

Abstract
Ovarian cancer is commonly diagnosed only after it has metastasized to the abdominal cavity (stage III). While the current standard of care of intraperitoneal (IP) administration of cisplatin and paclitaxel (PTX) combination chemotherapy has benefit, patient 5-year survival rates are low and have not significantly improved in the last decade. The ability to target chemotherapy selectively to ovarian tumors while sparing normal tissue would improve efficacy and decrease toxicities. We have previously shown that cisplatin-loaded nanoparticles (NPs) loaded within neural stem cells (NSCs) are selectively delivered to ovarian tumors in the abdominal cavity following IP injection, with no evidence of localization to normal tissue. Here we extended the capabilities of this system to also include PTX delivery. NPs that will be loaded into NSCs must contain a high amount of drug by weight but constrain the release of the drug such that the NSCs are viable after loading and can successfully migrate to tumors. We developed silica coated PTX nanocrystals (Si[PTX-NC]) meeting these requirements. Si[PTX-NC] were more effective than uncoated PTX-NC or Abraxane for loading NSCs with PTX. NSCs loaded with Si[PTX-NC] maintained their migratory ability and, for low dose PTX, were more effective than free PTX-NC or Si[PTX-NC] at killing ovarian tumors in vivo. This work demonstrates that NSC/NP delivery is a platform technology amenable to delivering different therapeutics and enables the pursuit of NSC/NP targeted delivery of the entire preferred chemotherapy regimen for ovarian cancer. It also describes efficient silica coating chemistry for PTX nanocrystals that may have applications beyond our focus on NSC transport.
Keywords: Neural Stem Cells, Ovarian Cancer, Targeted Drug Delivery, Silica Nanoparticles, Paclitaxel, Chemotherapy
Graphical Abstract
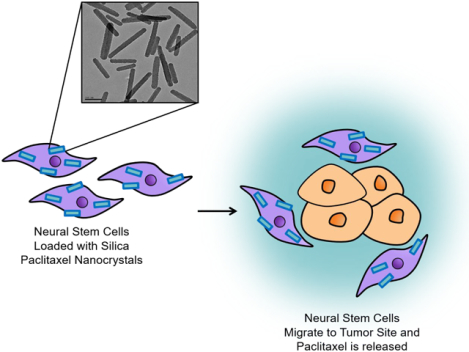
Introduction
Ovarian cancer is the leading cause of gynecologic cancer mortality for women in the US and each year ~20,000 women are diagnosed with it.1 The lack of validated screening programs and the absence of symptoms during the early stages of ovarian cancer contribute to the difficulty of early diagnosis. Because of this, many patients are diagnosed at an advanced stage of the disease with 60–70% of patients already at stage III or IV. Tumor debulking and IP chemotherapy using platinum- and taxane-based drugs are the current standard of treatment for patients, but the five-year survival rate remains at only 30%1–4. A targeted delivery system to concentrate chemotherapy specifically at ovarian tumor sites could substantially enhance both efficacy and reduce side effects for patients undergoing IP chemotherapy.
Cell carriers that have a propensity to migrate towards tumors or specific organs have shown promise for targeted therapy. Tumor tropic- or organotropic-cell carriers such as stem cells (mesenchymal stem cells; MSCs or neural stem cells; NSCs), T-cells, or macrophages, can be loaded with therapeutic nanoparticles (NPs) for targeted delivery5–10. We have previously shown that the clonal human HB1.F3.CD21 NSC line is tumor tropic, selectively migrates to a number of malignant solid tumors, including glioma, neuroblastoma, and metastatic breast carcinoma, and has shown safety in human glioma patients11–14. Recently, we demonstrated that IP administration of these NSCs can deliver both surface bound and internalized NPs to ovarian tumors with remarkable selectivity making them an attractive vehicle for drug targeting15. While NSCs can travel long distances and even cross the blood brain barrier to target tumors, we have generally found that NSCs most efficiently target tumors when they are administered in the locoregional area of the disease. Ie. Administering intracranially for brain tumors or intraperitoneally for ovarian cancer. We are currently focused on developing NSC/NPs as a treatment for ovarian cancer due to the frequency of the disease and the ability to administer IP to treat an IP disease. It is quite likely that such a therapeutic would be immediately applicable to other IP tumors and potentially to other locoregional cancer types such as bladder, brain, cervical, uterine and more.
For efficient drug transport, the NPs must be designed to contain a high amount of drug by weight to allow sufficient amounts of drug to be loaded into NSCs for effective tumor cell killing; however, the NPs must also prevent the drug cargo from affecting the NSCs during ex-vivo loading and during in vivo migration. We previously developed cisplatin-containing silica NPs (SiNP[Pt]) that met these criteria and demonstrated their targeted delivery to ovarian tumors by NSCs15. Here, we demonstrate the modularity of this platform technology and enable NSC/NP targeted delivery for the entire preferred chemotherapy regimen for ovarian cancer by developing a PTX-delivering NSC/NP variant.
Developing a PTX NSC/NP formulation presented substantially different challenges than our previous work with cisplatin. Cisplatin is a water-soluble compound where the active moiety is the platinum atom. It was thus possible to directly crosslink the Pt-drug into silica nanoparticles during synthesis and load these NPs into NSCs. On the other hand, PTX is an extremely hydrophobic, relatively large molecule that is prone to degradation in a variety of aqueous conditions16, 17. The most common formulations of PTX used in treating cancer are Taxol and Abraxane, which both rapidly release the drug upon injection18–20. Thus, in order to load NSCs, an experimental formulation of PTX was needed that allowed for high drug loading and delayed release.
While a number of NP formulations for PTX have been explored over the past several decades,1,2 nearly all have focused on finding improved excipients to solubilize the drug in aqueous environments, thus, not meeting our need for delayed release. We hypothesized that PTX nanocrystals would be a good starting point in addressing our requirements, as PTX nanocrystals are 100% drug by weight and have themselves shown slow dissolution in aqueous environments21, 22. Nanocrystals are generated by wet/dry milling or sonication of PTX23. They are typically stabilized by surfactants or block copolymers such as Pluronic F-127 (PEG-PPO-PEG). However, these nanocrystals are only temporarily stable and undergo fusion/aggregation over time24. Coatings to stabilize PTX nanocrystals have been explored, including β-LG (globular protein)25 and covalently-bound PEG26. However, we have previously found that NSCs have low uptake of PEGylated NPs and anticipated a similar problem for protein-coated NPs. Given our previous success in loading NSCs with silica NPs, we sought to develop silica coated PTX nanocrystals (Si[PTX-NC]).
Standard silica-coating conditions were unsuccessful in preparing the desired material, but by including an organosilane in the silica growth process, Si[PTX-NC]s were synthesized that met our requirements for NSC loading. They contain a high amount of drug by weight and show delayed release. Most importantly, these Si[PTX-NC]s enable PTX-loading of NSCs that is not possible with Abraxane or PTX-NCs. NSCs loaded with Si[PTX-NC]s remain viable and maintain their tumor tropic property. NSC/Si[PTX-NC]s are effective in eradicating ovarian tumor cells when co-cultured in vitro. When injected IP into an ovarian tumor model, we found that NSC/Si[PTX-NC]s bearing a low dose of PTX were more effective than PTX-NCs or Si[PTX-NC]s in reducing the growth of OVCAR-8 tumors in the IP cavity, demonstrating the promise of this approach for targeted cancer treatment. The novel Si[PTX-NC] formulation may also have applications beyond our focus on NSC transport.
Results and discussion
The first step in developing a method to produce Si[PTX-NC]s, was generating surfactant-stabilized PTX-NCs that were small and had modest polydispersity (Figure 1). PTX is insoluble in aqueous solutions and naturally exists as micron size, inhomogeneous crystals (Figure S1a). In order to form PTX-NCs, these long, heterogeneous crystals were dispersed in chloroform by vortexing in the presence of Pluronic F127. The ratio by weight of PTX to Pluronic F127 was first varied (1:1, 1:2.5, 1:5, and 1:10) in order to identify optimal conditions. Chloroform was then evaporated and the resulting thin film of PTX and Pluronic F127 was further dried under vacuum for one hour. Water was then added to the vial and the mixture was sonicated to produce rod-shaped PTX-NCs (Figure S1b, c). The 1:1 and 1:2.5 ratio (w/w) yielded polydisperse PTX nanocrystals. When PTX and Pluronic F127 were used at a 1:5 ratio (w/w), fairly monodisperse PTX nanocrystals were created. By further increasing the surfactant to a 1:10 ratio of PTX to Pluronic F127 (w/w), the critical micelle concentration was exceeded and micelles were visible in the background of the TEM image. Thus, the 1:5 ratio was used.
Figure 1.
