

Abstract
A new method for the preparation of 2,2-disubstituted indolines from 2-phenylethylamines was developed under Pd catalysis and PhI(OAc)2 as oxidant. Imines derived from 2-pyridinecarboxaldehyde were formed in situ to direct a C–H activation process. The resulting imines were also oxidized to the corresponding amides in the same Pd-catalyzed process to obtain the final indoline as a picolinamide.
A new method for the preparation of 2,2-disubstituted indolines from 2-phenylethylamines was developed under Pd catalysis and PhI(OAc)2 as oxidant.
Introduction
The development of selective methods for the direct conversion of carbon–hydrogen bonds into carbon–heteroatom and carbon–carbon bonds remains a critical challenge in organic chemistry. An interesting approach to address this issue involves the use of substrates that contain coordinating atoms (or directing groups) that bind to the metal center in a first step, and a further rearrangement allows the C–H bond activation. A variety of transition-metal compounds have been shown to be effective in this process. Palladium complexes are particularly attractive catalysts for such transformations because ligand-directed C–H functionalization at palladium centres can be used to obtain different types of C–Y bonds (Y being carbon, oxygen, nitrogen, sulphur, or halogen). Furthermore, palladium can activate C–H bonds both at sp2 and sp3 sites, and a wide range of catalytic processes have been described with different directing groups.1
The use of directing groups in C–H activation processes is a strategy that provides precise site selectivity. However, it often involves the introduction of functional groups in the structure of the substrates that remain in the skeleton of the products.2 A solution to this problem can be accomplished with the use of easily removable directing moieties.
In the last years, many efforts have been addressed to the use of transient directing groups in C–H activation.3 Among them, we explored the use of bidentate imine-derived groups as removable directing groups. We were interested in the functionalization of phenyl groups using palladium and strong oxidants following Sanford's works with Pd(ii)/Pd(iv) catalytic cycles.4 Our previous experience with primary amines as directing groups in C–H functionalization reactions prompted us to investigate the use of imines derived from 2-pyridinecarboxaldehyde in acetoxylation processes (Scheme 1).5
Scheme 1. C–H functionalization directed by imines derived from 2-pyridinecarboxaldehyde.

Results and discussion
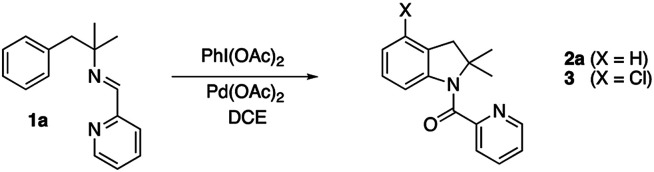
We initially tested imine 1a (Scheme 2) with several oxidants such as NBS for bromination or trimethylfluoropyridinium tetrafluoroborate for fluorination but complex mixtures were always obtained. Only when PhI(OAc)2 was used as an oxidant, several compounds could be isolated but none of them was the anticipated acetoxylated product. Instead, all the isolated compounds contained an indoline framework with a new C–N bond formed where the C–OAc bond was expected (Scheme 2).
Scheme 2. Indolines formation.

Indoline 4 structure was unequivocally determined by X-ray analysis (Fig. 1). The spectroscopic data of compounds 2a and 3 suggested analogous structures to compound 4.
Fig. 1. Crystal structure of indoline 4. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°): C2–C3 = 1.383(5), C2–C8 = 1.502(5), C8–C9 = 1.538(5), C9–N1 = 1.510(5), C3–N1 = 1.431(4), C10–O3 = 1.232(4), C10–N1 = 1.368(5), C3–C2–C8 = 110.5(3), C2–C3–N1 = 108.2(3), N1–C9–C8 = 101.3(3), C3–N1–C9 = 108.5(3), N1–C10–C11 = 118.9(3), C2–C8– C9 = 103.3(3), C3–C2–C8 = 110.5(3).

The unprecedented formation of the indoline framework involved the oxidation of an imine to an amide and an intramolecular C–H functionalization process but the detailed mechanism was unclear. Remarkably, several experiments showed that the imine derived from 2-pyridinecarboxaldehyde was necessary. Thus, similar imines derived from benzaldehyde or furfural, easily prepared from amine 5a by the same procedure to prepare imine 1a, did not afford the indoline structure under similar conditions (Scheme 3). Furthermore, unprotected amine 5a was just acetylated when the same process was applied.
Scheme 3. Alternative nitrogen atoms as directing groups.

The C–N bond formation in aromatic compounds via Pd-catalyzed C–H functionalization has been a topic of interest for the last years.6 The Pd-catalyzed synthesis of indolines from 2-phenylethylamines has been reported with picolinamides,7 sulfonamides,8 1,2,3-triazole-4-carboxamides,9 oxalyl amides,10 2-(methoxyimino)amides11 and 2-(tert-butoxycarbonylamino)-amides12 as directing groups in such C–H functionalization processes. Based on the above preliminary results and the importance of the indoline scaffold in natural and pharmaceutical bioactive compounds,13 we decided to explore the 2,2-disubstituted indoline formation using imines as directing groups. Due to the steric demand of the amino group, such indolines are usually formed from anilines and not by C–N bond formation as it is shown herein.13b
Synthesis of indolines from imines
The initial results encouraged us to explore different reaction conditions to improve the yield of indoline 2a (Table 1). An increase of equivalents of oxidant (entries 1 and 2) improved the yield and shortened the reaction time, being 1 hour the optimal conditions (entries 2–4). Once it was formed, the product 2a slowly decomposed under the reaction conditions (entries 2 and 3). A reduction of catalyst amount resulted in a drop of the yield (entry 5), being necessary the presence of the Pd(ii) catalyst to observe the reaction (entry 6). As expected, the removal of the oxidant was also adverse (entry 7). The amount of oxidant could be reduced up to 1.5 equiv. of PhI(OAc)2 without a decrease of the yield (entry 8). Reduction of the temperature to 60 °C required longer reaction times to achieved similar yields (entries 9 and 10). As expected, the formation of the chlorinated indoline 3 was only observed in DCE. Reaction in other solvents than DCE such as AcOH, acetonitrile or toluene did not afford 3 and followed the same behavior but lower yields of 2a were obtained: 20%, 40% and 50% respectively.
