

What you need to know.
Dystonia is a neurological condition characterised by abnormal postures and movements resulting from abnormal neural control of muscles
The most common forms of isolated dystonia in adults are focal, affecting the neck (cervical dystonia), eyes (blepharospasm), or associated with a task (eg, writer’s cramp)
Acute and tardive dystonia can occur as complications of medications such as dopamine receptor blockers
Neurophysiotherapy, botulinum toxin injections, and deep brain stimulation are effective treatments
Management of non-motor symptoms such as depression, anxiety, and associated pain are also important
Dystonia is an abnormality of movement and posture caused by the abnormal neural control of muscle contractions. It is a highly stigmatising long term condition associated with a reduced quality of life.1 Management is a multidisciplinary partnership between general practitioners, specialists, therapists, and patients and seeks to alleviate motor and non-motor symptoms and the wider psychosocial repercussions of dystonia. This practice pointer highlights the common forms of dystonia, flags rarer causes which can have significant repercussions if not recognised, and offers a clinical roadmap for patient management.
What is dystonia?
Dystonia is defined as “a movement disorder characterised by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both. Dystonic movements are typically patterned, twisting, and may be tremulous. Dystonia is often initiated or worsened by voluntary action and associated with overflow muscle activation.”2 A dystonic body part usually remains mobile and movements are often slow and effortful, with a reduced range. The most common forms of dystonia in adults are focal, affecting the neck (cervical dystonia), eyes (blepharospasm), or associated with a task (eg, writer’s cramp). However, dystonia can also present with a generalised pattern, which is more common with childhood onset forms. Dystonia is a subcategory of the hyperkinetic movement disorders in which too much or disordered movement is seen. Other subtypes include chorea, myoclonus, tics, and tremor. Pathophysiology and clinical descriptors of dystonia are outlined in boxes 1 and 2.2 3
Box 1. Pathophysiology of dystonia.
Dystonia was originally thought to be caused by disrupted basal ganglia function, as lesions in the basal ganglia (particularly the putamen) frequently resulted in dystonia.4 However, later studies showed that it is better conceptualised as a network disorder that also includes the cerebellum, thalamus, and cortex.5 Genetic mutations can cause generalised isolated dystonia (eg, TOR1A), focal isolated dystonia (eg, GNAL), and combined dystonia.6 7 Some evidence suggests that monogenic mutations converge on common molecular pathways.8 Other subtypes of dystonia, particularly focal dystonias, have stronger environmental influences (such as intensive stereotyped practice in musician’s dystonia9). Traditionally, isolated dystonia has been used to infer the essential mechanisms that underwrite dystonia. Classic neurophysiological markers of dystonia, such as disordered plasticity responses or reduced inhibition, have limited utility as they are variable across individuals and are not specific to dystonia.10 11
Box 2. Categories of key clinical descriptors for dystonia2 .
Age of onset
Infancy: birth to 2
Childhood: 3 to 12
Adolescence: 13 to 20
Early adulthood: 21 to 40
Late adulthood: over 40
Body distribution
Focal: one body region
Segmental: two or more contiguous body regions
Multifocal: two or more non-contiguous body regions
Generalised: two or more contiguous regions plus trunk
Hemidystonia: one side of the body
Temporal features
-
Disease course
Static
Progressive
-
Variability
Persistent
Diurnal
Task specific
Paroxysmal
Associated features
Isolated dystonia
-
Combined dystonia
Coexists with another movement disorder, or
Occurrence of other neurological or systemic manifestations
Confusingly, the term dystonia can be used interchangeably to refer to a range of related clinical entities. Dystonia can refer to a type of movement disorder (ie, pattern of abnormal movement, phenomenology), a clinical syndrome (eg, adult onset focal dystonia), and/or a specific diagnosis (eg, TOR1A-related dystonia).3 To move beyond such a non-specific use of the term dystonia, stratifying patients by clinical features often reveals likely causes and guides investigation.
