

Abstract
Prion diseases are rapidly progressive, incurable neurodegenerative disorders caused by misfolded, aggregated proteins known as prions, which are uniquely infectious. Remarkably, these infectious proteins have been responsible for widespread disease epidemics, including kuru in humans, bovine spongiform encephalopathy in cattle, and chronic wasting disease in cervids, the latter of which has spread across North America and recently appeared in Norway and Finland. The hallmark histopathologic features include widespread spongiform encephalopathy, neuronal loss, gliosis, and deposits of variably-sized aggregated prion protein ranging from small, soluble oligomers to long, thin, unbranched fibrils, depending on the disease. Here we explore recent advances, from the function of the cellular prion protein to the dysfunction triggering neurotoxicity, as well as mechanisms underlying prion spread between cells, and the effect of prion conformation on spreading pathways. We also highlight key findings that have revealed new therapeutic targets and consider unanswered questions for future research.
INTRODUCTION
Prion diseases are fatal neurodegenerative disorders of humans and animals and are remarkable due to their infectious nature. The infectious agent causing prion disease, known as PrPSc, is unusual as it lacks any specific nucleic acid; it is a pathogenic misfolded and aggregated form of the cellular prion protein, PrPC (1, 2). Following transmission to a naive host, prions seed the misfolding of host PrPC in an autocatalytic process, leading to an exponential increase in PrPSc in the brain and spinal cord that eventually leads to neuronal death (3). The primary amino acid sequence of PrPSc is determined by host PrPC, which is encoded by the prion gene, PRNP, on chromosome 20 in humans (4).
Prions are highly stable and accumulate in the central nervous system over months to years, eventually generating rampant spongiform degeneration and neuronal loss as well as activated astrocytes and microglia, and with a notable lack of peripheral inflammatory cells (5) (Figure 1). Although the incubation period may be years, the clinical phase is typically rapidly progressive (weeks to months) and may include behavior abnormalities, motor dysfunction, cognitive impairment, and ataxia, depending on the prion and species affected (6). No therapy is currently available beyond palliative care.
Figure 1.
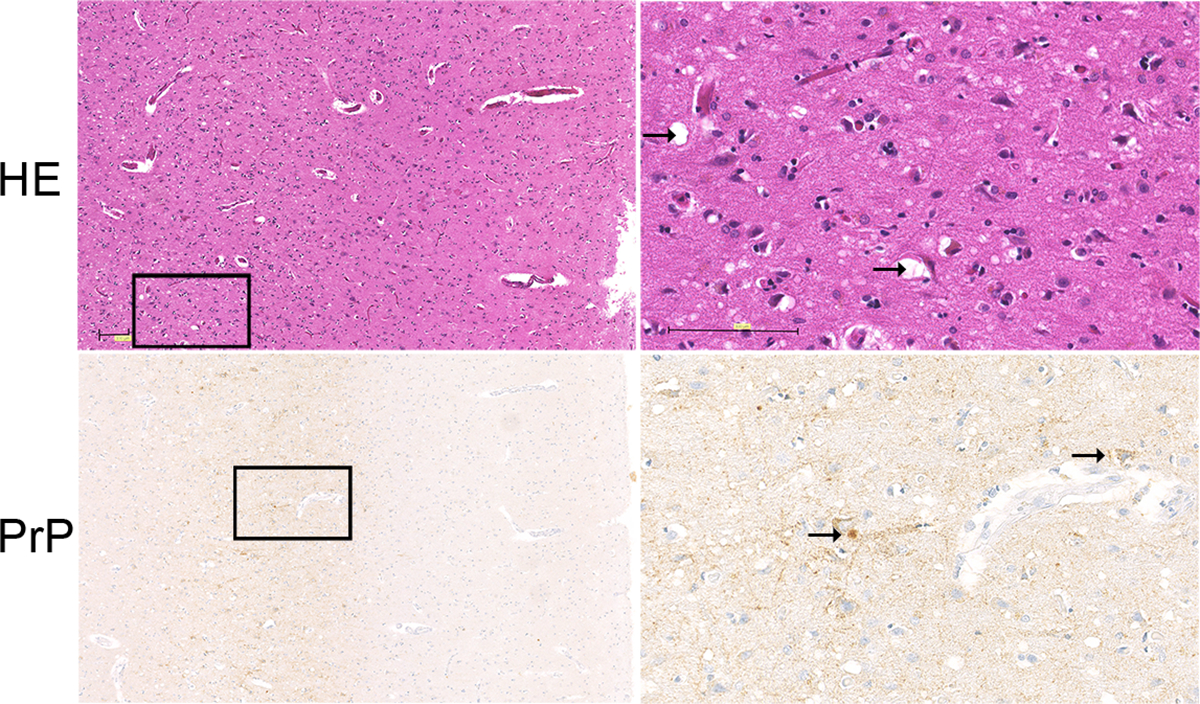
Hematoxylin and eosin and PrP immunostain of brain (frontal cortex) from a sCJD patient. Spongiosis is visible in the deep layers of the cortex (HE) (arrow indicates intaneuronal and parenchymal spongiform change, arrow) and synaptic, plaque-like, and perineuronal deposits of pathological prion protein (arrow indicates plaque-like and perineuronal deposits). The synaptic deposits of pathological prion protein are pronounced in the deep layers of the cortex. Scale bar = 100 μm.
In humans, prion diseases are categorized as sporadic, genetic, or acquired, with the majority of cases (~85%) being sporadic. Sporadic Creutzfeldt-Jakob disease (sCJD) has no known cause but has been hypothesized to be instigated by a somatic mutation in PRNP or the spontaneous conversion of PrPC to PrPSc (7). Genetic prion diseases have been classified by clinical symptoms and neuropathological features and consist of familial CJD (fCJD), fatal familial insomnia (FFI), and Gerstmann-Sträussler-Scheinker (GSS) disease. The mutations in PRNP are autosomal dominant, highly penetrant, and consist of missense mutations, insertions, and deletions, usually inciting disease onset in the 5th or 6th decade of life (6). Acquired prion diseases have been transmitted between individuals (kuru and iatrogenic CJD) and rarely from cattle to human (variant of Creutzfeldt-Jakob disease (vCJD)) (8, 9). Iatrogenic spread has occurred from prion-contaminated corneal and meningeal grafts, blood transfusions (10–13), human growth hormone (14, 15), and from prion-contaminated neurosurgical instruments and electrodes (16).
In addition to iatrogenic prion infection, prions have also precipitated large-scale, multi-species epidemics and even spread as a zoonosis. BSE was first described in 1986 (17) and within a decade, more than 180,000 cases of BSE were diagnosed in cattle with further expansion to zoo bovids, felids, and primates within Great Britain (18). By 1996, vCJD had targeted mainly young people (2nd decade) in the United Kingdom, likely from exposure to BSE-contaminated beef (9, 19), with 229 vCJD cases diagnosed to date (20). No recent cases have occurred, however secondary transmission of vCJD prions arose in transfusion recipients receiving blood or blood products originating from prion-infected donors (10–13).
Prion diseases in animals, including BSE, are largely acquired by ingestion, although atypical scrapie and BSE in aged sheep and cattle, respectively, may arise sporadically similar to sCJD (21–23). Classical scrapie affects sheep and goats nearly worldwide and has been recognized for more than 250 years (24). Chronic wasting disease (CWD) was first discovered in Colorado deer in 1967 (25) and affects free-ranging or captive deer, elk, reindeer, and moose (family Cervidae) in 25 U.S. states and two Canadian provinces, as well as ranched elk in South Korea (26) and most recently wild reindeer and moose in Norway (27) and Finland (28). Transmissible mink encephalopathy (TME) has been previously identified in farmed mink in the US, Canada, Russia, Finland and East Germany, and was thought to be due to dietary exposure to a prion-infected animal, although the origin of the epidemic remains unclear and no recent cases have been reported (29).
The complicated molecular mechanisms that govern how prions are converted and spread from extraneural entry sites into the brain, as well as how prions generate neurotoxic responses are the subject of this review focused on recent findings in prion pathogenesis. We also highlight new research linking prion conformation to disease phenotype.
Cellular Biology of the prion protein-- function and proteostasis
Prion protein synthesis and modification
The physiological (or cellular) form of the prion protein is glycosylphosphatidylinositol (GPI)-anchored and features two variably-occupied N-linked glycosylation sites (30). Mature PrPC consists of approximately 210 amino acids, arranged as a disordered N-terminal domain and a globular C-terminal domain composed of three α-helices and a short anti-parallel β-pleated sheet (31). In its mature form, PrPC is mainly present as a diglycosylated protein, located at the outer leaflet of the plasma membrane in lipid-enriched microdomains (32). Following internalization, PrPC is either recycled to the plasma membrane or the Golgi (retromer pathway) (33), or is transported to late endosomes, eventually residing in the pinched off intraluminal vesicles within multivesicular bodies (MVBs) for release as exosomes or for degradation in lysosomes (34, 35).
PrPC is subject to proteolytic cleavage, with α-cleavage and shedding of PrPC representing the two most important cleavage events (36). α-cleavage occurs in the middle of PrPC, releasing an unstructured N-terminal protein-fragment while leaving its C-terminal globular part attached to the membrane (37). This cleavage takes place during vesicular trafficking of PrPC within the secretory pathway (38). Initial reports identified the serine protease plasmin (39, 40) or ADAMs (proteins belonging to the a Disintegrin and Metalloproteinase family) (41) as potential proteases, yet recent data do not support this observation (42–44) and the exact nature of the responsible protease remains unclear (36).
A cleavage event occurring at the distal C-terminus of PrPC and releasing nearly full length PrP into the extracellular space is referred to as PrP-shedding (45, 46). PrP-shedding occurs only on the plasma membrane and ADAM10 is the only relevant PrP-sheddase, with diglycosylated PrPC representing the preferred substrate (47, 48).
