


Abstract
A key role of chromatin kinases is to phosphorylate histone tails during mitosis to spatiotemporally regulate cell division. Vaccinia-related kinase 1 (VRK1) is a serine–threonine kinase that phosphorylates histone H3 threonine 3 (H3T3) along with other chromatin-based targets. While structural studies have defined how several classes of histone-modifying enzymes bind to and function on nucleosomes, the mechanism of chromatin engagement by kinases is largely unclear. Here, we paired cryo-electron microscopy with biochemical and cellular assays to demonstrate that VRK1 interacts with both linker DNA and the nucleosome acidic patch to phosphorylate H3T3. Acidic patch binding by VRK1 is mediated by an arginine-rich flexible C-terminal tail. Homozygous missense and nonsense mutations of this acidic patch recognition motif in VRK1 are causative in rare adult-onset distal spinal muscular atrophy. We show that these VRK1 mutations interfere with nucleosome acidic patch binding, leading to mislocalization of VRK1 during mitosis, thus providing a potential new molecular mechanism for pathogenesis.
INTRODUCTION
Histone phosphorylation is a robust signaling platform that coordinates recruitment (1–3) or exclusion of downstream local chromatin regulators (4), alters the structure of individual nucleosomes (5,6), and facilitates chromatin condensation and decondensation during mitosis (7,8). Four residues of histone H3 (T3, S10, T11 and S28) are phosphorylated in a spatiotemporally coordinated fashion to regulate mitotic cell division (9–14). Vaccinia-related kinase 1 (VRK1) and haspin are the two known H3T3 kinases that contribute to global levels of phosphorylated H3T3 (H3T3ph). Haspin is thought to be the primary histone H3T3 kinase and haspin depletion leads to inappropriate alignment of chromosomes at the metaphase plate (15). VRK1 is a secondary H3T3 kinase, as VRK1 knockdown results in a significant loss of H3T3 phosphorylation, albeit to a lesser degree than depletion of the haspin kinase. Histone H3T3ph recruits the Aurora B kinase through direct recognition by survivin, an Aurora B adaptor protein, and stimulates Aurora B activity 8-fold (1,16). Phosphorylation of haspin by the Aurora B kinase is required for optimal activity of haspin towards H3T3 (17) and transcript levels of survivin are regulated by VRK1 (18). Thus, VRK1 may operate upstream of haspin in mitosis to phosphorylate H3T3 and trigger the feedback mechanism between the Aurora B kinase and haspin. Furthermore, the Aurora B kinase and VRK1 inhibit one another (19), indicating a delicately balanced network of self-regulation amongst chromatin kinases.
In addition to phosphorylation of H3T3, VRK1 has been shown to phosphorylate other histone residues, including H2AT120 (20), implicated in gene regulation, H3S10 (21) during mitosis, and H2A.XS139 (22), a key initiating event of the DNA damage response. Moreover, VRK1 phosphorylates transcription factors, including p53 (23), c-Jun (24) and ATF2 (25), as well as the DNA damage response protein 53BP1 (26) and the nuclear envelope component protein BAF (27). These activities underscore diverse roles for VRK1 in regulating transcription, nuclear structure, the cell cycle and the DNA damage response.
VRK1 is often overexpressed (28–31) but rarely mutated in human cancer (32). VRK1 overexpression is correlated with poor prognosis in breast cancer (33,34) and glioma (35). Interestingly, a variety of mutations in the VRK1 gene have been found in patients with developmental and degenerative motor neuron disorders including adult-onset distal spinal muscular atrophy (dSMA) (36–39), spinal muscular atrophy with pontocerebellar hypoplasia (SMA-PCH) (40) and amyotrophic lateral sclerosis (ALS) (36,41). These rare disorders are characterized by a loss of motor neurons in the spinal cord, leading to progressive muscular atrophy. VRK1 mutations in patients with motor neuron disorders have been found within the kinase domain (36,42), the nuclear localization sequence (43), at intron–exon splice junctions (44), and within the flexible C-terminal tail (37–39). The diversity of mutation sites within VRK1 suggests that insufficient protein levels, misfolding, mislocalization, or perturbations in catalytic activity can each contribute to neurodevelopmental diseases in humans.
Despite a wealth of information about the identity of VRK1 nuclear targets, little is known about how VRK1 engages these substrates, especially the nucleosome. Nucleosome-bound structures of many histone-modifying enzymes have been reported, for example structures containing histone methyltransferases and demethylases (45–52), histone acetyltransferases and deacetylases (53,54), and histone ubiquitylation and deubiquitylation machineries (55–57). Collectively, these structures reveal that chromatin enzymes engage nucleosomes through multivalent recognition of multiple nucleosomal surfaces. This orients the enzymes on the nucleosome surface to specify nucleosome-directed enzymatic activity. Despite a recent surge in structures of histone modifying enzyme-nucleosome complexes, little information exists to describe the molecular mechanisms governing nucleosome recognition by kinases.
Here, we use biochemistry, structural biology, and cell biology to define how VRK1 engages its nucleosomal substrate. VRK1 binds the nucleosome through two major interfaces, the nucleosome acidic patch and nucleosome-wrapped or extranucleosomal linker DNA. We identified an alternating arginine motif in the C-terminal tail of VRK1 that mediates acidic patch binding and used cryo-electron microscopy (cryo-EM) to solve a structure of this sequence in complex with the nucleosome. Mutation of the alternating arginine motif disrupts nucleosome binding and phosphorylation and prevents mitotic chromatin localization. Case reports have described homozygous mutations of VRK1 that truncate or mutate the VRK1 acidic patch binding motif, which are causative in adult-onset distal spinal muscular atrophy. We demonstrate that these mutations disrupt VRK1 chromatin localization, providing a likely molecular mechanism for this pathology.
MATERIALS AND METHODS
Preparation of nucleosomes
Consensus sequences for human histones H2A, H2B and H3.2 and Xenopus histone H4 were cloned into pST50 expression vectors, expressed in BL21(DE3)pLysS Escherichia coli, purified from inclusion bodies and reconstituted into H2A–H2B dimers or H3–H4 tetramers exactly as previously described (58). Histone H3 was purified from inclusion bodies, solubilized in 7 M guanidine and purified by size exclusion chromatography (Sephacryl S300, GE Healthcare) followed by Source S cation exchange chromatography as previously reported (59). After purification, the histone was lyophilized and resuspended in water with 1 mM DTT. Plasmids containing repeats of widom 601 sequences (60) were gifts from Song Tan (61). Plasmids were amplified in HB101 E. coli and purified by alkaline lysis. Nucleosomal DNA fragments were excised from plasmid DNA using EcoRV and purified by anion exchange chromatography with a Source Q resin (GE Healthcare). Nucleosomes and tetrasomes were prepared by salt gradient dialysis as previously described (45,58,59). FLAG-tagged histone H2A–H2B dimers were prepared by coexpression and PEI precipitation followed by cation exchange chromatography using a Source S resin (GE Healthcare) as previously described (58).
Preparation of BL21(DE3)pLysS-Lambda phosphatase
VRK1 was expressed in BL21(DE3)pLys E. coli also expressing Lambda phosphatase to minimize toxicity due to exogenous kinase activity. BL21(DE3)pLysS cells were first transformed with a plasmid encoding the Lambda phosphatase acquired as a gift from John Chodera, Nicholas Levinson and Markus Seeliger (Addgene plasmid #79748) and plated under spectinomycin and chloramphenicol selection. Competent cells were prepared as follows. Cultures were inoculated with 5–10 colonies and grown at 37°C at 200 rpm in 2XTY media supplemented with 10 mM MgCl2,10 mM MgSO4 and 2.5 mM KCl. Once cultures reached an OD600 of 0.4–0.8, culture flasks were cooled on ice for 10 min and then pelleted at 800 × g at 4°C for 7 min. Cells were then gently resuspended in transformation buffer (10 mM PIPES, 250 mM KCl, 15 mM CaCl2) prechilled on ice and DMSO was added to a final concentration of 13%. Cells were incubated for 10 min and then flash-frozen and stored at –80°C.
Expression and purification of proteins
Wild-type and mutant VRK1 proteins were expressed with dual N-terminal Strep-Tag II and 10xHis affinity tags using pST50 expression vectors (62). Constructs were transformed into BL21(DE3)pLysS/Lambda phosphatase competent cells under spectinomycin, chloramphenicol, and ampicillin selection. Protein expression was induced with 0.2 mM IPTG (UBP Bio) when cultures reached an OD600 of ∼0.6 and cells were harvested after 16–18 h induction at 18°C. Harvested cells were resuspended in sodium phosphate buffer (50 mM NaHPO4, pH 7.0, 300 mM NaCl, 5 mM 2-mercaptoethanol) and flash-frozen. Thawed cells were sonicated using a Branson Digital 450 Sonifier (Branson Ultrasonics) by six rounds of 14 pulses, 50% amplitude, 0.5 s on, 0.5 s off, on ice. Cleared cell lysate was applied to Talon resin (Takara) on an AKTA Start (GE Healthcare), resin was washed in phosphate buffer containing 20 mM imidazole, and protein was eluted with phosphate buffer containing 200 mM imidazole. Following Talon purification, N-terminal Strep-Tag II and 10xHis were cleaved by the addition of 1:100 molar equivalents of TEV protease. Protease cleavage was allowed to proceed overnight at room temperature during dialysis into H200 buffer (10 mM HEPES, pH 7.5, 200 mM NaCl,10 mM 2-mercaptoethanol). Protein was then diluted to a final salt concentration of 100 mM NaCl and further purified by cation exchange chromatography using a Source S resin (GE Healthcare), eluting over a salt gradient of 100–500 mM NaCl using an AKTA Pure 25M HPLC system (GE Healthcare). For some VRK1 mutants, a truncated band was observed by SDS-PAGE that could be removed using longer purification gradients. Following cation exchange chromatography, VRK1 was concentrated by centrifuge ultrafiltration (Vivaspin 20, 30 kD MWCO, Sartorius) and stored at –80°C after the addition of 20% glycerol. LANA peptides including residues 1–23 N-terminally tagged with AviTag-GST-6XHis were expressed in BL21(DE3) for 3 h at 37°C and purified as previously described (45).
The pET15B-BAF plasmid was acquired as a gift from Robert Craigie (Addgene plasmid #104152) and transformed into BL21(DE3) for expression. The BAF protein was expressed for 3 h at 37°C and cells were harvested and resuspended in T100 buffer (20 mM Tris–Cl, pH 8.0, 100 mM NaCl, 0.5 mM EDTA, 1 mM benzamidine, 10 mM 2-mercaptoethanol) prior to flash-freezing. After thawing at 30°C, cells were lysed on ice by four rounds of sonication at 70% amplitude, 10 × 0.5 s on, 0.5 s off. After sonication, all BAF protein was found in the pellet and washed once in Triton buffer (20 mM Tris–Cl, pH 8.0, 0.5 mM EDTA, 100 mM NaCl, 10 mM 2-mercaptoethanol, 1 mM benzamidine, 1% Triton X-100). The washed pellet was stored at –20°C. After thawing, the pellet was extracted with 1 mL DMSO and resuspended in phosphate buffer supplemented with 6 M guanidine (Gn-HCl) and purified by reversed-phase chromatography using a gradient of 40–80% acetonitrile on a 218P 10 μm C18 column (Vydac, Avantor) on a PrepLC 2000 Preparatory HPLC System (Waters). Purified BAF was flash-frozen, lyophilized and stored at –20°C. Lyophilized protein was resuspended in 20 mM HEPES, 150 mM NaCl, 6 M Gn-HCl, 1 mM DTT and refolding was attempted by both flash dilution into refolding buffer (100 mM potassium phosphate, pH 6.5, 5 mM EDTA, 200 mM NaCl, 1 mM DTT) and dialysis into 1 l of refolding buffer. Both methods yielded some soluble protein, which was pooled for use in kinase assays.