Indoline 2a formation.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Temp. (°C) | Time (h) | PhI(OAc)2 (equiv.) | Pd(OAc)2 (mol%) | 3a (%) | 2aa (%) |
| 1 | 90 | 24 | 1 | 10 | 9 | 26 |
| 2 | 90 | 3 | 2 | 10 | 7 | 37 |
| 3 | 90 | 1 | 2 | 10 | 16 | 61 |
| 4 | 90 | 0.5 | 2 | 10 | 10 | 40 |
| 5 | 90 | 1 | 2 | 5 | 12 | 40 |
| 6 | 90 | 1 | 2 | 0 | 0 | 0 |
| 7 | 90 | 1 | 0 | 10 | 0 | 0 |
| 8 | 90 | 1 | 1.5 | 10 | 8 | 66 |
| 9 | 60 | 1 | 2 | 10 | 0 | 27 |
| 10 | 60 | 24 | 2 | 10 | 8 | 60 |
Isolated yield.
Synthesis of indolines from imines formed in situ
The in situ formation of imines to promote the metal-catalyzed C–H activation has been recently used to functionalize sp2 and sp3 C–H bonds.14
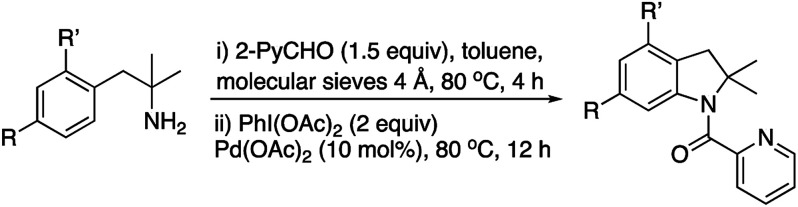
Accordingly, we next explored the possibility of the in situ preparation of the imine followed by the indoline formation reaction. Our first attempt using DCE and molecular sieves for the imine formation and subsequent addition of 2 equiv. of iodinane and 10 mol% of palladium acetate afforded only 25% yield of indoline 2a (Scheme 4).
Scheme 4. In situ imine 1a formation.

Since toluene gave also good yield in the indoline formation, we tried the two-step process in this solvent (Table 2, entry 1). Under these conditions, an overall yield of 54% was obtained which was comparable to the one obtained from pure imine 1a.
In situ indolines 2a–e formation.
| |||||
|---|---|---|---|---|---|
| Entry | Amine | R | R′ | Indoline | Yielda (%) |
| 1 | 5a | H | H | 2a | 54 |
| 2 | 5b | Me | H | 2b | 49 |
| 3 | 5c | H | Me | 2c | 64 |
| 4 | 5d | OMe | H | 2d | 52 |
| 5 | 5e | NO2 | H | 2e | 30 |
Isolated yield.
The palladium catalyst can be also used to accelerate the imine formation. Thus, the time of imine formation (step 1) could be reduced to 1 hour when the Pd(OAc)2 was added in this step without any loss of yield (55%). The amount of 2-pyridinecarboxaldehyde could also be reduced to 1 equiv. with similar overall yield (51%).
Once we confirmed that the imine could be prepared in situ using toluene as a solvent, we next studied the steric and electronic influence of substituents on the phenyl ring (Table 2).
Electron donating groups in para and ortho positions afforded similar yields (entries 2–4) whereas an electron withdrawing group lowered clearly the yield (entry 5).
The α-substitution of the amine is very critical. The absence of substituents α to the amine, such in amine 5f (Fig. 2), precluded the indoline formation. α-Monosubstituted amines such 1-phenyl-2-propanamine (5g) or methyl phenylglycinate (5h) did not afford the expected indolines 2f–h. Furthermore, the corresponding imines derived of 2-pyridinecarboxaldehyde of 5f–h amines did neither afforded the indolines. On the other hand, the use of either amine 5i or the corresponding imine, with an additional methylene group in the aliphatic chain, did not afford the expected isoquinoline type structure (2i). Thus, we were facing to some structural limitations rather than to problems due to the in situ formation of the imines.
Fig. 2. Non-α,α-disubstituted amines 5f–i.

However, indolines could be formed when the primary amine was α,α-disubstituted as in compounds 5j–l, (Scheme 5). Only, highly hindered α,α-diethyl-2-phenylethylamine afforded indoline 2l in low yield.
Scheme 5. α,α-Disubstitution effect in indoline formation.

To gain some mechanistic insight, the reaction between palladium acetate and imine 1a was studied (Scheme 6). When this reaction was carried out in toluene at room temperature the coordination compound 6 was obtained. In this complex the imine acts as a bidentate ligand, coordinated by both nitrogen atoms. This coordination compound experiences easily a metalation process. Treatment of this compound in acetic acid or toluene for 1 h at 90 °C, produced the cyclometalated derivative 7 in good yield. Complex 8 was obtained by reaction between 7 and LiBr at room temperature in acetone.
