

Abstract
Over the past two decades, mass spectrometric (MS)-based proteomics technologies have facilitated the study of signaling pathways throughout biology. Nowhere is this needed more than in plants, where an evolutionary history of genome duplications has resulted in large gene families involved in posttranslational modifications and regulatory pathways. For example, at least 5% of the Arabidopsis thaliana genome (ca. 1,200 genes) encodes protein kinases and protein phosphatases that regulate nearly all aspects of plant growth and development. MS-based technologies that quantify covalent changes in the side-chain of amino acids are critically important, but they only address one piece of the puzzle. A more crucially important mechanistic question is how noncovalent interactions—which are more difficult to study—dynamically regulate the proteome’s 3D structure. The advent of improvements in protein 3D technologies such as cryo-electron microscopy, nuclear magnetic resonance, and X-ray crystallography has allowed considerable progress to be made at this level, but these methods are typically limited to analyzing proteins, which can be expressed and purified in milligram quantities. Newly emerging MS-based technologies have recently been developed for studying the 3D structure of proteins. Importantly, these methods do not require protein samples to be purified and require smaller amounts of sample, opening the wider proteome for structural analysis in complex mixtures, crude lysates, and even in intact cells. These MS-based methods include covalent labeling, crosslinking, thermal proteome profiling, and limited proteolysis, all of which can be leveraged by established MS workflows, as well as newly emerging methods capable of analyzing intact macromolecules and the complexes they form. In this review, we discuss these recent innovations in MS-based “structural” proteomics to provide readers with an understanding of the opportunities they offer and the remaining challenges for understanding the molecular underpinnings of plant structure and function.
Instead of posttranslational modifications (e.g. phosphorylation), signaling pathways can be revealed using mass spectrometry to identify proteins undergoing a 3D conformational change in planta.
Introduction
Mass spectrometry has revolutionized the study of proteomes in all organisms. In 2000, the sequence for nearly all proteins encoded within the genome of the model plant Arabidopsis thaliana was published (The Arabidopsis Genome Initiative, 2000), paving the way for using MS-based analysis to identify and quantify any A. thaliana protein. In the subsequent two decades, the in silico predicted proteomes of many different plant species have expanded greatly, providing the essential framework for MS-based computational analyses of protein concentration, protein modifications and now, protein 3D structure. The shift from analyzing a protein’s primary sequence and its covalent modifications (i.e. posttranslational modifications) into the analysis of protein conformation, that is the secondary, tertiary, quaternary, and even the newly coined “quinary” structure, is an important but difficult transition that is just beginning. This very recent effort to develop and apply what can be called “conformational” or “structural” proteomics is the emphasis of this review. These methods are complementary to cryo-electron microscopy (cryo-EM), X-ray crystallography, and nuclear magnetic resonance (NMR), that is “traditional” methods for deciphering the 3D structure of a protein. However, unlike traditional methods, which require milligram amounts of pure protein, MS-based methods can be performed in complex mixtures of proteins in the microgram range. For this reason, mass spectrometry plays a critical role in enabling biologists to bridge the knowledge obtained from established single protein structures with the interactions and conformational shifts that allow these proteins to operate as parts of complex, multiprotein macromolecular machines packed tightly into the cytoplasm, nucleus, or other compartments of the cell.
ADVANCES BOX.
CL and XL MS-based methods are being developed and improved to analyze the 3D structure of proteins in solution.
These methods require less protein and can be performed with impure protein solutions, two disadvantages that plague traditional methods like cryo-EM, X-ray crystallography, and NMR.
Since a protein’s biochemical function depends on its 3D structure, a major motivation of this work is the expectation that observations of conformational changes may be more relevant than chemical changes in amino acid side-chains for studying biological function.
New higher mass range MS instruments for performing nondenaturing mass spectrometry at neutral pH are being used concurrently with CL- and XL-based MS methods for analyzing 3D structure.
The cytoplasm in all cells is predicted to be 100–450 mg·mL-1 protein concentration (Feig et al., 2017; Nawrocki et al., 2017), which is far higher than even the most concentrated solution of standard proteins we normally work with in the laboratory. This observation has recently emphasized the in vivo presence of a higher order of protein structure known as the quinary structure (just above the quaternary structure of individual proteins forming a binary complex) (Guin and Gruebele, 2019; Rickard et al., 2019; Song et al., 2019; Breindel et al., 2020; Gruebele, 2021; Ziegler et al., 2021). A quinary structure refers to the weak intermolecular interactions that are constantly in play within the tightly packed cytoplasm. Due to their weak and transient nature, they are difficult to study; however, they may well play a fundamental role in how proteins act collectively in the cell as part of their vital functions in growth and development. By using MS methods to study the interaction of amino acid side-chains in the proteins present in crude lysates and even intact cells, we gain a much greater understanding of how plants have evolved with proteins that use the three dimensions of space together with the single dimension of time to survive even the harshest of conditions.
Background
Although mass spectrometers have been utilized for many decades in biological research, high-throughput methods for routinely analyzing entire proteomes have become available only recently. The methods described herein are specialized uses for mass spectrometers and requires a basic knowledge of the theory and practice of mass spectrometry in order to understand them. For this reason, it is useful to begin with an overview of the basic methods by which mass spectrometers operate in studying proteins. The first applications of MS-based methods to the study of proteins emerged from the work of two Nobel laureates, John Fenn and Koichi Tanaka, who described two separate methods for forcing protein molecules into the gas phase, or as John Fenn once said, “[making] elephants fly”. Fenn’s approach is known as electrospray ionization (ESI) and Tanaka’s is termed matrix-assisted laser desorption ionization. Although these methods are quite different, both achieve the same end, that is adding positive or negative charges to a protein and removing most or all water and other noncovalent adducts, resulting in a naked protein molecule that can be studied in the gas phase. The reader is referred to many review papers (e.g. Kline and Sussman, 2010; from this author’s laboratory) and books describing in greater depth the details of these two ionization methods, as well as other fundamentals involved in the chemistry and instrumentation of analyzing and quantifying proteins in the gas phase. In essence, mass spectrometers are like the scale in the bathroom or at the doctor’s office, which measure the pull of gravity on the mass of our entire body. The difference here is that unlike scales that measure our entire body composed of many cells with incredibly diverse molecules, mass spectrometers measure the mass of individual molecules based on determining their mass and the number of charges they contain. These instruments do this by determining how fast or how far they travel in an imposed, controllable electric field. Thus, the most common output one sees from a mass spectrometer is the m/z value, or the molecular weight of the molecule divided by the number of charges it contains while flying inside the machine. In most cases, the molecule is flying in an orbital path (e.g. Orbitrap and ion trap mass analyzers) or a linear path (e.g. quadrupole or time of flight mass analyzers). Quadrupole- and ion-trap-based instruments use the principle of ion stability when trapped in the gas phase to determine m/z, whereas time-of-flight mass spectrometers measure the amount of time needed to move ions to determine m/z. Quadrupoles were the first mass analyzers developed and are still the workhorse for most of the pharmaceutical and research communities. Ion traps, including the now widely used very high resolving Orbitraps, are also employed, and “hybrid” instruments utilize both types of mass analyzers, usually in a sequential manner. Ion detectors are placed at the end of the road for the ions after they are separated, although, as discussed later in this review, new methods for using mass spectrometers as preparative rather than analytical instruments are now being explored. That is, “soft landing”, which refers to hydrated nondestructive surfaces on which the protein ions land, instead of the hard metal surfaces, which detect and either neutralize or degrade them (Cooks and Mueller, 2013). Recent instruments that operate without a vacuum use the resistance to travel caused by proteins bouncing against the air molecules to calculate the cross-sectional area of that molecule, much like the old Model E ultracentrifuges used to do for determining Svedberg units of proteins (here gravity pulled the molecules, not an electric field). In fact, the instrument used at airport security gates (the one where they rub a small piece of white paper on your baggage) is one such instrument, known as an ion mobility (IM) mass spectrometer. These days, IM is becoming more widely used as an orthogonal way to separate peptides independent of reversed-phase high-performance liquid chromatography (RP-HPLC) or capillary electrophoresis.