General approach
When a patient presents with a movement disorder consistent with dystonia, a targeted history can help pick out the key clinical features (box 2). Consider the age of onset, body distribution, course of disease, variability, and whether dystonia is associated with other features (box 2). For example, dystonia that begins in childhood is more likely to have a discoverable acquired cause and is more likely to progress from focal to generalised. In contrast, a typical adult onset focal dystonia is likely to be a subtype of isolated dystonia and will rarely spread. The family history reveals the likelihood of a genetic cause. Ask if patients can alleviate their dystonia by lightly touching surrounding body areas (“sensory trick”), a phenomenon that illustrates the dynamic nature of dystonia and the importance of sensory influences.12 Dystonia is often accentuated within a neurological examination that includes a range of postures and tasks. A latent abnormal posture (eg, in the head and neck) can be revealed by asking the patient to close their eyes and let position drift to where it feels most comfortable. If tremor accompanies dystonia it is usually jerky, variable in amplitude, and worsened by particular positions.
Types of dystonia
Isolated dystonia
In isolated dystonia, dystonia and/or tremor are the only symptoms and signs.3 These are chronic, relatively selective disorders of posture and movement that are non-degenerative and have no effect on lifespan. Adult onset focal isolated dystonia has a typical anatomical distribution of onset. Cervical dystonia, blepharospasm, and task specific dystonia are the most common forms (fig 1). Other subtypes include oromandibular dystonia with prominent jaw opening and closing. Laryngeal dystonia (also known as “spasmodic dysphonia”) affects the voice and either adduction or abduction of the muscles responsible for phonation is seen. The true prevalence of adult onset focal dystonia remains unknown and estimates range between 16 and 730 cases per 100 000 individuals.13 14 Adult onset focal forms rarely progress to generalised dystonia. By contrast, early onset generalised isolated dystonia often begins in childhood in a limb and then generalises over months. Early onset variants of dystonia have a lower prevalence of approximately 7 per 100 000 individuals.13
Fig 1.
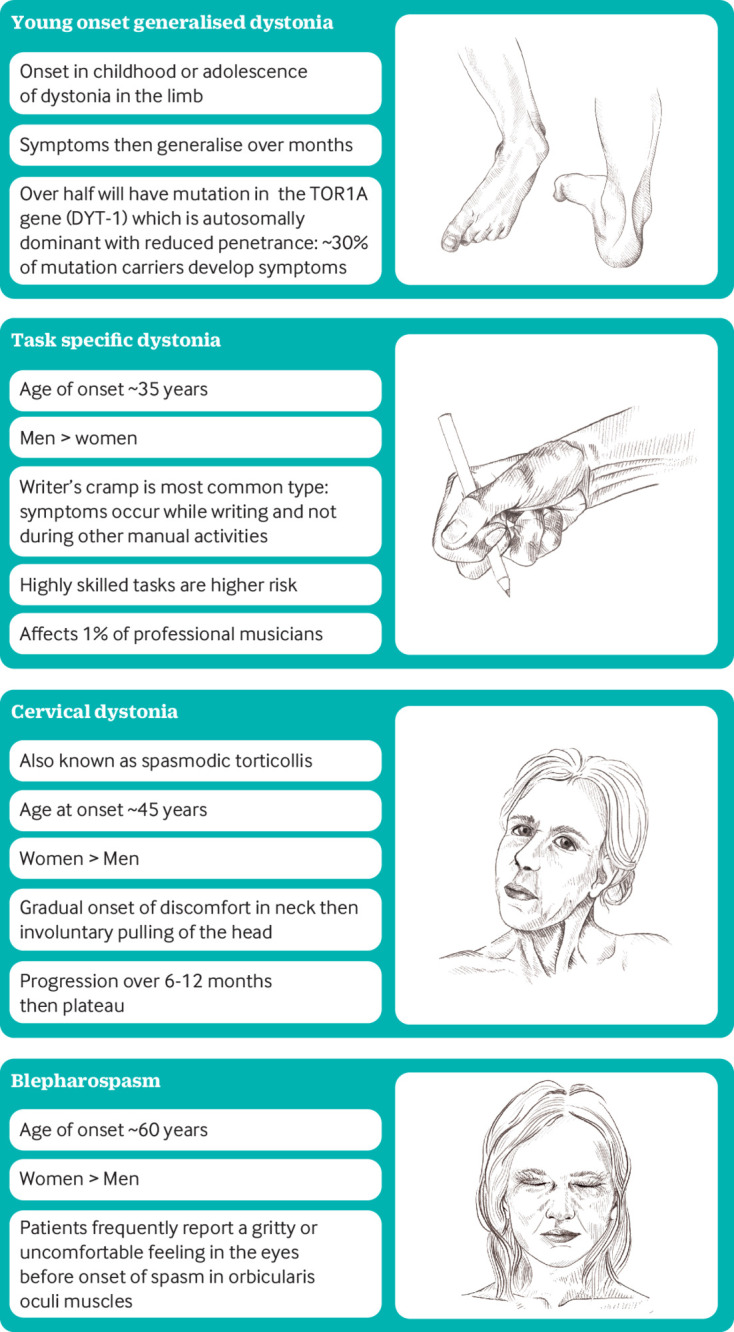
Different clinical patterns of isolated dystonia. Children and adolescents typically present with a generalised dystonia whereas presentation in adults is usually with a focal dystonia
Isolated dystonias can be subtle initially and diagnosis can be delayed for many months or years.15 Prompt recognition is often limited by a lack of knowledge among medical professionals.1 Part of the difficulty is that no reliable diagnostic test exists for dystonia, and routine brain imaging is unremarkable. In the future, kinematic, neurophysiological, and imaging tools are likely to be available to assist decisions about diagnosis.16
Combined dystonia
Dystonia with another movement disorder and/or occurrence of other neurological or systemic features is referred to as combined dystonia. As there are many potential causes, syndromic associations are used to narrow down diagnostic possibilities. Deciding which movement disorders or other features dominate is key. For example, foot dystonia and mild parkinsonism in a young adult is suggestive of a genetic cause of Parkinson’s disease (such as a LRRK2 mutation). Many centres now have panels of genetic tests that are performed according to the specific syndromic clustering.