Scheme 6. Stoichiometric metalation of imine 1a.

It is also remarkable that the metalation process can even occur in some extension in solid state. Thus, the 1H NMR spectrum of a sample of 6, containing initially less than 2% of cyclometalated derivative 7, shows an increase to an 8% of this complex after standing one week at room temperature in solid state. It has been previously reported that palladium(ii) complexes containing two acetate ligands in the coordination sphere are excellent starting materials to activate C–H bonds in a facile way, and also that these compounds experience the metalation in solid state in some extension.15
Three processes are mechanistically necessary for the indoline formation: (i) C–H functionalization of the phenyl ring, (ii) imine oxidation and (iii) C–N bond formation.
To answer whether the imine oxidation or C–H functionalization is the first step of the catalytic cycle, the reaction was performed with the imine derived of tert-butylamine (8). If the first step was imine oxidation, the reaction would afford the amide 9 since this imine cannot be C–H functionalized at the phenyl group. Under the standard reaction conditions, the structurally related imine 8 was not oxidized, and the imine was mainly recovered (Scheme 7).
Scheme 7. Oxidation of imine 8.

On the other hand, when the reaction was initiated in absence of oxidant PhI(OAc)2 and, once the palladacycle 7 was stoichiometrically formed, the oxidant was added, the indoline was formed.16 According to the above results and based on related processes,17 we propose a plausible mechanism based on a Pd(ii)/Pd(iv) cycle (Scheme 8).
Scheme 8. Indolines formation.

Initial coordination of the nitrogen atoms of the imine to Pd(OAc)2 would afford complex A that can be converted to palladacycle B upon heating. In our hands, such a complex can be formed under stoichiometric conditions in absence of oxidant. In presence of the oxidant PhI(OAc)2, complex B would be oxidized to a Pd(iv) complex (C). Acetate migration to the imine carbon would afford complex D.18 The cis disposition between one acetato ligand and the NCH(AcO) fragment in this compound permits the reduction of palladium in a process that involves the transference of the hydrogen atom in β position to the pendant carbonyl oxygen atom of the acetato ligand to form acetic acid. At this point, it is important the presence of the gem-dimethyl group, since only one hydrogen atom can be eliminated, and the process can be more selective. When this group was substituted by a methylene, a complex mixture was obtained under the reaction conditions, as it was the case with the imine derived of 2-pyridinecarboxaldehyde and the α,α-unsubstituted amine 5f. Pyridine-assisted deacetylation and oxidation of complex E by an additional equivalent of PhI(OAc)2 would form a Pd(iv) complex (F).
This complex could afford two different compounds by reductive elimination reactions. The formation of a C–N bond from this complex would afford the corresponding indoline 2a. In contrast the formation of a C–O (or a C–Cl bond when the reaction was performed in DCE)19 would afford an imine derivative ortho-acetato (or ortho-chloro) substituted, which by a sequence of metalation, oxidative addition and reductive elimination with formation of C–N bond would afford the minor compounds 4 or 3 respectively.
It should be noted that Sanford et al. have described that the distribution of different compounds obtained by competitive reductive elimination of C–Cl, C–C or C–N bonds form palladium(iv) complexes is highly sensitive to the reaction medium.20
The fact that similar imines derived from benzaldehyde or furfural did not afford the indoline structure under similar conditions can be explained because the coordination to the nitrogen atom of a pyridine fragment is a key point to afford the palladium(iv) derivatives. It should be noted that palladium catalyzed oxidative synthesis of pyrrolidine, indole and indoline derivatives needs the presence of the picolinamide fragment as a directing group.20,21
Conclusions
In summary, a new convenient route for 2,2-disubstituted indolines was developed via Pd-catalyzed C–H activation of imines derived from 2-pyridinecarboxaldehyde that can be formed in situ in one-pot synthesis from 2-phenylethylamines. An oxidant such as PhI(OAc)2 was necessary to promote a plausible Pd(ii)/Pd(iv) cycle with concomitant oxidation of the imine to an amide. The ability of the pyridine moiety to coordinate Pd seems to be essential for this catalytic cycle. The absence of hydrogens in the α-position of the amine is also a requirement for the formation of indolines.
Experimental
General considerations
Amines 5f–h are commercially available. Amines 5a–e and 5j–l were prepared according to known procedures.5a Amine 5i was obtained from 2-methyl-4-phenylbutan-2-ol by a Ritter–Jirgensons reaction.22 Elemental analysis C, H and N analyses were performed with an Eager 1108 microanalyzer. 1H (400 MHz) and 13C (101 MHz) nuclear magnetic resonance (NMR) spectra were registered using a Varian Mercury 400 MHz NMR spectrometer. CDCl3 (99.9%) was used as solvent while SiMe4 (TMS) was used as reference. The coupling constants (J) are expressed in Hz and the chemical shifts in part per million (ppm). The signals multiplicities are reported with the following abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and br (broad). More complicated signals are described as combination of the indicated abbreviations: e.g. dd (double of doublets), dt (double of triplets), etc. Two-dimensional experiments (NOESY, HSQC) were performed to confirm the signal assignments. ESI (+) spectra were acquired either on an LC/MSD-TOF instrument or on a ZQ mass spectrometer, utilizing a mixture of H2O : CH3CN (1 : 1, v/v) as the eluent. Crystallographic data for 4 were collected on a Bruker APEX II QUAZAR diffractometer at the Group of Magnetism and Functional Molecules (GMMF) of the Universitat de Barcelona, equipped with a microfocus multilayer monochromator with Mo Kα radiation (λ = 0.71073 Å). Data reduction and absorption corrections were performed by using SAINT23 and SADABS,23 respectively. The structure was solved using SHELXT24 and refined with OLEX2 suite.25
General procedure for the preparation of imines
Aldehyde (6.00 mmol) was added to a solution of amine 5a (900 mg, 6.00 mmol) in toluene (60 mL). The solution was heated to reflux overnight with a Dean–Stark apparatus. The crude mixture was cooled to room temperature, dried with MgSO4 and filtered. The solvent was eliminated at reduced pressure to afford the pure imine.