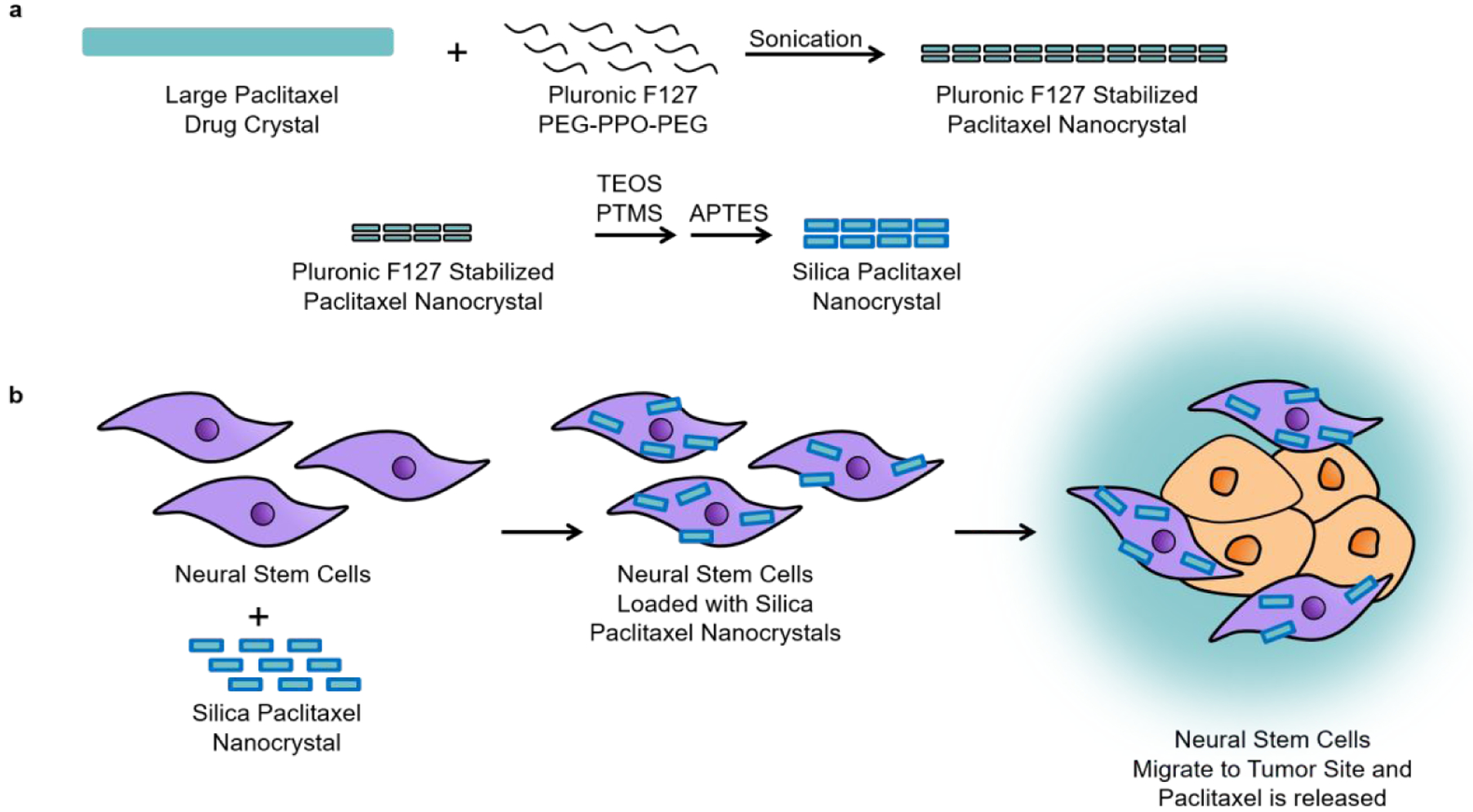
Scheme of Si[PTX-NC]s synthesis, NSC loading of Si[PTX-NC]s and ovarian cancer targeted delivery
After establishing temporary stabilization of PTX-NCs using PTX and Pluronic F127 at a 1:5 ratio (w/w), we attempted to grow a silica shell around the crystalline structures using a standard Stöber-type protocol by introducing tetraethyl orthosilicate (TEOS) and sodium hydroxide (NaOH) into the mixture. This resulted in spontaneous formation of silica networks with very little coating on the PTX-NCs (Figure S2). We hypothesized that this occurred because the nucleation rate of silica on the PTX-NCs was too slow, so a modified procedure was developed using a mixture of phenyltrimethoxy silane (PTMS) and TEOS. Since PTX is hydrophobic, it was thought that the phenyl group on the PTMS would have a high affinity for the hydrophobic surface of the PTX-NCs and rapidly displace the Pluronic F127, thus establishing a silane layer on the PTX-NC surface to template further silica growth. In addition, aminopropyltriethoxy silane (APTES) was used as both silica precursor and mild base catalyst for hydrolysis and condensation. Varying concentrations of TEOS, PTMS and APTES were tested and it was found that for optimal results, TEOS (4.8 mM) and PTMS (7.17 mM) should be added to the aqueous solution of PTX-NCs and allowed to stir for at least 4 hours. Hydrolyzed APTES (7.14 mM) was then added into the nanocrystal mixture and allowed to stir overnight for 16 hours. This particular formulation of silica nanocrystals is referred to as 1x Si[PTX-NC]. The nanocrystals were washed three times in H2O and then imaged by TEM. Because the particles were coated in silica (electron dense), they were not stained with uranyl acetate. By TEM, these particles have a uniform layer of silica on the surface of the PTX nanocrystal (Figure 2a). From the TEM images, the length was measured to be 397.1 ± 107.1 nm and the width was 70 ± 8.2 nm. In order to calculate shell thickness, the width of the PTX nanocrystals stabilized by Pluronic F127 (34 ± 7 nm) was subtracted from the width of the 1x Si[PTX-NC]s. The shell thickness was determined to be 36 ± 8 nm. Since APTES was added after TEOS and PTMS, the amine groups on the exterior of the silica shell caused the surface to be positively charged resulting in a zeta potential of +36.65 mV (Figure 2a). Dynamic light scattering (DLS) was also used to measure the Si[PTX-NC]s to confirm the length of the particles (Figure S3a, c). It was found that Si[PTX-NC]s were stable in water for 24 hours, with no aggregation or change in zeta potential (Figure S3 b, d). This is distinct from uncoated PTX nanocrystals which aggregated within several hours. The drug loading by weight of the 1x-Si[PTX-NC]s was determined to be 19%. The Si[PTX-NC]s showed delayed drug release in cell culture media (DMEM) alone and in DMEM supplemented with phosphate citrate. The addition of phosphate citrate lowered the pH to 4.5, which more closely models the endocytic environment after uptake in NSCs. Release was similar in both conditions and ~50% of PTX was released after 72 hours of incubation at 37 °C (Figure 2b).
Figure 2.
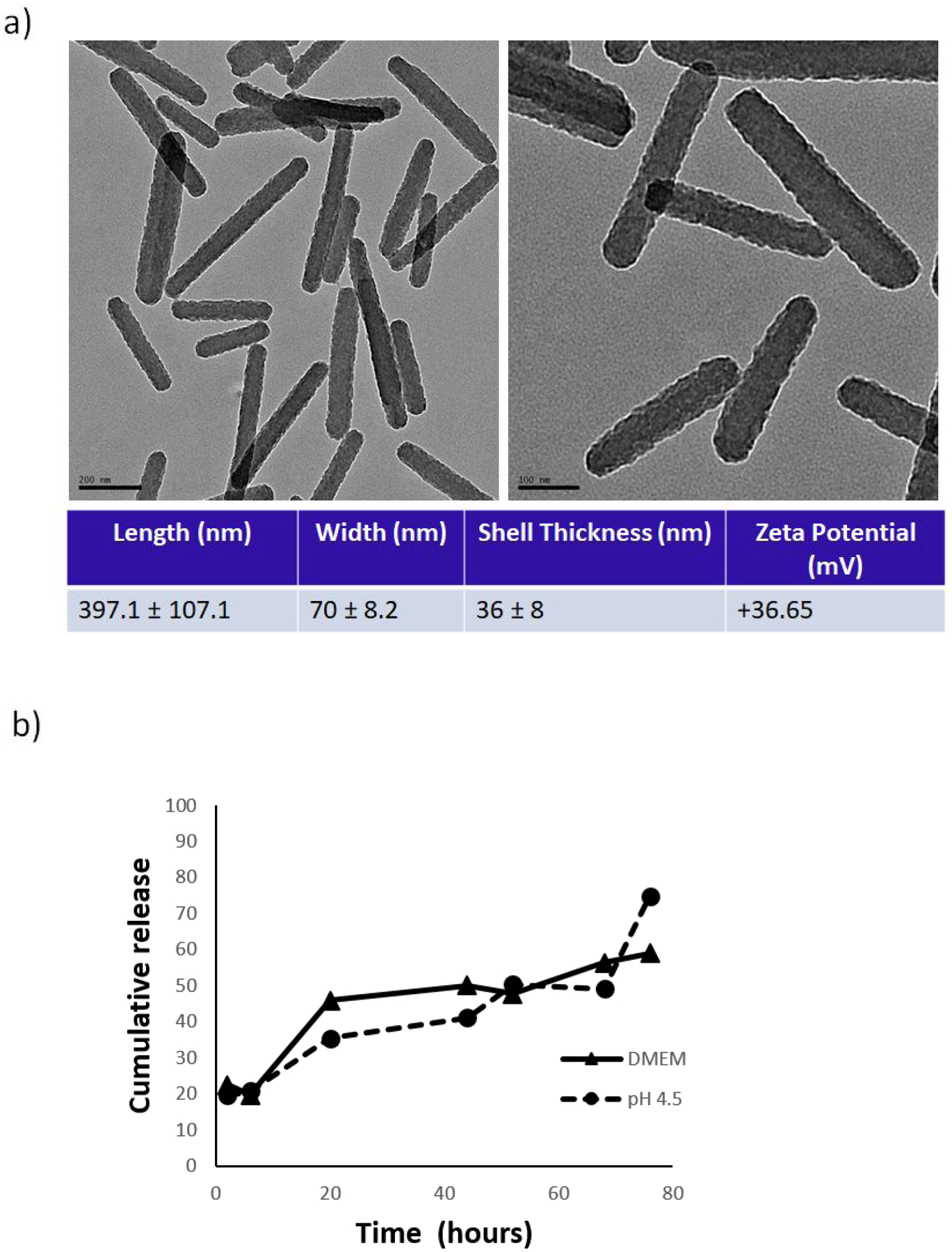
a) TEM image of Si[PTX-NC]s (left scale bar: 200 nm, right scale bar: 100 nm) 11000X; b) Cumulative release of PTX from Si[PTX-NC]s after incubation in DMEM medium with 10% FBS (solid line) and at pH 4.5 (DMEM-phosphate citrate) (dashed line), at 37 °C.
