

Abstract
METTL16 has recently been identified as an RNA methyltransferase responsible for deposition of N6-methyladenosine (m6A) in a few transcripts. Whether METTL16 methylates a large set of transcripts, similar to METTL3 and METTL14, remains unclear. Here we show that METTL16 exerts both methyltransferase activity-dependent and -independent functions in gene regulation. In cell nucleus, METTL16 functions as an m6A writer to deposit m6A into hundreds of its specific mRNA targets. In cytosol, METTL16 promotes translation in an m6A-independent manner. More specifically, METTL16 directly interacts with eukaryotic initiation factor 3a/b (eIF3a/b) and ribosomal RNAs (rRNAs) through its Mtase domain, thereby facilitating the assembly of the translation initiation complex (TIC) and promoting translation of over 4,000 mRNA transcripts. Moreover, we demonstrate that METTL16 is critical for the tumorigenesis of hepatocellular carcinoma. Collectively, our studies reveal previously unappreciated dual functions of METTL16 as an m6A writer and a translation initiation facilitator, which together contribute to its essential function in tumorigenesis.
Keywords: METTL16, N6-methyladenosine (m6A), methyltransferase, translation initiation, rRNAs, eIF3a/b, JUN, hepatocellular carcinoma (HCC)
Introduction
As the most prevailing internal decoration of mammalian mRNA, N6-methyladenosine (m6A) plays essential roles in normal biological processes and development via regulating the fate of target RNAs, and its dysregulation is associated with various types of diseases, including cancers1, 2. While the m6A methyltransferase complex (MTC), composed of METTL3 and METTL14, has been thought to be the main m6A writer3–7, depletion of METTL3 and METTL14 is known to cause ~60% decrease of m6A modification in some cell types, with a good portion of m6A marks in cellular do not map well with the binding sites of METTL3-METTL143, 8. These observations imply the existence of additional m6A methyltransferase. Human methyltransferase like proteins (METTL) belong to a superfamily composed of over 30 members with a conserved s-adenosylmethionine (SAM)-binding site9. However, the functions and related molecular mechanisms of most METTL proteins remain elusive10–12. More recently, METTL16 has also been identified as a m6A methyltransferase, but was only reported to add m6A to several non-coding RNAs including U6 snRNA, MALAT1 and XIST, and one mRNA target (MAT2A)13–18. The biological functions of METTL16 in normal development and diseases (e.g., cancers) have yet to be investigated, albeit a study reporting its role in mouse embryonic development17.
Being the most fundamental layer of gene expression, mRNA translation is a tightly coordinated process and accomplished by ribosome and its associated assembly factors19–21. Dysregulation of protein synthesis is associated with a wide spectrum of human diseases22, 23. Typically, mRNA translation occurs in four steps: initiation, elongation, termination, and ribosome recycling. Translation initiation, a rate-limiting phase, is the most regulated event in translation and begins with the formation of a 43S pre-initiation complex (PIC) on the basis of the 40S (small) ribosomal subunit24, 25. Subsequently, the 60S (large) ribosomal subunit is recruited, forming the 80S translation initiation complex (TIC). Translation initiation is governed by the availability and activity of eukaryotic translation initiation factors (eIFs). eIF3, including 13 members in mammals, encircles the 40S ribosome and serves as a scaffold for the recruitment and binding of other eIFs involved in mRNA binding, scanning and start codon recognition26. Aberrantly increased expression of eIF3 subunits frequently occurs in human cancers and overexpression of some unit can drive de-novo tumor formation via increasing protein synthesis27, 28.
In the present study, we show that, distinct from the dominant nuclear localization of METTL3/METTL14, METTL16 is preferentially distributed in cytosol; METTL16 deposits m6A into hundreds of mRNA targets as an m6A methyltransferase in nuclei, and promotes translation of thousands of mRNA transcripts in cytosol, through recruiting eIF3a/b and rRNAs to facilitate the formation of the 43S PIC and 80S TIC. The dual functions of METTL16, as an m6A writer and a translation facilitator, collectively devote to its broad cancer dependency and strong tumor-promoting effect.
Results
METTL16 deposits m6A into a small portion of METTL16-bound mRNA transcripts
In analysis of two independent large-scale genome-wide CRISPR-Cas9 knockout (KO) screening datasets29, 30, we found that, among the METTL family members, METTL16 is the most essential gene for the survival of the vast majority of cancer cells (Extended Data Fig. 1a, b), implying its functional importance in cancer. Immunofluorescence (IF) assays showed that METTL16 is localized in both nucleus and cytosol of normal (HEK293T) (Fig. 1a) and cancer cells (Huh7) (Extended Data Fig. 1c). Unlike METTL3/14 that are mainly located in nucleus, a large proportion of endogenous METTL16 is distributed in the cytosol (Fig. 1a–c and Extended Data Fig. 1c, d). Ectopically expressed wide-type or mutant (catalytic-dead mutations, F187G and PP185/186AA; catalytic-active mutation, R200Q) METTL16 also showed similar patterns (Extended Data Fig. 1e, f). As reported previously, m6A modifications are installed co-transcriptionally in the nucleus31–33. Thus, besides catalyzing the formation of m6A, METTL16 might exert additional function(s), particularly in the cytosol.
Fig. 1. Comparative analysis of subcellular distribution and m6A catalytic activity of METTL3, METTL14, and METTL16.
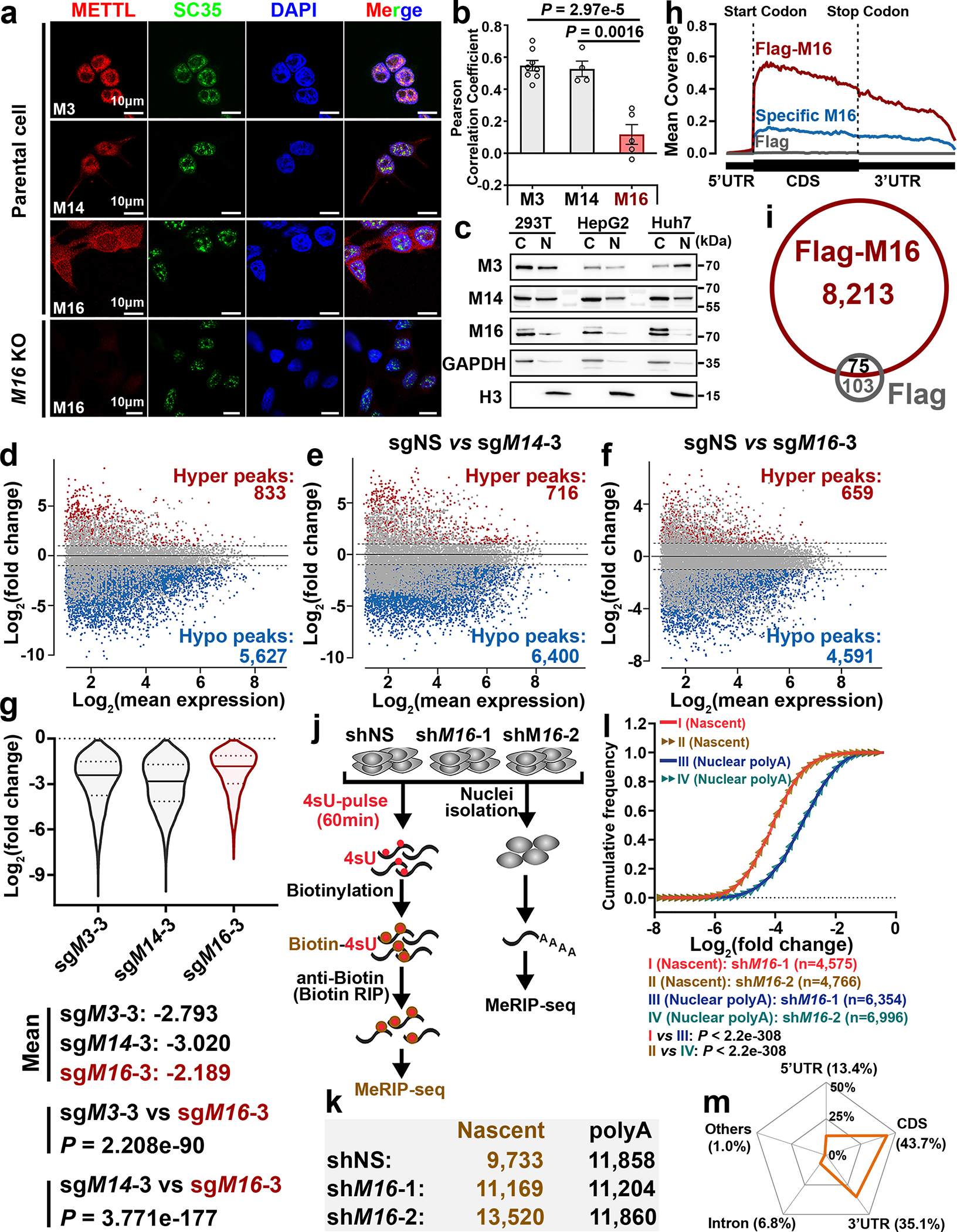
a, Subcellular localization of endogenous METTL3/14/16 in HEK293T cells. SC35 and DAPI, markers for nuclear speckle and nucleus, respectively. b, Pearson correlation analysis showing the extent of colocalization of the three METTL family members with nucleus in HEK293T cells. Data are mean ± s.e.m. Statistics: unpaired, two-tailed t-test. M3, n = 8; M14, n = 4; M16, n = 5 independent experiments. c, Expression levels of METTL3/14/16 in the cytoplasm (C) and nucleus (N) of the cells. Data shown represent 3 independent experiments. d-f, MA plots displaying the m6A-hyper and m6A-hypo peaks in poly(A) RNA upon METTL3 (d), METTL14 (e), or METTL16 (f) KO in HEK293T cells. The significantly increased (hyper) or decreased (hypo) m6A peaks are exhibited in red and blue, respectively (P < 0.01, calculated by exomePeak). g, Violin plots showing the fold changes of decreased m6A peaks upon METTL3, METTL14, or METTL16 KO. The three lines inside the violins were defined as the first quartile, median, and third quartile. Statistics: unpaired, two-tailed t-test. h, Metagene profile of enrichment of METTL16-bound transcripts (Flag-M16), specific METTL16-bound transcripts (Specific M16; i.e., the 3,206 transcripts in Extended Data Fig. 2k), and Flag-bound sites (Flag, background control) across the target transcripts. i, Venn diagram showing the overlap between METTL16-bound transcripts and Flag-bound transcripts. j, Schematic of m6A MeRIP-seq with nascent RNA and nuclear poly(A) RNA in HEK293T cells upon METTL16 KD. k, The number of m6A peaks in nascent RNA and poly(A) RNAs. l, Cumulative distribution function (CDF) plot depicting the fold changes of m6A-hypo peaks induced by METTL16 KD in nascent RNA and nuclear poly(A) RNA. Statistics: two-sided Wilcoxon and Mann–Whitney test. m, Radar chart showing the percentage of nascent RNA m6A peaks distributed at exons (including 5’UTR, CDS, 3’UTR), introns, and other regions in the control (shNS) group. The m6A MeRIP-seq have been performed with 2 biological replicates with similar results; the data shown in d-m represent a single representative experiment.
We then conducted m6A MeRIP-seq and triple-quadrupole-MS (QQQ-MS) assays to assess the methyltransferase activity of METTL16 and compare it with that of METTL3/14. The QQQ-MS data showed that, similar to the depletion of METTL3 or METTL14, METTL16 depletion also significantly decreased global m6A abundance in mRNAs; METTL16 depletion did not affect the expression of METTL3 and METTL14 (Extended Data Fig. 2a–c). Subsequent m6A MeRIP-seq results suggested METTL16 KO led to a transcriptome-wide decrease in m6A abundance in HEK293T cells, though to a degree relatively smaller than that caused by METTL3 or METTL14 KO (Fig. 1d–g, and Extended Data Fig. 2d–f). METTL16 KO caused a significant m6A abundance decrease in 3,075 transcripts, and 2,741 of those transcripts also showed reduced m6A abundance upon METTL3 KO and/or METTL14 KO (Extended Data Fig. 2g, left). Although METTL3/14 levels were not changed upon METTL16 depletion (see Extended Data Fig. 2b), the decreased m6A levels in these 2,741 shared target transcripts upon METTL16 KO could be attributed to the direct suppression of expression and methyltransferase activity of METTL16, and/or due to indirect suppression of the METTL3/METTL14 MTC activity owing to METTL16 KO-induced decrease of cellular SAM (methylation donor for METTL3/14/16) level14. Via the overlap analysis, we identified 334 METTL16-specific m6A targets, which are mainly involved in cell cycle, apoptotic process, RNA binding, and transcription regulator activity etc. (Extended Data Fig. 2g, right). Similar to m6A-hypo peaks of METTL3 and METTL14, those of METTL16 are also enriched in the vicinity of the stop codon (Extended Data Fig. 2h). In addition, we compared our m6A MeRIP-seq with other published datasets and demonstrated that >90% of METTL16-specific m6A targets we identified are not targets of METTL3 and METTL143 (Extended Data Fig. 2i), indicating those targets are truly METTL16-specific.
We performed RNA immunoprecipitation sequencing (RIP-seq) to identify the target transcripts bound by METTL16, METTL3 or METTL14 (Extended Data Fig. 2j). The METTL3-bound transcripts overlap very well with METTL14-bound transcripts, whereas almost half of METTL16-bound transcripts do not overlap with those bound by METTL3 or METTL14 (Extended Data Fig. 2k, left). Notably, there is a substantial enrichment of METTL16-binding sites in the vicinity of start codon of its target transcripts (Fig. 1h). By integrative analysis of METTL16 RIP-seq data and m6A MeRIP-seq data, we found that mRNA transcripts with m6A-hypo peaks upon METTL16 KO only account for a small proportion (23.8%; 1,953/8,213) of transcripts bound by METTL16 (Extended Data Fig. 2l, left). The top one binding motif of the 1,953 METTL16-bound transcripts with METTL16 KO-induced m6A-hypo peaks is “AGGACU”, analogous to the consensus m6A DRACH (D =A, G, or U; R = A or G; H = A, C, or U) motif (Extended Data Fig. 2l, right). In contrast, the top one binding motif of specific METTL16-bound transcripts is “UGAAGA” (Extended Data Fig. 2k, right), distinct from the conserved m6A motif, suggesting METTL16 unlikely deposits m6A to the majority of its binding sites.
We next conducted m6A MeRIP-seq with nascent RNA and nuclear poly(A) RNA (Fig. 1j, k) to assess the enzymatic activity of METTL16 towards newly transcribed RNAs and mature RNAs. Intriguingly, METTL16 appears to play a more robust role in depositing m6A modifications in nascent RNA than in nuclear poly(A) RNA, as the decreased trend of m6A modifications in nascent RNA upon METTL16 KD is much more evident than those of nuclear poly(A) RNA (Fig. 1l and Extended Data Fig. 2m). Notably, most of the METTL16-mediated m6A modification sites in nascent RNAs are also located in exons, not in introns (Fig. 1m). These data illustrate the spatial (mainly nuclei) and temporal (especially newly transcribed) installation of m6A mediated by METTL16, highlighting the dynamic changes of m6A modifications mediated by METTL16 during the maturation of mRNA transcripts.
Albeit the high overlap between METTL16’s targets and METTL3/14’s targets in our m6A MeRIP-seq (see Extended Fig. 2g), METTL3 and METTL14 fail to methylate the specific m6A target sites of METTL16, such as those located in MAT2A and BMP2 (Extended Data Fig. 3a, b). The top 3 signaling pathways suppressed by METTL16 KO as determined by GSEA34 are shown in Extended Data Fig. 3c. Moreover, forced expression of METTL3/14 can hardly reverse METTL16 KO-induced inhibitory effect on cell proliferation (Extended Data Fig. 3d, e). Since it was reported that METTL16 regulates expression of SAM synthetase MAT2A14, we sought to check whether METTL16 depletion causes a broad methylation suppression. However, we found that while METTL16 KO caused a significant decrease in m6A level (see Extended Data Fig. 2c), it did not significantly decrease other methylations, such as histone H3K36me3, H3K4me3, and H3K9me3 (Extended Data Fig. 3f), indicating that m6A is the main methylation affected by METTL16 KO.
METTL16 promotes translation efficiency globally
Given the high abundance of METTL16 in cytosol (see Fig. 1a–c and Extended Data Fig. 1c, d) and the low overlap between METTL16-bound transcripts and the transcripts with METTL16 KO-induced m6A-hypo peaks (see Extended Data Fig. 2l), we speculated that METTL16 also exerts methylation-independent activity, especially in cytosol, where translation occurs. To test the hypothesis, we conducted reporter tethering experiments with λN peptide-fused METTL proteins, which can tightly and specifically bind to 5 × BoxB RNA hairpins located in the 3’ UTR of firefly luciferase reporter (Fig. 2a). We chose METTL3 as a positive control as it was reported previously to enhance translation10, 35. Direct tethering of METTL16 proteins, either the wild type or catalytic (-dead or -active) mutants (Extended Data Fig. 4a), can significantly enhance the translation efficiency of luciferase, to an even higher degree than that caused by METTL3 (Fig. 2b). Conversely, genetic depletion of METTL16 substantially attenuates protein synthesis as determined by the Surface Sensing of Translation (SUnSET) assay36 and pulse labeling assay with methionine analogue homopropargylglycine (HPG)37, and METTL16 KO-induced translation suppression cannot be rescued by forced expression of METTL3/14 (Fig. 2c, e and Extended Data Fig. 4b–h); such translation suppression is not due to cell growth inhibition (Fig. 2d). Cap binding assays identified the localization of METTL16 in the 5’ cap region, indicating the potential involvement of METTL16 in translation initiation (Fig. 2f, g). Neither gain- nor loss-of-function mutations of METTL16 influence its enrichment in the m7G cap (Extended Data Fig. 4i), hinting that METTL16-mediated translational regulation might not rely on its methyltransferase activity.
Fig. 2. METTL16 enhances translation efficiency of target mRNAs.
