

Abstract
Brain organoids represent a powerful tool for studying human neurological diseases, particularly those impacting brain growth and structure. However, many diseases manifest with clear evidence of physiological and network abnormality in the absence of anatomical changes, raising the question of whether organoids possess sufficient neural network complexity to model these conditions. Here, we explore the network level functions of brain organoids using calcium sensor imaging and extracellular recording approaches that together reveal the existence of complex network dynamics reminiscent of intact brain preparations. We demonstrate highly abnormal and epileptiform-like activity in organoids derived from Rett syndrome patient induced pluripotent stem cells accompanied by transcriptomic differences revealed by single-cell analyses. We also rescue key physiological activities with an unconventional neuroregulatory drug, Pifithrin-α. Together, these findings provide an essential foundation for the utilization of brain organoids to study intact and disordered human brain network formation and illustrate their utility in therapeutic discovery.
INTRODUCTION
Brain organoids derived from human embryonic and induced pluripotent stem cells (hESCs and hiPSCs) recapitulate unique features of human brain development and are increasingly being used as model systems to gain novel insights into a variety of neurological diseases1–3. Organoids represent a significant advance in the toolkit available for understanding human brain function and disease mechanisms as much of our current knowledge is derived from studies of embryonic and adult animals, particularly rodents. While there is conservation in many mechanisms of brain development across evolution, it has become increasingly clear that the human brain nevertheless possesses distinct features4, 5, and it remains unclear how well animal models of neurological disease faithfully recapitulate human pathologies. Moreover, drugs shown to be effective in ameliorating neurological disease in animals frequently fail in clinical trials6, 7, emphasizing the need for human cell-based systems to evaluate drug efficacy.
Most brain organoid studies to date have capitalized on the anatomical and cytoarchitectural characteristics of organoids to model disorders that grossly impact human brain growth or organization such as microcephaly, macrocephaly, and lissencephaly1–3. However, the diverse functions of the human brain depend not only on its stereotyped anatomical structure, but also on the establishment and function of neural networks. Indeed, errors in the formation of these circuits or damage after their development are thought to underlie many neurological diseases ranging from autism and neuropsychiatric disorders to epilepsy and Alzheimer’s disease8, 9. Reliably assessing network activity becomes especially critical in situations in which there is clear clinical disease but no overt structural brain abnormality.
Despite evidence of the enhanced organizational complexity of brain organoids relative to other in vitro systems, the presence of sophisticated neural network activities has only recently been demonstrated in live whole-organoid preparations10, 11. A key feature of robust neural networks is the presence of distinct frequencies of oscillatory activity. Such activities are thought to depend on precisely tuned inhibitory-excitatory neuronal interactions and can be recorded from intact and sliced brain preparations, but are not readily achievable in two-dimensional culture systems12, 13. In addition, specific changes in oscillatory activity, such as the loss of gamma rhythms or the emergence of polymorphic low frequency activity, can be clear indicators of underlying neurological dysfunction14–16.
Here, we sought to develop and characterize brain organoid network activity, utilizing recent advances in organoid techniques17 to generate cerebral cortex-ganglionic eminence (Cx+GE) “fusion” organoids in which excitatory and inhibitory neurons functionally integrate18–20. We then used a combination of calcium sensor imaging and extracellular recordings of local field potentials to demonstrate the presence of intricate network-level activities including oscillatory rhythms. This work builds on prior approaches that analyzed network activity in cortical organoids using either calcium indicator imaging alone or plate-based multi-electrode recordings10, 11, 17, 21. The advances encompassed by the present techniques allowed us to clearly discern pathological network and oscillatory changes in fusion organoids containing mutations in the Methyl-CpG Binding Protein 2 (MECP2) gene associated with Rett syndrome22. Consistent with broadly similar patterns of clinical electroencephalographic abnormalities between MECP2 mutant patients as well as instances of between patient variability23, 24, we identified conserved physiological changes across organoids generated from multiple hiPSC lines derived from two patients as well as some physiological activities distinct to each patient. In both cases, neural network dysfunction was partially rescued by treatment with an unconventional neuroregulatory drug, Pifithrin-α. Collectively, these findings provide a framework for how brain organoids can be utilized to investigate network-level functions of the human brain and illustrate their utility in modeling neurological disorders and therapeutic testing.
RESULTS
Excitatory and inhibitory neuron integration within organoids
As cortical circuits in vivo contain a mixture of both excitatory and inhibitory connections, we sought to replicate this process using an organoid “fusion” technique to combine separately generated cortical and subcortical organoids and thereby create integrated structures (Fig. 1). Organoids derived from either H9 hESC or wild-type iPSC lines were directed towards cortex (Cx) or ganglionic eminence (GE) identities through the absence or presence of Sonic hedgehog (Shh) pathway agonists in the organoid differentiation scheme (Fig. 1a)17. Without Shh signaling, organoids exhibited cortical characteristics including expression of the apical and basal radial glial progenitor marker PAX6, the intermediate progenitor marker TBR2 (EOMES), deep cortical plate markers including TBR1, CTIP2 (BCL11B), and BHLHB5 (BHLHE22), and superficial layer markers such as SATB2, and BRN2 (POU3F2) (Fig. 1b, see also17, 25). Shh pathway-stimulated organoids by contrast expressed canonical GE progenitor and migratory interneuron markers such as NKX2.1, DLX1, DLX2, and OLIG2. Over time in culture, many neurons within GE organoids expressed GABAergic inhibitory neuron markers such as GAD65 (GAD2) and GABA along with a variety of interneuron subtype markers (Fig. 1b, see also17, 25).
Fig. 1 |. Generation and characterization of fusion brain organoids.
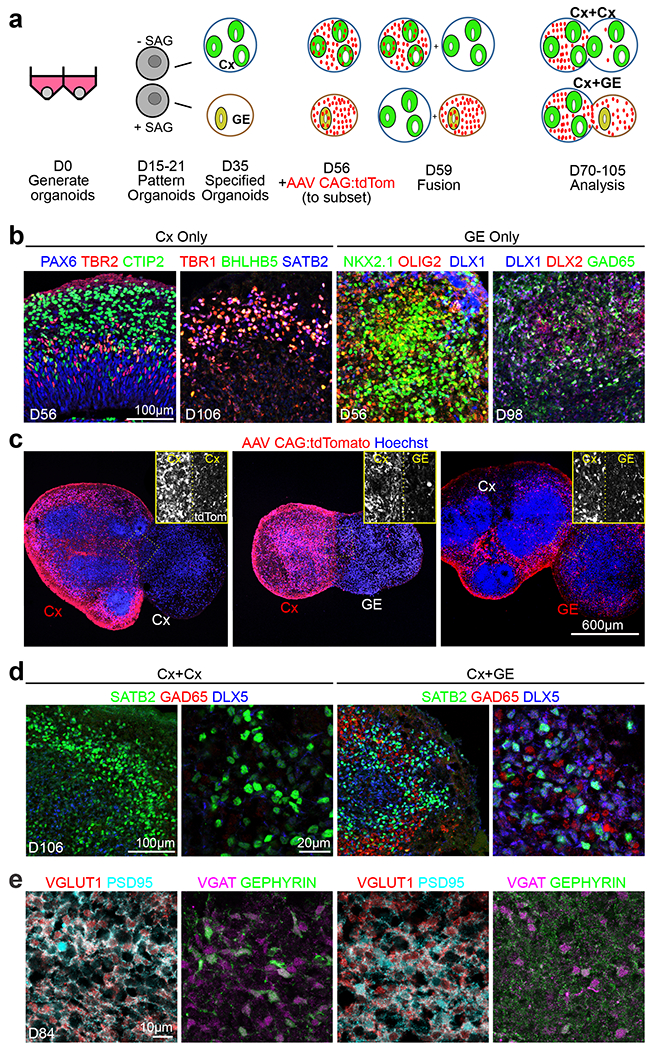
a, Schematic outlining the generation, patterning, and fusion of dorsal cortex (Cx) and ventral ganglionic eminence (GE) organoids. b, Immunohistochemical analysis of H9 hESC or non-mutant hiPSC-derived Cx and GE organoids prior to fusion at the indicated days (D) of differentiation in vitro. c, Prior to fusion, D56 Cx or GE organoids were infected with AAV1 CAG:tdTomato virus, allowing for tracking of cells emanating from each compartment. Two weeks after fusion, labeled Cx cells showed limited migration into adjacent Cx or GE structures (left and middle images) while labeled GE progenitors display robust migration and colonization of their Cx partner (right image). d, Immunohistochemical analysis showing intermingling of SATB2+ cortical neurons with DLX5+ GAD65+ inhibitory interneurons in the cortical compartment of D106 Cx+GE but not Cx+Cx fusion organoids. e, At day 84, Cx+Cx fusions (left panels) contain numerous excitatory synapses reflected by prominent colocalization of the pre- and post-synaptic markers VGLUT1 and PSD95, yet sparse numbers of inhibitory synapses detected by VGAT/GEPHYRIN costaining. Cx+GE fusions (right panels) on the other hand contain numerous VGLUT1+/PSD95+ excitatory and VGAT+/GEPHYRIN+ inhibitory synapses (right panels). All images in b-e are representative images from multiple experiments and represent one of at least 3 or more imaged sections. For specific details refer to Supplementary Table 4.
In the developing forebrain in vivo, GE-derived interneurons migrate tangentially into the adjacent cortex and functionally integrate into cortical neural networks, a process that can be recapitulated in vitro18–20. Using adeno-associated virus (AAV) CAG:tdTomato labeling of the GE organoid before Cx+GE fusion, we observed widespread migration of tdTomato+ cells from the GE and dispersion within the adjacent Cx such that at two weeks after fusion ~18% of Cx cells were tdTom+ (Figs. 1a,c and Extended Data Figs. 1a,d). Minimal tdTomato+ cell migration was seen in control Cx+Cx or Cx+GE fusions where Cx was pre-labeled with AAV-tdTomato. There was a high degree of reproducibility in the colonization of the Cx compartment by inhibitory interneurons which on average resulted in a final percentage of ~25% of cells being GABAergic (Figs. 1d and 3c,d), on par with the 20-30% density seen in the mammalian neocortex in vivo26, 27. This number also appeared consistent across organoid batches (Extended Data Figs. 1a,b).
Fig. 3 |. Rett syndrome fused and unfused organoids have similar cortical organization and cell type expression profiles.
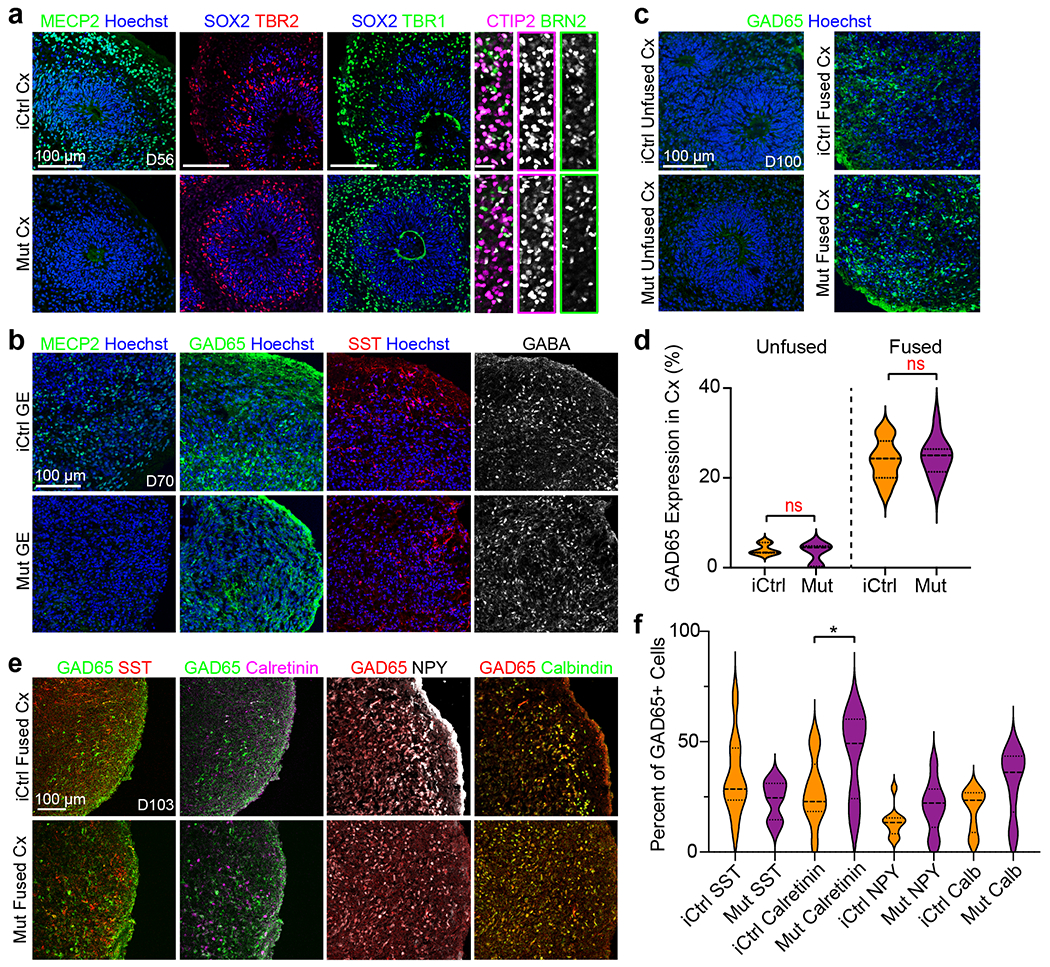
a,b, Generation and immunohistochemical analyses of isogenic Cx and GE organoids from Rett syndrome patient hiPSC that either contain (iCtrl) or lack (Mut) MECP2 expression (see also39). iCtrl and Mut Cx organoids exhibit comparable formation of neural progenitors (SOX2, TBR2), both deep and superficial layer neurons (CTIP2, BRN2), and inhibitory interneurons (GAD65, SST, and GABA) All images reflect representative images from at least 3 independently imaged sections, refer to supplementary table 4 for further details. c,d, D100 unfused iCtrl and Mut Cx organoids show minimal expression of GAD65, whereas ~20-25% of the cells in the Cx end of age matched Cx+GE organoids express GAD65, n = 3 independently generated organoids, 2631 cells, ns, not significant by a two-sided t-test. e,f, Immunohistochemical analysis of interneuron subtypes by the co-expression of GAD65 with SST, Calretinin, NPY or Calbindin in the Cx portion of day 100 iCtrl or Mut Cx+GE fusion organoids reveals the presence of multiple interneuron subtypes e. f, Cell counting reveals trends for all comparisons and a statistically significant difference between iCtrl and Mut samples with respect to the percentage of cells expressing calretinin, n = 3 fusion organoids per genotype, ≥ 980 cells for each sample counted, ns = not significant for all groups except calretinin. ANOVA P = 0.0003, F = 4.665, DF (between columns) = 7, followed by Sidak’s correction for multiple comparison between iCtrl and Mut for each marker; *P = 0.0391 for iCtrl vs Mut calretinin; ns for all other comparisons. Plots in d,f display the full distribution of individual data points with dotted lines indicating the median and quartile values.
Immunohistochemical analyses of the cortical aspect of Cx+GE fusions revealed intermingling of Cx-derived excitatory neurons, exemplified by SATB2 which is not expressed within GE organoids, and inhibitory interneurons identified by GAD65 and DLX5 costaining (Fig. 1d). By contrast, Cx+Cx fusions only expressed the neuronal marker SATB2 with few if any GAD65+ DLX5+ cells (Fig. 1d). Integration of excitatory and inhibitory interneurons within Cx+GE organoids was further confirmed by the prominence of both excitatory synapses, distinguished by apposed VGLUT1+ presynaptic and PSD95+ postsynaptic puncta, and inhibitory synapses visualized by VGAT and gephyrin staining (Fig. 1e). By comparison, Cx+Cx organoids mainly contained only excitatory synaptic puncta (Fig. 1e), indicating that most inhibitory synapses in the Cx+GE organoids are GE-derived.