Two treatable disorders are worth emphasising: Wilson’s disease and dopa responsive dystonia. Dystonia and liver abnormalities and/or psychiatric symptoms raise the possibility of Wilson’s disease.17 This autosomal recessive disorder of copper metabolism has a mean age of 18 for neurological presentation, often with a mixed movement disorder.18 Full blood count, liver function tests, and serum caeruloplasmin are useful initial investigations.17 Twenty four hour urine copper and slit lamp examination (for Kayser-Fleischer rings) can be organised if serum caeruloplasmin is low, neuro-imaging is abnormal, or there is a high index of suspicion for Wilson’s disease. First line treatments include copper chelating agents that increase urinary copper excretion or zinc salts that inhibit the intestinal absorption of copper. Dopa responsive dystonia encompasses a group of genetically and clinically heterogeneous disorders that often manifest with diurnal fluctuation of dystonia (worse at end of the day).19 Most patients have a deficiency in an enzyme involved in the biosynthesis of dopamine, and some forms are dramatically responsive to low doses of levodopa. The typical age of onset is in early childhood with a progressive limb dystonia. However, owing to the variability of presentation and difficulties making a laboratory diagnosis, many patients with young onset dystonia (<40 years) may receive a trial of levodopa to assess for the possibility of dopa responsive dystonia.
Acquired dystonia
Acquired dystonia occurs secondary to a precipitating event. Approximately half of children with dystonia have an acquired cause for their condition; causes include hypoxic brain injury at birth, preterm delivery, and kernicterus.20 Timing of the onset of dystonia can vary after a causal event. For example, in drug induced dystonia, acute reactions can be seen hours after the administration of antidopaminergic medications (eg, metoclopramide, prochlorperazine). One of the most common presentations is an oculogyric crisis, where the patient has upward, conjugate, tonic deviation of the eyes.21 Patients require urgent treatment, especially if laryngeal involvement compromises breathing, and supportive measures and intravenous anticholinergic usually have good effect. By contrast, tardive dystonia can occur many months or years after chronic exposure to a range of medications such as antipsychotics (eg, haloperidol). This is seen particularly in younger patients where a predominantly axial dystonia results in hyperextension of the spine and neck. Tardive dyskinesia (choreiform movements) is often seen in combination with the dystonia. Dystonia can unfortunately persist even after the causative drug is discontinued.22
Consider functional (psychogenic) dystonia if the patient shows an unusual distribution of dystonia, a fixed dystonia, and/or a reduction of symptoms with distraction.23 Functional dystonia is typically understood through a biopsychosocial framework of predisposing, precipitating, and perpetuating factors that cause abnormal brain processing of sensorimotor signalling. Patients can significantly improve if the syndrome is actively identified (functional dystonia should not be a diagnosis of exclusion) and a mechanistic explanation offered.24 Rehabilitation aimed at retraining movement (physiotherapy and/or multidisciplinary treatment) is increasingly available and other aspects of a treatment plan include management of pain and psychological input.23
Management
Patients with suspected dystonia should be referred to a neurologist specialising in movement disorders for further evaluation and to access treatments. Basic blood tests, including a full blood count, urea and electrolytes, liver function tests, and caeruloplasmin can be sent at this point. It is reasonable to leave brain imaging until neurological review, as imaging protocols are often adjusted to the patient’s clinical features. Treatment is tailored to the cause of dystonia. As a general rule, the first line treatment of focal dystonia is botulinum toxin injections, whereas generalised dystonia is treated with oral medications and surgery.25
Botulinum toxin injections