N-(2-Methyl-1-phenylpropan-2-yl)-1-(pyridin-2-yl)methan-imine (1a)
Brown oil, 1.43 g (6.00 mmol, 100% yield). 1H NMR (400 MHz, CDCl3): δ 8.63–8.61 (m, 1H), 8.13 (s, 1H), 8.09–8.06 (m, 1H), 7.78–7.73 (m, 1H), 7.32–7.28 (m, 1H), 7.23–7.13 (m, 5H), 2.92 (s, 2H), 1.28 (s, 6H). 13C NMR (101 MHz, CDCl3): δ 157.0, 155.4, 149.2, 138.3, 136.5, 130.7, 127.6, 126.1, 124.4, 120.8, 60.9, 49.6, 26.8. HRMS (ESI) (m/z) calcd for [M + H]+: 239.1543, found: 239.1542.
N-(2-Methyl-1-phenylpropan-2-yl)-1-phenylmethanimine (1b)
Brown oil, 1.42 g (6.00 mmol, 100% yield). 1H NMR (400 MHz, CDCl3): δ 7.99 (s, 1H), 7.75–7.70 (m, 2H), 7.42–7.38 (m, 3H), 7.23–7.13 (m, 5H), 2.91 (s, 2H), 1.26 (s, 6H). 13C NMR (101 MHz, CDCl3): δ 157.1, 138.1, 136.4, 131.1, 129.8, 128.9, 128.5, 127.1, 125.0, 60.9, 49.6, 26.8. HRMS (ESI) (m/z) calcd for [M + H]+: 238.1590, found: 238.1590.
1-(Furan-2-yl)-N-(2-methyl-1-phenylpropan-2-yl)methanimine (1c)
Brown oil, 1.36 g (6.00 mmol, 100% yield). 1H NMR (400 MHz, CDCl3): δ 7.77 (s, 1H), 7.53 (d, J = 1.8 Hz, 1H), 7.22–7.10 (m, 5H), 6.66 (d, J = 3.4 Hz, 1H), 6.47 (dd, J = 3.4, 1.8 Hz, 1H), 2.91 (s, 2H), 1.24 (s, 6H). 13C NMR (101 MHz, CDCl3): δ 152.2, 145.2, 144.4, 138.2, 130.8, 127.6, 126.0, 113.7, 111.4, 60.7, 49.3, 26.8. HRMS (ESI) (m/z) calcd for [M + H]+: 228.1383, found: 228.1383.
General procedure for the preparation of indoline 2a from imine 1a
The imine (0.50 mmol) was dissolved in the suitable solvent (3 mL), then Pd(OAc)2 (11 mg, 0.05 mmol) and PhI(OAc)2 (322 mg, 1.00 mmol) were added. The reaction mixture was stirred at 90 °C in a sealed vial then it was filtered through a pad of Celite. The solvent was evaporated at reduced pressure to give a crude mixture that was purified by flash silica gel chromatography (hexane/ethyl acetate).
(2,2-Dimethylindolin-1-yl)(pyridin-2-yl)methanone (2a)
Pale oil, 77 mg (0.31 mmol, 61%) from imine 1a in DCE for 1 h. 1H NMR (400 MHz, CDCl3): δ 8.60 (d, J = 4.5 Hz, 1H), 7.86–7.81 (m, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.42–7.38 (m, 1H), 7.13 (d, J = 7.7 Hz, 1H), 6.90–6.77 (m, 2H), 5.71 (br m, 1H), 3.05 (s, 2H), 1.70 (s, 6H). 13C NMR (101 MHz, CDCl3): δ 167.2, 155.0, 149.2, 142.1, 137.2, 130.5, 126.4, 125.3, 125.2, 123.4, 123.1, 115.7, 67.5, 45.5, 25.9. HRMS (ESI) (m/z) calcd for [M + H]+: 253.1335, found: 253.1339.
(4-Chloro-2,2-dimethylindolin-1-yl)(pyridin-2-yl)methanone (3)
Pale oil, 23 mg (0.08 mmol, 16%) from imine 1a in DCE for 1 h. 1H NMR (400 MHz, CDCl3): δ 8.58 (d, J = 4.8 Hz, 1H), 7.88–7.83 (m, 1H), 7.69–7.66 (m, 1H), 7.43–7.40 (m, 1H), 6.88 (d, J = 7.9 Hz, 1H), 6.77–6.63 (m, 1H), 5.64 (br s, 1H), 3.08 (s, 2H), 1.71 (s, 6H). 13C NMR (101 MHz, CDCl3): δ 167.3, 154.4, 149.1, 143.4, 137.3, 131.0, 129.0, 127.8, 125.5, 123.5, 122.9, 114.0, 67.6, 44.5, 26.0. HRMS (ESI) (m/z) calcd for [M + H]+: 287.0951, found: 287.0950.
