gastroesophageal junction cancer: interim results of a randomized, phase II trial (ATTRACTION-4). Ann Oncol, 2019. 30(2): p. 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fuchs CS, et al. , Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol, 2018. 4(5): p. e180013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shitara K, et al. , Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet, 2018. 392(10142): p. 123–133. [DOI] [PubMed] [Google Scholar]
- 165.Fuchs CS, et al. , Pembrolizumab versus paclitaxel for previously treated PD-L1-positive advanced gastric or gastroesophageal junction cancer: 2-year update of the randomized phase 3 KEYNOTE-061 trial. Gastric Cancer, 2022. 25(1): p. 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shitara K, et al. , Efficacy and Safety of Pembrolizumab or Pembrolizumab Plus Chemotherapy vs Chemotherapy Alone for Patients With First-line, Advanced Gastric Cancer: The KEYNOTE-062 Phase 3 Randomized Clinical Trial. JAMA Oncol, 2020. 6(10): p. 1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chung HC, et al. , Pembrolizumab versus paclitaxel for previously treated advanced gastric or gastroesophageal junction cancer (KEYNOTE-063): A randomized, open-label, phase 3 trial in Asian patients. Cancer, 2022. 128(5): p. 995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Moehler M, et al. , Phase III Trial of Avelumab Maintenance After First-Line Induction Chemotherapy Versus Continuation of Chemotherapy in Patients With Gastric Cancers: Results From JAVELIN Gastric 100. J Clin Oncol, 2021. 39(9): p. 966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Janjigian YY, et al. , First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet, 2021. 398(10294): p. 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kawazoe A, et al. , Clinicopathological features of programmed death ligand 1 expression with tumor-infiltrating lymphocyte, mismatch repair, and Epstein-Barr virus status in a large cohort of gastric cancer patients. Gastric Cancer, 2017. 20(3): p. 407–415. [DOI] [PubMed] [Google Scholar]
- 171.Le DT, et al. , PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med, 2015. 372(26): p. 2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Le DT, et al. , Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science, 2017. 357(6349): p. 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Marabelle A, et al. , Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol, 2020. 38(1): p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sahin U, et al. , Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin Cancer Res, 2008. 14(23): p. 7624–34. [DOI] [PubMed] [Google Scholar]
- 175.Rohde C, et al. , Comparison of Claudin 18.2 expression in primary tumors and lymph node metastases in Japanese patients with gastric adenocarcinoma. Jpn J Clin Oncol, 2019. 49(9): p. 870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Al-Batran S-E, et al. , FAST: An international, multicenter, randomized, phase II trial of epirubicin, oxaliplatin, and capecitabine (EOX) with or without IMAB362, a first-in-class anti-CLDN18.2 antibody, as first-line therapy in patients with advanced CLDN18.2+ gastric and gastroesophageal junction (GEJ) adenocarcinoma. Journal of Clinical Oncology, 2016. 34(18_suppl): p. LBA4001–LBA4001. [Google Scholar]
- 177.Sahin U, et al. , FAST: a randomised phase II study of zolbetuximab (IMAB362) plus EOX versus EOX alone for first-line treatment of advanced CLDN18.2-positive gastric and gastro-oesophageal adenocarcinoma. Ann Oncol, 2021. 32(5): p. 609–619. [DOI] [PubMed] [Google Scholar]
- 178.Morlock R, et al. , Health-related quality-of-life (HRQoL) results from the FAST study: A phase II trial of epirubicin, oxaliplatin, and capecitabine with or without IMAB362 in patients with advanced CLDN18.2+ gastric and gastroesophageal junction adenocarcinoma. Journal of Clinical Oncology, 2018. 36(4_suppl): p. 54–54. [Google Scholar]