Cx-GE organoids exhibit complex neural network oscillations
To determine the range of physiological activity in the fusion organoids, we utilized both live two-photon microscopy-based calcium indicator imaging of intact organoids and extracellular recordings of local field potentials (LFPs) (Fig. 1a). In accordance with existing ex vivo slice recording protocols, LFP recordings and calcium imaging were performed in the presence of low levels of kainate sufficient to induce oscillations yet not evoke seizure-like hyperexcitability28. We applied constrained non-negative matrix factorization extended (CNMF-E) methods for calcium signal processing to extract spiking dynamics from records29, 30. This enabled us to perform unbiased categorization of single-cell calcium dynamics into functional microcircuit clusters (Extended Data Fig. 2, Supplementary Fig. 1, and Supplementary Videos 1–3). In combination with LFP data, this approach allowed us to characterize brain organoid physiological activity at single-cell, microcircuit, and network levels.
After infection with AAV1 Syn:GCaMP6f virus, we measured spontaneous calcium activity as changes in GCaMP6f fluorescence (ΔF/F, Fig. 2b). Both Cx+Cx and Cx+GE fusions showed comparable baseline neural activities (Fig. 2c and Supplementary Fig. 1). However, when we assessed the role of inhibition by adding either the GABAA receptor antagonist bicuculline methiodide (BMI) or Gabazine, only Cx+GE fusions showed functional GABAergic interneuron-glutamatergic cell connectivity. Both drugs elicited repetitive waves of nearly complete synchronization of calcium transients in Cx+GE fusions (Figs. 2c,d), with no such effect in Cx+Cx fusions (Figs. 2c–e and Supplementary Videos 4–7). Hierarchical clustering revealed large groups of neurons with highly correlated activity in Cx+GE organoids following BMI treatment, while only small groups were observed in the Cx+Cx organoids (Fig. 2d).
Fig. 2 |. Cx+GE fusion organoids demonstrate complex neural network activities including oscillatory rhythms.
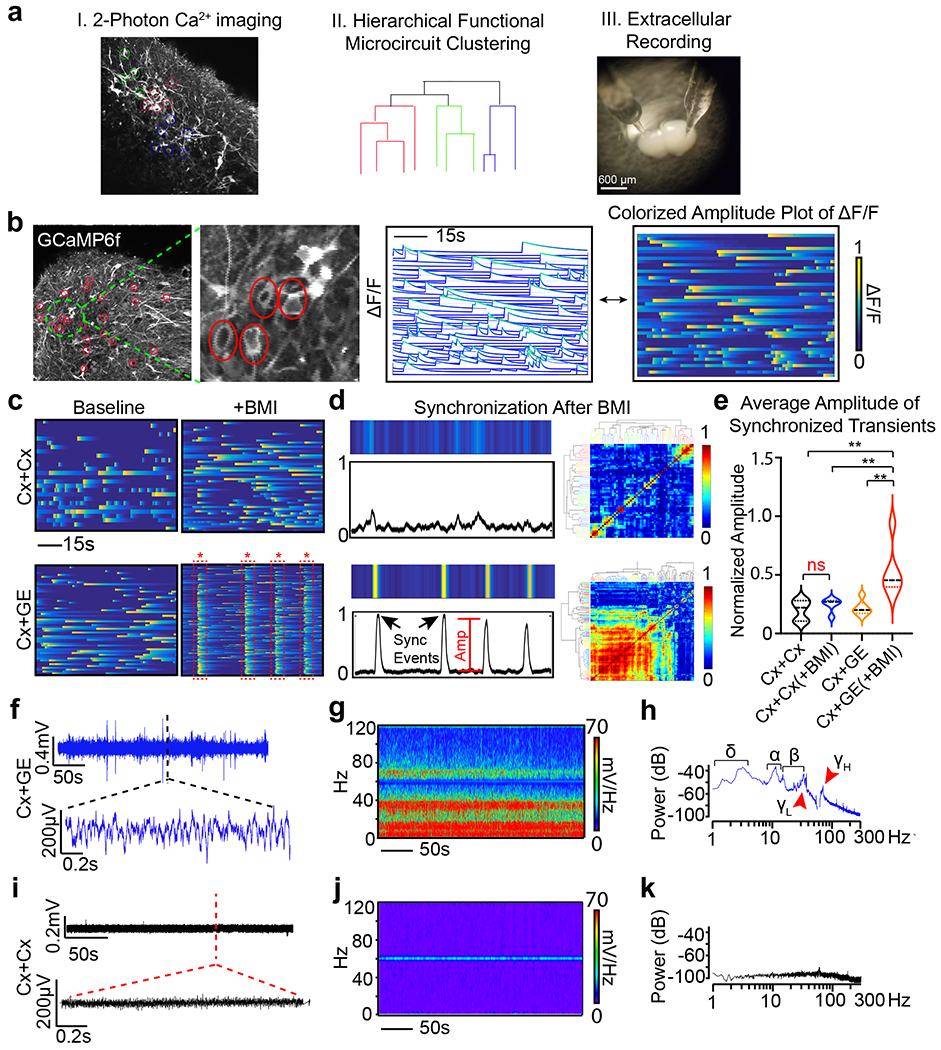
a, Schematic illustrating the identification of active neurons by virtue of their Ca2+ transients (I), representation of their network organization (II), and methods used to collect extracellular recordings (III). b, Example of live 2-photon microscopy imaging of an H9 hESC derived fusion organoid demonstrating acquisition of regions of interest (red circles) and the resulting activity profile shown as normalized change in fluorescence (ΔF/F), where each line is an individual neuron (middle plot) and representation of the same data as a colorized amplitude plot (right). c, Addition of 100 μM bicuculline methiodide (BMI) has a minimal effect on Cx+Cx fusions (top) yet elicits spontaneous synchronization of neural activities in Cx+GE organoids (bottom). d, These synchronizations can be transformed into a normalized amplitude versus time plot for quantitative analyses (left) and further visualized as a clustergram following hierarchical clustering of calcium spiking data (right). e, Pooled data of the amplitude measurements. Plots display the full distribution of individual data points with dotted lines indicating the median and quartile values. n = 3 independent experiments for Cx+Cx and Cx+GE. ANOVA P = 0.0011, F = 8.301, DF (between columns) = 3 followed by Tukey’s multiple comparison; **P = 0.0028 for Cx+Cx vs Cx+GE BMI; **P = 0.0100 for Cx+Cx BMI vs Cx+GE BMI; **P = 0.0031 for Cx+GE vs Cx+GE BMI. f-h, Local field potentials measured from a representative Cx+GE fusion reveal robust oscillatory activities at multiple frequencies during a 5-minute period, reflected in both raw traces f and spectrogram g. Spectral density analysis in h demonstrates the presence of multiple distinct oscillatory peaks ranging from ~1-100 Hz. i-k, Cx+Cx fusion organoids by contrast lack measurable oscillatory activities. Representative traces in (f-h) are taken from 3 independent experiments and in (i-k) from 4 independent experiments.
LFP measurements in untreated fusion organoids uncovered simultaneous sustained oscillations at multiple frequencies from 1-100 Hz in Cx+GE fusions (Figs. 2f–h), a hallmark of mature neural networks in vivo31. No discernible oscillatory activities were seen in Cx+Cx structures (Figs. 2i–k). These findings suggested that the presence of GE-derived inhibitory interneurons stimulates maturation of excitatory cortical networks, as has been shown in the rodent brain32, 33. Moreover, these data indicated that interneurons uniquely entrain the behavior of excitatory cells in Cx+GE fusions, and that the resultant networks could produce complex oscillations resembling those observed by extra- and intracranial recordings of the intact brain.
Generation of Cx+GE fusion organoids to model Rett Syndrome
We next used this platform to measure pathophysiological changes associated with human neurological disease. Rett syndrome is a neurodevelopmental disorder typically caused by de novo mutations in one copy of the MECP2 gene on the X chromosome, where affected females exhibit symptoms including motor delays, cognitive and neuropsychiatric disturbances, autism, and epilepsy as early as seven months of age34. However, cellular defects likely present well before clinical symptoms. For example, a recent hiPSC-based study has suggested that Rett may impact prenatal neurogenesis through microRNA-mediated alterations in AKT and ERK activity35. While neuroanatomical changes in dendritic arborization and spine density have been reported in multiple Rett models34, 36–39, gross structural brain abnormalities are less prevalent.
Due to random X-chromosome inactivation, female Rett patients are typically mosaic in their MECP2 status, with some cells expressing and others lacking a functional MECP2 allele. This feature permitted the isolation of isogenic hiPSC pairs from individual patients as hiPSC reprograming does not typically revert X-chromosome silencing39. We accordingly generated Cx and GE organoids from isogenic control (iCtrl) and MECP2-mutant (Mut) Rett syndrome patient hiPSCs harboring either a 705ΔG mutation which leads to a frameshift truncation after amino acid 236, or a 1461A>G missense mutation which alters the c-terminal end of the MECP2 protein39. The former patient was reported to have a history of electroencephalographic abnormalities and the latter had a documented history of overt seizures. We confirmed that the expected MECP2 positive or negative status of cells was retained following differentiation using immunohistochemistry and found no obvious differences in organoid cytoarchitecture or cell composition across samples (Figs. 3a–b; Extended Data Figs. 3 and 4a).
We next examined the interneuron composition of fusion organoids made from the first patient line (705ΔG mutation) in greater detail. The cortex region of both iCtrl and Mut Cx+GE fusions were indistinguishable with respect to migration of AAV1 CAG:tdTomato labeled GE-derived cells in the cortical compartment and contained similar percentages of GAD65+ interneurons (mean ~25% of all cells) (Figs. 3c–d; Extended Figs. 1a,b; Supplementary Figs. 2a,b). Immunohistochemical analyses revealed the presence of multiple interneuron subtypes in all fusion organoids including cells expressing SOMATOSTATIN (SST), CALRETININ (CALB2), NEUROPEPTIDE Y (NPY), and CALBINDIN (CALB1). The percentage of CALRETININ, CALBINDIN, and NPY-positive groups appeared slightly elevated in the Mut samples while SST was down; however, most changes were modest and did not achieve statistical significance except for CALRETININ (Figs. 3e,f).
Neurogenesis and fate changes in MECP2 mutant Cx+GE organoids
To obtain a more comprehensive profile of the cell composition within the Mut and iCtrl fusion organoids, we performed single-cell RNA sequencing (scRNA-seq) on pre-fusion day 56 Cx and GE organoids as well as day 70 and day 100 Cx+GE fusions. Multiple time points were selected to assess the progression of cells from progenitor to differentiated states. UMAP clustering revealed 9 clusters that expressed cell-type specific markers including ventricular radial glial cells (RGCs), outer radial glia cells (oRGs), excitatory neurons (upper layer callosal projection neurons, CPNs and deep layer corticofugal projection neurons, CFuPNs), and inhibitory interneurons (Figs. 4a,b and Supplementary Table 1). We plotted the expression of canonical markers associated with each of these clusters (e.g., GAD1 with interneurons or SATB2 with excitatory clusters) including many of the markers we had used for immunohistochemical analyses of neuronal subtypes (Fig. 4c and Supplementary Fig. 3). Genes associated with astrocyte progenitors and maturing astrocytes (e.g., GFAP, FGFR3, AQP4, and ALDH1L1 expression) overlapped with the oRG cluster, (Figs. 4a,c and Supplementary Fig. 3) but did not further separate into a distinct astrocyte cluster, reflecting the high degree of similarity in gene expression between these cell types.
Fig. 4 |. Single-cell transcriptomic analysis reveals the presence of diverse cellular populations in fusion organoids with a trend towards accelerated maturation and alterations in interneuron formation in MECP2 mutant samples.
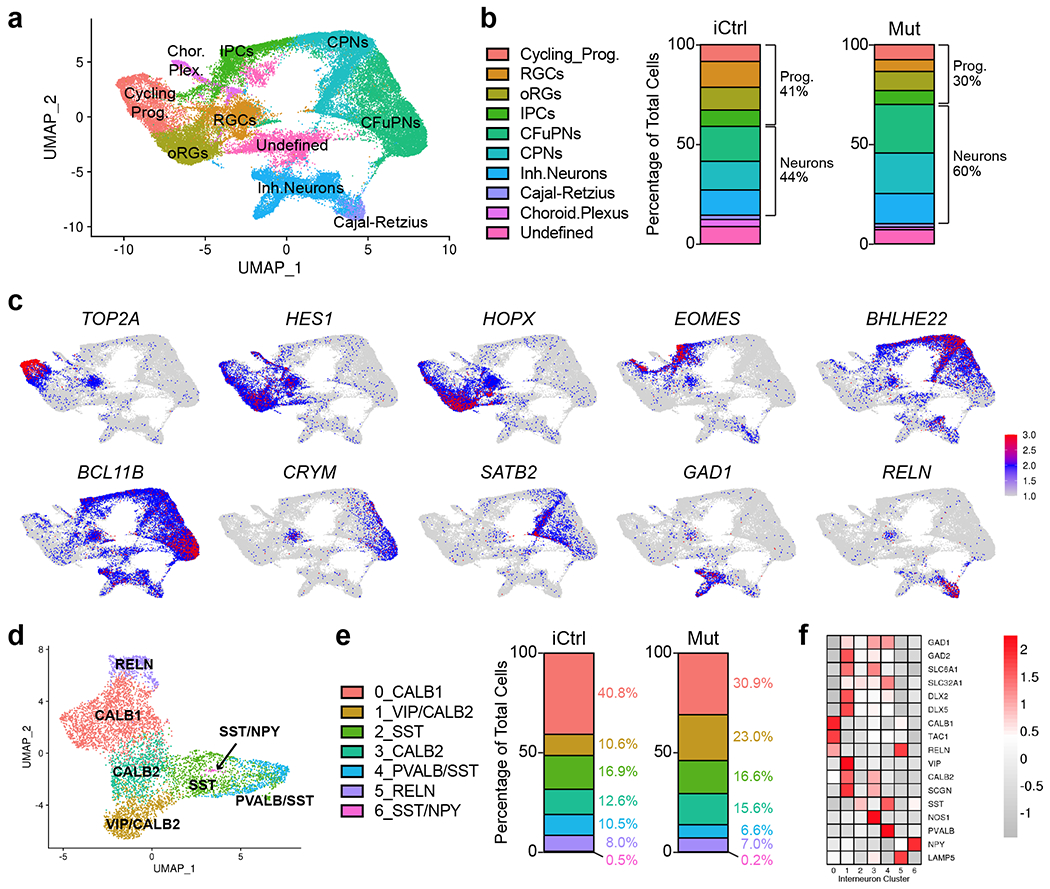
a, Uniform Manifold Approximation and Projection (UMAP) of combined iCtrl and Mut Cx and GE organoids. The plot includes cells from 3 D56 Cx and 3 GE organoids collected before fusion, 3 D70 Cx+GE fusion organoids, and 3 D100 Cx+GE fusion organoids. The total number of cells sequenced were as follows: D56 iCtrl, 9306; D56 Mut, 9186; D70 iCtrl, 10931; D70 Mut, 6260; D100 iCtrl, 7561; and D100 Mut, 6698 cells. b, Plots display the mean percentage of cells in the fusion organoids representing each of the clusters in a. Separation of the data by iCtrl and Mut status shows a trend of reduced progenitors and more differentiated neurons in Mut organoids compared to iCtrl samples. c, UMAPs of key genes associated with each of the major clusters identified in a. d, Re-clustered UMAP of the interneuron subset from a with interneuron subtype markers identifying each re-clustered subset. e, Percentage of cells for each of the clusters in d segregated by iCtrl and Mut reveals increased numbers of interneurons expressing PVALB/SST and CALB1 in iCtrl organoids and cells expressing VIP and CALB2 in Mut samples. f, Heat map with the relative expression of canonical interneuron-related genes within the re-clustered groups.
To identify the effects of MECP2 mutation on gene expression and cell fates, we performed differential expression analysis between all cells in the iCtrl and Mut organoids, and when separated by clusters. We found broadly overlapping gene expression profiles, with a trend towards increased percentage of cells exhibiting progenitor characteristics in iCtrl fusion organoids compared to Mut samples (Fig. 4b). Although both progenitor and mature cell types were seen at all time points, the data showed a shift from more immature cell type profiles in day 56 and 70 organoids, to more mature cell types such as CPNs in the day 100 samples. This shift appeared sooner in the MECP2 mutant samples. (Supplementary Fig. 3a).
Given the importance of interneuron function for neural oscillations in the fusion organoids, we isolated the “Inh.Neuron” population from Fig. 4a to further distinguish interneuron subtypes. We found 7 major subclusters defined by their distinct profile of interneuron-associated genes such as SST, NPY, CALB1, and CALB2, vasoactive intestinal polypeptide (VIP), and parvalbumin (PVALB), and variable expression of additional genes such as DLX2, DLX5, GAD2, SLC6A1, SLC32A1, LAMP5, SCGN, and TAC1 (Figs. 4d–f). While all interneuron groups were present in iCtrl and Mut fusion organoids, there were some modest yet notable differences in cell numbers. iCtrl samples showed an increased percentage of PVALB+/SST+ and CALB1+ neurons while the Mut mutant samples contained more VIP+ and CALB2+ cells (Fig. 4e).