Bottom-up versus top-down proteomics
The majority of proteomics publications to date have used so-called “bottom-up” methods for analyzing proteins. In short, this means that proteins in the sample are identified by computationally reassembling the sequences of ionized peptide fragments that were generated from those proteins during the experiment and spectrally matched to the master sequence using their m/z. This means that the first step in a typical bottom-up experiment is to digest the protein into peptides, typically with trypsin. Trypsin is widely used and preferred because it is very efficient and does not produce “ragged” edges, that is it almost exclusively cleaves C-terminally to lysines and arginines in a defined fashion. This is important for in silico analysis of the fragment ions detected in the mass spectrometer. It reduces the computational search space to peptides in which there is a lysine or arginine present at C terminus (and in the protein, one residue N-terminal to the identified sequence), and the prediction of the peptide sequence and side-chain modifications is more easily (i.e. quickly and accurately) performed. Furthermore, the charge state of a molecule in the gas phase in the mass spectrometers is very important in terms of how well it “flies” in response to the electric field. For this reason, trypsin is also beneficial because its reproducible cutting pattern ensures that at least one arginine or lysine (which are easily protonated to carry a +1 charge) is found on each peptide. The more highly charged a peptide is, the easier it is to measure since its m/z becomes smaller, allowing it to fit into the restricted m/z window of most mass analyzers. With large peptides and proteins, higher charge states can be a problem in that this creates a large number of possible co-existing states (e.g. +10, +9, +8 down to +1) that reduce the sensitivity since a single charge state has a higher spectral (signal) intensity than a population of molecules with many different charge states, diluting the intensity for any one charge state.
Another problem of size is that larger peptides and proteins have many isotopomers. In other words, ambient air is only 99.6% N14 and ∼0.4% N15 (likewise for the common isotope pair C12 and C13 and for O18 and O16, one isotope is much more abundant than the other but there remains a small amount of the minor one). This means each peptide or protein will actually be composed of a population of molecules with different isotopic compositions that reflect the natural abundance of each stable isotope, thus diluting again the spectral intensity compared to single 100% N14 or C12 or O16 abundances. This not only decreases the intensity of the major isotopomer, but it also creates a large number of different m/z values that must be computationally deconvoluted into a single value for the peptide or protein’s m/z.
Finally, the smaller peptides are also easier to separate on reverse phase HPLC columns since they are more homogenous in their chemical properties (hydrophobicity versus hydrophilicity) compared with large proteins. This bottleneck is actually becoming less of an issue these days as we learn to separate intact proteins better in the laboratory, but the two above issues have no obvious protein-level solution. Thus, it is generally easier and more routine to analyze a complex population of peptides from a single protein (“bottom-up”) than to analyze that intact protein itself (“top-down”).
However, top-down proteomics has a unique advantage over bottom-up: analyzing an intact protein preserves information about which modifications are associated with which proteins, whereas digesting the protein into proteolytically derived smaller peptide pieces destroys this information. Thus, the same single protein molecule may have five different phosphorylation events or four, three, two, one, and all combinations in between, and top-down proteomics preserves this information. The different states of phosphorylation (or other PTMs) of a single protein molecule are collectively referred to the “proteoforms” of that protein, and the importance of proteoforms remains a major motivation to improve top-down technologies (Aebersold et al., 2018; Smith and Kelleher, 2018). However, how soon top-down approaches become routine for studying complex proteomes remains to be seen and for now, nearly all proteomic approaches used in research laboratories and MS core facilities rely mainly on the bottom-up approach.
MS versus MSn
Acquiring a single mass spectrum on an intact protein or peptide (MS) is different than acquiring a tandem mass spectrum (MSn, most commonly either MS2 or MS3), which measures the m/z of fragment ions that are generated from fragmentation of the parent molecule (often referred to as the “precursor” ion, from an MS1 spectrum, in this context) (Figure 1).
Figure 1.

Principle by which peptides are sequenced via tandem mass spectrometry. First, a mass spectrum (MS1) is obtained to determine which intact masses are present in a sample as it is injected into the mass spectrometer (far left, size select). Next, each identified peptide is independently isolated using a mass filter and fragmented, generally via collision with inert gas molecules, within the instrument (middle, fragment). Finally, the fragment ion spectrum (MS2) is used as a template for matching in silico digested and fragmented proteomes to identify and annotate the sequence including posttranslational modifications (right, interpret sequence).
For small molecules (i.e. not polymers of amino acids), single sector instruments are most commonly used, although there can be fragmentation of covalent bonds as well, and it is these resultant fragment ions that are key to some of the most highly sensitive ways to perform targeted mass spectrometric (MS) measurements, known as selected reaction monitoring (SRM). SRM is generally performed with a triple quadrupole mass spectrometer, the workhorse of the pharmaceutical industry (although Orbitrap or TOF instruments are also capable of SRM-like analyses). It is also widely used in hospitals for drug analysis. These instruments are an excellent way to start learning about mass spectrometers because they illustrate all of the features used in general in all the instruments, but with different mass analyzers. Furthermore, SRM is now a routine method for analyzing the proteome with the greatest sensitivity, as will be described below.
In contrast to MS-based metabolomic work, proteomic work requires the use of tandem mass spectrometers, rather than single sector instruments. The reason for this is that to get the sequence of the peptide under study and, thus, the identity of the protein from which that sequence is derived, one must fragment the peptide bonds first. For this to occur, there is a “trap” where the ions have their energy raised, either via gas collision or via transfer of electrons from a source. This excitation results in an array of fragment ions that are derived from the breakage of peptide bonds joining the amino acids. Thus, one has a number of fragments of peptides that can have an intact amino terminus or an intact carboxy terminus. It is important to note that de novo amino acid sequencing is not completely feasible in MS-based methods on a proteome scale. Instead, it is best to have a complete in silico sequence of the proteome of the species under study as a search space constraining database. To generate this, a whole proteome is digested using trypsin in silico, and each resulting peptide is fragmented in silico to produce a possible fragment ion spectrum. Using these, the experimental fragment ion spectra are compared with the in silico fragmentation patterns, and best matches are used to confidently identify the peptides identified using MS. Thus, the development of modern search algorithms and software that automate the conversion of a MS/MS peptide fragmentation pattern into a real amino acid sequence was a critical development in the field of proteomics. There are also recent reports with success in creating de novo MS/MS sequencing methods (Peng et al., 2021), but since they may not be as reliable as having a bona fide transcriptome-derived proteome sequence to match up with the MS data, it is always helpful to have that information on hand to ensure accuracy. For plant research, this may be a bottleneck for using modern genomic approaches with the many thousands of potentially useful species of crops and plants found across the globe but this may be alleviated as the de novo sequencing methods improve.