Regular botulinum toxin injections are the mainstay of treatment for cervical dystonia (high quality evidence) and blepharospasm (moderate quality evidence), with randomised control trials showing that dystonia and quality of life are improved by injections (fig 2).26 The toxin prevents the release of the neurotransmitter acetylcholine from axon endings at the neuromuscular junction, and works by causing weakness in the dystonic muscle. Commercially available types of botulinum toxin have differences in potency, and doses across types are not equivalent. Treatment is often delivered via specialist botulinum toxin clinics within neurological or ears, nose, and throat services (spasmodic dystonia).
Fig 2.

Onset of effect of botulinum toxin injections is usually after 2-3 days and an effect is maintained for approximately 3 months. The optimal interval between injections depends on the type of dystonia and individual response. Initially repeated injections can have a cumulative effect with baseline gains in symptomatic control
Oral medications
Anticholinergic medications such as trihexyphenidyl can significantly reduce dystonic contractions in generalised dystonia.27 The dose of anticholinergics must be titrated up gradually to minimise side effects such as dry mouth, gastrointestinal upset, and confusion (particularly in older patients). Muscle relaxants, such as baclofen and benzodiazepines, are also used.
Surgery
The most common surgical approach to treat dystonia is deep brain stimulation, in which continuous stimulation is delivered to deep nuclei in the brain (mostly commonly basal ganglia and thalamic nuclei). A neurosurgical operation is required to implant permanent electrodes within the brain and these are connected via subcutaneously tunnelled leads in the neck to an implanted stimulator in the chest. The implanted stimulator can be programmed and checked by communicating hardware in clinic/at home. The precise mechanism of deep brain stimulation is debated but its long term efficacy is well documented in randomised controlled trials for isolated young onset generalised dystonia (such as TOR1A related dystonia) and refractory cervical dystonia.28 29 In the future, individually tailored surgical options will be available with novel stimulation sites and adaptive stimulation paradigms. Any patient with a neuromodulatory device should be under regular review by their specialist team. With certain models of deep brain stimulation, magnetic resonance imaging is only safe under certain conditions, and the types of surgical diathermy used at operations are limited. Most services operate help lines for queries from patients or clinicians.
Neurophysiotherapy
Access to specialist neurophysiotherapy for dystonia is variable. An emerging evidence base suggests that patients can regain motor control, improve dystonic contractions, and reduce subsequent disability with neurophysiotherapy.30 31 Neurophysiotherapy is best tailored to the specific subtype of dystonia.31 32
Non-motor symptoms
Although the treatment of dystonic muscle contractions is often given priority, depression and anxiety are common and can be just as disabling.33 Pain of the affected body part is also present in more than half of patients. Pain scores have been shown to improve with analgesia and neuropathic medications, effective treatment of dystonic contractions, and treatment of associated neuropsychiatric symptoms.33
Education into practice.
What categories can be used to describe the clinical features of a patient presenting with dystonia?
How would you summarise available treatments to a patient?
Resources for patients.
These organisations have a range of educational materials such as fact sheets, podcasts, and videos and work to connect people with dystonia, raise awareness of dystonia, and advance research initiatives:
https://www.neurosymptoms.org/en_GB/symptoms/fnd-symptoms/functional-dystonia/,
How this article was created.
We conducted a search of PubMed for evidence for dystonia until November 2021. AS wrote the first draft and then each author contributed to the manuscript until we had fully integrated our range of viewpoints. Our expertise bridges neurology, neurophysiotherapy, general practice, education, and patient advocacy.