Pulldowns
For all FLAG-nucleosome pulldowns, 20 μg FLAG-tagged recombinant nucleosomes were immobilized as bait on 20 μl Anti-FLAG M2 Magnetic beads slurry (Sigma) in BB75 buffer (20 mM HEPES, pH 7.5, 75 mM NaCl, 0.1 mM EDTA, 10% glycerol, 0.1% IGEPAL CA-630 (Sigma), 1 mM DTT). Wild-type or mutant VRK1 was added to a final concentration of 0.5–1 μM in a final volume of 100–125 μl BB75 and incubated with bait nucleosomes for 1 h at 4°C with rotation. Beads were rinsed twice with BB75 and then washed with BB75 for 30 min at 4°C with rotation. After washing, beads were transferred to a fresh tube for elution with 0.2 mg/ml 3X FLAG peptide (50 mM HEPES, pH 7.5, 150 mM NaCl) for 30 min on a shaking platform at 1000 rpm at 4°C. Competition pulldowns were performed in the presence of 10 μM AviTag-GST-HIS-tagged LANA (1–23) or AviTag-GST-HIS-tagged LANA (1–23, L8A, R9A, S10A) during the VRK1 binding phase and washed and eluted as in other pulldowns.
Kinase assays
Kinase assays were performed using recombinant VRK1 and recombinant nucleosomes in 1× Kinase Buffer (20 mM Tris–Cl, pH 7.5, 50 mM KCl, 1 mM MnCl2, 1 mM DTT, 100 μM ATP pH 7.0). Unless otherwise stated, substrate nucleosomes were added to a concentration of 2.5 μM and VRK1 was added to a concentration of 0.75 μM. Reactions were conducted at 30°C for 30 min and quenched with the addition of 1 μl 0.5M EGTA. Samples were centrifuged at 13.3 krpm for 5 min and added 1:1 to SDS gel loading buffer for analysis by immunoblotting as described below.
For P-32 kinase assays, BAF protein (0.5 μM) and wild-type or mutant VRK1 kinase (50 nM) were combined in kinase reaction buffer (20 mM Tris–Cl, pH 7.5, 50 mM KCl, 1 mM MnCl2, 1 mM DTT supplemented with 10 μCi γ-32P ATP (Perkin Elmer) and incubated at 30°C for 1 h. Assay samples were resolved on an 18% polyacrylamide gel and silver stained (Thermo). The stained gel was dried under a vacuum at 80°C and exposed to a blanked phosphor-screen overnight. The phosphor-screen was imaged on a Typhoon FLA 9500 instrument at 635 nm using the IP filter (GE Healthcare).
Electrophoretic mobility shift assay
Samples of varying concentrations of VRK1 and 1 μM 20N20 nucleosome were prepared in gel shift buffer (20 mM Tris–Cl, pH 7.5, 1 mM EDTA, 50 mM NaCl, 1 mM DTT) and incubated for 30 min at room temperature. Samples were supplemented with 5% sucrose for gel loading and run on a 10% Native gel in 0.5X TBE, 100 V for 80 min with an ice pack.
Immunoblotting
Samples were separated on 4–20% precast TGX gels (Bio-Rad). Following electrophoresis, gels were soaked in Towbin transfer buffer (25 mM Tris–Cl, pH 8.3, 193 mM glycine, 20% MeOH, 0.025% SDS) for 10 minutes prior to blotting. Proteins were transferred to methanol-activated PVDF membranes using the Bio-Rad Mini Trans-Blot Cell (BioRad) or Criterion Blotter (BioRad) at 350 mA for 60 min or 300 mA for 80 min, respectively. Membranes were soaked in methanol, dried, cut to size, reactivated in methanol, and washed three times for 5 min in 1× TBS with rotation. Membranes were blocked with TBS with 5% milk for 1 h at room temperature with rotation. Blocked membranes were washed three times with TBS-T and then incubated with primary antibodies diluted in 1× TBS-T with 5% milk. Following incubation for 2 h at room temperature or overnight at 4°C, membranes were washed three times with TBS-T and then incubated for 1 h at room temperature with a secondary antibody (LICOR). After incubation with secondary antibody, membranes were washed three times with TBS-T and three times with TBS. Imaging of Western blots with LICOR secondary antibodies was conducted using the LICOR Odyssey Imaging System and Image Studio software.
Primary antibodies used included: polyclonal anti-GFP (1:2000, Millipore, G1544), polyclonal alpha tubulin (1:250, Abcam, ab4074), monoclonal histone H3 (1:1000, Cell Signaling Technologies, 3638), monoclonal VRK1 (1:2000, R&D Biosciences, MAB5835), polyclonal VRK1 (1:1000, Abclonal, A7745), and monoclonal anti-phospho-Histone H3 (Thr3) clone JY325 (1:1000, Millipore, 04-746). Secondary antibodies used included: IRDye 800CW Goat anti-Rabbit IgG Secondary antibody (LI-COR) and IRDye 680RD Goat anti-Mouse IgG Secondary antibody (LI-COR).
Fluorescence quenching nucleosome binding assays
Site-specifically labeled nucleosomes for fluorescence quenching assays were prepared as previously described (63). Briefly, histone dimers containing H2AL65C mutations were incubated with two molar equivalents of Alexa Fluor 488 C5-Maleimide (Invitrogen) in labeling buffer (20 mM Tris–Cl, pH 7.5, 25 mM NaCl, 7 M guanidine hydrochloride, 0.2 mM TCEP) at 4°C, with rotation, for 2 h. A second two molar equivalents of fluorophore were added and the reaction was allowed to proceed for an additional 2 h. Labeled histones were assembled into nucleosomes as described above. Fluorescence quenching HIFI assays were performed essentially as previously described (63). VRK1 samples were diluted in 20 mM Tris–Cl, pH 7.0, 75 mM NaCl, 5mM DTT, 5% glycerol, 0.01% NP40, 0.01% CHAPS, 100 μg/ml BSA to concentrations of 50, 32, 16, 8, 4, 2, 1, 1, 0.5, 0.25, 0.125, 0.062, 0.031 and 0.016 μM. Identical buffer was used to dilute the labelled nucleosome to a concentration of 20 nM. 20 μl of VRK1 of each dilution was added to 20 μl of labelled nucleosome in triplicate in a LBS coated OptiPlate-384 (Perkin Elmer), with centrifugation for 1 min at 1000 rpm, mixing with a plate shaker and centrifugation. Plates were scanned with a EnVision 2103 Multilabel Plate Reader (Perkin Elmer, 485 nm excitation, 535 nm emission). Fluorescence intensity data was fit using a two-state binding model with ProFit7. Initial fitting showed Hill coefficients close to 1, suggesting lack of cooperativity. As a result, all data were fit with the Hill coefficient fixed at 1.
Cryo-electron microscopy and data analysis
Samples for cryo-EM grid preparation were prepared as discussed above for pulldowns with minor changes. VRK1 was pulled down with 150 μg nucleosome and eluted in 175 μl elution buffer containing 0.2 mg/ml 3× FLAG peptide at 4°C on a rotator. Subsequently, the complex was concentrated to 0.32 mg/ml (uncrosslinked sample) or 0.22 mg/ml (crosslinked sample). For the crosslinked sample, glutaraldehyde (Sigma-Aldrich) was added to 0.1% and the sample was incubated for 5 minutes at room temperature. To quench the crosslinking reaction, Tris–Cl at pH 7.5 was added to a final concentration of 20 mM and the sample was incubated for an additional 15 min at room temperature. Cryo-EM grids were prepared on a Vitrobot Mark IV (Thermo Fisher Scientific) set to 4°C and 100% humidity. A Tergeo EM plasma cleaner was used to plasma clean Quantifoil R1.2/1.3 copper 300 mesh grids for 60 s, after which 3 μl of sample was applied directly to the grids, blotted for 4 s with a –10 blot force, and plunge frozen into a 40/60% ethane/propane mixture. Grids were imaged on a Thermo Fisher Scientific 200kV TEM Talos Arctica G3 equipped with a Gatan K3 direct electron detector using SerialEM with a multi-shot record of a 3 × 3 (uncrosslinked) or 5 × 5 (crosslinked) regular pattern. Movies were collected in counting mode at 45 000 × nominal magnification, corresponding to a pixel size of 0.91 Å, with a defocus range of –0.5 to –3.0 μm, a dose rate of 14.7 (uncrosslinked) or 14.4 (crosslinked) e-/pixel/second for 60 frames, and a total dose of 54 (uncrosslinked) or 52 (crosslinked) e–/Å2. A total of 3072 (uncrosslinked) or 8000 (crosslinked) movies were collected for each sample. Movies were imported into RELION-3.1-beta in optics groupings corresponding to the beam-image shifts used for data collection and the RELION implementation of MotionCor2 was used for whole-frame motion correction. CTF estimation was carried out with CTFFIND-4.1, and only micrographs with an estimated resolution under 5 Å were included in subsequent analyses. Particles were picked using either or both of the RELION Laplacian-of-Gaussian and template-based picking methods, after which iterative rounds of 2D and 3D classification resulted in a final set of 53 704 (uncrosslinked) or 460 962 (crosslinked) particles. Following Bayesian particle polishing and CTF refinement, final reconstructions resolved to 4.1 Å (uncrosslinked) and 3.0 Å (crosslinked).
Four amino acid sequences representing all eight register and orientation combinations were modeled into the crosslinked cryo-EM map as described in the Supplementary Methods section. Models were refined by iterative model building in Coot (64) and real-space refinement in Phenix (65). Per residue model to map correlation coefficients were calculated using MolProbity in Phenix.
Molecular dynamics simulations
Initial models for eight candidate peptides were generated de novo using the cryo-EM map as discussed above and in the Supplementary Methods. All candidate peptides were extended to 7 residues to assess contributions from amino acids not visualized in the cryo-EM map. Models were simplified to include the L-chain of the VRK1 peptides and one H2A–H2B dimer and aligned to corresponding structures of the full nucleosome core particle (PDB ID: 1KX5) (66). The unstructured H2A–H2B tail regions were removed, which allowed for a reduction in simulation box size while maintaining the main globular histone-fold region of the dimer [H2A (16–108), H2B (32–121)]. VMD 1.94 (67) AutoPSF was used to recreate the side chains from the backbone of the VRK1 model fragments, as well as allow for visual inspection of the newly reconstructed VRK1 fragments.
Simulations were conducted utilizing GROMACS 2020.2 (Lindahl, Abraham, Hess and van der Spoel (2020) GROMACS 2020.2 Source code. 10.5281/zenodo.3773801) (68,69) with CHARMM 27 forcefield (70). Steepest descent minimization was calculated under vacuum until maximum force was less than 500 kJ/(mol*nm). GROMACS solvate was used to create a 10 nm in each direction dodecahedron box of TIP3P water molecules (71), and subsequently, GROMACS genion was used to neutralize and add 0.15 M KCl to the solvent. Steepest descent minimization was calculated with solvent until maximum force less than 500 kJ/(mol*nm). Molecular dynamic simulations utilized 2 fs time steps, verlet integrator, long range electrostatics by Particle Mesh Ewald (72), short range electrostatic and Van der Waals cutoffs of 1.0 nm and 1.2 nm, and a neighbor list cutoff of 1.0 nm. The system was heated slowly from 0 K to 300 K for 500 000 steps at a rate of 0.310 K per 500 steps by applying velocity rescaling of all particles to reach each desired temperature (73). The system was then equilibrated for 1 μs with a canonical (NVT) ensemble, in which the number of atoms, volume, and temperature were held constant. The simulations utilized the Berendsen pressure coupling. The system was then equilibrated for 2 μs with an isothermal-isobaric (NPT) ensemble, in which the number of atoms, pressure, and temperature were held constant. The MD production runs were simulated in triplicate for 5 μs in 1 μs segments.
Interaction energies were calculated using GROMACS. First, the water and ions were removed. Then, the frames were aligned using all H2A–H2B Cα atoms. Both the short range Coulombic and Lennard-Jones interaction energies of each individual amino acid of the VRK1 fragments were calculated in comparison to the H2A–H2B dimer.
Tissue culture
HAP1 cells (Horizon Discovery) were cultured in IMDM (Gibco) supplemented with 10% FBS (Seradigm) in standard conditions at 37°C, 5% CO2. 293T cells were cultured in DMEM (Gibco) supplemented with 10% FBS (Seradigm) in standard conditions at 37°C, 5% CO2.
Preparation of HAP1 cells stably expressing VRK1 and mutants
Lentiviruses bearing VRK1 and VRK1 mutants were prepared in 293T-X cells using second generation packaging vector psPAX2, pMD2.G envelope plasmid and the nLV Dual Promoter EF-1a-MCS-PGK-Puro gene delivery construct using described protocols (74). HAP1ΔVRK1 (Horizon Discovery) were infected with lentiviruses bearing constructs of interest and selected 3 days post-infection with 2 μg/ml puromycin.