Quantitative proteomics and the importance of understanding P-values and q-values
Whether one is measuring the concentration or the extent of modification with PTMs of all 30,000 plant proteins, it is absolutely essential to quantify these properties of proteins after isolation from plants following different chemical, environmental, or genetic perturbations for making conclusions about the mechanism by which that perturbation changes the plant phenotype. This need for quantitation brings special challenges to bear, compared with simple qualitative analyses of what sequences and modifications are present. Various methods have been developed and employed with plant tissues toward this goal, and they can be generally grouped into isotope-assisted and isotope-free methods. Regardless of which one is used, the most critical factor is the statistical treatment of the data. Thus, when one is dealing with hundreds and thousands of rows in an excel sheet with columns of data from replicate samples, the statistical methods that we normally were taught, that is the P-value, cannot be used without an understanding of its limitation with large datasets. There are many methods of performing corrections for statistical analyses with large data sets (cf. Krzywinski and Altman, 2014) but for this discussion we will focus on the q-value, and compare it to the P-value the reader is probably more familiar with. P-values are fine in experiments with smaller data sets. But when one is faced with an excel sheet containing thousands of rows, it is misleading. The reason for this is that when there are thousands of rows, at a P-value of 0.01, there will be a 1% chance that the treatment is showing a “significant” change, although this is totally by chance, rather than a real effect of the treatment. You can prove this to yourself by randomizing the columns of treatment and control data in the thousands of rows and then determining which proteins remain at the 1% level of P-value significance. This analysis is revealing because it reveals how large data sets are vulnerable to the multiple testing problem, that is the list of statistically significant proteins changes each time the sample is randomized.
In contrast, using the q-value is one of several ways to correct for this problem and in principle, should be used with large datasets. q-Values are essentially P-values that have been adjusted using a false discovery rate, which can be predicted for any possible distribution of P-values that emerge through statistical testing. In this way, the chance of encountering false positives in a large dataset is substantially reduced. However, while the q-value is much less prone to false positive artifacts, it can also reject some true positives. Thus, the choice of whether one uses a P-value or q-value to narrow the field down to a few “hits” is based on what kind of validation follow-up experiments are performed. If these follow-up experiments are not too timely and laborious, one can afford to use the P-value and avoid losing important true positives, while retaining the many false positives. On the other hand, if one uses the q-value, then there are fewer false positives to worry about, albeit at the loss of potentially important true positives. Thus, if the researcher wants to be certain about which proteins are potentially important biologically, one should use the q-value, but if there are enough validation experiments possible, this is not a hard and fast rule.
Deciphering the 3D structure of proteins with mass spectrometry
Having reviewed the fundamentals of mass spectrometry, the experimental approaches, and statistical treatments of MS data, we are now ready to encounter methods in mass spectrometry that are designed specifically for “structural proteomics.” There are three methods for studying the 3D structure of proteins using mass spectrometry, loosely divided into those that analyze covalent versus noncovalent modifications. The covalent bond-based methods are known as covalent labeling (CL) and crosslinking (XL), whereas the noncovalent method is called hydrogen–deuterium exchange (HDX). All of these methods work by labeling regions of the protein that are exposed to the bulk solvent, and these region-specific modifications can be detected at the peptide level in a bottom-up MS experiment. By analyzing which peptides are the most frequently modified in response to specific conditions, one can then map the regions of the protein that are undergoing conformational shifts or changes in interactions in response to those conditions. Such methods are widely used in both industry and academic labs. In the commercial arena, one of the major uses for these MS-based methods is in the identification of linear and 3D epitopes, toward engineering of monoclonal antibodies targeting specific diseases, resulting in the emergence of a rapidly expanding protein therapeutics industry. For example, Humira is an antibody that binds tumor necrosis factor in the blood and has proven effective in ameliorating arthritis and other inflammatory diseases. Another example is Herceptin, an antibody that binds to the ectopic domain of epidermal growth factor receptor, and has shown to have therapeutic value in the treatment of certain cancers. A crucial feature of improving the efficacy of these antibodies is identifying the few amino acids critical for the antibody–antigen recognition. Once identified, one can use deep landscape mutagenesis to create thousands of mutant antibodies. In this procedure, every possible mutation at four or five residues can be explored. When coupled to a suitable binding assay, this engineering strategy can provide marked and reiterative improvements in affinity that result in greater therapeutic value. Similarly, the recent SARS CoV-2 pandemic has created a great need for identifying which variants in the spike protein are involved in binding the antibodies we make naturally, as well as those being engineered in the laboratory. This is a critical area that is surprisingly still in its infancy considering that many issues, such as glycan shielding (due to glycosylated residues present in antibodies derived from mammalian or fungal expression systems) and the presence of dynamically moving domains (disordered domains), create great problems in using traditional structural methods such as cryo-EM, NMR, and X-ray crystallography. Another very important output of these MS-based methods is the identification of surfaces that are exposed or hidden when a protein attains a certain conformational state, or when a protein is interacting with another protein, RNA or DNA. Structural MS-based methods such as CL, XL, and HDX thus provide a rapid, orthogonal strategy for identifying epitopes and conformational changes without requiring the milligram quantities of pure protein needed by traditional methods in structural biology.
However, it is also important to note that the real power of these MS-based methods is that they can be used to study proteins that have been highly enriched and purified as well as proteins in very complex mixtures or environments such as cell lysates or living cells. The critical distinction here is that the MS-based methods can be used with complex protein solutions at the microgram level and are not restricted to proteins that are easy to express and purify, even though both applications are extensively used in the academic world.
In this context, an important difference between CL, XL, and HDX is that the former two methods probe changes in solvent accessibility of the amino acid side-chains, whereas the latter measures differences in solvent accessibility of the peptide backbone itself. In most cases, the two observations are similar, that is residues that are hidden show little exchange with protons at the peptide backbone as well as with modification of the side-chain and conversely, those that show peptide bond exposure are also generally exposed in terms of accessibility of reagents for the side-chain. Most CL reagents react with specific types of amino acids (e.g. carbodiimide-based reagents for the carboxylic acid containing amino acids, glutamic acid, and aspartic acid), but some highly reactive species, such as hydroxyl radicals, are capable of reacting with all 20 amino acids albeit with differing rates (for details, see two very comprehensive reviews by the founders of this field, Mark R. Chance at Case Western Reserve University and Michael L. Gross, at Washington University; Kiselar and Chance, 2018; Liu et al., 2020). Because of this high reactivity and its small size, the hydroxyl radical offers many advantages in these experiments and has been used previously to “footprint” the 3D structure of RNA. More recently, hydroxyl radical footprinting is one of the most widely used methods for investigating 3D structure of proteins as well, and various methods for producing this ROS, that is a reactive oxygen species, have been devised.