MECP2 mutant organoids show defects in synapse formation
We next performed differential gene expression analyses comparing iCtrl and Mut fusion organoids both in total and within each cell grouping (Supplementary Table 2). Genes upregulated in Mut versus iCtrl (total cells) were strongly enriched for autism risk (34% concordance with the SFARI Human Gene Module) and epilepsy (20% concordance with the DisGeNET Epilepsy set C0014544), and included four genes, MEF2C, GRIA2, SMC1A, and ZBTB18, where mutations have previously been connected to Rett syndrome-like neurodevelopmental phenotypes (Extended Data Figs. 5,6a)40–44. Genes downregulated in Mut versus iCtrl (total cells) were also enriched for epilepsy risk (17% concordance), but not autism (Extended Data Fig. 5).
Genes that were upregulated in MECP2 mutant organoids were notably enriched for gene ontology (GO) terms associated with neuronal projection, morphogenesis, and synaptic assembly, whereas genes that were downregulated were associated with mRNA catabolism, ER targeting, and protein translation (Fig. 5a and Supplementary Table 3). Most of these changes, particularly those associated with synapses, were most prominent in the CFuPN and CPN clusters, but not present in inhibitory neurons (Extended Data Fig. 7). Among the most upregulated genes in Mut organoids were known axonal and synapse-associated genes such as PCLO, ROBO2, EFNB2, and NRXN1 (Extended Data Figs. 5,6a and Supplementary Table 2). However, the most elevated gene in the absence of MECP2 function within all clusters was NNAT (Neuronatin), which has been implicated in the control of ER stress, neuronal excitability, receptor trafficking, and calcium-dependent signaling45, 46. NNAT was upregulated in all clusters but particularly high in the intermediate progenitor and inhibitory interneuron groups (Extended Data Fig. 6a).
Fig. 5 |. Gene ontology and synaptic staining analyses reveal defects in the balance of excitatory and inhibitory synapses in MECP2 mutant fusion organoids.
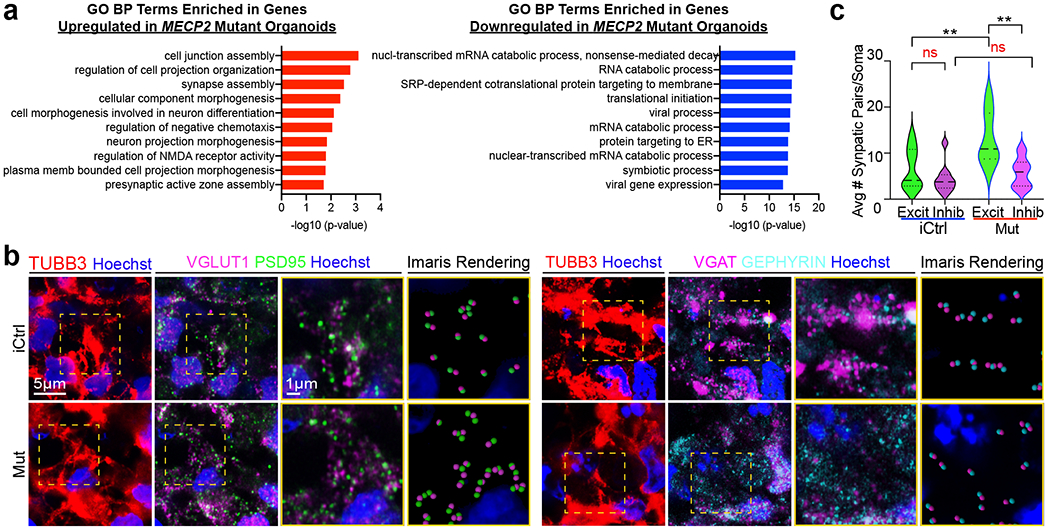
a, Top 10 most enriched Gene Ontology biological process (GO BP) terms associated with upregulated or downregulated differentially expressed genes when comparing Mut and iCtrl across all cells. b, Immunohistochemical analysis of excitatory (VGLUT1/PSD) and inhibitory (VGAT/GEPHYRIN) pre-/post-synaptic puncta reveals an increase in excitatory synapses in Mut Cx+GE fusion organoids. The yellow dotted boxes in the right most panels display representative TUBB3+ regions that were used for analyses. The adjacent two panels demonstrate the raw immunohistochemical image followed by Imaris software renderings of the colocalized pre- and post-synaptic markers. The final two panels are magnified versions of the boxed areas. c, Plots of the number of synapses per cell. Data were pooled from multiple organoids. VGLUT1/PSD95 (Excit), n = 3 organoids for both iCtrl Mut samples, 1180 cells; VGAT/GEPHYRIN (Inhibit), n = 4 organoids for iCtrl and Mut samples, 1654 cells. ANOVA P = 0.0002, F = 8.387, DF (between columns) = 3, followed by Tukey’s multiple comparison; **P = 0.0088 for Excit iCtrl vs Excit Mut; **P = 0.0014 for Excit Mut vs Inhib Mut; ns = not significant. Plots display the full distribution of individual data points with dotted lines indicating the median and quartile values.
Since we used kainate to stimulate neural oscillations in the fusion organoids, we surveyed the expression of kainate receptors between iCtrl and Mut fusion samples. While the predominant receptor GRIK5 as well as GRIK1, GRIK2, and GRIK4 were all comparably expressed, we did find a small upregulation of GRIK3 selectively in the CFuPN cluster within Mut organoids (Extended Data Fig. 6b). The significance of this elevation is unclear but may further predispose the Mut organoids to network hyperexcitability.
We lastly examined the status of synapse formation in the cortical compartment of iCtrl and Mut Cx+GE fusion organoids using VGLUT1/PSD95 costaining to distinguish pre-and post-synaptic components of excitatory synapses and VGAT/gephyrin costaining for inhibitory synapses. Consistent with the transcriptomic results, we found a significant increase in the density of excitatory puncta in Mut organoids compared to iCtrl, without any notable change in inhibitory synapses (Fig. 5b,c). Collectively, these results suggest that MECP2 deficiency results in changes in gene expression that ultimately alter the balance of excitatory vs. inhibitory synapses and may thereby impact neuronal network functions.
Hyperexcitability and hypersynchrony in MECP2 mutant organoids
Consistent with these synaptic data and even more striking, were activity differences revealed though GCaMP6f imaging. Mut Cx+GE fusions exhibited epochs of spontaneously synchronized calcium transients (Figs. 6a–c and Supplementary Videos 8,9) reminiscent of the synchronizations observed following administration of GABAA receptor antagonists to control samples (Figs. 2c–e and Supplementary Videos 4,5) and the epileptiform changes seen in murine Rett syndrome models47. These results were notably consistent across different batches of organoids tested and two independent Mut cell lines generated from the same Rett patient (Extended Data Figs. 1b,d). Previous reports have indicated increased activity and diminished size of neural microcircuits in mouse chemoconvulsant epilepsy models48. We similarly observed that increased synchronization of calcium transients in Mut Cx+GE organoids was accompanied by reductions in both the size of microcircuit clusters and the number of neurons within each cluster (Extended Data Fig. 8a).
Fig. 6 |. Rett syndrome fusion organoids display GE dependent hypersynchronous neural network activity.
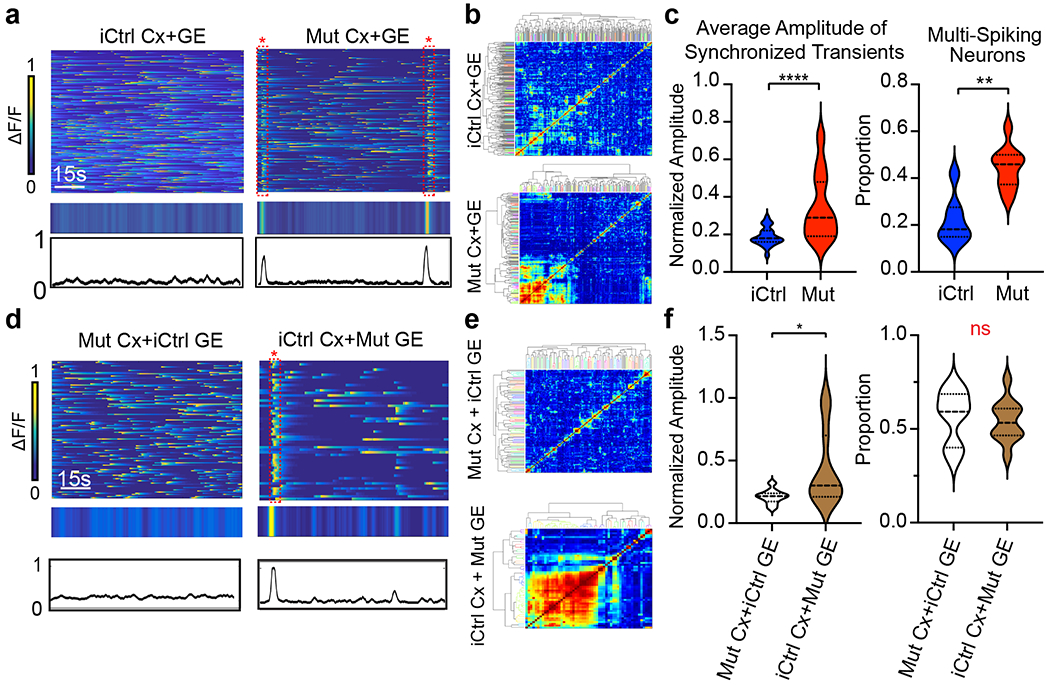
a, Mut Cx+GE fusions exhibit spontaneous synchronized Ca2+ transients that are not seen in iCtrl Cx+GE, reflected in the raw ΔF/F colorized amplitude plot (top), synchronization amplitude plot (bottom), and clustergram b. c, Pooled data quantifications, n = 12 independently generated iCtrl and n = 7 independently generated Mut fusion organoids, ****P < 0.0001 for the average amplitude of synchronized transients; **P = 0.0012 for multi-spiking neurons, significance assessed by two-sided Mann-Whitney U. d, Mixed fusions with iCtrl Cx and Mut GE exhibit spontaneously synchronized calcium transients, whereas as mixed fusions with Mut Cx and iCtrl GE do not, as seen in the raw ΔF/F colorized amplitude plot (top), synchronization amplitude plot (middle), and clustergram e. f, Pooled data quantifications, n = 10 independently generated iCtrl Cx+Mut GE and n = 11 Mut Cx+iCtrl GE, *P =0.0308, DF = 19, t = 2.334, two tailed student’s t-test. Plots in c,f display the full distribution of individual data points with dotted lines to indicate the median and quartile values.
To extend these results further, we generated Cx+GE fusion organoids from the second group of Rett patient hiPSC lines (hereafter referred to as iCtrl-II and Mut-II; two independent lines of Mut-II were utilized). GCaMP6f imaging revealed decorrelated activity in iCtrl-II organoids reminiscent of the results from prior H9 hESC and iCtrl-I hiPSC Cx+GE experiments (Extended Data Figs. 4b, top traces; compare to Figs. 2c and 6a). By contrast, Mut-II organoids exhibited multiple instances of individual neurons firing at a rapid and persistent rate (Extended Data Fig. 4b, bottom red boxed regions; see also Supplementary Videos 10,11), though we did not observe hypersynchronous bursts as seen in Mut-I organoids (Extended Data Figs. 4b,c, compare to Figs. 6a,b). The rapid GCaMP6f activity in Mut-II organoids resulted in a significant decrease in the mean and median interspike interval and, as was the case with Mut-I organoids, an increase in the proportion of multi-spiking neurons (Extended Data Fig. 4d). Consonant with the lack of visible hypersynchronicity, there was no effect on the amplitude of synchronized transients (Extended Data Fig. 4d). These results were consistent with respect to organoid batches and cell lines used within the iCtrl-II vs. Mut-II groupings, yet significantly different between genotypes (Extended Data Figs. 1b,c). Collectively, these data demonstrate a high degree of consistency in aspects of the neural network dysfunction phenotype in MECP2 Mut organoids associated with hyperexcitability, but also demonstrate distinct characteristics based on their patient origins.
MECP2 mutant organoids exhibit aberrant neural oscillations
LFP recordings of iCtrl-I Cx+GE fusions demonstrated infrequent spikes and sustained low frequency and gamma oscillations with few epochs of higher frequency oscillations (> 100 Hz), like the profile exhibited by H9 hESC-derived fusion organoids (Figs. 7a–e, compare to Figs. 2f–h). By contrast, Mut-I Cx+GE organoids lacked low frequency and gamma oscillations (Figs. 7a–c “Mut”) and instead exhibited recurring epileptiform-appearing spikes and high frequency oscillations (HFOs, ~200-500 Hz; Figs. 7a–g, and Extended Data Fig. 9). These findings concurred with calcium indicator data which showed rare but large high amplitude calcium synchronizations or high frequency firing in Mut fusion organoids that could result in spikes or HFOs, as opposed to the numerous small synchronizations seen in iCtrl samples which likely generate sustained lower frequency oscillations without epileptiform events (Fig. 6). Hypersynchrony, HFOs, and spikes seen in Mut Cx-GE organoids are all consistent with the electrographic changes observed in human epilepsy49, 50. Indeed, electroencephalographic abnormalities or overt epilepsy were documented in the Rett patients whose hiPSCs were used in this study39.
Fig. 7 |. Rett syndrome fusion organoids display GE-dependent epileptiform changes.
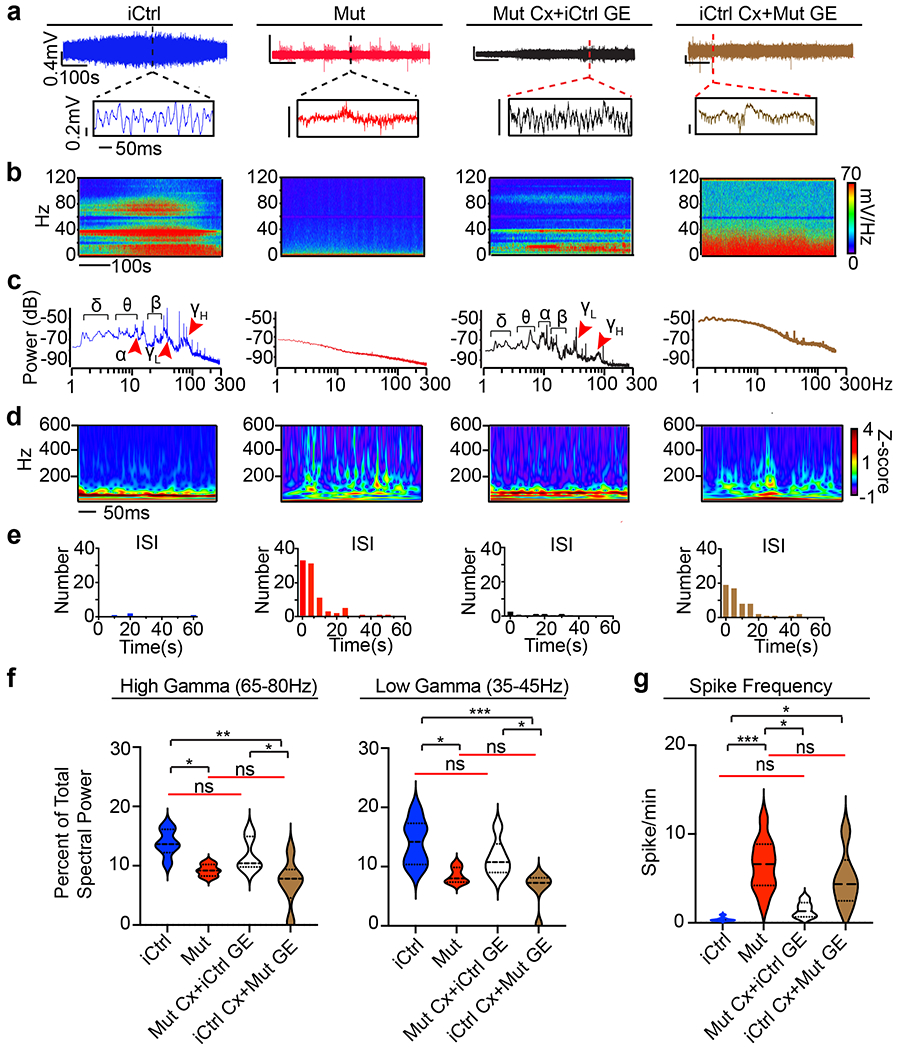
a, Raw trace of a representative 10-minute LFP recording (top) and time expanded window (bottom) from unmixed Mut or iCtrl Cx+GE fusion organoids and Mut/iCtrl mixed Cx+GE fusions. b,c, Spectrograms and periodograms derived from the entire recordings shown in a. d, Morlet plot showing high frequency activity associated with the time expanded segments shown in a. e, Frequency histogram of interspike intervals derived from the raw trace in a. f, Quantification of high and low gamma spectral power from LFP recordings demonstrates a significant decrease of gamma power in Mut Cx+GE fusions and mixed fusions with a Mut GE. High gamma; Ordinary ANOVA, overall P = 0.0020, F = 7.089, DF (between columns) = 3, Tukey’s multiple comparisons, **P = 0.0018, *P = 0.0353 iCtrl vs Mut and *P = 0.0345 Mut Cx + iCtrl GE vs iCtrl Cx + Mut GE. Low gamma; Ordinary ANOVA, overall P = 0.0174, F = 8.038, DF (between columns) = 3, Tukey’s multiple comparisons, ***P = 0.0009, *P = 0.0174 iCtrl vs Mut, *P = 0.0309 Mut Cx + iCtrl GE vs iCtrl Cx + Mut GE. g, Spike frequency across multiple independent experiments Kruskal-Wallis test, overall P = 0.0002, Dunn’s multiple comparisons ***P = 0.002, *P = 0.0159 iCtrl vs iCtrl GE+ Mut Cx and *P = 0.0416 Mut vs Mut Cx + iCtrl GE. n = 6 independently generated organoids for each condition (iCtrl and Mut) in f and g. Plots in f,g display the full distribution of individual data points with dotted lines to indicate the median and quartile values.