Live-cell fluorescence imaging of mitotic cells
HAP1 transduced with eGFP-VRK1 constructs were seeded at a density of 80 000 cells/well in glass dishes (1.5 H–N, CellVis) coated with 2.5 μg/ml fibronectin. After 16 h, cells were stained with 1 μg/ml Hoechst 33342 (Thermo) for 5 min, rinsed twice gently with DPBS, and incubated in pre-warmed Fluorobrite DMEM media, 10% FBS, 1× glutamine for imaging.
Imaging was performed using a Nikon Ti Eclipse inverted widefield microscope with a Plan-Apochromat dry objective lens 40× (NA 0.95). Images were captured using an Andor Zyla 4.2 sCMOS detector (12 bit) and NIS-Elements AR software. Fluorescence filter sets were from Chroma: Hoechst – 395/25; 425; 460/50 nm (excitation; beam splitter; emission filter), GFP – 470/40; 495; 525/50 nm. Autofocus was provided by the Nikon Perfect Focus System (PFS) and a live-cell imaging enclosure (Okolabs) was used to maintain constant temperature (37°C) and a humidified atmosphere (5% CO2).
Image analysis was performed using Python 3.7.10 with Numpy (1.20.3), Pandas (1.3.3) and Matplotlib (3.4.3) libraries. Characterization of VRK1 mitotic localization was based on two independent imaging experiments. Images were acquired automatically in random positions on a plate, pooled together and their order was mixed to allow for blind selection of cells for the analysis. Cells in metaphase were selected manually, based on the distribution of the Hoechst signal. The position of the metaphase plate (two ending points) in each selected cell was defined manually. Based on the defined metaphase plate orientation, the Hoechst and VRK1 signals were quantified along a 30 pixel (∼4.8 μm) wide line perpendicular to the metaphase plate. Traces from individual cells were included in the analysis if they showed a high contrast in Hoechst signal (narrow metaphase plate) and above the threshold VRK1 signal.
Chromatin fractionation
Fractionation was performed as previously described (75). Briefly, 15 cm dishes of HEK293T were transfected with 25 μg of plasmid and media was changed 16–20 hours post-transfection. Cells were harvested 48 hours after transfection and snap-frozen. Cells were thawed on ice for 10 minutes and resuspended in 1 mL of hypotonic buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.1 mM PMSF, 0.5 mM DTT) with a wide-bore tip. Cells were lysed by incubation on ice for 30 minutes and manual Dounce homogenization with 40 strokes on ice using a 2 mL tissue grinder (Kimble) and SC pestle. Following manual homogenization, the nuclear fraction was separated by centrifugation at 1500 × g for 5 min at 4°C. After decanting the cytosolic fraction, nuclei were resuspended in ice-cold Buffer III.A (10 mM Tris–Cl, pH 7.4, 2 mM MgCl2, 0.1 mM PMSF, 5 mM CaCl2) by gentle pipetting with a wide-bore tip. Chromatin was digested using 4 units/μl of MNase (New England Biolabs) and incubated at 37°C for 30 min with manual agitation every 10 min to resuspend settled chromatin. Digestion was quenched at 30 min with the addition of 25 μl ice-cold 0.1 M EDTA. Chromatin was centrifuged at 400 × g for 10 min at 4°C to isolate chromatin from MNase soluble fraction. Following normalization of the amount of chromatin in each sample by Bradford assay of MNase soluble fractions, chromatin was resuspended in ice-cold Buffer III.B (10 mM Tris–Cl, pH 7.4, 2 mM MgCl2, 0.1 mM PMSF) and centrifuged again at 400 × g for 10 min at 4°C. Chromatin was subsequently extracted in increasingly stringent salt concentration buffers (10 mM Tris, pH 7.4, 2 mM MgCl2, 2 mM EDTA, 0.1% Triton X-100, 70, 140, 290, 590 mM NaCl, 0.1 mM PMSF) by resuspending in the appropriate buffer and incubating at 4°C for 30 minutes with rotation. Each fraction was isolated by centrifugation at 400 × g for 10 min at 4°C. Fractions were analyzed by immunoblotting as described above.
RESULTS
H3T3 phosphorylation by VRK1 is stimulated by extranucleosomal linker DNA
VRK1 has been shown to colocalize with chromatin (76) and co-immunoprecipitate with histones (21,77). As many chromatin-modifying enzymes use multivalent interactions with multiple nucleosome components for optimal activity (78), we first sought to determine if H3 is sufficient for H3T3 phosphorylation by VRK1, or whether VRK1 requires a nucleosome substrate. To establish the substrate preferences of VRK1, we performed kinase assays using recombinant histone H3, a reconstituted histone H3–H4 tetramer complex, a mix of H3–H4 tetramer and H2A–H2B dimer (histone mix), a reconstituted nucleosome with 20 base pairs of linker DNA on each side of the nucleosome core (20N20 Nuc, where N represents the 145 bp of DNA wrapped around the histone octamer), or a 20N20 tetrasome lacking both H2A–H2B dimers. We detected little to no H3T3 phosphorylation by VRK1 on H3 alone, an H3–H4 tetramer, or the histone mix using a site- and phosphorylation-specific antibody (Figure 1A). However, VRK1 robustly phosphorylated both a 20N20 tetrasome (composed of an H3–H4 tetramer and DNA) and the 20N20 nucleosome, suggesting that nucleosomal DNA promotes kinase activity.
Figure 1.

VRK1 phosphorylation of nucleosomes is stimulated by extranucleosomal linker DNA. (A) Immunoblots and quantitation (n = 3 technical replicates on same blot with standard deviations indicated) of in vitro kinase assay using histone H3, H3–H4 tetramer, mix of H3–H4 tetramer and H2A–H2B dimer (histone mix), tetrasome, or 20N20 nucleosome substrate. 20N20 nomenclature denotes a nucleosome including 145 base pairs of DNA wrapping the histone octamer with 20 base pairs of linker DNA on both sides. (B) Model of 20N20 nucleosome created by merging DNA from PDBID: 6VYP with histones from PDBID: 1KX5 to depict location of H3 N-terminal tail relative to linker DNA. (C) Immunoblots and quantitation (n = 3 technical replicates on same blot with standard deviations indicated) of kinase assay using panel of nucleosomes bearing different lengths of symmetric and asymmetric linker DNA. (D) Coomassie stained gels of FLAG-IP of recombinant VRK1 (0.5 μM) by FLAG-tagged 1N1 and 20N20 nucleosomes in 75 mM NaCl, top, and flowthrough, bottom. Flowthrough panel contrast enhanced to visualize low concentration samples. Uncropped blots are shown in Supplementary Figure S7.
The H3 N-terminal tails emerge from the nucleosome disk by threading between the DNA gyres adjacent to the sites of DNA entry and exit from the nucleosome core particle (Figure 1B). We hypothesized that the nucleosome preference of VRK1 may in part result from simultaneous engagement of the H3 N-terminal tail and extranucleosomal linker DNA. To test this hypothesis, we assembled 20N20 and 1N1 nucleosomes with symmetric 20 bp and 1 bp linkers, respectively. While both nucleosomes were modified by VRK1, H3T3 phosphorylation was stimulated by extranucleosomal linker DNA (Figure 1C). We next examined whether linker DNA enhances VRK1-mediated H3T3 phosphorylation due to improved substrate binding. We performed pulldowns of VRK1 using 20N20 and 1N1 nucleosomes assembled with N-terminally FLAG-tagged H2A. While VRK1 immunopurified with both nucleosomes, inclusion of linker DNA enriched VRK1 binding (Figure 1D). These results suggest that while VRK1 can bind to and phosphorylate nucleosome core particles that lack linker DNA, enhanced nucleosome binding facilitated by linker DNA interactions stimulates VRK1-mediated H3T3 phosphorylation.
To further explore the dependence of VRK1 on linker DNA length, we tested VRK1 activity on nucleosomes with incrementally extended symmetric linker DNA. While VRK1 exhibited low-level H3T3 phosphorylation of 1N1 nucleosomes, we observed progressive increases in phosphorylation of 10N10, 20N20 and 31N31 nucleosomes (Figure 1C and Supplementary Figure S1). Such activity enhancements could result from VRK1 interacting with one or both linker DNAs and/or from altered positioning or dynamics of the H3 tail. If binding to both linker DNAs is required for VRK1 activity, we would expect that removal of one linker DNA would lead to reversal of all linker DNA-mediated activity gains. VRK1 could also bind one linker DNA, leading to increased phosphorylation of one or both copies of H3. We would expect that removal of one linker DNA would lead to loss of roughly half of linker DNA-mediated activity in the former case but may cause no change in the latter case. At similar linker lengths, VRK1 phosphorylated nucleosomes with asymmetrical linkers roughly equally to symmetrical linkers (Figure 1C and Supplementary Figure S1), suggesting that only one linker is required for phosphorylation of both copies of histone H3. We performed electrophoretic mobility shift assays to better understand the stoichiometry of the VRK1-nucleosome complex given that the nucleosome has a pseudo-two-fold symmetry and two copies of H3. At low concentrations of VRK1, we observed a single shifted band for the VRK1-nucleosome complex (Supplementary Figure S2). Higher concentrations of VRK1 resulted in band smearing and sample that was unable to enter the gel suggesting heterogenous binding and aggregation. Altogether, our results favor the hypothesis that VRK1 forms a functional 1:1 complex with the nucleosome capable of fully phosphorylating the nucleosome, while higher stoichiometries lead to aggregation.
Cryo-EM analysis of the VRK1-nucleosome complex demonstrates heterogeneous linker DNA binding and acidic patch interaction
We next attempted to solve a structure of the VRK1-nucleosome complex by cryo-electron microscopy (cryo-EM). Our initial efforts to reconstitute the complex in solution by adding VRK1 to 20N20 nucleosomes followed by purification using size exclusion chromatography were unsuccessful due to precipitation of the complex under all conditions tested. Inspired by our FLAG-nucleosome pulldowns, we wondered whether complex formation would be possible on a bead, potentially separating complexes and preventing inter-complex aggregation. We immobilized FLAG-H2A 20N20 nucleosomes on M2 anti-FLAG magnetic beads, followed by incubation with VRK1 and elution with 3xFLAG peptide. Eluted samples concentrated without aggregation, allowing us to proceed to cryo-EM sample preparation. We collected over 3000 micrographs of the concentrated, uncrosslinked VRK1-nucleosome complex. Two-dimensional classification of ∼475 000 particles showed an orientation preference for top (disk) views and many class averages lacked density for VRK1 (Figure 2A, Supplementary Figure S3A, and Table S1). These 2D class averages included 84% of all identified particles and are either unbound or, if bound, VRK1 density cannot be clearly visualized. However, we also observed multiple top view class averages with clear evidence of the VRK1 kinase domain in proximity to either nucleosomal or extra-nucleosomal linker DNA (Figure 2A). Linker DNA–proximal positions of VRK1 could be inferred confidently from three of these class averages and represent 2% of all particles (Figure 2A, B). Particles with VRK1 bound to wrapped nucleosomal DNA were also visualized, but the DNA binding positions could not be assigned unambiguously due to the inability to distinguish between interaction with the top or bottom gyres of nucleosomal DNA in top view 2D projections. Approximately 3% of particles were assigned to three classes that showed VRK1 binding near the DNA entry/exit site corresponding to either an SHL 2 position and/or the underlying SHL –6 position (Figure 2A, B). An additional 2D class with 1% of total particles placed VRK1 at the SHL ±4 position, opposite the dyad. This analysis demonstrated interactions of the VRK1 kinase domain with linker and nucleosome wrapped DNA, but such heterogeneity, especially when paired with orientational bias, only resulted in a 4.1 Å structure of an unbound nucleosome (Supplementary Figure S3A and Table S1). We envision that linker-DNA binding by the VRK1 kinase domain positions the active site in range of the H3 N-terminal tails for efficient phosphorylation (Figure 1B). Additional binding partners may also help stabilize VRK1 for nucleosome phosphorylation.
Figure 2.