While the 1x formulation was selected as optimal for our studies, the silica shell (Si-shell) thickness could also be adjusted by varying the concentrations of TEOS, PTMS, and APTES. Reducing the concentration of PTMS, TEOS, and APTES resulted in aggregation and incomplete silica coverage around the PTX nanocrystals (Figure S4a). Increasing the concentration of PTMS, TEOS, and APTES resulted in a controlled increase in Si-shell thickness; the 2x, 4x, and 8x formulations had shell thicknesses of 49.4 ± 13, 64.3 ± 8, and 113.1 ± 12 nm, respectively (Figure S4b). However, all of these syntheses also resulted in the formation of drug-free spherical Si nanoparticles. While the Si[PTX-NCs] could be purified from the empty Si spheres using sucrose density centrifugation, this step was unnecessary for the 1x formulation. Thus, the 1x synthesis was considered optimal and used for all of the studies described herein. It is likely that the other formulation would have resulted in altered cell loading and drug release, but this was not explored. It is also possible that since the 1x synthesis includes a relatively polydisperse collection of nanocrystals that there is a size dependence as to which crystals load into the NSCs, but we were unable to measure this directly.
While the drug loading and release profile were favorable for the Si[PTX-NC]s, a key concern was if the silica coating would reduce the efficacy of the drug in eradicating ovarian cancer cells. OVCAR8 cells were treated with Si[PTX-NC]s and in vitro efficacy was evaluated. It was found that Si[PTX-NC]s and PTX-NCs had extremely similar OVCAR8 cell killing (Figure 3). Little to no cell killing was observed when cells were treated with empty silica nanospheres at an equivalent concentration of silica to that used in the Si[PTX-NC]s treatments (Figure S6).
Figure 3.
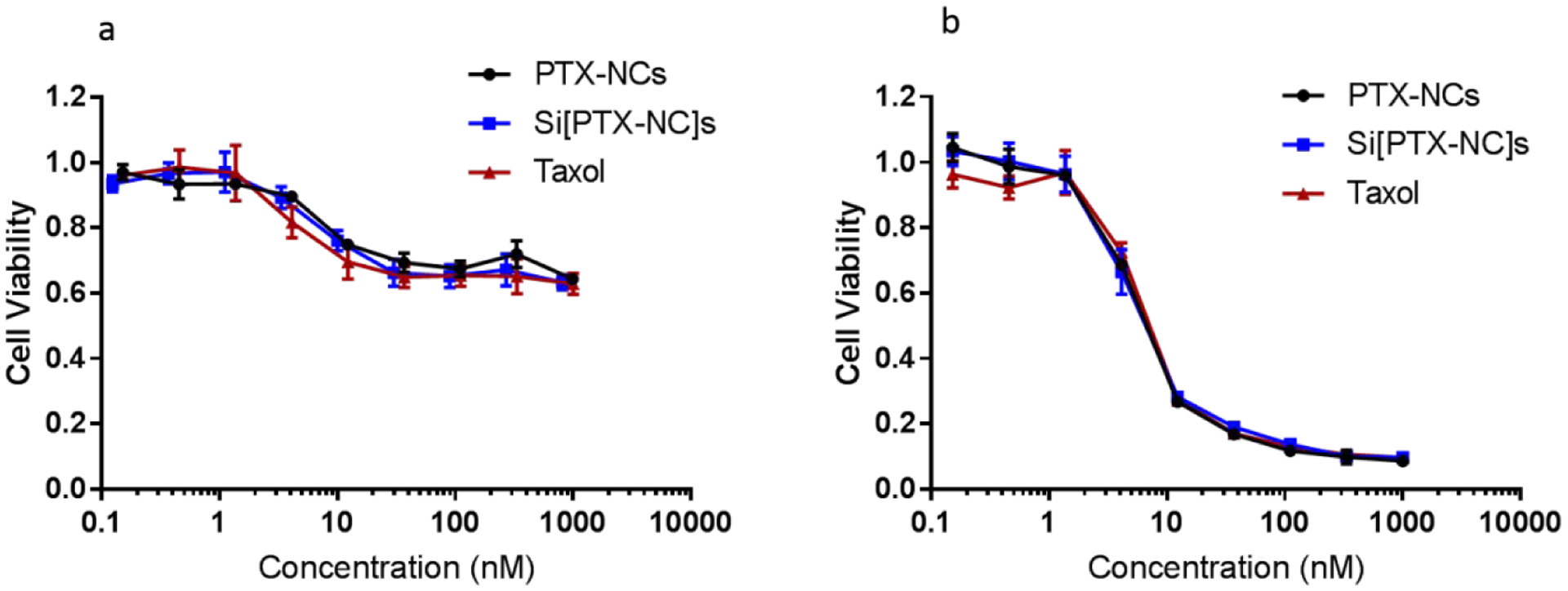
OVCAR-8 viability after a) 24 hours and b) 72 hours incubation with different concentrations of PTX-NCs, Si[PTX-NC]s and Taxol.
Having observed that the Si[PTX-NC]s were cytotoxic to ovarian cancer cells, we proceeded to evaluate the viability and migratory abilities of NSCs loaded with these NPs. NSCs were treated with Si[PTX-NC]s, PTX-NCs or Abraxane for one hour. The amounts of PTX added to the cells were matched (25 μg/mL) in all treatment conditions. After removing the incubation media containing the different treatments, the cells were rinsed with PBS and then collected. The PTX content of the cells was measured by HPLC. NSCs loaded with Si[PTX-NC]s resulted in in higher PTX levels (3.16 μg PTX/ million NSCs) relative to NSCs treated with PTX-NCs (0.26 μg PTX/ million NSCs). PTX was not detected in cells when NSCs were treated with Abraxane (Figure 4a). When the initial concentration of Si[PTX-NC]s was increased, this resulted in a modest increase in drug loading within the cells ranging from 2.57 – 3.81 μg PTX/ million NSCs (Figure 4b). Based on our previous work, NSCs accumulated at ovarian tumors after IP injection within 24 hours15, so NSCs loaded with Si[PTX-NC]s that are viable and migratory for 24 hrs are good candidates for in vivo efficacy. When the initial PTX loading concentration exceeded 40 μg/mL, NSC viability was compromised after 24 hrs (Figure 4c). Thus, we chose a loading concentration of 25 μg/mL for PTX in Si[PTX-NC]s for all further studies to prevent toxicity. In a separate experiment, we found that at this concentration of PTX NSCs remain viable whether treated with PTX-NCs, Si[PTX-NCs] or Abraxane (Figure S6c). We proceeded to loaded the NSCs with Si[PTX-NCs] using the 25 μg/mL concentration for PTX and analyzed the release kinetics of PTX from NSC/Si[PTX-NC]s. It was found that PTX release was slightly faster for NSC/Si[PTX-NC]s (59% in 48 hrs, Figure 4d) compared to Si[PTX-NC]s alone (48% in 48 hrs, Figure 2b). While these values are relatively similar, it is likely that acidic cell culture media does not fully capture the environment within the endosomes in the NSCs and additional factors in vitro accelerate degradation of the silica coating, leading to faster drug release.
Figure 4.
