

Abstract
Mutations in NMNAT1, a key enzyme involved in the synthesis of NAD+ in the nucleus, lead to an early onset severe inherited retinal degeneration (IRD). We aimed to understand the role of nuclear NAD+ in the retina and to identify the molecular mechanisms underlying NMNAT1-associated disease, using a mouse model that harbors the p.V9M mutation in Nmnat1 (Nmnat1V9M/V9M). We identified temporal transcriptional reprogramming in the retinas of Nmnat1V9M/V9M mice prior to retinal degeneration, which begins at 4 weeks of age, with no significant alterations in gene expression at 2 weeks of age and over 2600 differentially expressed genes by 3 weeks of age. Expression of the primary consumer of NAD+ in the nucleus, PARP1, an enzyme involved in DNA damage repair and transcriptional regulation, as well as 7 other PARP family enzymes, was elevated in the retinas of Nmnat1V9M/V9M. This was associated with elevated levels of DNA damage, PARP-mediated NAD+ consumption and migration of Iba1+/CD45+ microglia/macrophages to the subretinal space in the retinas of Nmnat1V9M/V9M mice. These findings suggest that photoreceptor cells are especially sensitive to perturbation of genome homeostasis, and that PARP-mediated cell death may play a role in other genetic forms of IRDs, and potentially other forms of neurodegeneration.
Graphical Abstract
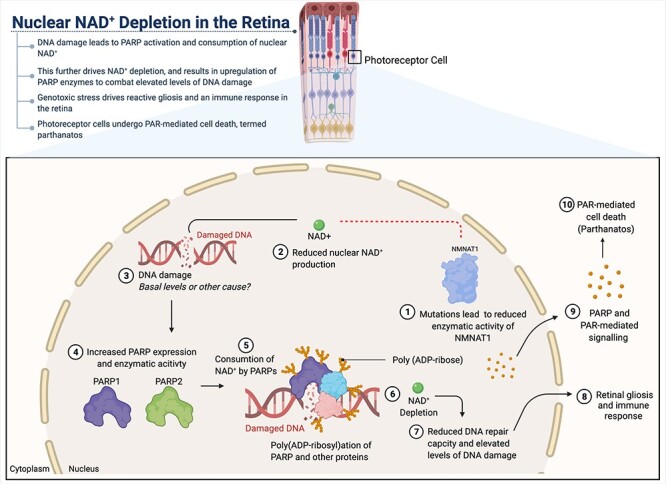
Mechanisms of photoreceptor degeneration due to reduced NMNAT1 activity. Mutations in the nuclear NMNAT, NMNAT1, lead to reduced enzymatic activity (1), resulting in decreased production of nuclear NAD+ (2). Reduced nuclear NAD+ availability leads to photoreceptor-specific DNA damage (3), increased expression and activity of PARP enzymes (4), which drives enhanced consumption of nuclear NAD+ by PARPs (5). This process leads to nuclear NAD+ depletion (6), further elevation of DNA damage (7) and activation of retinal gliosis and an immune response (8). We hypothesize that these processes result in PAR-mediated cell death of the photoreceptors, a process known as parthanatos (9, 10).
Introduction
Nicotinamide nucleotide adenylyltransferases (NMNATs) are enzymes that convert the nicotinamide adenine dinucleotide (NAD+) precursor nicotinamide mononucleotide (NMN) and ATP into NAD+. There are thee different NMNAT enzymes, encoded by different genes, that are characterized by their subcellular localization. These include NMNAT1, which is localized to the nucleus, NMNAT2, which is localized to the cytoplasm/Golgi, and NMNAT3, which is localized to the mitochondria. Although NMNAT1 is expressed ubiquitously, mutations in this gene lead to inherited retinal degeneration (IRD), with few reports of other systemic effects (1–5). Due to its nuclear localization, NMNAT1 is essential for providing NAD+ as a cofactor for nuclear processes, including DNA repair by poly (ADP-ribose) polymerases (PARPs) and regulation of gene expression by PARPs and Sirtuins (6,7). Knockout of NMNAT1 is embryonic lethal, indicating that at least some activity of this enzyme is critical for proper development and survival (8).
Although it is known that mutations in NMNAT1 lead to retina-specific disease, the mechanisms of disease pathogenesis remain poorly understood. Our lab has previously characterized a mouse model of NMNAT1-associated disease that harbors the p.Val9Met mutation in Nmnat1 (9). Nmnat1V9M/V9M mice recapitulate many aspects of NMNAT1-associated disease, including early-onset retinal degeneration, with loss of retinal photoreceptor cells beginning at 4 weeks of age. We have previously shown that the p.Val9Met mutation results in a 63% reduction in enzymatic activity of purified recombinant human NMNAT1, suggesting that reduced enzymatic function is sufficient to drive disease pathology (1). Using the Nmnat1V9M/V9M mouse model, we have also shown that there is a retina-specific reduction in the levels of NAD+ (9). Other studies using a mouse model with conditional knockout of Nmnat1 have suggested that NMNAT1 plays an essential role in inhibiting sterile alpha and TIR motif-containing 1 (SARM1), an NADase involved in mediating axon degeneration (10,11). These findings suggest that in the setting of complete loss of NMNAT1 function, reduction of NAD+ may be due to consumption by SARM1, which results in photoreceptor cell death.
In the present study, we aimed to better understand how reduced nuclear NAD+ drives retinal pathology and whether SARM1 plays a role in human disease, which is associated with reduced but not complete loss of NMNAT1 function (1–3). In the Nmnat1V9M/V9M mouse model, which has a hypomorphic mutation that better represents human disease, we did not observe elevated levels of cyclic ADP-ribose (cADPR), a marker of SARM1 activation (12), at any age examined (2, 3 and 4 weeks of age). This suggests that SARM1 does not play a significant role in disease where some enzymatic function of NMNAT1 is retained. Further, metabolomic studies did not identify significant alterations in the levels of glycolytic or TCA-related metabolites, suggesting that total cellular metabolism is for the most part unaltered in Nmnat1V9M/V9M mice.
Since reduced NMNAT1 function did not activate SARM1 or notably alter cellular metabolism, we focused further studies on the nuclear-specific effects of reduced NAD+ in the retina. We found that reduced nuclear NAD+ leads to transcriptional reprogramming prior to retinal degeneration. This included increased expression of PARP enzymes, which was associated with increased PARP enzymatic activity and elevated levels of DNA damage. This enhanced activity of PARPs likely contributes to the reduction of the nuclear NAD+ pool in the retina, further reducing the ability of PARPs to synthesize poly-ADP ribose (PAR) chains and initiate DNA repair processes (13). We found that the DNA damage is greatest in the photoreceptor cells, suggesting that nuclear NAD+ is essential for proper photoreceptor function due to the requirement of nuclear NAD+ for PARP consumption to maintain genomic homeostasis. Based on the data from the transcriptome analyses, it appears that the genotoxic stress associated with reduced NAD+ levels and defective DNA repair results in an immune response and reactive gliosis in the retina.
These findings support genotoxic stress and PARP overactivation as a mechanism of cell death in NMNAT1-associated disease. Other studies have shown that PARP overactivation leads to PARP-mediated cell death, termed parthanatos, in other models of IRDs (14–16), as well in a variety of neurodegenerative conditions such as lethal seizures (17), amyotrophic lateral sclerosis (ALS) (18) and Alzheimer’s disease (19). Our findings suggest that the vulnerability of photoreceptor cells to genotoxic stress is likely to underlie other genetic forms of IRDs, and that parthanatos may be a general cell death mechanism in photoreceptors, as well as in other neurodegenerative conditions. These findings provide critical insight into the essential role of nuclear NAD+ in the photoreceptors and may help identify potential therapies to prevent neurodegeneration in a spectrum of diseases.
Results
SARM1 is not activated prior to or at the onset of retinal degeneration in Nmnat1V9M/V9M mice
Previous studies have shown that SARM1, an NADase that mediates axon degeneration (10), is activated following conditional knockout of Nmnat1 in the mouse retina, resulting in subsequent retinal degeneration (11). To examine whether SARM1 activation plays a role in retinal degeneration in the mouse model of NMNAT1-associated disease due to the p.V9M mutation (a hypomorphic mutation), we measured the levels of cyclic ADP-ribose (cADPR) in the retinas of Nmnat1V9M/V9M and control animals. cADPR is produced when SARM1 is activated and cleaves NAD+ and is a biomarker of SARM1 activity (12). We measured the levels of cADPR in the neural retinas of wild-type (WT) and Nmnat1V9M/V9M mice via liquid chromatography-mass spectrometry (LC–MS) at 2, 3 and 4 weeks of age to assess SARM1 activation prior to (2 and 3 weeks of age) and at the onset of retinal degeneration (4 weeks of age). There were no statistically significant differences in the levels of cADPR between the WT and Nmnat1V9M/V9M mice at any age examined (Fig. 1A). These results suggest that SARM1-mediated axon degeneration does not mediate photoreceptor degeneration in the Nmnat1V9M/V9M model of NMNAT1-associated disease.
Figure 1.
Altered steady state levels of metabolites in the neural retina, kidney and retinal pigment epithelium (RPE) of Nmnat1V9M/V9M mice. (A) Cyclic ADP-ribose (cADPR) was measured in the retina by LC–MS at 2, 3 or 4 weeks of age. (n = 8 WT and 8 Nmnat1V9M/V9M mice per age, two-way ANOVA, multiple comparisons, P  0.1560, error bars represent the standard error of the mean (SEM)). (B) Steady state metabolite levels in the neural retina (top), posterior eyecup (including the RPE) (middle) or kidney (bottom), at 2, 3 or 4 weeks of age measured by LC–MS/GC–MS. Statistically significant differences in the Nmnat1V9M/V9M tissue as compared to WT are noted by red asterisks (n
0.1560, error bars represent the standard error of the mean (SEM)). (B) Steady state metabolite levels in the neural retina (top), posterior eyecup (including the RPE) (middle) or kidney (bottom), at 2, 3 or 4 weeks of age measured by LC–MS/GC–MS. Statistically significant differences in the Nmnat1V9M/V9M tissue as compared to WT are noted by red asterisks (n 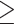 6 WT and n
6 WT and n 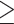 6 Nmnat1V9M/V9M mice for each tissue at each age, two-way ANOVA, multiple comparisons, P < 0.05, error bars represent the SEM). (C) Quantification of western blots measuring the levels of pyruvate kinase M1 (PKM1) (P = 0.385), pyruvate kinase M2 (PKM2) (P = 0.324), phospho-PKM2 (p-PKM2) (P = 0.701) and hexokinase II (HKII) (P = 0.231) in the retinas of 3-week-old mice (n = 4 per genotype, paired t-test, error bars represent the SEM). Abbreviations: 2-methylbutyroylcarnitine (2-MBC), 3-phosphoglyceric acid (3PG), inosine monophosphate (IMP), N-aspartic acid (N-Asp), NMN, PEP.