We also performed LFP recordings on Cx+GE organoids produced from the second Rett patient iPSC lines. iCtrl-II organoids displayed infrequent spikes and sustained low frequency and gamma oscillations with few epochs of higher frequency oscillations (> 100 Hz) like iCtrl-I samples (Extended Data Figs. 10a–c “iCtrl-II” compare to Figs. 2f–h and 7a–e). By contrast, Mut-II Cx+GE organoids lacked low frequency and gamma oscillations and instead exhibited recurring epileptiform-appearing spikes and high frequency oscillations (Extended Data Figs. 10a–c “Mut-II”). Quantification of these data revealed a significant loss of low gamma power in Mut-II as compared to iCtrl-II, a substantial but non-significant loss of high gamma power in Mut-II relative to iCtrl-II, and a significant increase in spike frequency in Mut-II (Extended Data Figs. 10d,e). We again observed consistency in these measurements across organoid batches, independent cell lines from the same patient, and within genotypes, with marked differences across genotypes (Extended Data Figs. 1b,d).
MECP2 deficiency in interneurons drives neural network dysfunction
Epilepsy is present in 60-80% of Rett patients and thought to arise primarily from interneuron dysfunction51, 52, though MECP2 loss in other cell types has also been implicated22. As our Cx and GE organoids are respectively enriched in excitatory vs. inhibitory interneurons, we generated “mixed” fusions in which either the Cx or GE half of the fused structure was Mut-I while the other half was iCtrl-I, as a means of determining the compartment and cell type in which MECP2 deficiency matters most. Mut-I Cx+iCtrl-I GE mixed-fusion organoids displayed calcium transients nearly identical to unmixed iCtrl Cx+GE fusions and did not show any evidence of hypersynchrony. By contrast, iCtrl-I Cx+Mut -I GE organoids exhibited hypersynchronous calcium transients like unmixed Mut-I Cx+GE fusions, although an increase in the proportion of multispiking neurons was not detected (Figs. 6d–f and Supplementary Videos 12,13).
We likewise found that Mut-I Cx+iCtrl-I GE organoids displayed an LFP profile nearly identical to unmixed iCtrl-I Cx+GE fusions (Figs. 7a–e and Extended Data Fig. 9). By contrast, iCtrl-I Cx+Mut-I GE organoids demonstrated frequent spikes and HFOs along with deficits in distinct lower frequency oscillations and gamma activity, like unmixed Mut-I Cx+GE fusions (Fig. 7a–e and Extended Data Fig. 9). Together, these data suggest that MECP2 deficiency in GE-derived interneurons is the primary driver of the neural network dysfunction within the fusion organoids.
Among these observations, changes in spike frequency and gamma oscillations may be of particular relevance as both human and murine in vivo studies have shown an inverse relationship between gamma band power and epileptiform discharges14, 15. In addition, gamma oscillations are thought to require complex inhibitory-excitatory network interactions that are highly prone to disruption by epileptic or interictal discharges14. Strong gamma peaks were consistently present in in unmixed iCtrl-I Cx+GE and mixed Mut-I Cx+iCtrl GE organoids, and absent or reduced in amplitude in Mut-I Cx+GE and iCtrl-I Cx+Mut GE structures. Consistent with these conclusions, quantification of gamma spectral power revealed a significant and comparable loss of both low and high gamma spectral power in the unmixed Mut-I and mixed iCtrl-I Cx+Mut GE organoids as well as significantly increased spike frequency in these groups relative to unmixed iCtrl-I Cx+GE samples (Fig. 7f,g).
Neural oscillation defects can be restored by Pifithrin-α
A heralded, but largely unrealized, application of patient-derived hiPSCs is in personalized medicine. We therefore sought to determine the utility of the epileptiform phenotype observed in Rett syndrome organoids for drug testing. First, we used a broad-spectrum anti-seizure medication, sodium valproate (VPA), commonly used to treat epilepsy resulting from Rett Syndrome53. We also tested a putative TP53 target inhibitor, Pifithrin-α, based on previous studies showing that MECP2 deficiency leads to over-activation of the TP53 pathway and premature neuronal senescence39, 54. Consistent with its known spike suppressant properties, exposure to VPA for 48 h significantly reduced spiking activity in the Mut-I Cx+GE organoids (Figs. 8a,c), although it did not reduce HFOs and had a modest effect restoring lower frequency oscillations (Figs. 8a–c). Pifithrin-α similarly reduced spike frequency but remarkably also suppressed HFOs and resulted in the re-emergence of gamma oscillations (Fig. 8a–c). A nearly identical response was also seen with Mut-II Cx+GE fusion organoids (Extended Data Fig. 10a–e). We combined all LFP experiments and plotted spike rate vs. gamma spectral power for both low gamma and high gamma and observed the expected inverse relationship for both patient lines (Figs. 7f,g and Extended Data Fig. 10f). This inverse relationship is consistent with what has previously been described in both mouse models of neurological disease and human epilepsy patients14, 15.
Fig. 8 |. Partial restoration of gamma oscillations and suppression of HFOs in Rett syndrome fusion organoids by administration of Pifithrin-α.
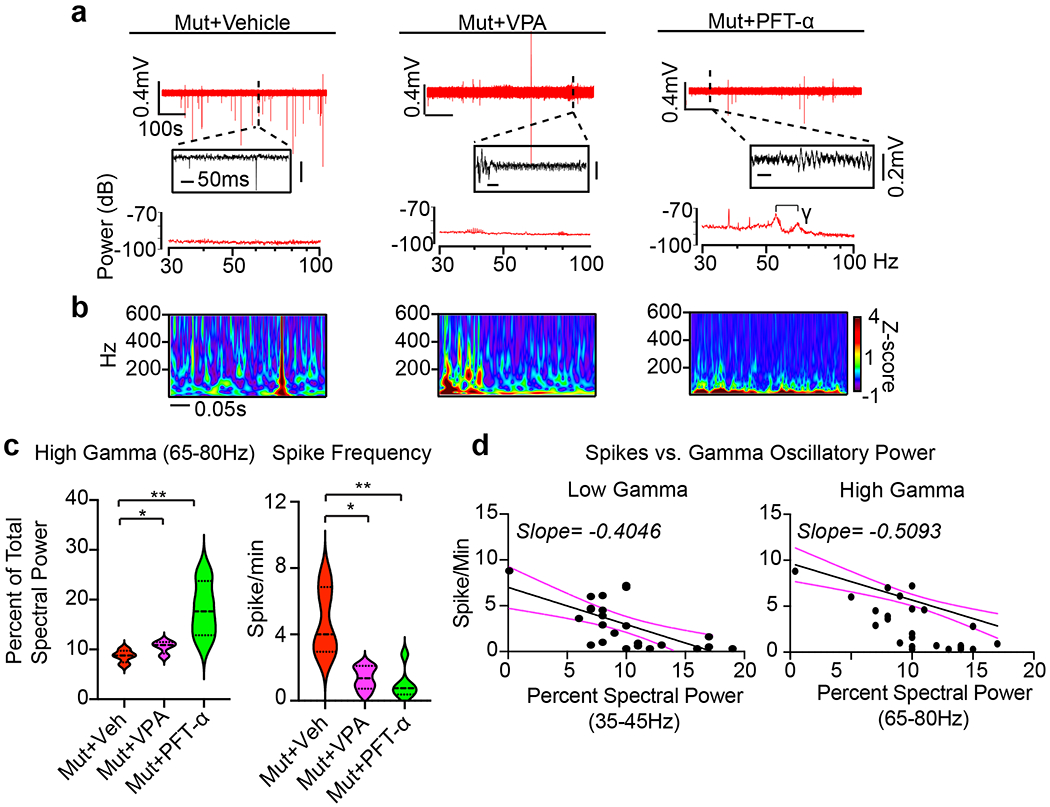
a, Raw trace (top), time expanded window (middle), and periodogram (bottom) from representative Mut Cx+GE fusion organoids treated for 48 h with vehicle (DMSO, Veh), 2 mM sodium valproate (VPA), or 10 μM Pifithrin-α (PFT). b, Morlet plot derived from the time expanded segment in a. c, Quantification of high gamma oscillations and spike frequency in Mut Cx+GE shows a highly significant rescue of both high gamma spectral power and a reduction in spike frequency following treatment with PFT and more modest, but significant, rescue in both measures following VPA treatment. High gamma quantification; Ordinary ANOVA, overall P = 0.0085, F = 8.476, DF (between columns) = 2, Tukey’s Multiple comparisons, **P = 0.0093, *P = 0.0299, n = 4 independently generated organoids for each of the 3 conditions (Veh, VPA, and PFT). Spike Frequency following drug addition; Kruskal-Wallis test, overall P = 0.0020, Dunn’s multiple comparisons **P = 0.0042, *P = 0.0420. Plot displays the full distribution of individual data points with dotted lines to indicate the median and quartile values. d, Plots of high and low gamma spectral power versus spike frequency demonstrates an inverse relationship between gamma power and spiking. The solid black line is the best fit following simple linear regression, and the dotted magenta lines indicate 95% confidence intervals for the estimated line of best fit. The slope of the line of best fit is indicated at the top of the graph. The calculated slope is significantly different from zero with P < 0.0001 for high gamma and P = 0.0007 for low gamma,
To determine if drug effects were associated with changes in cell death, activating cleavage of caspase3 was assessed by immunohistochemistry but did not reveal any significant differences (Supplementary Fig. 4). We also did not observe any specific effects of Pifithrin-α on iCtrl-I Cx+GE fusion LFP activity or cell death (Supplementary Fig. 5). Together, these results suggest that while VPA largely reduced neuronal hyperexcitability, Pifithrin-α may additionally modulate more upstream excitatory-inhibitory physiologic interactions resulting in a more global restoration of network-level functions. These findings further illustrate the potential value of the fusion organoid modeling approach in personalized drug discovery.
DISCUSSION
Collectively, these experiments demonstrate the existence of highly sophisticated physiological activities within Cx+GE organoids, congruent with their cytoarchitectural and cellular complexity. These results stand in agreement with findings recently reported by Trujillo et al, who have also observed the emergence of neural oscillations in cortical organoids cultured for prolonged time periods using a different organoid protocol and recording approach11. Our studies further demonstrate that the emergence of higher order network activities such as multi-frequency oscillations requires functional integration of inhibitory interneurons into the excitatory network framework as permitted by the organoid fusion technique, since no oscillations were apparent without them. Critically, this approach also allowed us to identify striking electrophysiological phenotypes in MECP2 mutant Cx+GE organoids despite their cytoarchitectural similarity to iCtrl samples. In addition, the use of two Rett patient derived hiPSCs, each harboring unique MECP2 mutations, allowed us to more confidently identify the physiological signatures of neural network dysfunction that arise in Rett syndrome brain organoids. There appeared to be a high degree of reproducibility and consistency in the results across multiple experiments, organoid batches, cell lines, and modes of analysis, further confirming the validity of the fusion organoid model.
Although we did not observe overt differences between Mut and iCtrl fusion organoids by immunohistochemical analyses or scRNAseq with respect to major cell types, we did see trends that may contribute to the marked physiologic phenotypes we have documented. Perhaps most notably, and consistent with our finding that the interneuron-enriched GE compartment primarily accounts for the physiologic changes in Mut fusion organoids, was a relative enrichment of PVALB interneurons in iCtrl and VIP interneurons in Mut samples. Together with SST interneurons, these two subtypes provide crucial inhibitory input to pyramidal (excitatory) cells that, in turn, mediate neural oscillations55. Specifically, PVALB neuron activity is thought to mediate 20-80 Hz oscillations including gamma activity, whereas SST interneurons, which are directly inhibited by VIP, mediate oscillations in the 5-30 Hz frequency range55, 56. The consistent loss of lower frequency oscillations in Mut fusion organoids may arise from VIP mediated reductions in SST activity, whereas reductions in PVALB cells may contribute to the loss of gamma oscillations. Significant reductions in PVALB+ interneurons have been seen in the prefrontal cortex of autistic individuals57.
Of note, and consistent with our LFP findings, we also observed a relative decrease in SST expression in Mut organoids by immunohistochemistry, although this was not observed in our scRNAseq data. We also saw a modest increase in excitatory but not inhibitory synaptic puncta in Mut organoids, likely related to the transcriptomic changes revealed by our single-cell sequencing analysis. Genes that were differentially expressed in MECP2 mutant organoids showed strong associations with axonal growth, synapse formation, autism risk, and epilepsy. The relative increase in excitatory input may further contribute to disruptions in neural oscillations and predispose the organoids towards hyperexcitability.
In addition to our mechanistic studies, we sought to exploit our system as a novel platform for drug testing. We were intrigued to discover a more complete rescue of the Mut phenotype with a novel drug, Pifithrin-α, than was seen with a traditional anti-seizure medication, VPA. Pifithrin-α is a TP53 inhibitor shown previously to counteract cellular stress and senescence responses seen in MECP2 deficient neurons in vitro39. These data merit future investigations into both the neuroregulatory actions of Pifithrin-α as well as the role of MECP2 in the repression of TP53-mediated senescence pathways.
In summary, these findings illustrate the potential of brain organoids both as a unique platform for characterizing human neural networks and for personalized drug discovery and research. A remaining challenge is to delineate the precise microcircuit and cell type-specific perturbations that underlie both the oscillatory and pathological epileptiform-like changes revealed in these studies. Important clues in the pursuit of this endeavor have already been revealed by the transcriptomic analyses we have completed. The fusion organoid system that we employed is highly amenable to such detailed cellular and circuit analyses and provides unprecedented opportunities for modeling neural network dysfunction associated with a variety of human neurological disorders.
METHODS
hESC and hiPSC culture and organoid generation.
All hPSC experiments were conducted following prior approval from the University of California Los Angeles (UCLA) Embryonic Stem Cell Research Oversight Committee (ESCRO) and Institutional Review Board. Cortex (Cx) and ganglionic eminence (GE) organoids were generated from the H9 hESC line58 or Rett hiPSCs39 as described previously17 and outlined in schematic form in Fig 1a. Fusion was performed with minor modifications as previously reported19. Cx and GE Organoids were cut at day 56 and two halves (e.g. Cx+GE or Cx+Cx) were combined in a microcentrifuge tube containing 300 μl of N2B27 media17 and placed in a hyperoxic incubator containing 5% CO2 and 40% O2 for 72 hours. Fused structures were then carefully transferred to 24-well oxygen permeable dishes (Lumox, Sarstedt) and maintained in a hyperoxic environment with media changes every other day until their use. Neuron migration experiments were conducted by infection of either a Cx or GE organoid with 5 μl of ~1.98×1013 ml−1 AAV1-tdTomato (pENN.AAV.CAG.tdTomato.WPRE.SV40, a gift of Dr. James M. Wilson, University of Pennsylvania Vector Core AV-1-PV3365) on day 56 and fusion was performed as described 3 days after infection.