Traditionally, hydroxyl radicals can be produced by the Fenton reaction using Fe to break apart hydrogen peroxide, much like enzymes that create hydroxyls as part of the reactive oxygen species do, in living cells. The use of hydroxyls for mapping the 3D structure of biopolymers such as RNA originally used a synchrotron to perform radiolysis, that is very high energy ionizing radiation was used to create hydroxyl radicals directly from water. This same approach was first explored by Mark Chance for protein footprinting, and his laboratory remains a leader in the field (cf. Chance et al., 2020). It is important to note that the Fenton reaction is rarely used, even in a rapid liquid pulse apparatus for protein structural studies, because proteins show dynamic motion that can be in micro and millisecond timescales and thus, creating hydroxyls with a stability spanning nanoseconds or microseconds is best to avoid over-reacting the protein rather than getting a snapshot. Since hydroxyl radicals have an in-solution lifespan on the order of nanoseconds, methods to create such brief pulses are preferred. Besides the synchrotron, two other methods have been devised to create such brief pulses to “paint” the surface accessible amino acid side-chains in a protein: using an electronically generated plasma to create the radicals directly from water (called plasmalysis, cf. Minkoff et al., 2017) developed recently in the authors’ laboratory, or using pulses of light from a laser to create hydroxyls in a solution containing hydrogen peroxide (cf. Liu et al., 2020, a very comprehensive recent review). Many of these laboratories are also exploring the use of their pulsed technology to create radicals from other molecules, such as sulfate or nitrate, to increase the versatility and reactivity with all 20 amino acids. In addition, any reagent which reacts with a particular amino acid (e.g. sulfhydryl or amino reagents that attack cysteine and lysine, as well as carbodiimide-based reagents that attack the carboxyl acid containing amino acids, glutamic, and aspartic acids; cf. Liu et al., 2018) can also be used to probe for changes in conformation within proteins, although the potential issue of reactive promiscuity during extended lifetimes of the reagent always needs to be considered.
The unifying parameter utilized in these experiments is the solvent-accessible surface area (SASA) value of a molecule or part of the molecule. This can be calculated from the amino acid sequence in proteins for which a high-resolution 3D structure is known, via cryo-EM, NMR, or X-ray studies. For these experiments, one calculates the SASA by computationally rolling a tiny ball the size of a solvent molecule (usually water) along the protein surface and measuring how far it penetrates. For the purpose of this discussion, let us consider the 3D structure of the plant plasma membrane proton pump (abbreviated as AHA for Arabidopsis H+-ATPase), a ∼1,000 amino acid long polytopic membrane protein (10 transmembrane domains) whose structure for the first 900 amino acids (i.e. lacking the C terminal regulatory domain) was determined over a decade ago (Figure 2A; Pedersen et al., 2007). Although there are 12 genes encoding AHA in the A. thaliana genome, AHA isoforms 1, 2, and 3 are the most highly expressed and are essential for generating the protonmotive force, composed of an electric potential plus a chemical gradient of protons, that is used to drive the secondary transport of most solutes across the plasma membrane. In addition to this fundamental role in plant and fungal nutrition, in land plants especially, this enzyme is regulated by signaling pathways that alter the phosphorylation status of several serine and threonine amino acids within the C-terminal regulatory domain of this pump, thereby causing an increase or decrease of the cell wall pH, which in turn increases or decreases the rate of cell elongation in response to changes in auxin, light, gravity, and the presence or absence of pathogens (Haruta et al., 2015; Falhof et al., 2016; Figure 2B). Interestingly, although the 3D structure of this enzyme was reported over a decade ago using X-ray crystallography, it did not show the position of amino acids in the important C terminal 100 amino acids, presumably because this regulatory region was not locked into a specific conformation prior to the formation of the crystals (Figure 2C, Post-Albers scheme).
Figure 2.
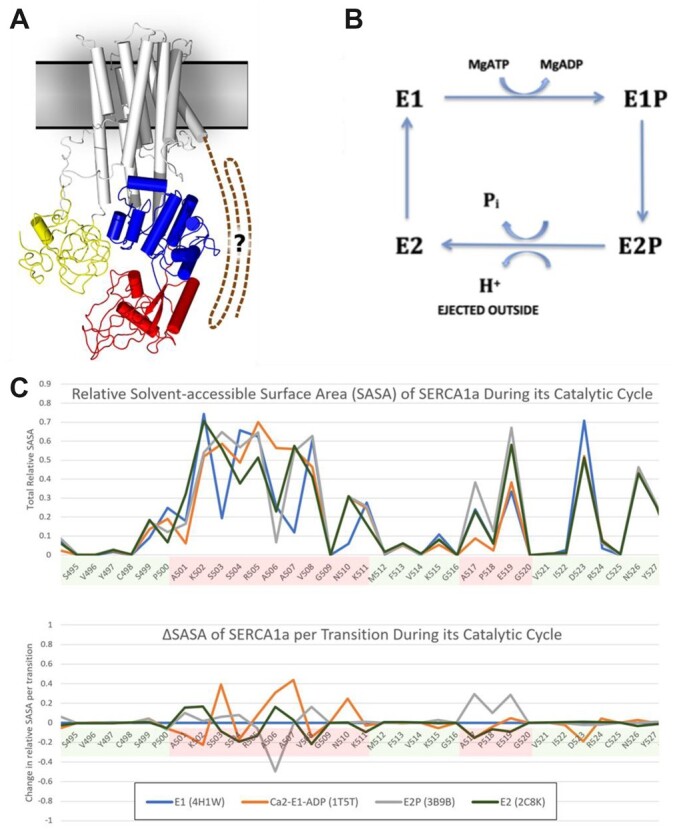
The plant plasma membrane proton pump is a P-type ATPase with a 3D structure, catalytic cycle and conformational changes similar to other well-known P-type ATPases including the calcium pump found in the sarcoplasmic reticulum (SERCA) of animals. A, 3D structure of AHA2 predicted from X-ray crystal structural data after expression and purification in yeast. Yellow, red, and blue domains are the actuator, nucleotide binding, and phosphorylation domains, respectively, in the catalytic portion of the protein. The regulatory C-terminal domain is shown with a question mark since there is no structural data available from the crystals on this domain although it plays a critical role in plant physiology since it regulates changes in catalytic activity in response to auxin, blue light, and other effectors. In (B), the Post-Albers Catalytic Cycle Scheme for the four conformations predicted to be present in AHA2 as it passes through its cycle of ATP hydrolysis and proton transport. E denotes the enzyme and P denotes the form of the enzyme which is phosphorylated at an aspartyl residue. In (C), the SASA values (solvent accessibility surface area) for the side-chains of amino acids in a portion of the SERCA mammalian calcium pump that, like AHA2, undergoes the Post Albers Scheme of conformational changes. Using publicly available software, we calculated the SASA for each of these SERCA conformations and presented this on the Y-axis, as a function of amino acid position in the protein shown on the X-axis. Green shading corresponds to no/low changes in solvent exposure, whereas Red indicates amino acids whose solvent exposure changed significantly.