6 Nmnat1V9M/V9M mice for each tissue at each age, two-way ANOVA, multiple comparisons, P < 0.05, error bars represent the SEM). (C) Quantification of western blots measuring the levels of pyruvate kinase M1 (PKM1) (P = 0.385), pyruvate kinase M2 (PKM2) (P = 0.324), phospho-PKM2 (p-PKM2) (P = 0.701) and hexokinase II (HKII) (P = 0.231) in the retinas of 3-week-old mice (n = 4 per genotype, paired t-test, error bars represent the SEM). Abbreviations: 2-methylbutyroylcarnitine (2-MBC), 3-phosphoglyceric acid (3PG), inosine monophosphate (IMP), N-aspartic acid (N-Asp), NMN, PEP.
Steady state levels of metabolites are not notably altered in the neural retina, retinal pigment epithelium and kidney of Nmnat1V9M/V9M mice
Since we identified that activation of SARM1 does not play a role in degeneration in the Nmnat1V9M/V9M mice, we hypothesized that degeneration may be due to altered levels of key metabolites in the retina. We have previously measured the levels of NAD+ pathway-related metabolites in a variety of tissues (i.e. retina, kidney, skin, liver, lung, brain and heart) in WT and Nmnat1V9M/V9M mice at 3 weeks of age, which is prior to retinal degeneration, and at 6 weeks of age, which is after retinal degeneration has begun (20). Although the NAD+ precursor, NMN, was elevated in several tissues, including the retina, liver and brain, we observed a retina-specific reduction in the levels of NAD+ (20). The retina is one of the most metabolically active tissues in the body (21), and proper function of major metabolic pathways that utilize NAD+, including glycolysis (22–24) and the citric acid cycle (TCA) (25), is essential for maintenance and survival of the retina. To determine whether the steady state levels of metabolites in critical metabolic pathways, other than those in the NAD pathway, are impacted in the retina, we collected neural retina, posterior eyecup (which includes the retinal pigment epithelium (RPE)), and kidney tissue from WT and Nmnat1V9M/V9M mice. Kidney tissue was used as a negative control tissue, as no differences were observed in the levels of metabolites between WT and Nmnat1V9M/V9M mice in the NAD+ panel performed previously (20).
We assessed the steady state levels of over 140 metabolites in these tissues, collected prior to retinal degeneration (2 and 3 weeks of age) or after the onset of retinal degeneration (4 weeks of age). Although we did observe elevated levels of several metabolites in the retina, eyecup, and kidney at the three ages examined, these changes were transient (Fig. 1B). For example, slightly increased levels of pyruvate, a glycolytic metabolite that is important for photoreceptor metabolism (22,24), were detected in the retinas of Nmnat1V9M/V9M mice at 3 weeks of age. This change was minor, with a 1.7-fold increase, and altered pyruvate levels were not detected at other time points or in other tissues. The level of phosphoenolpyruvate (PEP) was increased in the kidneys of Nmnat1V9M/V9M mice at 2 and 3 weeks of age, but other related metabolites were not altered (Fig. 1B). A full list of the metabolites measured and their relative abundance in each tissue can be found in Supplementary Material, Tables S1-S3, and significantly altered metabolites over time (longitudinal analysis) can be found in Table S4.
Based on these findings, we sought to determine if the protein levels of metabolic enzymes associated with glycolysis are altered in the retinas of the Nmnat1V9M/V9M mice using western blotting. PKM1 and PKM2 are the predominate pyruvate kinase isoforms expressed in the retina, where PKM2 is primarily expressed in the photoreceptors and PKM1 is expressed in the inner retina (26). PKM2 is phosphorylated at tyrosine 105 (p-PKM2), which inhibits its activity (26). We did not see any statistically significant differences in the levels of PKM1, PKM2, or P-PKM2 protein between the WT and Nmnat1V9M/V9M mice (Fig. 1C, Supplementary Material, Fig. S2). We also assessed the levels of hexokinase and did not observe any differences in the levels between WT and Nmnat1V9M/V9M mice (Fig. 1C, Supplementary Material, Fig. S2). These results suggest that there are no differences in expression of glycolytic enzymes in the retina at 3 weeks of age.
Overall, we did not observe major alterations in the levels of glycolytic or TCA metabolites in the retina, suggesting that there is no disruption in cellular metabolism due to reduced levels of nuclear NAD+. Consistent with the fact that we observed no alterations in the steady state levels of TCA metabolites, we also did not note any alterations in mitochondrial morphology in the RPE or in the photoreceptor inner segment, as visualized by TEM (Supplementary Material, Fig. S1), at 2 or 3 weeks of age. These findings support the hypothesis that the nuclear NAD+ pool is particularly affected in NMNAT1-associated disease.
Retina-specific temporal transcriptional reprogramming in Nmnat1V9M/V9M mice
We next aimed to identify retina-specific alterations in gene expression that may give insight about how reduced NMNAT1 activity leads to retina-specific disease. To this end, we performed RNA sequencing (RNA-seq) on retina and kidney samples from WT and Nmnat1V9M/V9M mice at 2 weeks and 3 weeks of age. We used normalized gene expression profiles, distance matrices, and hierarchical clustering to determine which samples have similar gene expression patterns. At two weeks of age, there was no distinct clustering between the WT and Nmnat1V9M/V9M retina or kidney samples, indicating limited differences in gene expression due to the Nmnat1 p.Val9Met mutation at this timepoint (Supplementary Material, Fig. S3).
At 3 weeks of age, there were no differences in gene expression between the kidneys of WT and Nmnat1V9M/V9M mice, with no clustering of the expected genotypic groupings (Supplementary Material, Fig. S3A). However, there was distinct clustering of the Nmnat1V9M/V9M and WT retinas at 3 weeks of age (Supplementary Material, Fig. S3A). We performed differential gene expression (DGE) analysis to determine if there were any high confidence differentially expressed genes (DEGs) in the retinas of Nmnat1V9M/V9M mice as compared to WT mice at 3 weeks of age. DGE analysis showed that there were over 2600 DEGs in the retinas of Nmnat1V9M/V9M mice as compared to those of WT mice (Fig. 2A). These consisted of 986 (38%) genes that were significantly downregulated and 1632 (62%) genes that were significantly upregulated (Fig. 2A, left). Significantly downregulated genes included two uncharacterized zinc finger transcription factors Zfp990 and Zfp979, as well as Glb1l3 (Fig. 2A, right), a gene whose expression has been shown to be significantly reduced both prior to and during the retinal degenerative process (27). Significantly upregulated genes included genes associated with retinal stress and neuroprotection, such as Stat3, Gfap, NPY4R and Gadd45β (28–32) (Fig. 2A, right). Other significantly upregulated genes include Cebpd, which is involved in regulation of transcription, and Lad1, a gene that encodes a relatively uncharacterized cytoskeletal factor, ladinin-1 (Fig. 2A, right). These genes have also been shown to have elevated expression during the retinal degenerative process, although their roles in this process are poorly understood (33,34).
Figure 2.
RNA-seq temporal transcriptional reprogramming in retinas of Nmnat1V9M/V9M mice. (A) Differential gene expression in the retinas of Nmnat1V9M/V9M mice at 3 weeks of age. (Left) Pie chart displaying total number of differentially expressed genes (DEGs) and percent of DEGs that were upregulated (red) or downregulated (blue). Volcano plots display log2(fold change) on the x-axis and the -log10(adjusted P-value) on the y-axis (right). (B-D) Gene ontology analysis of 3-week retina RNAseq data showing significantly enriched GO terms for molecular function (B), cellular component (C) or biological processes (D). (E) Pathways that were significantly enriched in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis at 3 weeks of age in the retina. (F) Transcription factor binding sites that were enriched in the DEGs in the 3-week retina RNAseq dataset. The number of upregulated (red) or downregulated (blue) genes that contain each specific transcription factor binding site are plotted.
To confirm this temporal transcriptional reprogramming, retina and kidney samples were collected in the same manner as those collected for RNA-seq, from an independent set of mice aged 2, 3, or 4 weeks. Using qRT-PCR, we measured the expression of several genes that were highly enriched in the RNA-seq dataset from the retinas of Nmnat1V9M/V9M mice at 3 weeks of age. These included Gadd45β, which encodes growth arrest and DNA damage inducible beta, Lad1 and Lif, which encodes leukemia inhibitory factor, a neuroprotective cytokine whose expression is increased in response to retinal stressors (28). At two weeks of age, there were no differences in the expression of Gadd45β, Lad1, or Lif between the Nmnat1V9M/V9M and WT mice in the retina or kidney (Supplementary Material, Fig. S3B). However, at 3 weeks of age, expression of all three genes was significantly increased in the retinas of Nmnat1V9M/V9M mice, with a 10-fold increase in the expression of Gadd45β (P < 0.0001), a 200-fold increase in the expression of Lad1 (P < 0.0001), and a 10-fold increase in the expression of Lif (P = 0.0184) in the retinas of Nmnat1V9M/V9M mice as compared to those of WT mice (Supplementary Material, Fig. S3B). Upregulation of these genes persisted at 4 weeks of age (P < 0.0001 for Gadd45β, P = 0.0032 for Lad1 and P = 0.004 for Lif). Although these genes were all significantly upregulated in the retinas of Nmnat1V9M/V9M mice at 3 and 4 weeks of age, there were no significant differences in their expression in the kidney at any age examined (Supplementary Material, Fig. S3B). These findings confirm the temporal reprogramming observed by the RNA-seq, with major differences in gene expression between the WT and Nmnat1V9M/V9M retinas occurring at 3 weeks, but not 2 weeks of age. These findings also confirm the lack of DEGs observed in the kidney samples. Upregulation of Gadd45β and Lif in the retina are consistent with a neuroprotective response (28–31,33). Since we observed significant upregulation of neuroprotective factors, we next aimed to assess other pathways that may be significantly enriched in the RNA-seq dataset.
GO, KEGG and transcription factor analysis show enrichment for immune response-related pathways
We used gene ontology (GO) analysis on the 3-week retina RNA-seq data to group the DEGs identified in the retinas of the Nmnat1V9M/V9M mice based on their functional characteristics (Supplementary Material, Table S5). Of note, the molecular function group of the GO analysis was enriched for NAD+ ADP-ribosyltransferase activity, as well as other nucleotide binding terms, DNA-binding terms and several cytokine binding terms (Fig. 2B). The NAD+ ADP-ribosyltransferase activity includes PARP enzymes. The cellular component functional group was enriched for extracellular matrix, photoreceptor cell cilium and photoreceptor outer segment (Fig. 2C). The biological process functional group was enriched for the term immune system processes and cell death related terms, including regulation of cell death, programmed cell death and apoptotic process (Fig. 2D).
We also performed Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis on the 3-week retina RNA-seq data to determine which pathways were significantly enriched in the DEGs detected in the retinas of the Nmnat1V9M/V9M mice. Significantly enriched pathways included several immune-related pathways, such as cytokine-cytokine receptor interaction, NF- κB signaling and the TNF signaling pathway (Fig. 2E, Supplementary Material, Table S6). Other enriched pathways included apoptosis, necroptosis, phototransduction and cytosolic DNA sensing pathways (Fig. 2E). These findings suggest that the DEGs are significantly enriched for immune-related pathways, cell death pathways and photoreceptor-specific pathways.