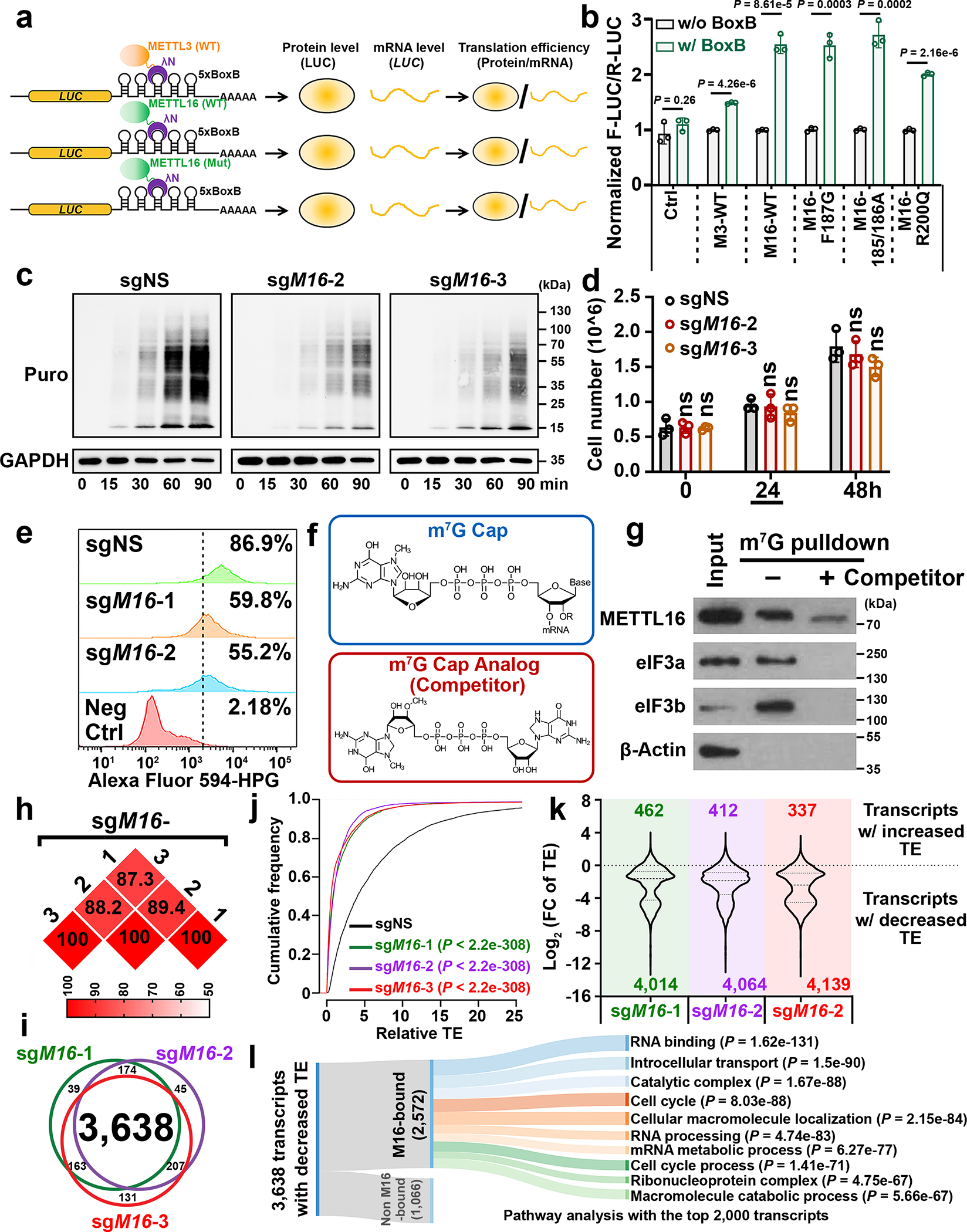
a, Experimental schedule of RNA tethering experiments to determine translation efficiency. b, Translation efficiency as determined by the tethering assays. F-Luc, Firefly luciferase; R-Luc, Renilla luciferase, internal control. Data are mean ± s.d. (n = 3 independent experiments). Statistics: unpaired, two-tailed t-test. c, Representative Western blotting images of SUnSET assays to quantify the amount of nascent (puromycin-labelled) peptides in HEK293T cells with METTL16 knockout (KO). To make the data comparable, all the samples were treated equally. Data shown represent 3 independent experiments. d, The cell number in sgNS, sgM16-2, and sgM16-3 groups. The samples were collected at 24 hours for the SUnSET assay shown in Fig. 2c. Data are mean ± s.d. (n = 3 independent experiments). Statistics: unpaired, two-tailed t-test. ns, not significant (P ≥ 0.05). e, Nascent protein synthesis upon METTL16 KO as determined by the pulse labeling assay via incorporation of L-homopropargylglycine (HPG). f, Structures of m7G cap and its analog. Data shown represent 2 independent experiments. g, Western blotting of m7G pulldown assays with indicated antibodies. eIF3a and eIF3b, positive controls. β-Actin, negative control. Data shown represent 2 independent experiments. h, The reproducibility of Ribo-seq with three diverse gRNAs against METTL16. Here, we presented the percentages of overlapping transcripts with decreased translation efficiency (TE) caused by the three gRNAs mediated METTL16 KO in HEK293T. i, Venn diagram showing high overlap of the transcripts with inhibited TE across the three gRNA groups. j, CDF plot depicting the transcriptome-wide inhibition of translation efficiency (TE) upon METTL16 KO. Statistics: two-sided Wilcoxon and Mann–Whitney test. k, Violin plots showing the Log2(FC of TE) upon METTL16 KO. FC, fold change. l, Sankey diagram presenting the subsequent pathway analysis of the transcripts whose translation initiation is facilitated by METTL16. Among the 3,638 transcripts with suppressed TE upon METTL16 KO, 2,572 are directly bound by METTL16 protein. Among the 2,572 targets, we selected the top 2,000 candidates for pathway analysis. Here, we showed the top 10 enriched pathways.
To probe METTL16-mediated translation regulation at the whole transcriptome level, ribosome profiling (Ribo-seq) was conducted (Extended Data Fig. 4j, k). Three distinct gRNAs against METTL16 unanimously induced translation inhibition of over 4,000 transcripts each (with 3,638 shared transcripts) (Fig. 2h–k). The majority of such transcripts are METTL16-bound transcripts, and are enriched in RNA binding, RNA processing, cell cycle, and mRNA metabolic process pathways (Fig. 2l).
METTL16 facilitates translation initiation via interaction with eIF3a/b in cytosol
To elucidate the mechanism underlying the effect of METTL16 on translation promotion, we sought to investigate its involvement and role in translation initiation, because the initiation, rather than elongation or termination, is often the most tightly orchestrated rate-limiting step of translation24, 25, and METTL16 localizes in the 5’ cap region. As the largest initiation factor, eIF3 plays a pivotal role via acting as a scaffold for recruitment of mRNA, 40S subunit, and other eIFs, and is indispensable to stimulate nearly all steps of translation initiation38, 39. The far-Western blotting with eIF3 complex implied that METTL16 may directly and specially bind to eIF3a, b, and/or c (Fig. 3a, b, and Extended Data Fig. 5a). Subsequent Co-IPs verified the direct interaction of METTL16 with eIF3a/b, but not with eIF3c (Fig. 3c, d). Moreover, their interactions are direct and RNA-independent (Extended Data Fig. 5b–f). In situ proximity ligation assay (PLA) further validated the physical interactions between METTL16 and eIF3a/b (Fig. 3e). As expected, most of the in situ PLA spots are located in cytoplasm, rather than in nucleus (Fig. 3f, g). Analogous to METTL16 KO, eIF3a and eIF3b KO remarkably suppress translation efficiency (Fig. 3h, i). On sucrose density gradients, METTL16 was strongly enriched in the 40S/60S/80S ribosomes together with eIF3a/b (Fig. 3j), indicating the co-occurrence of METTL16 and eIF3a/b, and their involvement in translation initiation. Additionally, around half of the eIF3a- or eIF3b-bound transcripts40 are also directly bound by METTL16; over 40% transcripts with inhibited translation efficiency upon METTL16 KO are also directly bound by eIF3a and eIF3b (Fig. 3k, l). Catalytic gain- or loss-of-function mutations of METTL16 did not affect its interaction with eIF3a/b (Extended Data Fig. 5g), consistent with our observation (see Fig. 2a, b, and Extended Data Fig. 4i) that METTL16-mediated translation promotion is independent of its methyltransferase activity.
Fig. 3. METTL16 facilitates translation initiation via direct interaction with eIF3a and eIF3b.
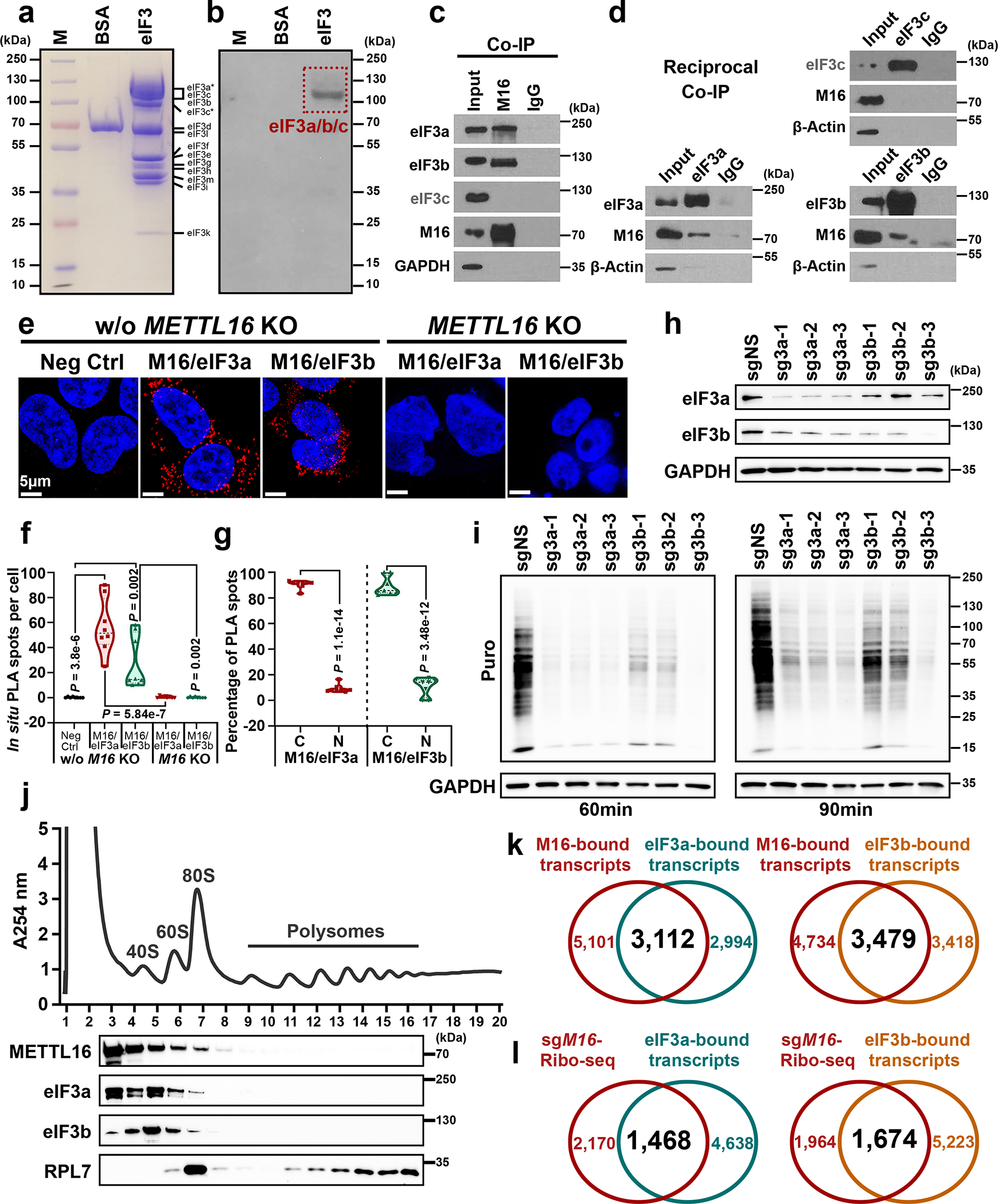
a, Coomassie brilliant blue staining of protein markers, BSA (negative control), and eIF3 family members. b, Far-Western blotting for the direct interaction between METTL16 and eIF3 family members. Images in a and b are representative of three biologically independent experiments with consistent results. c, Co-IP of METTL16 with endogenous eIF3a, eIF3b, and eIF3c. d, Reciprocal Co-IP of endogenous eIF3a, eIF3b, and eIF3c with METTL16. Data shown represent 2 independent experiments. e, In situ detection of METTL16-eIF3a and METTL16-eIF3b interaction via proximity ligation assays (PLA) in HEK293T cells with or without METTL16 KO. f, Quantification of in situ PLA puncta from the five groups. Statistics: unpaired, two-tailed t-test. n = 8 independent experiments. g, The distribution of in situ PLA spots from METTL16-eIF3a and METTL16-eIF3b in cytoplasm (C) and nuclear (N) of HEK293T cells without METTL16 KO. Statistics: unpaired, two-tailed t-test. n = 8 independent experiments. h, The KO efficiency of eIF3a and eIF3b in HEK293T cells. i, Representative Western blotting images of SUnSET assays to quantify the amount of nascent peptides in HEK293T cells with eIF3a and eIF3b KO. Data shown represent 3 independent experiments. j, Polysome profiles of HEK293T cells as determined by sucrose density gradient ultracentrifugation. The localization of eIF3a, eIF3b, METTL16, and RPL7 proteins were validated by Western blotting. Data shown represent 3 independent experiments. k, Venn diagram showing the overlap between METTL16-bound transcripts and eIF3a-bound transcripts (left), or between METTL16-bound transcripts and eIF3b-bound transcripts (right). Both the eIF3a- and eIF3b-bound transcripts were downloaded from the POSTAR3 (http://postar.ncrnalab.org/). l, Venn diagram showing the overlap between the transcripts with decreased translation efficiency in METTL16 KO cells and the transcripts directly bound by eIF3a (left) or eIF3b (right).
METTL16-mediated translation enhancement is Mtase domain dependent
We next sought to characterize which domain of METTL16 is essential for its binding with eIF3a/b and thereby enhancing translation efficiency. Among all the domains of METTL16 protein, only the Mtase domain is highly evolutionarily conserved (Extended Data Fig. 6a). Co-IPs were performed with the full-length (FL) and 8 truncated METTL16 proteins (Extended Data Fig. 6b). The results demonstrate that it is the Mtase domain, especially the 79–289 a.a. fragment, that directly interacts with eIF3a/b (Fig. 4a–f and Extended Data Fig. 6c–w). Further tethering experiments proved that the Mtase domain, but not the other domains, is indispensable for METTL16-mediated promotion of translation efficiency (Fig. 4g). Consistently, it is Mtase domain, but not the others, that display strong binding to the 5’ cap region (Fig. 4h). Both Gain- and loss-of-function mutants can reverse METTL16 KO-mediated translation suppression (Fig. 4i, j), highlighting that METTL16-induced translation regulation does not rely on its enzymatic activity. We also generated a METTL16 mutant with exclusive cytoplasmic localization via disturbing its canonical nuclear localization signal (NLS), and found that the NLS mut could restore METTL16 KO-mediated translation suppression and also largely reverse the growth inhibition (Fig. 4i, k–m), demonstrating that METTL16 exerts its major biological role in cytosol. Collectively, our data demonstrate that the Mtase domain is required for the interactions of METTL16 with eIF3a/b and 5’ cap region and is necessary for the function of METTL16 in promoting translation initiation.
Fig. 4. Both the Mtase domain and the cytoplasmic localization of METTL16 are necessary for translation regulation.
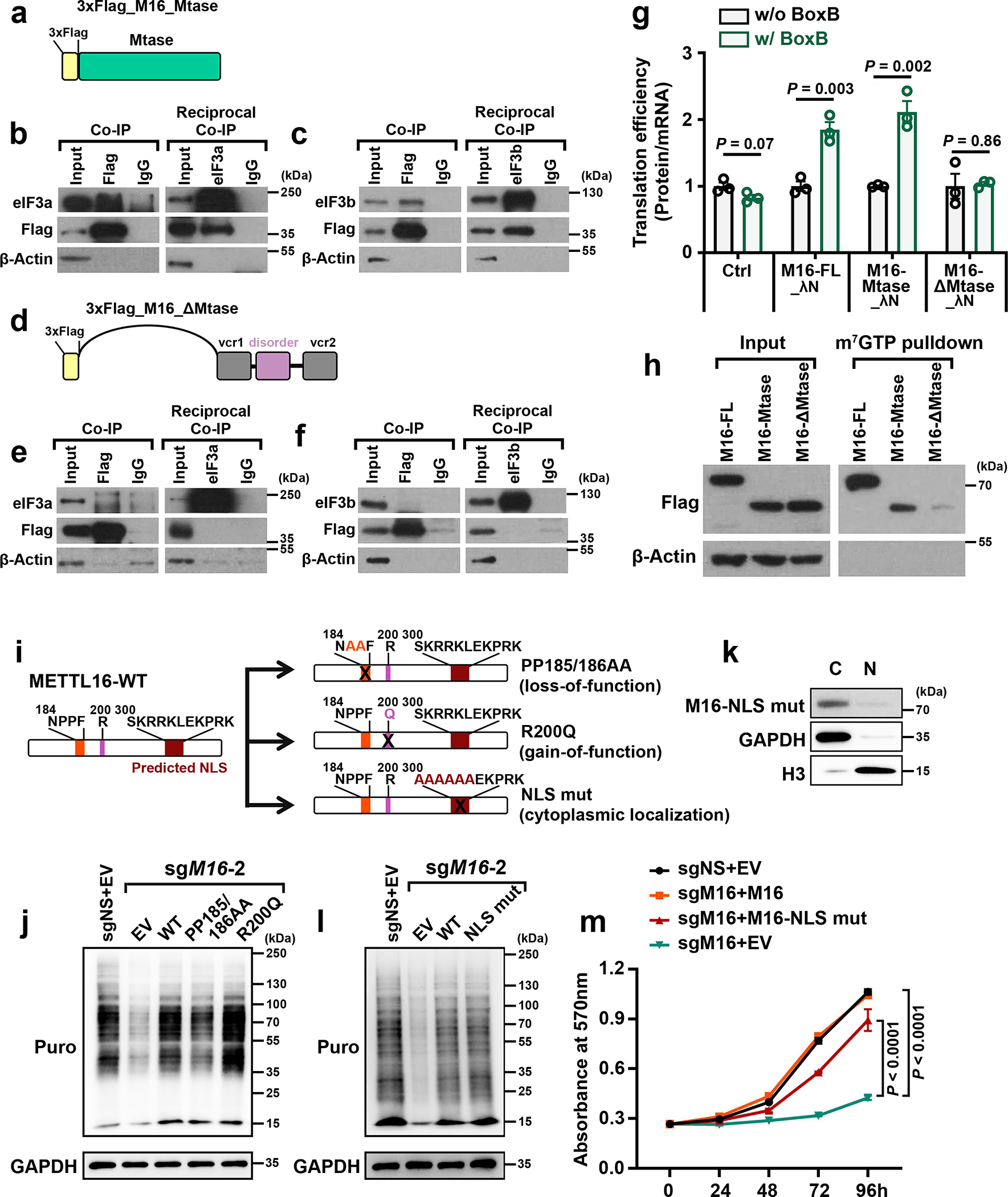
a, Schematic of truncated METTL16 that contains just the Mtase domain, with 3 × Flag tag attached at the N-terminal. b, c, Co-IP (left) and reciprocal Co-IP (right) showing the direct interaction between METTL16_Mtase (Flag) and eIF3a (b) or eIF3b (c). β-Actin was used as negative control. d, Schematic of another truncated METTL16 that contains vcr1-disorder-vcr2 (ΔMtase domain), with 3 × Flag tag attached to the N-terminal. e, f, Co-IP (left) and reciprocal Co-IP (right) showing no interaction between METTL16_ΔMtase (Flag) and eIF3a (e) or eIF3b (f). g, RNA tethering experiments depicting the translation efficiency with forced expression of full length (FL), Mtase, and ΔMtase. The 3 × Flag tag was fused to the N-terminal, while λN peptide was fused to the C-terminal of the constructs. Data are mean ± s.d. (n = 3 independent experiments). Statistics: unpaired, two-tailed t-test. h, m7GTP pull-down assays illustrating the robust binding of full length and Mtase domain of METTL16, but not the ΔMtase, in the cap region. i, Schematic of three METTL16 mutants, including catalytic-dead METTL16 (PP185/186AA), catalytic-active METTL16 (R200Q), and exclusive cytoplasmic METTL16 (NLS mut). j, The rescue effects of PP185/186AA and R200Q on METTL16 KO-induced translation inhibition in HEK293T cells. k, Subcellular localization of METTL16 NLS mut in HEK293T cells. l, The rescue effects of exclusive cytoplasmic METTL16 overexpression on METTL16 KO-induced translation inhibition in HEK293T cells. For b, c, e, f, h, j, k, and l, data shown represent 2 independent experiments with similar results. m, The rescue effects of wildtype METTL16 and exclusive cytoplasmic METTL16 overexpression on METTL16 KO-induced cell proliferation suppression in HEK293T cells. Data are mean ± s.d. (n =3 independent experiments). Statistics: two-way ANOVA.