The fact that proteins undergo conformational transitions is a critical part of studying their structure as it relates to function and this is well illustrated with the plasma membrane calcium pump present on the sarcoplasmic reticulum of animal cells (called SERCA). This enzyme has been well studied because it is very abundant, easily purified, and importantly, has four well-known different conformers, that is conformational states, since they are dependent on the concentration of MgATP and calcium. While conformations of the AHA’s are also dependent on MgATP, the concentration of protons is harder to control, and since there are nonspecific effects of changing the pH on any protein’s structure, the AHA structure is difficult to study. However, for the purposes of this illustration, we took four 3D structures reflecting the major Post-Albers catalytic conformers of the calcium pump and calculated the SASA for the side-chain of each of the protein’s amino acids. A representative plot of these SASA values for a portion of the protein is provided in Figure 2. Although only a brief portion is shown for illustrative purposes, it is evident that there are substantial 3D changes during catalytic transitions, which are observable at the level of secondary structure as reflected in the calculated SASA. Such transitions at distinct sites of the protein can thus be studied and quantified during the protein’s dynamic changes in structure as it undergoes its catalytic cycle. Although hydroxy radical footprinting is currently one of the most commonly used CL method to measure changes in SASA, other reagents that target a more select group of amino acids are often used as well. For example, in Liu et al. (2018), the author’s laboratory analyzed the binding of a plant peptide hormone to its receptor via footprinting with GEE/EDC, reagents that preferentially label surface exposed carboxyl groups in glutamic acid and aspartic acid.
In general, one would expect that there is a high degree of correlation between the computed SASA values and the reactivity found during MS experiments with covalent (CL and XL) and noncovalent (HDX) reagents. In practice, due to the unknown degree of dynamic motion and the effects of neighboring residues on the reactivity of particular amino acids in the sequence (i.e. chemical context), this is not always the case. It is also not widely appreciated that even small tryptic peptides can attain a conformation, that is a folded state that is observed during NMR or IM MS experiments. Therefore, there is generally a high degree of consistency of MS-based CL, XL, and HDX observations with those expected based on the static conformations observed in traditional methods, but it is important to view these MS data as reflecting a population of microstates that a protein is undergoing as it dynamically moves within the same overall biological conformation. While NMR-based methods are probably the least prone to such problems (i.e. HDX with mass spectrometry and HDX with NMR should show identical results), unknown changes in protein structure that occur when it leaves a solution and enters the vitrification state with detergents present on the grids during cryo-EM or the crystal state with X-ray methods (particularly since crystal contacts can artifactually enforce a conformation), can complicate the interpretation, but these are generally minor issues in most experiments.
Another method widely used to probe protein 3D structure via mass spectrometry is crosslinking. In the author’s laboratory, we have explored two methods which can be performed both in vivo and in vitro depending on the reagents used. One uses an interesting innovation developed in the Schultz laboratory (cf. Noren et al., 1989), where a single amino acid in a protein is replaced by a genetically encoded, unnatural amino acid, benzoyl phenylalanine (BPA). BPA is a photoaffinity reagent that reversibly converts to a highly reactive carbene intermediate in the presence of ultraviolet light, and this intermediate can enter the C–H bond of any side-chain in amino acids close to the position of the BPA, forming a new carbon–carbon bond that is irreversible once formed. We used this method to identify the points of contact between the regulatory C-terminal domain and the various cytoplasmic domains of the plasma membrane proton pump AHA2 (Nguyen et al., 2018; Figure 3A). A key advantage of this method is that since only low doses of long wavelength UV light (∼360 nm, which does not damage proteins) are needed, one can incorporate the reagent at single amino acid locations and activate the XL in vivo, thus eliminating artifacts caused by altering the conformation, for example by isolating membranes and solubilizing the protein with a detergent. The second method we have used is chemical XL with the MS-cleavable crosslinker, DSSO. We also used this to study the dynamic interactions of AHA2’s C-terminus with its other domains (Figure 3), and were able to distinguish between intermolecular interactions in oligomeric AHA2 and intramolecular interactions by employing isotopic labeling of the protein before XL (Nguyen et al., 2020). While DSSO’s reactivity is restricted largely to primary amines (lysine residues and the protein’s N-terminus) because of its N-hydroxysuccinimide chemistry, it has several key advantages: it does not need to be genetically encoded into a protein and instead may be added directly in-solution, its linker arm length allows it to capture intramolecular as well as intermolecular interactions, and it can be cleaved inside the mass spectrometer using high-energy collision. This extra level of fragmentation always produces a set of diagnostic peptides with a well-characterized mass-shift, which means that crosslinked peptides can be detected and selected for further fragmentation of the joined peptides separately. In a typical MS experiment, the more a peptide can be fragmented, the more information can be gained from it. Therefore, this boosts the utility of using DSSO as a crosslinker for rapidly scanning many interactions in a single MS experiment.
Figure 3.
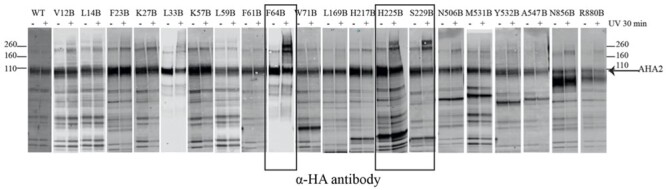
Cross-linking of AHA2 by BPA using site-directed mutagenesis to produce an enzyme containing BPA at various locations within the protein. The three boxed lanes on this sodium dodecyl sulfate–polyacrylamide gel electrophoresis show positions of BPA in which a prominent crosslinked product is formed after photolysis of intact cells. Figure is taken from Nguyen et al. (2018).
Thermal proteome profiling
For many years, it has been recognized that each protein has a characteristic temperature at which it unfolds, known as its melting temperature (Tm). In addition, it has also been recognized that when a protein binds its ligand (e.g. ATP binding enzymes), the protein undergoes an increase in stability with respect to thermal denaturation reflected in a detectable increase in the protein’s Tm while in the ligand-bound state. The Tm value is calculated with a pure protein by identifying the percentage of protein molecules in a solution, which retain their folded state, using various methods for distinguishing folded from denatured protein. Precipitation upon heating is one method which has been exploited widely in combination with centrifugation and sodium dodecyl sulfate–polyacrylamide gel electrophoresis, for example, to quantify the percentage of protein that precipitates out after treatment for a few minutes at any temperature above (or below) room temperature. Savitski et al. (2014) reported a method that utilizes this same feature but instead of working with a single pure protein, he demonstrated that one could start with a complex proteome, present in the intact cell or in a crude lysate, and by coupling this analysis to a multiplexed tandem mass spectrometry method for identifying and quantifying any protein that remains in the supernatant after heat denaturation, one could readily determine the Tm values for thousands of proteins simultaneously (see top scheme outlining the procedure in Figure 4A). Pilot experiments demonstrated that this ligand-induced shift in Tm could be unambiguously observed for hundreds of proteins in a complex proteome in a crude cell extract of A. thaliana containing tens of thousands of proteins simply by adding MgATP to the lysate.