To better understand which transcription factors (TFs) may be mediating the temporal transcriptional reprogramming that we observed, we performed enrichment analysis of transcription factor binding sites (TFBs) to determine which TFBs the DEGs are enriched for. Significantly enriched TFs included NF-κB, which regulates inflammation, immunity and cell survival (35), STAT3, which activates proliferative, migratory and inflammatory processes (36), and several IRF family TFs, which activate immune responses (37) (Fig. 2F, Supplementary Material, Table S7). These findings suggest that the TFBs enriched in the DEGs are associated with activation of an immune response. We also examined whether the DEGs that contain these TFBs were upregulated (red) or downregulated (blue) and found that many TFBs were associated with upregulation of gene expression (Fig. 2F). Interestingly, we noted that KEGG, TF and GO analyses were all enriched for immune-related pathways, suggesting that reduced nuclear NAD+ in the Nmnat1V9M/V9M mice is triggering immune activation.
Increased expression of PARPs and elevated levels of DNA damage in the retinas of Nmnat1V9M/V9Mmice
The NAD+ ADP-ribosyltransferase activity GO term was significantly enriched in the retina at 3 weeks of age, and the DEGs in this category included several Parp genes. PARP enzymes are the main consumer of nuclear NAD+ due to their ADP-ribosyltransferase activity and formation of poly-ADP ribose chains. There are 17 different PARP enzymes, which are involved in a variety of essential cellular processes, including DNA repair, transcriptional regulation and cell death (38). Of these PARPs, 16 catalyze the transfer of ADP-ribose to protein (39), DNA (40), or RNA (41) targets. We have also previously shown that poly-ADP ribosylation is elevated in the retinas of Nmnat1V9M/V9M mice (20). Therefore, we aimed to assess whether PARP expression or activity is altered in the retinas of Nmnat1V9M/V9M mice using the RNA-seq dataset. We found that out of the 17 genes encoding PARP enzymes, 8 were significantly upregulated in the retinas of Nmnat1V9M/V9M mice, including Parp1, Parp3, Tiparp (Parp7), Parp9, Parp12, Zc3hav1 (Parp13), Parp14 and Parp16 (Fig. 3A, Supplementary Material, Fig. S4). Importantly, there were no differences in the expression level of Sarm1 (Supplementary Material, Fig. S4), consistent with the levels of cADPR.
Figure 3.
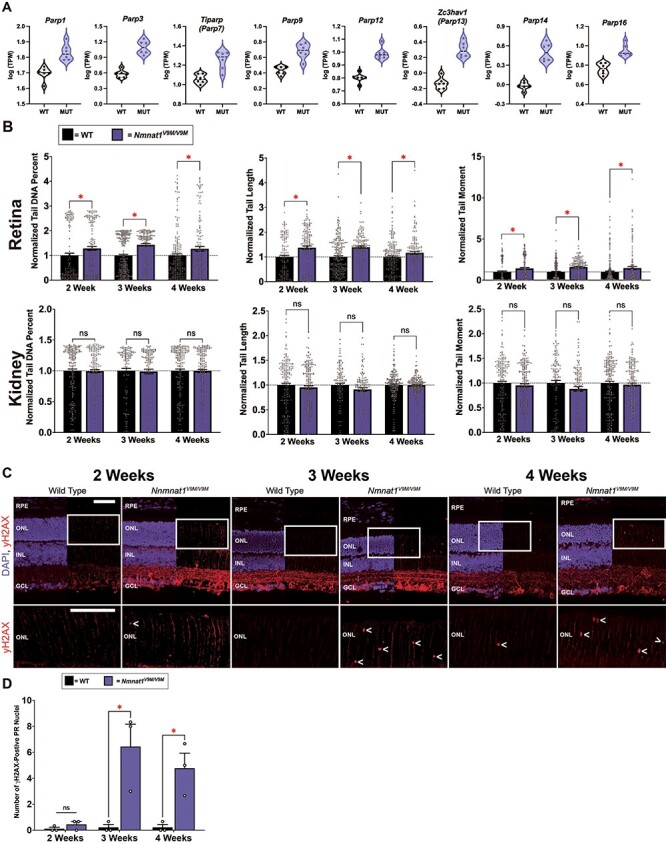
Increased expression of Parps and elevated levels of DNA damage in the retinas of Nmnat1V9M/V9M mice. (A) Violin plots showing the expression of PARP enzymes in the retina at 3 weeks of age. Graphs plot the logarithmic (base10) transcripts per million (TPM) in WT and Nmnat1V9M/V9M (MUT) mice as measured via RNAseq. The 8 PARP enzymes shown all had significantly greater expression in the Nmnat1V9M/V9M retinas. Dots represent individual TPM values per sample. (B) Analysis of DNA damage in WT and Nmnat1V9M/V9M retina and kidney tissue using the comet assay. Top row: retina samples, bottom row: kidney samples. Statistically significant differences in the Nmnat1V9M/V9M tissue as compared to WT are noted by red asterisks (n  4 WT and n = 4 Nmnat1V9M/V9M mice for each tissue at each age, two-way ANOVA, multiple comparisons, P < 0.05, error bars represent the SEM, points represent the respective value for each individual cell). (C) Retinal frozen sections stained with an antibody for γH2AX (red) and DAPI to label the nuclei (blue, DAPI channel is overlayed on half of the image) from mice aged 2 weeks (left), 3 weeks (middle) or 4 weeks (right). (Continued) The white rectangle represents an enlarged area of the ONL (bottom row). The < symbol indicates γH2AX-positive ONL nuclei. Scale bars indicate 20 μm. RPE = retinal pigment epithelium, ONL = photoreceptor outer nuclear layer, INL = inner nuclear layer, GCL = ganglion cell layer. (D) Quantification of γH2AX-positive ONL nuclei from IHC images. Age is plotted on the x-axis. The red asterisks indicate statistical significance (n
4 WT and n = 4 Nmnat1V9M/V9M mice for each tissue at each age, two-way ANOVA, multiple comparisons, P < 0.05, error bars represent the SEM, points represent the respective value for each individual cell). (C) Retinal frozen sections stained with an antibody for γH2AX (red) and DAPI to label the nuclei (blue, DAPI channel is overlayed on half of the image) from mice aged 2 weeks (left), 3 weeks (middle) or 4 weeks (right). (Continued) The white rectangle represents an enlarged area of the ONL (bottom row). The < symbol indicates γH2AX-positive ONL nuclei. Scale bars indicate 20 μm. RPE = retinal pigment epithelium, ONL = photoreceptor outer nuclear layer, INL = inner nuclear layer, GCL = ganglion cell layer. (D) Quantification of γH2AX-positive ONL nuclei from IHC images. Age is plotted on the x-axis. The red asterisks indicate statistical significance (n  3 WT and n = 3 Nmnat1V9M/V9M mice at each age, two-way ANOVA, multiple comparisons, P < 0.05, error bars represent the SEM).
3 WT and n = 3 Nmnat1V9M/V9M mice at each age, two-way ANOVA, multiple comparisons, P < 0.05, error bars represent the SEM).
Since one of the major functions of PARP1 is to respond to DNA damage, and activation of PARP1 can reduce NAD+ levels by as much as 60% (13), we aimed to assess whether there is elevated DNA damage in the retinas of Nmnat1V9M/V9M mice as compared to WT. To this end, we performed the comet assay, a method that uses single cell gel electrophoresis to measure DNA strand breaks (42). This is measured by assessing the length of the comet tail produced after gel electrophoresis, the percent of DNA present in the comet tail, and the tail moment, which represents the tail length multiplied by the percent of DNA in the tail. To measure both single strand and double strand DNA breaks, as well as alkali liable sites, the assay was run under alkaline conditions (43). Using kidney tissue as a control, we found that the retinas of Nmnat1V9M/V9M mice had significantly greater levels of DNA damage at 2, 3 and 4 weeks of age as compared to those from age-matched WT mice. There were also no differences in the levels of DNA damage between the kidney samples of Nmnat1V9M/V9M or WT mice at any age examined (Fig. 3B).
To determine the cell types in the retina that have elevated levels of DNA damage, we stained retina sections with an antibody against γH2AX, the phosphorylated form of histone H2AX. Histone H2AX becomes phosphorylated at Ser139 in response to DNA double strand breaks (44). At 2 weeks of age, we noticed a small number of distinct foci in the photoreceptor nuclear layer, the outer nuclear layer (ONL), of both WT and Nmnat1V9M/V9M retinas (Fig. 3C and D). However, by 3 weeks of age, there were significantly more γH2AX-positive photoreceptor nuclei in the Nmnat1V9M/V9M retinas as compared to the WT retinas, which persisted at 4 weeks of age (Fig. 3C and D). To confirm that the γH2AX-positive cells are not infiltrating macrophages, we co-stained with antibodies for γH2AX and CD45, and confirmed that there is no colocalization (Supplementary Material, Fig. S4). These findings suggest that reduced nuclear NAD+ results in elevated levels of genotoxic stress in the retina and that this imbalance in genomic integrity is photoreceptor specific.
Increased PARP enzymatic activity in the photoreceptors of Nmnat1V9M/V9M mice
Since we have previously observed reduced NAD+ specifically in the retina of Nmnat1V9M/V9M mice and increased expression of many of the PARP enzymes, we aimed to assess whether PARP enzymatic activity is elevated in the Nmnat1V9M/V9M retina as well. We performed staining with 6-fluoro-NAD+, a fluorescent NAD+ analog that binds to enzymes with available NAD+-binding sites, in unfixed retinal sections from 2-, 3- or 4-week-old WT and Nmnat1V9M/V9M retinas, as described previously (45). To specifically assess binding of 6-fluoro-NAD+ to PARP enzymes, a subset of slides were pretreated with olaparib, an FDA-approved drug used to treat metastatic breast cancer and potent PARP inhibitor. Olaparib inhibits PARP by binding in the NAD+ binding pocket of PARP1, with some leakiness for other PARP enzymes (46).
At 2 weeks of age, we observed several 6-fluoro-NAD+-positive foci in the nuclei of cells in the inner nuclear layer (INL) of both WT and Nmnat1V9M/V9M retinas, with no significant differences between the number of foci between the two groups (P = 0.9950, Fig. 4A and B). However, by 3 weeks of age there were few 6-fluoro-NAD+-positive foci in the INL, but an increase in the number of 6-fluoro-NAD+ foci in the ONL in Nmnat1V9M/V9M retinas (P = 0.0909, Fig. 4C). This increase in the number of 6-fluoro-NAD+ foci in the ONL of Nmnat1V9M/V9M mice reached statistical significance by 4 weeks of age (P < 0.0001, Fig. 4C). To assess whether the 6-fluoro-NAD+ is binding PARP enzymes, we pretreated a subset of slides with olaparib. There was no staining of 6-fluoro-NAD+ in the olaparib-treated slides (Fig. 4A), suggesting that the binding observed was specific to PARP enzymes. There was also no 6-fluoro-NAD+ signal in slides treated with the reaction mixture only (data not shown). These findings suggest that in addition to increased PARP expression in the retina, there are photoreceptor-specific increases in PARP enzymatic activity.
Figure 4.