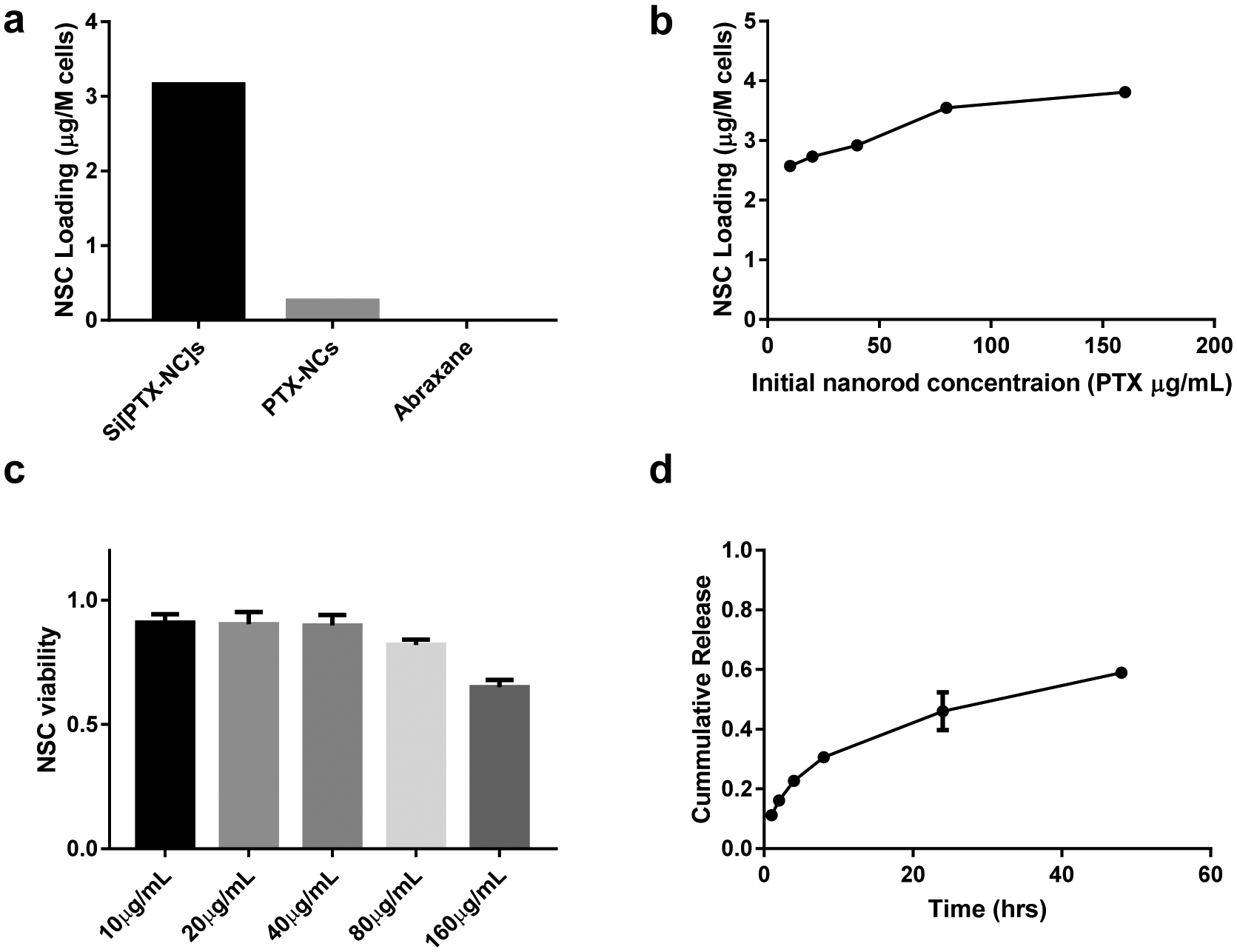
a) PTX loading into NSCs with different PTX formulation at 25 μg/mL for 1 hour, b) Correlation of PTX loading into NSCs with increasing concentration of Si[PTX-NC]s incubated with NSCs; c) NSC viability 24 hours after Si[PTX-NC] loading; d) Cumulative release of PTX from NSC/Si[PTX-NC] hybrids.
PTX is a very potent microtubule stabilizer and can inhibit cell migration at doses well below their IC5027. In order to evaluate if this was a concern, the migration capability of NSCs loaded with Si[PTX-NC]s was investigated using a Boyden Chamber migration assay. Untreated NSCs and NSCs incubated with different PTX formulations were seeded on a membrane placed above either 5% BSA (negative control) or SKOV-3 tumor-conditioned media as a model ovarian tumor condition media. After 4 hours, cells that had migrated to the bottom of the membrane were quantified and the migration ratio was calculated as cells migrated vs cells seeded. As is shown in Figure 5a, NSCs loaded with Si[PTX-NC]s did show some reduction in migration efficiency (48% vs 74% for unloaded NSCS). However, this was far superior to NSCs exposed to PTX-NCs or Abraxane which both showed minimal migration (10%). It is interesting that the NSCs showed little difference in viability when treated with the different formulations (Figure S6c) but a dramatic difference in migratory capacity. It is unclear what leads to this dramatic difference; one possibility is that it is because migration is compromised at a lower dose than viability and if the viability study was extended out or run at higher concentrations of PTX a similar result would be obtained.
Figure 5.
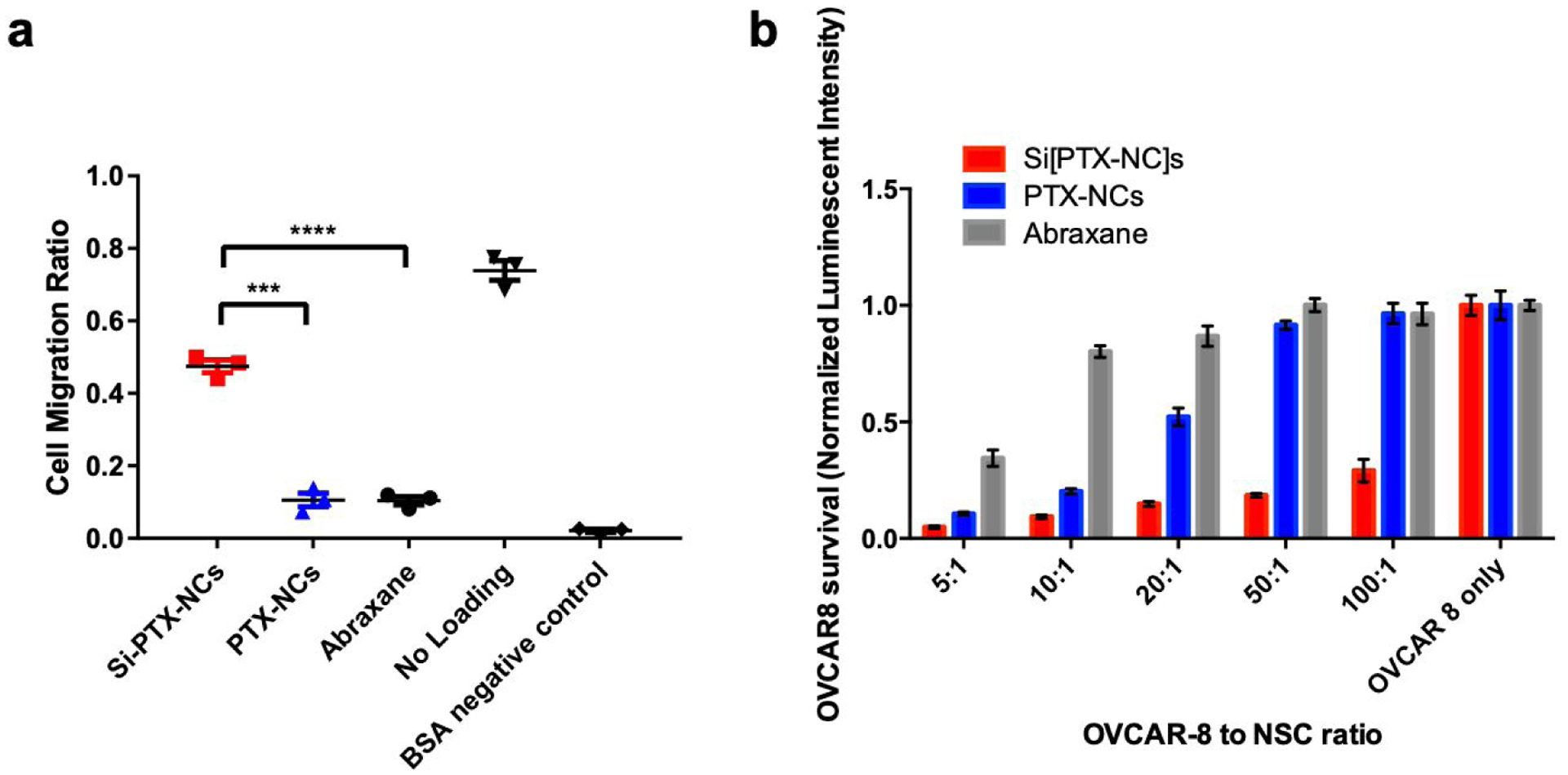
a) NSCs migration after loading with Si[PTX-NC]s, PTX-NCs and Abraxane by Boyden chamber assay; b) OVCAR-8 viability after co-culture with different ratios of PTX formulations by luciferase luminescence assay. c) OVCAR-8 viability after 96 hours of treatments with equivalent concentration of PTX-NC, Si[PTX-NC] and NSC/Si[PTX-NC].