Interactions of METTL16 with both eIF3a/b and rRNAs facilitate the formation of the 80S TIC
Strikingly, we found that METTL16 also directly interacts with rRNAs, to a degree comparable to its interaction with its known targets; such association does not happen to other METTL members, such as METTL3 even though it also enhances translation (Extended Data Fig. 7a–h). CLIP-qPCR with truncated METTL16 mutants demonstrates that the Mtase domain is also crucial for the interaction of METTL16 with rRNAs (Fig. 5a–d). Accordingly, we presume that the interactions of METTL16 with both eIF3a/b and 18S rRNA (the core of the 40S ribosomal subunit) will facilitate the binding between eIF3a/b and 18S rRNA and thereby the interaction of the eIF3 complex with the 40S ribosomal subunit. Indeed, we showed that METTL16 KD dramatically inhibited the interaction between eIF3a/b and 18S rRNA, whereas restoration of METTL16 could sufficiently rescue the inhibited associations (Fig. 5e–h). The binding ability of METTL16 with rRNAs in translation initiation fraction is much stronger than that in polysomes (Fig. 5i–k), highlighting the involvement of METTL16 in translation initiation complex. Moreover, we presumed that the interactions of METTL16 with 28S/5.8S rRNAs (the core of the 60S ribosomal subunit) may further recruit the 60S subunit to the eIF3/40S-subunit complex (i.e., the 43S PIC) and thereby facilitate the formation of the 80S TIC. Indeed, our ribosome profiles showed that METTL16 KO substantially reduced the formation of 80S ribosome (Fig. 5l–n). In vitro translation experiment directly verified that METTL16 enhanced translation capacity (Fig. 5o). It was reported that METTL16 can enhance splicing of retained intron of some of targets (e.g., MAT2A) in nucleus14 (also validated by us (see Extended Data Fig. 7i)), which in turn could protect the targets from decay and facilitate their transportation into the cytoplasm, leading to enhanced translation of the targets. However, only 296 transcripts showed significantly (P < 0.05) increased intron retention in METTL16-depleted cells based on the data generated by Pendleton et al.14 (see Extended Data Fig. 7j). Among the 3,638 transcripts with decreased translation efficiency in METTL16-depleted cells, merely 117 were accompanied with increased intron retention, suggesting that METTL16-mediated translation promotion is largely unrelated to its role in intron splicing (Extended Data Fig. 7k). Taken together, METTL16-eIF3a/b and METTL16–18S rRNA (the 40S ribosomal subunit) interactions stimulate the interplay between the eIF3 complex and the 40S ribosomal subunit, and thereby the assembly of the 43S PIC; the additional METTL16–28S/5.8S rRNAs (the 60S subunit) interactions subsequently facilitate the formation of the 80S TIC and therefore promote translation initiation (Fig. 5p).
Fig. 5. METTL16 facilitates the formation of TIC by interactions with both eIF3a/b and rRNAs.
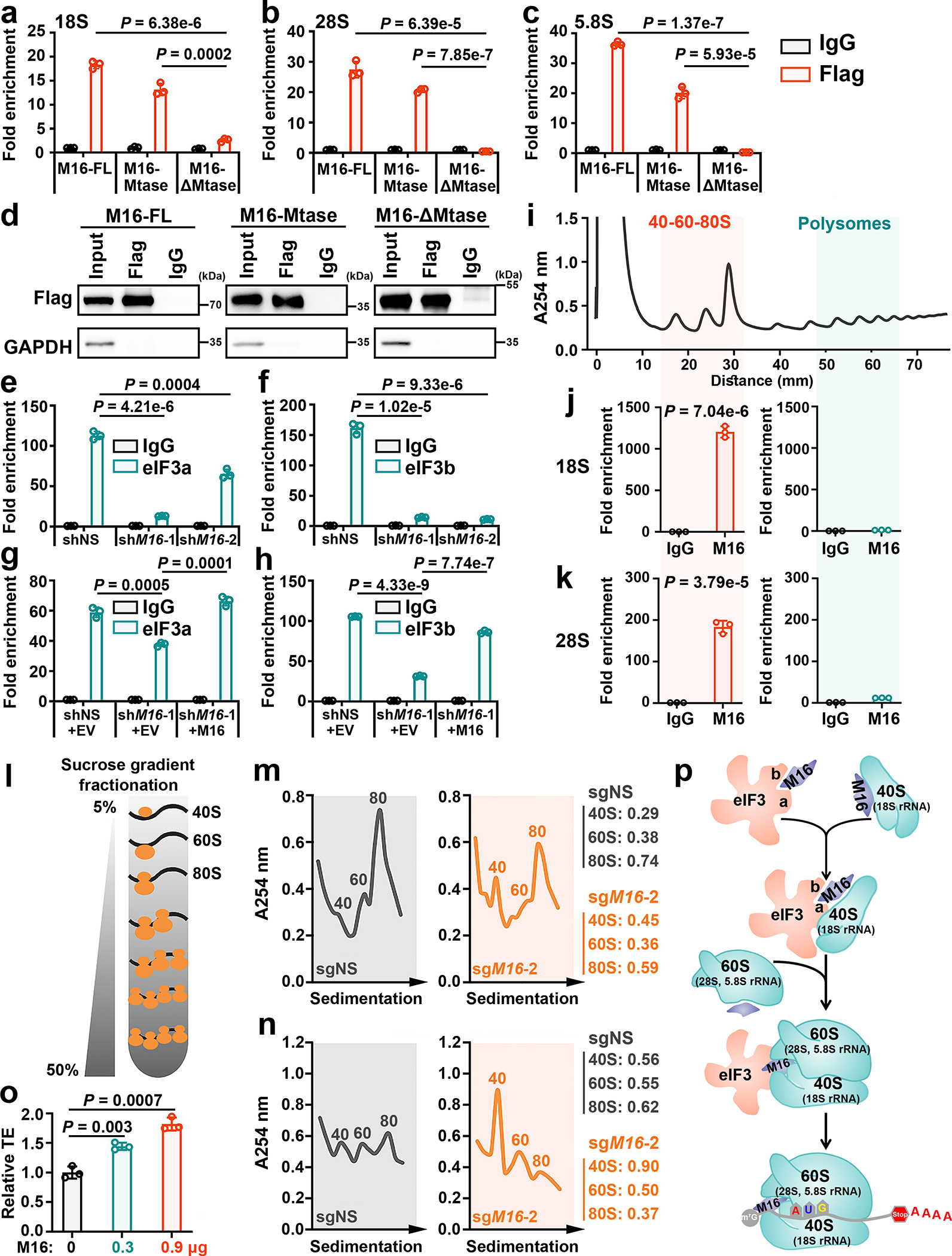
a, The binding of full length (FL), Mtase domain, and ΔMtase domain of METTL16 on 18S (a), 28S (b), and 5.8S (c) rRNAs in the cytoplasm fraction as determined by CLIP-qPCR. d, The comparable pulldown efficiency among METTL16-FL, Mtase, and ΔMtase groups. Data shown represent 3 independent experiments. e, f, CLIP-qPCR analysis showing the decreased association between 18S rRNA and eIF3a (e) or eIF3b (f) upon METTL16 KD. g, h, Rescue assay showing the rescued binding of eIF3a (g) or eIF3b (h) with 18S rRNA upon manipulating the expression of METTL16. i, Isolation of translation initiation fraction (40–60-80S) and polysome fraction for RIP according to polysome profiling. Data shown represent 3 independent experiments. j, k, The interactions between METTL16 and 18S rRNA (j) or 28S rRNA (k) in the translation initiation fraction (left) and polysome fraction (right). HEK293T cells were used in all the above studies. l, Schematic of sucrose gradient fractionation. m, n, Sucrose density gradient profiles showing the abundance of 40S, 60S, and 80S ribosomes from HEK293T (m) and HepG2 (n) cells upon METTL16 KO. Data shown represent 3 independent experiments. o, Effect of ectopic METTL16 protein on translation efficiency of luciferase mRNA as determined by in vitro translation. p, Schematic model of how METTL16 enhances translation initiation. The direct interactions of METTL16 with eIF3a/b and rRNAs drive the eIF3/40S ribosomal subunit (with 18S rRNA as the core) interaction and the subsequent assembly of 40S ribosomal subunit/60S ribosomal subunit (with 5.8S and 28S rRNAs as the core) into the 80S TIC, leading to stimulated translation initiation. For a-c, e-h, j, k, and o, data are represented as mean ± s.d. (n = 3 independent experiments). Statistics: unpaired, two-tailed t-test. For j and k, the P values indicate the binding of METTL16 with 18S rRNA (j) and 28S rRNA (k) in 40–60-80S vs Polysomes.
METTL16 plays a crucial tumor-promoting role in hepatocellular carcinoma (HCC)
As METTL16 appears to be essential for the survival of the vast majority types of cancer cells tested in two large-scale CRISPR-Cas9 screenings (see Extended Data Fig. 1a, b), we sought to determine its role in cancer and investigate whether its pathological role is attributed to its methyltransferase activity and/or translation-promotion function. First, we examined METTL16 expression patterns and prognostic impact of its expression levels in patients of all cancer types available in the TCGA datasets. We found METTL16 is aberrantly overexpressed in HCC and its increased expression correlates with a poor prognosis in HCC patients (Fig. 6a–c and Extended Data Fig. 8a–c). Depletion of METTL16 substantially decreased global m6A abundance and inhibited translation efficiency in HCC cells (Fig. 6d, e and Extended Data Fig. 8d–f). Further, METTL16 KO, as well as eIF3a and eIF3b KO, significantly suppressed the proliferation of HCC cells (Fig. 6f–i, and Extended Data Fig. 8g–i). To figure out which function(s) of METTL16 (i.e., RNA methylation or translational regulation) is more important to the survival/growth of HCC cells, we conducted rescue assays with wild-type METTL16 and catalytic inactive mutant METTL16 (PP185/186AA) in METTL16 KO HCC cells. While forced expression of wild-type METTL16 completely rescued METTL16 KO-induced inhibitory effect on HepG2 cell survival/growth (Fig. 6h, i, and Extended Data Fig. 8i), ectopic expression of catalytic dead METTL16 rescued to the ~60% level (Fig. 6h, i). To further evaluate whether the Mtase domain is necessary for the oncogenic role of METTL16 in HCC, we overexpressed Mtase and ΔMtase domains in METTL16 KO cells. Ectopic expression of Mtase domain could partially reversed the METTL16 KO-mediated growth inhibition, with a significantly stronger rescue effect than does ΔMtase (Fig. 6j). Adding additional domains such as the “vcr1” and “disorder” domains to Mtase can further enhance its rescue effect (Fig. 6j), suggesting that while the Mtase domain is critical for METTL16’s function, other domains likely also contribute to the overall function of METTL16. In addition to cell proliferation, METTL16 depletion also significantly suppressed migration and invasion of HCC cells (Fig. 6k, l and Extended Data Fig. 8j). Similarly, forced expression of Mtase displayed more robust role than ΔMtase in restoring METTL16 KO-induced migration suppression (Fig. 6m). To investigate whether METTL16 represents a potential safe therapeutic target in treating HCC, we assessed its role in two normal human liver epithelial cells and showed that METTL16 is dispensable for their survival (Fig. 6n, o and Extended Data Fig. 8k). Our xenograft mouse models showed that METTL16 KO dramatically suppressed the formation and progression of HCC tumor in vivo (Fig. 6p–r and Extended Data Fig. 8l, m). Furthermore, METTL16 is significantly positively correlated with eIF3a and eIF3b in expression (Extended Data Fig. 8n, o), and high expression level of eIF3b is also associated with poor prognosis in HCC patients (Extended Data Fig. 8p). Collectively, our data demonstrate the oncogenic role of METTL16 in tumorigenesis.
Fig. 6. The crucial tumor-promoting role of METTL16 in hepatocellular carcinoma (HCC).
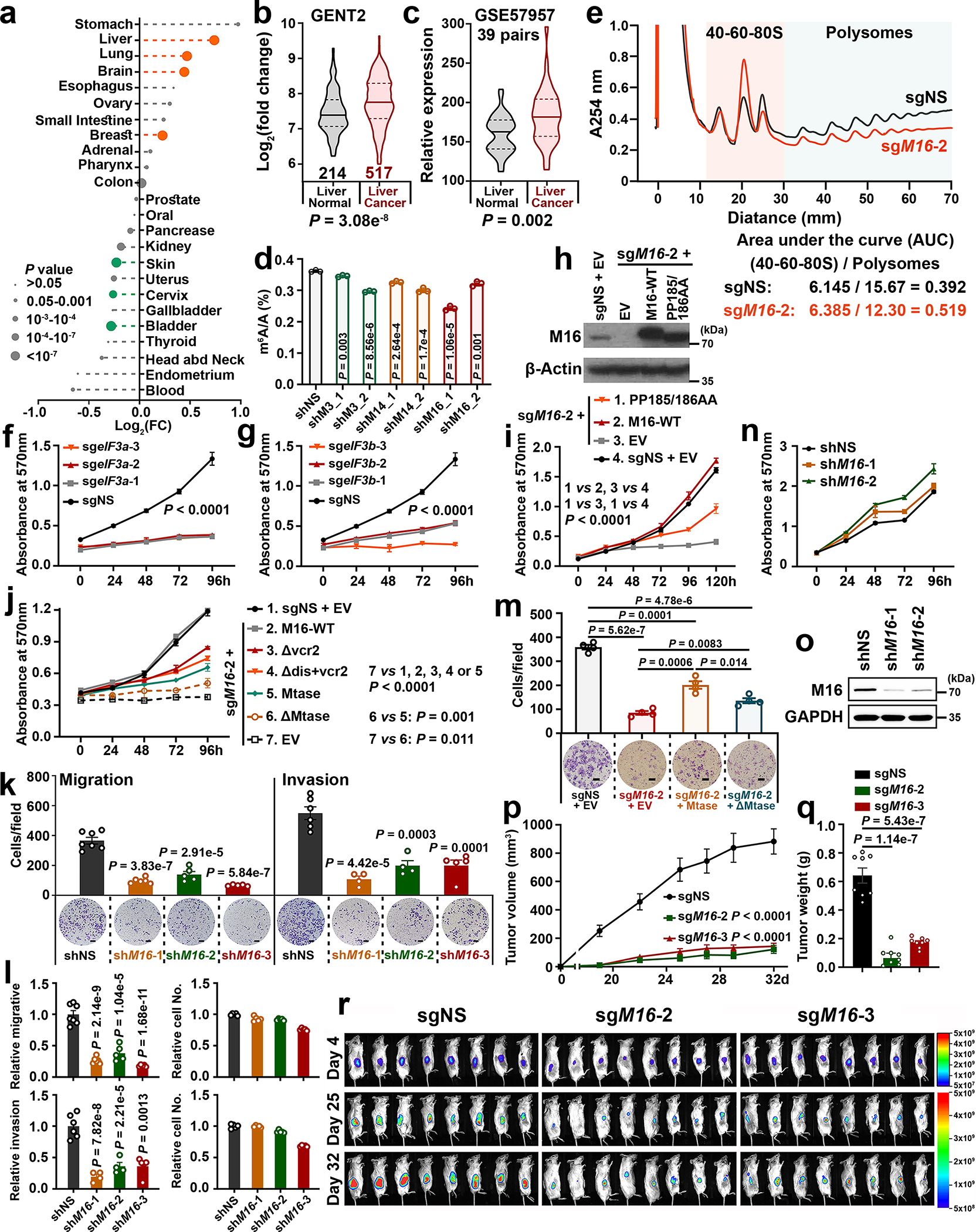
a, Relative expression levels of METTL16 in the 24 cancer types. FC represents the ratio of METTL16 levels in a specific subtype of tumors and the corresponding healthy control. Statistics: unpaired, two-tailed t-test. b, c, Comparison of METTL16 expression levels between liver cancers and normal controls. Statistics: two-tailed t-test, unpaired (b) or paired (c). d, Global m6A abundance in poly(A) RNA isolated from HepG2 cells upon METTL3/14/16 KD (n = 3). e, Representative polysome profiles of HepG2 cells upon METTL16 KO (n = 3). f, g, The effect of eIF3a (f) or eIF3b (g) KO on proliferation in HepG2 cells. h, i, Rescued expression (h, n = 3) and effects (i) of wild type and mutant METTL16 in HepG2 cells upon METTL16 KO. j, Rescued effects of wildtype and truncated mutants of METTL16 on cell proliferation of HepG2 cells upon METTL16 KO. k, Effects of METTL16 KD on migration and invasion of SUN449 cells. Migration: n = 7 (shNS), 6 (shM16-1), 5 (shM16-2, and shM16-3); Invasion: n = 6 (shNS), 4 (shM16-1 and shM16-2), 5 (shM16-3). l, The relative migration/invasion, and relative cell number (n = 5) in the SUN449 cells. The P values, unpaired two-sided t-test, represented the difference between relative migration (or invasion) and relative cell number in each group. m, Rescued effects of forced expression of METTL16-Mtase and ΔMtase on migration in HepG2 upon METTL16 KO (n = 4). n, o, Effects of METTL16 KD on cell proliferation (n) and its KD efficacy (o) in CL-48 cells. p, Average growth curves of liver tumors. Data are presented as mean ± s.e.m. (n = 8); two-way ANOVA. q, The weights of tumors at endpoints. Data are mean ± s.d. (n = 8); two-sided t-test. r, Bioluminescence imaging of xenografted mice. Unit: photons/second/cm2/steradian. For d, k, l, and m, data are mean ± s.d.; statistics: unpaired, two-tailed t-test. For f, g, i, and j, data are mean ± s.d. (n = 3); statistics: two-way ANOVA. All the “n” indicates the number of independent experiments.
Discussion
As a recently identified m6A methyltransferase, METTL16 has been reported to target only a few transcripts, including 3 noncoding RNAs (U6 snRNA, MALAT1 and XIST) and one mRNA (MAT2A), which contain specific bulge structures with a consensus sequence AC(m6)AGAGA13–18. In the present study, we identified 1,953 mRNA transcripts as potential targets of METTL16 in m6A deposition, as evidenced by the binding of METTL16 with these transcripts and the significant decrease in m6A abundance of the transcripts upon METTL16 depletion. Nevertheless, because METTL16 depletion also reduces SAM levels through suppressing SAM synthetase MAT2A14, the METTL16 depletion-induced m6A decrease in part of these 1,953 transcripts could be (at least partially) due to indirect inhibition of the enzymatic activity of the METTL3/METTL14 complex due to SAM reduction41. Notably, among the transcripts with m6A-hypo peaks, 334 appear to be METTL16-specific targets in m6A deposition as their m6A level decreased only upon METTL16 KO, but not upon METTL3 or METTL14 KO, suggesting that METTL16 does deposit m6A in a set of specific mRNA transcripts. METTL16 KD caused a much more evident m6A abundance decrease in nascent RNAs than in poly(A) RNA, implying that METTL16 adds many more m6A modifications in nascent RNAs than in mature mRNA transcripts; many m6A modifications in nascent RNAs likely disappear during pre-mRNA maturation. Consistently, pre-mRNAs were reported to be potential targets of METTL1615. Since most of the METTL16-mediated m6A modification sites in nascent RNAs are located in exons, not in introns, they might be involving in RNA splicing, as it was reported that m6A modification affects RNA splicing42, 43 and METTL16 indeed regulates MAT2A splicing14. Thus, METTL16 may play a broad role in regulating the splicing of many pre-mRNA transcripts, which warrants further systematic studies.
More interestingly, consistent with the preferential distribution of METTL16 in cytosol rather than in nuclei, METTL16 binds to over 4,000 mRNA transcripts and the majority of them do not have METTL16-mediated m6A modification. This implies that METTL16 likely exerts methylation-independent activity, especially in the cytosol. Indeed, we show that METTL16 promotes RNA translation efficiency via directly interacting with eIF3a/b and rRNAs and facilitating the assembly of the 40S and 60S subunits into the 80S TIC. Such effect is independent of its methyltransferase activity. While METTL3 was also reported to promote translation10, 35, 44, it utilizes distinct mechanisms than does METTL16, by (i) promoting RNA circularization via binding with m6A modified target mRNA 3’UTR and eIF3h10, 35; and (ii) promoting translation through an eIF4F-independent mechanism44. Clearly, distinct from METTL3, the function of METTL16 in promoting translation is neither methyltransferase activity-dependent nor m6A-dependent, and instead is attributed to its unique interactions with both eIF3a/b and rRNAs. In addition, another METTL protein, METTL13 was reported as an eEF1A methyltransferase that promotes translation via methylating of eEF1A45, 46, which is also distinct from the mechanism of METTL16 in translation promotion.