Elevated PARP enzymatic activity in the photoreceptors of Nmnat1V9M/V9M mice. (A) 6-fluoro-NAD+ staining in unfixed frozen retinal sections. Asterisks indicate 6-fluoro-NAD-positive cells (top row). White boxes indicate areas of the ONL that are magnified in the center row. < symbols indicate 6-fluoro-NAD-positive cells. Bottom row shows staining in slides pretreated with olaparib. Scale bars indicate 20 μm. RPE = retinal pigment epithelium, ONL = photoreceptor outer nuclear layer, INL = inner nuclear layer, GCL = ganglion cell layer. (B) Quantification of 6-fluoro-NAD-positive INL nuclei at 2, 3 or 4 weeks of age (two-way ANOVA, multiple comparisons, P  0.1071, error bars represent the SEM, n = 3–4 mice per group per age, points represent the average quantification from 3 images per mouse). (C) Quantification of 6-fluoro-NAD-positive photoreceptor ONL nuclei at 2, 3 or 4 weeks of age (two-way ANOVA, multiple comparisons, P-values are indicated, error bars represent the standard error of the mean (SEM), n = 4 mice per group per age, points represent the average quantification from 3 images per mouse).
0.1071, error bars represent the SEM, n = 3–4 mice per group per age, points represent the average quantification from 3 images per mouse). (C) Quantification of 6-fluoro-NAD-positive photoreceptor ONL nuclei at 2, 3 or 4 weeks of age (two-way ANOVA, multiple comparisons, P-values are indicated, error bars represent the standard error of the mean (SEM), n = 4 mice per group per age, points represent the average quantification from 3 images per mouse).
Reduced NMNAT1 enzymatic activity leads to immune activation in the retina
In addition to enrichment of NAD+-ribosylation activity and increased expression of PARP enzymes, we also noted from the RNA-seq data and the GO and KEGG pathway analysis that the 3-week retina dataset was enriched for genes associated with immune-related pathways. To further examine the immune response in the retina, we stained retinal sections with an antibody against glial fibrillary acidic protein (GFAP), a protein whose expression is increased in retinal astrocytes and Müller glial cells in response to retinal stress via regulation by cytokines. Gfap expression was also highly upregulated in our RNA-seq dataset. At two weeks of age, there were no major differences in GFAP staining between the WT and Nmnat1V9M/V9M retinas, with staining localized near the ganglion cell layer (Fig. 5A, left panel). However, by 3 weeks of age, there was a significant increase in GFAP staining in Nmnat1V9M/V9M mice (Fig. 5A, middle panel), which was further increased by 4 weeks of age (Fig. 5A, right panel).
Figure 5.
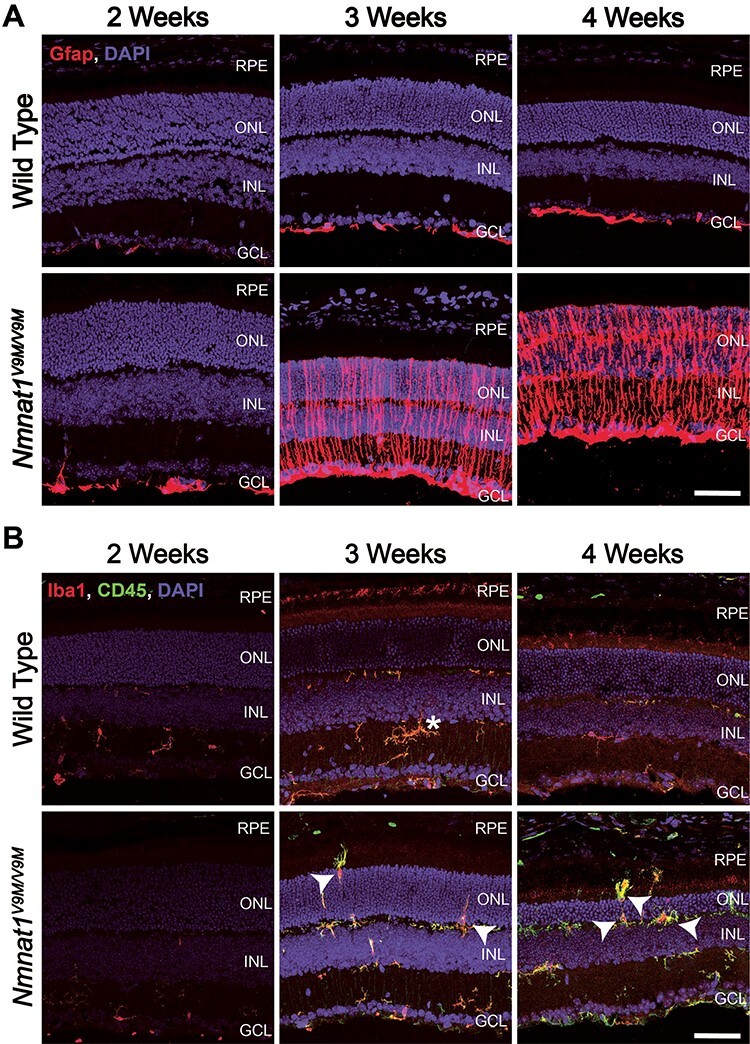
Reactive gliosis and immune response in the retinas of Nmnat1V9M/V9M mice. (A) Retina sections from mice aged 2, 3 or 4 weeks of age stained with an antibody against Gfap (red) and DAPI to label nuclei (blue). (B) Staining for Iba1 (red), CD45 (green) and DAPI (blue). Iba1 and CD45-positive cells with amoeboid morphology that are migrating into the ONL and subretinal space are indicated with white arrows. The white asterisk indicates ramified Iba1 and CD45-positive cell. Scale bar indicates 20 μm. RPE = retinal pigment epithelium, ONL = photoreceptor outer nuclear layer, INL = inner nuclear layer, GCL = ganglion cell layer.
To further characterize the immune response in the Nmnat1V9M/V9M retinas, we stained for Iba1 and CD45, which are markers of microglia/macrophages. At 2 weeks of age, Iba1 and CD45 staining were localized to the inner retina and displayed ramified morphology, in both the Nmnat1V9M/V9M and WT retinas (Fig. 5B). However, by 3 weeks of age, the Iba1+/CD45+ cells in the Nmnat1V9M/V9M mice displayed amoeboid morphology, with staining present in the photoreceptor nuclear layer, which persisted at 4 weeks of age (Fig. 5B). These findings suggest that mutations in NMNAT1 drive reactive gliosis and microglia/macrophage recruitment to the subretinal space, likely as a response to the genotoxic stress due to PARP-mediated nuclear NAD+ depletion.
Discussion
Although NMNAT1 is expressed ubiquitously, mutations in the NMNAT1 gene lead to retina-specific disease. To assess why mutations in NMNAT1 uniquely affect the retina, and to develop a better understanding of the role of nuclear NAD+ in the retina in general, we studied the mechanism of retinal degeneration in Nmnat1V9M/V9M mice, a model of NMNAT1-associated disease (9). Our findings suggest that nuclear NAD+ is essential for photoreceptor cell survival, mainly via PARP-mediated consumption to maintain genome homeostasis. In the Nmnat1V9M/V9M model, we did not observe elevated levels of cADPR at any age examined (2, 3 and 4 weeks of age), suggesting that SARM1 does not play a significant role in disease where some enzymatic function of NMNAT1 is retained. Further, cellular metabolism is largely not significantly altered in the Nmnat1V9M/V9M mice, suggesting that reduced NAD+ due to mutations in NMNAT1 has nuclear-specific effects. We hypothesize that genotoxic stress and over-activation of PARP activity drive immune and gliotic cascades, and ultimately result in death of photoreceptor cells. These findings suggest that parthanatos, a PARP1-dependent mode of cell death (14–16), is the mechanism of cell death in NMNAT1-associated disease, and that this pathway may be a general cell death mechanism in IRDs.
Our findings suggest that nuclear NAD+ is essential in photoreceptors primarily to support PARP-mediated maintenance of genomic integrity. Using RNA-seq and GO analysis, we identified enrichment of the NAD+ ADP-ribosyl transferase GO term and significantly increased gene expression of PARP enzymes in the retinas of Nmnat1V9M/V9M mice just prior to the onset of retinal degeneration. As one of the major functions of PARP enzymes is to respond to DNA damage, we assessed whether there were increased levels of DNA damage in the retinas of Nmnat1V9M/V9M mice. Using the comet assay, we identified that levels of DNA damage were significantly elevated in the retinas of Nmnat1V9M/V9M mice at 2, 3 and 4 weeks of age. Since we do not observe NAD+ depletion in the retina until 3 weeks of age (20), these data suggest that the DNA damage occurs prior to reduction of NAD+. The DNA damage observed is likely not due to elevated levels of oxidative stress, as we have previously shown that the retinas of Nmnat1V9M/V9M mice do not have evidence of DNA, protein, or lipid oxidation and the ratio of GSH:GSSG is not altered prior to retinal degeneration (20). In addition, the RNA-seq dataset is not enriched for upregulation of antioxidant enzymes, suggesting that photoreceptor-specific DNA damage is mediated via other mechanisms. Although we have not been able to identify the driving force of the DNA damage, future studies will aim to identify the cause of the retina-specific increases in the levels of DNA damage.
Staining for the double strand DNA break marker γH2AX and assessment of binding of the NAD+ fluorescent analog 6-fluoro-NAD+ indicate that the DNA damage associated with reduced NMNAT1 enzyme activity is mainly photoreceptor-specific and that there are photoreceptor-specific increases in PARP enzymatic activity. We have also previously shown that PAR polymers are elevated in the retinas of Nmnat1V9M/V9M mice, specifically in the photoreceptors (20). Elevated levels of DNA damage have been observed in the retinas of WT C57BL/6 mice during retinal development (47). However, these findings and the data from the present study suggest that this developmental DNA damage is likely isolated to the inner retina, rather than the photoreceptors. Our data suggest that photoreceptors have a unique requirement for nuclear NAD+ to support PARP-mediated prevention of DNA damage and suggest that NAD+-depletion is mediated by PARP activity that is photoreceptor-specific.
Studies have shown that PARP activity is increased in other rodent models of retinal degeneration, including mice with loss of function mutations in the Pde6b gene, the dominant p.Pro23His mutation in the Rho gene, and rats with a p.Ser334ter mutant Rho transgene (14–16). Interestingly, knockout of Parp1 does not affect retinal function in WT mice but can protect against retinal degeneration in Pde6b mutant mice (16). These findings suggest that PARP1 plays an important role in mediating retinal degeneration in a cell death pathway that is common to multiple retinal degenerations via parthanatos, a PARP1-dependent mode of cell death (14–16). Interestingly, genome instability has also been shown to play a role in a variety of other neurodegenerative conditions, including lethal seizures (17), amyotrophic lateral sclerosis (ALS) (18) and Alzheimer’s disease (19). The data presented in the present study support the hypothesis that photoreceptor cell death in NMNAT1-associated retinal degeneration is mediated by parthanatos due to failure to meet the demand for nuclear NAD+ for PARP consumption to maintain genomic homeostasis.