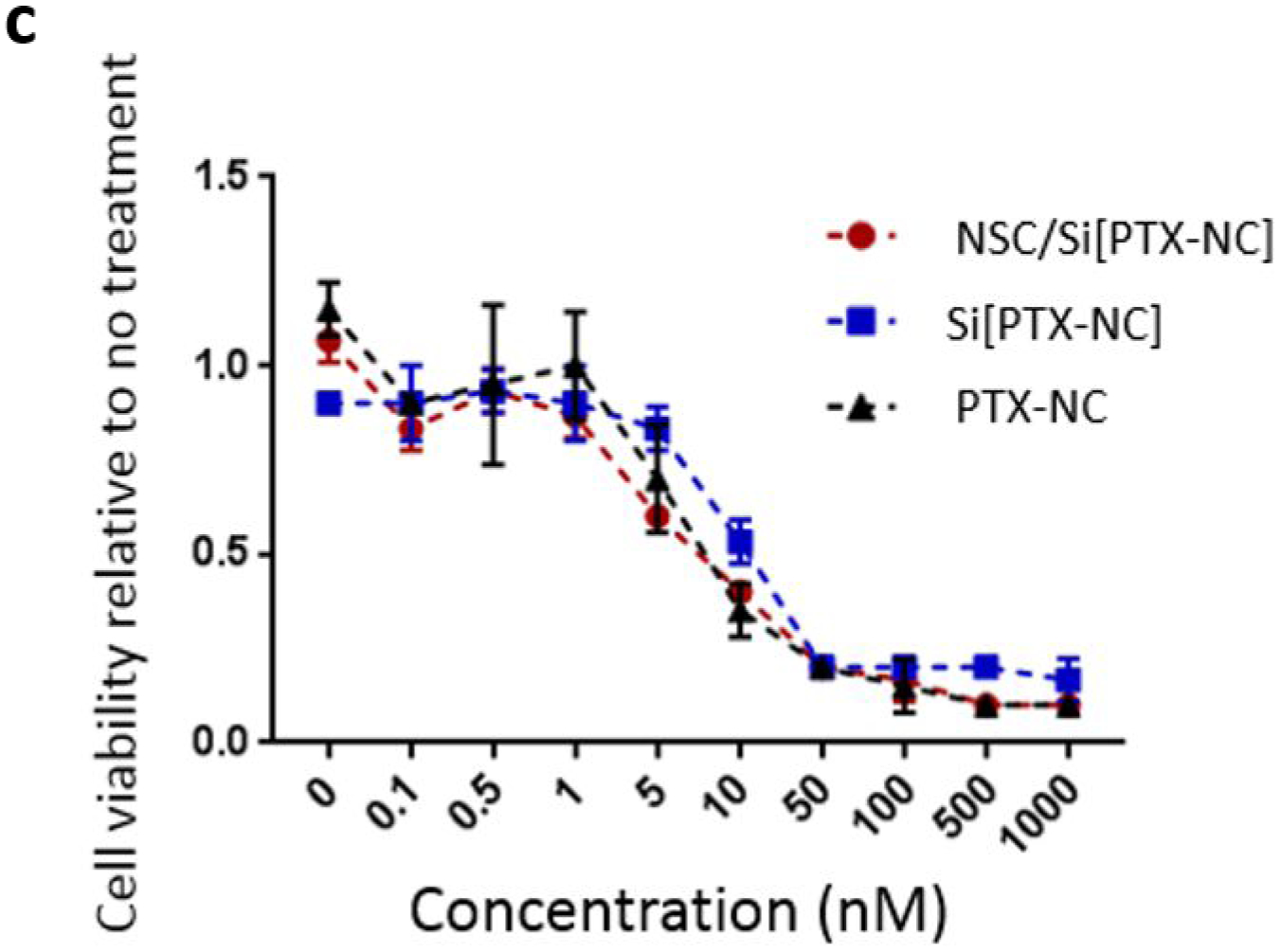
Confident that the PTX remains active in the Si[PTX-NC] formulation and could be transported by NSC/Si[PTX-NC]s, we next evaluated if the PTX remained effective at killing cancer cells after loading into NSCs. NSCs were first incubated with Si[PTX-NC]s or PTX-NCs for 1 hour and then washed, trypsinized and replated in varying ratios into wells containing luciferase expressing OVCAR8 cells. OVCAR8 cells were selected so that the model is relevant to high-grade serous ovarian cancer, which is the most frequent histotype of the patients diagnosed with stage III peritoneal disseminated disease.15 OVCAR8 viability was determined by measuring luciferase activity after 72 hours, normalized relative to the untreated OVCAR8 monoculture. NSCs loaded with Si[PTX-NC]s were found to be the most effective at killing OVCAR8 cells, eradicating most of the cancer cells even at a ratio of 1:100 (NSC:OVCAR8) (Figure 5b). The relative cell killing of NSC/Si[PTX-NC]s and NSC/PTX-NCs directly matched their relative loading amounts, as NSCs loaded with Si[PTX-NC]s contained 12x more PTX and also demonstrated 12x more potent OVCAR8 cell killing. The NSCs exposed to Abraxane did show minor OVCAR8 cell killing, which suggests that there may be some PTX-loading in these conditions that is just too low for our analysis. This direct relationship between PTX content in the cells and efficacy suggested that there was no decrease in relative PTX efficacy after loading into NSC/Si[PTX-NC]s. This was confirmed in a separate experiment where the OVCAR8 cell number was held constant and the dose of the NSC[PTX-NC]s was varied and compared to matched concentrations of PTX-NCs and Si[PTX-NCs] (Figure 5C).
After demonstrating the effectiveness of NSC/Si[PTX-NC]s in the in vitro co-culture model, we moved on to in vivo testing using a nude mouse model of metastatic ovarian cancer. Mice were intraperitoneally implanted with OVCAR8 cells to establish the orthotopic model of stage III ovarian cancer. Prior to treatment, the mice were imaged to assess tumor growth and divided into groups with equal average luminescent signal (mice with little to no signal were excluded from treatment). In order to determine a PTX-dose range where the PTX-NCs alone were not sufficiently effective, an in vivo dosing study was performed by injecting only free PTX-NCs IP 3 weeks after tumor cell implantation at increasing doses: 0, 0.0625 mg/kg, 0.25mg/kg, 1 mg/kg and 2 mg/kg. A dose response was seen in the tumor burden after 3 weeks of treatment, with 2 mg/kg proving quite effective and 0.25 mg/kg only slightly outperforming the PBS control (Figure S7). Thus, we selected a dose of 0.5 mg/kg for comparison of PTX-NC, Si[PTX-NC] and NSC/Si[PTX-NC] in order to test in a range where the free drug is minimally effective and the impact of targeted delivery would be straightforward to observe.
For this next in vivo efficacy study, 3 weeks after tumor injection, the mice were imaged to assess tumor growth and then, 30 mice with luminescent tumor signal were repartitioned into 5 groups of 6 mice each (images of pre-treatment before repartition of groups are shown in Fig S9). Each group of mice was treated with either PBS (control), NSC only (control), PTX-NCs, Si[PTX-NC]s, or NSC/Si[PTX-NC]s twice a week for 3 weeks. In order to ensure equal dosing for each group (0.5 mg/kg), PTX-loading was measured in NSC/Si[PTX-NSC]s by HPLC and the amount of NSCs corresponding to a dose of 0.5 mg/kg (in 200 μL of PBS) was injected into the NSC/Si[PTX-NC] group. PTX-NC and Si [PTX-NC] treatment groups were also matched to 0.5 mg/kg. The number of NSCs in the NSCs only group was matched to the number of NSCs in the NSC/Si[PTX-NC] group. The mice were subjected to live luminescent imaging one day before the first injection of every treatment week. All mice were imaged 4 days after the final treatment and one day later they were sacrificed. Tumors larger than 1 mm3 were collected and weighed. Two mice from the free drug group (PTX-NC) and one mouse from the Si[PTX-NC] had to be imaged and sacrificed two days before the rest of the mice as they showed deteriorating body conditions. As shown in Figure 6, mice treated with NSC/Si[PTX-NC] had significantly reduced luminescent signals as well as reduced tumor burden compared to all other treatment groups. This experiment was also repeated with an earlier onset of treatment (12 days), and a similar outcome was observed (Figure S8). In this earlier treatment setting, it was also found that, as expected, the effect of targeting is obscured when the PTX concentration is increased to the point that the free PTX-NCs is highly effective (Figure S9).
Figure 6.