Our observations and previous studies7, 47 showed that METTL3/14 are predominantly located in cell nucleus, in contrast to the preferential localization of METTL16 in cytosol. Thus, METTL16 may play a more potent role than METTL3 in promoting translation in the cytosol. Indeed, we found that METTL16 promotes translation efficiency to a higher degree than does METTL3. Consistently, two large-scale CRISPR-Cas9 screening data suggest that METTL16 is a more essential gene than METTL3, METTL14, and any other METTL genes for the survival of various types of human cancer cells. In the present study, we show that METTL16 plays a critical tumor-promoting role in liver cancer, in which METTL16 is overexpressed and its increased expression is associated with a poor prognosis. Genetic depletion of METTL16 significantly inhibited human liver cancer cell growth, migration and invasion, and robustly suppressed tumor growth in vivo, accompanied with significantly decreased global m6A and translation. We found that around 50% of METTL16’s targets in translation regulation are also potential targets of eIF3a/3b, and eIF3a/b also play potent oncogenic role in HCC. Thus, targeting the METTL16/eIF3a/eIF3b axis represents new therapeutic strategy for cancer (e.g., HCC) treatment. Since the Mtase domain of METTL16 is critical for its both methylation-dependent and -independent activities, development of effective inhibitors targeting the Mtase domain would be promising and hold great therapeutic potential for cancer treatment.
Methods
Cell culture.
The following cells were purchased from ATCC: HepG2, Hep3B, PLC/PRF/5, HEK293T, SNU449, SNU475, K562, CL-48, and THLE-2. Huh7 was purchased from ECACC. MHCC97H was kindly provided by Dr. Wendong Huang. All these cells were cultured in the growth medium with Penicillin-Streptomycin (15–140-122, Thermo Fisher Scientific) and Plasmocin prophylactic (ant-mpp, InvivoGen) as specified by the manufacturers and maintained in an incubator with 5% CO2 at 37°C. All the cell lines were confirmed without mycoplasma contamination before further study.
Animal studies.
NOD-SCID IL2Rγ-null (NSG, Jackson 005557) mice were originally purchased from the Jackson Laboratory and bred at a certified animal facility of City of Hope in accordance with standard procedures. All mice were grouped housed on a 12 hours:12hours light-dark cycle with ad libitum to food and water. For each experiment, similar age- (5–8 weeks) mice were used and randomly assigned to each group. All experiments were compliant with federal and state government guidelines and Institutional Animal Care and Use Committee (IACUC) protocols approved by City of Hope.
The single Cas9 clone of HepG2 cells was established and selected with 5 μg/ml blasticidin, and infected with pLenti-CMV-Puro-LUC (17477, Addgene) to stably express luciferase via selection with 2 μg/ml puromycin. Then, the Cas9-LUC-HepG2 cells were infected with pLenti-sgMETTL16-hygromycin or scramble control and selected with 1 mg/ml hygromycin. The stable cell lines (1 × 106) were injected subcutaneously into 5–8 weeks old NSG mice. The size of tumor was measured using a caliper and the volume was calculated as (width × width × length / 2) mm3. In vivo bioluminescence imaging was applied to monitor the tumor progression as previously described48. All of the mice were euthanized at the same time when tumor volume exceeded 1000 mm3.
Immunofluorescence imaging.
The cells were seeded onto the sterile slides and placed in the 6 well plates with the cell density of 1 × 105 cells per well. After incubation for 24h, the cells were gently rinsed by phosphate-buffered saline (PBS) 3 times followed by fixed with 4% paraformaldehyde for 15min. Then, the cells were washed with PBS three times followed by permeabilized by 0.1% Triton X-100 in PBS for 15 min. To block the nonspecific binding, the cells were incubated with 2% bovine serum albumin (BSA) in PBS for 1h. The cells were subsequently incubated with primary antibodies against METTL16 (1:100, HPA020352, Sigma-Aldrich), METTL3 (1:100, ab195352, Abcam), METTL 14 (1:100, HPA038002, Sigma-Aldrich) and SC35 (1:500, ab11826, Abcam) overnight at 4°C, respectively. After washing with PBS three times, the corresponding fluorescence-labeled secondary antibodies (1:200, goat anti-rabbit IgG(H+L) Alexa Fluor 555: 4413S, Cell Signaling Technology; 1:200, goat anti-mouse IgG(H+L) Alexa Fluor 488: 4408S, Cell Signaling Technology) were applied to stain the cells and kept incubating for 1h. The nuclei were counterstained by mounting the cells in DAPI (F6057, Sigma-Aldrich). The immunofluorescent signals were photographed by a confocal laser scanning microscope (CLSM) (Carl Zeiss LSM 880).
Protein extraction and Western blotting.
Whole cells were lysed in RIPA buffer (Sigma-Aldrich) with 1% protease inhibitor cocktail (Thermo Fisher Scientific) and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Western blotting analysis was performed as previously described49. All Western blotting was performed with mini electrophoresis system from Bio-Rad. The primary and senondary antibodies were listed in the Reporting Summary.
RNA isolation, cDNA synthesis, and qPCR.
Total RNA was isolated with the miRNeasy kit (217004, Qiagen) according to the manufacturer’s protocol. PolyATract mRNA isolation system IV (Z5310, Promega) was applied to enrich the mRNA from total RNA. The RNA concentration was measured by a NanoDrop spectrometer. For the synthesis of cDNA, 0.1–0.5 μg of total RNA was used for reverse transcription followed the protocol of QuantiTect Reverse Transcription Kit (205314, Qiagen). The quantitative real-time PCR (qPCR) was performed using SYBR Green qPCR Master Mix (FERK0253, Thermo Fisher) and a QuantStudio (TM) 7 Flex Real-Time PCR system (Applied Biosystem). GAPDH or β-Actin was set as endogenous control and each reaction was performed in triplicate. All the primers used for qPCR analysis were listed in Supplementary Table 1.
UHPLC-QQQ-MS/MS.
The levels of N6-methyladenosine (m6A) and adenosine (A) were measured by ultra-high pressure liquid chromatography coupled with triple-quadrupole tandem mass spectrometry (UHPLC-QQQ-MS/MS). The RNA samples were enriched and isolated as previously described49. The nucleosides were separated by reverse phase ultra-performance liquid chromatography on a C18 column (00A-4475-AN, Phenomenex) with online mass spectrometry detection using an Agilent 6490 triple-quadrupole LC mass spectrometer. The total run time was 5 min at a flow rate of 0.4 mL/min with 0.1% formic acid (FA) in ddH2O as Solvent A and 0.1% FA in acetonitrile as Solvent B. The following gradient elution was performed: 0.01 min, 100% A, 2.2 min, 90% A, 2.7 min, 3% A, 4 min, 100% A. Quantification was performed by comparison with a standard curve obtained from pure adenosine and N6-methyladenosine standards. The ratio of m6A to A was calculated according to the calibrated concentrations.
Sucrose density gradient ultracentrifugation.
The cells in 150-mm dishes were treated with 100 μg/ml cycloheximide (Sigma-Aldrich) at 37°C for 10 minutes. Subsequently, the cells were washed twice with ice-cold PBS containing 100 μg/ml cycloheximide and harvested in 1 ml lysis buffer (20 mM Tris-HCL pH7.5, 100 mM KCl, 5 mM MgCl2, 1% Triton-X100, 100 μg/ml cycloheximide) supplemented with 1 × protease inhibitor cocktail (Thermo Scientific) and 40 U/ml RiboLock RNase inhibitor (Thermo Scientific). The samples were centrifuged at 16,000 g for 15 minutes at 37°C. Supernants were collected and the absorbance at 260 nm was measured. The sucrose gradient was generated with Biocomp Gradient Station. Equal amounts of OD260 from each group were layered onto 11 ml sucrose gradient and centrifuged at 220,000g in a Beckman SW-41 Ti rotor for 2.5 hours at 4°C. After centrifugation, the fractions were collected and determined at absorbance 254 nm with BioComp’s Piston Gradient Fractionator. Then, the fractions were subjected to Western blotting to evaluate protein localization, and RNA immunoprecipitation with Flag antibody to assess the interaction between METTL16 and rRNAs.
Cross-linking immunoprecipitation and qPCR (CLIP-qPCR).
CLIP was performed as previously described50 with some modifications. Briefly, the cells at 80% confluence were washed with ice-cold PBS, cross-linked by 254 nM UV (150 mJ/cm2, ×3), and harvested by trypsinization. The cytoplasm and nuclear fractions were separated by nuclear isolation buffer (1.28 M sucrose, 40 mM Tris-HCl pH 7.5, 20 mM MaCl2, 4% Triton X-100). The lysates were sonicated, clarified by centrifugation at 12,000 g for 10 min at 4°C and pre-cleared by Protein A/G Magnetic Beads (Thermo Fisher Scientific). 2–8 μg anti-FLAG antibody (F3165, Sigma-Aldrich), anti-eIF3a antibody (3411S, Cell Signaling Technology), anti-eIF3b (1:1000, sc-137214, Santa Cruz Biotechnology), and IgG (12–371, Millipore Sigma) were conjugated to Protein A/G Magnetic Beads by gently rotation at 4°C for 4 h, followed by washing 3 times with RIP buffer (150 mM KCl, 25 mM Tris-HCl pH 7.4, 5 mM EDTA, 0.5 mM DTT, 0.5% NP40, 100 U/mL RNase inhibitor, 1 × protease inhibitor). Then, the antibody-conjugated beads were incubated with pre-cleared lysates with rotation at 4°C overnight. After washing with RIP buffer for three times, the beads were dispersed in 80 μL PBS, followed by DNase I (EN0521, Thermo Fisher Scientific) digestion and Proteinase K (E00492, Thermo Fisher Scientific) treatment at 37°C for 15 min. One tenth of fragmented RNA was saved as input control. Both input and co-immunoprecipitated RNAs were recovered by RNeasy mini kit (74104, Qiagen) and applied for reverse transcription and qPCR analyses using primers in Supplementary Table 1.
In vitro translation.
The in vitro translation assay was conducted with Flexi rabbit reticulocyte lysate system according to the manufacturer’s instructions. Briefly, the 50 μl non-radioactive luciferase control reactions with 0, 0.3, or 0.9 μg METTL16 protein (81085, Active motif) were incubated at 30°C for 1 hour. Then, 2.5 μl product from each group was added into a luminometer tube containing 50 μl luciferase assay reagent, and the luciferase level in each group was determined with Promega GloMax luminometer.
Proximity ligation assay (PLA).
The cells were seeded onto sterile coverslips (CLS-1760–015, Fisher Scientific) in a 12-well tissue culture plate supplemented with 1 mL complete culture media and incubated at 37°C with 5% CO2. After incubation for 36 h, the supernatant was removed and the cells were rinsed three times with 1mL PBS, followed by fixed with 4% paraformaldehyde for 15 minutes. After washed three times with PBS, the cells were then incubated with 1 × Permeabilization Buffer (00–8333-56, Thermo Fisher Scientific) for 15 minutes and rinsed by PBS for three times. The cells were then blocked with Duolink block solution for 1h at room temperature and incubated overnight at 4°C with the following combination of antibodies, (1) mouse anti-FLAG antibody (F3165, Sigma-Aldrich) & rabbit anti-eIF3a antibody (3411S, Cell Signaling Technology); (2) rabbit anti-METTL16 antibody (HPA020352, Sigma-Aldrich) & mouse anti-eIF3b antibody (sc-137214, Santa Cruz Biotechnology); (3) mouse anti-FLAG antibody (F3165, Sigma-Aldrich) & mouse anti-eIF3b antibody (sc-137214, Santa Cruz Biotechnology). Subsequently, the cells were washed with large volume of PBS with 5% BSA for twice and incubated in the mixture of PLA probes (anti-rabbit PLUS (Sigma-Aldrich, DUO92002) and anti-mouse MINUS (DUO92004, Sigma-Aldrich)) for 1 h at 37°C. The cells were then washed with 1× Duolink In Situ Wash Buffer A (DUO82049, Sigma-Aldrich) twice and incubated with ligation mix at 37°C for 30 min. After that, the cells were washed with 1× Duolink In Situ Wash Buffer A for twice and incubated with amplification mix (DUO92008, Sigma-Aldrich) at 37°C for 100 min. In the end, the cells were washed twice with 1× Duolink In Situ Wash Buffer B, washed once with 0.01× Buffer B, and mounted with Duolink In Situ Mounting Medium with DAPI (DUO82040, Sigma-Aldrich). The pictures were captured under Zeiss LSM 880 with Airyscan confocal microscope (Zeiss, Germany).
RNA tethering experiment and dual luciferase reporter assay.
The pGL3-BoxB (with 5 × BoxB after firefly luciferase) was purchased from Addgene (92004). The pGL3 control vector without BoxB was used as control and purchased from Promega (E1741). The λN peptide sequence (MDAQTRRRERRAEKQAQWKAAN) was fused to the C-terminal of METTL3 and METTL16, and all these sequences were inserted into pmiRNA1 vector via In-Fusion clone. All the primers used to contruct these plasmids were listed in Supplementary Table 1. These plasmids were extracted by Plasmid Mini Kit (12125, Qiagen). The reporter plasmids, including pGL3-BoxB (300 ng), pGL3 control (300 ng), and pRL-TK (10 ng, E2241, Promega), and the effecter plasmids (METTL16_ λN and METTL3_ λN in pmiRNA1, 300 ng) were transfected into HEK293T cells with Effectene transfection reagent (301427, Qiagen) in 24-well plates. The relative luciferase activities (protein level) were assessed at 48h upon transfection with Dual-Luciferase Reporter Assay Systems (E1910, Promega). Firefly luciferase (F-Luc) activity was normalized to the Renilla luciferase (R-Luc) activity. The RNA samples were collected, digested with DNase, and then subjected to qPCR to determine the expression of F-Luc and R-Luc at RNA levels. Finally, the F-Luc activity was normalized by R-Luc to evaluate translation efficiency (protein/RNA). The normalized F-Luc activity in the pGL3 control was set to 1.
Surface sensing of translation (SUnSET) assay.
SUnSET was performed as previously reported36 to monitor protein synthesis by pausing with puromycin in cellulo. Briefly, the cells were seeded in 12-well plate with a density of 3 × 105 cells per well and incubated in the culture medium supplemented with puromycin (1–2μg/ml) for indicated time points. The protein samples were extracted from the cells and Western blotting was conducted with anti-puromycin (MABE343, EMD Millipore).
Pulse labeling assay.
The pulse labeling assay with L-homopropargylglycine (HPG; the analog of methionine) was conducted as previously to evaluate abundance newly synthesized proteins37. The experiment was exerted with Click-iT HPG Alexa Fluor 594 Protein Synthesis Assay Kit according to the manufacturer’s instructions. Briefly, the cells were incubated with 1 × Click-iT working solution for 30 min in the CO2 incubator. Then the cells were collected and subjected to fixation with 3.7% formaldehyde for 15 min and permeabilization with 0.5% Triton in PBS for 20 min. After washed twice with 3% BSA in PBS, all the following steps were protected from light. The cells were suspended in 100 μl Click-iT reaction cocktail for 30 min, washed once with rinse buffer, stained with 100 μl 1 × NuclearMask blue stain working solution for another 30 min at room temperature. Eventually, the cells were collected, washed twice with PBS, and the levels of nascent proteins were evaluated by flow cytometry.
Far-Western blotting.
Far-Western blotting was performed by referring to a published literature51 with minor modifications. The eIF3 protein complex (30 μg) in rabbit reticulocytes (P1318, BioVision) was separated by a 10% SDS-PAGE and transferred to PVDF membrane. An equal amount of BSA was also loaded as a negative control. The gel was first stained with coomassie blue to ensure the proteins had been successfully transferred and then washed by wash buffer (5% ethanol and 10% acetic acid in ddH2O) to remove the dye. Afterward, the proteins on the membrane were denatured and renatured in AC buffer (100 mM NaCl, 20 mM Tris-HCl pH 7.6, 0.5 mM EDTA, 10% glycerol, 0.1% Tween-20, 2% non-fat milk and 1 mM DTT) by gradually reducing the guanidine-HCl concentration. After blocking the membrane with 5% non-fat milk in 1 × PBST buffer at RT for 1 h, the membrane was incubated with 1 μg/ml recombinant METTL16 protein (81085, Active Motif) at 4°C overnight. After that, the membrane was washed with 1 × PBST buffer to remove the unbound protein and incubated with anti-METTL16 antibody (1:1000, HPA020352, Millipore Sigma) in the PBST buffer containing 3% milk at RT for 1h. Finally, the HRP conjugated goat anti-rabbit secondary antibody (1:5000, ab6721, Abcam) was incubated for another 1h and chemiluminescent detection was performed.
Generation of 3rd generation lentivirus and establishment of stable cells.
The 3rd generation of lentivirus particles were packaged in HEK293T cells and utilized to infect human cells as previously described48, 49. Briefly, 1.5μg pMD2.G (12259, Addgene), 0.9μg pMDLg/pRRE (12251, Addgene), 2.1μg pRSV-Rev (12253, Addgene) and 5.4μg target plasmids were transfected into HEK293T cells in 100 mm cell culture plate at the same time with Effectene transfection reagent (301427, Qiagen). The lentivirus particles were collected at 48 and 72 h post transfection followed by transduced into target cells for overexpression, or knockdown with the aid of 4 μg/ml polybrene. After two rounds of infection, cells infected with pCDH or shRNAs were selected for at least three passages with 1 μg/ml puromycin. For the cells infected with pmiRNA1, the GFP positive cells were selected for further experiments. The TRC lentiviral vectors encoding shRNAs against METTL3, METTL14, and METTL16, and the non-specific control were purchased from GE Dharmacon and Sigma Aldrich, and the detailed information was listed in Supplementary Table 1.
CRISPR/Cas9 mediated knockout of METTL16.
The 2nd generation of lentivirus particles, including the one expressing Cas9 and the sgRNAs, were packaged in HEK293T cells and used to infect human cells as previously described48. The lentiCas9-Blast plasmid (52962, Addgene) was used to overexpress Cas9 protein. The sgRNAs against METTL16 within lentiGuide-Puro (52963, Addgene) were used to mediate knockout of METTL16 for all the experiments except for SUnSET assay. To avoid the potential effects of lentiGuide-Puro in the SUnSET, the sgRNAs against METTL16, METTL3, METTL14, eIF3a, and eIF3b were inserted into lenti-sgRNA hygro (104991, Addgene). The sequence of those sgRNAs were listed in Supplementary Table 1 and cloned into lentiGuide-Puro or lenti-sgRNA hygro via In-Fusion cloning (638916, Takara). Both the Cas9- and sgRNA expressing lentivirus were generated using the second-generation package system. In brief, 3μg expression plasmid, 0.75μg pMD2.G (12259, Addgene), and 2.25μg psPAX2 (12260, Addgene) were mixed well and co-transfected into HEK293T cells in 60mm cell culture dish with Effectene Transfection Reagent. The Cas9-expressing lentivirus particles were collected 48 and 72 h post transfection and directly added into the target cells with the existence of 4 μg/ml polybrene. After two rounds of infection, the single clones were selected with 10 μg/ml blasticidin (ant-bl-1, InvivoGen) and allowed to grow for 3–4 weeks. Then the single clones were infected with sgRNA-expressing lentivirus and the sgRNA-infected cells were selected with 2 μg/ml puromycin (for lentiGuide-Puro) or 100–500 μg/ml hygromycin (10687010, Thermo Fisher Scientific).