Previous work has demonstrated that inhibition of SARM1, an NADase that has been shown to mediate axon degeneration, is sufficient to prevent retinal degeneration in a mouse model with inducible knockout of Nmnat1 in the retina (11). In those studies, the level of cADPR, which is indicative of SARM1 activity (12), was elevated in the retinas of Nmnat1 knockout mice, while the levels of NAD+ were decreased (11). Combined knockout of Sarm1 and Nmnat1 reduced levels of cADPR and restored levels of NAD+, suggesting that SARM1 may play a role in NAD+ depletion and cell death associated with complete loss of NMNAT1 function. We aimed to determine whether activation of SARM1 mediates retinal degeneration in the Nmnat1V9M/V9M model, which still retains some NMNAT1 function, and thus is a better model of human disease, as patients with mutations in NMNAT1 do not have complete loss of function of the enzyme (1). In the Nmnat1V9M/V9M model, we did not observe elevated levels of cADPR at any age examined (2, 3 and 4 weeks of age), suggesting that SARM1 does not play a significant role in disease where some enzymatic function of NMNAT1 is retained. Interestingly, we have previously published that the levels of NAD+ are decreased in the retinas of Nmnat1V9M/V9M mice (20), which suggests that an enzyme other than SARM1 is responsible for NAD+ depletion in this model. We detect reduced NAD+ at 3 weeks of age (20), which is consistent with the elevated PARP expression, which also occurred at 3 weeks of age. These data suggest that NAD+ depletion is mediated by PARP, indicating a critical role for nuclear NAD+ in the retina to support PARP activity. We hypothesize that the upregulation of PARP exacerbates NAD+ depletion, resulting in eventual PAR-mediated cell death, known as parthanatos.
In addition to its role in maintaining genomic homeostasis, PARP1 has also been shown to act as a transcriptional cofactor for NF-κB (48–50), a transcription factor that promotes expression of pro-inflammatory genes (51,52). We observed significant enrichment for a variety of immune-related pathways in the RNA-seq dataset, and the identified DEGs were enriched for genes with NF-κB binding sites. Since we observed both upregulation of PARP1 and enrichment of NF-κB-mediated pathways, this suggests that PARP1 may also play a role in mediating the inflammatory processes in Nmnat1V9M/V9M mice. Staining for CD45 and Iba1, markers of microglia/macrophages, demonstrated ‘activation’ of these cells beginning at 3 weeks of age, consistent with the significant alterations in gene expression in the retinas of Nmnat1V9M/V9M mice that were also observed at this age. This included morphological changes in the CD45/Iba1-positive cells, with a transition from a ramified structure to an amoeboid structure, and migration of these cells into the photoreceptor nuclear layer and subretinal space. Disruption of photoreceptor homeostasis in other models has been shown to lead to migration of microglia and monocyte-derived cells to the subretinal space (53,54). Migration of microglia to the subretinal space has been characterized in several other models of retinal degeneration (53,55–57). We hypothesize that the genotoxic stress and activation of PARP is driving the reactive gliosis and immune signaling cascades in the Nmnat1V9M/V9M mice.
Using steady state metabolomic analysis, we found that levels of metabolites in key metabolic processes in the retina, including glycolysis and the TCA cycle, were not altered in the Nmnat1V9M/V9M mice, suggesting that reduced NAD+ due to mutations in NMNAT1 has nuclear-specific effects. We did observe that several purine nucleosides, including adenosine, inosine and hypoxanthine, were significantly elevated in the retinas of 2-week-old Nmnat1V9M/V9M mice, although this effect was transient. Previous studies have shown that the levels of purines are highly sensitive to the metabolic status of the retina, and that most of the purine metabolites in the retina are in the photoreceptor cells (58,59). Other studies have reported that purine nucleotide levels are increased in the retinas of rats after ischemia (60) and in the retinas of mice prior to retinal degeneration in the Rd10 model of IRD (61). It is possible that these metabolites are elevated as a neuroprotective or anti-inflammatory response. In addition, these metabolites may be elevated as a compensatory mechanism to increase levels of NAD+. A recent study in yeast has shown that depletion of adenine results in upregulation of de novo NAD+ synthesis genes, while increased levels of adenine lead to activation of the ATP-dependent pyridine salvage route. This salvage route increases levels of NAD+ and relies on activity of Nma1, the yeast homolog of human NMNAT (62). This suggests that nucleosides may play a role in regulation of NAD+ synthesis.
The findings in the present study suggest that nuclear NAD+ depletion drives photoreceptor-specific DNA damage, increased expression and activity of PARPs, an inflammatory signaling cascade, and reactive gliosis in the retina, ultimately resulting in photoreceptor cell death. This highlights the critical role of nuclear NAD+ in the retina and suggests that photoreceptors are uniquely affected by nuclear NAD+ depletion. The genomes of photoreceptor cells appear more vulnerable to DNA damage than those of other cell types, although the specific stresses, which drive the demand for PARP-mediated DNA repair in photoreceptor cells remain to be identified (15,16,63,64). This is possibly a more general mechanism of retinal degeneration than previously recognized (14–16). We hypothesize that DNA damage, PARP activation and perhaps NAD+ depletion, could be a broad mechanism common to inherited retinal degenerations. The role of these processes should be systematically addressed in future studies. The data in the present study provide insight about the mechanisms of NMNAT1-mediated retinal degeneration. Identification of other genetic forms of IRD in which DNA damage, PARP activation, NAD+ depletion and parthanatos plays a role may help identify potential therapies to prevent vision loss in these genetic forms of IRD.
Materials and Methods
Mouse lines
The Nmnat1V9M/V9M mouse line was derived from a founder identified during an N-ethyl-N-nitrosourea (ENU) mutagenesis screen (9). This line is maintained on the C57Bl/6 J background with breeders heterozygous for the p.V9M mutation (Nmnat1V9M/WT). This allows for the use of littermate Nmnat1V9M/V9M and Nmnat1WT/WT mice for experiments. The Nmnat1V9M/WT mice produced from this cross were only used for breeding. For experiments, equal numbers of male and female mice were used, and all samples were collected at the same time of the day (morning).
Genotyping
Genotyping for the p.V9M mutation was performed on toe or tail biopsies using Sanger sequencing of a portion of the Nmnat1 gene amplified using PCR, as described previously (9). Mice were screened for potential variants that can affect retinal function, including Rd1 (65), Rd8 (66) and RPE65 (67) variants, and were confirmed to be WT for all. Mice were also confirmed to be WT for a variant in nicotinamide nucleotide transhydrogenase (NNT) (68), which is an enzyme involved in NADPH production in the matrix of mitochondria, which has been identified in some C57Bl/6 J lines.
Steady state metabolomic analysis
Equal numbers of male and female mice were euthanized by cervical dislocation (59). Retina and kidney samples were harvested, with two retinas and two kidneys in each tube, respectively, and immediately flash frozen. Samples were stored at −80°C until use. Metabolite extraction, LC–MS/gas chromatography–mass spectrometry (GC–MS), and analysis were performed as described previously (25,69). Values were normalized to the average abundance in WT samples.
Transmission electron microscopy
Transmission electron microscopy (TEM) tissue preparation and imaging was performed as described previously (70). In brief, upon enucleation, globes were placed in ½ Karnovsky’s fixative (2% formaldehyde (v/v) + 2.5% glutaraldehyde (v/v), in 0.1 M sodium cacodylate buffer, pH 7.4) for the removal of the cornea and lens. Posterior eyecups were fixed for 24 h. After fixation, samples were rinsed with 0.1 M sodium cacodylate buffer, post-fixed with 2% osmium tetroxide (v/v) in 0.1 M sodium cacodylate buffer for 1.5 h, en bloc stained with 2% gadolinium triacetate (v/v) in 0.05 M sodium maleate buffer (pH 6) for 30 min, then dehydrated with graded ethyl alcohol solutions, transitioned with propylene oxide and infiltrated in tEPON-812 epoxy resin (Tousimis, Rockville, Maryland, USA) using an automated EMS Lynx 2 EM tissue processor (Electron Microscopy Sciences, Hatfield, Pennsylvania, USA). The processed samples were oriented in tEPON-812 epoxy resin in flat molds and polymerized using an oven. Semi-thin and ultrathin sections were obtained using a Leica UC7 ultramicrotome (Leica Microsystems, Buffalo Grove, Illinois, USA) and diamond knives (Diatome, Hatfield, Pennsylvania, USA). Semi-thin sections were cut at 1 μm thickness and stained with 1% toluidine blue in 1% sodium tetraborate aqueous solution for assessment by light microscopy. A region containing longitudinally aligned photoreceptor outer and inner segments ~ 1 mm from the optic nerve was selected from each sample from the semi-thin toluidine blue stained sections and block face trimmed to < 1 × 0.5 mm for ultramicrotomy. Ultrathin sections on grids were stained with aqueous 2.5% gadolinium triacetate (v/v) and modified Sato’s lead citrate. Grids were imaged using a FEI Tecnai G2 Spirit transmission electron microscope (FEI, Hillsboro, Oregon, USA) at 80 kV interfaced with an AMT XR41 digital CCD camera (Advanced Microscopy Techniques, Woburn, Massachusetts, USA) for digital TIFF file image acquisition. TEM digital images were captured at a 2 × 2 k pixel 16-bit resolution.
RNA extraction
Mice were euthanized by cervical dislocation and retina and kidney tissue were immediately collected. Two retinas and two kidneys were placed in each tube, respectively. Samples were immediately flash frozen and stored at −80°C until use. For retina samples, samples were thawed on ice, and 500 μL of a solution containing Buffer RLT Plus (Qiagen, 1 053 393), supplemented with 0.5% (v/v) antifoaming Reagent DX (Qiagen, 19 088) and 1% (v/v) β-mercaptoethanol (BME) (Sigma Aldrich, M6250), was added to each tube. Retina samples were homogenized using a rotor homogenizer (Bel-Art, F65000–0000) and disposable RNase-free pestles (Bel-Art, F65000–0002). For kidney samples, a sterile scalpel (Fisher Scientific, NC9999403) was used to slice a small section from one kidney from each mouse. Each section was placed in a 2 mL tube (Qiagen, 990 381), each containing a 5 mm stainless steel bead (Qiagen, 69 989) and 1000 μL of the Buffer RLT Plus solution described above. Kidney samples were homogenized for 30 s at 30 Hz using the Tissue Lyser II (Qiagen, 85 300) system. Samples were then centrifuged at 13 000 g for 1 min at 4°C. Homogenized retina and kidney samples were flash frozen and stored at −80°C until further use. RNA extraction was performed using the QIAsymphony RNA kit (Qiagen, 931 636), as described by the manufacturer, using the RNA CT 400 protocol for the retina samples and the RNA CT 800 protocol for the kidney samples. The quality and quantity of the extracted RNA was assessed using the Quant-iT RNA Assay Kit (Molecular Probes, Q33140) with the Qubit 2.0 fluorometer (ThermoFisher Scientific, Q32866), the Nanodrop (ThermoFisher), and High Sensitivity RNA ScreenTape (Agilent, 5067–5579) with the TapeStation (Agilent).