General procedure for the preparation of indolines from amines
2-Pyridinecarboxaldehyde (70 μL, 0.75 mmol) was added to a solution of amine (0.50 mmol) in dry toluene (3 mL) in the presence of 4 Å molecular sieves. The solution was stirred for 4 hours at 80 °C in a sealed vial, then Pd(OAc)2 (11 mg, 0.05 mmol) and PhI(OAc)2 (322 mg, 1 mmol) were added. The suspension was stirred overnight at 80 °C in the sealed vial. The crude mixture was filtered through a pad of Celite, the solvent was eliminated at reduced pressure and the mixture was purified by flash silica gel chromatography (hexane/ethyl acetate) to afford the indoline.
Pyridin-2-yl(2,2,6-trimethylindolin-1-yl)methanone (2b)
Pale oil, 66 mg (0.25 mmol, 49%) from amine 5b (81 mg). 1H NMR (400 MHz, CDCl3): δ 8.62–8.60 (m, 1H), 7.86–7.82 (m, 1H), 7.64 (d, J = 7.8 Hz, 1H), 7.43–7.39 (m, 1H), 7.01 (d, J = 7.5 Hz, 1H), 6.71 (d, J = 7.5 Hz, 1H), 5.51 (br s, 1H), 3.00 (s, 2H), 2.00 (s, 3H), 1.68 (s, 6H). 13C NMR (101 MHz, CDCl3): δ 167.1, 155.1, 149.1, 142.2, 137.2, 136.1, 127.5, 125.0, 124.8, 123.9, 123.4, 116.7, 67.7, 45.2, 25.9, 21.6. HRMS (ESI) (m/z) calcd for [M + H]+: 267.1492, found: 267.1491.
Pyridin-2-yl(2,2,4-trimethylindolin-1-yl)methanone (2c)
Pale oil, 85 mg (0.32 mmol, 64%) from amine 5c (81 mg). 1H NMR (400 MHz, CDCl3): δ 8.58 (d, J = 4.6 Hz, 1H), 7.82–7.81 (m, 1H), 7.64 (d, J = 7.8 Hz, 1H), 7.40–7.38 (m, 1H), 6.73–6.68 (m, 2H), 5.51 (br m, 1H), 2.97 (s, 2H), 2.20 (s, 3H), 1.72 (s, 6H). 13C NMR (101 MHz, CDCl3): δ 167.2, 155.0, 149.1, 141.7, 137.2, 134.6, 129.2, 126.5, 125.2, 124.1, 123.4, 113.2, 67.1, 44.3, 26.1, 18.6. HRMS (ESI) (m/z) calcd for [M + H]+: 267.1492, found: 267.1494.
(6-Methoxy-2,2-dimethylindolin-1-yl)(pyridin-2-yl)methanone (2d)
Pale oil, 73 mg (0.26 mmol, 52%) from amine 5d (90 mg). 1H NMR (400 MHz, CDCl3): δ 8.65 (d, 1H, J = 4.8 Hz), 7.86–7.82 (m, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.42–7.38 (m, 1H), 7.01 (d, J = 8.2 Hz, 1H), 6.44 (dd, J = 8.2, 2.2 Hz, 1H), 5.30 (br s, 1H), 3.43 (s, 3H), 2.98 (s, 2H), 1.69 (s, 6H). 13C NMR (101 MHz, CDCl3): δ 167.1, 158.5, 155.0, 149.2, 143.1, 137.3, 125.4, 125.2, 123.4, 122.6, 108.9, 102.6, 68.4, 55.1, 44.8, 25.9. HRMS (ESI) (m/z) calcd for [M + H]+: 283.1441, found: 283.1442.
(2,2-Dimethyl-6-nitroindolin-1-yl)(pyridin-2-yl)methanone (2e)
Pale oil, 35 mg (0.12 mmol, 30%) from amine 5e (75 mg). 1H NMR (400 MHz, CDCl3): δ 8.60–8.56 (m, 1H), 7.98–7.94 (m, 1H), 7.80–7.77 (m, 2H), 7.52–7.49 (m, 1H), 7.27 (d, J = 7.7 Hz, 1H), 6.58 (br s, 1H), 3.13 (s, 2H), 1.72 (s, 6H). 13C NMR (101 MHz, CDCl3): δ 167.4, 153.5, 149.1, 147.2, 143.3, 137.9, 137.9, 126.2, 125.2, 123.9, 118.4, 110.8, 68.8, 45.1, 25.9. HRMS (ESI) (m/z) calcd for [M + H]+: 298.1186, found: 298.1189.
Diethyl 1-picolinoylindoline-2,2-dicarboxylate (2j)
Pale oil, 39 mg (0.15 mmol, 57%) from amine 5j (50 mg). 1H NMR (400 MHz, CDCl3, 50 °C): δ 8.49 (br m, 1H), 8.05 (br m, 1H), 7.87–7.82 (m, 1H), 7.40–7.35 (m, 1H), 7.15 (d, J = 7.7 Hz, 1H), 7.05 (br m, 1H), 7.04–6.98 (m, 1H), 4.22–4.08 (br m, 4H), 3.74 (s, 2H), 1.34–1.02 (br m, 6H). 13C NMR (101 MHz, CDCl3, 45 °C): δ 168.1, 165.5, 152.9, 147.2, 137.2, 127.8, 127.3, 125.4, 124.2, 124.1, 116.9, 75.2, 62.0, 40.7, 13.8. HRMS (ESI) (m/z) calcd for [M + H]+: 369.1445, found: 369.1450.