How patients were involved in the creation of this article.
One of the authors is a patient and patients’ advocate (with Dystonia Europe, Polish Dystonia Society) and has been fully involved with the planning and writing of the article.
Contributorship and the guarantor: All authors wrote and reviewed the article and the guarantor is Anna Sadnicka.
Competing interests: none declared.
We thank Victoria Devenport, illustrator (vikki.devenport@gmail.com) for the pencil sketches of isolated dystonia.
Provenance and peer review: commissioned; externally peer reviewed.
References
- 1. Smit M, Albanese A, Benson M, et al. Collaborative Working Group . Dystonia management: what to expect from the future? The perspectives of patients and clinicians within DystoniaNet Europe. Front Neurol 2021;12:646841. 10.3389/fneur.2021.646841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord 2013;28:863-73. 10.1002/mds.25475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fung VS, Jinnah HA, Bhatia K, Vidailhet M. Assessment of patients with isolated or combined dystonia: an update on dystonia syndromes. Mov Disord 2013;28:889-98. 10.1002/mds.25549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marsden CD, Quinn NP. The dystonias. BMJ 1990;300:139-44. 10.1136/bmj.300.6718.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neychev VK, Gross RE, Lehéricy S, Hess EJ, Jinnah HA. The functional neuroanatomy of dystonia. Neurobiol Dis 2011;42:185-201. 10.1016/j.nbd.2011.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ozelius LJ, Hewett JW, Page CE, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 1997;17:40-8. 10.1038/ng0997-40 [DOI] [PubMed] [Google Scholar]
- 7. Fuchs T, Saunders-Pullman R, Masuho I, et al. Mutations in GNAL cause primary torsion dystonia. Nat Genet 2013;45:88-92. 10.1038/ng.2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gonzalez-Latapi P, Marotta N, Mencacci NE. Emerging and converging molecular mechanisms in dystonia. J Neural Transm (Vienna) 2021;128:483-98. [DOI] [PubMed] [Google Scholar]
- 9. Altenmüller E, Ioannou CI, Lee A. Apollo’s curse: neurological causes of motor impairments in musicians. Prog Brain Res 2015;217:89-106. 10.1016/bs.pbr.2014.11.022 [DOI] [PubMed] [Google Scholar]
- 10. Sadnicka A, Hamada M. Plasticity and dystonia: a hypothesis shrouded in variability. Exp Brain Res 2020;238:1611-7. 10.1007/s00221-020-05773-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kassavetis P, Sadnicka A, Saifee TA, et al. Reappraising the role of motor surround inhibition in dystonia. J Neurol Sci 2018;390:178-83. 10.1016/j.jns.2018.04.015 [DOI] [PubMed] [Google Scholar]
- 12. Patel N, Hanfelt J, Marsh L, Jankovic J, members of the Dystonia Coalition . Alleviating manoeuvres (sensory tricks) in cervical dystonia. J Neurol Neurosurg Psychiatry 2014;85:882-4. 10.1136/jnnp-2013-307316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Steeves TD, Day L, Dykeman J, Jette N, Pringsheim T. The prevalence of primary dystonia: a systematic review and meta-analysis. Mov Disord 2012;27:1789-96. 10.1002/mds.25244 [DOI] [PubMed] [Google Scholar]
- 14. Müller J, Kiechl S, Wenning GK, et al. The prevalence of primary dystonia in the general community. Neurology 2002;59:941-3. 10.1212/01.WNL.0000026474.12594.0D [DOI] [PubMed] [Google Scholar]
- 15. Bertram KL, Williams DR. Delays to the diagnosis of cervical dystonia. J Clin Neurosci 2016;25:62-4. 10.1016/j.jocn.2015.05.054 [DOI] [PubMed] [Google Scholar]