Library preparation and sequencing
RNA was extracted from both the retinas and kidneys of 8 mice (4 males and 4 females) per genotype per age. Supplementary Material, Table S8 lists the biological replicates, their assigned unique intrinsic sample name and ID, and the RIN number. All samples had RNA integrity numbers (RIN) between 8.6 and 9.7. Spike-in RNA standards, sequins, were used as a reference in all samples (71). Exactly 1 μL of RNA sequins (diluted 1:25 in sterile RNase-free H2O) was added to 50 ng of RNA to a total volume of 50 μL for each sample. Two different sets of sequin mixtures were used, one for Nmnat1V9M/V9M samples and one for WT samples. The NEBNext Ultra II Directional RNA Library Prep with Sample Purification Beads (New England Biolabs, E7765S), NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs, E7490S), and NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1) (New England Biolabs, E7600) kits were used to generate stranded paired-end (PE) libraries, as described in the manufacturer’s protocol. High Sensitivity ScreenTape (Agilent, 5067–5585) was used on the TapeStation (Agilent) to assess library quality. For each genotype, tissue and age, the six best library preps were selected for sequencing, based on library quantity and percent adapter or primer dimer. Libraries were quantified using the KAPA Library Quantification Kit for Illumina Platforms (Roche, KK4875, 07960336001). Each library was normalized to 1.5 nM, pooled and 1% PhiX was added. Libraries were sequenced for 101 cycles (2 × 101bp (PE)) using the NovaSeq 6000 (Illumina) at the Ocular Genomic Institute, MEE, Boston, MA (USA).
RNA-Seq analysis
Read and sample level QC was performed. Read quality was assessed with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and MultiQC (72). An in-house Perl script was used to check and filter out reads with presence of adapter and ambiguous character, ‘N’. The Bowtie2 aligner (73) was used to identify ribosomal contamination. Reads aligned to rRNA reference sequences were dropped from all downstream analysis using SAMtools (74). The STAR aligner (75) was used to align PE reads to a mouse reference in two-pass mode within the sample and across replicates for each sample set. To identify possible sample mixing/mismatch, data normalization and hierarchical clustering was performed with DESeq2 (71) and further confirmed using V9M mutation as a surrogate, post alignment with IGV. featureCounts from the Subread package, was used to generate gene expression matrix with the following non-default settings: reads must be paired, both the pairs must be mapped, use only uniquely mapped reads, multi-mapped reads are not counted, chimeric reads are not counted and strand specificity turned on (76). Anaquin was used to further evaluate alignment sensitivity and gene expression (77). Here sensitivity indicates the fraction of annotated regions covered by alignment. The Picard tools (http://broadinstitute.github.io/picard/) and RSeQC (78) were used to calculate mean fragment length. The approach implemented in Kallisto (79) was used to covert raw reads to TPM values. An average TPM of the lowest Sequins between test and control samples was calculated and used as cutoff. DESeq2 (80) was used for differential gene expression analysis. RNA-seq analysis is described in detail in Mehrotra et al. (81). g:Profiler (Ref, version e101_eg48_p14_baf17f0) (82) with a custom background gene set was used for gene set enrichment analysis (GSEA), pathway enrichment and transcription factor binding site analysis.
Quantitative real-time PCR
RNA was exacted from retina and kidney samples as described above. 8 mice of each genotype, including 4 male and 4 female mice, were used for each age. All samples had RNA integrity numbers (RIN) between 8.6 and 9.7. Reverse transcription was performed using 1000 ng RNA and iScript Reverse Transcription Supermix (Bio-Rad, 1 708 840), according to the manufacturer’s instructions. Real-time PCR was performed using Fast SYBR Green Master Mix (ThermoFisher, 4 385 612) and the QuantStudio 3 real-time PCR system (ThermoFisher Scientific). PCR conditions were as follows: 95°C for 3 min, 95°C for 10 s, 60°C for 30 s, repeat step 2–3 40 times, 95°C for 5 s. Primer sequences are listed in Supplementary Material, Table S9. Relative gene expression was determined using the 2-ΔΔCT method (83), with normalization to Gapdh and to the WT control.
Western blotting
Retina and kidney samples were collected and immediately flash frozen. Upon use, samples were thawed on ice and submerged in a solution containing RIPA buffer (Sigma, R0278), 1X sodium orthovanadate (NEB, P0758S) and 1X protease inhibitor cocktail (Roche, 4 693 132 001). Samples were incubated on ice for 30 min and subsequently sonicated. Samples were centrifuged at 13 500 g for 30 min at 4°C. The supernatant was collected, and the protein concentration was quantified using the Pierce BCA assay (Thermo Scientific, 23 225), according to the manufacturer’s instructions. About, 25 μg of protein was combined with 4X sample buffer (LI-COR, 928–40 004) containing 10% (v/v) BME (Sigma Aldrich, M6250). Samples were incubated at 95°C for 5 min and then loaded into 4–20% Tris–Glycine gels (ThermoFisher, XP04205) and run using 1X Novex Tris–Glycine SDS Running Buffer (ThermoFisher, LC2675) at 120 mV for 1.5 to 2 h. The protein was transferred to a membrane using the iBlot system (ThermoFisher, IB21001), according to the manufacturer’s instructions. The membrane was then blocked for 1 h in Intercept Blocking Buffer (LI-COR, 927–60 001) and incubated with the respective antibodies overnight at 4°C (see Supplementary Material, Table S10). After washing 3 times, for 5 min each wash, in 1X Tris-buffered saline (TBS), blots were incubated with secondary antibodies for 1 h at room temperature (IRDye 800CW Goat anti-Rabbit IgG Secondary Antibody, LI-COR, 926–32 211; IRDye 680RD Goat anti-Mouse IgG Secondary Antibody, LI-COR, 926–68 070). The membrane was then washed 3 times with 1X TBS and imaged using the Odyssey CLx imaging system (LI-COR). For quantification of the bands, the Odyssey Empiria Studio acquisition software (LI-COR) was used.
Immunohistochemistry
After euthanasia, eyes were enucleated using curved forceps and placed in freshly made 4% (v/v) paraformaldehyde (PFA) (Fisher Scientific, RSOF0010250) for 4 min. Eyes were transferred to PBS, and the cornea was removed. The eyes were then fixed in 4% PFA for an additional 20 min. Eyes were washed in PBS and transferred to 30% sucrose (w/v) mixed in 1X PBS overnight. The lens was then removed from the eyecup, and the eyecup was transferred to a disposable base mold containing optical cutting temperature (OCT) medium (Tissue-Tek, 4583). Molds were cryofrozen on ethanol-soaked dry ice and stored at −80°C until cryosectioning. Retinal sections of 10 μm thickness were prepared using a cryostat (Leica, CM 1950) and stored at −20°C until use. Upon use, slides were thawed at room temperature for 10 min. Slides were adhered to Sequenza coverplates (Electron Microscopy Sciences, 71 407–02) using several drops of 1X PBS. Slides were washed with 1X PBS containing 0.5% (v/v) Triton X-100 (0.5% PBS-T) (Millipore Sigma, X100) 3 times, for 5 min each wash. Slides were blocked with 10% normal horse serum (v/v) (Vector Laboratories, S-2000) diluted in 0.5% PBS-T for 1 h at room temperature and subsequently incubated with primary antibodies diluted in fresh blocking solution (see Supplementary Material, Table S3) for 2 h at room temperature. Slides were washed 3 times in 0.5% PBS-T for 5 min each wash, incubated with DAPI for 5 min at room temperature and washed 3 more times in 1X PBS. Slides were mounted in 40% glycerol diluted in 1X PBS (v/v) and cover slipped.
Microscope image acquisition
Images were taken using the Leica SP8 confocal microscope. Images were taken at 60× at room temperature. The Leica LasX software was used for image acquisition and to export images.
Image quantification
Quantification of γH2AX- or 6-fluoro-NAD+-positive nuclei was performed on coded images by a masked observer. The number of foci plotted for each mouse is based on the average number of foci from 3 images of different areas of the retina from the same mouse.
Comet assay
Retina and kidney samples were harvested from mice and immediately flash frozen and stored at −80°C until use. Two retinas and two kidneys were pooled in each tube, respectively. To obtain a single cell suspension, samples were thawed on ice, suspended in a solution containing HBSS (Ca++ and Mg++-free), 10% DMSO (v/v) and 20 mM EDTA, and homogenized using a rotor homogenizer (Bel-Art, F65000–0000) and disposable RNase- and DNase-free pestles (Bel-Art, F65000–0002). The solution was then passed through a 40 μM cell strainer. The single cell suspensions were diluted to a concentration of 1 × 105 cells. Samples were then loaded on a CometChip (43) (Trevigen, 4260–096-CSK) in triplicate, and the comet assay was performed under alkaline conditions, as described in the manufacturer’s protocol. After staining, the chip was imaged using a fluorescent scope (Nikon, Eclipse Ti). Three images of different areas of each well were taken at 10X magnification. Comets were analyzed using the OpenComet plugin for ImageJ (42,84). Comet parameters were averaged between the two groups and compared using unpaired t-tests.
Assessing NAD + -consuming enzymatic activity with 6-Fluoro-NAD +
6-fluoro-NAD+ staining was performed as described previously (45). In brief, after euthanasia, retinas were collected in PBS. The cornea and lens were removed, and unfixed posterior eyecups were embedded in OCT media (Tissue-Tek, 4583) in disposable base molds. OCT blocks were flash frozen and stored at −80°C until use. Frozen blocks were sectioned as described above. Unfixed sections were either (1) preincubated with olaparib and treated with 6-fluoro-NAD+: 100 nM olaparib (Cell Signaling Technology, 93852S) in 100 mM Tris buffer with 0.2% Triton X-100 (v/v), pH 8.0 for 30 min at room temperature + 50 μM 6-Fluo-10-NAD+ diluted in 100 mM Tris buffer with 0.2% Triton X-100 (v/v), pH 8.0 for 2.5 h at 37°C, (2) incubated with 6-fluoro-NAD+: 50 μM 6-Fluo-10-NAD+ diluted in 100 mM Tris buffer with 0.2% Triton X-100 (v/v), pH 8.0 for 2.5 h at 37°C, or (3) incubated with reaction mixture only: 100 mM Tris buffer with 0.2% Triton X-100 (v/v), pH 8.0 for 2.5 h at 37°C.
Graphs and images
GraphPad Prism (Version 9.0) was used to produce graphs and figures. The graphical abstract was adapted from ‘Poly(ADP-Ribose) Metabolism in Trypanosoma Cruzi’, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
Statistics
To assess differences between two groups, unpaired t-tests were used. To assess differences between more than two groups, one-way ANOVA was used. A P-value of 0.05 was considered statistically significant. GraphPad Prism (Version 9.0) was used for statistical analysis.
Study approval
Mice were housed in the Schepens Eye Research Institute Animal Care Facility for the duration of these studies. Mice were given a 4% fat rodent diet and water ad libitum and kept in a room with 12 h cyclic lighting conditions. All procedures were conducted according to the ARVO Statement for Use of Animals in Ophthalmic and Vision Research and the National Institutes of Health Guide for the Care and Use of Laboratory animals (NIH Publications No. 8023, revised 1978) and were approved by the Schepens Eye Research Institute Animal Care and Use Committee.
Abbreviations
- NMNAT1
nicotinamide nucleotide adenylyltransferase 1
- PARP
Poly (ADP-ribose) polymerase
- SARM1
Sterile alpha and TIR motif containing 1
- LC–MS
Liquid chromatography–mass spectrometry
- GC–MS
gas chromatography–mass spectrometry
- IRD
inherited retinal degeneration
Supplementary Material
Acknowledgements
The authors wish to acknowledge Philip Seifert and the Schepens Eye Research Institute Morphology Core for their assistance with the TEM imaging. The authors also wish to acknowledge Daniela Pignatta and the OGI core for their assistance with the RNA-seq. Conflict of Interest statement. The authors have declared that no conflict of interest exists.