Prion protein function
A detailed explanation of all of the functions attributed to PrPC would go beyond the scope of this review. In fact, we (MG and colleagues) have recently proposed to list PrPC among the expanding group of “multifunctional” proteins, in which several functions are attributed to just one protein (36). An incomplete list of PrPC functions would include its role in neural development (49), cell adhesion (50), axon guidance, synapse formation (51), neuroprotection (52, 53), regulation of circadian rhythm (54), myelin maintenance (55, 56) maintenance of ion homeostasis (57, 58), and signaling (59, 60).
Interestingly, some of the best described functions are not accredited to PrPC in its membrane-bound, GPI-anchored form but rather to soluble PrPC fragments, which can only be generated by regulated proteoloysis such as alpha-cleavage and shedding. This is true for the recently described function of soluble PrP in the maintenance of myelin homeostasis (56), or for the role of soluble versions of PrP in inducing neurite outgrowth (61). For myelin maintenance, binding of the flexible N-terminal part of soluble PrP acts as agonistic ligand to a G-protein-coupled receptor (GPCR) expressed on Schwann cells, Adgrg6 (Gpr126) (56), whereas the molecular details for the neurite outgrowth-promoting role of soluble PrP are not understood. Interestingly in this instance, membrane-bound PrPC itself may act as a receptor via homophilic interactions (61). In both instances, it is obvious that regulated proteolysis would be an elegant mode of functional regulation to transmit information to distant sites. This is reminiscent of functions attributed to proteolytic cleavage fragments from the amyloid precursor protein (APP) (62, 63). Yet while insights into the processing of APP and its biological and pathogenic consequences are vast, relatively little is known about the physiological roles of PrPC cleavage fragments.
Prion protein malfunction: Mechanisms of neurodegeneration in prion disease
Loss of function of PrPC vs toxic gain of function of PrPSc?
A key event in the pathophysiology of prion diseases is the PrPSc template-directed misfolding of PrPC into a pathogenic, conformationally altered, β-sheet-rich version of itself. This conversion process lies at the root of the now widely accepted prion hypothesis, which states that the infectious agent for prion diseases (the “prion”) is entirely made up of proteins and devoid of specific nucleic acids (64). Today we know that a pathogenic, conformationally altered version of PrPC is a key component of the infectious agent responsible for transmission of prion diseases. This disease-associated version of PrPC can be partially resistant against protease-digestion and is designated as PrPSc (where Sc stands for the pathogenic version of the protein found in sheep suffering from “scrapie”, a prion disease found only in sheep and goats). Originally, only highly protease-resistant forms were termed PrPSc but it is now accepted that there are also pathogenic PrP conformers that are mildly protease-resistant, referred to here as sPrPSc, and since these versions are just as infectious as PrPSc, a biochemical definition of protease-resistance is not adequate (65). Thus one has to include protease-sensitive disease associated PrP species into the pool of conformationally-altered versions of PrP able to induce prion disease. Alternatively, the term “PrPSc” is still widely used to describe disease associated PrP-species and for the sake of clarity, we will use this term in this review when referring to pathogenic, conformationally-altered versions of PrP.
The PrPC to PrPSc conversion process involves a massive structural rearrangement of the primarily α-helical protein into a highly β-sheet-rich structure (approximately 47% β-sheet) (66). The mechanism that underlies PrPC conversion into PrPSc remains unknown. One hypothesis is that short segments of PrPSc interact with PrPC in a “steric zipper”, in which complementary amino acid side chains from two β-sheets tightly interdigitate and effectively stabilize growing fibrils, largely through hydrogen bonds (67, 68). Sequence differences within steric zipper segments have been shown to block prion conversion between species (69, 70).
PrPC is converted to PrPSc on the plasma membrane or within the endocytic pathway, and a recent study by Greene and colleagues suggests that prion conversion occurs primarily within MVBs and not on the plasma membrane, as preventing MVB maturation sharply reduced PrPSc production (34).
The generation and progressive accumulation of PrPSc is of key importance for the development of clinical prion disease, although there are rare instances, such as subclinical disease in prion-infected mice, where the presence of PrPSc does not lead to neurodegeneration (71). It is conceivable that the partial loss of some of the physiological functions of PrPC may contribute to prion-associated neurodegeneration. A key argument against loss of function playing a role in prion disease is that loss of PrPC function in PrP knockout mice does not lead to neuronal death (72). On the other hand, we have only begun to understand how PrPC functions on a molecular level, with PrPC, or its proteolytic cleavage products, acting as receptor or ligand or both, most likely in concert with many binding partners (73). Thus a certainly recurring redundancy in this system may compensate loss of function phenotypes, which may only become apparent once additional stressors are active (36, 56).
Mechanisms underlying the toxic gain of function of PrPSc
The evidence for direct or indirect neurotoxicity of PrPSc is compelling and there is no doubt that cerebral accumulation of misfolded PrPSc plays a key role in the pathophysiology of prion diseases, but how?
Disturbed protein homeostasis in prion disease
Neuronal proteostasis, which is the interplay of protein synthesis (including correct protein folding, trafficking, and processing) and protein degradation, is essential for correct neuronal function (74). Disturbed proteostasis occurs in prion disease at multiple levels. PrPSc disturbs the ubiquitin/proteasome system already at early disease states leading to impaired function of this protein degradation pathway, enhancing the buildup of PrPSc (75). There is also mounting evidence that buildup of PrPSc affects the autophagy–lysosome pathway responsible for degradation of aggregated proteins, although in one study temporal analysis indicates that this is a consequence of protein buildup and not causally involved in disease initiation (76). Additionally there is evidence that exhaustion of unfolded protein response (UPR) pathways occurs early in prion disease (77–79). UPR is a cellular stress response aiming to protect the endoplasmic reticulum regarding its function in protein synthesis and sorting. PrPSc stresses the ER and sets off a vicious cycle resulting in disturbed PrPC trafficking, impaired PrPC functions, and translational shutdown that weakens the neurons, causing synaptic loss and ultimately inducing cell death pathways (80). Interestingly, restoring the disturbed protein translation has been shown to be neuroprotective (80).
PrPSc mediated toxicity at the neuronal membrane
PrPSc aggregation occurs in a highly ordered fashion, and oligomeric, rather than fibrillar forms of PrPSc-aggregates, are thought to be more neurotoxic (81). Morphological studies have shown the close relationship of PrPSc-deposits and the neuronal plasma membrane (82). How this translates into neurotoxicity is not fully understood but two lines of thought have emerged. In the first scenario, PrPSc-aggregates lead to major membrane disturbance, possibly by corrupting the function of neuronal receptors such as the NMDA receptor and thus altering plasma membrane permeability (83). GPI-anchored PrPC is able to efficiently transduce neurotoxicity and prion disease is accelerated in mice where PrPC shedding is impaired, both of which support this line of thought (84, 85). In the second scenario, membrane-bound PrPC itself may act as a receptor of prion toxicity. Indeed, a direct interaction between PrPSc and PrPC induces neurotoxicity similar to a mechanism first described in Alzheimer’s disease, where oligomeric species of Aβ bind membrane PrPC complexed to metabotropic glutamate receptor (mGluR5), activating intracellular Fyn kinase and ultimately leading to synaptotoxicity (86–90).
PrPC has also been incriminated in neurotoxic responses, as antibody binding to the C-terminal globular domain leads to toxic signal generation through the N-terminus, inducing calpain activation and ROS production (91). PrPSc has been found to cause a similar toxic signaling cascade, again with calpain activation and ROS generation (92). In cultured primary neurons expressing a mutant PrP lacking residues in a central region (105–125), abnormal ion channel currents occurred, sensitizing neurons to glutamate-induced excitotoxicity. These abnormal currents may represent very early toxic signaling events in affected cells and underlie early neurodegeneration (93). Nevertheless, the sequence of events leading to receptor-mediated neurotoxicity are not yet completely defined and GPI-anchored PrPC would need a co-receptor to enable intraneuronal signal transduction.
Prion spread into the CNS – an update
Similar to rare neurotropic infectious agents such as rabies virus, prions have managed to access the CNS from extraneural entry sites. The initial prion replication site in the CNS can often be linked to the entry site by peripheral nerves, incriminating retrograde axonal transport as a possible mechanism of prion transit. For example, feeding prions to hamsters leads to early prion deposition in enteric and autonomic ganglia as well as vagus and splanchnic nerves, and subsequently in the thoracic spinal cord and dorsal motor nucleus of the vagus in the brainstem, consistent with retrograde prion spread along autonomic PNS pathways into the CNS (94). BSE and CWD prions in cattle and deer, respectively, are also first detected in the CNS within the vagal nucleus, consistent with prion entry through the GI tract and transit via the vagal nerve into the brain (95, 96). Similarly, prion exposure of the mouse eye induces prion deposition along the optic nerve and tract, followed by the contralateral superior colliculus to which it projects, further suggesting prion spread via neural circuitry (97). Additional support for prion transit in nerves was provided by studies manipulating sympathetic innervation to the prion-infected spleen, which markedly affected prion entry into the CNS (98, 99). Interestingly, prion conformation also plays a role in prion neuroinvasion, as fibril-forming prions spread poorly to the brain as compared to oligomeric or subfibrillar prions (100–103). Nevertheless, since prions circulate in blood within minutes post-inoculation (104), additional non-neural pathways of prion entry into the CNS, such as passage across the blood brain barrier, cannot be excluded.
Prion spread from the gastrointestinal tract to the brain
Prion spread following ingestion is similar to that used by other infectious agents exploiting entry portals to invade the host. Transepithelial prions transit through the intestinal epithelium by way of M cells, as M cell depletion reduces oral susceptibility to prion disease (105); additional studies by multiple laboratories support M cells as key players that passage prions across the mucosal barrier (105–109). Enteritis may heighten susceptibility to oral prion infection, potentially by enabling prion passage through the mucosa (110). Once subepithelial, neurotropic prions, such as BSE, are thought to spread by retrograde axonal transport along autonomic PNS pathways into the brainstem (111, 112). Lymphotropic prions, such as sheep scrapie, deer CWD, and likely vCJD, also rapidly spread (within hours) to Peyer’s patches and draining lymph nodes, potentially transported by classical dendritic cells (95, 113, 114), as depletion of dendritic cells impedes the early replication of prions in lymph nodes (115, 116). Interestingly, lymphotropic prions also accumulate within inflamed organs harboring lymphoid follicles, such as kidney or mammary gland, leading to prion excretion or secretion into urine or milk, respectively (117–120).