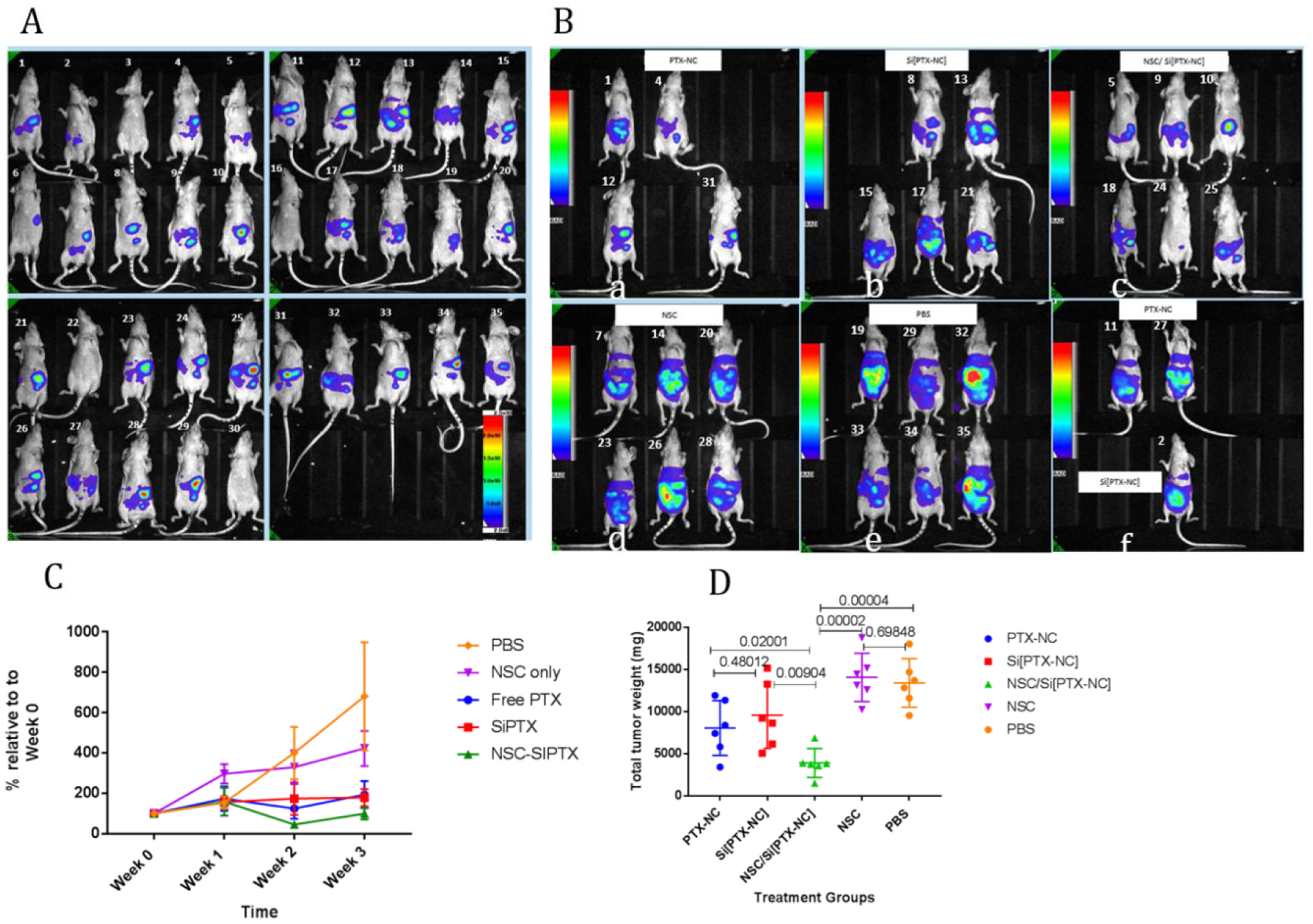
Luminescent imaging of mice (A) before and (B) after treatment with (a) PTX-NC, (b) Si[PTX-NC], (c) NSC/Si[PTX-NC], (d) NSC, (e) PBS, (f) 2 first mice are from the PTX-NC and third mouse is from Si[PTX-NC], imaged 2 days earlier than the rest of mice. (C) Quantified analysis of images represented as a percentage of intensity after each week of treatment (Week 1, week2, week3 ) relative to the pre-treatment (Week0), (D) total tumor weight collected after sacrificing mice. Dose of PTX injected is 0.5 mg/kg for all PTX treatment groups. The NSC number in NSC group is matched to the Number in NSC/Si[PTX-NC]. All Treatments here started 3 weeks after IP injection of 2.10E6 OVCAR-8 cells, and was performed for 3 weeks, 2 times a week. After the pre-treatment imaging (in A), all mice were repartitioned into groups based on the mean intensities (with exclusion of mice with low to no signal) and then treated randomly with the mentioned treatments.
CONCLUSION
Here, we have demonstrated the modularity of NSC/NP hybrids for drug delivery by enabling the delivery of PTX. This required developing a novel technique for coating PTX-NCs with a silica shell. The silica coating was essential for obtaining loading of the PTX into NSCs resulting in greater drug loading relative to other formulations and also preserving NSC viability and migration. In an in vitro setting, NSC/Si[PTX-NC]s were effective at eliminating ovarian tumor cells even at a ratio of 1:100 (NSC : OVCAR8). It was also demonstrated, in two separate experiments with two different onsets of treatment, that a low dose of PTX in NSC/Si[PTX-NC]s was more effective than free Si[PTX-NC]s or PTX-NCs at reducing tumor burden in a mouse model of metastatic ovarian cancer. The significant improvement using a low dose of chemotherapy supports future studies loading the NSC/NPs with more toxic payloads, like monomethyl auristatin, that cannot be administered as free drugs and are highly effective at low doses.
MATERIALS AND METHODS
Materials.
All organic and inorganic compounds and solvents were purchased from Sigma-Aldrich, unless otherwise mentioned. Paclitaxel (PTX) was purchased from Ark Pharm. Abraxane was purchased from Celegene. The human, v-myc-immortalized, HB1.F3.CD21 NSC line was obtained from Dr. Seung Kim (University of British Columbia, Canada), and clone 21 selected for characterization and clinical use at City of Hope).
Instrumentation.
Dynamic light scattering (DLS) and ζ potential (ZP) measurements were performed on a Brookhaven 90 Plus/BI-MAS Instrument (Brookhaven Instruments). DLS measurements were obtained by performing 5 runs at 30 s per run, and the ZP values were obtained by measuring 10 runs involving 30 cycles per run. TEM images were obtained with an FEI Tecnai T12 transmission electron microscope at an accelerating voltage of 120 keV, and images were taken with a Gatan Ultrascan 2K CCD camera. NPs dispersed in water at an optimal concentration were drop-cast onto glow-discharged, 300 mesh carbon–Formvar coated grids and allowed to dry before imaging. HPLC analysis was performed by Agilent 1100 Series with UV detector at 220 nm, using a 2.6 μm-C18-100 A° Column and a flow rate of 0.9 mL /min, sample injection volume was 10 μL. Solvents were 0.1% TFA in Water (Solvent A) and 0.1% TFA in Acetonitrile (Solvent B) and the gradient elution method was: 0 min = 75 % Solvent A and 25 % Solvent B, 2 min = 66 % solvent A and 34 % solvent B, 6 min = 39.6 % solvent A and 60.4 % solvent B, 6.01 min to 10 min = 0% solvent A and 100 % solvent B, 10.01 min of 75 % (A) and 25% solvent B. Standard curve was made by measuring the area under PTX peak (at 5 min) at different PTX concentrations.
Synthesis of PTX-NC
Paclitaxel (4 mg) and Pluronic F127 (20 mg) were both weighed and combined in a scintillation vial. Chloroform (1 mL) was added to the scintillation vial and vortexed to completely solubilize PTX and Pluronic F127. A steady stream of nitrogen was used to evaporate the chloroform. This formed a dry film on the bottom of the vial. The vial was placed in a vacuum for 10 min, followed by adding 14 mL MilliQ H2O (14 mL). This was placed on a shaker for 20 minutes and vortexed. Qsonica cup sonicator (amplitude=100) was used to sonicate the mixture to form the nanosuspension. Pulse-on time was 5 minutes and pulse-off time was set to 10 minutes and this was repeated two more times and the sample was removed after the third pulse-on time for a total of 35 minutes of process time. To verify formation of nanocrystals, the solution was imaged by TEM. Four μL of the solution was removed and placed on a 300-mesh copper grid. After 30 seconds, it was blotted off and stained with 2% uranyl acetate. After 30 seconds, uranyl acetate was blotted off and allowed to dry.
Synthesis of Si[PTX-NC]
After formation of the nanosuspension described above, TEOS and PTMS were added to the nanosuspension to get a final concentration of 4.80 mM (TEOS) and 7.17 mM (PTMS) and stirred for 3 hours at 1500 rpm at RT. A solution of hydrolyzed APTES was made by diluting APTES (70 μL) in water (37.5 μL) to have a final concentration of 2.8M. A total 37 μL of this hydrolyzed APTES solution was immediately added to the nanosuspension mixture to get a final concentration of 7.14 mM. The mixture was allowed to stir overnight for 16 hours at RT, 1500 rpm. The Si[PTX-NC]s were then centrifuged at 20 000 × g for 10 minutes and washed with milliQ H2O three times. After each centrifugation, a dense pellet is observed at the bottom of the conical tube and a slurry is coating it. The slurry coating contains many empty, spherical SiNPs. In order to obtain pure Si[PTX-NC]s, after each centrifugation, the slurry is carefully removed by pipette. In order to visualize the silica shell grown around the nanocrystals, TEM was used. The grid was charged initially before pipetting 4 μL of the nanosuspension onto the grid. It was dried in an oven at 60°C for 5 minutes before imaging. Other concentrations of silica precursor were also explored (0.5x, 2x, 4x, and 8x) to optimize the synthesis of Si[PTX-NC] (See Figure S4). Empty silica NPs were synthesized with similar method, except no PTX was added to the initial mixture and 4x TEOS/PTMS/APTES concentration was used.