Generation of Rett hiPSCs.
Rett iPSCs were derived from fibroblast lines GM07982 and GM17567 obtained from the National Institute of General Medical Sciences Cell Repository maintained at the Coriell Institute for Medical Research, and generated by lentiviral transduction of the cells with the Yamanaka factors (Oct4, Klf4, Sox2, and cMyc) as previously described39. GM07982 cells were isolated from a 25-year-old female noted to have electroencephalographic abnormalities, and found to contain a frameshift mutation, 705delG, in the MECP2 gene resulting in a premature stop codon and protein truncation (E235fs). GM17567 cells were isolated from a 5-year-old female with a history of significantly abnormal electroencephalograms and seizures and found to harbor an A>G missense mutation at nucleotide 1461 (1461A>G), resulting in a substitution of a tryptophan in place of the stop codon at codon 487 (X487W). As Rett females are typically heterozygous for the MECP2 mutation, the collected fibroblasts are mosaic in their MECP2 protein status with approximately half of the cells expressing the non-mutant allele. Unlike murine cells, the inactive X chromosome in human cells remains inactive after reprograming to pluripotency59, allowing the generation MECP2 mutant (Mut) and isogenic control (iCtrl) hiPSCs from the same patient fibroblasts. Confirmation of MECP2 control or mutant status was achieved though immunostaining and immunoblotting analyses of the iPSC lines and differentiated organoids.
Immunohistochemistry.
Organoids were immersion fixed in 4% paraformaldehyde, cryoprotected in 30% sucrose, frozen in Tissue-Tek Optimal Cutting Temperature (O.C.T., Sakura) media, and cryosectioned. Immunostaining was performed using previously published laboratory protocols17, 60. Primary antibodies used include the following: goat anti-BRN2 (POU3F2; Santa Cruz Biotechnology sc-6029), 1:4000; mouse anti-CALBINDIN (Clone CB-955, Sigma-Aldrich C9848), 1:5000; rabbit anti-CALRETININ (EMD Millipore AB5054), 1:2000; 1:500; rabbit anti-Cleaved Caspase-3 (Asp175) (Cell Signaling 9661), rat anti-CTIP2 (BCL11B; Abcam ab18465), 1:1000; rabbit anti-DLX161 (generous gift of Drs. Soo Kyung Lee and Jae Lee), 1:3000; guinea pig anti-DLX262 (generous gift of Drs. Kazuaki Yoshikawa and Hideo Shinagawa), 1:3000; guinea pig anti-DLX562 (generous gift of Drs. Kazuaki Yoshikawa and Hideo Shinagawa), 1:3000; rabbit anti-FOXG1 (Abcam ab18259), 1:1000; rabbit anti-GABA (Sigma-Aldrich A2052), 1:10000; mouse anti-GAD65 (BD Biosciences 559931), 1:200; mouse anti-GEPHYRIN (Synaptic Systems 147021), 1:500; goat anti-LHX2 (C-20, Santa Cruz Biotechnology sc-19344), 1:1000; rabbit anti-MECP2 (Diagenode C15410052), 1:1000; mouse anti-N-CADHERIN (CDH2, BD Biosciences 610920), 1:800; rabbit anti-Neuropeptide Y (EMD Millipore, AB10980),1:1000; mouse anti-NKX2.1 (Novocastra NCL-L-TTR-1), 1:500; mouse anti-PAX6 (Developmental Studies Hybridoma Bank), 1:100; rabbit anti-PAX6 (MBL International PD022), 1:1000; mouse anti-PSD95 (Millipore MAB1598), 1:1000; mouse anti-SATB2 (Abcam ab51502), 1:100; goat anti-SOX2 (Santa Cruz Biotechnology sc-17320, 1:100; rat ant-SOMATOSTATIN (SST, EMD Millipore MAB354), 1:100; rabbit anti-TBR1 (Abcam ab31940), 1:2000; chicken anti-TBR2 (EOMES; EMD Millipore AB15894), 1:1000; rabbit anti-tubulin B3 (TUBB3, BioLegend, 802001), 1:1000; guinea pig anti-VGAT (Synaptic Systems 131004), 1:1000; guinea pig anti-VGLUT1 (SLC17A7; EMD Millipore AB5905), 1:1000. Secondary antibody staining was conducted using Dylight 405-, FITC-, Alexa 488-, Cy3-, Alexa 594-, Cy5-, Alexa 647, Dylight 647-conjugated donkey anti-species-specific IgG or IgM antibodies (Jackson ImmunoResearch or Invitrogen/Molecular Probes) at a 1:1000 dilution. Nuclei were often counterstained using Hoechst 33258 added to the secondary antibody mix at a final concentration of 1 μg ml−1. Single plane and tiled composite images were obtained using 10X or 20X objectives on a Zeiss LSM 800 (running Zen 2.3 Blue edition software) or LSM 700 (running Zen 2011 Black edition software) confocal microscope except for synaptic puncta, which were imaged using a 63X objective on a Zeiss LSM 880 confocal microscope equipped with Airyscan technology (running Zen 2.3 Black edition software). All images that were directly compared were obtained with identical laser power settings. Image adjustments were limited to brightness, contrast, and level and were applied equally to all images in a set being compared. ImageJ/Fiji (versions 1 and 263 and Adobe Photoshop (2019, 2020, and 2021) software was used for image adjustments. Composite figures were assembled using Adobe Illustrator (2019, 2020, and 2021).
Sample Preparation for Single-cell Sequencing.
Papain dissociation reagents were prepared according to manufacturer recommendations for the Papain Dissociation System (Worthington, #LK003150), with a slight modification. Papain was resuspended in 5 ml Hibernate E medium (Brainbits, #HE) containing N2 and B27 supplements (Life Technologies, #17502048 & #12587010) (HNB) to yield a final concentration of 20U Papain/ml, to negate the need for 95% O2/5% CO2 equilibration. DNAse was resuspended in EBSS as recommended and mixed gently to avoid shearing before being added to the papain solution. The final papain/DNAse solution was then incubated at 37°C for 10 minutes prior to use to ensure complete solubilization. Three unfused Cx and GE (for day 56) or 3 fused organoids (for day 70 and 100) were combined into a single tube for each dissociation. To dissociate, organoids were washed twice with PBS (Fisher Scientific, #SH3002802) in a 1.5 ml microcentrifuge tube before being transferred to a 10 cm dish containing fresh PBS. Organoids were gently diced into small chunks using a single edge razor blade (Fisher Scientific, #12-640), and then transferred to a 15 ml conical tube and pelleted to remove the PBS. Organoid chunks were subsequently resuspended in 2 ml of papain/DNAse solution at a final concentration of 20U/ml. Organoids were incubated at 37°C with constant agitation for 30 minutes. After 30 minutes, the organoids were manually triturated 5 times using a 5 ml pipette to break up clumps, then placed at 37°C for a further 15 minutes. After 15 minutes, organoids were very gently titrated 10 times with a P1000 tip and placed for a further 15 minutes at 37°C. In total, organoids were incubated in papain for 1h to obtain a single-cell solution. The suspension was then filtered through a 40 μM strainer (Fished Scientific, #08-771-1) into a fresh 15 ml conical tube and centrifuged at 300 x g for 10 minutes. The papain/DNAse solution was removed and cells resuspended in HNB lacking papain/DNase and centrifuged again. This process was repeated once more to completely remove papain and most cell debris. Finally, cells were resuspended in 1 ml of PBS containing 0.04% bovine serum albumin and counted using Trypan blue staining and a Countess II Automated Cell Counter (Thermo Fisher, #AMQAX1000). The resultant cell solution used for single-cell sequencing contained >90% live cells and was adjusted to a cell concentration of 1000 cells/μl before loading onto the 10X Genomics chip.
Single-Cell RNAseq data processing.
FASTQ files for each sample were processed using the cellranger 4.0.0 pipeline, and counts were generated with function ‘cellranger count’ with the provided annotation refdata-gex-GRCh38-2020-A (10X Genomics). The data were combined into a Seurat object containing the six samples using Seurat v3.2.064, 65. The data were filtered for cells with nFeatures_RNA > 500, nFeatures_RNA < mean+3*(standard deviation), and percent mitochondria genes < 10%. Data integration and batch correction was performed using the R package Linked Inference of Genomic Experimental Relationships (LIGER)66 . The data were normalized with the default parameters using Seurat functions NormalizeData() and FindVariableFeatures(). Data were then scaled using ScaleData(datExpr, split.by = “orig.ident”, do.center = FALSE). Next, data integration/batch correction was performed with the Seurat-wrapper functions for LIGER including RunOptimizeALS(datExpr, k = 20, lambda = 5, split.by = “orig.ident”), and RunQuantileNorm(datExpr, split.by = “orig.ident”) were performed. The data were then clustered using FindNeighbors(datExpr, reduction = “iNMF”, dims = 1:20) and FindClusters(datExpr, resolution = 0.3). UMAP visualization was performed using RunUMAP(datExpr, dims = 1:ncol(datExpr[[“iNMF”]]), reduction = “iNMF”). Cluster marker genes were determined using differential expression between each cluster and the other clusters using the function FindAllMarkers(object = datExpr). Cluster assignments were manually performed referencing the calculated marker genes and common cell type marker genes from literature sources67–72. To estimate the uncertainty in cluster assignments, bootstrapped confidence intervals for cell-type proportions were generated using the R package single-cell differential composition (scDC) with the function scDC_noClustering(cellTypes, subject, calCI = TRUE, calCI_method = “percentile”), where cellTypes were the cluster assignments and subject were the cell genotypes73. Differential expression between Mut and iCtrl samples overall and within each cluster was determined using FindMarkers(datExpr, ident.1=‘Cluster#_Rett’, ident.2=‘Cluster#_Ctrl’) and then filtered for genes with a false discovery rate (FDR) < 0.05. Up and downregulated genes that passed FDR correction were ordered by fold change and gene ontology enrichment analysis was performed using the gost() function in gprofiler2_0.2.0 with the following parameters. Background genes were restricted to genes expressed in the dataset using custom_bg = background_genes, organism = “hsapiens”, ordered_query = TRUE, user_threshold = 0.05, correction_method = “bonferroni”, and sources = c(“GO:BP”,”GO:MF”,”GO:CC”,”HP”)74, 75 . For representative GO plots (e.g., Fig. 5a and Extended Data Fig. 7) term size was restricted to 1000 and the top 10 terms by -log10(p-value) were plotted with exclusion of successive terms containing identical evidence codes. For gene list enrichment shown in Extended Data Fig. 5, the ASD associated gene list was downloaded from https://gene.sfari.org/database/human-gene/ and Epilepsy list (C0014544) from https://www.disgenet.org/search. The gene lists were reduced to genes expressed in the single-cell organoid dataset and compared for overlap with the up/downregulated genes between Mut and iCtrl when comparing all cells. To test for enrichment, Fisher’s Exact test was performed using the function fisher.test() and then the corresponding p-values were adjusted for multiple comparisons using p.adjust(p, method=‘bonferroni’,n=length(p)).
Cell and synaptic puncta quantification.
All cell quantifications were obtained using at least 9 images per sample consisting of 3 non-contiguous regions per image and at least 3 images obtained from independent experiments. For GAD65 quantification, tiled images of fusion or unfused organoids were visualized in Photoshop (Adobe), a box of equal size was used to demarcate regions of interest on the outer edges of organoids, and total numbers of GAD65+ cells and Hoechst+ nuclei within the boxed region were manually tabulated. Synaptic puncta were identified and colocalized using Bitplane Imaris version 9.3 or 9.5 image processing software using the “spots” identifier, set to detect identically sized objects surrounding TUBB3+ cellular processes and thresholded against Hoechst staining to exclude any nuclear overlap. The native “colocalization” function on Imaris was used to identify overlapping puncta.
Live organoid calcium imaging.
The genetically encoded calcium indicator GCaMP6f was introduced into organoids between day 88-95 via infection with 5 μl of 1.98x1013 GC ml−1 pAAV1.Syn.GCaMP6f.WPRE.SV40 virus76, a gift from Dr. Douglas Kim & the GENIE Project (Addgene viral prep # 100837-AAV1 or UPENN Vector core AV-1-PV2822). All imaging was performed 12-14 days after infection using a Scientifica two-photon microscope with a Coherent Chameleon tunable laser. Calcium transients were recorded at an excitation of 920 nm using a 20X 0.8NA water-immersion objective (Nikon) and at a frame rate of 31 Hz with 512 × 512-pixel resolution and 0.5 × 0.5 mm field of view. Recording was performed in artificial cerebrospinal fluid (aCSF) as described below with additional 10mM HEPES to maintain a pH of 7.3-7.4 in the absence of O2/CO2 perfusion (see Extracellular Recordings below for details). Following initial imaging in the absence of drugs, organoids were then subjected to 1-minute incubation with the GABAA receptor antagonist gabazine (25 μM) or bicuculline methiodide (100 μM) and the identical fields were re-imaged after drug exposure.
Microcircuit identification.
Raw Ca2+ imaging files in tiff format were processed to identify fluorescence transients (ΔF/F0) and spike estimation in MATLAB (Mathworks Inc.) using the constrained non-negative matrix factorization-extended (CNMF-E) methodology29, 30. Following CNMF-E based data extraction neuronal microcircuit clusters were identified by performing hierarchical clustering on the correlation matrix of neuronal Ca2+ spiking data and analyzed based on Muldoon et al.48. The correlation between all pair-wise combinations of the time course of spikes for all neurons identified by CNMF-E was calculated to generate a correlation matrix. Following generation of the correlation matrix, linkage analysis was performed using the MATLAB linkage function from the statistics toolbox (with ‘complete’/furthest distance). The generated hierarchical clustering was input into the dendrogram function from the MATLAB statistics toolbox with ‘Color Threshold’ fixed at 1.5 for all experimental groups. By then assigning each neuron to a cluster determined by its assigned color in the dendrogram, clusters were created in which there was high correlation between all neurons in the cluster. To calculate the number of pairs of neurons that were significantly correlated within each dataset we first generated 1000 shuffled time courses for each neuron using MATLAB’s randperm function. Pairwise correlations for the randomly shuffled time courses were calculated in the same way as the original data, and a pair of neurons were considered correlated if their correlation coefficient in the original data was significantly different to the 1000 shuffled datasets with P < 0.05. To determine the threshold of simultaneous firing, the synchronization of the time shuffled data was calculated, and the threshold was set at the 99th percentile of synchronization in the shuffled data. Synchronization above this threshold was considered “synchronized”. These data were then plotted on a normalized y-axis ranging from 0 (no synchronizations) to 1 (100% synchronization) and time on the x-axis. The total number of synchronizations, average synchronization amplitude, and average synchronization duration were then tabulated.
Extracellular recordings.
Organoids were recorded between age day ~100-107. Live organoids were perfused with 500nM kainic acid in aCSF (containing in mM: NaCl 126, D-glucose 10, MgCl2 1.2, CaCl2 2, KCl 5, NaH2PO4 1.25, Na Pyruvate 1.5, L-Glutamine 1, NaHCO3 26, pH 7.3-7.4 when bubbled with 95% O2, 5% CO2) to initiate oscillatory network activity. LFP activity was recorded using a patch pipette filled with aCSF connected with a head stage to a field amplifier (A-M Systems Inc., model 3000), and band pass filtered between 0.1 and 1000 Hz by to an instrumentation amplifier (Brownlee BP Precision, model 210A). Field potentials were digitized at 4096 Hz with a National Instruments A/D board using EVAN (custom-designed LabView-based software from Thotec) and analyzed with custom procedures (Wavemetrics, Igor Pro 8). Lower frequency activity was visualized for 10-minute epochs using power spectral densities (PSDs), which were calculated using the “dsperiodogram” function of Igor Pro, and spectrograms, which were generated using the Gabor method on Igor Pro. High frequency activity up to 650 Hz was visualized by generating Morlet wavelet plots of 0.5-1.0 second epochs of the raw traces used for low frequency analyses. Inter-spike intervals and spike frequencies were tabulated on Igor Pro using the identical 10-minute epochs used above.