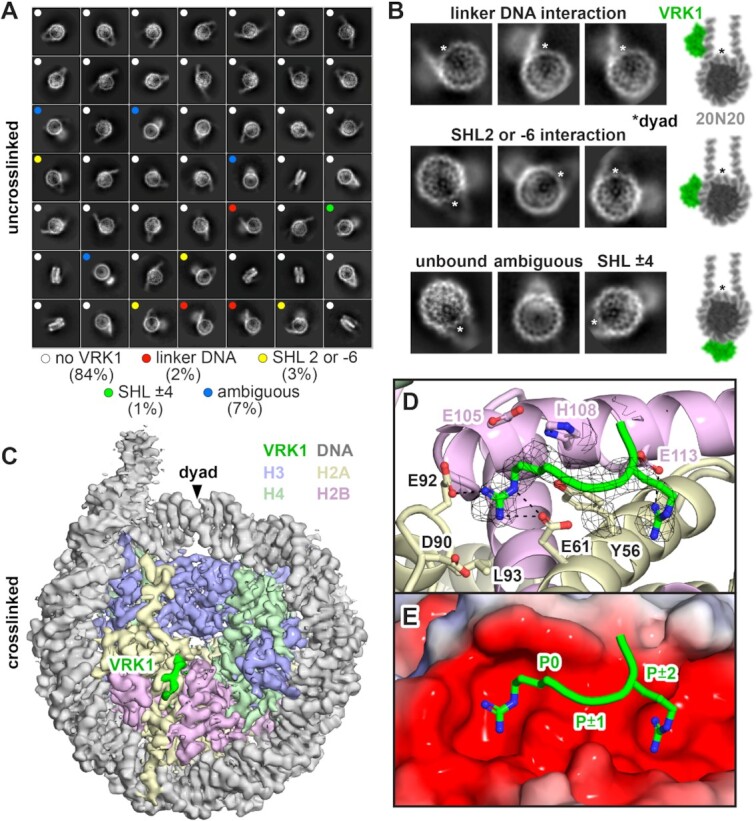
Cryo-EM analysis of VRK1-nucleosome complex. (A) 2D class averages of uncrosslinked VRK1-nucleosome complex displayed in class distribution order, representing 97% of all selected particles. Colored dots indicate presence and location of VRK1 kinase domain density. (B) Zoomed view of select 2D class averages highlighting specific VRK1 binding locations, left. The nucleosome dyad is indicated by an asterisk for orientation. Models of VRK1 kinase domain (green) superimposed on 20N20 nucleosome (grey). (C) Cryo-EM density map of crosslinked VRK1-nucleosome complex showing density from VRK1 (green) bound to the nucleosome disk face. (D) A generic peptide including two arginines separated by one amino acid modeled into the VRK1 cryo-EM density. (E) Electrostatic representation of the nucleosome acidic patch using APBS (87) overlaid with the VRK1 peptide. The canonical arginine anchor is labeled P0 for orientation and placing the other residues into the P ± 1 and P ± 2 positions.
In an effort to improve complex stability and allow visualization of more bound particles, we crosslinked our VRK1-nucleosome complex with glutaraldehyde prior to sample vitrification. Based on 2D classification, crosslinking destabilized kinase domain-DNA interactions as VRK1 kinase domain-DNA binding was no longer clearly observed in class averages, despite some blurry density above the nucleosome disk in some classes. However, 3D classification clearly showed EM density bound to the H2A–H2B acidic patch, a major hot spot for nucleosome interactions (Figure 2C, Supplementary Figure S3B, and Table S1). This extra density was not observed in the uncrosslinked reconstruction at similar map thresholds. Chromatin protein-nucleosome complex structures show that proteins almost universally bind to the nucleosome acidic patch using one or more arginines, the most common being the arginine anchor which inserts into the H2A E61/D90/E92 cavity. The arginine anchor interactions are often supported by variant arginines that engage neighboring locations within the acidic patch. Iterative refinement of our cryo-EM data led to a 3.0 Å reconstruction with density for at least three amino acids bound to the acidic patch, including a putative arginine anchor (P0), an adjacent residue with a small side chain (P ± 1), and a third residue with a longer side chain, highly suggestive of an additional variant-type arginine (P ± 2) (Figure 2D, E). The short length of the VRK1 acidic patch-binding density made it difficult to unambiguously model a specific VRK1 sequence, so we turned to biochemical characterization and computational modeling to further guide model building.
VRK1 interacts directly with the nucleosome acidic patch
We previously demonstrated that immobilized nucleosomes can pull down VRK1 from nuclear lysates and that the interaction is either directly or indirectly dependent on the H2A–H2B acidic patch (58). While VRK1 was just below the significance threshold for acidic patch dependence in our nucleosome affinity proteomics screen, pulldowns from nuclear lysates followed by immunoblots for VRK1 showed significantly reduced binding to an acidic-patch-neutralized nucleosome. Our cryo-EM reconstruction showing extra density in the region of the acidic patch confirms that VRK1 binds directly to the acidic patch. Indeed, a severe mutation of the acidic patch (H2A E61A, E64S, N68A, D72S, N89A, D90A, E91S) (Figure 3A, B) eliminated VRK1 binding to 1N1 nucleosomes and depleted VRK1 binding, albeit to a lesser degree, to 20N20 nucleosomes (Figure 3B). To further establish a role of direct acidic patch binding by VRK1, we used a known nucleosome acidic patch binding peptide from the Latency-Associated Nuclear Antigen (LANA) protein in competitive pulldowns (79). The LANA peptide binds the nucleosome acidic patch using an arginine anchor and therefore closely overlaps with the VRK1 density observed in our reconstructed cryo-EM map. If the observed VRK1-acidic patch interaction is critical for nucleosome binding by VRK1, we predicted that the LANA peptide would compete VRK1 off of an immobilized nucleosome. Inclusion of a recombinant tagged-LANA in VRK1 pulldowns with FLAG-H2A nucleosomes resulted in a decrease in nucleosome-bound VRK1 and an increase in VRK1 in the flowthrough fraction (Figure 3C). Some residual VRK1 binding in the presence of the LANA peptide was expected due to the multivalent nature of the VRK1-nucleosome interface established by our cryo-EM studies. Importantly, a mutant version of LANA that is unable to bind the nucleosome (LANA mut: L8A, R9A, S10A) failed to compete with VRK1 for nucleosome binding (Figure 3C). Because the small footprint of the LANA peptide on the nucleosome is sufficient to disrupt VRK1 binding, we anticipate that the LANA peptide and VRK1 occupy similar binding positions on the nucleosome acidic patch consistent with our cryo-EM reconstruction.
Figure 3.
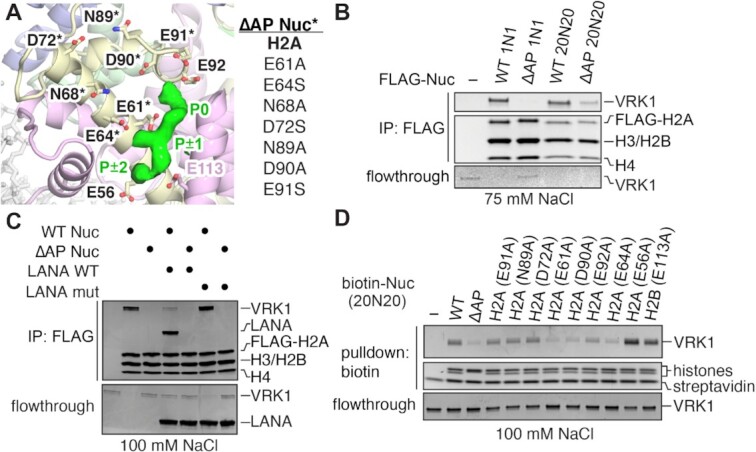
VRK1 directly interacts with the nucleosome acidic patch. (A) Refined structure of nucleosome from crosslinked VRK1-nucleosome complex together with VRK1 density to illustrate design of the acidic-patch mutant (ΔAP) nucleosome. (B) Coomassie stained gels of FLAG-IP of VRK1 (0.5 μM) using FLAG-tagged 1N1 and 20N20 wild-type (WT) and ΔAP mutant nucleosomes performed at 75 mM NaCl, top, and flowthrough, bottom. Flowthrough panel contrast enhanced to visualize low concentration samples. (C) Coomassie stained gels of FLAG-IP of VRK1 (0.5 μM) with WT and ΔAP 1N1 nucleosomes in the presence of 20x acidic patch competitor LANA WT (1–23) and negative control competitor LANA mut (1–23, L8A, R9A, S10A) performed at 100 mM NaCl, top, and flowthrough, bottom. (D) Coomassie stained gels of VRK1 (1 uM) pulldown by biotinylated 20N20 nucleosomes with individual acidic patch mutants performed at 100 mM NaCl, top, and flowthrough, bottom.
To identify key acidic patch residues that contribute to the interaction with VRK1, we prepared biotinylated nucleosomes with single alanine point mutants of H2A and H2B that sample regions within the nucleosome acidic patch. Mutation of H2A residues E61, D90 and E92, which constitute the canonical arginine anchor-binding cavity, depleted VRK1 from nucleosomes relative to the wild-type nucleosome (Figure 3D). Interestingly, while arginine anchors typically interact with all three of these acidic side chains, the H2A E92A mutation was slightly less deleterious than either the E61A or D90A mutations. H2A E64 was equally important for nucleosome binding, which may reflect interactions with the small P ± 1 side chain, but the effect of the E64A mutation was larger than we expected based on the typical dominant role for arginines in acidic patch binding. Mutation of residues H2A E56, D72, N89, E91 or H2B E113 to alanine had no effect on binding (Figure 3D). Such a result was expected for H2A residues D72, N89 and E91, which are not in close proximity to the observed VRK1 density (Figure 3A). However, we were surprised by the ability of VRK1 to bind H2A E56 and H2B E113A nucleosomes similarly to wild-type nucleosomes given the proximity of the mutations to the P ± 2 position in the VRK1 density. Overall, our results suggest that the canonical arginine anchor in the P0 position is more important for nucleosome acidic patch binding than the other arginine in the P ± 2 position.
VRK1 binds nucleosomes using an arginine-rich C-terminal tail
We next wanted to identify the sequence of VRK1 that engages the acidic patch. VRK1 possesses a canonical kinase fold with an additional αC4 helix linking the N- and C-lobes of the kinase (80). A basic-acidic-basic motif that has been linked to autoregulation of the kinase extends from the C-lobe and has been shown by NMR studies to transiently interact with the catalytic site of the kinase (81) (Figure 4A). Given that the globular kinase domain appears to interact with DNA in our non-crosslinked cryo-EM dataset, we wondered whether an arginine anchor in VRK1 is located within this C-terminal basic-acidic-basic motif. Similar to mutation of the nucleosome acidic patch, truncation of the VRK1 protein to residues 1–364 (VRK1(1–364)), which removes the acidic and second basic regions of this motif but leaves the first basic sequence that interacts with the kinase domain intact, eliminates interaction with nucleosomes (Figure 4B). This narrows the location of the putative arginine anchor observed in our cryo-EM structure to the C-terminal basic sequence. This sequence contains an alternating arginine motif with four arginines interspersed with polar or charged residues. Several registers of this sequence could be consistent with our interpretation of the observed VRK1 acidic patch binding density including arginines in the P0 and P ± 2 positions. This R-X-R sequence motif occurs only two other times in the amino acid sequence of VRK1 (residues 101–103, 277–279), both within alpha helices, and therefore inconsistent with the extended peptide backbone of the observed cryo-EM density (82). Multiple sequence alignment from a variety of higher eukaryotes reveals that this region is conserved generally, but strong conservation is maintained for at least two positively charged residues aligning to R387 and R393 in the human VRK1 sequence (Figure 4C). Mutation of VRK1 R387, R389, R391 and R393 to alanine (hereafter referred to as VRK1(4RA)) abolished all VRK1 interaction with the nucleosome (Figure 4D). The VRK1(4RA) mutant H3T3 kinase activity was reduced by more than 50% relative to wild-type VRK1 (Figure 4E), despite having normal levels of intrinsic kinase activity toward another substrate, BAF (Supplementary Figure S4).
Figure 4.