Isolation of Si[PTX-NC]s using a sucrose gradient
In order to isolate empty or spherical particles from either 2x or 4x Si[PTX-NC], a sucrose gradient was used. The gradient consisted of five different layers, each containing 2 mL of 18%, 21%, 24%, 27%, and 30% sucrose solution in a 15 mL conical tube. In order to isolate the 8x Si[PTX-NC], a sucrose gradient consisting of 10%, 20%, 30%, 40%, and 50% sucrose was used. Si[PTX-NC] (750 μL) was added to the top layer slowly. The gradient was centrifuged at 3600 × rpm for 35 minutes. The top layer (500 μL) consisted of spherical silica NPs and small Si[PTX-NC]. The second layer (800 μL) contained the nanocrystals of interest (200–500 nm). The other layers were discarded since they contain aggregated nanocrystals or nanocrystals that are larger than 500 nm. Si[PTX-NC] were centrifuged at 14,000 × g for 20 minutes and washed with milliQ H2O three times. Si[PTX-NC] were imaged by TEM to ensure complete isolation from spherical silica NPs and aggregates. In order to measure shell thickness, the width of the PTX nanocrystals stabilized by Pluronic F127 was subtracted from the width of the silica-coated nanocrystals (ImageJ).
Quantification and release kinetic of PTX in Si[PTX-NC]s
To quantify drug-loading, the Si[PTX-NC]s were weighed and HPLC was used to measure the PTX content. To determine a dry mass, a water suspension containing around 2–3mg of Si[PTX-NC] (1mL) was freeze-dried by lyophilizer (Labconco) and weighed by balance. To determine PTX-loading, another sample from the same Si[PTX-NC] water suspension (10 μL) was dissolved using hydrofluoric acid (2 μL, 48%) for 15 minutes and neutralized with sodium bicarbonate (3 mg) and calcium chloride (2 mg). 90 μL acetonitrile was then added to the mixture and filtered by 0.4 μm syringe filter for HPLC analysis. PTX loading capacity was calculated by (weight of PTX (determined by HPLC)*100)/(total weight of the Si[PTX-NC] (weight of the dry powder)). For PTX release study, Si[PTX-NC] containing 40 μg PTX was suspended in 1 mL of DMEM cell culture medium supplemented with 10% fatal bovine serum (FBS). At different time points, the suspension was centrifuged at 10000 g and the supernatant was extracted by 1 mL of ethyl acetate. The extracts were dried under vacuum and dissolved in 100 μL acetonitrile for HPLC analysis.
Quantification of ethyl acetate extract efficiency.
To quantify the ethyl acetate extract efficiency, Taxol with known amount of PTX (50 μg) was dissolved in 1 mL of DMEM cell culture medium with 10% fatal bovine serum (FBS) and extracted by 1 mL of ethyl acetate. The extracts were dried under vacuum and dissolved in 100 μL acetonitrile for HPLC analysis. The extract efficiency is calculated by dividing PTX amount determined by HPLC with PTX amount initially dissolved and the extract efficiency is 97%.
Cell Culture
All cells were cultured and maintained at 37 °C in a humidified incubator (Thermo Electron Corporation) containing 5% CO2. Neural stem cells and OVCAR-8 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (Gemini Bio), 1% L-glutamine (Invitrogen), and 1% penicillin–streptomycin (Invitrogen). SKOV-3 cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (Gemini Bio), 1% L-glutamine (Invitrogen), and 1% penicillin–streptomycin (Invitrogen). When the cells reached 80% confluency, they were passaged using a 0.25% trypsin-ethylenediaminetetraacetic (trypsin EDTA) acid solution (Invitrogen); media was changed every 2 – 3 days. SKOV-3 cells were used to generate ovarian tumor-conditioned media by replacing culture media with serum-free media when cells were 80 – 100% confluent, followed by a 48 h incubation period.
Viability of OVCAR-8 treated with various concentrations of Si[PTX-NC]
OVCAR-8 cells were seeded in 96-well plates (4000 cells/well, 100 μL medium). They were incubated at 37° C for 16 hours before treatment. All dilutions of PTX-F127 nanocrystal and Si[PTX-NC] were done in media. After treatment, cells were incubated for 24 and 72 hours. At each time point 20 μL of MTS solution (Promega) was added to each `well and cell viability was measured by microplate reader at 490 nm absorbance wavelength after incubation at 37° C for two hours.
Viability and release of PTX from NSCs loaded with Si[PTX-NC]
NSCs loading with Si[PTX-NC] was achieved by incubating NSCs (5×105 cells/well in 6-well plates) for 1 h with 10, 20, 40, 80, 160 μg/mL suspension of Si[PTX-NC]s in PBS at 37 °C for loading efficiency experiment and 25 μg/mL suspension of Si[PTX-NC] for further experiments. Any unloaded Si[PTX-NC] were removed by repetitive PBS washing (3 times). The Si[PTX-NC]-loaded NSCs were then trypsinized and pelleted via centrifugation at 1500 rpm for 3min. The cells were first counted then an aliquot is span down at 5000 ×g for 5 min and the pellet is ruptured using hydrofluoric acid under continuous sonication for 20 min and homogenizing via pipetting in order to fully digest the cell pellet and collect the PTX. The hydrofluoric acid is then neutralized by sodium bicarbonate and calcium chloride and Acetonitrile was added to ruptured cells and filtered by 0.4 μm syringe filter for HPLC analysis to determine PTX loading in NSCs. To measure PTX released from Si[PTX-NC]-loaded NSCs, cell pellets were suspended in 1 mL of medium and the supernatant was collected at different time points. PTX was extracted using 1 mL of ethyl acetate. The extracts were dried under vacuum and dissolved in 100 μL acetonitrile for HPLC analysis. To determine the NSCs viability after loading, Si[PTX-NC]-loaded NSCs (4000 cells/well) were re-plated onto 96-well plates and viability was measured after 24 hours by MTS assay and normalized to the data for the unloaded NSCs.
Viability of NSCs treated with the various formulations
To determine the NSCs viability after loading with different formulation, NSCs (5×105 cells/well in 6-well plates) were incubated with different formulations in PBS suspensions for 1h at 37 °C at PTX equivalent concentration of 30 μM. Cells were washed 3 times with PBS and cells were replated onto 96 well plates at a density of 4000 cells/well. NSC viability was measured after 24 hours by MTS assay and normalized to the data for the unloaded NSCs.
Migration of NSCs
NSCs’ migration capability was measured using the in vitro transwell Boyden chamber assay. NSCs were incubated with Si[PTX-NC], PTX-F127 nanocrystals or Abraxane (25 μg/mL PTX in PBS) for 1 hour. In a 24-well tissue culture plate, 500 μL of target media (either containing only BSA as a negative control, or derived from the culture of SKOV-3 cells) was added to each well. At a density of 1 × 105 cells/well, PTX loaded NSCs in DMEM and 5% w/v BSA were placed in the transwell chambers and incubated at 37 °C for 4 hours. After the incubation period, the transwell chambers were placed in a new 24-well tissue culture plate containing accutase and incubated 10 mins at 37 °C. Detached cells were then transferred to a 96-well v-bottom plate, centrifuged at 1,500 rpm for 5 mins, and re-suspended in 1:1 media to ViaCount. NSC migration to conditioned media of PTX treated and non-treated cells was assessed using Guava EasyCyte technology.
Tumor killing effect in vitro by co-culturing Si[PTX-NC]-loaded NSCs and OVCAR-8 ovarian cancer cells
OVCAR-8 cells were seeded in 96-well plates at 4000 cells/well. They were incubated at 37 °C for 16 hours before treatment. NSCs were loaded with Si[PTX-NC], PTX-NC and Abraxane as previously described. After a 1 hour incubation period, NSCs were repetitively washed 3 times by PBS to remove unloaded Si[PTX-NC], PTX-NC or Abraxane and were detached with trypsin-EDTA. PTX loaded NSCs were transferred to the wells containing OVCAR-8 cells at ratios of 1:5, 1:10, 1:20, 1:50, and 1:100 by using serial dilutions. Each condition and ratio was done in triplicates. Cells were co-cultured for 72 hours and then OVCAR-8 viability was determined by adding 10 μL D-luciferin solution (3 mg/mL) to each well and measuring luciferase luminescent intensity by microplate reader.
In Vivo Orthotopic Ovarian Cancer Model, bioluminescent imaging and tumor collection
7 week old female athymic nude mice (Charles River/NCI) were inoculated with 2M OVCAR-8.eGFP.ffluc human ovarian cancer cells via IP injection. For the treatment at high doses (results in SI, Fig S7), the tumors were allowed to grow 12 days and bioluminescent imaging was conducted on day 12 (before treatment), day 19, day 26 and day 33 (before sacrificing). Mice were injected with 200 μL 20mg/mL D-luciferin IP and anesthetized with isoflurane. The images were taken by Lagos X, Spectral Instruments Imaging at F stop 4 exposure time 15s. On day 14, 16, 21, 23, 28 and 30. Mice were treated with different doses of NSC/Si[PTX-NC]s, Si[PTX-NC]s, PTX-NCs, NSCs or PBS. Total drug administered was normalized to the amount of PTX in NSC/Si[PTX-NC]s after drug loading. The drug loading amount was quantified by HPLC analysis as described in quantification of PTX in Si[PTX-NC]s section. The result of quantification is shown below: 118.0 μg/mouse on day 14, 83.4 μg/mouse on day 16, 68.4 μg/mouse on day 21, 105.1 μg/mouse on day 23, 58.6 μg/mouse on day 28, 75.08 μg/mouse on day 30. Mice were sacrificed on day 34 and tumors larger than 1 mm3 were collected in 50 mL conical tubes and weighed on balance.