Within the lymphoid tissue, PrPSc accumulates within the germinal centers of lymphoid follicles, both on the plasma membrane of follicular dendritic cells (FDCs) and within tingible body macrophages (121), where it persists throughout the infection (95). FDCs trap antigens on their plasma membrane for display to B cells (122, 123) and have proven highly capable of replicating prions (124), sustaining lymphoid prion infections for months to years (124). On the surface of FDCs, the CD21/35 receptor is thought to bind a PrPSc - complement complex, as both soluble C1q and regulatory protein factor H bind PrPSc (125–128), and CD21/35 receptor knockout mice show low attack rates after a peripheral prion infection (128). Together these studies indicate a crucial role for complement receptors in prion replication and spread to the CNS.
This peripheral phase of prion replication has been exploited for developing prevention strategies to block prion spread to the CNS. FDCs require B cell signalling through tumor necrosis factor and lymphotoxin to develop and maintain a mature state (122), and blocking lymphotoxin signaling induces FDC dedifferentiation and prevents prion accumulation in lymphoid tissue. This prevention strategy has worked very effectively in mice treated with lymphotoxin β-receptor agonists or anti-receptor antibodies (129), abolishing splenic prion replication and prolonging disease following an intraperitoneal challenge (129). Preventing disease by this strategy must begin early, however, as nerve entry occurs quickly after a prion exposure, within 14 days post oral challenge in mice (130).
Lymphoid tissues may also serve as a source of new prion strains. Cross-species prion transmission has generated new prion strains within lymphoid tissues, suggesting that lymphoid tissues may be more promiscuous than CNS in replicating prions having a different PrP sequence (131). The mechanism underlying this reduced threshold for prion replication is unclear, however the PrPSc glycan sialation levels influence capture by complement receptors and replication in lymphoid tissue, and the glycans on PrPSc are more sialated in the lymphoid tissue than in brain (132). Highly sialated PrPSc has been postulated to contribute to the permissiveness of lymphoid tissue to prion replication (132, 133).
Prion conversion within the CNS
Once within the brain and spinal cord, prions are converted by neurons and astrocytes. Astrocytes are highly susceptible to prion infection in vitro and can readily transfer prions to neurons (134, 135). On the other hand, microglia do not have a major role in replication, but are instead critical for prion clearance; depletion of microglia accelerates disease in vivo and increases PrPSc accumulation in organotypic brain slices (136). In contrast, oligodendrocytes lack any known significant contribution to prion replication or spread through the CNS (124). Although much is known about the cells that replicate prions in the brain, a pressing research need is to better understand how protein aggregates spread through the brain, from neuron-to-neuron (137–139) and between neurons and astrocytes (140).
Cell-to-cell prion spread through the CNS
Once in the brain, prions spread through neuroanatomically connected brain regions by poorly understood mechanisms (141–144). In vitro, prions spread cell-to-cell via (i) exosomes and (ii) tunneling nanotubes (138, 145–147), with yet-to-be-tested other possible mechanisms including synaptosomes, GPI-painting, microvesicles, or PrPSc cleavage from the plasma membrane (Figure 2).
Figure 2.
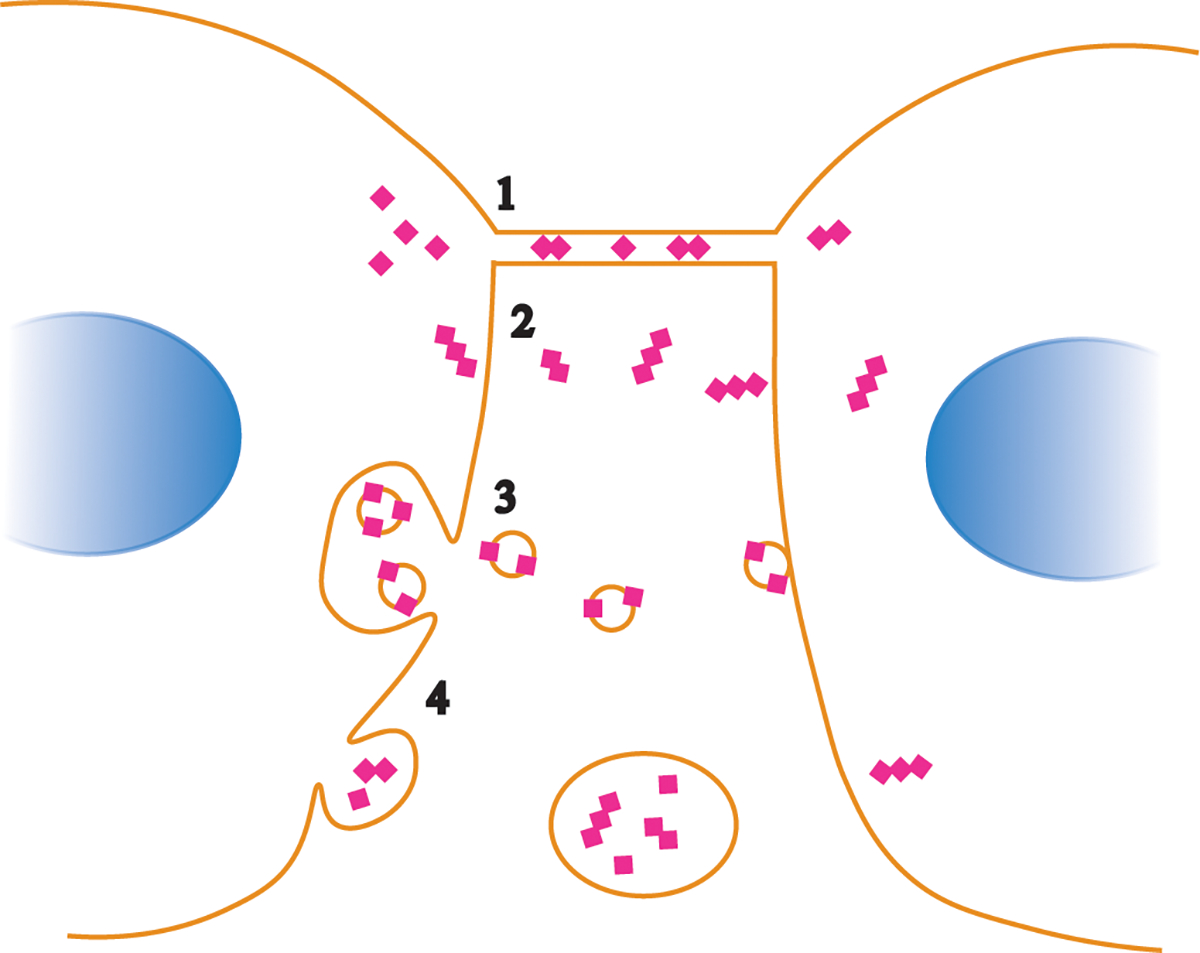
Possible pathways of prion spread from cell-to-cell. Prion aggregates may spread through transport in tunneling nanotubes (1), GPI painting, by which GPI-anchored proteins transfer from one cell surface to a neighboring cell surface (2), trafficking within exosomes (3), or from membrane budding and transport within vesicles (4).
PrPC and PrPSc were both shown to sort into MVBs for release within exosomes, 40–100 nm extracellular vesicles (EVs) that arise within multivesicular bodies (MVB) (147–149)[add Greene]. The extent to which prions are released within exosomes varies depending on the prion strain, as certain strains traffic more extensively into exosomes (145).
Further supporting the importance of EVs in prion transport, Saa and colleagues showed that vCJD prion-infected mice harbored EVs containing infectious prions in plasma starting at preclinical disease stages (150), which suggests that vesicles may transport prions long-range. Nevertheless, whether exosomes or other EVs are the most relevant mechanism for prion spread through the CNS is not yet clear. Recent methodological advances for isolating exosomes and other EVs from the brain is expected to shed light on the role of exosomes for prion spread in vivo (151).
Another possible route for direct cell-to-cell spread of prions is through tunneling nanotubes. Tunnelling nanotubes are thin membranous tubes that connect cells and serve as a mechanism for cell-to-cell communication, as organelles including lysosomes and mitochondria can be transported in nanotubes (139, 152). In addition to organelles, PrPSc was transferred to naïve cells via nanotubes, including transfer from primary dendritic cells to neurons as well as from neuron-to-neuron (138). Tunnelling nanotubes may be induced by cell stress.
Lysosomes may also be involved in the cell-to-cell transport of prions, either through transfer within tunnelling nanotubes or through lysosomal exocytosis, as observed for amyloid-β and α-synuclein (153, 154).
PrPSc conformation impacts disease phenotype
In experimental prion disease of rodents, a wide range of incubation periods and brain targets have been reported (155), depending on the prion conformation, or strain. Much work has been done to examine the relationship between the biochemical properties of PrPSc and the survival time. Studies of yeast prions (Sup35) indicated that the rate of prion propagation is inversely proportional to the aggregate stability, and suggested that more fibril fragmentation, or a higher “frangibility”, would produce new free ends for prion formation and accelerate prion propagation (156). Consistent with this notion, murine prion strains with shorter incubation periods typically have a lower PrPSc stability compared to that of longer incubation period strains (157–159). In contrast however, hamster prion strains with short incubation periods had a relatively high PrPSc stability compared to long incubation period strains (160, 161). Similar to the hamster prion model, patients with sporadic CJD accumulating stable PrPSc had a shorter, more rapidly progressive clinical disease, potentially due instead to faster PrPC conversion (162, 163).
Protease-sensitive forms of PrPSc, sPrPSc, have been implicated in disease pathogenesis (164), and factoring in these species may also help explain the above discrepancies in PrPSc stability and incubation period. The relative ratio of sPrPSc to PK-resistant PrPSc is strain-specific, and evidence suggests that these small sPrPSc oligomers can influence the prion conversion rate (165, 166). However, some groups have suggested that the abundance of sPrPSc does not exceed 10% of the total amount of PrPSc and, therefore, downplay the relative contributions of these species to disease (167). Overall, the relationship between the biochemical properties of PrPSc and the outcome of disease is still poorly understood. This may be due, in part, to the many other factors that contribute to the incubation period of disease in vivo, including the various clearance mechanisms. The use of protein misfolding cyclic amplification (PMCA), which recapitulates prion conversion in vitro (168), continues to provide useful information on factors that influence the rate of PrPSc formation.