An alternating arginine motif at the VRK1 C-terminus is required for nucleosome binding and phosphorylation. (A) Schematic representation of VRK1 protein containing a kinase domain and a flexible C-terminal basic-acidic-basic motif (BAB). (B) Coomassie stained gels of FLAG-IP of indicated VRK1 proteins (0.5 μM) using FLAG-tagged 1N1 nucleosomes, top, and flowthrough, bottom. (C) Multiple-sequence alignment of VRK1 in higher eukaryotes using ClustalW. Acidic residues are indicated in red and basic residues in blue. Boxes indicate conserved positively charged positions corresponding to residues 387 and 393 in human VRK1. (D) Coomassie stained gels of FLAG-IP of indicated VRK1 proteins (1 μM) with FLAG-tagged 1N1 WT or ΔAP nucleosomes, top, and flowthrough, bottom. VRK1(4RA) = VRK1(R387A, R389A, R391A, R393A). (E) Immunoblots and quantitation (n = 3 technical replicates on same blot with standard deviations indicated, *denotes P < 0.05) of kinase assay with indicated VRK1 proteins using 20N20 nucleosome substrate. (F) Representative fluorescence microscopy images of live HAP1 cells transduced with wild-type (WT) or mutant eGFP-VRK1 (green) and stained with Hoechst (magenta) during metaphase. Colocalized pixels are shown in white. (G) Quantification of the mean fluorescence intensity of eGFP, top, and Hoechst staining, bottom, perpendicular to the metaphase plate of HAP1 transduced with eGFP-VRK1 WT (208 cells) and eGFP-VRK1(4RA) (245 cells). Means and standard deviations are shown. Uncropped blots are shown in Supplementary Figure S7.
To extend our in vitro observations to a native chromatin context, we visualized the localization of eGFP-tagged VRK1 in transduced HAP1 cells during mitotic chromatin condensation. By comparing the localization of eGFP-tagged VRK1 with Hoechst-stained chromatin in live cells, we determined that wild-type VRK1 is enriched on chromatin during mitosis and in particular at the metaphase plate (Figure 4F, G and Supplementary Movies 1–3). Strikingly, chromatin enrichment of VRK1(4RA) during mitosis is lost, indicating that the alternating arginine nucleosome acidic patch-binding motif is required for the VRK1-chromatin interaction (Figure 4F, G and Supplementary Movies 4-6). These conclusions are supported by salt-gradient chromatin fractionation of eGFP-tagged VRK1 in asynchronous 293T cells, which demonstrated that VRK1(4RA) is largely lost from the chromatin fraction at a sub-physiological 80 mM salt, whereas wild-type VRK1 exhibited much greater salt stability within the chromatin fraction (Supplementary Figure S5).
VRK1 R387 is critical for nucleosome binding
Our cryo-EM data and mutational analysis implicate the C-terminal alternating arginine motif in nucleosome acidic patch binding. In order to interrogate this interface at amino acid resolution and identify the VRK1 arginine anchor observed in our cryo-EM reconstruction, we needed to be able to detect subtle differences in nucleosome binding between VRK1 mutants. We therefore adapted a quantitative fluorescence quenching nucleosome-binding assay that measures environmental changes due to a chromatin factor binding at a specific location on the nucleosome disk face (63) (Figure 5A). Scanning of positions for fluorophore attachment to the nucleosome revealed a 14% fluorescence quenching of Alexa Fluor-488 maleimide conjugates of H2A L65C nucleosomes with saturating levels of VRK1. The H2A L65 location is close enough to the VRK1 acidic patch binding site to elicit fluorescence changes, but distant enough from the arginine anchor cavity that it would not be expected to interfere with binding. The observed fluorescence quenching was used to calculate localized apparent affinity constants for VRK1 and VRK1 mutants at the acidic patch. In this assay, VRK1 bound to 1N1 nucleosomes (used here to emphasize acidic patch-dependent binding) with an apparent KD (KD,app) of 0.20 ± 0.03 μM. The VRK1(4RA) mutant showed severe defects in binding to nucleosomes, with a KD,app value 50 times higher than the wild-type VRK1. We speculate that residual fluorescence quenching by VRK1(4RA) is due to low-affinity DNA binding at SHL 2/–6 as observed in 2D averages of uncrosslinked VRK1-nucleosome complexes or aggregation of saturated VRK1-nucleosome complexes at high protein concentrations as observed during initial attempts to reconstitute the complex for structural analysis. In an attempt to identify the VRK1 arginine anchor, we next measured nucleosome-binding affinities of alanine mutations of each of the four arginines in the VRK1 C-terminal alternating arginine motif. Our pulldown experiments demonstrated that the arginine anchor position is most important for a stable VRK1-acidic patch interaction. The largest increase in KD,app was observed for R387A, with a 10-fold increase to 2.0 μM ± 0.7 (Figure 5B). However, the R389A, R391A and R393A point mutations each resulted in at least a 3-fold reduction in binding affinity with KD,app between 0.7 and 1 μM. All individual arginine mutations showed defects in nucleosome binding by pulldown assay with the degree of binding loss closely correlated with the calculated KD,app values (Supplementary Figure S6A) even though there was no measurable change in H3T3 phosphorylation (Supplementary Figure S6B). Based on these results, we conclude that among the four arginines in the C-terminal alternating arginine motif, VRK1 R387 is most important for the VRK1–nucleosome interaction, but that each of the four arginines participates in nucleosome binding.
Figure 5.
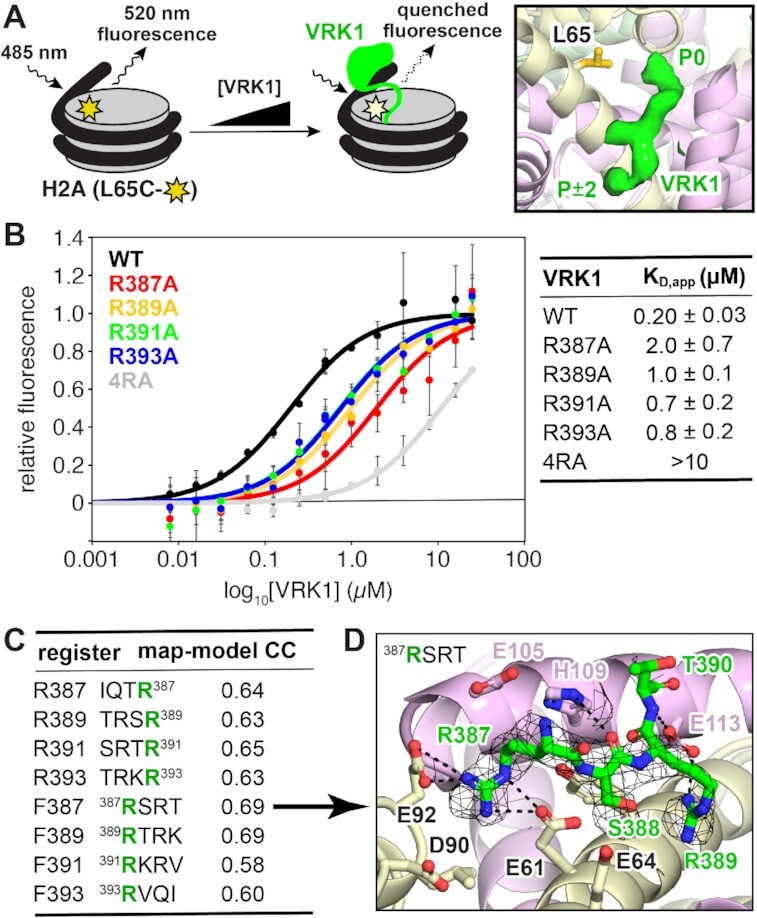
Fluorescence quenching binding assay reveals that arginine mutants are defective in binding at the nucleosome acidic patch. (A) Schematic representation of fluorescence quenching nucleosome binding assay, left. Nucleosomes are modified by conjugation of Alexa Fluor 488 to H2A L65C. Binding of VRK1 to the nucleosome alters the local chemical environment around the fluorophore, leading to partial quenching. Location of H2A L65 relative to VRK1 density, right. (B) Nucleosome binding data and fits for indicated VRK1 proteins. n = 3 technical replicates conducted on a single plate, with means and standard deviations indicated. (C) Map to model correlation coefficients for eight models with each of four VRK1 arginines in the arginine anchor position (green) in both the C- to N-terminal (top 4, reverse (R)) and N- to C-terminal (bottom 4, forward (F)) orientations. (D) Preferred model including the VRK1 R387 arginine anchor in the N- to C-terminal orientation.
VRK1 C-terminus recognizes acidic patch dynamically
To test the hypothesis that R387 functions as an arginine anchor against our cryo-EM data, we performed systematic structural modeling of different registers and orientations of the VRK1 alternating arginine motif into the map. We generated eight models, each inserting one of the four arginines into the arginine anchor position (P0) and extended the peptide in either the N- to C-terminal (forward) or C- to N-terminal (reverse) direction (Supplemental Methods). Generally, models with arginines in the P0 and P ± 2 positions separated by a small amino acid in the P ± 1 position yielded the highest map to model correlations (Figure 5C). Among this subset, two models in the N- to C-terminal orientation correlated best with the map. One of these models places VRK1 R387 and R389 in the arginine anchor and variant arginine positions, respectively, while the other model places R389 and R391 in those positions. Given the clear importance of VRK1 R387 for nucleosome binding, we favor the model with the R387 arginine anchor (Figure 5D). In this model, VRK1 R387 makes charged hydrogen bonding interactions with both H2A E61 and E92. VRK1 R389 makes an additional charged hydrogen bonding interaction with H2B E113. These side chain interactions are supported by several VRK1 main chain to histone side chain hydrogen bonds.
Two inconsistencies exist between our cryo-EM and our biochemical data. Firstly, all four arginines in the C-terminal tail of VRK1 contribute to nucleosome binding, but only two arginines are visible in our cryo-EM map. Secondly, our pulldown assays revealed a dependence on the H2A E64 position, which had no interaction with an arginine in our cryo-EM density (Figure 3D). A recent review of structures of proteins in complex with the nucleosome defined two positions within the acidic patch where variant arginines support arginine anchor binding (83). Type 1 variant arginines bind in the location occupied by the P ± 2 arginine in our cryo-EM models. Type 2 variant arginines bind next to the arginine anchor in the H2A E61/E64/L65 cavity. Given that a cryo-EM map is reconstructed from an average of a subset of particles, we hypothesized that VRK1 may use all four arginines to dynamically sample additional regions of the acidic patch (e.g. H2A E64) that are not visualized due to particle selection and/or averaging. To explore this possibility, we performed molecular dynamics simulations of the same eight orientation and register combinations that we modeled into our cryo-EM map in complex with an H2A–H2B histone-fold dimer. We extended the peptides to a length of seven amino acids to capture additional interactions with the acidic patch that were not visualized in cryo-EM averages (Figure 6A). Interestingly, we found a moderate correlation (R2 = 0.60) between the number of arginines and the total VRK1-acidic patch interaction energy, indicating that presenting more arginines at the acidic patch provides an overall benefit to binding, despite our observation of only two arginines by cryo-EM (Figure 6B). This is consistent with our finding that mutation of each arginine to some degree disfavors nucleosome binding. Surprisingly, we find that P ± 1 residues contribute very little to the overall interaction energy despite the clear importance of the underlying H2A E64 side chain (Figure 6C). Per residue interaction energies reinforced the importance of arginines; with few exceptions, arginines contributed more interaction energy than all other residues including lysine (Figure 6C).
Figure 6.

Molecular dynamics simulations demonstrate dynamic arginine binding. (A) Seven residue sequences of VRK1 modeled onto H2A–H2B dimer for molecular dynamics simulations. (B) Total interaction energy plotted versus number of arginines for each register/orientation combination. Means (n = 3) and standard deviations shown. (C) Per residue interaction energies indicated under sequences aligned by arginine anchor (P0) position. (D) Overlay of arginine CZ position for indicated arginines for all register/orientation combinations for 500 timepoints (every 10 ns) over triplicate simulations (5 μs total per replicate simulation). Top and side views as indicated at left. Right panels show arginine anchor (R), and variant arginine type 1 (V1) and type 2 (V2) positions.