Contributor Information
Emily E Brown, Ocular Genomics Institute, Department of Ophthalmology, Massachusetts Eye and Ear, Harvard Medical School, Boston, MA, 02114, USA.
Michael J Scandura, Ocular Genomics Institute, Department of Ophthalmology, Massachusetts Eye and Ear, Harvard Medical School, Boston, MA, 02114, USA.
Sudeep Mehrotra, Ocular Genomics Institute, Department of Ophthalmology, Massachusetts Eye and Ear, Harvard Medical School, Boston, MA, 02114, USA.
Yekai Wang, Department of Ophthalmology and Visual Sciences, West Virginia University, Morgantown, WV, 26506, USA; Department of Biochemistry, West Virginia University, Morgantown, WV, 26506, USA.
Jianhai Du, Department of Ophthalmology and Visual Sciences, West Virginia University, Morgantown, WV, 26506, USA; Department of Biochemistry, West Virginia University, Morgantown, WV, 26506, USA.
Eric A Pierce, Ocular Genomics Institute, Department of Ophthalmology, Massachusetts Eye and Ear, Harvard Medical School, Boston, MA, 02114, USA.
Funding
The National Eye Institute [EY012910 (E.A.P), EY032462, EY031324 (J.D.), P30EY003790 (Core Grant)]
Author Contributions
E.E.B. designed research studies, conducted experiments, acquired data, analyzed data and wrote the manuscript. M.J.S. designed research studies, conducted experiments, acquired data, analyzed data and contributed to writing the manuscript. S.M. designed research studies, acquired data, analyzed data and edited the manuscript. Y.W. conducted experiments, acquired data and analyzed data. J.D. conducted experiments, acquired data, analyzed data and assisted in editing the manuscript. E.A.P. designed research studies and edited the manuscript.
References
- 1. Falk, M.J., Zhang, Q., Nakamaru-Ogiso, E., Kannabiran, C., Fonseca-Kelly, Z., Chakarova, C., Audo, I., Mackay, D.S., Zeitz, C., Borman, A.D. et al. (2012) NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet., 44, 1040–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perrault, I., Hanein, S., Zanlonghi, X., Serre, V., Nicouleau, M., Defoort-Delhemmes, S., Delphin, N., Fares-Taie, L., Gerber, S., Xerri, O. et al. (2012) Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat. Genet., 44, 975–977. [DOI] [PubMed] [Google Scholar]
- 3. Koenekoop, R.K., Wang, H., Majewski, J., Wang, X., Lopez, I., Ren, H., Chen, Y., Li, Y., Fishman, G.A., Genead, M. et al. (2012) Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet., 44, 1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chiang, P.W., Wang, J., Chen, Y., Fu, Q., Zhong, J., Chen, Y., Yi, X., Wu, R., Gan, H., Shi, Y. et al. (2012) Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat. Genet., 44, 972–974. [DOI] [PubMed] [Google Scholar]
- 5. Abad-Morales, V., Wert, A., Ruiz Gomez, M.A., Navarro, R. and Pomares, E. (2021) New insights on the genetic basis underlying SHILCA syndrome: characterization of the NMNAT1 pathological alterations due to compound heterozygous mutations and identification of a novel alternative isoform. Int. J. Mol. Sci., 22, 2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chambon, W., Weill, J.D., Doly, J., Strosser, M.T. and Mandel, P. (1966) On the formation of a novel adenylic compound by enzymatic extracts of liver nuclei. Biochem. Biophys. Res. Commun., 25, 638–643. [Google Scholar]
- 7. Tanny, J.C. and Moazed, D. (2001) Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: evidence for acetyl transfer from substrate to an NAD breakdown product. Proc. Natl. Acad. Sci. U. S. A., 98, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Conforti, L., Janeckova, L., Wagner, D., Mazzola, F., Cialabrini, L., di Stefano, M., Orsomando, G., Magni, G., Bendotti, C., Smyth, N. and Coleman, M. (2011) Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration. FEBS J., 278, 2666–2679. [DOI] [PubMed] [Google Scholar]
- 9. Greenwald, S.H., Charette, J.R., Staniszewska, M., Shi, L.Y., Brown, S.D., Stone, L., Liu, Q., Hicks, W.L., Collin, G.B., Bowl, M.R. et al. (2016) Mouse models of NMNAT1-Leber congenital Amaurosis (LCA9) recapitulate key features of the human disease. Am. J. Pathol., 186, 1925–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gerdts, J., Brace, E.J., Sasaki, Y., DiAntonio, A. and Milbrandt, J. (2015) SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science, 348, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sasaki, Y., Kakita, H., Kubota, S., Sene, A., Lee, T.J., Ban, N., Dong, Z., Lin, J.B., Boye, S.L., DiAntonio, A. et al. (2020) SARM1 depletion rescues NMNAT1-dependent photoreceptor cell death and retinal degeneration. elife, 9, e62027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sasaki, Y., Engber, T.M., Hughes, R.O., Figley, M.D., Wu, T., Bosanac, T., Devraj, R., Milbrandt, J., Krauss, R. and DiAntonio, A. (2020) cADPR is a gene dosage-sensitive biomarker of SARM1 activity in healthy, compromised, and degenerating axons. Exp. Neurol., 329, 113252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu, L., Su, X., Quinn, W.J., 3rd, Hui, S., Krukenberg, K., Frederick, D.W., Redpath, P., Zhan, L., Chellappa, K., White, E. et al. (2018) Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab., 27, 1067–1080.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kaur, J., Mencl, S., Sahaboglu, A., Farinelli, P., van Veen, T., Zrenner, E., Ekstrom, P., Paquet-Durand, F. and Arango-Gonzalez, B. (2011) Calpain and PARP activation during photoreceptor cell death in P23H and S334ter rhodopsin mutant rats. PLoS One, 6, e22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paquet-Durand, F., Silva, J., Talukdar, T., Johnson, L.E., Azadi, S., van Veen, T., Ueffing, M., Hauck, S.M. and Ekstrom, P.A. (2007) Excessive activation of poly(ADP-ribose) polymerase contributes to inherited photoreceptor degeneration in the retinal degeneration 1 mouse. J. Neurosci., 27, 10311–10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sahaboglu, A., Tanimoto, N., Kaur, J., Sancho-Pelluz, J., Huber, G., Fahl, E., Arango-Gonzalez, B., Zrenner, E., Ekström, P., Löwenheim, H., Seeliger, M. and Paquet-Durand, F. (2010) PARP1 gene knock-out increases resistance to retinal degeneration without affecting retinal function. PLoS One, 5, e15495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Komulainen, E., Badman, J., Rey, S., Rulten, S., Ju, L., Fennell, K., Kalasova, I., Ilievova, K., McKinnon, P.J., Hanzlikova, H. et al. (2021) Parp1 hyperactivity couples DNA breaks to aberrant neuronal calcium signalling and lethal seizures. EMBO Rep., 22, e51851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kok, J.R., Palminha, N.M., Dos Santos Souza, C., El-Khamisy, S.F. and Ferraiuolo, L. (2021) DNA damage as a mechanism of neurodegeneration in ALS and a contributor to astrocyte toxicity. Cell. Mol. Life Sci. in press, 78, 5707–5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ainslie, A., Huiting, W., Barazzuol, L. and Bergink, S. (2021) Genome instability and loss of protein homeostasis: converging paths to neurodegeneration? Open Biol., 11, 200296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greenwald, S.H., Brown, E.E., Scandura, M.J., Hennessey, E., Farmer, R., Du, J., Wang, Y. and Pierce, E.A. (2021) Mutant NMNAT1 leads to a retina-specific decrease of NAD+ accompanied by increased poly(ADP-ribose) in a mouse model of NMNAT1-associated retinal degeneration. Hum. Mol. Genet. in press, 30, 644–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ames, A., 3rd, Li, Y.Y., Heher, E.C. and Kimble, C.R. (1992) Energy metabolism of rabbit retina as related to function: high cost of Na+ transport. J. Neurosci., 12, 840–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang, R., Shen, W., Du, J. and Gillies, M.C. (2020) Selective knockdown of hexokinase 2 in rods leads to age-related photoreceptor degeneration and retinal metabolic remodeling. Cell Death Dis., 11, 885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kanow, M.A., Giarmarco, M.M., Jankowski, C.S., Tsantilas, K., Engel, A.L., Du, J., Linton, J.D., Farnsworth, C.C., Sloat, S.R., Rountree, A. et al. (2017) Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. elife, 6, 28899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petit, L., Ma, S., Cipi, J., Cheng, S.Y., Zieger, M., Hay, N. and Punzo, C. (2018) Aerobic glycolysis is essential for normal rod function and controls secondary cone death in retinitis Pigmentosa. Cell Rep., 23, 2629–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grenell, A., Wang, Y., Yam, M., Swarup, A., Dilan, T.L., Hauer, A., Linton, J.D., Philp, N.J., Gregor, E., Zhu, S. et al. (2019) Loss of MPC1 reprograms retinal metabolism to impair visual function. Proc. Natl. Acad. Sci. U. S. A., 116, 3530–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rajala, R.V., Rajala, A., Kooker, C., Wang, Y. and Anderson, R.E. (2016) The Warburg effect mediator pyruvate kinase M2 expression and regulation in the retina. Sci. Rep., 6, 37727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Le Carre, J., Schorderet, D.F. and Cottet, S. (2011) Altered expression of beta-galactosidase-1-like protein 3 (Glb1l3) in the retinal pigment epithelium (RPE)-specific 65-kDa protein knock-out mouse model of Leber's congenital amaurosis. Mol. Vis., 17, 1287–1297. [PMC free article] [PubMed] [Google Scholar]
- 28. Joly, S., Lange, C., Thiersch, M., Samardzija, M. and Grimm, C. (2008) Leukemia inhibitory factor extends the lifespan of injured photoreceptors in vivo. J. Neurosci., 28, 13765–13774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu, B., Sun, X., Suyeoka, G., Garcia, J.G. and Leiderman, Y.I. (2013) TGFbeta signaling induces expression of Gadd45b in retinal ganglion cells. Invest. Ophthalmol. Vis. Sci., 54, 1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu, B., Suyeoka, G., Papa, S., Franzoso, G. and Neufeld, A.H. (2009) Growth arrest and DNA damage protein 45b (Gadd45b) protects retinal ganglion cells from injuries. Neurobiol. Dis., 33, 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kang, S., Larbi, D., Andrade, M., Reardon, S., Reh, T.A. and Wohl, S.G. (2020) A comparative analysis of reactive Muller glia gene expression after light damage and microRNA-depleted Muller glia-focus on microRNAs. Front. Cell Dev. Biol., 8, 620459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Santos-Carvalho, A., Ambrosio, A.F. and Cavadas, C. (2015) Neuropeptide Y system in the retina: from localization to function. Prog. Retin. Eye Res., 47, 19–37. [DOI] [PubMed] [Google Scholar]
- 33. Samardzija, M., Wariwoda, H., Imsand, C., Huber, P., Heynen, S.R., Gubler, A. and Grimm, C. (2012) Activation of survival pathways in the degenerating retina of rd10 mice. Exp. Eye Res., 99, 17–26. [DOI] [PubMed] [Google Scholar]