PrPC is the major host factor that controls the tempo of prion formation. Genetic ablation of Prnp renders animals resistant to prion infection and agent replication (169–172). Conversely, increasing PrPC expression results in a reduction in the incubation period (173, 174). Consistent with these in vivo studies, in vitro experiments have shown that the abundance of PrPC positively correlates to conversion efficiency (175). Interestingly, recent work has shown that as the prion disease progresses, the PrPC level is reduced (44). The reduction in PrPC levels may contribute to a decline in the rate of prion conversion and/or slow the onset of neurodegeneration (176). Additional PrPC factors that influence conversion include the post-translational modifications of PrPC. Specifically, the sialylation status of the N-links glycans impacts prion conversion in a strain dependent manner (177). Consistent with this observation, removal of sialylation can increase the efficiency of prion formation (133, 178).
Host cellular co-factors also influence the rate of prion formation. Removal of RNA significantly reduced PrPSc formation, whereas RNA supplementation restored PrPSc formation in a PMCA reaction (179). Interestingly, the extent of reduction induced by RNA depletion was strain dependent as was the composition of nucleic acid that restored PrPSc formation (180). Phosphatidylethanolamine (PE) also supported the formation of both mouse and hamster PrPSc in vitro (181). Importantly, PMCA conversion of three separate prion strains with PE as a co-factor resulted in the three strains converging into a single strain (181). Recent evidence suggests that strain specific co-factors may not be the only mechanism responsible for prion tissue tropism. For example, if the relative rate of PrPSc clearance exceeds PrPSc formation, infection is not established (182). Overall, the distribution of convertible PrPC and host cellular co-factors, in combination with the relative rates of prion formation and clearance, may influence the strain-specific pace and tropism of disease (Figure 3).
Figure 3.
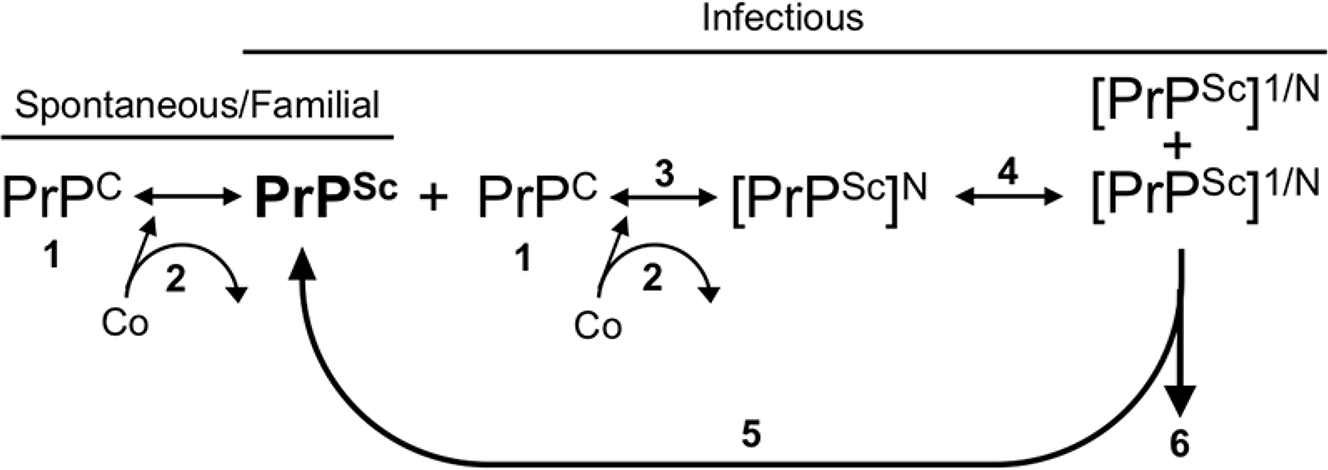
Strain-specific factors in prion formation. Prion formation is dependent on the presence of PrPC (1). For the conversion of PrPC to PrPSc in spontaneous, familial, or infectious etiologies, cofactors (Co) may participate in the formation of PrPSc, although it is unknown if they are incorporated into the growing polymer or simply used as a structural scaffold (2). The rate of PrPSc formation (3) is dictated by the incoming prion strain (PrPSc), the level of PrPC (1), and the cofactors present (2). PrPSc fragmentation can result in newly fragmented PrPSc serving as a seed for conversion (5) or PrPSc clearance from the cell (6). The rate of prion formation (3) must be greater than the rate of clearance (6) to establish a productive infection. Strain-specific PrPSc conformations may utilize specific subpopulations of PrPC, cofactors, and clearance mechanisms that may all contribute to strain-specific cellular and tissue tropism.
Multiple prion strains can coexist in a host
A co-existence of prion subtypes is commonly found in sCJD-affected patients (183), and interestingly, the subtypes have different rates of PrPSc formation in vitro (184). The relative percentage of sCJD cases that contain both PrPSc subtypes is not agreed upon (185–187). Differences in estimates of the co-occurrence of PrPSc subtypes may be explained by incomplete PK digestion of PrPSc that may allow for an overestimation (188), or sampling a limited number of brain regions or employing a limited number of anti-PrP antibodies, which may lead to an underestimation. Overall, it is clear that in human prion disease, mixtures of prion subtypes occur. The effect of these subtype mixtures on disease development and transmission in natural cases of prion disease are unclear.
Prion strains can interfere with conversion when present in mixtures. Prion strain interference occurs when a slowly replicating (long incubation period) strain interferes with the replication of a relatively quickly replicating (shorter incubation period) prion strain. The relative onset of replication of the blocking and superinfecting strain dictates the outcome of strain emergence (189). Consistent with this observation, replication of the blocking strain is required for strain interference to occur (190, 191). Interestingly, in animals infected with two strains under conditions where strain interference does not occur, PrPSc levels of both strains are altered (192). This is consistent with the hypothesis that prions have properties of a quasispecies, hypothesized to be populations of similar, but not identical, conformations of PrPSc (193). Altering the prion conversion environment in vitro can also alter the strain properties (194–197), and the selection of drug resistant prions that revert to a drug sensitive phenotype once the drug is removed is consistent with this hypothesis (194). Overall, prion strains are highly dynamic mixtures regardless of incubation period or clinical outcome of disease, and must be considered in the development of therapies that may target specific prion conformations.
Therapeutic Implications
Prions cause toxicity in the central nervous system and yet the underlying mechanisms remain incompletely defined. Neuronal PrPC is part of a key pathway inciting neurodegeneration, as an elegant study from Mallucci and colleagues showed that depleting neuronal PrPC in transgenic mice 8 weeks post-inoculation reverses early spongiform degeneration and the progression to clinical scrapie (198). Such remarkable findings, together with a rich body of research that indicates the requirement of PrPC in prion-induced neurodegeneration, indicate that reducing PrPC expression may be a key therapeutic intervention.
Prion activation of the unfolded protein response leads to a decrease in protein translation associated with synaptic failure and neuronal loss in prion-diseased mice (80), and restoring protein translation is neuroprotective (80). Thus as a second possible therapy, pharmacologic restoration of protein translation may aid neuronal survival (199). Additional potential therapeutic strategies may rely on increasing the clearance of prion aggregates, blocking the cell-to-cell spread of prions, and directly inhibiting prion conversion using mutated full length or peptide fragments of PrPC that bind PrPSc and block fibril growth. Taking these studies t(85)ogether, the essential role of PrPC in mediating neuronal toxicity is clear, and much has been learned in recent years about the mechanisms of toxicity, yet a complete understanding of how neurodegeneration develops remains to be elucidated.
Future Directions
Although much has been discovered in recent years on the mechanisms of prion conversion, transmission, and pathogenesis, basic structural and mechanistic questions in the prion disease field remain unresolved. How are the multiple functions of PrPC executed and how do PrPC proteolytic cleavage products contribute to the purported functions? What is the structure of PrPSc and how widely do PrPSc molecules from different strains vary in structure? How does the structure of PrPSc impact neural cell targeting and influence neuronal toxicity? What are the pathways of prion-induced neuronal toxicity? How do prions spread through the brain? What are the major prion clearance pathways? Prion disease investigation has led the way in dementia research in recent years and answers to questions raised above are within reach. Additionally, answers to these basic questions will enable the rationale design of new therapeutic strategies, which may also help to restore lost confidence regarding novel therapeutic approaches in the wider field of dementia research.