Molecular dynamics simulations revealed a diversity of VRK1 binding poses over the simulated timeline for all registers and orientations. To visualize these binding poses and identify major clusters of arginine-acidic patch interaction, we first grouped arginine residues into categories based on their positions relative to the arginine anchor (P0) and orientation (forward or reverse) and tracked the position of all arginine CZ atoms over 500 timepoints during triplicate simulations (Figure 6D). We found little differentiation between the CZ positions in the forward and reverse orientations, indicating that the C-terminal sequence is likely capable of interacting with the acidic patch in either orientation. Compared to other positions, the arginine anchor (R) was nearly invariant, always clustering tightly within the H2A E61/D90/E92 cavity. The two arginines on one side of the arginine anchor (Forward P2/Reverse P-2 and Forward P4/Reverse P-4) were more dynamic, yet occupied two major positional clusters closely aligning with the variant type 1 (V1) and variant type 2 (V2) positions identified by structural analysis. The arginine on the opposite side of the arginine anchor (Forward –2/Reverse 2) was also dynamic and frequently occupied the V2 position. These simulations paired with our binding experiments suggest that while the arginine anchor (P0) interacts with the H2A E61/D90/E92 cavity, the other arginines can dynamically sample both the V1 and V2 positions to stabilize acidic patch binding. Notably, arginine binding to the V2 position may help explain the critical importance of H2A E64 for VRK1 acidic patch binding.
Disease mutants of VRK1 lose mitotic chromatin localization
Recent case studies utilizing whole-exome sequencing have identified homozygous mutations of VRK1 to be causative in adult-onset distal hereditary motor neuropathies (36–39). Interestingly, two pathogenic mutations of VRK1 occur in the C-terminal region that is critical for VRK1 to bind to the nucleosomes. One case identified a homozygous c.1124G > A nonsense mutation that prematurely truncates VRK1 prior to W375 (W375*) (37) and consequently removes all four arginine residues in the alternating arginine motif. Another case described a homozygous c.1160G > A missense mutation resulting in a VRK1(R387H) substitution of the R387 residue that is most crucial for nucleosome binding (39). We hypothesized that the corresponding gene products would be defective in their interactions with individual nucleosomes, thus preventing chromatin association in cells.
We examined the ability of the R387H and W375* VRK1 mutants to bind and phosphorylate nucleosomes. VRK1(R387H) was defective in nucleosome binding using our fluorescence quenching assay, with a KD,app of 1.6 ± 0.6 μM, 8-fold higher than wild-type VRK1 (Figure 7A). We expect that despite the substitution for another positively charged residue at the R387 position, the topology of the nucleosome acidic patch does not accommodate a histidine in the narrow canonical arginine anchor cavity. The VRK1(W375*) nonsense mutant was even more defective in nucleosome binding, with a KD,app of 3.1 ± 0.8 μM, similar to VRK(1–364). Nucleosome pulldown assays confirm the relative loss in nucleosome binding by VRK1(R387H) and VRK1(W375*), with minimal binding or no binding observed, respectively (Figure 7B). Like VRK1(4RA), VRK1(W375*) was defective in phosphorylating nucleosomes at H3T3 (Figure 7C). VRK1(R387H) exhibited wild-type levels of H3T3 phosphorylation, indicating that acidic patch binding defects of this mutant are insufficient to grossly alter histone phosphorylation in vitro (Figure 7C). Both VRK1 disease mutants phosphorylated BAF at wild-type levels (Supplementary Figure S4).
Figure 7.

Distal spinal muscular atrophy mutants of VRK1 are defective in nucleosome binding and fail to properly associate with chromatin at the metaphase plate. (A) Nucleosome binding data and fits for indicated VRK1 proteins. n = 3 technical replicates conducted on a single plate, with means and standard deviations indicated. (B) FLAG-IP of indicated VRK1 proteins (1 μM) with FLAG-tagged 20N20 nucleosomes at 100 mM salt, top, and flowthrough, bottom. Beads only FLAG-IP shown as negative control. (C) Immunoblot and quantitation (n = 3–4 technical replicates on same blot with standard deviations indicated, *denotes P < 0.05, **denotes P < 0.01, ns = not significant) of kinase assays performed on 20N20 nucleosomes with indicated VRK1 proteins. Significance determined using two-tailed unpaired t-test. (D) Representative fluorescence microscopy images of live HAP1 cells transduced with indicated mutant eGFP-VRK1 (green) and stained with Hoechst (magenta) during metaphase. Colocalized pixels are shown in white. (E) Quantification of the mean fluorescence intensity of eGFP, top, and Hoechst staining, bottom, perpendicular to the metaphase plate of HAP1 transduced with WT eGFP-VRK1 (208 cells), eGFP-VRK1 (R387H) (290 cells), or eGFP-VRK1 (W375*) (249 cells). Means and standard deviations are shown. Uncropped blots are shown in Supplementary Figure S7.
We next examined the chromatin localization of VRK1(R387H) and VRK1(W375*) in mitotic cells using fluorescence microscopy. eGFP-VRK1(W375*) failed to enrich at the metaphase plate, similar to VRK1(4RA) (Figure 7D, E and Supplementary Movies 7–8). Consistent with more modest losses in binding to reconstituted nucleosomes, VRK1(R387H) showed partial enrichment at the metaphase plate as compared to the wild-type VRK1 and stronger than either VRK1(W375*) or VRK1(4RA) mutants (Figure 7D, E and Supplementary Movies 9–11). Overall, we find clear concordance between in vitro binding of reconstituted nucleosomes and mitotic chromatin localization across all VRK1 mutants examined. Based on this, we anticipate that nucleosome acidic patch binding is the major contributor to mitotic chromatin localization of VRK1 and that loss of chromatin localization contributes to disease phenotypes.
DISCUSSION
Chromatin kinases regulate key cellular processes including mitosis by coordinating the phosphorylation of histones and chromatin-bound proteins. In this study, we show that a paradigmatic chromatin kinase, VRK1, engages individual nucleosomes in a high-affinity, multivalent binding interaction in order to catalyze histone H3T3 phosphorylation. Based on cryo-EM data and in vitro nucleosome binding assays, it is clear that VRK1 binds both DNA and histone components of the nucleosome. Our cryo-EM analysis of uncrosslinked VRK1-20N20 nucleosome complexes yielded 2D averages that demonstrated that VRK1 interacts heterogeneously with either nucleosome-wrapped or extranucleosomal linker DNA. Consistent with this observation, a recent UV-crosslinking based proteomics screen identified VRK1 as a DNA-binding protein in cells, detecting a DNA crosslinked sequence on the C-lobe of the kinase (residues 322–329) located spatially nearby to an arginine and lysine-rich patch that may serve as the DNA-binding region of the kinase domain (84). Several VRK1-DNA binding locations, especially in the linker region or near SHL 2 or –6, place the kinase domain of VRK1 in proximity to the H3 N-terminal tail and provide a mechanistic basis for our observation that the addition of linker DNA promotes nucleosome phosphorylation. Given the robust activation of H3T3 phosphorylation in the presence of linker DNA, we expect that among the VRK1-DNA binding locations we observed by cryo-EM, the linker DNA-bound form is the most catalytically active. Removal of one linker DNA has little to no effect on H3T3 phosphorylation. This leads us to the conclusion that VRK1 binding to one linker DNA can promote phosphorylation of either copy of H3 in the nucleosome.
In addition to binding DNA, we show that VRK1 interacts with the acidic patch on the nucleosome disk face. The acidic patch is an established hot-spot for nucleosome binding. Nearly all acidic patch binding proteins engage the acidic patch with one or more arginines (83). Our cryo-EM reconstructions of crosslinked VRK1-nucleosome complexes show density for a three amino acid region of VRK1 binding to the acidic patch, including the arginine anchor and a second arginine separated by one intervening residue. In order to identify the acidic patch binding sequence of VRK1, we performed truncation and mutation analysis of the VRK1 C-terminal unstructured region. We found that mutation of four arginines in an alternating arginine motif spanning residues 387–393 near the C-terminus of VRK1 impairs nucleosome binding and H3T3 phosphorylation. Molecular dynamics simulations of the nucleosome-binding region of VRK1 reveal dynamic acidic patch recognition wherein a canonical arginine anchor stably binds the acidic patch and the other nearby arginine residues participate in dynamic recognition of type 1 and type 2 variant arginine positions. This observation provides clarity to the mechanism of redundant acidic patch recognition in proteins that bind with an extended peptide including multiple arginines, such as the nuclear pore complex protein ELYS (58,85). We anticipate that proteins that recognize the acidic patch in this way are abundant but understudied due to the limitations of analyzing dynamic structures by traditional structural techniques commonly employed in chromatin structural biology.
This nucleosome binding defect incurred by mutation of arginines in VRK1 residues 387–393 was also observed in cells through the loss of VRK1 metaphase plate localization during cell division in HAP1 cells. Interestingly, the mitotic chromatin localization we observed for VRK1 differs from the results of fixed-cell imaging of the kinase in HeLa and U2OS cells (19,21). It has been reported that cellular fixation causes an artifact that erroneously excludes transcription factors from chromatin during mitosis (86). Indeed, when we imaged paraformaldehyde-fixed cells, wild-type VRK1 was excluded from the metaphase plate (data not shown). Glutaraldehyde fixation of cryo-EM samples similarly destabilized VRK1-DNA interactions (Figure 2 and Supplementary Figure S3). Our results indicate that the observation that fixation induces loss of chromatin localization in mitosis extends to other chromatin factors, whose localization should be determined in live cells, and corrects this misunderstanding specifically for VRK1.
Previously, NMR studies of VRK1 revealed an interaction between the VRK1 C-terminal tail and its catalytic loop, with residues 341–361 looping back towards the N-lobe (81). If residues 341–361 of the VRK1 C-terminal tail interact with the catalytic loop of the protein, this leaves an additional 26 amino acid of low complexity sequence prior to the alternating arginine motif to span the distance from the kinase domain on linker DNA to the nucleosome acidic patch. With a conservative average amino acid length of 3.4 Å per amino acid, the linker could reach 88 Å across the nucleosome disk face. Given that the nucleosome acidic patch is about 45 Å from the emergence of the linker DNA, we anticipate that the alternating arginine motif should be able to access the nucleosome acidic patch with the catalytic domain simultaneously engaged with extranucleosomal linker DNA. In an effort to identify the arginine anchor from the four arginines in the alternating arginine motif, we mutated each arginine individually. The VRK1 R387A mutation caused the greatest defect in nucleosome binding. We suspect that R387 is the primary arginine anchor but that the other arginines can contribute redundantly to acidic patch binding potentially through an ensemble of binding poses.
We observed a strong nucleosome substrate preference for VRK1 over H3 alone or an H3–H4 tetramer. This can be rationalized due to VRK1 binding to both DNA and the nucleosome acidic patch that is presented by histones H2A and H2B. In contrast, VRK1 phosphorylation of native H3 has been previously reported using P-32 ATP (21). Such a discrepancy may result from modifications present in native but not recombinant histones, due to modification of non-H3T3 sites, or merely a consequence of the heightened sensitivity of a radioactivity-based readout. Despite preference of the kinase for a nucleosomal substrate over free histones, we observed weak kinase activity towards nucleosomes in general, especially compared to another substrate, BAF. A 1:3 ratio of kinase to substrate nucleosome was necessary to observe robust activity at H3T3 by western blot. This indicates that although VRK1 is capable of phosphorylating nucleosomes, another accessory factor may be necessary for efficient H3T3 phosphorylation. Alternatively, VRK1 may be more active toward other nuclear substrates. Regardless, we hypothesize that chromatin localization constrains VRK1 in the proximity of other cell-cycle related chromatin-based substrates. Unlike VRK1, the other major H3T3 kinase, haspin, has been demonstrated to robustly phosphorylate both free H3 and nucleosomes (15).
Myriad recessive VRK1 mutations have been shown to be causative in rare cases of late onset distal spinal muscular atrophy. These mutations disrupt VRK1 activity through kinase destabilization, decreasing intrinsic kinase activity, or mislocalization to the cytoplasm. Recently, two new mutations of VRK1 have been linked to the disease, the R387H missense mutation (39) and the W375* nonsense mutation (37). Notably, both of these mutations disrupt the nucleosome binding alternating arginine motif. R387H and W375* mutations resulted in impaired chromatin binding during mitotic chromosome condensation. Moreover, our kinase assays revealed that W375* decreases H3T3 phosphorylation. While we observed no significant H3T3 phosphorylation defect due to the R387H mutation, we expect that loss of chromatin localization of VRK1 would impair phosphorylation of many other non-nucleosomal chromatin-bound substrates. Given the late onset of pathology, it is not surprising that the mutations may have more subtle phenotypes during in vitro and cellular assays. Overall, our studies suggest a novel mechanism in which VRK1 mutations may lead to distal spinal muscular atrophy through loss of chromatin association due to defective nucleosome acidic patch binding.