Ethyl 2-methyl-1-picolinoylindoline-2-carboxylate (2k)
Pale oil, 91 mg (0.29 mmol, 59%) from amine 5k (103 mg). 1H NMR (400 MHz, CDCl3): δ 8.62 (br m, 1H), 7.83 (td, J = 7.7, 1.7 Hz, 1H), 7.63 (br m, 1H), 7.42 (dd, J = 7.2, 5.3 Hz, 1H), 7.14 (d, J = 7.7 Hz, 1H), 6.95–6.75 (m, 2H), 5.63 (br m, 1H), 4.19 (br m, 2H), 3.53 (d, J = 15.7 Hz, 1H), 3.02 (d, J = 15.7 Hz, 1H), 1.79 (s, 3H), 1.21 (br s, 3H). 13C NMR (101 MHz, CDCl3): δ 172.5, 166.0, 153.7, 149.5, 137.3, 129.4, 127.1, 125.4, 125.2, 123.8, 123.2, 114.7, 69.7, 61.5, 41.7, 21.6, 14.1. HRMS (ESI) (m/z) calcd for [M + H]+: 311.1390, found: 311.1385.
(2,2-Diethylindolin-1-yl)(pyridin-2-yl)methanone (2l)
Pale oil, 38 mg (0.14, mmol, 27%) from amine 5l (88 mg). 1H NMR (400 MHz, CDCl3): δ 8.62 (d, 1H, J = 4.6 Hz), 7.85–7.80 (m, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.41–7.38 (m, 1H), 7.12 (d, J = 7.3 Hz, 1H), 6.87–6.71 (m, 2H), 5.54 (br m, 1H), 3.10 (s, 2H), 2.54–2.40 (m, 2H), 1.88–1.78 (m, 2H), 0.93 (t, J = 7.4 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 167.1, 155.0, 149.4, 143.4, 137.2, 131.0, 126.4, 125.0, 124.9, 123.1, 122.9, 114.9, 73.6, 37.9, 30.4, 8.3. HRMS (ESI) (m/z) calcd for [M + H]+: 281.1648, found: 281.1649.
Synthesis of coordination compound 6
Palladium acetate (112 mg, 0.50 mmol) was added to a solution of imine 1a (120 mg, 0.50 mmol) in toluene (20 mL), the resulting solution was stirred 2 h at room temperature and then filtered to obtain 6 as yellow crystals (135 mg, 0.32 mmol, 60% yield). 1H NMR (400 MHz, CDCl3): δ 8.42 (d, J = 5.2, 1H), 8.05 (td, J = 7.8, 1.5 Hz, 1H), 7.65 (d, J = 7.5 Hz, 2H), 7.58–7.55 (m, 2H), 7.44 (s, 1H), 7.28 (t, J = 7.5 Hz, 2H), 7.19 (t, J = 7.5 Hz, 1H), 3.27 (s, 2H), 2.13 (s, 3H), 2.08 (s, 3H), 1.5 (s, 6H). MS (ESI) (m/z) calcd for [M–2AcO]+: calcd for 344.04, found: 344.04; calcd for [M–2AcO + CH3CN]+: 385.07, found: 385.07.
Synthesis of metalated compound 7
200 mg (0.43 mmol) of 6 were dissolved in toluene and heated at 90 °C for 1 hour. The resulting solution was filtered, the solvent was evaporated, and the residue was treated with diethyl ether to obtain 7, as yellow crystals, 120 mg, (0.30 mmol, 70% yield). 1H NMR (400 MHz, CDCl3): δ 8.60 (d, J = 5.1 Hz, 1H), 8.34 (s, 1H), 7.94 (td, J = 7.8, 1.6 Hz, 1H), 7.67 (d, J = 7.7 Hz, 2H), 7.57 (ddd, J = 7.8, 5.1, 1.3 Hz, 1H), 7.35 (m, 1H), 6.92 (m, 2H), 6.79 (m 1H), 2.95 (br s, 2H), 2.15 (br s, 3H), 1.40 (br s, 6H). EA (calc. for C18H20N2O2Pd): C: 53.4% (53.68%); H: 5.1% (5.01%); N: 6.6% (6.95%). MS (ESI) (m/z) calcd for [M–AcO]+: 343.04, found: 343.04.
Synthesis of metalated compound 8
A mixture of 200 mg (0.50 mmol) of 7 and 130 mg of LiBr (1.50 mmol) was stirred in acetone at room temperature for 1 hour and then filtered to obtain 8 as yellow crystals (165 mg, 0.40 mmol, 80% yield). 1H NMR (400 MHz, CDCl3): δ 9.50 (d, J = 5.5 Hz, 1H), 8.51 (s, 11H), 8.06 (dd, J = 7.8, 1.2 Hz, 1H), 8.0 (td, J = 7.8, 2.0 Hz, 1H), 7.74 (d, J = 7.7, 1H), 7.66–7.62 (dd, J = 7.8, 5.5 Hz, 1H), 6.97 (td, J = 7.2, 1.3 Hz, 1H), 6.90–6.85 (m, 2H), 3.15 (br s, 2H), 1.45 (br s, 6H). EA (calc. for C16H17BrN2Pd): C: 45.4% (45.36%); H: 4.1% (4.04%); N: 6.4% (6.61%). MS (ESI) (m/z) calcd for [M–Br]+: 343.04, found: 343.04; calcd for [M–Br + CH3CN]+: 384.07, found: 384.07.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the Generalitat de Catalunya (2017SGR474 and 2014SGR107) and the Spanish Ministerio de Economia y Competitividad (PGC2018-098630-B-I00, CTQ2015-65040-P, SAF2014-52223-C2-1-R and fellowship to A. M.).