Statistical information.
Graphical plots and statistical analyses were performed using Graphpad Prism 9 software, unless otherwise indicated. All samples were subject to Shapiro-Wilk and Kolmogorov-Smirnov, normality testing. If the data passed normality by one of these tests, then the data were assumed to be normally distributed. Non-normal samples were analyzed by a two-tailed Mann-Whitney U-test or Kruskal-Wallis test followed by Dunn’s multiple comparison test. Normally distributed data were analyzed by a two-tailed Student’s t-test or ANOVA with post hoc Tukey’s multiple comparison test. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications10, 11, 17–20. Organoids were generated from hiPSCs or hESCs in batches of 96 organoids per plating. Following quality control checks including visual inspection during early differentiation (days 6, 18, and 35), individual organoids were randomly selected from the remaining pool for experiments. Data collection and analysis were not performed blind to the conditions of the experiments. No data points were excluded from the analyses for any reason. Violin plots display the full distribution of individual data points with dotted lines to indicate the median and quartile values. Figure legends specify sample numbers and P values for all statistical tests. Each n represents an independent experiment. Supplementary Table 4 specifies the number of non-contiguous sections imaged prior to selection of representative images. The n for all other representative images is specified in the figure legends.
Extended Data
Extended Data Fig. 1. Plots and table of batch and patient line variability for key experimental measures.
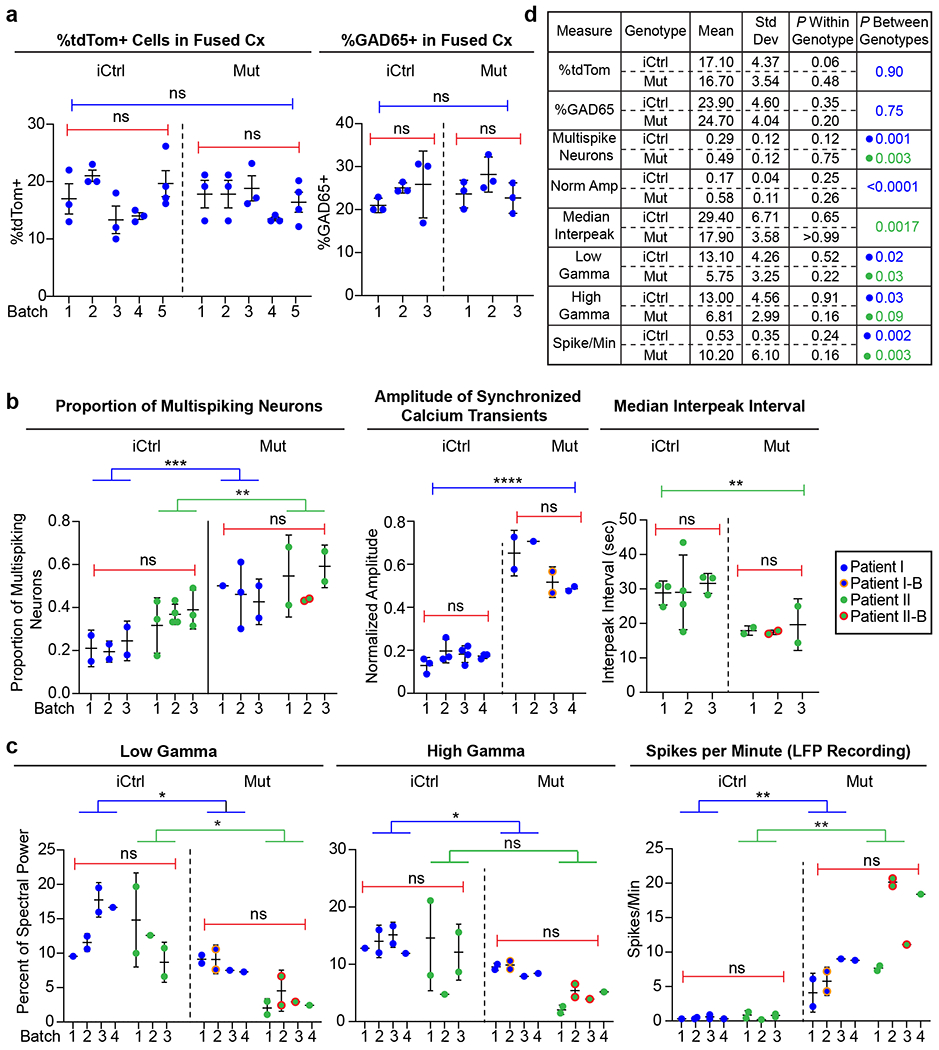
a, Plots of experimental results from different batches of iCtrl and Mut Cx+GE fusion organoids analyzed for the percentage of cells in the cortical compartment that expressed tdTomato (tdTom) after the GE portion was labeled with AAV1-CAG:tdTom virus (left panel) or GAD65 antibodies (right panel). Each dot represents an individual organoid section used for analysis and numbered elements on the x-axis represent individual experiments. No significant within or across genotype differences were noted for either percentage of Cx expressing tdTom or GAD65. b,c, Plots of individual experimental results from iCtrl and Mut Cx+GE fusion calcium indicator and LFP experiments. Each dot represents results from an independent experiment, numbered elements on the x-axis represent independent organoid batches. Blue dots represent hiPSC line I (Rett patient with a 705delG frameshift mutation), green dots represent hiPSC line II (Rett patient with 1461A>G missense mutation), and orange and red circles indicate independently isolated hiPSC lines from the same patient. For calcium indicator and LFP data, plots were generated for all experiments in which significant differences between Mut and iCtrl Cx+GE fusions were reported. In all cases in which the same measure resulted in statistically significant differences between Mut and iCtrl in both hiPSC patient lines, the two patient lines were combined for within genotype statistical analyses (e.g., proportion of multispiking neurons). d, Table with mean, standard deviation (Std Dev), within genotype P value, and between genotype P value for all measures shown in a-c. The results show relatively low Std Dev within genotypes as reflected in non-significant P values, yet highly significant differences between the iCtrl and Mut groups in nearly all functional measurements. All between batch statistical analyses were by ANOVA. All between genotype analyses by ANOVA with correction for multiple comparisons by Tukey’s test, unless otherwise specified in the main text.
Extended Data Fig. 2. Constrained non-negative matrix factorization (CNMF) based Ca2+ data extraction workflow and output.

a, Raw image of an GCaMP6f infected Cx+GE organoid (left) and CNMF based identification of fluorescently active (spiking) GCaMP regions of interest (right). b-d, Identification and analysis of individual neuronal Ca2+ spiking data. b, Changes in GCaMP6f fluorescence (normalized ΔF/F) for each neuron in a displayed as individual spike trains (left) or the same data displayed as a colorized amplitude plot (right). Individual spiking data are then used to determine various measures of spiking behavior including overall synchronicity based on a threshold level determined following spike shuffling c and calculation of interspike intervals d. e, Simultaneous to b-d, Ca2+ spiking data are categorized into neuronal microcircuits (clusters) based on correlations between individual Ca2+ spikes. f, during initial analyses, alternative clustering approaches including cross-correlation was used and the neural microcircuits resulting from multiple approaches were compared to determine the optimal clustering paradigm.
Extended Data Fig. 3. Immunohistochemical analyses reveal similar cell composition in iCtrl and Mut fusion organoids.
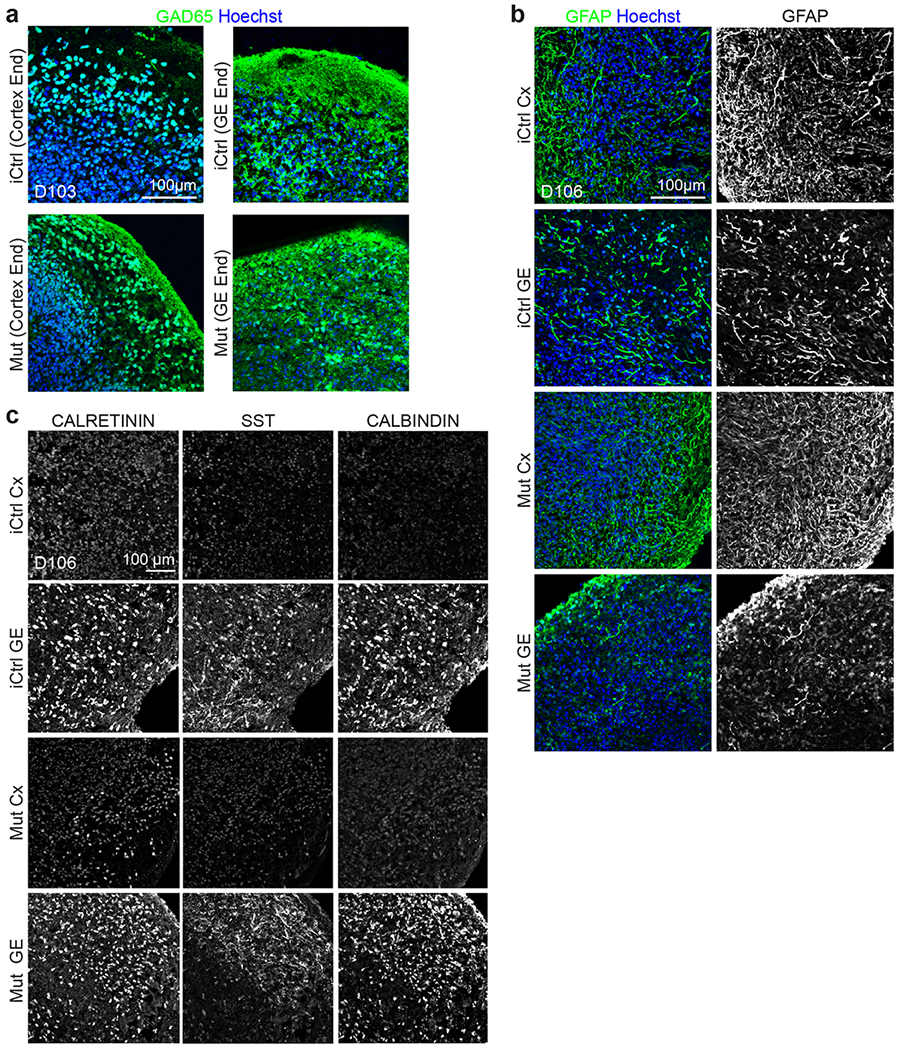
a, Day ~100 iCtrl and Mut Cx+GE fusion organoids have comparable numbers of GAD65+ positive cells in both the GE and Cx end (quantification in Fig. 3c). b, Both unfused Mut and unfused iCtrl day ~100 GE organoids contain multiple interneuron subtypes including CALRETININ, CALBINDIN, and SOMATOSTATIN (SST) expressing cells. c, Mut and iCtrl Day ~100 GE and Cx organoids also contain GFAP+ astrocytes. All images are representative examples from 3 or more independently imaged sections. See Supplementary Table 4 for additional details.
Extended Data Fig. 4. Rett syndrome fusion organoids from a second patient hiPSC line exhibit neural network irregularities in calcium indicator measurements.
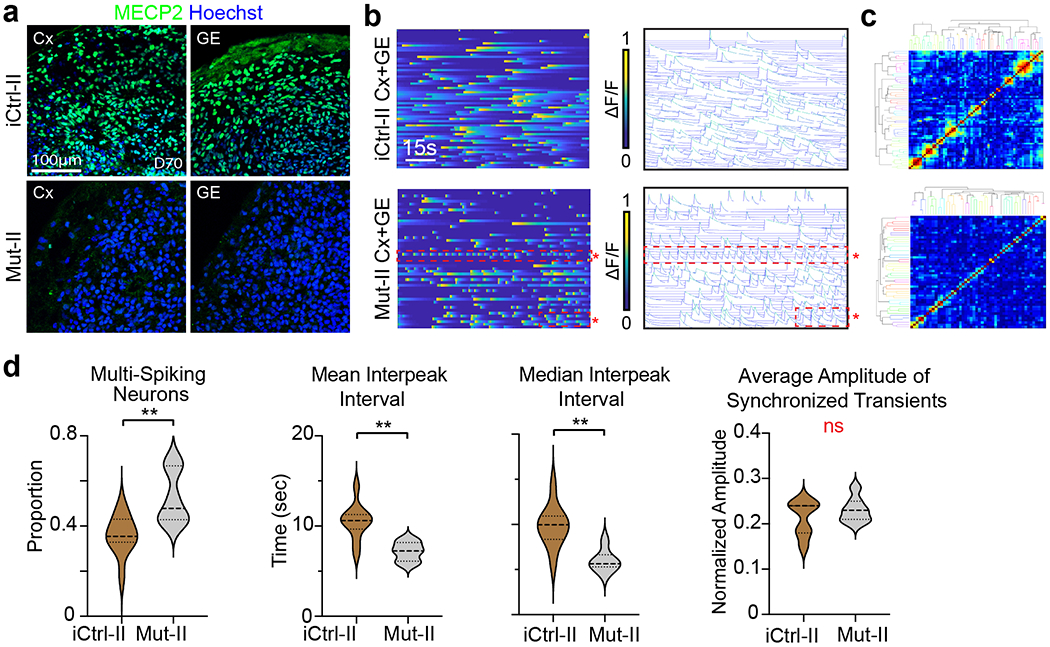
a, Immunohistochemical analyses of isogenic Cx and GE organoids from a second Rett syndrome patient hiPSC line (harboring a 1461A>G missense mutation, indicated by “II”) reveals either the presence (iCtrl-II) or absence (Mut-II) of MECP2 expression. Representative images from n=2 independent experiments and 6 imaged sections. b, Mut-II Cx+GE fusions contain hyperexcitable neurons as indicated by the red boxed regions in the bottom ΔF/F colorized amplitude plot and spike plot. These plots show trains of repeatedly firing Ca2+ transients with short interspike intervals that are not present in iCtrl-II Cx+GE (top plots). c, There is no discernible change in synchronization of calcium transients between Mut and iCtrl as reflected in the clustergrams. d, The hyperexcitable phenotype in Mut-II Cx+GE fusions is reflected in the pooled data both by significant increases in multispiking neurons and decreases in mean and median interpeak intervals. Pooled data quantifications, n = 10 iCtrl-II and n =6 Mut-II fusion organoids, where each n is an independently generated organoid. Two-sided Mann-Whitney tests were used, *P = 0.0071 for the proportion of multispiking neurons, **P = 0.0047 for the mean interspike interval, **P = 0.0017 for the median interspike interval, ns = not significant. Plot in d displays the full distribution of individual data points with dotted lines indicating the median and quartile values.
Extended Data Fig. 5. Enrichment of autism and epilepsy risk genes in up/downregulated genes in MECP2 mutant and isogenic control organoids.

a, Overlap of differentially expressed genes in MECP2 mutant organoids (all cell groups) with SFARI autism spectrum disorder (ASD) gene categories 1-3 and DisGeNET epilepsy Gene-Disease Association list (CUI: C0014544). Overlaps between data are indicated by red and green shading and displayed as Venn diagrams in b. c, Two-sided Fisher’s Exact Test was used to determine if Up/Downregulated genes show enrichment for genes in SFARI and epilepsy gene lists. Odds ratio from the test are displayed along with Bonferroni-corrected P values. Up/Epilepsy: ***P = 0.0016, Down/Epilepsy: ****P =1.81×10−5, Up/ASD: ****P = 5.72×10−9, Down/ASD: P = 1.00.
Extended Data Fig. 6. UMAP representation of select genes associated with synaptogenesis and kainate responsivity.