DATA AVAILABILITY
Cryo-EM maps have been deposited in the EMDB with accession numbers EMD-25778 (uncrosslinked) and EMD-25777 (crosslinked) and the preferred cryo-EM model of the VRK1-nucleosome complex has been deposited in the PDB with accession number 7TAN.
Supplementary Material
ACKNOWLEDGEMENTS
Cryo-EM data collection was completed at UNC Cryo-EM core facility under the direction of J.D.S with the assistance of Jared Peck and supported by the National Cancer Institute of the National Institutes of Health under award number P30CA016086. Sarah Clinkscales, Xiaokang Yan and Nate Hathaway provided expertise for lentivirus production. VRK1_HUMAN_D0 and Lambda Phosphatase were gifts from John Chodera, Nicholas Levinson & Markus Seeliger (Addgene plasmid #79684; Addgene plasmid #79748). nLV Dual Promoter EF-1a-MCS-PGK puro, Md2G, and pspax2 were gifts from Nate Hathaway. Plasmids used to prepare nucleosomal DNA were provided by Song Tan. We thank members of the McGinty lab for project discussions and manuscript revision. We would also like to thank Jean Cook, members of the Center for Integrative Chemical Biology and Drug Discovery (CICBDD), and the UNC Chromatin Interest Group for guidance and advice.
Author contributions: G.R.B., Y.Z. and D.N.A. designed and conducted experiments. C.J.S. and J.D.S. performed cryo-EM grid preparation, data acquisition and analysis. K.M.K. aided in design of microscopy experiments, imaged and analyzed data for fluorescence microscopy. M.R.W. performed molecular dynamics simulations. Y.K., A.P.C., A.S., H.C.S. and E.C.A. performed supporting experiments and prepared nucleosomes for experiments. G.R.B. and R.K.M. prepared figures and wrote the manuscript which was edited by all authors. R.K.M. and D.K. provided financial support for the study.
Contributor Information
Gabrielle R Budziszewski, Department of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, NC, USA.
Yani Zhao, Division of Chemical Biology and Medicinal Chemistry, Center for Integrative Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Chapel Hill, NC, USA.
Cathy J Spangler, Department of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, NC, USA.
Katarzyna M Kedziora, Bioinformatics and Analytics Research Collaborative, University of North Carolina, Chapel Hill, NC, USA.
Michael R Williams, Division of Chemical Biology and Medicinal Chemistry, Center for Integrative Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Chapel Hill, NC, USA.
Dalal N Azzam, Division of Chemical Biology and Medicinal Chemistry, Center for Integrative Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Chapel Hill, NC, USA.
Aleksandra Skrajna, Division of Chemical Biology and Medicinal Chemistry, Center for Integrative Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Chapel Hill, NC, USA.
Yuka Koyama, Division of Chemical Biology and Medicinal Chemistry, Center for Integrative Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Chapel Hill, NC, USA.
Andrew P Cesmat, Division of Chemical Biology and Medicinal Chemistry, Center for Integrative Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Chapel Hill, NC, USA.
Holly C Simmons, Division of Chemical Biology and Medicinal Chemistry, Center for Integrative Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Chapel Hill, NC, USA.
Eyla C Arteaga, Division of Chemical Biology and Medicinal Chemistry, Center for Integrative Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Chapel Hill, NC, USA.
Joshua D Strauss, Department of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, NC, USA.
Dmitri Kireev, Division of Chemical Biology and Medicinal Chemistry, Center for Integrative Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Chapel Hill, NC, USA.
Robert K McGinty, Department of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, NC, USA; Division of Chemical Biology and Medicinal Chemistry, Center for Integrative Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Chapel Hill, NC, USA; Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, NC, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [T32GM119999 to G.R.B.]; NIH [F99CA253730 to C.J.S.]; NIH [R01GM132299 to D.K.]; NIH [R35GM133498 to R.K.M.]; American Cancer Society Postdoctoral Fellowship [PF-18-153-01-DMC to A.S.]; Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation [2020-225716 to K.M.K.]. Funding for open access charge: NIH [R35GM133498].
Conflict of interest statement. None declared.
REFERENCES
- 1. Kelly A.E., Ghenoiu C., Xue J.Z., Zierhut C., Kimura H., Funabiki H.. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase aurora B. Science. 2010; 330:235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang F., Dai J., Daum J.R., Niedzialkowska E., Banerjee B., Stukenberg P.T., Gorbsky G.J., Higgins J.M.G.. Histone H3 thr-3 phosphorylation by haspin positions aurora b at centromeres in mitosis. Science. 2010; 330:231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paull T.T., Rogakou E.P., Yamazaki V., Kirchgessner C.U., Gellert M., Bonner W.M.. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000; 10:886–895. [DOI] [PubMed] [Google Scholar]
- 4. Fischle W., Tseng B.S., Dormann H.L., Ueberheide B.M., Garcia B.A., Shabanowitz J., Hunt D.F., Funabiki H., Allis C.D.. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005; 438:1116–1122. [DOI] [PubMed] [Google Scholar]
- 5. North J.A., Šimon M., Ferdinand M.B., Shoffner M.A., Picking J.W., Howard C.J., Mooney A.M., van Noort J., Poirier M.G., Ottesen J.J.. Histone H3 phosphorylation near the nucleosome dyad alters chromatin structure. Nucleic Acids Res. 2014; 42:4922–4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brehove M., Wang T., North J., Luo Y., Dreher S.J., Shimko J.C., Ottesen J.J., Luger K., Poirier M.G.. Histone core phosphorylation regulates DNA accessibility. J. Biol. Chem. 2015; 290:22612–22621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilkins B.J., Rall N.A., Ostwal Y., Kruitwagen T., Hiragami-Hamada K., Winkler M., Barral Y., Fischle W., Neumann H.. A cascade of histone modifications induces chromatin condensation in mitosis. Science. 2014; 343:77–80. [DOI] [PubMed] [Google Scholar]
- 8. Nikalayevich E., Ohkura H.. The NuRD nucleosome remodelling complex and NHK-1 kinase are required for chromosome condensation in oocytes. J. Cell Sci. 2015; 128:566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paulson J.R., Taylor S.S.. Phosphorylation of histones 1 and 3 and nonhistone high mobility group 14 by an endogenous kinase in hela metaphase chromosomes. J. Biol. Chem. 1982; 257:6064–6072. [PubMed] [Google Scholar]
- 10. Hendzel M.J., Wei Y., Mancini M.A., Van Hooser A., Ranalli T., Brinkley B.R., Bazett-Jones D.P., Allis C.D.. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997; 106:348–360. [DOI] [PubMed] [Google Scholar]
- 11. Goto H., Tomono Y., Ajiro K., Kosako H., Fujita M., Sakurai M., Okawa K., Iwamatsu A., Okigaki T., Takahashi T.et al.. Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J. Biol. Chem. 1999; 274:25543–25549. [DOI] [PubMed] [Google Scholar]
- 12. Polioudaki H., Markaki Y., Kourmouli N., Dialynas G., Theodoropoulos P.A., Singh P.B., Georgatos S.D.. Mitotic phosphorylation of histone H3 at threonine 3. FEBS Lett. 2004; 560:39–44. [DOI] [PubMed] [Google Scholar]
- 13. Preuss U., Landsberg G., Scheidtmann K.H.. Novel mitosis-specific phosphorylation of histone H3 at thr11 mediated by Dlk/ZIP kinase. Nucleic Acids Res. 2003; 31:878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shoemaker C.B., Chalkley R.. An H3 histone-specific kinase isolated from bovine thymus chromatin. J. Biol. Chem. 1978; 253:5802–5807. [PubMed] [Google Scholar]
- 15. Dai J., Sultan S., Taylor S.S., Higgins J.M.G.. The kinase haspin is required for mitotic histone H3 thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 2005; 19:472–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bolton M.A., Lan W., Powers S.E., McCleland M.L., Kuang J., Stukenberg P.T.. Aurora b kinase exists in a complex with survivin and INCENP and its kinase activity is stimulated by survivin binding and phosphorylation. Mol. Biol. Cell. 2002; 13:3064–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang F., Ulyanova N.P., van der Waal M.S., Patnaik D., Lens S.M.A., Higgins J.M.G.. A positive feedback loop involving haspin and aurora b promotes CPC accumulation at centromeres in mitosis. Curr. Biol. 2011; 21:1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Santos C.R., Rodríguez-Pinilla M., Vega F.M., Rodríguez-Peralto J.L., Blanco S., Sevilla A., Valbuena A., Hernández T., van Wijnen A.J., Li F.et al.. VRK1 signaling pathway in the context of the proliferation phenotype in head and neck squamous cell carcinoma. Mol. Cancer Res. 2006; 4:177–185. [DOI] [PubMed] [Google Scholar]
- 19. Moura D.S., Campillo-Marcos I., Vázquez-Cedeira M., Lazo P.A.. VRK1 and AURKB form a complex that cross inhibit their kinase activity and the phosphorylation of histone H3 in the progression of mitosis. Cell. Mol. Life Sci. 2018; 75:2591–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aihara H., Nakagawa T., Mizusaki H., Yoneda M., Kato M., Doiguchi M., Imamura Y., Higashi M., Ikura T., Hayashi T.et al.. Histone H2A T120 phosphorylation promotes oncogenic transformation via upregulation of cyclin d1. Mol. Cell. 2016; 64:176–188. [DOI] [PubMed] [Google Scholar]
- 21. Kang T.-H., Park D.-Y., Choi Y.H., Kim K.-J., Yoon H.S., Kim K.-T.. Mitotic histone H3 phosphorylation by vaccinia-related kinase 1 in mammalian cells. Mol. Cell. Biol. 2007; 27:8533–8546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salzano M., Sanz-García M., Monsalve D.M., Moura D.S., Lazo P.A.. VRK1 chromatin kinase phosphorylates H2AX and is required for foci formation induced by DNA damage. Epigenetics. 2015; 10:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lopez-Borges S., Lazo P.A.. The human vaccinia-related kinase 1 (VRK1) phosphorylates threonine-18 within the mdm-2 binding site of the p53 tumour suppressor protein. Oncogene. 2000; 19:3656–3664. [DOI] [PubMed] [Google Scholar]
- 24. Sevilla A., Santos C.R., Barcia R., Vega F.M., Lazo P.A.. c-Jun phosphorylation by the human vaccinia-related kinase 1 (VRK1) and its cooperation with the N-terminal kinase of c-Jun (JNK). Oncogene. 2004; 23:8950–8958. [DOI] [PubMed] [Google Scholar]
- 25. Sevilla A., Santos C.R., Vega F.M., Lazo P.A.. Human vaccinia-related kinase 1 (VRK1) activates the ATF2 transcriptional activity by novel phosphorylation on thr-73 and ser-62 and cooperates with JNK. J. Biol. Chem. 2004; 279:27458–27465. [DOI] [PubMed] [Google Scholar]
- 26. Sanz-García M., Monsalve D.M., Sevilla A., Lazo P.A.. Vaccinia-related kinase 1 (VRK1) is an upstream nucleosomal kinase required for the assembly of 53BP1 foci in response to ionizing radiation-induced DNA damage. J. Biol. Chem. 2012; 287:23757–23768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nichols R.J., Wiebe M.S., Traktman P.. The vaccinia-related kinases phosphorylate the N' terminus of BAF, regulating its interaction with DNA and its retention in the nucleus. Mol. Biol. Cell. 2006; 17:2451–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin K.J., Patrick D.R., Bissell M.J., Fournier M.V.. Prognostic breast cancer signature identified from 3D culture model accurately predicts clinical outcome across independent datasets. PLoS One. 2008; 3:e2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Valbuena A., Suárez-Gauthier A., López-Rios F., López-Encuentra A., Blanco S., Fernández P.L., Sánchez-Céspedes M., Lazo P.A.. Alteration of the VRK1-p53 autoregulatory loop in human lung carcinomas. Lung Cancer. 2007; 58:303–309. [DOI] [PubMed] [Google Scholar]