Figure 4.
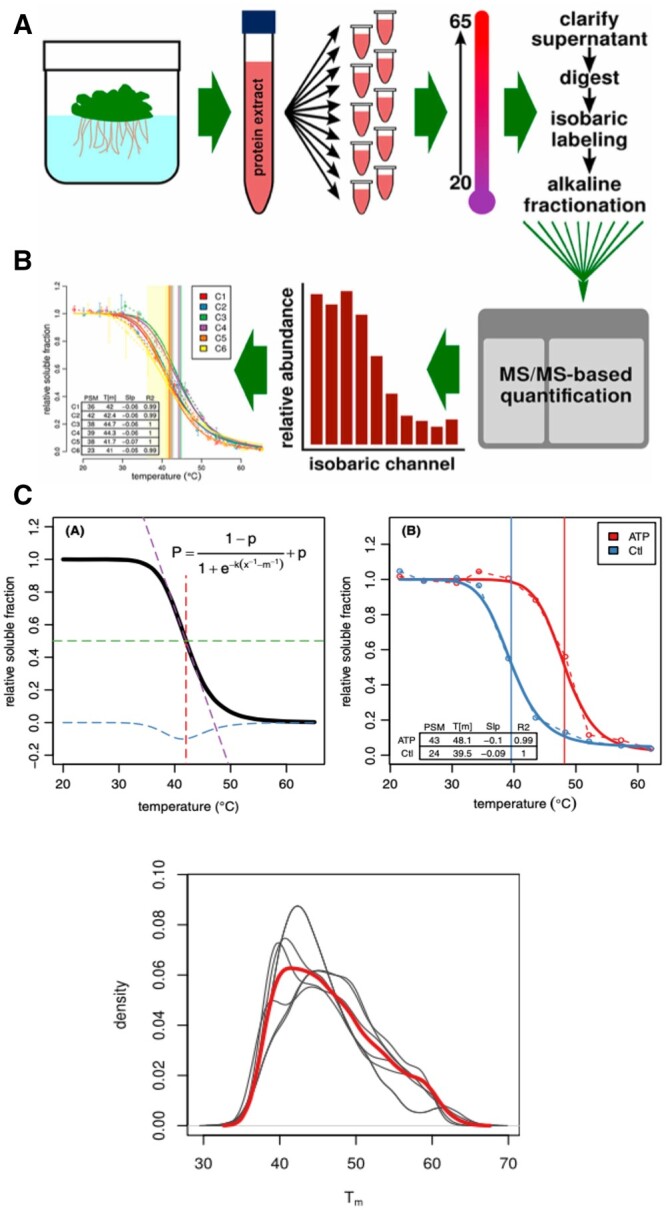
TPP of A. thaliana crude cell lysates. In (A), a summary of the overall scheme for measuring the thermal denaturation temperature for thousands of proteins in the A. thaliana proteome simultaneously. On the left side of (B) is shown the profile for a model protein, with the equation used to predict the thermal denaturation temperature. On the right of (B) is shown experimental data for one protein after lysate is treated with or without MgATP. The increased Tm denotes an increase in stability and thermal denaturation temperature required to unfold a protein after its ligand (here, MgATP) is bound to the protein. In (C), the overall TPP temperatures for thousands of proteins isolated from plants grown under varying conditions. Data, figures, abbreviations, and units of measurement in figures are taken from Volkening et al. (2019).
Recently, while this method has been extensively utilized in animal and microbial systems, there has been only one report on its use in plants so far. In 2019, the author’s laboratory (Volkening et al., 2019) reported that crude lysates of A. thaliana behaved much like those of animals and microbes in terms of thermal proteome profiling (TPP) applications and showed that with MgATP as a positive control, that is those proteins annotated as having an ATP binding site were enriched in the group that altered their Tm when MgATP was added compared with a control lacking added MgATP (Figure 4B). While this system offers great promise for proteome wide conformational studies, it has some limitations. First, the computational analysis requires a substantial percentage of the population of any one protein sequence to be altered in its Tm. Thus, for those enzymes in which the phosphorylation status only affects less than a majority of the molecules (i.e. the stoichiometry is less than 1.0 phosphates per protein), there is insufficient resolution to clearly delineate the identity of that protein. Furthermore, even when a protein can be identified with an altered Tm, the data do not provide any information on the specific domains or amino acids that are involved within that protein. Therefore, TPP may best be used in conjunction with other structural proteomics methods.
Limited proteolysis
Another MS-based method that reports on changes in solvent accessibility involves susceptibility to limited protease digestion (Kaur et al., 2018; Ma et al., 2018; Meng et al., 2018; Cabrera et al., 2020; Pepelnjak et al., 2020; Cappelletti et al., 2021; Wiebelhaus et al., 2021). Thus, it is well known that when proteins have their conformation altered, their susceptibility to digestion with proteases is altered; this property has allowed the development of proteolytic methods for generating peptides in response to solvent exposure. For example, early work studying the inhibitory properties of the C-terminal domain of the Neurospora plasma membrane proton pump utilized limited proteolysis (Mandala and Slayman, 1988). However, modern mass spectrometry methods allow detection of these unique cleavage events at the peptide level, especially when one uses highly promiscuous proteases which cleave nonspecifically throughout a peptide backbone (such as proteinase K). Controlled digestion of proteins during this “limited” proteolysis step (LiP) can then be arrested at defined timepoints, after which the peptides can be completely digested with trypsin or LysC in preparation for analysis by LC-MS/MS. At the peptide level, “nontryptic” cleavages that emerged during the LiP step can then be distinguished from standard tryptic cleavages, and be associated with specific conformational changes. Because this method detects conformational shifts occurring in the peptide backbone, it fits into the category of techniques which measure changes in SASA of the peptide bond (like HDX) rather than the amino acid side-chain. Like TPP, it is amenable to multiplexing using isobaric isotope-encoded MS reagents or with isotope-free methods for quantification. Although there have been no reports on its use in plant systems, its utility for proteome wide analyses with crude mixtures is well documented and it is expected that its use will become more widespread in the plant community as access to the hardware and software required for its use becomes more widespread among the biological community. In general, one must appreciate that the study of phosphorylation or ubiquitinylation as PTMs in plants (and all organisms) did not become a widespread technique until a reliable method for enrichment of these PTMs became available (e.g. titanium dioxide columns for phosphorylation or antibody-based system for ubiquitinylation). In the same respect, MS methods that study conformational changes in crude mixtures require a means of enriching for those particular proteins that become altered by these methods. Methods aimed at this goal are being explored in the limited proteolysis area, as well as the others, including, in particular, HDX, and once these are available, it is expected that such experiments looking at changes in protein 3D structure in intact tissues will be as common as PTM studies.