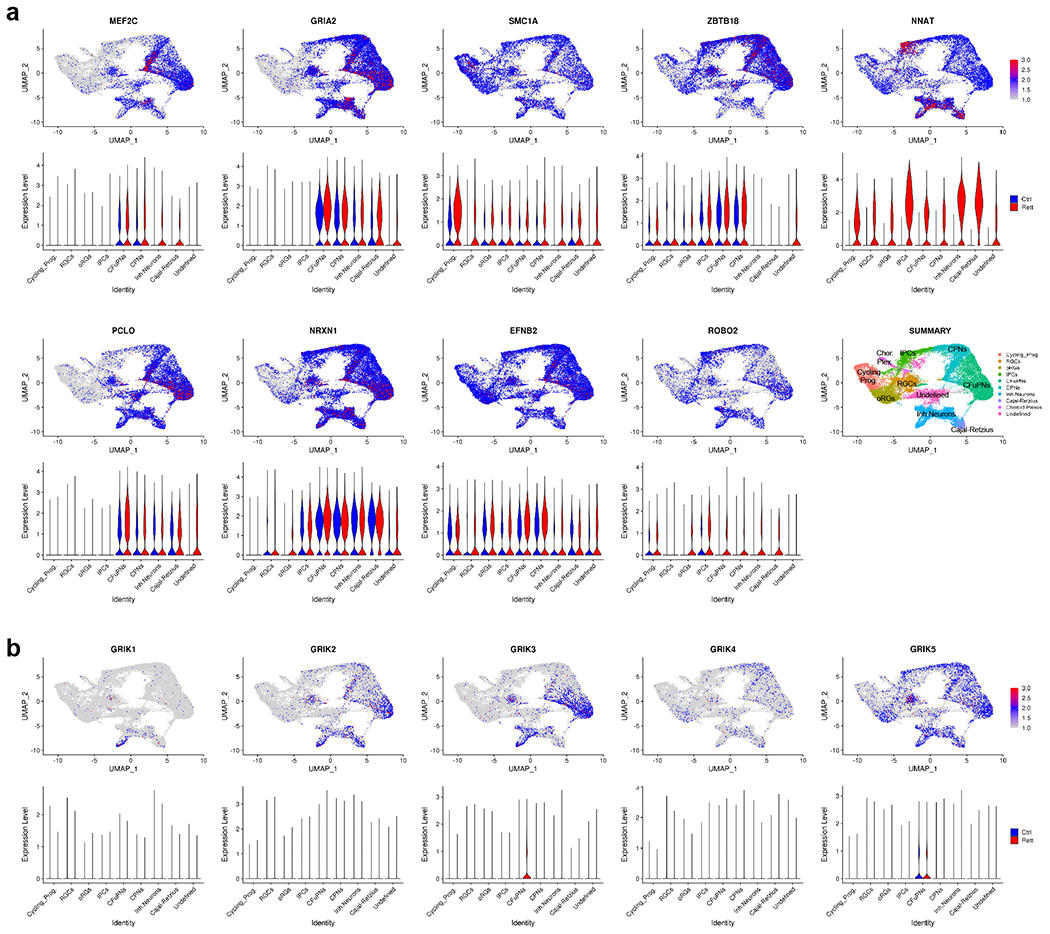
a, UMAP representation of select genes associated with axonal projections and synaptogenesis found to be upregulated in MECP2 mutant Cx+GE fusion organoids. Violin plots display the relative expression level of each gene across the indicated cell clusters. b, UMAP representation of kainate receptor gene expression within the Cx+GE fusion organoids.
Extended Data Fig. 7. Gene ontology analysis of neuronal subtype clusters.
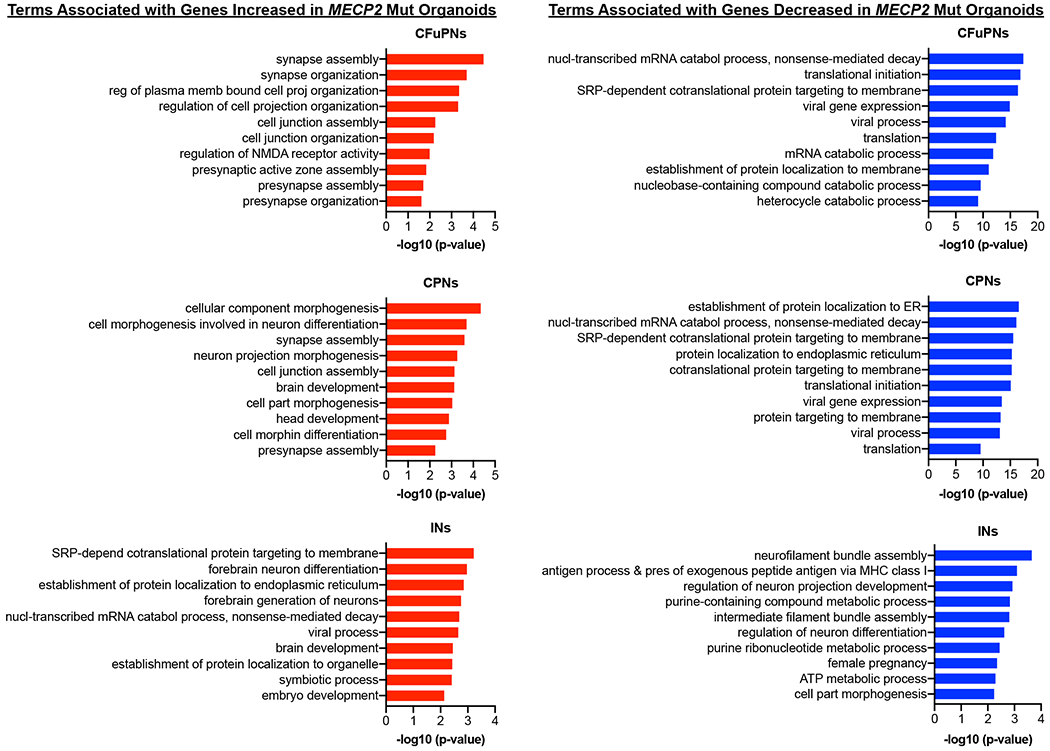
Top 10 most enriched Gene Ontology biological process (GO BP) terms associated with upregulated or downregulated differentially expressed genes when comparing Mut and iCtrl within the main excitatory (CPN and CFuPN) and interneuron (IN) clusters. Upregulated genes in the excitatory clusters are highly enriched for terms associated with synaptogenesis and axonal morphogenesis while downregulated genes are associated with mRNA catabolism and translation. In contrast, synaptogenesis terms are absent among the upregulated genes in the IN cluster, with this set populated by terms associated with forebrain differentiation and axonal morphogenesis. Downregulated genes in the IN cluster are enriched for metabolism and cellular cytoskeleton associated terms.
Extended Data Fig. 8. Spatially restricted microcircuit clusters and fewer synchronous events in MECP2 Mut Cx+GE organoids.

a, Pooled data for neuronal clusters derived here using Ca2+ activity correlations, reveal spatially restricted (smaller) microcircuit clusters with fewer average neurons per cluster in Mut compared to iCtrl. b, Pooled data of synchronous events demonstrates significantly fewer events (but with each event having a significantly higher amplitude, see Fig. 3) in Mut compared to iCtrl. Synchronous events have similar overall duration in both conditions (n = 6 for iCtrl, n = 7 for Mut and represents independently generated organoids, *P = 0.0436 for Pairwise Distances, *P = 0.0203 for Cluster Circumference, *P = 0.0321 for Cluster Area, **P = 0.0089 for Neurons per Cluster, and *P = 0.0180 for Number of Synchronized Transients). Plots display the full distribution of individual data points with dotted lines to indicate the median and quartile values. Following a normality test, statistical significance was determined using a two-sided Mann-Whitney U-test.
Extended Data Fig. 9. Additional independent examples of local field potential recordings.
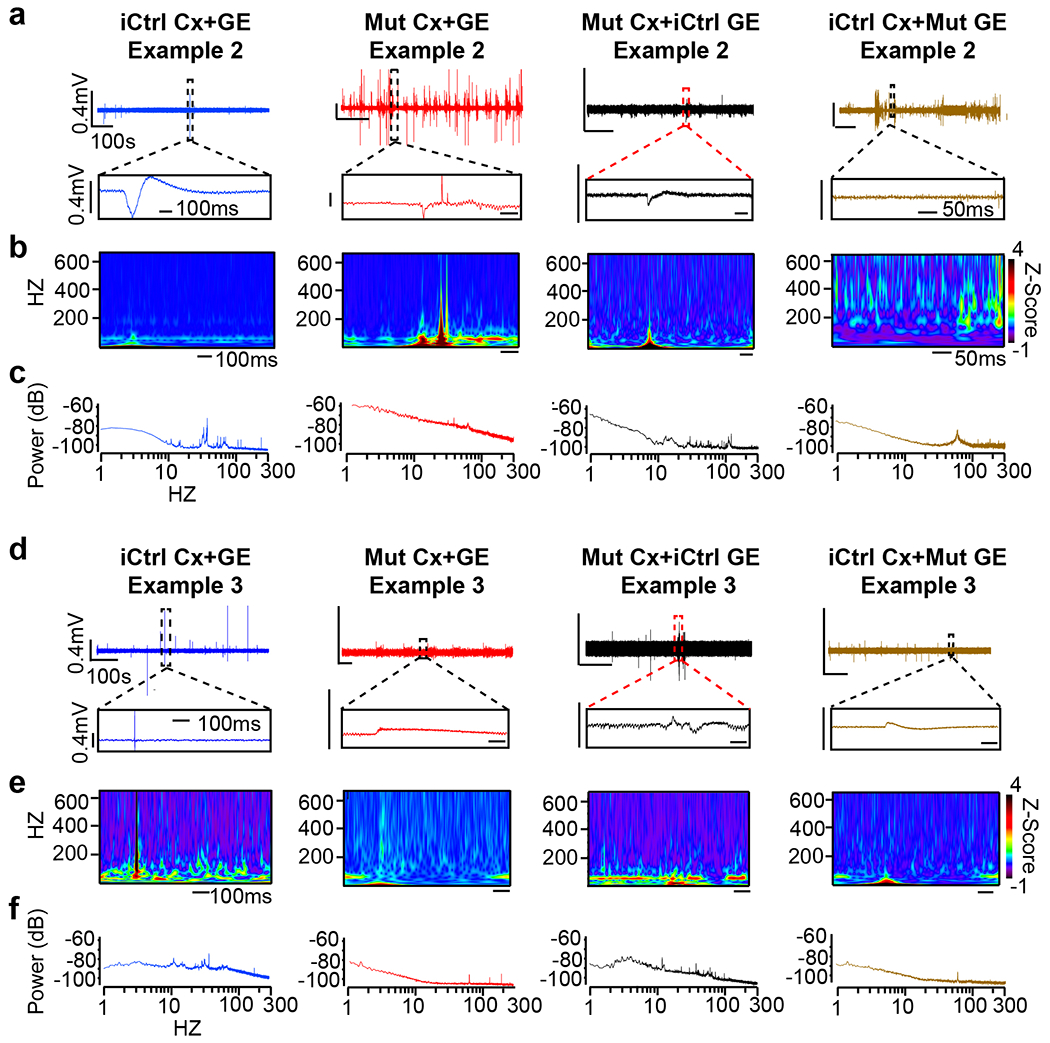
a,d, Representative raw 10-minute LFP traces (top) and time expanded segments (bottom) from either unmixed iCtrl or Mut Cx+GE fusion organoids, or Mut Cx+iCtrl GE or iCtrl Cx+Mut GE mixed fusion organoids. b,e, Morlet plots derived from the time expanded segments shown in a,d. c, f, Periodogram derived from the entire 10 min traces shown in a,d.
Extended Data Fig. 10. Rett syndrome fusion organoids from a second patient hiPSC line demonstrate epileptiform changes in extracellular recordings.

a, Raw trace of a representative 10-minute LFP recording (top) and time expanded window (bottom) from iCtrl-II, Mut-II, or Mut-II +PFT-α Cx+GE fusion organoids. b, Morlet plots showing high frequency activity associated with the time expanded segments shown in (a). (c) Periodograms derived from the entire recordings shown in a. d, Quantification of high and low gamma spectral power from LFP recordings demonstrates a significant decrease of low gamma power and a sizeable but non-significant loss of high gamma power in Mut-II Cx+GE fusions. PFT-α treatment of Mut-II Cx+GE fusions results in a statistically significant rescue of both low and high gamma oscillatory power. Low gamma; Ordinary ANOVA, overall P = 0.0024, Tukey’s Multiple comparisons, *P =0.0313 iCtrl II vs Mut II, *P=0.0211 Mut II vs Mut II +PFT. High gamma; Ordinary ANOVA, overall P = 0.0091, Tukey’s multiple comparisons, *P = 0.0243 Mut II vs Mut II + PFT, P = 0.09 between iCtrl-II and Mut. e, Spike frequency across multiple independent experiments Kruskal-Wallis test, overall P = 0.0003, Dunn’s multiple comparisons **P = 0.0028, *P = 0.0276. For d and e, n = 5 for iCtrl-II and Mut-II +PFT-α, n = 6 for Mut-II (total n = 16). f, Plots of high and low gamma spectral power versus spike frequency demonstrates an inverse relationship between gamma power and spiking. The solid black line is the best fit following linear regression, and the dashed magenta lines indicate 95% confidence intervals for the estimated line of best fit. The slope of the line of best fit is indicated above each graph. Plots in d and e display the full distribution of individual data points with dotted lines to indicate the median and quartile values.
Supplementary Material
Supplementary Tables 1-3 | Each table and subsection is indicated in the tab name.
Supplementary Video 1 | Live two-photon microscopic imaging of neural activities within a representative Cx+GE fusion organoid. Day 90 H9 hESC-derived Cx+GE fusion organoids were infected with AAV1-GCaMP6f and imaged 12 days later (Day 102) using two-photon confocal microscopy.
Supplementary Video 3 | Post hoc clustering of calcium activity. An example demonstrating the segregation of neurons based on calcium activity into microcircuit clusters. The clusters in this example are based on correlated calcium transients between individual neurons. The rows of neurons on the right represent most neurons in each cluster and are color-coded. Each cluster’s calcium activity (color matched to the right panel) is plotted as ΔF/F on the left. This example is from the same H9 hESC-derived Cx+GE fusion organoid shown in movies S1 and S2.
Supplementary Video 4 | Activity profile of a representative Cx+GE fusion organoid before exposure to bicuculline. This video displays the baseline GCaMP6f activity profile of a day 99 H9 hESC-derived Cx+GE fusion organoid immediately prior to addition of 100μM of the GABAA receptor antagonist bicuculline methiodide.
Supplementary Video 2 | Constrained non-negative matrix factorization extended (CNMF-E)-based evaluation of calcium activities within a Cx+GE fusion organoid. The video demonstrates real time translation of changes in GCaMP6f fluorescence shown in Supplementary Video 1 into peaks of activity used in subsequent analyses. Neurons demonstrating activity are identified and numbered on the left and normalized ΔF/F values for each neuron are plotted on the right.
Supplementary Video 5 | Calcium transient synchrony after bicuculline administration to a representative Cx+GE fusion organoid. This video displays changes in the neural network activities of the Cx+GE fusion organoid shown in Supplementary Video 4 approximately one minute after the addition of 100 μM bicuculline methiodide. Note the repeated synchronization of calcium transients across the organoid.
Supplementary Video 6 | Neural network activity of a representative Cx+GE fusion organoid immediately before the addition of gabazine. This video displays the baseline calcium transients present in a day 98 H9 hESC-derived Cx+GE fusion organoid immediately prior to addition of 25 μM of the GABAA receptor antagonist gabazine.
Supplementary Video 7 | Neural network activity of a representative Cx+GE fusion organoid immediately after the addition of gabazine. This video displays the prominent synchronization of neuronal activities seen in the day 98 Cx+GE fusion organoid shown in Supplementary Video 6 approximately one minute after addition of 25 μM gabazine.
Supplementary Video 8 | Calcium activity in an iCtrl Cx+GE fusion organoid. A representative example of live two-photon confocal imaging of GCaMP6f fluorescence from a day 103 iCtrl hiPSC-derived Cx+GE fusion organoid.
Supplementary Video 9 | Spontaneous synchronizations of calcium transients in an MECP2 mutant Cx+GE fusion organoid. A representative example of the abnormal synchronizations of calcium transients seen in day 103 Mut hiPSC-derived Cx+GE fusion organoids.
Supplementary Video 10 | Calcium activity in an iCtrl Cx+GE fusion organoid created from Rett patient II hiPSC. A representative example of live two-photon confocal imaging of GCaMP6f fluorescence within a day 100 iCtrl hiPSC-derived Cx+GE fusion organoid generated from hiPSC derived from a second Rett syndrome patient.
Supplementary Video 11 | Hyperexcitable calcium indicator activity in an MECP2 mutant Cx+GE fusion organoid created from Rett patient II hiPSC. A representative example of hyperexcitable calcium indicator activity seen in day 103 Mut hiPSC-derived Cx+GE fusion organoids. The yellow arrows indicate examples of hyperexcitable neurons resulting in reduced interpeak intervals between activations. This is representative of the calcium indicator activity seen in Mut Cx+GE organoids generated from Rett patient II hiPSC.