- 30. Kim I.-J., Quigley D., To M.D., Pham P., Lin K., Jo B., Jen K.-Y., Raz D., Kim J., Mao J.-H.et al.. Rewiring of human lung cell lineage and mitotic networks in lung adenocarcinomas. Nat. Commun. 2013; 4:1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hennig E.E., Mikula M., Rubel T., Dadlez M., Ostrowski J.. Comparative kinome analysis to identify putative colon tumor biomarkers. J. Mol. Med. (Berl.). 2012; 90:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E.et al.. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019; 47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mon A.M., MacKinnon A.C., Traktman P.. Overexpression of the VRK1 kinase, which is associated with breast cancer, induces a mesenchymal to epithelial transition in mammary epithelial cells. PLoS One. 2018; 13:e0203397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fournier M.V., Martin K.J., Kenny P.A., Xhaja K., Bosch I., Yaswen P., Bissell M.J.. Gene expression signature in organized and growth-arrested mammary acini predicts good outcome in breast cancer. Cancer Res. 2006; 66:7095–7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ben Z., Gong L., Qiu Y.. High expression of VRK1 is related to poor prognosis in glioma. Pathol. Res. Pract. 2018; 214:112–118. [DOI] [PubMed] [Google Scholar]
- 36. Stoll M., Teoh H., Lee J., Reddel S., Zhu Y., Buckley M., Sampaio H., Roscioli T., Farrar M., Nicholson G.. Novel motor phenotypes in patients with VRK1 mutations without pontocerebellar hypoplasia. Neurology. 2016; 87:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feng S.-Y., Li L.-Y., Feng S.-M., Zou Z.-Y.. A novel VRK1 mutation associated with recessive distal hereditary motor neuropathy. Ann. Clin. Transl. Neurol. 2019; 6:401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li N., Wang L., Sun X., Lu Z., Suo X., Li J., Peng J., Peng R.. A novel mutation in VRK1 associated with distal spinal muscular atrophy. J. Hum. Genet. 2019; 64:215–219. [DOI] [PubMed] [Google Scholar]
- 39. Greenbaum L., Barel O., Nikitin V., Hersalis-Eldar A., Kol N., Reznik-Wolf H., Dominissini D., Pras E., Dori A.. Identification of a homozygous VRK1 mutation in two patients with adult-onset distal hereditary motor neuropathy. Muscle Nerve. 2019; 70:1491. [DOI] [PubMed] [Google Scholar]
- 40. Vinograd-Byk H., Sapir T., Cantarero L., Lazo P.A., Zeligson S., Lev D., Lerman-Sagie T., Renbaum P., Reiner O., Levy-Lahad E. The spinal muscular atrophy with pontocerebellar hypoplasia gene VRK1 regulates neuronal migration through an amyloid-β precursor protein-dependent mechanism. J. Neurosci. 2015; 35:936–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nguyen T.P., Biliciler S., Wiszniewski W., Sheikh K.. Expanding phenotype of VRK1 mutations in motor neuron disease. J. Clin. Neuromuscul. Dis. 2015; 17:69–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gonzaga-Jauregui C., Lotze T., Jamal L., Penney S., Campbell I.M., Pehlivan D., Hunter J.V., Woodbury S.L., Raymond G., Adesina A.M.et al.. Mutations in VRK1 associated with complex motor and sensory axonal neuropathy plus microcephaly. JAMA Neurol. 2013; 70:1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Renbaum P., Kellerman E., Jaron R., Geiger D., Segel R., Lee M., King M.-C., Levy-Lahad E.. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am. J. Hum. Genet. 2009; 85:281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sedghi M., Moslemi A.-R., Olive M., Etemadifar M., Ansari B., Nasiri J., Emrahi L., Mianesaz H.-R., Laing N.G., Tajsharghi H.. Motor neuron diseases caused by a novel VRK1 variant - A genotype/phenotype study. Ann. Clin. Transl. Neurol. 2019; 6:2197–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anderson C.J., Baird M.R., Hsu A., Barbour E.H., Koyama Y., Borgnia M.J., McGinty R.K.. Structural basis for recognition of ubiquitylated nucleosome by Dot1L methyltransferase. Cell Rep. 2019; 26:1681–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Worden E.J., Hoffmann N.A., Hicks C.W., Wolberger C.. Mechanism of cross-talk between H2B ubiquitination and H3 methylation by dot1L. Cell. 2019; 176:1490–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hsu P.L., Shi H., Leonen C., Kang J., Chatterjee C., Zheng N.. Structural basis of H2B ubiquitination-dependent H3K4 methylation by COMPASS. Mol. Cell. 2019; 76:712–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Worden E.J., Zhang X., Wolberger C.. Structural basis for COMPASS recognition of an H2B-ubiquitinated nucleosome. eLife. 2020; 9:e53199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim S.-A., Zhu J., Yennawar N., Eek P., Tan S.. Crystal structure of the LSD1/CoREST histone demethylase bound to its nucleosome substrate. Mol. Cell. 2020; 78:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bilokapic S., Halic M.. Nucleosome and ubiquitin position set2 to methylate H3K36. Nat. Commun. 2019; 10:3795–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li W., Tian W., Yuan G., Deng P., Sengupta D., Cheng Z., Cao Y., Ren J., Qin Y., Zhou Y.et al.. Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature. 2021; 590:498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ho C.-H., Takizawa Y., Kobayashi W., Arimura Y., Kimura H., Kurumizaka H.. Structural basis of nucleosomal histone H4 lysine 20 methylation by SET8 methyltransferase. Life Sci. Alliance. 2021; 4:e202000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee J.-H., Bollschweiler D., Schäfer T., Huber R.. Structural basis for the regulation of nucleosome recognition and HDAC activity by histone deacetylase assemblies. Sci. Adv. 2021; 7:eabd4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu P., Li C., Chen Z., Jiang S., Fan S., Wang J., Dai J., Zhu P., Chen Z.. The nua4 core complex acetylates nucleosomal histone H4 through a double recognition mechanism. Mol. Cell. 2016; 63:965–975. [DOI] [PubMed] [Google Scholar]
- 55. McGinty R.K., Henrici R.C., Tan S.. Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature. 2014; 514:591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Witus S.R., Burrell A.L., Farrell D.P., Kang J., Wang M., Hansen J.M., Pravat A., Tuttle L.M., Stewart M.D., Brzovic P.S.et al.. BRCA1/BARD1 site-specific ubiquitylation of nucleosomal H2A is directed by BARD1. Nat. Struct. Mol. Biol. 2021; 28:268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Morgan M.T., Haj-Yahya M., Ringel A.E., Bandi P., Brik A., Wolberger C.. Structural basis for histone H2B deubiquitination by the SAGA DUB module. Science. 2016; 351:725–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Skrajna A., Goldfarb D., Kedziora K.M., Cousins E.M., Grant G.D., Spangler C.J., Barbour E.H., Yan X., Hathaway N.A., Brown N.G.et al.. Comprehensive nucleosome interactome screen establishes fundamental principles of nucleosome binding. Nucleic Acids Res. 2020; 48:9415–9432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Luger K., Rechsteiner T.J., Richmond T.J.. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999; 304:3–19. [DOI] [PubMed] [Google Scholar]
- 60. Lowary P.T., Widom J.. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998; 276:19–42. [DOI] [PubMed] [Google Scholar]
- 61. Kim S.-A., Chatterjee N., Jennings M.J., Bartholomew B., Tan S.. Extranucleosomal DNA enhances the activity of the LSD1/CoREST histone demethylase complex. Nucleic Acids Res. 2015; 43:4868–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tan S., Kern R.C., Selleck W.. The pST44 polycistronic expression system for producing protein complexes in escherichia coli. Protein Expression Purif. 2005; 40:385–395. [DOI] [PubMed] [Google Scholar]
- 63. Winkler D.D., Luger K., Hieb A.R.. Quantifying chromatin-associated interactions: the HI-FI system. Methods Enzymol. 2012; 512:243–274. [DOI] [PubMed] [Google Scholar]
- 64. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Afonine P.V., Poon B.K., Read R.J., Sobolev O.V., Terwilliger T.C., Urzhumtsev A., Adams P.D.. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol. 2018; 74:531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Davey C.A., Sargent D.F., Luger K., Maeder A.W., Richmond T.J.. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9Å resolution. J. Mol. Biol. 2002; 319:1097–1113. [DOI] [PubMed] [Google Scholar]
- 67. Humphrey W., Dalke A., Schulten K.. VMD: visual molecular dynamics. J Mol Graph. 1996; 14:38–38. [DOI] [PubMed] [Google Scholar]
- 68. Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E.. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015; 1-2:19–25. [Google Scholar]
- 69. Berendsen H., van der Spoel D, van Drunen R.. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995; 91:43–56. [Google Scholar]
- 70. Brooks B.R., Brooks C.L., Mackerell A.D., Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S.et al.. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009; 30:1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926. [Google Scholar]
- 72. Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G.. A smooth particle mesh ewald method. J. Chem. Phys. 1995; 103:8577. [Google Scholar]
- 73. Bussi G., Donadio D., Parrinello M.. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007; 126:014101. [DOI] [PubMed] [Google Scholar]
- 74. Tiscornia G., Singer O., Verma I.M.. Production and purification of lentiviral vectors. Nat. Protoc. 2006; 1:241–245. [DOI] [PubMed] [Google Scholar]
- 75. Herrmann C., Avgousti D.C., Weitzman M.D.. Differential salt fractionation of nuclei to analyze Chromatin-associated proteins from cultured mammalian cells. Bio Protoc. 2017; 7:e2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shiio Y., Eisenman R.N., Yi E.C., Donohoe S., Goodlett D.R., Aebersold R.. Quantitative proteomic analysis of chromatin-associated factors. J. Am. Soc. Mass. Spectrom. 2003; 14:696–703. [DOI] [PubMed] [Google Scholar]
- 77. Lee N., Kim D.-K., Han S.H., Ryu H.G., Park S.J., Kim K.-T., Choi K.Y.. Comparative interactomes of VRK1 and VRK3 with their distinct roles in the cell cycle of liver cancer. Mol. Cells. 2017; 40:621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McGinty R.K., Tan S.. Recognition of the nucleosome by chromatin factors and enzymes. Curr. Opin. Struct. Biol. 2016; 37:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Barbera A.J., Chodaparambil J.V., Kelley-Clarke B., Joukov V., Walter J.C., Luger K., Kaye K.M.. The nucleosomal surface as a docking station for kaposi's sarcoma herpesvirus LANA. Science. 2006; 311:856–861. [DOI] [PubMed] [Google Scholar]
- 80. Couñago R.M., Allerston C.K., Savitsky P., Azevedo H., Godoi P.H., Wells C.I., Mascarello A., de Souza Gama F.H., Massirer K.B., Zuercher W.J.et al.. Structural characterization of human vaccinia-related kinases (VRK) bound to small-molecule inhibitors identifies different P-loop conformations. Sci. Rep. 2017; 7:7501–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shin J., Chakraborty G., Bharatham N., Kang C., Tochio N., Koshiba S., Kigawa T., Kim W., Kim K.-T., Yoon H.S.. NMR solution structure of human vaccinia-related kinase 1 (VRK1) reveals the C-terminal tail essential for its structural stability and autocatalytic activity. J. Biol. Chem. 2011; 286:22131–22138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ngow Y.S., Rajan S., Ye H., Yoon H.S.. Crystal structure of human vaccinia-related kinase 1 in complex with AMP-PNP, a non-hydrolyzable ATP analog. Protein Sci. 2019; 28:524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. McGinty R.K., Tan S.. Principles of nucleosome recognition by chromatin factors and enzymes. Curr. Opin. Struct. Biol. 2021; 71:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Stützer A., Welp L.M., Raabe M., Sachsenberg T., Kappert C., Wulf A., Lau A.M., David S.-S., Chernev A., Kramer K.et al.. Analysis of protein-DNA interactions in chromatin by UV induced cross-linking and mass spectrometry. Nat. Commun. 2020; 11:5250–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kobayashi W., Takizawa Y., Aihara M., Negishi L., Ishii H., Kurumizaka H.. Structural and biochemical analyses of the nuclear pore complex component ELYS identify residues responsible for nucleosome binding. Commun. Biol. 2019; 2:163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Teves S.S., An L., Hansen A.S., Xie L., Darzacq X., Tjian R.. A dynamic mode of mitotic bookmarking by transcription factors. Elife. 2016; 5:e22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L.E., Brookes D.H., Wilson L., Chen J., Liles K.et al.. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018; 27:112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM maps have been deposited in the EMDB with accession numbers EMD-25778 (uncrosslinked) and EMD-25777 (crosslinked) and the preferred cryo-EM model of the VRK1-nucleosome complex has been deposited in the PDB with accession number 7TAN.