Plasmid construction.
The plasmids used in the current study were constructed by In-Fusion cloning (638916, Takara) according to the manufacturer’s instruction. First, the pmiRNA1 or pCDH vectors were linearized by XbaI (FD0684, Thermo Fisher Scientific) and NotI (FD0594, Thermo Fisher Scientific) and recovered by QIAquick Gel Extraction Kit (28706, Qiagen). Dr. Nicholas K. Conrad kindly provided his pcDNA3-METTL16, METTL16-PP185/186AA and METTL16-F187G14. Dr. Yunsun Nam kindly provided her pcDNA3-METTL16-R200Q18. These plasmids were used as templates to generate the following PCR fragments using CloneAmp HiFi PCR premix: 3×Flag-METTL16, 3×Flag-METTL16-PP185/186AA, 3×Flag-METTL16-F187G, 3×Flag-METTL16-R200Q, 3×Flag-METTL16-λN, 3×Flag-METTL16-PP185/186AA-λN, 3×Flag-METTL16-F187G-λN, and 3×Flag-METTL16-R200Q-λN. These CDS fragments were in-fused into pmiRNA1. The pcDNA3/Flag-METTL3 (53739, Addgene) was used as the template to generate 3×Flag-METTL3 and 3×Flag-METTL3-λN and the two PCR products were also infused into pmiRNA1. The pcDNA3/Flag-METTL14 (53740, Addgene) was used as the template to generate 3×Flag-METTL14 and the PCR products were also infused into pmiRNA1. The fragments of truncated METTL16s with 3×Flag at N-terminal, including Δvcr2, Δdis+vcr2, ΔMtase, Mtase, Mtase (1–149), Mtase (150–289), Mtase (1–78), Mtase (79–289), Mtase-λN, and ΔMtase-λN, were amplified based on pmiRNA1–3×Flag-METTL16, and inserted into pmiRNA1. The pCDH-M16(Mut1) (resistant to shMETTL16-1), pCDH-M16–2 (resistant to sgMETTL16-2) and pCDH-M16–3 (resistant to sgMETTL16-3) were constructed based on pCDH-3×Flag-METTL16 with In-Fusion cloning. The pmiRNA1-M16–2 (resistant to sgMETTL16-2) was constructed based on pmiRNA1–3×Flag-METTL16. The pmiRNA1-M16-NLS, pmiRNA1-M16-PP185/186AA, pmiRNA1-M16-R200Q, pmiRNA1-M16-Mtase, pmiRNA1-M16-ΔMtase, pmiRNA1-M16-Δvcr2, and pmiRNA1-M16-Δdis+vcr2 were developed based on pmiRNA1-M16–2 through the same protocol and used for rescue assay in METTL16 KO (sgMETTL16-2) cells. The CDSs of eIF3a, eIF3b, eIF3c with 3×HA tag at C-terminal were amplified from MGC fully sequenced human eIF3a cDNA (MHS6278–211690028, Dharmacon), eIF3b cDNA (MHS6278–202759939, Dharmacon), and eIF3c cDNA (MHS6278–202755752, Dharmacon), respectively. All the In-Fusion PCR primers were shown in Supplementary Table 1. The plasmids were extracted with Plasmid Mini kit (12125, Qiagen).
Co-immunoprecipitation (Co-IP).
The cells with 80% confluence in 15 mm cell culture dishes were collected and lysed within 1ml RIPA buffer containing 1 × protease inhibitor cocktail and 1 × phosphatase inhibitor cocktail for 20 min on ice. The supernatant with protein were obtained by centrifugation at 13,000g for 15 min at 4oC. One-tenth volume of lysate was taken out as Input and the left part was mixed with 50 μl of Protein A/G magnetic beads (88803, Thermo Fisher Scientific) and gently rotated at 4oC for 1 h to reduce non-specific binding and background. Then, 500–1,000 μg pre-cleared samples were incubated with antibodies against FLAG (F3165, Sigma-Aldrich), eIF3a (3411S, Cell Signaling Technology), eIF3b (sc-137214, Santa Cruz Biotechnology), and IgG (12–371, Millipore) for 1h under gentle agitation followed by incubation with 25μl of pre-washed Protein A/G magnetic beads under rotation at 4°C overnight. The proteins associated with Protein A/G magnetic beads were collected by placing the tubes into a magnetic stand, washed 3 times with IP washing buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 1 mM EDTA, 150 mM NaCl, 1% Triton-X, 0.2 mM sodium orthovanadate) and then subjected to Western blotting. For the Co-IP assays with RNase digestion, the lysates were digested with RNase A/T1 mix (EN0551, Thermo Scientific) at RT for 30 min and the RNA was extracted to validate digestion efficacy before IP.
Cap binding assay.
The cells at 80% confluence were digested, washed by PBS, and then lysed by lysis buffer (0.1 M NaCl, 20 mM pH7.4 Tris-HCl, 1% protease inhibitor, 1% phosphatease inhibitor) at 4°C for 2 h. Subsequently, 10% of the cell extracts were taken out as Input and the other extracts were incubated with γ-aminophenyl-m7GTP agarose C10-linked beads (AC-155S, Jena Bioscience) with gentle agitation for 2 h at 4°C. Meanwhile, an equal portion of cell extracts was incubated with m7GTP-agarose beads together with cap analog (m7G(5’)ppp(5’)G) (AM8048, Invitrogen). Then, the beads were washed three times with 1 × Tris-buffered Saline (TBS) buffer and subjected to Western blotting analysis with FLAG (Sigma-Aldrich), eIF3a (Cell Signaling Technology) and eIF3b (Santa Cruz Biotechnology).
Isolation of nascent RNA.
The newly transcribed RNAs were isolated based on metabolic labeling with 4-thiouridine (4sU) as previously described52 with minor modification. Briefly, the cells at 80% confluency were labeled with 400 μM 4sU (T4509, Sigma Aldrich) for 60 min and total RNAs were extracted with TRIzol reagents immediately at the end of labeling period. The next step is biotinylation of total RNA, linking biotin specifically to the thiol group of 4sU labeled transcripts. Biotinylation was conducted by assembling a 500 μl reaction containing 100 μg total RNA, 200 μl Biotin-HPDP (21341, Thermo Fisher Scientific; 1mg/ml in N,N-Dimethylformamide, D4551, Sigma Aldrich), 50 μl 10× Biotinylation buffer (100 mM Tris-HCl pH 7.4, 10 mM EDTA). The reaction was incubated for 1 h at RT in the dark with gentle agitation. The biotinylated RNAs were recovered by isopropanol/ethanol precipitation and resuspended with RNase-free water at approximately 1 μg/μl. To separate the labeled and unlabeled RNA, 100 μg of biotinylated RNAs were mixed with 100 μl μMACS MicroBeads (130–074-101, Miltenyi Biotec) and incubated at room temperature with rotation for 15 min. The labeled RNAs were isolated with μMAC columns according to the instruction, recovered by ethanol precipitation, and subjected to MeRIP seq directly.
Determination of cell proliferation via MTT assay.
In vitro assessment of cell proliferation was measured by CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTT, G400, Promega). The cells were seeded into 96-well tissue culture plates at a cell density of 5 × 103 cells per well, respectively. At each time point, 15 μl of MTT solution was added into each well. After incubation at 37°C for 3–4 hours, 100 μl Solubilization/Stop Solution was added to solubilize the formazan product. Finally, the absorbance at 570 nm was recorded using a microplate reader.
Transwell migration and invasion assay.
Transwell assays were carried out using Corning transwell permeable inserts (pore size, 8 μm, 07–200-150, Fisher Scientific) in a 24-well plate. The cells were dissociated and resuspended in serum-free medium. For the migration assay, 5 × 104 cells were inoculated in the upper chamber in 200μL serum-free medium and 600μl completed medium with 10% FBS was added to the lower chamber. For the invasion assay, the permeable inserts were carefully pre-coated with 70μL 1:8 diluted Matrigel (356234, Corning), and incubated at 37°C for three hours. Then 1 × 105 cells in serum-free medium were carefully layered onto the Matrigel matrix. The plates were incubated in a humidified tissue culture incubator at 37°C, 5% CO2 atmosphere. After incubation of 24 hours for migration and 48 hours for invasion, the non-migrated cells were wiped and the upper chambers were immersed in 5% formaldehyde solution for 5 minutes. Each insert was stained with 0.1% crystal violet for 10 minutes and then rinsed with PBS. Random fields were captured under a microscope and cells were counted.
Ribo-seq (Ribosome profiling).
Ribo-seq was conducted as previously described53 with minor modifications. The cells at 70–80% confluency were treated with 100 μg/ml cycloheximide (C1988, Sigma-Aldrich) for 5 min at 37 °C to prevent translation before lysed in ice-cold lysis buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 5mM MgCl2, 1 mM DTT, 100 μg/ml cycloheximide, 1% v/v Triton X-100, and 25 U/ml Turbo DNase) for 10 min on ice. The lysates were collected by centrifugation at 20,000 g for 10 min at 4 °C, and 10% of the cell lysate was reserved as Input and prepared for RNA seq after depletion of rRNA. The RNA concentration of cell lysate was determined by Qubit RNA HS assay kit (Q32852, Thermo Fisher Scientific) according to the manufacturer’s instructions, and then the lyates with 30 μg total RNA were diluted to 200 μl with polysome buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 5mM Mg2Cl, 1 mM DTT, and 100 μg/ml cycloheximide) plus 1.5 μl RNase I (N6910K, Epicentre) and incubated for 45 min at room temperature with gentle agitation. The nuclease digestion was quenched by the addition of 10 μl SUPERase In RNase Inhibitor (AM2694, Invitrogen) and loaded onto a 0.9 ml sucrose cushion (1 M sucrose and 20 U/ml SUPERase In RNase Inhibitor in polysome buffer) in the polypropylene tube (344625, Beckman Coulter). Then, the pellet ribosomes were collected by centrifugation in a TLA-120.2 rotor at 220,000g, 4°C for 2 hours, and RNA samples were recovered from the ribosomal pellet via QIAzol (79306, Qiagen). The RNA samples were separated on a 15% polyacrylamide TBE-Urea gel (EC68852BOX, Invitrogen) and the gel band between 17–34 nt was cut and collected. The footprinting RNA was recovered from gel by addition of 400 μl RNA gel extraction buffer (300 nM NaOAc pH 5.5, 1 mM EDTA, 0.25% v/v SDS, and 0.1 U/ml SUPERase In RNase Inhibitor). After frozen on dry ice for 30 min, the samples were allowed to thaw overnight at RT with gentle rotation, and the footprint fragments were purified by isopropanol precipitation. Before library construction, the end structures of the RNA fragments were repaired by T4 polynucleotide kinase (T4 PNK, M0201S, New England Biolabs): (1) 3’ end healing reaction (10 μl): 7 μl RNA sample, 1 μl 10×T4 PNK buffer, 1 μl T4 PNK, and 1 μl SUPERase In RNase Inhibitor were mixed and incubated for 1 h at 37 °C; (2) 5’-end phosphorylation: 1 μl 10 mM ATP and 1 μl extra T4 PNK were added to the reaction and the mixture was kept for 30 min at 37 °C. The RNA was purified by ethanol precipitation. The libraries were constructed using the NEBNext small RNA prep kit (E7330S, New England BioLabs) after depletion of rRNA, and sequenced on Illumina Hiseq2500 with single end 51-bp read length.
MeRIP-seq (m6A-seq).
For m6A-seq with poly(A) RNA, we followed our protocol as previously described49. Briefly, total RNA was extracted with QIAzol and poly(A) RNA was isolated with GenElute mRNA miniprep kit (MRN10–1KT, Sigma-Aldrich). RNA fragmentation reagent (AM8740, Invitrogen) was used to randomly fragment RNA and anti-m6A antibody (202003, Synaptic Systems) was applied for m6A immunoprecipitation. For m6A-seq with nascent RNA, the nascent RNAs were directly subjected to fragmentation and RNA immunoprecipitation with m6A antibody. Both input and m6A IP samples were prepared for sequencing.
RNA immunoprecipitation (RIP)-seq.
The RIP experiment was employed according to the protocol from Abcam (https://www.abcam.com/epigenetics/rna-immunoprecipitation-rip-protocol) with minor modifications. Briefly, the confluent cells at 150 mm dish were cross-linked by 0.75 % formaldehyde (F8775, Sigma-Aldrich) with gentle rotation at RT for 10 min and the reaction was quenched by 125 nM glycine with shaking for 5 min. The cell pellets were collected, lysed in 1 ml M-PER buffer (78501, Thermo Fisher Scientific) with 100 U/ml RNase inhibitor and 1 × protease inhibitor on ice for 20 min, and sonicated in a Bioruptor Pico device (Diagenode) with 30s on/30s off for 10 cycles. The lysate was collected by centrifugation at 13,000g, 4 °C for 10 min and the protein concentration was determined by Bio-Rad protein assay (Bio-Rad). 10% of the lysate was reserved as Input. For immunoprecipitation, 8 μg FLAG (Sigma-Aldrich) was conjugated to Protein A/G Magnetic Beads by gentle rotation at 4°C for 4 h, followed by washing 3 times with RIP buffer (150 mM KCl, 25 mM Tris-HCl pH 7.4, 5 mM EDTA, 0.5 mM DTT, 0.5% NP40, 100 U/mL RNase inhibitor, 1 × protease inhibitor). Then, the FLAG-conjugated beads were incubated with 1–2 mg lysates in RIP buffer with rotation at 4°C overnight, and then washed with RIP buffer for 3 times. The co-precipitated RNAs were recovered from the beads with RNeasy mini kit (Qiagen). Both Input RNA and RIP RNA were subjected to digestion with DNase I (EN0521, Thermo Fisher Scientific) and depletion of rRNA. Then the libraries were constructed with KAPA Stranded mRNA-Seq Kit (Illumina Platforms) (KR0960m Kapa Biosystems) with 10 cycles of PCR amplification and sequenced on Illumina Hiseq2500.
Ribo-seq analysis.
For data analysis of Ribo-seq, the reads were quality-based trimmed and the adaptors were removed using Cutadapt54. The reads with minimum-length 16 nts were mapped to GRCh38 reference genome using STAR55. Then the ribosome profiling was analyzed by RSEM (RNA-Seq by Expectation Maximization)56. Only the genes with expression ≥ 0.01 in sgNS group were selected for further analysis. Translation efficiency (TE) was calculated by the ratio of (normalized ribosome profiling abundance+1)/(normalized input RNA abundance+1). The changes of TE were calculated as log2(sgMETTL16/sgNS).
MeRIP-seq analysis.
For data analysis of m6A-seq with poly(A) RNA and nascent RNA, all the reads from both input and m6A IP samples were aligned to the GRCh38 reference genome using STAR55. A gene annotation GTF file was built with nascent RNA coordinates, then passed to exomePeak to call m6A peaks57. The differentially methylated m6A peaks (P < 0.05) were also identified by exomePeak. The distribution of m6A peaks was generated using MetaPlotR58 and deepTools59. Integrative Genomics Viewer (IGV) were used to visualize the m6A peaks60.
RIP-seq analysis.
For data analysis of RIP-seq, all the reads from both input and IP samples were aligned to the GRCh38 reference genome using STAR55. The RNA-protein binding locations were calculated using exomePeak with default parameters57. The potential targets were also identified using normalized both input and IP RNA abundance and enriched using Molecular Signatures Database (MSigDB)61. To calculate the transcriptome wide distribution of transcript regions, the normalized reads counts of 5’ UTR, start codon (0.8 < rel_location < 1.2), cds, stop codon (1.8 < rel_location < 2.2) and 3’ UTR were generated using metaPlotR58. The different gene expression was determined by RSEM56. The RIP targets of METTL16 were defined as expression > 0.1, RIP/Input >2.
Statistics and reproducibility.
Representative data from ≥ 3 independent experiments are presented as mean ± SD, or mean ± SEM. For all the violin plots, median values, 25th percentage, 75 percentage, minimum, maximum values are shown. Statistical significance for two-sample comparisons was determined by Student’s two-tailed t-test. While two-way ANOVA was conducted to assess the significance between two groups. Two-sided Wilcoxon and Mann–Whitney test was employed to compare the cumulative distribution functions of two sample sets. Statistical significance for survival was calculated by the log-rank test. Pearson’s correlation analyses were performed with R packages MASS, ggplot2 and ggpubr. P < 0.05 was considered statistically significant. NS, not significant. All the Western blotting, Co-IP, m7G pulldown, PLA, and immunofluorescence were repeated at least two biologically independent times with similar results.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All the sequencing data generated by this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) under accession number GSE156798. The correlation analyses between METTL16 and eIF3a/eIF3b were derived from GTEx program (https://www.gtexportal.org/). The raw data for determining the genetic dependence of METLL family members in cancers were derived from https://depmap.org/portal/ and https://score.depmap.sanger.ac.uk/. The expression data of METTL16 in cancers and the corresponding control were derived from GENT2 (http://gent2.appex.kr/gent2/). The survival analyses of METTL16 in TCGA cancers were derived from GEPIA2 (http://gepia2.cancer-pku.cn/#index) and TCGA Research Network (http://cancergenome.nih.gov/). The Sankey diagram was built by SnakeyMatic (https://sankeymatic.com/). Three published data-sets, GSE103948 (UV-CRAC-seq data-set and PAR-CRAC-seq data-set), GSE90914 (m6A-seq data-set, input group) and GSE46705 (m6A-seq data-set), have also been re-analyzed in the present study. The eIF3a- and eIF3b-bound transcripts were downloaded from POSTAR3 (http://postar.ncrnalab.org/). Unprocessed blot and statistical source data are provided with this article. All other data supporting the fundings of this study are available from the corresponding authors on reasonable request.
Code availability
The intron retention was evaluated with IRFinder62 (https://github.com/williamritchie/IRFinder). All the softwares and packages used in this article have been listed in the Reporting Summary and are publicly available.
Extended Data
Extended Data Fig. 1. The dependency of METTL16 in human cancers and its preferential localization in cytoplasm.