For the treatment with increasing doses of Free PTX-NC (results in SI, Fig S8), the tumors were allowed to grow for 3 weeks instead of 12 days and the treatment schedule was identical to the previous study (2 treatments per week, for 3 weeks). The bioluminescent imaging was performed one day before the first treatment of each treatment week and the final imaging occurred at the end of the 3 weeks of treatment (4 days after the last treatment) followed by sacrificing the mice and weighing the tumors the following day. For the treatment with 0.5 mg/kg, we performed two experiments with treatments occurring at 10 days and at 3 weeks after tumor injection. Treatment and Imaging schedules were the same for both time points and the dose was kept constant at each injection (0.5mg/kg of PTX).
Supplementary Material
ACKNOWLEGEMENTS
We gratefully acknowledge: Marcia Miller, Zhuo Li and Ricardo Zerda for assistance with sample preparation and TEM imaging, and the Animal Resource Center core for assistance with animal work. Research reported in this publication included work performed in the Animal Resource Center supported by the National Cancer Institute of the National Institutes of Health under award number P30CA33572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Studies were supported by generous funding from STOP Cancer, The Rosalinde and Arthur Gilbert Foundation, the Alvarez Family Foundation, The Anthony F. & Susan M. Markel Foundation, Jeanne and Bruce Nordstrom, and NIH/NCI R01CA197359.
Footnotes
Supporting Information
The supporting information contains TEM images of PTX nanocrystals prepared using various amounts of Pluronic F127 and initial attempts at silica growth using TEOS and NaOH. It also contains further characterization of 1x Si[PTX-NC]s, specifically DLS and zeta potential measurements, silica shell growth of Si[PTX-NC]s, sucrose gradient separation of 2x, 4x, and 8x Si[PTX-NC]s, and viability of OVCAR-8 cells treated with blank silica NPs (empty SiNPs with no PTX). Lastly, it contains in vivo results of the treatment at a dose around 4 mg/ kg as well as the dosing study performed with free drug at increasing doses and the treatment with lower dose 0.5 mg/kg 10 days after tumor injection.
These materials are available free of charge via the Internet at http://pubs.acs.org.
DISCLOSURE
KSA is an officer and board member of TheraBiologics, Inc., a clinical stage biopharmaceutical company supporting development of NSC-mediated cancer treatments. The remaining authors declare no conflict of interest.
REFERENCES:
- (1).Lengyel E (2010) Ovarian Cancer Development and Metastasis. The American Journal of Pathology 177, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Webber K, and Friedlander M Chemotherapy for epithelial ovarian, fallopian tube and primary peritoneal cancer. Best Practice & Research Clinical Obstetrics & Gynaecology. [DOI] [PubMed] [Google Scholar]
- (3).Bookman MA (2016) Optimal primary therapy of ovarian cancer. Annals of Oncology 27, i58–i62. [DOI] [PubMed] [Google Scholar]
- (4).Group, U. S. C. S. W. (2016) (Atlanta: U.S. Department of Health and Human Services, C. f. D. C. a. P. a. N. C. I., Ed.).
- (5).Huang B, Abraham WD, Zheng Y, Bustamante López SC, Luo SS, and Irvine DJ (2015) Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Science Translational Medicine 7, 291ra94–291ra94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Stephan MT, Moon JJ, Um SH, Bershteyn A, and Irvine DJ (2010) Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med 16, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Choi M-R, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D, Badve S, Sturgis J, Robinson JP, Bashir R, et al. (2007) A Cellular Trojan Horse for Delivery of Therapeutic Nanoparticles into Tumors. Nano Letters 7, 3759–3765. [DOI] [PubMed] [Google Scholar]
- (8).Li L, Guan Y, Liu H, Hao N, Liu T, Meng X, Fu C, Li Y, Qu Q, Zhang Y, et al. (2011) Silica Nanorattle–Doxorubicin-Anchored Mesenchymal Stem Cells for Tumor-Tropic Therapy. ACS Nano 5, 7462–7470. [DOI] [PubMed] [Google Scholar]
- (9).Mooney R, Weng Y, Garcia E, Bhojane S, Smith-Powell L, Kim SU, Annala AJ, Aboody KS, and Berlin JM (2014) Conjugation of pH-responsive nanoparticles to neural stem cells improves intratumoral therapy. Journal of Controlled Release 191, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cheng Y, Morshed R, Cheng S-H, Tobias A, Auffinger B, Wainwright DA, Zhang L, Yunis C, Han Y, Chen C-T, et al. (2013) Nanoparticle-Programmed Self-Destructive Neural Stem Cells for Glioblastoma Targeting and Therapy. Small 9, 4123–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Aboody KS, Najbauer J, Metz MZ, D’Apuzzo M, Gutova M, Annala AJ, Synold TW, Couture LA, Blanchard S, Moats RA, et al. (2013) Neural Stem Cell–Mediated Enzyme/Prodrug Therapy for Glioma: Preclinical Studies. Science Translational Medicine 5, 184ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Aboody KS, Najbauer J, and Danks MK (2008) Stem and progenitor cell-mediated tumor selective gene therapy. Gene therapy 15, 739–52. [DOI] [PubMed] [Google Scholar]
- (13).Zhao D, Najbauer J, Annala AJ, Garcia E, Metz MZ, Gutova M, Polewski MD, Gilchrist M, Glackin CA, Kim SU, et al. (2012) Human neural stem cell tropism to metastatic breast cancer. Stem cells 30, 314–25. [DOI] [PubMed] [Google Scholar]
- (14).Portnow J, Synold TW, Badie B, Tirughana R, Lacey SF, D’Apuzzo M, Metz MZ, Najbauer J, Bedell V, Vo T, et al. (2017) Neural Stem Cell–Based Anticancer Gene Therapy: A First-in-Human Study in Recurrent High-Grade Glioma Patients. Clinical Cancer Research 23, 2951–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cao P, Mooney R, Tirughana R, Abidi W, Aramburo S, Flores L, Gilchrist M, Nwokafor U, Haber T, Tiet P, et al. (2017) Intraperitoneal Administration of Neural Stem Cell-Nanoparticle Conjugates Targets Chemotherapy to Ovarian Tumors. Bioconjugate chemistry 28, 1767–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tian J, and Stella VJ (2010) Degradation of paclitaxel and related compounds in aqueous solutions III: Degradation under acidic pH conditions and overall kinetics. Journal of pharmaceutical sciences 99, 1288–98. [DOI] [PubMed] [Google Scholar]
- (17).MacEachern-Keith GJ, Wagner Butterfield LJ, and Incorvia Mattina MJ (1997) Paclitaxel Stability in Solution. Analytical Chemistry 69, 72–77. [Google Scholar]
- (18).Ma P, and Mumper RJ (2013) Paclitaxel Nano-Delivery Systems: A Comprehensive Review. Journal of nanomedicine & nanotechnology 4, 1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Stinchcombe TE (2007) Nanoparticle albumin-bound paclitaxel: a novel Cremphor-EL-free formulation of paclitaxel. Nanomedicine (London, England) 2, 415–23. [DOI] [PubMed] [Google Scholar]
- (20).Bernabeu E, Helguera G, Legaspi MJ, Gonzalez L, Hocht C, Taira C, and Chiappetta DA (2014) Paclitaxel-loaded PCL–TPGS nanoparticles: In vitro and in vivo performance compared with Abraxane®. Colloids and Surfaces B: Biointerfaces 113, 43–50. [DOI] [PubMed] [Google Scholar]
- (21).Liu Y, Huang L, and Liu F (2010) Paclitaxel Nanocrystals for Overcoming Multidrug Resistance in Cancer. Molecular pharmaceutics 7, 863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xiaoqing M, Wuwei Y, Tao F, Jing L, and Peng H (2018) Drug nanocrystals for cancer therapy. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology 10, e1499. [DOI] [PubMed] [Google Scholar]
- (23).Lu Y, Chen Y, Gemeinhart RA, Wu W, and Li T (2015) Developing nanocrystals for cancer treatment. Nanomedicine (London, England) 10, 2537–52. [DOI] [PubMed] [Google Scholar]
- (24).Deng J, Huang L, and Liu F (2010) Understanding the structure and stability of paclitaxel nanocrystals. International journal of pharmaceutics 390, 242–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Li Y, Wu Z, He W, Qin C, Yao J, Zhou J, and Yin L (2015) Globular protein-coated Paclitaxel nanosuspensions: interaction mechanism, direct cytosolic delivery, and significant improvement in pharmacokinetics. Mol Pharm 12, 1485–500. [DOI] [PubMed] [Google Scholar]
- (26).Zhang H, Hu H, Zhang H, Dai W, Wang X, Wang X, and Zhang Q (2015) Effects of PEGylated paclitaxel nanocrystals on breast cancer and its lung metastasis. Nanoscale 7, 10790–10800. [DOI] [PubMed] [Google Scholar]
- (27).Ganguly A, Yang H, and Cabral F (2011) Class III β-Tubulin Counteracts the Ability of Paclitaxel to Inhibit Cell Migration. Oncotarget 2, 368–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.