Native mass spectrometry and soft landing: Two MS methods at the bleeding edge
Approximately a decade ago it became clear that proteins move in the electric field within the vacuum of a mass spectrometer mainly as naked molecules, that is without any noncovalently bound ligands or even water molecules. The reason for this is that in order to obtain sufficient charge on each protein so that an m/z value could be obtained that were within the mass range of the instruments (generally under 5,000 m/z, despite the average protein having a molecular weight in the order of 50,000 Da or higher) it was necessary to run the samples under highly acidic conditions. Thus, samples were routinely run under ESI in 0.1% formic acid. Further denaturation was obtained by using organic solvents such as acetonitrile at concentrations of 5% and higher in the sample solvent as well as in the RP-HPLC solutions used to elute the proteins from the reversed phase solid supports. As the field matured, it was observed that one could also apply protein samples at pHs more akin to those which maintain native 3D structure, that is pH 7, and that the proteins would still be emitted during electrospray as charged individual molecules that would move as normal in the electric field. In order to make this method more amenable to typical protein analysis, it was necessary to develop mass analyzers that worked at higher m/z. To that end, ion traps and time of flight instruments capable of detecting m/z values of 100,000 and more, without loss of resolution, were produced, with the ultra-high mass range hybrid quadrupole–orbitrap mass spectrometer (Q-Exactive UHMR) being a notable example. This field has now matured extensively and culminated in more routine observations of very large molecular weight folded proteins and macromolecular complexes that can be observed in the mass spectrometers without denaturation. A good example of this is the recent determination of the molecular weight of a single ribosome, that is a complex of dozens of proteins and RNA molecules that fly in the instrument as a single analyte at a discrete m/z (van de Waterbeemd et al., 2017). Interestingly, on the higher m/z shoulder of the peak, one observes ribosomes containing sub-stoichiometric loosely associated proteins whose identity was known based on wet work performed with other technologies in the laboratory. A very attractive aspect of this work, within the general context of assembling an atlas of the cell’s proteome, is that it suggests the feasibility of observing the loss of individual noncovalent ligands—be they small molecules or even associated peptides or proteins—by performing controlled denaturation in the mass spectrometer, which can be done by carefully altering conditions such as the concentration of organic solvent or the collision energy. These technological advances offer more promise for bridging the gap between “live” proteins and enzymes and the dead proteins and proteolyzed pieces that we normally use during MS proteomics experiments.
Another promising application of nondestructive mass spectrometry is the concept that it may be used as a preparative method for purifying proteins for downstream use in other experiments, rather than simply destroying these analytes. One such preparative application for studying biological macromolecules is a technology known as “soft landing,” in which a hydrated surface is used under conditions where the macromolecule can land onto the surface without being destroyed (instead of analyzing the protein molecule as an electronic event after it hits the metal surface of the detector). In 1999, Richard Smith demonstrated the use of soft landing combined with PCR to use the mass spectrometer as a preparative instrument rather than an analytical instrument for separating and purifying DNA molecules based on their different molecular weights (Feng et al., 1999). In essence, this is the ultimate size exclusion chromatography column since the speed and resolution of working in the gas phase greatly exceeds that which can be done in any other fashion using normal metal or glass chromatography or centrifuge tubes. Graham Cook took this one step further and demonstrated that one can “soft land” protein molecules. In other words, he was able to use nondenaturing mass spectrometer to inject and elute intact protein molecules onto a soft surface, allowing them to retain their enzymatic activity despite being run in the gas phase and collected on hydrated surfaces rather than a surface used for detection (Ouyang et al., 2003). While this field is still in its infancy it has interesting potential for future advances, offering a rapid way to separate important biomolecules in a preparative fashion so that other methods of analysis can be performed after they leave a mass spectrometer.
Concluding remarks
In this review, we have described the latest technologies available for studying the 3D structure of proteins in living cells. Just as most modern mass spectrometers have a working life of only 5–10 years before they are made obsolete by new instruments, the manner in which these technologies are used by biologists also undergoes rapid evolution. Thus, the communication between disciplines, that is between the technologists who develop new methodologies and the plant biologists who can make best use of them, is essential to ensure that new understanding, and not just a collection of data, is created. These new technologies provide enormous opportunities for deciphering how a proteome operates in vivo. However, there are also many challenges ahead to ensure that technology is being “pulled” in the right directions by biologists attempting to solve the most fundamental questions left in our understanding of the mechanisms by which plant cells function.
OUTSTANDING QUESTIONS BOX.
While it was recently shown that ribosome and membrane proteolipid macroassemblies could be analyzed using nondenaturing mass spectrometry, what level of affinity is needed to identify proteins that are more loosely bound but highly important for function?
Do XL and CL methods performed in vitro always replicate the in vivo situation, for example in a very crowded cytoplasm or intracellular organelle?
Can university core facilities become more adept at providing these newly emerging technologies to the biologists with the pressing questions that require new ways of analysis?
Will the use of mass spectrometers as preparative instruments via “soft landing” provide a new way to analyze single protein molecules via cryo-EM or other sensitive imaging methods?
Acknowledgments
Figures 1 and 2c were created with BioRender.com.
Funding
The research (Figure 2) and writing of this manuscript was supported by grants to MRS from the Department of Energy (Biological Energy Sciences, Grant No. DEFG02-88ER13938) and the National Science Foundation (Molecular and Cellular Biochemistry Grant No. 1943816 and Plant Genome Research Program Grant No. 2010789) and are gratefully acknowledged.
Conflict of interest statement. None declared.
Contributor Information
Matthew R Blackburn, Department of Biochemistry and Center for Genomic Science Innovation, University of Wisconsin, Madison, Wisconsin 53706, USA.
Benjamin B Minkoff, Department of Biochemistry and Center for Genomic Science Innovation, University of Wisconsin, Madison, Wisconsin 53706, USA.
Michael R Sussman, Department of Biochemistry and Center for Genomic Science Innovation, University of Wisconsin, Madison, Wisconsin 53706, USA.
M.R.B. designed and performed the research in Figure 2. B.B.M. contributed mass spectrometric tools and helped analyze data. M.R.S. wrote the paper and M.R.B. and B.B.M. assisted in the editing.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plphys/pages/General-Instructions) is: Michael R. Sussman (msussman@wisc.edu).