REFERENCES
- 1.Bolton DC, McKinley MP, Prusiner SB. 1982. Identification of a protein that purifies with the scrapie prion. Science 218: 1309–11 [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216: 136–44 [DOI] [PubMed] [Google Scholar]
- 3.Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. 1998. Prion protein biology. Cell 93: 337–48 [DOI] [PubMed] [Google Scholar]
- 4.Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C. 1986. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 46: 417–28 [DOI] [PubMed] [Google Scholar]
- 5.DeArmond SJ, Prusiner SB. 1995. Etiology and pathogenesis of prion diseases. Am J Pathol 146: 785–811 [PMC free article] [PubMed] [Google Scholar]
- 6.Takada LT, Geschwind MD. 2013. Prion diseases. Semin Neurol 33: 348–56 [DOI] [PubMed] [Google Scholar]
- 7.Prusiner SB. 1989. Creutzfeldt-Jakob disease and scrapie prions. Alzheimer Dis Assoc Disord 3: 52–78 [DOI] [PubMed] [Google Scholar]
- 8.Scott MR, Will R, Ironside J, Nguyen HO, Tremblay P, DeArmond SJ, Prusiner SB. 1999. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci U S A 96: 15137–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. 1997. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent Nature 389: 498–501 [DOI] [PubMed] [Google Scholar]
- 10.Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. 2004. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364: 527–9 [DOI] [PubMed] [Google Scholar]
- 11.Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. 2004. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 363: 417–21 [DOI] [PubMed] [Google Scholar]
- 12.Wroe SJ, Pal S, Siddique D, Hyare H, Macfarlane R, Joiner S, Linehan JM, Brandner S, Wadsworth JD, Hewitt P, Collinge J. 2006. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet 368: 2061–7 [DOI] [PubMed] [Google Scholar]
- 13.Peden A, McCardle L, Head MW, Love S, Ward HJ, Cousens SN, Keeling DM, Millar CM, Hill FG, Ironside JW. 2010. Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia 16: 296–304 [DOI] [PubMed] [Google Scholar]
- 14.Gibbs CJ Jr., Joy A, Heffner R, Franko M, Miyazaki M, Asher DM, Parisi JE, Brown PW, Gajdusek DC. 1985. Clinical and pathological features and laboratory confirmation of Creutzfeldt-Jakob disease in a recipient of pituitary-derived human growth hormone. N Engl J Med 313: 734–8 [DOI] [PubMed] [Google Scholar]
- 15.Brown P 1998. Transmission of spongiform encephalopathy through biological products. Dev Biol Stand 93: 73–8 [PubMed] [Google Scholar]
- 16.Brown P, Preece M, Brandel JP, Sato T, McShane L, Zerr I, Fletcher A, Will RG, Pocchiari M, Cashman NR, d’Aignaux JH, Cervenakova L, Fradkin J, Schonberger LB, Collins SJ. 2000. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology 55: 1075–81. [DOI] [PubMed] [Google Scholar]
- 17.Wells GA, Scott AC, Johnson CT, Gunning RF, Hancock RD, Jeffrey M, Dawson M, Bradley R. 1987. A novel progressive spongiform encephalopathy in cattle. Vet.Rec. 121: 419–20 [DOI] [PubMed] [Google Scholar]
- 18.Anderson RM, Donnelly CA, Ferguson NM, Woolhouse ME, Watt CJ, Udy HJ, MaWhinney S, Dunstan SP, Southwood TR, Wilesmith JW, Ryan JB, Hoinville LJ, Hillerton JE, Austin AR, Wells GA. 1996. Transmission dynamics and epidemiology of BSE in British cattle. Nature 382: 779–88 [DOI] [PubMed] [Google Scholar]
- 19.Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, Doey LJ, Lantos P. 1997. The same prion strain causes vCJD and BSE [letter] Nature 389: 448–50 [DOI] [PubMed] [Google Scholar]
- 20.Diack AB, Head MW, McCutcheon S, Boyle A, Knight R, Ironside JW, Manson JC, Will RG. 2014. Variant CJD. 18 years of research and surveillance. Prion 8: 286–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baron T, Biacabe AG, Arsac JN, Benestad S, Groschup MH. 2007. Atypical transmissible spongiform encephalopathies (TSEs) in ruminants. Vaccine 25: 5625–30 [DOI] [PubMed] [Google Scholar]
- 22.Benestad SL, Arsac JN, Goldmann W, Noremark M. 2008. Atypical/Nor98 scrapie: properties of the agent, genetics, and epidemiology. Vet Res 39: 19. [DOI] [PubMed] [Google Scholar]
- 23.Biacabe AG, Morignat E, Vulin J, Calavas D, Baron TG. 2008. Atypical bovine spongiform encephalopathies, France, 2001–2007. Emerg Infect Dis 14: 298–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Detwiler LA. 1992. Scrapie. Rev Sci Tech 11: 491–537 [DOI] [PubMed] [Google Scholar]
- 25.Williams ES, Young S. 1980. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis 16: 89–98 [DOI] [PubMed] [Google Scholar]
- 26.Lee YH, Sohn HJ, Kim MJ, Kim HJ, Lee WY, Yun EI, Tark DS, Cho IS, Balachandran A. 2013. Strain characterization of the Korean CWD cases in 2001 and 2004. J Vet Med Sci 75: 95–8 [DOI] [PubMed] [Google Scholar]
- 27.Benestad SL, Mitchell G, Simmons M, Ytrehus B, Vikoren T. 2016. First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet Res 47: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.2018. Moose dies of CWD for 1st time in Finland. In Daily Finland [Google Scholar]
- 29.Marsh RF, Hadlow WJ. 1992. Transmissible mink encephalopathy. Rev Sci Tech 11: 539–50 [DOI] [PubMed] [Google Scholar]
- 30.Mayor S, Riezman H. 2004. Sorting GPI-anchored proteins. Nat Rev Mol Cell Biol 5: 110–20 [DOI] [PubMed] [Google Scholar]
- 31.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wüthrich K. 1996. NMR structure of the mouse prion protein domain PrP (121–231). Nature 382: 180–82 [DOI] [PubMed] [Google Scholar]
- 32.Shyng SL, Heuser JE, Harris DA. 1994. A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. J Cell Biol 125: 1239–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. 2003. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J 22: 3591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yim YI, Park BC, Yadavalli R, Zhao X, Eisenberg E, Greene LE. 2015. The multivesicular body is the major internal site of prion conversion. J Cell Sci 128: 1434–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo BB, Bellingham SA, Hill AF. 2015. The neutral sphingomyelinase pathway regulates packaging of the prion protein into exosomes. J Biol Chem 290: 3455–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linsenmeier L, Altmeppen HC, Wetzel S, Mohammadi B, Saftig P, Glatzel M. 2017. Diverse functions of the prion protein - Does proteolytic processing hold the key? Biochim Biophys Acta 1864: 2128–37 [DOI] [PubMed] [Google Scholar]
- 37.Mange A, Beranger F, Peoc’h K, Onodera T, Frobert Y, Lehmann S. 2004. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol Cell 96: 125–32 [DOI] [PubMed] [Google Scholar]
- 38.Walmsley AR, Watt NT, Taylor DR, Perera WS, Hooper NM. 2009. alpha-cleavage of the prion protein occurs in a late compartment of the secretory pathway and is independent of lipid rafts. Mol Cell Neurosci 40: 242–8 [DOI] [PubMed] [Google Scholar]
- 39.Praus M, Kettelgerdes G, Baier M, Holzhutter HG, Jungblut PR, Maissen M, Epple G, Schleuning WD, Kottgen E, Aguzzi A, Gessner R. 2003. Stimulation of plasminogen activation by recombinant cellular prion protein is conserved in the NH(2)-terminal fragment PrP23–110. Thromb Haemost 89: 812–9 [PubMed] [Google Scholar]
- 40.Kornblatt JA, Marchal S, Rezaei H, Kornblatt MJ, Balny C, Lange R, Debey MP, Hui Bon Hoa G, Marden MC, Grosclaude J. 2003. The fate of the prion protein in the prion/plasminogen complex. Biochem Biophys Res Commun 305: 518–22 [DOI] [PubMed] [Google Scholar]
- 41.Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, Grassi J, Lopez-Perez E, Checler F. 2001. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem 276: 37743–6 [DOI] [PubMed] [Google Scholar]
- 42.Barnewitz K, Maringer M, Mitteregger G, Giese A, Bertsch U, Kretzschmar HA. 2006. Unaltered prion protein cleavage in plasminogen-deficient mice. Neuroreport 17: 527–30 [DOI] [PubMed] [Google Scholar]
- 43.Wik L, Klingeborn M, Willander H, Linne T. 2012. Separate mechanisms act concurrently to shed and release the prion protein from the cell. Prion 6: 498–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mays CE, Coomaraswamy J, Watts JC, Yang J, Ko KW, Strome B, Mercer RC, Wohlgemuth SL, Schmitt-Ulms G, Westaway D. 2014. Endoproteolytic processing of the mammalian prion glycoprotein family. Febs J 281: 862–76 [DOI] [PubMed] [Google Scholar]
- 45.Borchelt DR, Rogers M, Stahl N, Telling G, Prusiner SB. 1993. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology 3: 319–29 [DOI] [PubMed] [Google Scholar]
- 46.Harris DA, Huber MT, van Dijken P, Shyng SL, Chait BT, Wang R. 1993. Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry 32: 1009–16 [DOI] [PubMed] [Google Scholar]
- 47.Taylor DR, Parkin ET, Cocklin SL, Ault JR, Ashcroft AE, Turner AJ, Hooper NM. 2009. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem 284: 22590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altmeppen HC, Prox J, Puig B, Kluth MA, Bernreuther C, Thurm D, Jorissen E, Petrowitz B, Bartsch U, De Strooper B, Saftig P, Glatzel M. 2011. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol Neurodegener 6: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. 2006. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci U S A 103: 3416–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, Crossin KL, Edelman GM, DeArmond SJ, Cohen FE, Prusiner SB. 2001. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol 314: 1209–25 [DOI] [PubMed] [Google Scholar]
- 51.Santuccione A, Sytnyk V, Leshchyns’ka I, Schachner M. 2005. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol 169: 341–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roucou X, Gains M, LeBlanc AC. 2004. Neuroprotective functions of prion protein. J Neurosci Res 75: 153–61 [DOI] [PubMed] [Google Scholar]
- 53.Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. 2009. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 284: 35973–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tobler I, Gaus SE, Deboer T, Achermann P, Fischer M, Rülicke T, Moser M, Oesch B, McBride PA, Manson JC. 1996. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 380: 639–42 [DOI] [PubMed] [Google Scholar]
- 55.Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J, Aguzzi A. 2010. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci 13: 310–8 [DOI] [PubMed] [Google Scholar]