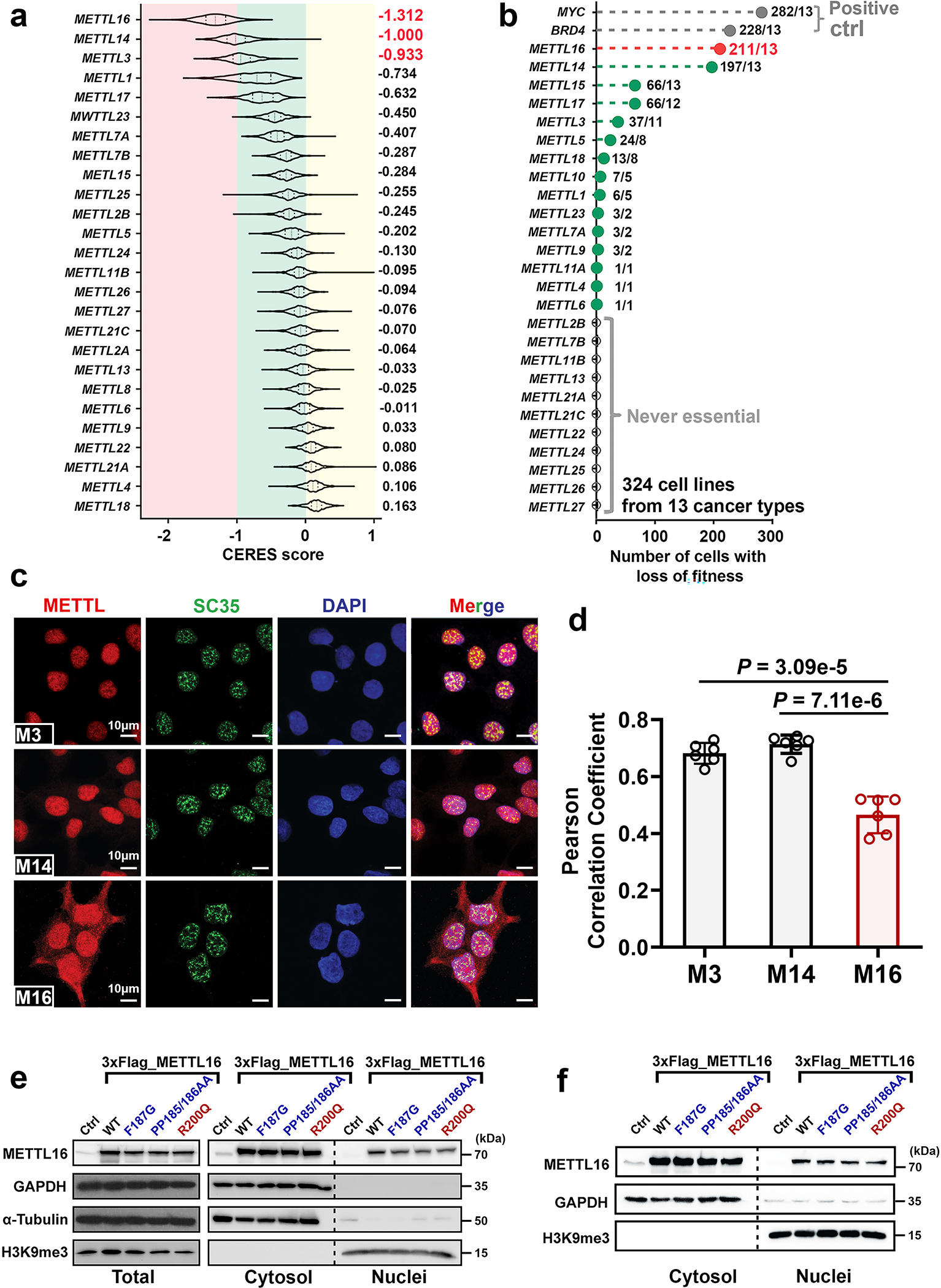
a, CERES scores of a set of METTL family members from genome-scale CRISPR-Cas9 essentiality screens across 769 human cancer cell lines. The raw data were downloaded from DepMap (https://depmap.org/portal/). As the CERES scores, 0 and −1 represent the median effects of nonessential genes and common core essential genes, respectively. The lower CERES score indicates the higher cancer dependency of the specific gene. For each violin, the minimum, first quartile, median, third quartile, and maximum were displayed. The average of CERES scores for each METTL family member was shown. b, METTL16 was also detected by another genome-scale CRISPR-Cas9 screen with 324 cancer cell lines as a common essential gene in majority of human cancer cell lines in all 13 cancer types. The raw data were derived from https://score.depmap.sanger.ac.uk/. In this screen, 324 cancer cell lines from 13 cancer types were included. MYC and BRD4 were shown as positive controls, which represent the appealing cancer therapeutic targets. The number of cells with loss-of-fitness effects and the number of cancer types were highlighted for each gene. For example, METTL16 (211/13) indicates that knockout (KO) of METTL16 exhibits loss-of-fitness effects (essential function) in 211 of the 324 cancer cell lines and the 211 cancer cell lines cover all the 13 cancer types. c, Subcellular localization of endogenous METTL3, METTL14, and METTL16 in Huh7 cells as determined by immunofluorescence. SC35 worked as a nuclear speckle marker. In line with the trend in HEK293T cells, a much higher percentage of METTL16 is located in the cytoplasm than METTL3 and METTL14. Data shown represent 3 independent experiments. d, Pearson correlation coefficients showing the extent of colocalization of the three METTL family members with nuclear (DAPI) in Huh7 cells. The Pearson correlation coefficients were determined by the ZEN software. Again, both METTL3 and METTL14, but not METTL16, are predominantly localized in nuclear. Data are mean ± s.e.m. Statistics: unpaired, two-tailed t-test. n = 6 independent experiments. e, f, Western blotting showing the overexpression efficacy of wild type (WT) METTL16 and its three mutants, and their subcellular distributions in HEK293T (e) and HepG2 (f) cells. Here, F187G and PP185/186AA represent loss-of-function mutation of METTL16 methyltransferase activity, while R200Q represents a gain-of-function mutation. 3 × Flag was infused to the N-terminal of WT or mutant METTL16. GAPDH and α-Tubulin were used as loading control of total protein and cytoplasmic protein samples, and H3K9me3 was selected as loading control of nuclear protein.
Extended Data Fig. 2. Transcriptome-wide analysis of METTL16 methyltransferase activity.
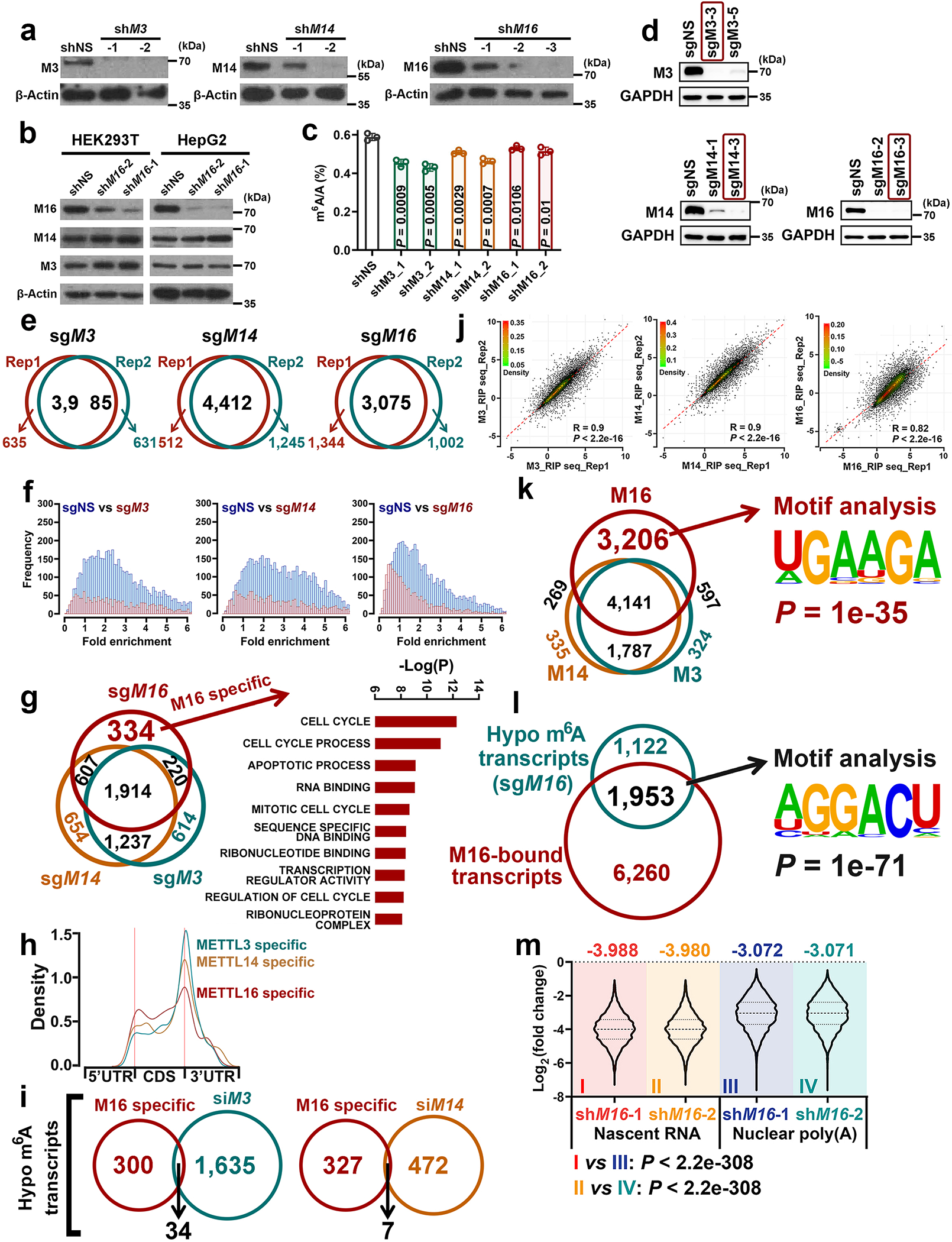
a, Validate the knockdown (KD) efficiency of the shRNAs against METTL3, METTL14, or METTL16 via Western blotting. Images are representative of three biologically independent experiments with similar results. b, Expression of METTL3 and METTL14 in HEK293T (left) and HepG2 (right) cells upon METTL16 KD. Images are representative of two biologically independent experiments with similar results. c, Global changes of m6A abundance upon METTL3, METTL14, or METTL16 KD in HEK293T cells. Data are mean ± s.d. Statistics: unpaired, two-tailed t-test. n = 3 independent experiments. d, Validation of the KO efficiency of the gRNAs against METTL3, METTL14, or METTL16 via Western blotting in HEK293T cells. The RNA samples from sgMETTL3-3, sgMETTL14-3, and sgMETTL16-3 cells, along with the control cells, were collected for subsequent m6A MeRIP-Seq. The 2 independent experiments have been performed with similar results. e, Venn diagram showing the overlap of the transcripts with decreased m6A levels between the two biological replicates of sgMETTL3-3, sgMETTL14-3, and sgMETTL16-3. f, The frequency distributions of the m6A-hypo peaks caused by the KO of METTL3, METTL14, or METTL16 as detected by m6A MeRIP-Seq with poly(A) RNA from HEK293T cells. Only the significantly decreased m6A peaks (P < 0.01) were classified as m6A-hypo peaks and shown in the plot. g, Venn diagram showing the overlap analysis of the m6A-hypo transcripts induced by the KO of METTL3, METTL14, and/or METTL16 (left panel). Gene set enrichment analysis (GSEA) of the 334 METTL16-specific targets was performed, and the top 10 enriched pathways were shown (right panel). h, Global distribution of specific m6A-hypo peaks induced by the KO of METTL3, METTL14, and/or METTL16. i, Venn diagram showing the overlap between the 334 METTL16-specific targets and the transcripts with m6A-hypo peaks in METTL3 KD (left panel) or METTL14 KD (right panel) cells. j, Scatterplot showing the high reproducibility of the RIP-seq replicates of METTL3, METTL14, and METTL16. The Pearson correlation coefficients (R) of the normalized RIP-seq reads across the two replicates were calculated and displayed in the plots. A smoother regression line and 2D kernel density contour bands were also presented. P values were determined by Pearson’s correlation test. k, Venn diagram showing the overlap among METTL3, METTL14, and METTL16-bound transcripts (left panel) and the top one binding motif of the 3,206 specific METTL16-bound transcripts (right panel). l, Venn diagram showing the overlap between the METTL16-bound transcripts (RIP-seq) and the METTL16 KO-mediated m6A-hypo transcripts (MeRIP-seq) (left panel). Both seq analyses were conducted with poly(A) RNA from HEK293T cells. The top one consensus binding motif identified in METTL16-bound transcripts with METTL16 KO-induced m6A-hypo peaks was shown (right panel). m, Violin plots showing the significant m6A-hypo peaks induced by METTL16 KD in nascent RNA and nuclear poly(A) RNA from HEK293T cells. For each violin, the minimum, first quartile, median, third quartile, and maximum were presented. The average value of Log2(fold change) from each group was also displayed. The P values were calculated by unpaired two-sided t-test.
Extended Data Fig. 3. METTL16 specifically deposits m6A RNA methylation in a set of mRNA targets and its biological role in cell proliferation/growth and translation promotion can’t be substituted by METTL3 and METTL14.
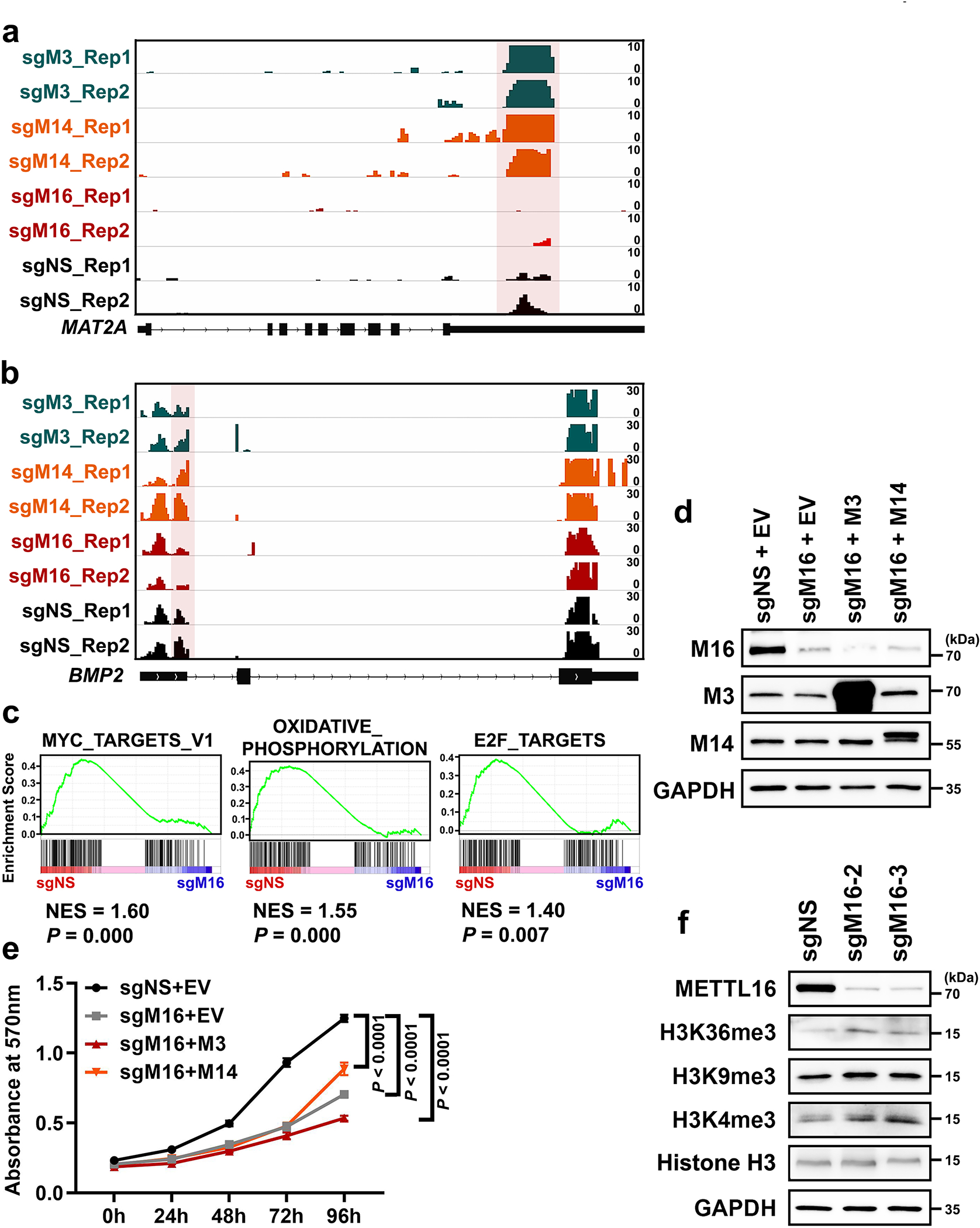
a, b, Integrative Genomics Viewer (IGV) tracks displaying the m6A read distribution and changes (based on the m6A MeRIP-seq data) in MAT2A (a) and BMP2 (b) mRNA upon the KO of METTL3, METTL14, or METTL16 in HEK293T cells. The m6A decorations in MAT2A and BMP2 mRNA are METTL16-dependent, and METTL3/14-independent. c, The top 3 signaling pathways suppressed by METTL16 KO (sgMETTL16-3) as determined by GSEA. d, Overexpression efficacy of METTL3 and METTL14 in HEK293T cells with endogenous METTL16 KO (sgMETTL16-2). Images are representative of 2 biologically independent experiments with similar results. e, Effects of METTL3 and METTL14 overexpression on cell growth in HEK293T cells with endogenous METTL16 KO (sgMETTL16-2). Data are plotted as mean ± s.d. (n=3 independent experiments). Statistics: two-way ANOVA. f, Assessment of the global level changes of histone methylations, including H3K36me3, H3K9me3, and H3K4me3, in HEK293T cells upon endogenous METTL16 KO by Western blotting. Images are representative of 2 biologically independent experiments with similar results.
Extended Data Fig. 4. METTL16 enhances translation efficiency.
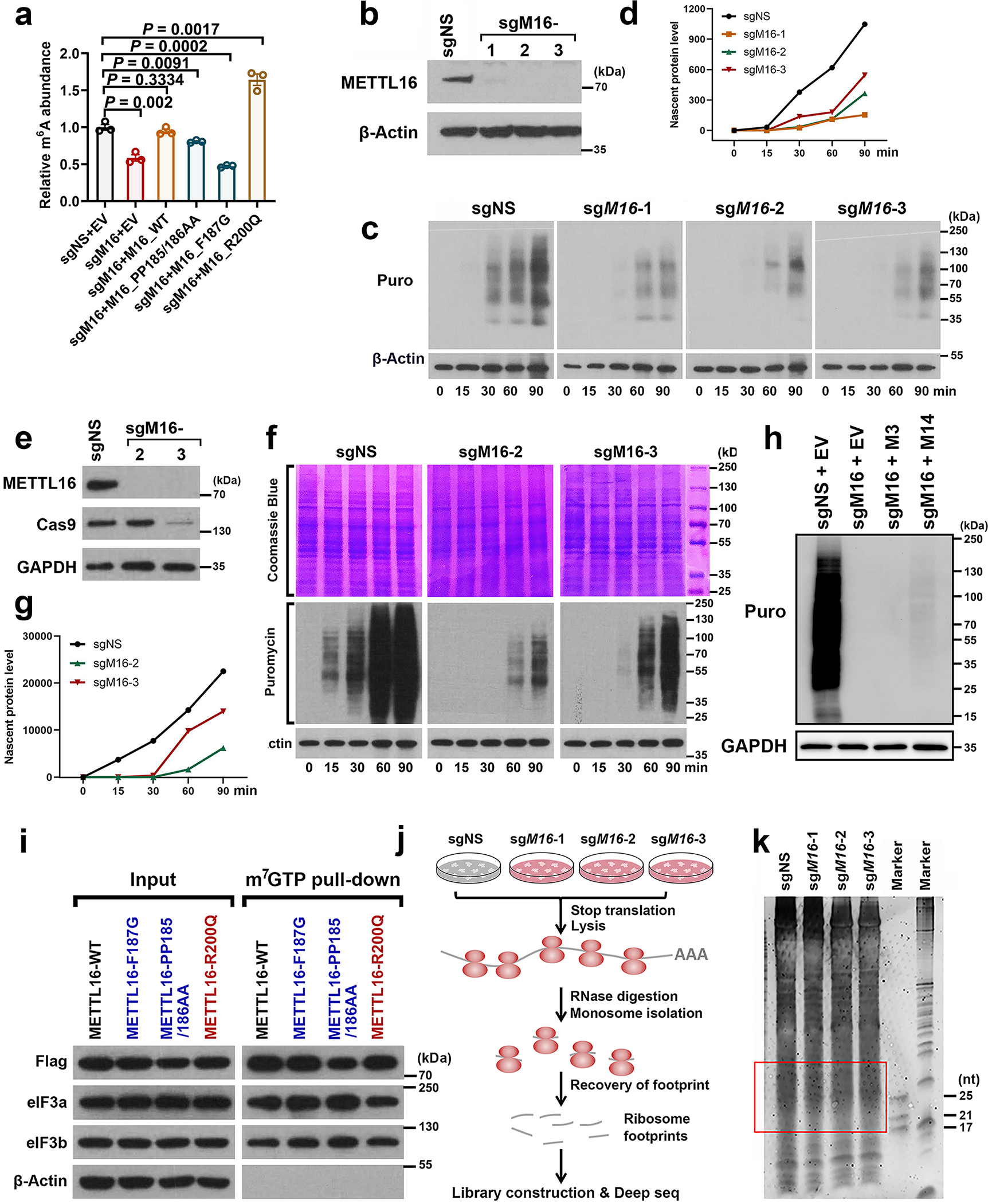
a, Relative m6A level changes in U6 snRNA in METTL16 KO (sgMETTL16-2) HEK293T cells upon ectopic expression of METTL16 WT and mutants (F187G, PP185/186AA, and R200Q) as determined by gene-specific m6A qPCR. Data are mean ± s.e.m. (n = 3 independent experiment). Statistics: unpaired, two-sided t-test. b, Western blotting confirming the KO efficacy of the sgRNAs against METTL16 in HEK293T cells. Images are representative of three biologically independent experiments with similar results. c, d, Representative WB images of SUnSET assays (c) and the quantitative SUnSET data (d) showing the effects of METTL16 KO on translation efficiency (based on the levels of nascent protein levels at given time periods) in HEK293T cells. β-Actin was used as loading controls. To make the data comparable, all the conditions for each sample are the same, including the total amount of loaded protein, concentration of antibodies, the time to run SDS-gel, transfer membrane, and develop signals. Representative images from three independent experiments with similar results were displayed. e, Western blotting showing the efficiency of METTL16 KO in K562 cells. f, g, Representative WB images of SUnSET assays (f) and the quantitative SUnSET data (g) showing the translation efficiency in K562 cells upon METTL16 deletion. Both Coomassie blue staining and β-Actin were used as loading controls. h, Effects of METTL3 and METTL14 overexpression on translation efficiency in HEK293T cells with endogenous METTL16 KO (sgMETTL16-2). All the cells were pulsed with puromycin for 60 minutes. Images are representative of three biologically independent experiments with similar results. i, Western blotting of the m7G pulldown assays to determine the potential binding of METTL16 WT and mutants in the 5’ cap region. Representative images from two independent m7G pulldown assays with similar results were presented. j, Experimental workflow of ribosome profiling with HEK293T cells upon METTL16 KO. k, The image of RNA gel with the ribosome footprint samples. The footprints within 17–34 nt were recovered for library construction and deep sequencing.