Supplementary Video 13 | Spontaneous synchronizations of calcium transients in a mixed iCtrl Cx and MECP2 mutant GE (iCtrl Cx+ Mut GE) fusion organoid. A representative example of the abnormal synchronizations of calcium transients seen in ~day 100 mixed iCtrl Cx + Mut GE hiPSC-derived fusion organoids. This example is from a 102-day old fusion organoid.
Supplementary Video 12 | Calcium activity in a mixed Mut Cx and iCtrl GE (Mut Cx + iCtrl GE) fusion organoid. A representative example of live two-photon confocal imaging of GCaMP6f fluorescence from a day 101 mixed Mut Cx + iCtrl GE hiPSC-derived fusion organoid.
ACKNOWLEDGEMENTS
We thank Samantha Butler, Tom Carmichael, and members of the Novitch lab for helpful discussions and comments on the manuscript; Neda Vishlaghi, and Felix Turcios-Hernandez for technical assistance, and Jae Lee, Soo-Kyung Lee, Hideo Shinagawa, and Kazuaki Yoshikawa for valuable reagents. We also thank the UCLA Eli and Edythe Broad Stem Cell Research Center (BSCRC) and Intellectual and Developmental Disabilities Research Center microscopy cores for access to imaging facilities. This work was supported by grants from the California Institute for Regenerative Medicine (CIRM) (DISC1-08819 to B.G.N.), the National Institute of Health (R01NS089817, R01DA051897, and P50HD103557 to B.G.N.; K08NS119747 to R.A.S.; K99HD096105 to M.W.; R01MH123922, R01MH121521, and P50HD103557 to M.J.G; R01GM099134 to K.P.; R01NS103788 to W.E.L; R01NS088571 to J.M.P.; R01NS030549 and R01AG050474 to I.M.), and research awards from the UCLA Jonsson Comprehensive Cancer Center and BSCRC Ablon Scholars Program (B.G.N.), the BSCRC Innovation Program (B.G.N, K.P, and W.E.L.), the UCLA BSCRC Steffy Brain Aging Research Fund (B.G.N. and W.E.L.), and the UCLA Clinical and Translational Science Institute (B.G.N.), Paul Allen Family Foundation Frontiers Group (K.P. and W.E.L.), the March of Dimes Foundation (W.E.L.), and the Simons Foundation Autism Research Initiative Bridge to Independence Program (R.A.S. and M.J.G.). R.A.S. was also supported by the UCLA/NINDS Translational Neuroscience Training Grant (R25NS065723), a Research and Training Fellowship from the American Epilepsy Society, a Taking Flight Award from CURE Epilepsy, and a Clinician Scientist training award from the UCLA BSCRC. J.E.B. was supported by the UCLA BSCRC Rose Hills Foundation Graduate Scholarship Training Program. M.W. was supported by postdoctoral training awards provided by the UCLA BSCRC and the Uehara Memorial Foundation. O.A.M. and A.K. were supported in part by the UCLA-California State University Northridge CIRM-Bridges training program (EDUC2-08411). We also acknowledge the support of the IDDRC Cells, Circuits and Systems Analysis, Microscopy and Genetics and Genomics Cores of the Semel Institute of Neuroscience at UCLA, which are supported by the NICHD (U54HD087101 and P50HD10355701). We lastly acknowledge support from a Quantitative and Computational Biosciences Collaboratory Postdoctoral Fellowship to S.M. and the Quantitative and Computational Biosciences Collaboratory community, directed by Matteo Pellegrini.
Footnotes
COMPETING INTERESTS
The use of pifithrin compounds to treat Rett Syndrome and fusion organoids to screen for preclinical efficacy is covered by a patent application filed by the UC Regents with R.A.S, W.E.L., and B.G.N. as inventors. The remaining authors declare no competing interests.
CODE AVAILABILITY
CNMF/CNMF-E has been previously published29, 30 and the original version of CNMF_E is publicly available on Github at https://github.com/zhoupc/CNMF_E. Additional code used in this study is available at https://github.com/SiFTW/CNMFeClustering/.
DATA AVAILABILITY
Raw and processed single-cell RNA sequencing data were deposited at the Gene Expression Omnibus with accession number GSE165577. The authors declare that all other data supporting the findings of this study are available within the paper and its supplementary information files.
REFERENCES
- 1.Di Lullo E & Kriegstein AR The use of brain organoids to investigate neural development and disease. Nat Rev Neurosci 18, 573–584 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amin ND & Pasca SP Building Models of Brain Disorders with Three-Dimensional Organoids. Neuron 100, 389–405 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Qian X, Song H & Ming GL Brain organoids: advances, applications and challenges. Development 146 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rakic P Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci 10, 724–735 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Molnar Z, et al. Evolution and development of the mammalian cerebral cortex. Brain Behav Evol 83, 126–139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Worp HB, et al. Can animal models of disease reliably inform human studies? PLoS Med 7, e1000245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dawson TM, Golde TE & Lagier-Tourenne C Animal models of neurodegenerative diseases. Nat Neurosci 21, 1370–1379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stam CJ Modern network science of neurological disorders. Nat Rev Neurosci 15, 683–695 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Palop JJ & Mucke L Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci 17, 777–792 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun AX, et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science 366, 1486–1492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trujillo CA, et al. Complex Oscillatory Waves Emerging from Cortical Organoids Model Early Human Brain Network Development. Cell Stem Cell 25, 558–569 e557 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buzsaki G & Wang XJ Mechanisms of gamma oscillations. Annu Rev Neurosci 35, 203–225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Headley DB & Pare D Common oscillatory mechanisms across multiple memory systems. NPJ Sci Learn 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verret L, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsumoto JY, et al. Network oscillations modulate interictal epileptiform spike rate during human memory. Brain 136, 2444–2456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Dellen E, et al. Local polymorphic delta activity in cortical lesions causes global decreases in functional connectivity. Neuroimage 83, 524–532 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Watanabe M, et al. Self-Organized Cerebral Organoids with Human-Specific Features Predict Effective Drugs to Combat Zika Virus Infection. Cell Rep 21, 517–532 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bagley JA, Reumann D, Bian S, Levi-Strauss J & Knoblich JA Fused cerebral organoids model interactions between brain regions. Nat Methods 14, 743–751 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birey F, et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiang Y, et al. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell Stem Cell 21, 383–398 e387 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakaguchi H, et al. Self-Organized Synchronous Calcium Transients in a Cultured Human Neural Network Derived from Cerebral Organoids. Stem Cell Reports (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lyst MJ & Bird A Rett syndrome: a complex disorder with simple roots. Nat Rev Genet 16, 261–275 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Garofalo EA, Drury I & Goldstein GW EEG abnormalities aid diagnosis of Rett syndrome. Pediatr Neurol 4, 350–353 (1988). [DOI] [PubMed] [Google Scholar]
- 24.Operto FF, Mazza R, Pastorino GMG, Verrotti A & Coppola G Epilepsy and genetic in Rett syndrome: A review. Brain Behav 9, e01250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe M, et al. TGFβ superfamily signaling regulates the state of human stem cell pluripotency and competency to create telencephalic organoids. bioRxiv, 875773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hendry SH, Schwark HD, Jones EG & Yan J Numbers and proportions of GABA-immunoreactive neurons in different areas of monkey cerebral cortex. J Neurosci 7, 1503–1519 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sahara S, Yanagawa Y, O’Leary DD & Stevens CF The fraction of cortical GABAergic neurons is constant from near the start of cortical neurogenesis to adulthood. J Neurosci 32, 4755–4761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferando I & Mody I In vitro gamma oscillations following partial and complete ablation of delta subunit-containing GABAA receptors from parvalbumin interneurons. Neuropharmacology 88, 91–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pnevmatikakis EA, et al. Simultaneous Denoising, Deconvolution, and Demixing of Calcium Imaging Data. Neuron 89, 285–299 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou P, et al. Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buzsaki G & Draguhn A Neuronal oscillations in cortical networks. Science 304, 1926–1929 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Wang DD & Kriegstein AR GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J Neurosci 28, 5547–5558 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang DD & Kriegstein AR Blocking early GABA depolarization with bumetanide results in permanent alterations in cortical circuits and sensorimotor gating deficits. Cereb Cortex 21, 574–587 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leonard H, Cobb S & Downs J Clinical and biological progress over 50 years in Rett syndrome. Nat Rev Neurol 13, 37–51 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Mellios N, et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry 23, 1051–1065 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Armstrong DD, Dunn K & Antalffy B Decreased dendritic branching in frontal, motor and limbic cortex in Rett syndrome compared with trisomy 21. J Neuropathol Exp Neurol 57, 1013–1017 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Belichenko PV, et al. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J Comp Neurol 514, 240–258 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Marchetto MC, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohashi M, et al. Loss of MECP2 Leads to Activation of P53 and Neuronal Senescence. Stem Cell Rep 10, 1453–1463 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.D’Haene E, et al. A neuronal enhancer network upstream of MEF2C is compromised in patients with Rett-like characteristics. Hum Mol Genet 28, 818–827 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, et al. Novel MEF2C point mutations in Chinese patients with Rett (-like) syndrome or non-syndromic intellectual disability: insights into genotype-phenotype correlation. BMC Med Genet 19, 191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salpietro V, et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat Commun 10, 3094 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huisman S, et al. Phenotypes and genotypes in individuals with SMC1A variants. Am J Med Genet A 173, 2108–2125 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Lopes F, et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J Med Genet 53, 190–199 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Oyang EL, Davidson BC, Lee W & Poon MM Functional characterization of the dendritically localized mRNA neuronatin in hippocampal neurons. PLoS One 6, e24879 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma J, et al. Neuronatin-mediated aberrant calcium signaling and endoplasmic reticulum stress underlie neuropathology in Lafora disease. J Biol Chem 288, 9482–9490 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu H, et al. Loss and Gain of MeCP2 Cause Similar Hippocampal Circuit Dysfunction that Is Rescued by Deep Brain Stimulation in a Rett Syndrome Mouse Model. Neuron 91, 739–747 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feldt Muldoon S, Soltesz I & Cossart R Spatially clustered neuronal assemblies comprise the microstructure of synchrony in chronically epileptic networks. Proc Natl Acad Sci U S A 110, 3567–3572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bragin A, Engel J Jr., Wilson CL, Fried I & Buzsaki G High-frequency oscillations in human brain. Hippocampus 9, 137–142 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Bragin A, Wilson CL, Almajano J, Mody I & Engel J Jr. High-frequency oscillations after status epilepticus: epileptogenesis and seizure genesis. Epilepsia 45, 1017–1023 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Ito-Ishida A, Ure K, Chen H, Swann JW & Zoghbi HY Loss of MeCP2 in Parvalbumin-and Somatostatin-Expressing Neurons in Mice Leads to Distinct Rett Syndrome-like Phenotypes. Neuron 88, 651–658 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krajnc N Management of epilepsy in patients with Rett syndrome: perspectives and considerations. Ther Clin Risk Manag 11, 925–932 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vignoli A, et al. Effectiveness and tolerability of antiepileptic drugs in 104 girls with Rett syndrome. Epilepsy Behav 66, 27–33 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Squillaro T, et al. Reduced expression of MECP2 affects cell commitment and maintenance in neurons by triggering senescence: new perspective for Rett syndrome. Mol Biol Cell 23, 1435–1445 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee B, Shin D, Gross SP & Cho KH Combined Positive and Negative Feedback Allows Modulation of Neuronal Oscillation Frequency during Sensory Processing. Cell Rep 25, 1548–1560 e1543 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Chen G, et al. Distinct Inhibitory Circuits Orchestrate Cortical beta and gamma Band Oscillations. Neuron 96, 1403–1418 e1406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hashemi E, Ariza J, Rogers H, Noctor SC & Martinez-Cerdeno V The Number of Parvalbumin-Expressing Interneurons Is Decreased in the Prefrontal Cortex in Autism. Cereb Cortex 27, 1931–1943 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables 1-3 | Each table and subsection is indicated in the tab name.
Supplementary Video 1 | Live two-photon microscopic imaging of neural activities within a representative Cx+GE fusion organoid. Day 90 H9 hESC-derived Cx+GE fusion organoids were infected with AAV1-GCaMP6f and imaged 12 days later (Day 102) using two-photon confocal microscopy.
Supplementary Video 3 | Post hoc clustering of calcium activity. An example demonstrating the segregation of neurons based on calcium activity into microcircuit clusters. The clusters in this example are based on correlated calcium transients between individual neurons. The rows of neurons on the right represent most neurons in each cluster and are color-coded. Each cluster’s calcium activity (color matched to the right panel) is plotted as ΔF/F on the left. This example is from the same H9 hESC-derived Cx+GE fusion organoid shown in movies S1 and S2.
Supplementary Video 4 | Activity profile of a representative Cx+GE fusion organoid before exposure to bicuculline. This video displays the baseline GCaMP6f activity profile of a day 99 H9 hESC-derived Cx+GE fusion organoid immediately prior to addition of 100μM of the GABAA receptor antagonist bicuculline methiodide.
Supplementary Video 2 | Constrained non-negative matrix factorization extended (CNMF-E)-based evaluation of calcium activities within a Cx+GE fusion organoid. The video demonstrates real time translation of changes in GCaMP6f fluorescence shown in Supplementary Video 1 into peaks of activity used in subsequent analyses. Neurons demonstrating activity are identified and numbered on the left and normalized ΔF/F values for each neuron are plotted on the right.
Supplementary Video 5 | Calcium transient synchrony after bicuculline administration to a representative Cx+GE fusion organoid. This video displays changes in the neural network activities of the Cx+GE fusion organoid shown in Supplementary Video 4 approximately one minute after the addition of 100 μM bicuculline methiodide. Note the repeated synchronization of calcium transients across the organoid.
Supplementary Video 6 | Neural network activity of a representative Cx+GE fusion organoid immediately before the addition of gabazine. This video displays the baseline calcium transients present in a day 98 H9 hESC-derived Cx+GE fusion organoid immediately prior to addition of 25 μM of the GABAA receptor antagonist gabazine.
Supplementary Video 7 | Neural network activity of a representative Cx+GE fusion organoid immediately after the addition of gabazine. This video displays the prominent synchronization of neuronal activities seen in the day 98 Cx+GE fusion organoid shown in Supplementary Video 6 approximately one minute after addition of 25 μM gabazine.
Supplementary Video 8 | Calcium activity in an iCtrl Cx+GE fusion organoid. A representative example of live two-photon confocal imaging of GCaMP6f fluorescence from a day 103 iCtrl hiPSC-derived Cx+GE fusion organoid.
Supplementary Video 9 | Spontaneous synchronizations of calcium transients in an MECP2 mutant Cx+GE fusion organoid. A representative example of the abnormal synchronizations of calcium transients seen in day 103 Mut hiPSC-derived Cx+GE fusion organoids.
Supplementary Video 10 | Calcium activity in an iCtrl Cx+GE fusion organoid created from Rett patient II hiPSC. A representative example of live two-photon confocal imaging of GCaMP6f fluorescence within a day 100 iCtrl hiPSC-derived Cx+GE fusion organoid generated from hiPSC derived from a second Rett syndrome patient.
Supplementary Video 11 | Hyperexcitable calcium indicator activity in an MECP2 mutant Cx+GE fusion organoid created from Rett patient II hiPSC. A representative example of hyperexcitable calcium indicator activity seen in day 103 Mut hiPSC-derived Cx+GE fusion organoids. The yellow arrows indicate examples of hyperexcitable neurons resulting in reduced interpeak intervals between activations. This is representative of the calcium indicator activity seen in Mut Cx+GE organoids generated from Rett patient II hiPSC.
Supplementary Video 13 | Spontaneous synchronizations of calcium transients in a mixed iCtrl Cx and MECP2 mutant GE (iCtrl Cx+ Mut GE) fusion organoid. A representative example of the abnormal synchronizations of calcium transients seen in ~day 100 mixed iCtrl Cx + Mut GE hiPSC-derived fusion organoids. This example is from a 102-day old fusion organoid.
Supplementary Video 12 | Calcium activity in a mixed Mut Cx and iCtrl GE (Mut Cx + iCtrl GE) fusion organoid. A representative example of live two-photon confocal imaging of GCaMP6f fluorescence from a day 101 mixed Mut Cx + iCtrl GE hiPSC-derived fusion organoid.
Data Availability Statement
Raw and processed single-cell RNA sequencing data were deposited at the Gene Expression Omnibus with accession number GSE165577. The authors declare that all other data supporting the findings of this study are available within the paper and its supplementary information files.