- 56.Kuffer A, Lakkaraju AK, Mogha A, Petersen SC, Airich K, Doucerain C, Marpakwar R, Bakirci P, Senatore A, Monnard A, Schiavi C, Nuvolone M, Grosshans B, Hornemann S, Bassilana F, Monk KR, Aguzzi A. 2016. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 536: 464–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar H. 1997. The cellular prion protein binds copper in vivo. Nature 390: 684–7 [DOI] [PubMed] [Google Scholar]
- 58.Watt NT, Taylor DR, Kerrigan TL, Griffiths HH, Rushworth JV, Whitehouse IJ, Hooper NM. 2012. Prion protein facilitates uptake of zinc into neuronal cells. Nat Commun 3: 1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. 2000. Signal transduction through prion protein. Science 289: 1925–8. [DOI] [PubMed] [Google Scholar]
- 60.Lewis V, Hooper NM. 2011. The role of lipid rafts in prion protein biology. Front Biosci (Landmark Ed) 16: 151–68 [DOI] [PubMed] [Google Scholar]
- 61.Amin L, Nguyen XT, Rolle IG, D’Este E, Giachin G, Tran TH, Serbec VC, Cojoc D, Legname G. 2016. Characterization of prion protein function by focal neurite stimulation. J Cell Sci 129: 3878–91 [DOI] [PubMed] [Google Scholar]
- 62.Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S, Hornburg D, Evans LD, Moore S, Daria A, Hampel H, Muller V, Giudici C, Nuscher B, Wenninger-Weinzierl A, Kremmer E, Heneka MT, Thal DR, Giedraitis V, Lannfelt L, Muller U, Livesey FJ, Meissner F, Herms J, Konnerth A, Marie H, Haass C. 2015. eta-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526: 443–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andrew RJ, Kellett KA, Thinakaran G, Hooper NM. 2016. A Greek Tragedy: The Growing Complexity of Alzheimer Amyloid Precursor Protein Proteolysis. J Biol Chem 291: 19235–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prusiner SB. 2013. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 47: 601–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Collinge J 2016. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 539: 217–26 [DOI] [PubMed] [Google Scholar]
- 66.Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. 1991. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry 30: 7672–80 [DOI] [PubMed] [Google Scholar]
- 67.Apostol MI, Wiltzius JJ, Sawaya MR, Cascio D, Eisenberg D. 2011. Atomic structures suggest determinants of transmission barriers in mammalian prion disease. Biochemistry 50: 2456–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, Madsen AO, Riekel C, Eisenberg D. 2007. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 447: 453–7 [DOI] [PubMed] [Google Scholar]
- 69.Kurt TD, Bett C, Fernandez-Borges N, Joshi-Barr S, Hornemann S, Rulicke T, Castilla J, Wuthrich K, Aguzzi A, Sigurdson CJ. 2014. Prion transmission prevented by modifying the beta2-alpha2 loop structure of host PrPC. J Neurosci 34: 1022–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kurt TD, Jiang L, Fernandez-Borges N, Bett C, Liu J, Yang T, Spraker TR, Castilla J, Eisenberg D, Kong Q, Sigurdson CJ. 2015. Human prion protein sequence elements impede cross-species chronic wasting disease transmission. J Clin Invest [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hill AF, Joiner S, Linehan J, Desbruslais M, Lantos PL, Collinge J. 2000. Species-barrier-independent prion replication in apparently resistant species. Proc Natl Acad Sci U S A 29: 10248–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mallucci GR, Ratte S, Asante EA, Linehan J, Gowland I, Jefferys JG, Collinge J. 2002. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. Embo J 21: 202–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Linden R 2017. The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front Mol Neurosci 10: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rubinsztein DC. 2006. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443: 780–6 [DOI] [PubMed] [Google Scholar]
- 75.McKinnon C, Goold R, Andre R, Devoy A, Ortega Z, Moonga J, Linehan JM, Brandner S, Lucas JJ, Collinge J, Tabrizi SJ. 2016. Prion-mediated neurodegeneration is associated with early impairment of the ubiquitin-proteasome system. Acta Neuropathol 131: 411–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu Y, Tian C, Wang SB, Xie WL, Guo Y, Zhang J, Shi Q, Chen C, Dong XP. 2012. Activation of the macroautophagic system in scrapie-infected experimental animals and human genetic prion diseases. Autophagy 8: 1604–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Soto C, Satani N. 2011. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol Med 17: 14–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hetz C, Mollereau B. 2014. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 15: 233–49 [DOI] [PubMed] [Google Scholar]
- 79.Torres M, Matamala JM, Duran-Aniotz C, Cornejo VH, Foley A, Hetz C. 2015. ER stress signaling and neurodegeneration: At the intersection between Alzheimer’s disease and Prion-related disorders. Virus Res 207: 69–75 [DOI] [PubMed] [Google Scholar]
- 80.Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR. 2012. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 485: 507–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Caughey B, Baron GS, Chesebro B, Jeffrey M. 2009. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 78: 177–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wegmann S, Miesbauer M, Winklhofer KF, Tatzelt J, Muller DJ. 2008. Observing fibrillar assemblies on scrapie-infected cells. Pflugers Arch 456: 83–93 [DOI] [PubMed] [Google Scholar]
- 83.Chiesa R 2015. The elusive role of the prion protein and the mechanism of toxicity in prion disease. PLoS Pathog 11: e1004745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Altmeppen HC, Prox J, Krasemann S, Puig B, Kruszewski K, Dohler F, Bernreuther C, Hoxha A, Linsenmeier L, Sikorska B, Liberski PP, Bartsch U, Saftig P, Glatzel M. 2015. The sheddase ADAM10 is a potent modulator of prion disease. Elife 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. 2005. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308: 1435–9 [DOI] [PubMed] [Google Scholar]
- 86.Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Muller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, Multhaup G, Winklhofer KF, Tatzelt J. 2011. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J 30: 2057–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. 2009. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457: 1128–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Solforosi L, Bellon A, Schaller M, Cruite JT, Abalos GC, Williamson RA. 2007. Toward molecular dissection of PrPC-PrPSc interactions. J Biol Chem 282: 7465–71 [DOI] [PubMed] [Google Scholar]
- 89.Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. 2012. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 15: 1227–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, Strittmatter SM. 2013. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79: 887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sonati T, Reimann RR, Falsig J, Baral PK, O’Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, Swayampakula M, Rahman MH, Das D, Kav N, Riek R, Liberski PP, James MN, Aguzzi A. 2013. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 501: 102–6 [DOI] [PubMed] [Google Scholar]
- 92.Herrmann US, Sonati T, Falsig J, Reimann RR, Dametto P, O’Connor T, Li B, Lau A, Hornemann S, Sorce S, Wagner U, Sanoudou D, Aguzzi A. 2015. Prion infections and anti-PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog 11: e1004662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Biasini E, Unterberger U, Solomon IH, Massignan T, Senatore A, Bian H, Voigtlaender T, Bowman FP, Bonetto V, Chiesa R, Luebke J, Toselli P, Harris DA. 2013. A mutant prion protein sensitizes neurons to glutamate-induced excitotoxicity. J Neurosci 33: 2408–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McBride PA, Schulz-Schaeffer WJ, Donaldson M, Bruce M, Diringer H, Kretzschmar HA, Beekes M. 2001. Early spread of scrapie from the gastrointestinal tract to the central nervous system involves autonomic fibers of the splanchnic and vagus nerves. J Virol 75: 9320–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fox KA, Jewell JE, Williams ES, Miller MW. 2006. Patterns of PrPCWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus). J Gen Virol 87: 3451–61 [DOI] [PubMed] [Google Scholar]
- 96.Kaatz M, Fast C, Ziegler U, Balkema-Buschmann A, Hammerschmidt B, Keller M, Oelschlegel A, McIntyre L, Groschup MH. 2012. Spread of Classic BSE Prions from the Gut via the Peripheral Nervous System to the Brain. Am J Pathol 181: 515–24 [DOI] [PubMed] [Google Scholar]
- 97.Fraser H 1982. Neuronal spread of scrapie agent and targeting of lesions within the retino-tectal pathway. Nature 295: 149–50 [DOI] [PubMed] [Google Scholar]
- 98.Glatzel M, Heppner FL, Albers KM, Aguzzi A. 2001. Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron 31: 25–34. [DOI] [PubMed] [Google Scholar]
- 99.Prinz M, Heikenwalder M, Junt T, Schwarz P, Glatzel M, Heppner FL, Fu YX, Lipp M, Aguzzi A. 2003. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 425: 957–62 [DOI] [PubMed] [Google Scholar]
- 100.Bett C, Joshi-Barr S, Lucero M, Trejo M, Liberski P, Kelly JW, Masliah E, Sigurdson CJ. 2012. Biochemical properties of highly neuroinvasive prion strains. PLoS Pathogens 8: e1002522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bett C, Kurt TD, Lucero M, Trejo M, Rozemuller AJ, Kong Q, Nilsson KP, Masliah E, Oldstone MB, Sigurdson CJ. 2013. Defining the conformational features of anchorless, poorly neuroinvasive prions. PLoS Pathog 9: e1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bett C, Lawrence J, Kurt TD, Orru C, Aguilar-Calvo P, Kincaid AE, Surewicz WK, Caughey B, Wu C, Sigurdson CJ. 2017. Enhanced neuroinvasion by smaller, soluble prions. Acta Neuropathol Commun 5: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Aguilar-Calvo P, Bett C, Sevillano AM, Kurt TD, Lawrence J, Soldau K, Hammarstrom P, Nilsson KPR, Sigurdson CJ. 2018. Generation of novel neuroinvasive prions following intravenous challenge. Brain Pathol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Elder AM, Henderson DM, Nalls AV, Hoover EA, Kincaid AE, Bartz JC, Mathiason CK. 2015. Immediate and Ongoing Detection of Prions in the Blood of Hamsters and Deer following Oral, Nasal, or Blood Inoculations. J Virol 89: 7421–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Donaldson DS, Kobayashi A, Ohno H, Yagita H, Williams IR, Mabbott NA. 2012. M cell-depletion blocks oral prion disease pathogenesis. Mucosal Immunol 5: 216–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heppner FL, Christ AD, Klein MA, Prinz M, Fried M, Kraehenbuhl JP, Aguzzi A. 2001. Transepithelial prion transport by M cells. Nat Med 7: 976–7 [DOI] [PubMed] [Google Scholar]