- 34. Barben, M., Ail, D., Storti, F., Klee, K., Schori, C., Samardzija, M., Michalakis, S., Biel, M., Meneau, I., Blaser, F., Barthelmes, D. and Grimm, C. (2018) Hif1a inactivation rescues photoreceptor degeneration induced by a chronic hypoxia-like stress. Cell Death Differ., 25, 2071–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oeckinghaus, A. and Ghosh, S. (2009) The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol., 1, a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Diallo, M. and Herrera, F. (2021) The role of understudied post-translational modifications for the behavior and function of signal transducer and activator of transcription 3. FEBS J., 16116. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 37. Mancino, A. and Natoli, G. (2016) Specificity and function of IRF family transcription factors: insights from genomics. J. Interf. Cytokine Res., 36, 462–469. [DOI] [PubMed] [Google Scholar]
- 38. Hottiger, M.O., Hassa, P.O., Luscher, B., Schuler, H. and Koch-Nolte, F. (2010) Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci., 35, 208–219. [DOI] [PubMed] [Google Scholar]
- 39. Cohen, M.S. and Chang, P. (2018) Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat. Chem. Biol., 14, 236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Munnur, D. and Ahel, I. (2017) Reversible mono-ADP-ribosylation of DNA breaks. FEBS J., 284, 4002–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Munnur, D., Bartlett, E., Mikolcevic, P., Kirby, I.T., Rack, J.G.M., Mikoc, A., Cohen, M.S. and Ahel, I. (2019) Reversible ADP-ribosylation of RNA. Nucleic Acids Res., 47, 5658–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gyori, B.M., Venkatachalam, G., Thiagarajan, P.S., Hsu, D. and Clement, M.V. (2014) OpenComet: an automated tool for comet assay image analysis. Redox Biol., 2, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sykora, P., Witt, K.L., Revanna, P., Smith-Roe, S.L., Dismukes, J., Lloyd, D.G., Engelward, B.P. and Sobol, R.W. (2018) Next generation high throughput DNA damage detection platform for genotoxic compound screening. Sci. Rep., 8, 2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rogakou, E.P., Pilch, D.R., Orr, A.H., Ivanova, V.S. and Bonner, W.M. (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem., 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- 45. Belhadj, S., Rentsch, A., Schwede, F. and Paquet-Durand, F. (2021) Fluorescent detection of PARP activity in unfixed tissue. PLoS One, 16, e0245369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fong, P.C., Boss, D.S., Yap, T.A., Tutt, A., Wu, P., Mergui-Roelvink, M., Mortimer, P., Swaisland, H., Lau, A., O'Connor, M.J. et al. (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med., 361, 123–134. [DOI] [PubMed] [Google Scholar]
- 47. Martin-Oliva, D., Martin-Guerrero, S.M., Matia-Gonzalez, A.M., Ferrer-Martin, R.M., Martin-Estebane, M., Carrasco, M.C., Sierra, A., Marin-Teva, J.L., Calvente, R., Navascues, J. et al. (2015) DNA damage, poly(ADP-ribose) polymerase activation, and phosphorylated histone H2AX expression during postnatal retina development in C57BL/6 mouse. Invest. Ophthalmol. Vis. Sci., 56, 1301–1309. [DOI] [PubMed] [Google Scholar]
- 48. Mishra, M. and Kowluru, R.A. (2017) Role of PARP-1 as a novel transcriptional regulator of MMP-9 in diabetic retinopathy. Biochim. Biophys. Acta Mol. basis Dis., 1863, 1761–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chiarugi, A. and Moskowitz, M.A. (2003) Poly(ADP-ribose) polymerase-1 activity promotes NF-kappaB-driven transcription and microglial activation: implication for neurodegenerative disorders. J. Neurochem., 85, 306–317. [DOI] [PubMed] [Google Scholar]
- 50. Hassa, P.O. and Hottiger, M.O. (1999) A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol. Chem., 380, 953–959. [DOI] [PubMed] [Google Scholar]
- 51. Morishita, S., Oku, H., Horie, T., Tonari, M., Kida, T., Okubo, A., Sugiyama, T., Takai, S., Hara, H. and Ikeda, T. (2014) Systemic simvastatin rescues retinal ganglion cells from optic nerve injury possibly through suppression of astroglial NF-kappaB activation. PLoS One, 9, e84387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dong, N., Chang, L., Wang, B. and Chu, L. (2014) Retinal neuronal MCP-1 induced by AGEs stimulates TNF-alpha expression in rat microglia via p38, ERK, and NF-kappaB pathways. Mol. Vis., 20, 616–628. [PMC free article] [PubMed] [Google Scholar]
- 53. O'Koren, E.G., Yu, C., Klingeborn, M., Wong, A.Y.W., Prigge, C.L., Mathew, R., Kalnitsky, J., Msallam, R.A., Silvin, A., Kay, J.N. et al. (2019) Microglial function is distinct in different anatomical locations during retinal homeostasis and degeneration. Immunity, 50, 723–737.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yu, C., Roubeix, C., Sennlaub, F. and Saban, D.R. (2020) Microglia versus monocytes: distinct roles in degenerative diseases of the retina. Trends Neurosci., 43, 433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. O'Koren, E.G., Mathew, R. and Saban, D.R. (2016) Fate mapping reveals that microglia and recruited monocyte-derived macrophages are definitively distinguishable by phenotype in the retina. Sci. Rep., 6, 20636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Reyes, N.J., O'Koren, E.G. and Saban, D.R. (2017) New insights into mononuclear phagocyte biology from the visual system. Nat. Rev. Immunol., 17, 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ma, W., Zhang, Y., Gao, C., Fariss, R.N., Tam, J. and Wong, W.T. (2017) Monocyte infiltration and proliferation reestablish myeloid cell homeostasis in the mouse retina following retinal pigment epithelial cell injury. Sci. Rep., 7, 8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Du, J., Rountree, A., Cleghorn, W.M., Contreras, L., Lindsay, K.J., Sadilek, M., Gu, H., Djukovic, D., Raftery, D., Satrustegui, J. et al. (2016) Phototransduction influences metabolic flux and nucleotide metabolism in mouse retina. J. Biol. Chem., 291, 4698–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhu, S., Yam, M., Wang, Y., Linton, J.D., Grenell, A., Hurley, J.B. and Du, J. (2018) Impact of euthanasia, dissection and postmortem delay on metabolic profile in mouse retina and RPE/choroid. Exp. Eye Res., 174, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Roth, S., Rosenbaum, P.S., Osinski, J., Park, S.S., Toledano, A.Y., Li, B. and Moshfeghi, A.A. (1997) Ischemia induces significant changes in purine nucleoside concentration in the retina-choroid in rats. Exp. Eye Res., 65, 771–779. [DOI] [PubMed] [Google Scholar]
- 61. Weiss, E.R., Osawa, S., Xiong, Y., Dhungana, S., Carlson, J., McRitchie, S. and Fennell, T.R. (2019) Broad spectrum metabolomics for detection of abnormal metabolic pathways in a mouse model for retinitis pigmentosa. Exp. Eye Res., 184, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pinson, B., Ceschin, J., Saint-Marc, C. and Daignan-Fornier, B. (2019) Dual control of NAD(+) synthesis by purine metabolites in yeast. elife, 8, 43808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pan, Y.R., Song, J.Y., Fan, B., Wang, Y., Che, L., Zhang, S.M., Chang, Y.X., He, C. and Li, G.Y. (2020) mTOR may interact with PARP-1 to regulate visible light-induced parthanatos in photoreceptors. Cell Commun. Signal, 18, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sahaboglu, A., Barth, M., Secer, E., Amo, E.M., Urtti, A., Arsenijevic, Y., Zrenner, E. and Paquet-Durand, F. (2016) Olaparib significantly delays photoreceptor loss in a model for hereditary retinal degeneration. Sci. Rep., 6, 39537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chang, B., Hawes, N.L., Pardue, M.T., German, A.M., Hurd, R.E., Davisson, M.T., Nusinowitz, S., Rengarajan, K., Boyd, A.P., Sidney, S.S. et al. (2007) Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vis. Res., 47, 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mattapallil, M.J., Wawrousek, E.F., Chan, C.C., Zhao, H., Roychoudhury, J., Ferguson, T.A. and Caspi, R.R. (2012) The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest. Ophthalmol. Vis. Sci., 53, 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wenzel, A., Reme, C.E., Williams, T.P., Hafezi, F. and Grimm, C. (2001) The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J. Neurosci., 21, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ronchi, J.A., Figueira, T.R., Ravagnani, F.G., Oliveira, H.C., Vercesi, A.E. and Castilho, R.F. (2013) A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic. Biol. Med., 63, 446–456. [DOI] [PubMed] [Google Scholar]
- 69. Li, B., Zhang, T., Liu, W., Wang, Y., Xu, R., Zeng, S., Zhang, R., Zhu, S., Gillies, M.C., Zhu, L. and du, J. (2020) Metabolic features of mouse and human retinas: rods versus cones, macula versus periphery, retina versus RPE. iScience, 23, 101672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Seifert, P. (2017) Modified Hiraoka TEM grid staining apparatus and technique using 3D printed materials and gadolinium triacetate tetrahydrate, a non-radioactive uranyl acetate substitute. J. Hist. Dent., 40, 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hardwick, S.A., Chen, W.Y., Wong, T., Deveson, I.W., Blackburn, J., Andersen, S.B., Nielsen, L.K., Mattick, J.S. and Mercer, T.R. (2016) Spliced synthetic genes as internal controls in RNA sequencing experiments. Nat. Methods, 13, 792–798. [DOI] [PubMed] [Google Scholar]
- 72. Ewels, P., Magnusson, M., Lundin, S. and Kaller, M. (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Langmead, B. and Salzberg, S.L. (2012) Fast gapped-read alignment with bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liao, Y., Smyth, G.K. and Shi, W. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–930. [DOI] [PubMed] [Google Scholar]
- 77. Wong, T., Deveson, I.W., Hardwick, S.A. and Mercer, T.R. (2017) ANAQUIN: a software toolkit for the analysis of spike-in controls for next generation sequencing. Bioinformatics, 33, 1723–1724. [DOI] [PubMed] [Google Scholar]
- 78. Wang, L., Wang, S. and Li, W. (2012) RSeQC: quality control of RNA-seq experiments. Bioinformatics, 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- 79. Bray, N.L., Pimentel, H., Melsted, P. and Pachter, L. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol., 34, 525–527. [DOI] [PubMed] [Google Scholar]
- 80. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mehrotra, S. (2020) Evaluating methods for differential gene expression and alternative splicing using internal synthetic controls. bioRxiv in press, August 06, 2020. 10.1101/2020.08.05.238295Arxiv biorxiv;2020.08.05.238295v1 preprint: not peer reviewed. [DOI] [Google Scholar]
- 82. Raudvere, U., Kolberg, L., Kuzmin, I., Arak, T., Adler, P., Peterson, H. and Vilo, J. (2019) G:profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res., 47, W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Livak, K.J. and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 84. Schneider, C.A., Rasband, W.S. and Eliceiri, K.W. (2012) NIH image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.