Electronic supplementary information (ESI) available. CCDC 1883868. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra05670j
Notes and references
- (a) Gensch T. Hopkinson M. N. Glorius F. Wencel-Delord J. Chem. Soc. Rev. 2016;45:2900–2936. doi: 10.1039/C6CS00075D. [DOI] [PubMed] [Google Scholar]; (b) Hartwig J. F. J. Am. Chem. Soc. 2016;138:2–24. doi: 10.1021/jacs.5b08707. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rouquet G. Chatani N. Angew. Chem., Int. Ed. 2013;52:11726–11743. doi: 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]; (d) Zhu R.-Y. Farmer M. E. Chen Y.-Q. Yu J.-Q. Angew. Chem., Int. Ed. 2016;55:10578–10599. doi: 10.1002/anie.201600791. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chen Z. Wang B. Zhang J. Yu W. Liu Z. Zhang Y. Org. Chem. Front. 2015;2:1107–1295. doi: 10.1039/C5QO00004A. [DOI] [Google Scholar]; (f) Lyons T. W. Sanford M. S. Chem. Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Baudoin O. Chem. Soc. Rev. 2011;40:4902–4911. doi: 10.1039/C1CS15058H. [DOI] [PubMed] [Google Scholar]; (h) Hickman A. J. Sanford M. S. Nature. 2012;484:177–185. doi: 10.1038/nature11008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Wencel-Delord J. Droge T. Liu F. Glorius F. Chem. Soc. Rev. 2011;40:4740–4761. doi: 10.1039/C1CS15083A. [DOI] [PubMed] [Google Scholar]
- (a) Sambiagio C. Schönbauer D. Blieck R. Dao-Huy T. Pototschnig G. Schaaf P. Wiesinger T. Zia M. F. Wencel-Delord J. Besset T. Maes B. U. W. Schnürch M. Chem. Soc. Rev. 2018;47:6603–6743. doi: 10.1039/C8CS00201K. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li Y. Xu S. Chem.–Eur. J. 2018;24:16218–16245. doi: 10.1002/chem.201800921. [DOI] [PubMed] [Google Scholar]; (c) Ping L. Chung D. S. Bouffard J. Lee S. Chem. Soc. Rev. 2017;46:4299–4328. doi: 10.1039/C7CS00064B. [DOI] [PubMed] [Google Scholar]
- For reviews, see: ; (a) Bhattacharya T. Pimparkar S. Maiti D. RSC Adv. 2018;8:19456–19464. doi: 10.1039/C8RA03230K. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhao Q. Poisson T. Pannecoucke X. Besset T. Synthesis. 2017;49:4808–4826. doi: 10.1055/s-0036-1590878. [DOI] [Google Scholar]; (c) Yadav M. R. Rit R. K. Shankar M. Sahoo A. K. Asian J. Org. Chem. 2015;4:846–864. doi: 10.1002/ajoc.201500105. [DOI] [Google Scholar]; (d) Wang C. Huang Y. Synlett. 2013;24:145–149. [Google Scholar]; For recent examples, see: ; (e) Liu Y. Ge H. Nat. Chem. 2017;9:26–32. doi: 10.1038/nchem.2606. [DOI] [Google Scholar]; (f) Chen X.-Y. Sorensen E. J. J. Am. Chem. Soc. 2018;140:2789–2792. doi: 10.1021/jacs.8b00048. [DOI] [PubMed] [Google Scholar]
- Neufeldt S. R. Sanford M. S. Acc. Chem. Res. 2012;45:936–946. doi: 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]; and references therein
- (a) Mancinelli A. Alamillo C. Albert J. Ariza X. Etxabe H. Farràs J. Garcia J. Granell J. Quijada F. J. Organometallics. 2017;36:911–919. doi: 10.1021/acs.organomet.6b00944. [DOI] [Google Scholar]; (b) Rodríguez A. Albert J. Ariza X. Garcia J. Granell J. Farràs J. La Mela A. Nicolás E. J. Org. Chem. 2014;79:9578–9585. doi: 10.1021/jo501658s. [DOI] [PubMed] [Google Scholar]; (c) Albert J. Ariza X. Calvet T. Font-Bardia M. Garcia J. Granell J. Lamela A. López B. Martinez M. Ortega L. Rodriguez A. Santos D. Organometallics. 2013;32:649–659. doi: 10.1021/om301140t. [DOI] [Google Scholar]; (d) López B. Rodriguez A. Santos D. Albert J. Ariza X. Garcia J. Granell J. Chem. Commun. 2011;47:1054–1056. doi: 10.1039/C0CC03478A. [DOI] [PubMed] [Google Scholar]
- (a) Timsina Y. N. Gupton B. F. Ellis K. C. ACS Catal. 2018;8:5732–5776. doi: 10.1021/acscatal.8b01168. [DOI] [Google Scholar]; (b) Park Y. Kim Y. Chang S. Chem. Rev. 2017;117:9247–9301. doi: 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]; (c) Rit R. K. Shankar M. Sahoo A. K. Org. Biomol. Chem. 2017;15:1282–1293. doi: 10.1039/C6OB02162J. [DOI] [PubMed] [Google Scholar]; (d) Zhou Y. Yuan J. Yang Q. Xiao Q. Peng Y. ChemCatChem. 2016;8:2178–2192. doi: 10.1002/cctc.201600079. [DOI] [Google Scholar]; (e) Nack W. A. Chen G. Synlett. 2015;26:2505–2511. doi: 10.1055/s-0034-1381051. [DOI] [Google Scholar]