Extended Data Fig. 5. The direct interaction between METTL16 and eIF3a/b in HEK293T cells.
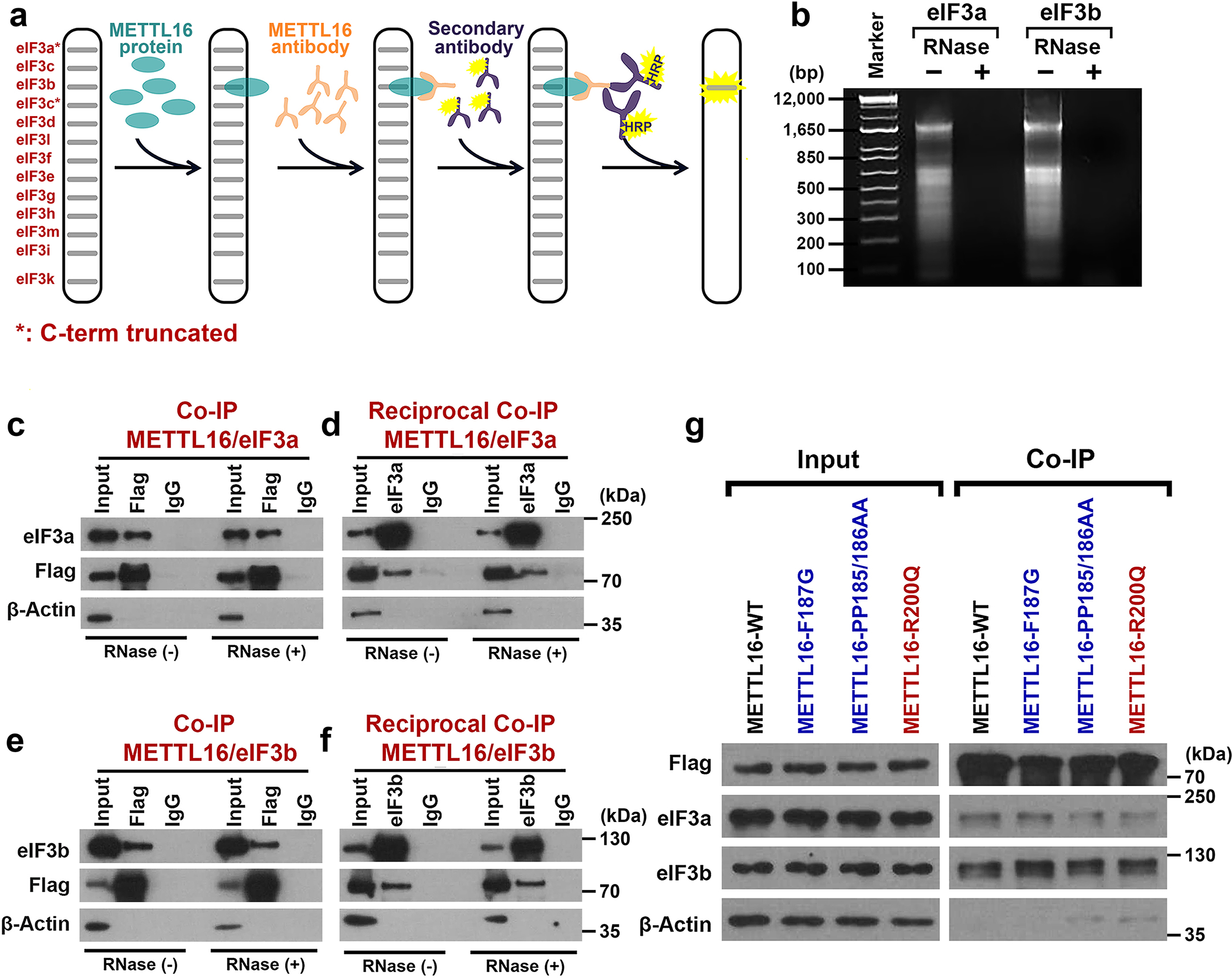
a, The experimental scheme for Far-Western blotting showing how to detect the potential direct interaction between METTL16 protein and eIF3 family members. Note: the * indicates the C-term truncated eIF3 family member. b, Agarose gel electrophoresis of the RNA samples recovered from Co-IP samples with or without RNase digestion. Samples of eIF3a were used for Extended Data Fig. 5c and d; while samples of eIF3b were used for Extended Data Fig. 5e and f. c, d, Co-IP (c) and reciprocal Co-IP (d) showing the potent interaction between eIF3a and METTL16 is RNA independent. Representative images from two biological replicates with similar results were presented. e, f, Co-IP (e) and reciprocal Co-IP (f) showing the robust association between eIF3b and METTL16 is also RNA independent. Representative images from two biological replicates with similar results were displayed. g, Western blotting of Co-IP assays to test the binding of METTL16 wild type (WT) and mutants (F187G, PP185/186AA, and R200Q) with eIF3a and eIF3b. The 3 × Flag tag was fused to the N-terminal of WT and mutant METTL16 proteins. Representative images from two independent Co-IP assays with similar results were presented.
Extended Data Fig. 6. Co-IP and reciprocal Co-IP with truncated METTL16 proteins to determine which domain is crucial for its association with eIF3a and eIF3b.
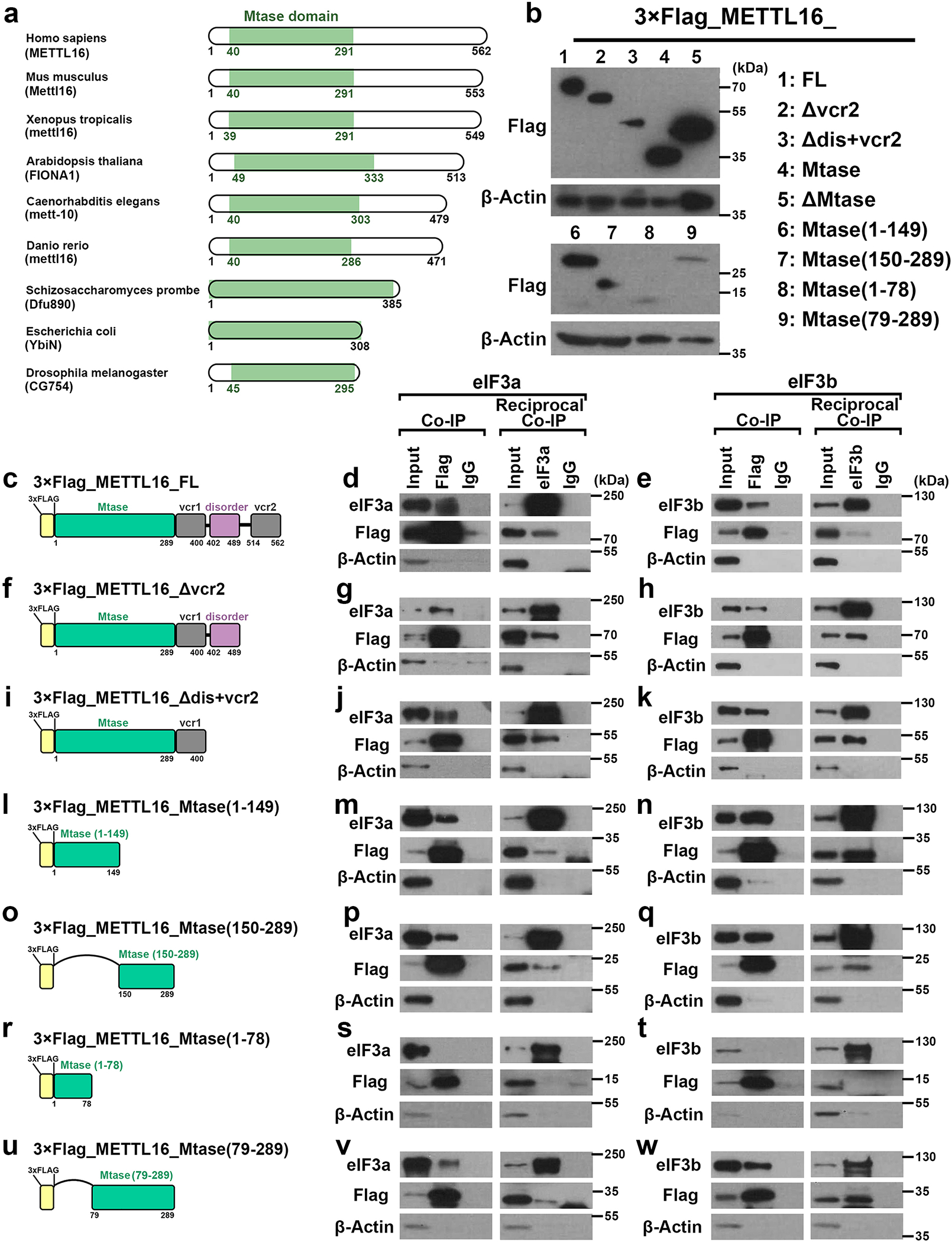
a, Diagrams and respective names of METTL16 homologs from various organisms. Among all the 9 species, the Mtase domain is highly conserved. b, Western blotting showing the expression of truncated METTL16 proteins in HEK293T cells. The 3 × Flag tag was attached into the N-terminal of all the truncated mutants. c, Schematic description of full length METTL16 with 3 × Flag tag at N-terminal. d, Co-IP and reciprocal Co-IP showing the interaction between eIF3a and full length METTL16. HEK293T cells with stable expression of METTL16 and eIF3a were used. e, Co-IP and reciprocal Co-IP showing the interaction between eIF3b and full length METTL16. HEK293T cells with stable expression of METTL16 and eIF3b were used. f, Schematic description of truncated METTL16_Δvcr2 (depletion of vcr2 domain) with 3 × Flag tag at N-terminal. g, Co-IP and reciprocal Co-IP showing the interaction between eIF3a and METTL16_Δvcr2. HEK293T cells with stable expression of METTL16_Δvcr2 and eIF3a were used. h, Co-IP and reciprocal Co-IP showing the interaction between eIF3b and METTL16_Δvcr2. HEK293T cells with stable expression of METTL16_Δvcr2 and eIF3b were used. i, Schematic description of truncated METTL16_Δdis+vcr2 (depletion of disorder domain plus vcr2 domain) with 3 × Flag tag at N-terminal. j, Co-IP and reciprocal Co-IP showing the interaction between eIF3a and METTL16_Δdis+vcr2. HEK293T cells with stable expression of METTL16_Δdis+vcr2 and eIF3a were used. k, Co-IP and reciprocal Co-IP showing the interaction between eIF3b and METTL16_Δdis+vcr2. HEK293T cells with stable expression of METTL16_Δdis+vcr2 and eIF3b were used. l, Schematic description of truncated METTL16_Mtase (1–149) with 3 × Flag tag at N-terminal. m, Co-IP and reciprocal Co-IP showing the interaction between eIF3a and METTL16_Mtase (1–149). HEK293T cells with stable expression of METTL16_Mtase (1–149) and eIF3a were used. n, Co-IP and reciprocal Co-IP showing the interaction between eIF3b and METTL16_Mtase (1–149). HEK293T cells with stable expression of METTL16_Mtase (1–149) and eIF3b were used. o, Schematic description of truncated METTL16_Mtase (150–289) with 3 × Flag tag at N-terminal. p, Co-IP and reciprocal Co-IP showing the interaction between eIF3a and METTL16_Mtase (150–289). HEK293T cells with stable expression of METTL16_Mtase (150–289) and eIF3a were used. q, Co-IP and reciprocal Co-IP showing the interaction between eIF3b and METTL16_Mtase (150–289). HEK293T cells with stable expression of METTL16_Mtase (150–289) and eIF3b were used. r, Schematic description of truncated METTL16_Mtase (1–78) with 3 × Flag tag at N-terminal. s, Co-IP and reciprocal Co-IP showing the lack of interaction between eIF3a and METTL16_Mtase (1–78). HEK293T cells with stable expression of METTL16_Mtase (1–78) and eIF3a were used. t, Co-IP and reciprocal Co-IP showing the lack of interaction between eIF3b and METTL16_Mtase (1–78). HEK293T cells with stable expression of METTL16_Mtase (1–78) and eIF3b were used. u, Schematic description of truncated METTL16_Mtase (79–289) with 3 × Flag tag at N-terminal. v, Co-IP and reciprocal Co-IP showing the interaction between eIF3a and METTL16_Mtase (79–289). HEK293T cells with stable expression of METTL16_Mtase (79–289) and eIF3a were used. w, Co-IP and reciprocal Co-IP showing the interaction between eIF3b and METTL16_Mtase (79–289). HEK293T cells with stable expression of METTL16_Mtase (79–289) and eIF3b were used. For all the IP experiments, representative images from two biological replicates with similar results were presented.
Extended Data Fig. 7. The robust binding of METTL16 with rRNAs and the effect of METTL16 on intron retention.
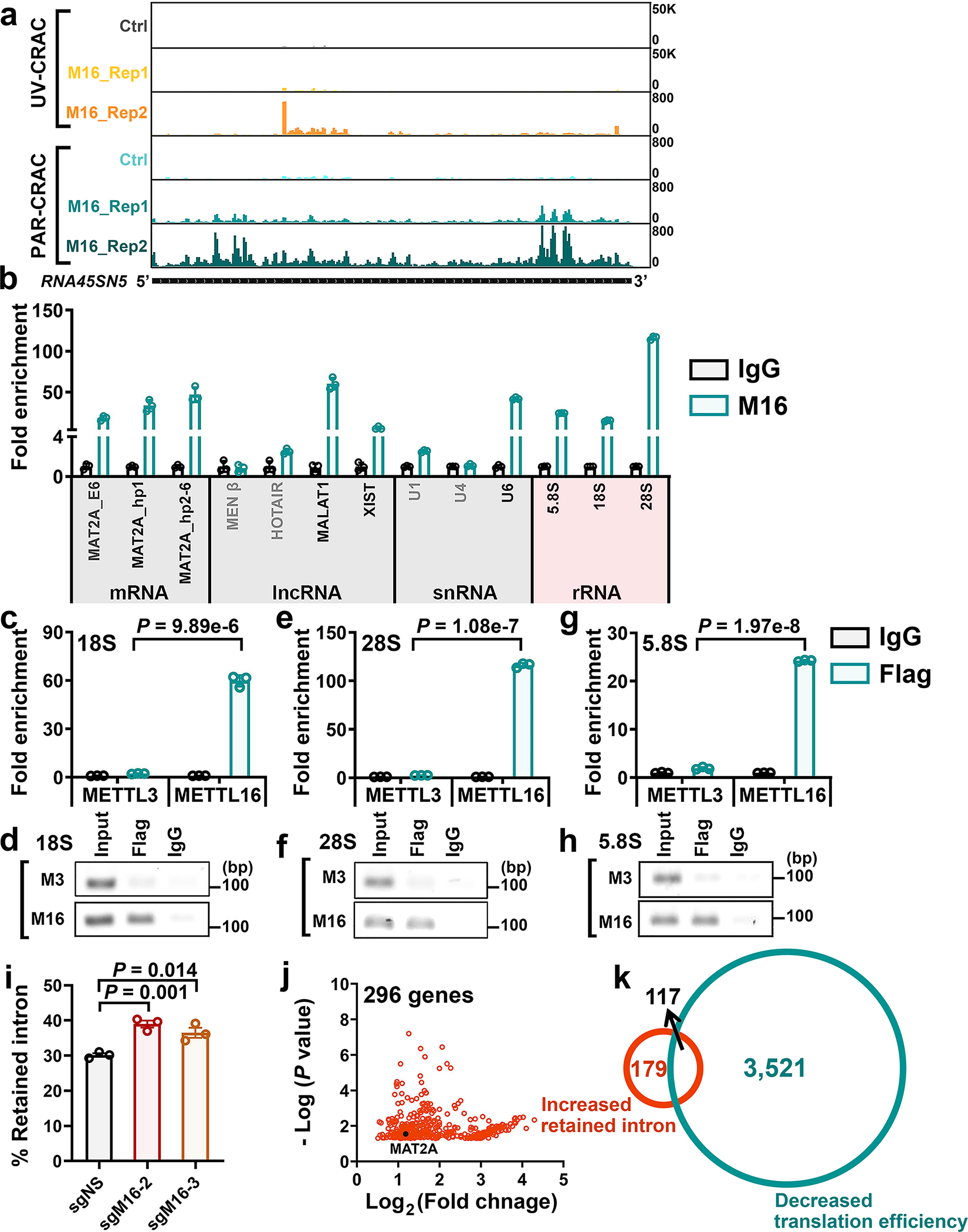
a, IGV tracks showing the binding pattern of METTL16 on ribosomal 45S RNA clusters (Here we exhibited RNA45SN5). 45S RNA is further processed into 18S rRNA, 5.8S rRNA and 28S rRNA. UV-CRAC, UV crosslinking and analysis of cDNA; PAR-CRAC, photoactivatable ribonucleoside enhanced crosslinking and analysis of cDNA. b, The binding of METTL16 with mRNA (MAT2A), lncRNAs, snRNAs, and rRNAs in HEK293T cells. Data are mean ± s.e.m. Data shown represent 3 independent experiments. MAT2A, MALAT1, XIST, and U6 were included as positive control targets of METTL16 for the test. MEN β, HOTAIR, U1, and U4 were included as negative controls. The results indicated our crosslinking RNA immunoprecipitation and qPCR (CLIP-qPCR) works well. c, d, The quantitative (c) and semi-quantitative (d) analysis of the enrichments of METTL3 and METTL16 on 18S rRNA in the cytoplasm fraction. e, f, The quantitative (e) and semi-quantitative (f) analysis of the enrichments of METTL3 and METTL16 on 28S rRNA in the cytoplasm fraction. g, h, The quantitative (g) and semi-quantitative (h) analysis of the enrichments of METTL3 and METTL16 on 5.8S rRNA in the cytoplasm fraction. For c-h, The 3 × Flag tag was infused to the N-terminal of METTL3 and METTL16, and the IP assays were conducted with Flag antibody; IgG serves as a negative control. The quantitative results were derived from qPCR; while the semi-quantitative results were derived from RT-PCR and the products were run on an 2% agarose gel. For c, e, and g, data are mean ± s.e.m. Data shown represent 3 independent experiments. Statistics: unpaired, two-sided t-test (METTL16 vs. METTL3). i, The effect of METTL16 KO on intron retention of MAT2A mRNA in HEK293T cells. Data are mean ± s.e.m. Data shown represent 3 independent experiments. Statistics: unpaired, two-sided t-test. j, The transcripts with significantly (P < 0.05) increased intron retention in HEK293T cells upon METTL16 depletion. Herein, the transcriptome-wide RNA-seq data was from GSE90914 and the intron retention was detected with IRFinder. k, Venn diagram showing the overlap between the transcripts with increased intron retention and the transcripts with decreased translation efficiency in HEK293T cells upon METTL16 depletion.