- 107.Donaldson DS, Sehgal A, Rios D, Williams IR, Mabbott NA. 2016. Increased Abundance of M Cells in the Gut Epithelium Dramatically Enhances Oral Prion Disease Susceptibility. PLoS Pathog 12: e1006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Miyazawa K, Kanaya T, Takakura I, Tanaka S, Hondo T, Watanabe H, Rose MT, Kitazawa H, Yamaguchi T, Katamine S, Nishida N, Aso H. 2010. Transcytosis of murine-adapted bovine spongiform encephalopathy agents in an in vitro bovine M cell model. J Virol 84: 12285–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Takakura I, Miyazawa K, Kanaya T, Itani W, Watanabe K, Ohwada S, Watanabe H, Hondo T, Rose MT, Mori T, Sakaguchi S, Nishida N, Katamine S, Yamaguchi T, Aso H. 2011. Orally administered prion protein is incorporated by m cells and spreads into lymphoid tissues with macrophages in prion protein knockout mice. Am J Pathol 179: 1301–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sigurdson CJ, Heikenwalder M, Manco G, Barthel M, Schwarz P, Stecher B, Krautler NJ, Hardt WD, Seifert B, Macpherson AJ, Corthesy I, Aguzzi A. 2008. Bacterial Colitis Increases Susceptibility to Oral Prion Disease. J Infect Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hoffmann C, Ziegler U, Buschmann A, Weber A, Kupfer L, Oelschlegel A, Hammerschmidt B, Groschup MH. 2007. Prions spread via the autonomic nervous system from the gut to the central nervous system in cattle incubating bovine spongiform encephalopathy. J Gen Virol 88: 1048–55 [DOI] [PubMed] [Google Scholar]
- 112.Masujin K, Matthews D, Wells GA, Mohri S, Yokoyama T. 2007. Prions in the peripheral nerves of bovine spongiform encephalopathy-affected cattle. J Gen Virol 88: 1850–8 [DOI] [PubMed] [Google Scholar]
- 113.Sigurdson CJ, Williams ES, Miller MW, Spraker TR, O’Rourke KI, Hoover EA. 1999. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J Gen Virol 80: 2757–64 [DOI] [PubMed] [Google Scholar]
- 114.Heggebo R, Press CM, Gunnes G, Lie KI, Tranulis MA, Ulvund M, Groschup MH, Landsverk T. 2000. Distribution of prion protein in the ileal Peyer’s patch of scrapie- free lambs and lambs naturally and experimentally exposed to the scrapie agent. J Gen Virol 81 Pt 9: 2327–37. [DOI] [PubMed] [Google Scholar]
- 115.Raymond CR, Aucouturier P, Mabbott NA. 2007. In vivo depletion of CD11c+ cells impairs scrapie agent neuroinvasion from the intestine. J Immunol 179: 7758–66 [DOI] [PubMed] [Google Scholar]
- 116.Cordier-Dirikoc S, Chabry J. 2008. Temporary depletion of CD11c+ dendritic cells delays lymphoinvasion after intraperitonal scrapie infection. J Virol 82: 8933–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Heikenwalder M, Zeller N, Seeger H, Prinz M, Klohn PC, Schwarz P, Ruddle NH, Weissmann C, Aguzzi A. 2005. Chronic lymphocytic inflammation specifies the organ tropism of prions. Science 307: 1107–10 [DOI] [PubMed] [Google Scholar]
- 118.Seeger H, Heikenwalder M, Zeller N, Kranich J, Schwarz P, Gaspert A, Seifert B, Miele G, Aguzzi A. 2005. Coincident scrapie infection and nephritis lead to urinary prion excretion. Science 310: 324–6 [DOI] [PubMed] [Google Scholar]
- 119.Ligios C, Cancedda MG, Carta A, Santucciu C, Maestrale C, Demontis F, Saba M, Patta C, DeMartini JC, Aguzzi A, Sigurdson CJ. 2011. Sheep with scrapie and mastitis transmit infectious prions through the milk. J Virol 85: 1136–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ligios C, Sigurdson CJ, Santucciu C, Carcassola G, Manco G, Basagni M, Maestrale C, Cancedda MG, Madau L, Aguzzi A. 2005. PrP(Sc) in mammary glands of sheep affected by scrapie and mastitis. Nat Med 11: S. [DOI] [PubMed] [Google Scholar]
- 121.Sigurdson CJ, Barillas-Mury C, Miller MW, Oesch B, van Keulen LJ, Langeveld JP, Hoover EA. 2002. PrP(CWD) lymphoid cell targets in early and advanced chronic wasting disease of mule deer. J Gen Virol 83: 2617–28 [DOI] [PubMed] [Google Scholar]
- 122.Endres R, Alimzhanov MB, Plitz T, Futterer A, Kosco-Vilbois MH, Nedospasov SA, Rajewsky K, Pfeffer K. 1999. Mature follicular dendritic cell networks depend on expression of lymphotoxin beta receptor by radioresistant stromal cells and of lymphotoxin beta and tumor necrosis factor by B cells. J Exp Med 189: 159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Aydar Y, Sukumar S, Szakal AK, Tew JG. 2005. The influence of immune complex-bearing follicular dendritic cells on the IgM response, Ig class switching, and production of high affinity IgG. J Immunol 174: 5358–66 [DOI] [PubMed] [Google Scholar]
- 124.McCulloch L, Brown KL, Bradford BM, Hopkins J, Bailey M, Rajewsky K, Manson JC, Mabbott NA. 2011. Follicular dendritic cell-specific prion protein (PrP) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathog 7: e1002402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Klein MA, Kaeser PS, Schwarz P, Weyd H, Xenarios I, Zinkernagel RM, Carroll MC, Verbeek JS, Botto M, Walport MJ, Molina H, Kalinke U, Acha-Orbea H, Aguzzi A. 2001. Complement facilitates early prion pathogenesis. Nat Med 7: 488–92. [DOI] [PubMed] [Google Scholar]
- 126.Mabbott NA, Bruce ME, Botto M, Walport MJ, Pepys MB. 2001. Temporary depletion of complement component C3 or genetic deficiency of C1q significantly delays onset of scrapie. Nat Med 7: 485–7 [DOI] [PubMed] [Google Scholar]
- 127.Michel B, Ferguson A, Johnson T, Bender H, Meyerett-Reid C, Pulford B, von Teichman A, Seelig D, Weis JH, Telling GC, Aguzzi A, Zabel MD. Genetic Depletion of Complement Receptors CD21/35 Prevents Terminal Prion Disease in a Mouse Model of Chronic Wasting Disease. J Immunol 189: 4520–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zabel MD, Heikenwalder M, Prinz M, Arrighi I, Schwarz P, Kranich J, von Teichman A, Haas KM, Zeller N, Tedder TF, Weis JH, Aguzzi A. 2007. Stromal complement receptor CD21/35 facilitates lymphoid prion colonization and pathogenesis. J Immunol 179: 6144–52 [DOI] [PubMed] [Google Scholar]
- 129.Montrasio F, Frigg R, Glatzel M, Klein MA, Mackay F, Aguzzi A, Weissmann C. 2000. Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science 288: 1257–9 [DOI] [PubMed] [Google Scholar]
- 130.Mabbott NA, Young J, McConnell I, Bruce ME. 2003. Follicular dendritic cell dedifferentiation by treatment with an inhibitor of the lymphotoxin pathway dramatically reduces scrapie susceptibility. Journal of Virology 77: 6845–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Beringue V, Herzog L, Jaumain E, Reine F, Sibille P, Le Dur A, Vilotte JL, Laude H. 2012. Facilitated cross-species transmission of prions in extraneural tissue. Science 335: 472–5 [DOI] [PubMed] [Google Scholar]
- 132.Srivastava S, Makarava N, Katorcha E, Savtchenko R, Brossmer R, Baskakov IV. 2015. Post-conversion sialylation of prions in lymphoid tissues. Proc Natl Acad Sci U S A 112: E6654–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Katorcha E, Makarava N, Savtchenko R, Baskakov IV. 2015. Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci Rep 5: 16912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cronier S LH, Peyrin JM. 2004. Prions can infect primary cultured neurons and astrocytes and promote neuronal cell death. Proc. Natl. Acad. Sci. U.S.A. 101: 12271–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cronier S, Carimalo J, Schaeffer B, Jaumain E, Beringue V, Miquel MC, Laude H, Peyrin JM. 2012. Endogenous prion protein conversion is required for prion-induced neuritic alterations and neuronal death. Faseb j 26: 3854–61 [DOI] [PubMed] [Google Scholar]
- 136.Zhu C, Herrmann US, Falsig J, Abakumova I, Nuvolone M, Schwarz P, Frauenknecht K, Rushing EJ, Aguzzi A. 2016. A neuroprotective role for microglia in prion diseases. J Exp Med 213: 1047–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kanu N, Imokawa Y, Drechsel DN, Williamson RA, Birkett CR, Bostock CJ, Brockes JP. 2002. Transfer of scrapie prion infectivity by cell contact in culture. Curr Biol 12: 523–30 [DOI] [PubMed] [Google Scholar]
- 138.Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chenouard N, de Chaumont F, Martino A, Enninga J, Olivo-Marin JC, Mannel D, Zurzolo C. 2009. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 11: 328–36 [DOI] [PubMed] [Google Scholar]
- 139.Zhu S, Victoria GS, Marzo L, Ghosh R, Zurzolo C. 2015. Prion aggregates transfer through tunneling nanotubes in endocytic vesicles. Prion 9: 125–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Victoria GS, Arkhipenko A, Zhu S, Syan S, Zurzolo C. 2016. Astrocyte-to-neuron intercellular prion transfer is mediated by cell-cell contact. Sci Rep 6: 20762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. 2006. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: 389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Braak H, Braak E. 2000. Pathoanatomy of Parkinson’s disease. J Neurol 247 Suppl 2: Ii3–10 [DOI] [PubMed] [Google Scholar]
- 143.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. 2003. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: 197–211 [DOI] [PubMed] [Google Scholar]
- 144.Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. 2015. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16: 109–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Arellano-Anaya ZE, Huor A, Leblanc P, Lehmann S, Provansal M, Raposo G, Andreoletti O, Vilette D. 2015. Prion strains are differentially released through the exosomal pathway. Cell Mol Life Sci 72: 1185–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vella LJ, Sharples RA, Lawson VA, Masters CL, Cappai R, Hill AF. 2007. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J Pathol 211: 582–90 [DOI] [PubMed] [Google Scholar]
- 147.Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G. 2004. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A 101: 9683–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Coleman BM, Hanssen E, Lawson VA, Hill AF. 2012. Prion-infected cells regulate the release of exosomes with distinct ultrastructural features. Faseb j 26: 4160–73 [DOI] [PubMed] [Google Scholar]