- (a) He G. Zhao Y. Zhang S. Lu C. Chen G. J. Am. Chem. Soc. 2012;134:3–6. doi: 10.1021/ja210660g. [DOI] [PubMed] [Google Scholar]; (b) Nadres E. T. Daugulis O. J. Am. Chem. Soc. 2012;134:7–10. doi: 10.1021/ja210959p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) He G. Lu C. Zhao Y. Nack W. A. Chen G. Org. Lett. 2012;14:2944–2947. doi: 10.1021/ol301352v. [DOI] [PubMed] [Google Scholar]
- (a) Mei T.-S. Leow D. Xiao H. Laforteza B. N. Yu J.-Q. Org. Lett. 2013;15:3058–3061. doi: 10.1021/ol401246u. [DOI] [PubMed] [Google Scholar]; (b) Mei T.-S. Wang X. Yu J.-Q. J. Am. Chem. Soc. 2009;131:10806–10807. doi: 10.1021/ja904709b. [DOI] [PubMed] [Google Scholar]; (c) Li J.-J. Mei T.-S. Yu J.-Q. Angew. Chem., Int. Ed. 2008;47:6452–6455. doi: 10.1002/anie.200802187. [DOI] [PubMed] [Google Scholar]; For analogous Cu-catalyzed functionalization, see: ; (d2) Takamatsu K. Hirano K. Satoh T. Miura M. J. Org. Chem. 2015;80:3242–3249. doi: 10.1021/acs.joc.5b00307. [DOI] [PubMed] [Google Scholar]
- Ye X. He Z. Ahmed T. Weise K. Akhmedov N. G. Petersen J. L. Shi X. Chem. Sci. 2013;4:3712–3716. doi: 10.1039/C3SC51211H. [DOI] [Google Scholar]
- Wang C. Chen C. Zhang J. Han J. Wang Q. Guo K. Liu P. Guan M. Yao Y. Zhao Y. Angew. Chem., Int. Ed. 2014;53:9884–9888. doi: 10.1002/anie.201404854. [DOI] [PubMed] [Google Scholar]
- He Y.-P. Zhang C. Fan M. Wu Z. Ma D. Org. Lett. 2015;17:496–499. doi: 10.1021/ol503514t. [DOI] [PubMed] [Google Scholar]
- Zheng Y. Song W. Zhu Y. Wei B. Xuan L. Org. Biomol. Chem. 2018;16:2402–2405. doi: 10.1039/C8OB00207J. [DOI] [PubMed] [Google Scholar]
- See for example: ; (a) Liu D. Zhao G. Xiang L. Eur. J. Org. Chem. 2010:3975–3984. doi: 10.1002/ejoc.201000323. [DOI] [Google Scholar]; and references therein. For recent examples of 2,2-disubstituted indolines see: ; (b1) Ni Y. Yu Q. Liu Q. Zuo H. Yu H.-B. Wei W.-J. Liao R.-Z. Zhong F. Org. Lett. 2018;20:1404–1408. doi: 10.1021/acs.orglett.8b00176. [DOI] [PubMed] [Google Scholar]; and references therein
- St John-Campbell S. Bull J. A. Org. Biomol. Chem. 2018;16:4582–4595. doi: 10.1039/C8OB00926K. [DOI] [PubMed] [Google Scholar]
- Font H. Font-Bardia M. Gómez K. González G. Granell J. Macho I. Martínez M. Dalton Trans. 2014;43:13525–13536. doi: 10.1039/C4DT01463D. [DOI] [PubMed] [Google Scholar]
- Compared with the Pd-catalyzed reaction, these stoichiometric conditions gave lower yield (20%)
- (a) Topczewski J. J. Sanford M. S. Chem. Sci. 2015;6:70–76. doi: 10.1039/C4SC02591A. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Desai L. V. Stowers K. J. Sanford M. S. J. Am. Chem. Soc. 2008;130:13285–13293. doi: 10.1021/ja8045519. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Deprez N. R. Sanford M. S. Inorg. Chem. 2007;46:1924–1935. doi: 10.1021/ic0620337. [DOI] [PubMed] [Google Scholar]
- Intermolecular addition of the acetate group to complex B or C might also occur
- Botla V. Akudari A. Malapaka C. Tetrahedron Lett. 2019;60:115–119. doi: 10.1016/j.tetlet.2018.11.071. [DOI] [Google Scholar]
- Whitfield S. R. Sanford M. S. J. Am. Chem. Soc. 2007;129:15142–15143. doi: 10.1021/ja077866q. [DOI] [PubMed] [Google Scholar]
- Karnakanti S. Zang Z.-L. Zhao S. Shao P.-L. Hu P. He Y. Chem. Commun. 2017;53:11205–11208. doi: 10.1039/C7CC06448A. [DOI] [PubMed] [Google Scholar]
- Jirgensons A. Kauss V. Kalvinsh I. Gold M. R. Synthesis. 2000:1709–1712. doi: 10.1055/s-2000-8208. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M., SAINT and SADABS, Bruker AXS Inc., Madisson, Wisconsin, USA, 2012 [Google Scholar]
- (a) Sheldrick G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]; (b) Sheldrick G. M., SHLEXTL
- Dolomanov O. V. Bourhis L. J. Gildea R. J. Howard J. A. K. Puschmann H. J. Appl. Crystallogr. 2009;42:339–341. doi: 10.1107/S0021889808042726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.