References
- Aebersold R, Agar JN, Amster IJ, Baker MS, Bertozzi CR, Boja ES, Costello CE, Cravatt BF, Fenselau C, Garcia BA, et al. (2018) How many human proteoforms are there? Nat Chem Biol 14: 206–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815 [DOI] [PubMed] [Google Scholar]
- Breindel L, Yu J, Burz DS, Shekhtman A (2020) Intact ribosomes drive the formation of protein quinary structure. PLoS ONE 15: e0232015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera A, Wiebelhaus N, Quan B, Ma R, Meng H, Fitzgerald MC (2020) Comparative analysis of mass-spectrometry-based proteomic methods for protein target discovery using a one-pot approach. J Am Soc Mass Spectrom 31: 217–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappelletti V, Hauser T, Piazza I, Pepelnjak M, Malinovska L, Fuhrer T, Li Y, Dörig C, Boersema P, Gillet L, et al. (2021) Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ. Cell 184: 545–559.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance MR, Farquhar ER, Yang S, Lodowski DT, Kiselar J (2020) Protein footprinting: Auxiliary engine to power the structural biology revolution. J Mol Biol 432: 2973–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooks RG, Mueller T (2013) Through a glass darkly: Glimpses into the future of mass spectrometry. Mass Spectrom (Tokyo) 2: S0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falhof J, Pedersen JT, Fuglsang AT, Palmgren M (2016) Plasma membrane H(+)-ATPase regulation in the center of plant physiology. Mol Plant 9: 323–337 [DOI] [PubMed] [Google Scholar]
- Feig M, Yu I, Wang PH, Nawrocki G, Sugita Y (2017) Crowding in cellular environments at an atomistic level from computer simulations. J Phys Chem B 121: 8009–8025 doi:10.1021/acs.jpcb.7b03570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Wunschel DS, Masselon CD, Pasa-Tolic L, Smith RD (1999) Retrieval of DNA using soft-landing after mass analysis by ESI-FTICR for enzymatic manipulation. J Am Chem Soc 121: 8961–8962 [Google Scholar]
- Gruebele M (2021) Protein folding and surface interaction phase diagrams in vitro and in cells. FEBS Lett 595: 1267–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guin D, Gruebele M (2019) Weak chemical interactions that drive protein evolution: Crowding, sticking, and quinary structure in folding and function. Chem Rev 119: 10691–10717 [DOI] [PubMed] [Google Scholar]
- Haruta M, Gray WM, Sussman MR (2015) Regulation of the plasma membrane proton pump (H(+)-ATPase) by phosphorylation. Curr Opin Plant Biol 28: 68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur U, Meng H, Lui F, Ma R, Ogburn RN, Johnson JHR, Fitzgerald MC, Jones LM (2018) Proteome-wide structural biology: An emerging field for the structural analysis of proteins on the proteomic scale. J Proteome Res 17: 3614–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselar J, Chance MR (2018) High-resolution hydroxyl radical protein footprinting: Biophysics tool for drug discovery. Annu Rev Biophys 47: 315–333. doi: 10.1146/annurev-biophys-070317-033123 [DOI] [PubMed] [Google Scholar]
- Kline KG, Sussman MR (2010) Protein quantitation using isotope-assisted mass spectrometry. Annu Rev Biophys 39: 291–308 [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Altman N (2014) Comparing samples—part II. Nat Methods 11: 355–356 [Google Scholar]
- Liu P, Haruta M, Minkoff BB, Sussman MR (2018) Probing a plant plasma membrane receptor kinase's three-dimensional structure using mass spectrometry-based protein footprinting. Biochemistry 57: 5159–5168 [DOI] [PubMed] [Google Scholar]
- Liu XR, Zhang MM, Gross ML (2020) Mass spectrometry-based protein footprinting for higher-order structure analysis: Fundamentals and applications. Chem Rev 120: 4355–4454. doi:10.1021/acs.chemrev.9b00815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma R, Meng H, Wiebelhaus N, Fitzgerald MC (2018) Chemo-selection strategy for limited proteolysis experiments on the proteomic scale. Anal Chem 90: 14039–14047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandala SM, Slayman CW (1988) Identification of tryptic cleavage sites for two conformational states of the Neurospora plasma membrane H+-ATPase. J Biol Chem 263: 15122–15128 [PubMed] [Google Scholar]
- Meng H, Ma R, Fitzgerald MC (2018) Chemical denaturation and protein precipitation approach for discovery and quantitation of protein–drug interactions. Anal Chem 90: 9249–9255 [DOI] [PubMed] [Google Scholar]
- Minkoff BB, Blatz JM, Choudhury FA, Benjamin D, Shohet JL, Sussman MR (2017) Plasma-generated OH radical production for analyzing three-dimensional structure in protein therapeutics. Sci Rep 7: 12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki G, Wang PH, Yu I, Sugita Y, Feig M (2017) Slow-down in diffusion in crowded protein solutions correlates with transient cluster formation. J Phys Chem B 121: 11072–11084 doi:10.1021/acs.jpcb.7b08785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TT, Blackburn MR, Sussman MR (2020) Intermolecular and intramolecular interactions of the Arabidopsis plasma membrane proton pump revealed using a mass spectrometry cleavable cross-linker. Biochemistry 59: 2210–2225 [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Sabat G, Sussman MR (2018) In vivo cross-linking supports a head-to-tail mechanism for regulation of the plant plasma membrane P-type H(+)-ATPase. J Biol Chem 293: 17095–17106 doi:10.1074/jbc.RA118.003528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG (1989) A general method for site-specific incorporation of unnatural amino acids into proteins. Science 244: 182–188 [DOI] [PubMed] [Google Scholar]
- Ouyang Z, Takáts Z, Blake TA, Gologan B, Guymon AJ, Wiseman JM, Oliver JC, Davisson VJ, Cooks RG (2003) Preparing protein microarrays by soft-landing of mass-selected ions. Science 301: 1351–1354 [DOI] [PubMed] [Google Scholar]
- Pedersen BP, Buch-Pedersen MJ, Morth JP, Palmgren MG, Nissen P (2007) Crystal structure of the plasma membrane proton pump. Nature 450: 1111–1114 [DOI] [PubMed] [Google Scholar]
- Peng W, Pronker MF, Snijder J (2021) Mass spectrometry-based de novo sequencing of monoclonal antibodies using multiple proteases and a dual fragmentation scheme. J Proteome Res 20: 3559–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepelnjak M, de Souza N, Picotti P (2020) Detecting protein-small molecule interactions using limited proteolysis-mass spectrometry (LiP-MS). Trends Biochem Sci 45: 919–920 [DOI] [PubMed] [Google Scholar]
- Rickard MM, Zhang Y, Gruebele M, Pogorelov TV (2019) In-cell protein-protein contacts: Transient interactions in the crowd. J Phys Chem Lett 10: 5667–5673 [DOI] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, Martinez Molina D, Jafari R, Dovega RB, Klaeger S, et al. (2014) Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346: 1255784. [DOI] [PubMed] [Google Scholar]
- Smith LM, Kelleher NL (2018) Proteoforms as the next proteomics currency. Science 359: 1106–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Lv T, Chen J, Wang J, Yao L (2019) Characterization of residue specific protein folding and unfolding dynamics in cells. J Am Chem Soc 141: 11363–11366 [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd M, Fort KL, Boll D, Reinhardt-Szyba M, Routh A, Makarov A, Heck AJ (2017) High-fidelity mass analysis unveils heterogeneity in intact ribosomal particles. Nat Methods 14: 283–286 [DOI] [PubMed] [Google Scholar]
- Volkening JD, Stecker KE, Sussman MR (2019) Proteome-wide analysis of protein thermal stability in the model higher plant Arabidopsis thaliana. Mol Cell Proteomics 18: 308–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiebelhaus N, Zaengle-Barone JM, Hwang KK, Franz KJ, Fitzgerald MC (2021) Protein folding stability changes across the proteome reveal targets of Cu toxicity in E. coli. ACS Chem Biol 16: 214–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler SJ, Mallinson SJB, St John PC, Bomble YJ (2021) Advances in integrative structural biology: Towards understanding protein complexes in their cellular context. Comput Struct Biotechnol J 19: 214–225 [DOI] [PMC free article] [PubMed] [Google Scholar]