- 149.Guo BB, Bellingham SA, Hill AF. 2016. Stimulating the Release of Exosomes Increases the Intercellular Transfer of Prions. J Biol Chem 291: 5128–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Saa P, Castilla J, Soto C. 2005. Cyclic amplification of protein misfolding and aggregation. Methods Mol Biol 299: 53–65 [DOI] [PubMed] [Google Scholar]
- 151.Papadopoulos VE, Nikolopoulou G, Antoniadou I, Karachaliou A, Arianoglou G, Emmanouilidou E, Sardi SP, Stefanis L, Vekrellis K. 2018. Modulation of beta-Glucocerebrosidase Increases alpha-Synuclein secretion and Exosome release in Mouse Models of Parkinson’s Disease. Hum Mol Genet [DOI] [PubMed] [Google Scholar]
- 152.Wang X, Gerdes HH. 2015. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ 22: 1181–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, Olivo-Marin JC, Melki R, Zurzolo C. 2016. Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. Embo j 35: 2120–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Annunziata I, Patterson A, Helton D, Hu H, Moshiach S, Gomero E, Nixon R, d’Azzo A. 2013. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-beta secretion via deregulated lysosomal exocytosis. Nat Commun 4: 2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dickinson AG. 1976. Scrapie in sheep and goats. Front Biol 44: 209–41 [PubMed] [Google Scholar]
- 156.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. 2004. Conformational variations in an infectious protein determine prion strain differences. Nature 428: 323–8 [DOI] [PubMed] [Google Scholar]
- 157.Colby DW, Prusiner SB. 2011. Prions. Cold Spring Harb Perspect Biol 3: a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wille H, Bian W, McDonald M, Kendall A, Colby DW, Bloch L, Ollesch J, Borovinskiy AL, Cohen FE, Prusiner SB, Stubbs G. 2009. Natural and synthetic prion structure from X-ray fiber diffraction. Proc Natl Acad Sci U S A 106: 16990–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ghaemmaghami S, Watts JC, Nguyen HO, Hayashi S, DeArmond SJ, Prusiner SB. 2011. Conformational transformation and selection of synthetic prion strains. J Mol Biol 413: 527–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Peretz D, Scott MR, Groth D, Williamson RA, Burton DR, Cohen FE, Prusiner SB. 2001. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci 10: 854–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC. 2011. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog 7: e1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Safar JG, Xiao X, Kabir ME, Chen S, Kim C, Haldiman T, Cohen Y, Chen W, Cohen ML, Surewicz WK. 2015. Structural determinants of phenotypic diversity and replication rate of human prions. PLoS Pathog 11: e1004832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cescatti M, Saverioni D, Capellari S, Tagliavini F, Kitamoto T, Ironside J, Giese A, Parchi P. 2016. Analysis of Conformational Stability of Abnormal Prion Protein Aggregates across the Spectrum of Creutzfeldt-Jakob Disease Prions. J Virol 90: 6244–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nazor KE, Kuhn F, Seward T, Green M, Zwald D, Purro M, Schmid J, Biffiger K, Power AM, Oesch B, Raeber AJ, Telling GC. 2005. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J 24: 2472–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. 1998. Eight prion strains have PrPSc molecules with different conformations. Nature Medicine 4: 1157–65 [DOI] [PubMed] [Google Scholar]
- 166.Kim EJ, Cho SS, Jeong BH, Kim YS, Seo SW, Na DL, Geschwind MD, Jeong Y. 2012. Glucose metabolism in sporadic Creutzfeldt-Jakob disease: a statistical parametric mapping analysis of (18) F-FDG PET. Eur J Neurol 19: 488–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Saverioni D, Notari S, Capellari S, Poggiolini I, Giese A, Kretzschmar HA, Parchi P. 2013. Analyses of protease resistance and aggregation state of abnormal prion protein across the spectrum of human prions. J Biol Chem 288: 27972–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shikiya RA, Eckland TE, Young AJ, Bartz JC. 2014. Prion formation, but not clearance, is supported by protein misfolding cyclic amplification. Prion 8: 415–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Büeler HR, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. 1993. Mice devoid of PrP are resistant to scrapie. Cell 73: 1339–47 [DOI] [PubMed] [Google Scholar]
- 170.Büeler HR, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. 1992. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356: 577–82 [DOI] [PubMed] [Google Scholar]
- 171.Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M, Burton D, Yang SL, DeArmond SJ. 1993. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci U S A 90: 10608–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 1994. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 8: 121–7 [DOI] [PubMed] [Google Scholar]
- 173.Prusiner SB, Scott M, Foster D, Pan KM, Groth D, Mirenda C, Torchia M, Yang SL, Serban D, Carlson GA, et al. 1990. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63: 673–86 [DOI] [PubMed] [Google Scholar]
- 174.Manson JC, Clarke AR, McBride PA, McConnell I, Hope J. 1994. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 3: 331–40 [PubMed] [Google Scholar]
- 175.Mays CE, Titlow W, Seward T, Telling GC, Ryou C. 2009. Enhancement of protein misfolding cyclic amplification by using concentrated cellular prion protein source. Biochem Biophys Res Commun 388: 306–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mays CE, van der Merwe J, Kim C, Haldiman T, McKenzie D, Safar JG, Westaway D. 2015. Prion Infectivity Plateaus and Conversion to Symptomatic Disease Originate from Falling Precursor Levels and Increased Levels of Oligomeric PrPSc Species. J Virol 89: 12418–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Katorcha E, Makarava N, Savtchenko R, D’Azzo A, Baskakov IV. 2014. Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLoS Pathog 10: e1004366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Srivastava S, Katorcha E, Daus ML, Lasch P, Beekes M, Baskakov IV. 2017. Sialylation Controls Prion Fate in Vivo. J Biol Chem 292: 2359–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Deleault NR, Lucassen RW, Supattapone S. 2003. RNA molecules stimulate prion protein conversion. Nature 425: 717–20 [DOI] [PubMed] [Google Scholar]
- 180.Saa P, Sferrazza GF, Ottenberg G, Oelschlegel AM, Dorsey K, Lasmezas CI. 2012. Strain-specific role of RNAs in prion replication. J Virol 86: 10494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. 2012. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci U S A 109: 8546–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Shikiya RA, Langenfeld KA, Eckland TE, Trinh J, Holec SA, Mathiason CK, Kincaid AE, Bartz JC. 2017. PrPSc formation and clearance as determinants of prion tropism. PLoS Pathog 13: e1006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F. 1999. Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrP(Sc) in the same brain Neurology 53: 2173–6 [DOI] [PubMed] [Google Scholar]
- 184.Haldiman T, Kim C, Cohen Y, Chen W, Blevins J, Qing L, Cohen ML, Langeveld J, Telling GC, Kong Q, Safar JG. 2013. Co-existence of distinct prion types enables conformational evolution of human PrPSc by competitive selection. J Biol Chem 288: 29846–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A. 2005. Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol 4: 805–14 [DOI] [PubMed] [Google Scholar]
- 186.Schoch G, Seeger H, Bogousslavsky J, Tolnay M, Janzer RC, Aguzzi A, Glatzel M. 2006. Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med 3: e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Notari S, Capellari S, Langeveld J, Giese A, Strammiello R, Gambetti P, Kretzschmar HA, Parchi P. 2007. A refined method for molecular typing reveals that co-occurrence of PrP(Sc) types in Creutzfeldt-Jakob disease is not the rule. Lab Invest 87: 1103–12 [DOI] [PubMed] [Google Scholar]
- 188.Notari S, Capellari S, Giese A, Westner I, Baruzzi A, Ghetti B, Gambetti P, Kretzschmar HA, Parchi P. 2004. Effects of different experimental conditions on the PrPSc core generated by protease digestion: Implications for strain typing and molecular classification of CJD. J Biol Chem [DOI] [PubMed] [Google Scholar]
- 189.Bartz JC, Kramer ML, Sheehan MH, Hutter JA, Ayers JI, Bessen RA, Kincaid AE. 2007. Prion interference is due to a reduction in strain-specific PrPSc levels. J Virol 81: 689–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kimberlin RH, Walker CA. 1985. Competition between strains of scrapie depends on the blocking agent being infectious. Intervirology 23: 74–81 [DOI] [PubMed] [Google Scholar]
- 191.Taylor DM, Dickinson AG, Fraser H, Marsh RF. 1986. Evidence that transmissible mink encephalopathy agent is biologically inactive in mice. Neuropathol Appl Neurobiol 12: 207–15 [DOI] [PubMed] [Google Scholar]
- 192.Langenfeld KA, Shikiya RA, Kincaid AE, Bartz JC. 2016. Incongruity between Prion Conversion and Incubation Period following Coinfection. J Virol 90: 5715–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Collinge J, Clarke AR. 2007. A general model of prion strains and their pathogenicity. Science 318: 930–6 [DOI] [PubMed] [Google Scholar]
- 194.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. 2010. Darwinian evolution of prions in cell culture. Science 327: 869–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mahal SP, Browning S, Li J, Suponitsky-Kroyter I, Weissmann C. 2010. Transfer of a prion strain to different hosts leads to emergence of strain variants. Proc Natl Acad Sci U S A 107: 22653–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gonzalez-Montalban N, Lee YJ, Makarava N, Savtchenko R, Baskakov IV. 2013. Changes in prion replication environment cause prion strain mutation. Faseb j 27: 3702–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Makarava N, Baskakov IV. 2013. The evolution of transmissible prions: the role of deformed templating. PLoS Pathog 9: e1003759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. 2003. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302: 871–4 [DOI] [PubMed] [Google Scholar]
- 199.Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR. 2013. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 5: 206ra138. [DOI] [PubMed] [Google Scholar]