Extended Data Fig. 8. The tumor-promoting role of METTL16 in hepatocellular carcinoma (HCC) and its positive correlations with eIF3a/b in expression.
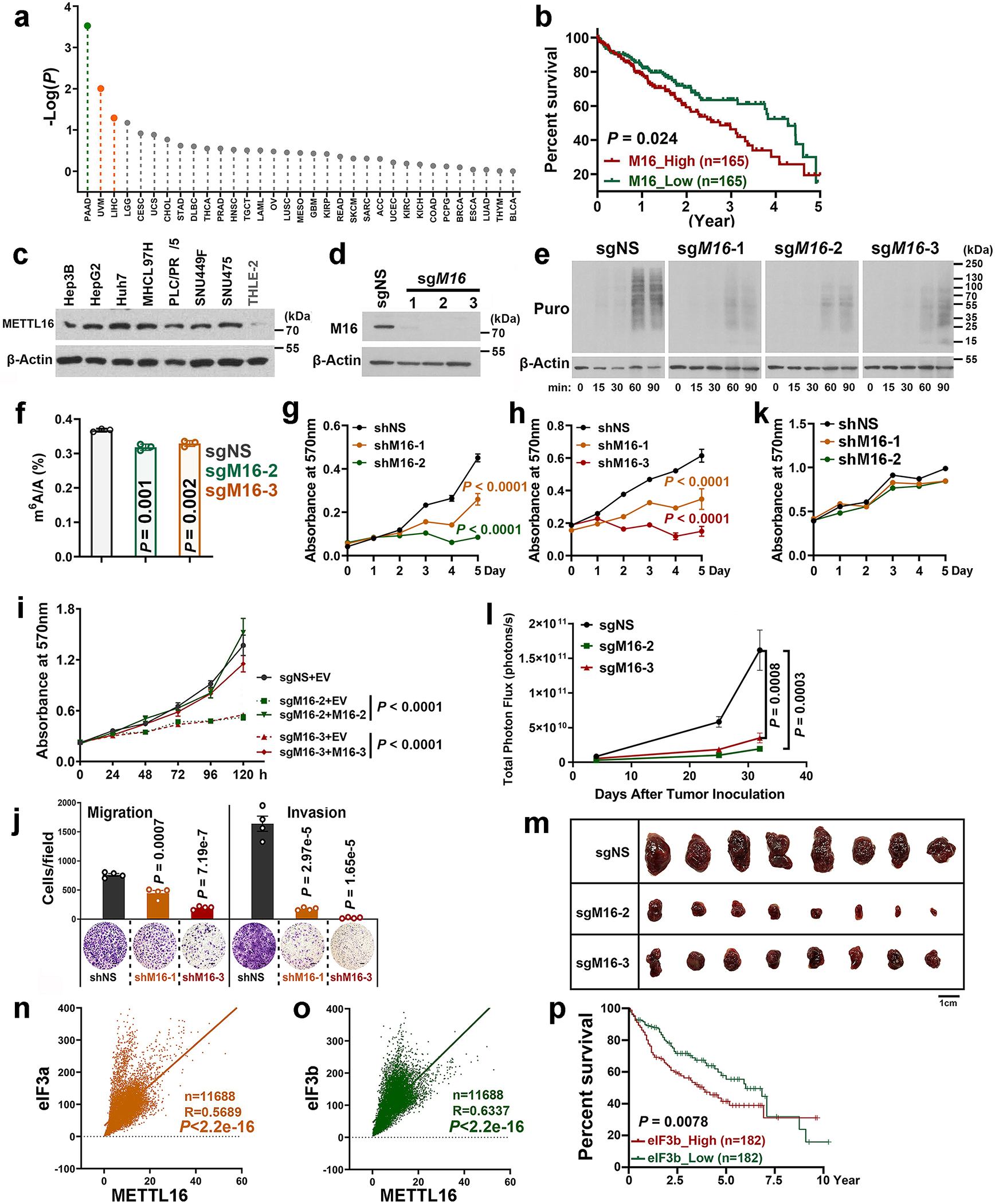
a, The prognostic impacts of METTL16 expression levels in the TCGA cancer patient datasets, which were downloaded from GEPIA2 (http://gepia2.cancer-pku.cn/#index). In each cancer type, the overall survivals between patients with high METTL16 expression levels (the top 50%) and those with low levels (the low 50%) were compared and the P value was calculated by the log-rank test, and then the -Log(P) value was presented. In PAAD (Pancreatic adenocarcinoma), high expression of METTL16 significantly (P < 0.05) correlates with better survival; while in UVM (Uveal melanoma) and LIHC (Liver hepatocellular carcinoma), high expression of METTL16 significantly (P < 0.05) correlates with poorer survival. METTL16 expression levels are not significantly associated with prognosis in other cancer types. b, Overall survival analysis of METTL16 in HCC from the TCGA dataset. The P value is determined by logrank test. c, Expression of METTL16 in liver cancer cell lines (Hep3B, HepG2, Huh7, MHCC97H, PLC/PRF/5, SUN449, and SUN475) and healthy liver cell line (THLE-2). Representative images from two independent replicates with similar results were presented. d, Validation of METTL16 KO efficiency via Western blotting in HepG2 Cas9 single clone. e, The Western blotting images of SUnSET assays showing the effects of METTL16 KO on translation efficiency in HepG2 liver cancer cells. To make the data comparable, all the conditions for each sample are the same, including the total amount of loaded protein, concentration of antibodies, the time to run SDS-gel, transfer membrane, and develop signals. f, Determination of m6A levels in mRNAs of HepG2 cells with or without METTL16 KO via QQQ-MS. Data are mean ± s.d. Data shown represent 3 independent experiments. Statistics: unpaired, two-sided t-test. g, h, Effects of METTL16 KD on cell growth in SNU449 (g) and SNU475 (h) HCC cells. Data are plotted as mean ± s.d. (n=3 independent experiments). Statistics: two-way ANOVA. i, Effects of METTL16 expression changes on the growth of HepG2 cells. METTL16 KO (sgM16–2 + EV; sgM16–3 + EV) drastically suppressed cell proliferation as determined by MTT assays; restored expression of METTL16 (sgM16–2 + M16–2; sgM16–3 + M16–3) could rescue/reverse the inhibitory effect of METTL16 KO. Here, M16–2 and M16–3 represent the synonymous mutations of METTL16 ORF that can’t be targeted by sgM16-2 and sgM16-3, respectively. Data are plotted as mean ± s.d. n = 4 (sgM16–2 + M16–2, 120 h; and sgM16–3 + M16–3, 120h); n =5 (sgM16–3 + M16–3, 72h); n = 6 (all other groups) independent experiments. Statistics: two-way ANOVA. j, Effects of METTL16 KD on the migration and invasion of SUN475 liver cancer cells. Data are mean ± s.d. (n=3 independent experiments). Statistics: unpaired two-sided t-test. k, The effects of METTL16 KD on cell proliferation in THLE-2 cells (a primary human normal liver cell line). Data are plotted as mean ± s.e.m. (n=3 independent experiments). l, Total photon flux of the mice xenografted with HepG2 cells with or without METTL16 KO. Data are plotted as mean ± s.e.m. (n=8 independent mice). Statistics: two-way ANOVA. m, Tumor images at the endpoint of the xenograft models implanted with HepG2 liver cancer cells with or without METTL16 KO. n, o, The correlation between METTL16 and eIF3a (n), or eIF3b (o) in expression as detected by Pearson’s correlation analysis. All the raw data were downloaded from GTEx (Genotype-Tissue Expression, https://www.gtexportal.org/home/). The number of samples, R value, and P value were displayed. Statistics: Pearson correlation. p, Overall survival analysis of eIF3b in HCC from the TCGA dataset. The P value is determined by logrank test.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
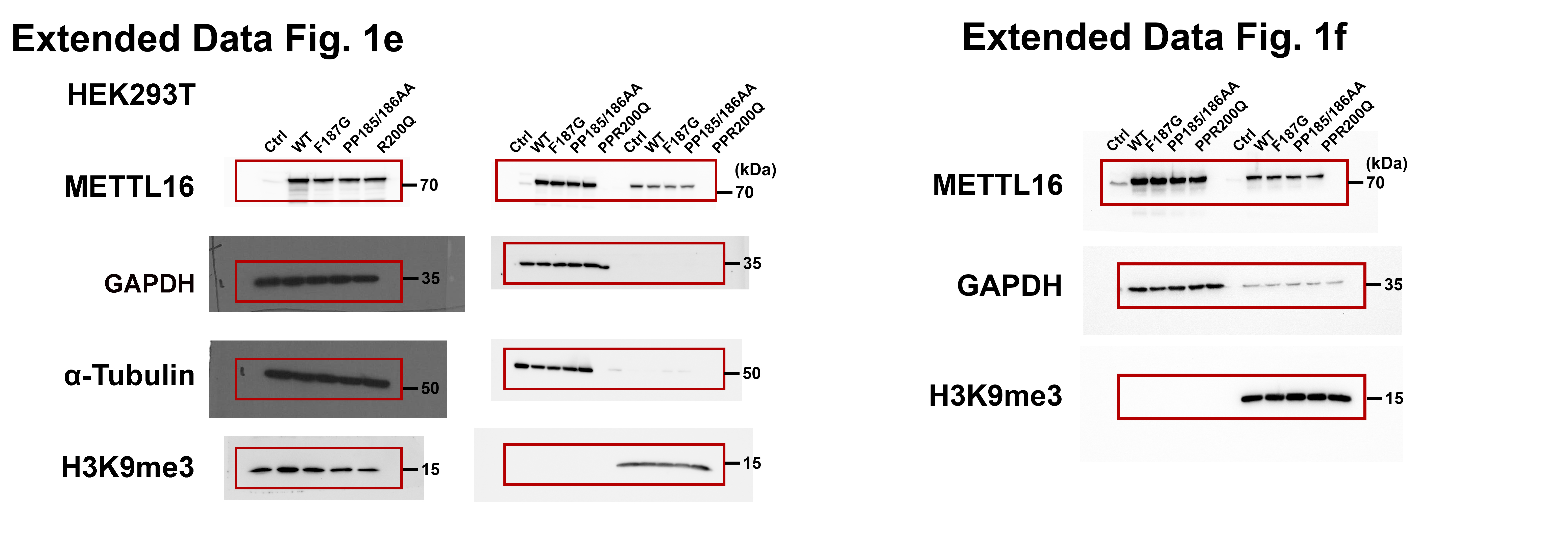{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Dr. Nicholas K. Conrad for kindly providing pcDNA3-METTL16, pcDNA3-METTL16-PP185/186AA, and METTL16-F187G. This work was supported in part by the U.S. National Institutes of Health (NIH) grants CA243386 (J.C.), R01 CA214965 (J.C.), R01 CA236399 (J.C.), R01 CA211614 (J.C.), R01 DK124116 (J.C), RM1 HG008935 (C.H.), The Simms/Mann Family Foundation (J.C.), The Margaret Early Medical Research Trust (R. S), and R01 CA139158 (W.H). J.C. is a Leukemia & Lymphoma Society (LLS) Scholar. C.H. is an investigator of the Howard Hughes Medical Institute.
Abbreviations:
- m6A
N6-methyladenosine
- MTC
methyltransferase complex
- METTL
methyltransferase like proteins
- SAM
s-adenosylmethionine
- PIC
pre-initiation complex
- TIC
translation initiation complex
- eIF
eukaryotic translation initiation factor
- KO
knockout
- KD
knockdown
- QQQ-MS
triple-quadrupole-MS
- SUnSET
Surface Sensing of Translation
- Ribo-seq
ribosome-sequencing
- lncRNA
long non-coding RNA
- HCC
hepatocellular carcinoma
Footnotes
Competing interests
C.H. is a scientific founder and a scientific advisory board member of Accent Therapeutics, Inc., and holds equities with the company. J.C. is a scientific advisory board member of Race Oncology. The remaining authors declare no competing interests.
References
- 1.Frye M, Harada BT, Behm M & He C RNA modifications modulate gene expression during development. Science 361, 1346–1349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang H, Weng H & Chen J m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 37, 270–288 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu J et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10, 93–95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534, 575–578 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Wang P, Doxtader KA & Nam Y Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell 63, 306–317 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng X et al. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res 28, 507–517 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ping XL et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24, 177–189 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz S et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep 8, 284–296 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ignatova VV, Jansen P, Baltissen MP, Vermeulen M & Schneider R The interactome of a family of potential methyltransferases in HeLa cells. Sci Rep 9, 6584 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choe J et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 561, 556–560 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weng H et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell 22, 191–205 e199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol 20, 1074–1083 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown JA, Kinzig CG, DeGregorio SJ & Steitz JA Methyltransferase-like protein 16 binds the 3’-terminal triple helix of MALAT1 long noncoding RNA. Proc Natl Acad Sci U S A 113, 14013–14018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pendleton KE et al. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 169, 824–835 e814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warda AS et al. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep 18, 2004–2014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shima H et al. S-Adenosylmethionine Synthesis Is Regulated by Selective N(6)-Adenosine Methylation and mRNA Degradation Involving METTL16 and YTHDC1. Cell Rep 21, 3354–3363 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Mendel M et al. Methylation of Structured RNA by the m(6)A Writer METTL16 Is Essential for Mouse Embryonic Development. Mol Cell 71, 986–1000 e1011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doxtader KA et al. Structural Basis for Regulation of METTL16, an S-Adenosylmethionine Homeostasis Factor. Mol Cell 71, 1001–1011 e1004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu J, Cargnello M, Topisirovic I & Pelletier J Translation Initiation Factors: Reprogramming Protein Synthesis in Cancer. Trends Cell Biol 26, 918–933 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Bhat M et al. Targeting the translation machinery in cancer. Nat Rev Drug Discov 14, 261–278 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Silvera D, Formenti SC & Schneider RJ Translational control in cancer. Nat Rev Cancer 10, 254–266 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Tahmasebi S, Khoutorsky A, Mathews MB & Sonenberg N Translation deregulation in human disease. Nat Rev Mol Cell Biol 19, 791–807 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Kapur M & Ackerman SL mRNA Translation Gone Awry: Translation Fidelity and Neurological Disease. Trends Genet 34, 218–231 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonenberg N & Hinnebusch AG Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hershey JW, Sonenberg N & Mathews MB Principles of translational control: an overview. Cold Spring Harb Perspect Biol 4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.des Georges A et al. Structure of mammalian eIF3 in the context of the 43S preinitiation complex. Nature 525, 491–495 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomes-Duarte A, Lacerda R, Menezes J & Romao L eIF3: a factor for human health and disease. RNA Biol 15, 26–34 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee AS, Kranzusch PJ & Cate JH eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 522, 111–114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyers RM et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49, 1779–1784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Behan FM et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568, 511–516 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Slobodin B et al. Transcription Impacts the Efficiency of mRNA Translation via Co-transcriptional N6-adenosine Methylation. Cell 169, 326–337 e312 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ke S et al. m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31, 990–1006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang H et al. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 567, 414–419 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin S, Choe J, Du P, Triboulet R & Gregory RI The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell 62, 335–345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt EK, Clavarino G, Ceppi M & Pierre P SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6, 275–277 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Dieterich DC et al. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci 13, 897–905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hinnebusch AG eIF3: a versatile scaffold for translation initiation complexes. Trends Biochem Sci 31, 553–562 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Erzberger JP et al. Molecular Architecture of the 40SeIF1eIF3 Translation Initiation Complex. Cell 159, 1227–1228 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu Y et al. POSTAR2: deciphering the post-transcriptional regulatory logics. Nucleic Acids Res 47, D203–D211 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sigitas Mikutis MG, Erdem Sendinc, Madoka E. Hazemi, Hannah Kiely-Collins, Demetrios Aspris, George S. Vassiliou, Yang Shi, Konstantinos Tzelepis, and Gonçalo J. L. Bernardes meCLICK-Seq, a Substrate-Hijacking and RNA Degradation Strategy for the Study of RNA Methylation. ACS Central Science (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao W et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell 61, 507–519 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Zhao X et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell research 24, 1403–1419 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coots RA et al. m(6)A Facilitates eIF4F-Independent mRNA Translation. Mol Cell 68, 504–514 e507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu S et al. METTL13 Methylation of eEF1A Increases Translational Output to Promote Tumorigenesis. Cell 176, 491–504 e421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jakobsson ME et al. The dual methyltransferase METTL13 targets N terminus and Lys55 of eEF1A and modulates codon-specific translation rates. Nat Commun 9, 3411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scholler E et al. Interactions, localization, and phosphorylation of the m(6)A generating METTL3-METTL14-WTAP complex. RNA 24, 499–512 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su R et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 38, 79–96 e11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su R et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 172, 90–105 e123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramanathan M et al. RNA-protein interaction detection in living cells. Nat Methods 15, 207–212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Y, Li Q & Chen XZ Detecting protein-protein interactions by Far western blotting. Nat Protoc 2, 3278–3284 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Garibaldi A, Carranza F & Hertel KJ Isolation of Newly Transcribed RNA Using the Metabolic Label 4-Thiouridine. Methods Mol Biol 1648, 169–176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGlincy NJ & Ingolia NT Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Journal Vol 17, NO 1 (2011). [Google Scholar]
- 55.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meng J et al. A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods 69, 274–281 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olarerin-George AO & Jaffrey SR MetaPlotR: a Perl/R pipeline for plotting metagenes of nucleotide modifications and other transcriptomic sites. Bioinformatics 33, 1563–1564 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramirez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robinson JT, Thorvaldsdottir H, Wenger AM, Zehir A & Mesirov JP Variant Review with the Integrative Genomics Viewer. Cancer Res 77, e31–e34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liberzon A et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Middleton R et al. IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol 18, 51 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the sequencing data generated by this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) under accession number GSE156798. The correlation analyses between METTL16 and eIF3a/eIF3b were derived from GTEx program (https://www.gtexportal.org/). The raw data for determining the genetic dependence of METLL family members in cancers were derived from https://depmap.org/portal/ and https://score.depmap.sanger.ac.uk/. The expression data of METTL16 in cancers and the corresponding control were derived from GENT2 (http://gent2.appex.kr/gent2/). The survival analyses of METTL16 in TCGA cancers were derived from GEPIA2 (http://gepia2.cancer-pku.cn/#index) and TCGA Research Network (http://cancergenome.nih.gov/). The Sankey diagram was built by SnakeyMatic (https://sankeymatic.com/). Three published data-sets, GSE103948 (UV-CRAC-seq data-set and PAR-CRAC-seq data-set), GSE90914 (m6A-seq data-set, input group) and GSE46705 (m6A-seq data-set), have also been re-analyzed in the present study. The eIF3a- and eIF3b-bound transcripts were downloaded from POSTAR3 (http://postar.ncrnalab.org/). Unprocessed blot and statistical source data are provided with this article. All other data supporting the fundings of this study are available from the corresponding authors on reasonable request.