- 16. Valeriani D, Simonyan K. A microstructural neural network biomarker for dystonia diagnosis identified by a DystoniaNet deep learning platform. Proc Natl Acad Sci U S A 2020;117:26398-405. 10.1073/pnas.2009165117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shribman S, Poujois A, Bandmann O, Czlonkowska A, Warner TT. Wilson’s disease: update on pathogenesis, biomarkers and treatments. J Neurol Neurosurg Psychiatry 2021;92:1053-61. 10.1136/jnnp-2021-326123 [DOI] [PubMed] [Google Scholar]
- 18. Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol 2015;14:103-13. 10.1016/S1474-4422(14)70190-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wijemanne S, Jankovic J. Dopa-responsive dystonia--clinical and genetic heterogeneity. Nat Rev Neurol 2015;11:414-24. 10.1038/nrneurol.2015.86 [DOI] [PubMed] [Google Scholar]
- 20. Lin JP, Lumsden DE, Gimeno H, Kaminska M. The impact and prognosis for dystonia in childhood including dystonic cerebral palsy: a clinical and demographic tertiary cohort study. J Neurol Neurosurg Psychiatry 2014;85:1239-44. 10.1136/jnnp-2013-307041 [DOI] [PubMed] [Google Scholar]
- 21. Barow E, Schneider SA, Bhatia KP, Ganos C. Oculogyric crises: Etiology, pathophysiology and therapeutic approaches. Parkinsonism Relat Disord 2017;36:3-9. 10.1016/j.parkreldis.2016.11.012 [DOI] [PubMed] [Google Scholar]
- 22. Kiriakakis V, Bhatia KP, Quinn NP, Marsden CD. The natural history of tardive dystonia. A long-term follow-up study of 107 cases. Brain 1998;121:2053-66. 10.1093/brain/121.11.2053 [DOI] [PubMed] [Google Scholar]
- 23. Nielsen G, Stone J, Matthews A, et al. Physiotherapy for functional motor disorders: a consensus recommendation. J Neurol Neurosurg Psychiatry 2015;86:1113-9. 10.1136/jnnp-2014-309255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stone J, Burton C, Carson A. Recognising and explaining functional neurological disorder. BMJ 2020;371:m3745. 10.1136/bmj.m3745 [DOI] [PubMed] [Google Scholar]
- 25. Balint B, Mencacci NE, Valente EM, et al. Dystonia. Nat Rev Dis Primers 2018;4:25. 10.1038/s41572-018-0023-6 [DOI] [PubMed] [Google Scholar]
- 26. Simpson DM, Hallett M, Ashman EJ, et al. Practice guideline update summary: Botulinum neurotoxin for the treatment of blepharospasm, cervical dystonia, adult spasticity, and headache: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2016;86:1818-26. 10.1212/WNL.0000000000002560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jankovic J. Medical treatment of dystonia. Mov Disord 2013;28:1001-12. 10.1002/mds.25552 [DOI] [PubMed] [Google Scholar]
- 28. Volkmann J, Mueller J, Deuschl G, et al. DBS study group for dystonia . Pallidal neurostimulation in patients with medication-refractory cervical dystonia: a randomised, sham-controlled trial. Lancet Neurol 2014;13:875-84. 10.1016/S1474-4422(14)70143-7 [DOI] [PubMed] [Google Scholar]
- 29. Kupsch A, Kuehn A, Klaffke S, et al. Deep brain stimulation in dystonia. J Neurol 2003;250(Suppl 1):I47-52. 10.1007/s00415-003-1110-2 [DOI] [PubMed] [Google Scholar]
- 30. van den Dool J, Visser B, Koelman JH, Engelbert RH, Tijssen MA. Long-term specialized physical therapy in cervical dystonia: outcomes of a randomized controlled trial. Arch Phys Med Rehabil 2019;100:1417-25. 10.1016/j.apmr.2019.01.013 [DOI] [PubMed] [Google Scholar]
- 31. De Pauw J, Van der Velden K, Meirte J, et al. The effectiveness of physiotherapy for cervical dystonia: a systematic literature review. J Neurol 2014;261:1857-65. 10.1007/s00415-013-7220-8 [DOI] [PubMed] [Google Scholar]
- 32. Sadnicka A, Rosset-Llobet J. A motor control model of task-specific dystonia and its rehabilitation. Prog Brain Res 2019;249:269-83. 10.1016/bs.pbr.2019.04.011 [DOI] [PubMed] [Google Scholar]
- 33. van den Dool J, Tijssen MA, Koelman JH, Engelbert RH, Visser B. Determinants of disability in cervical dystonia. Parkinsonism Relat Disord 2016;32:48-53. 10.1016/j.parkreldis.2016.08.014 [DOI] [PubMed] [Google Scholar]