


Abstract
Imbalance in the finely orchestrated system of chromatin-modifying enzymes is a hallmark of many pathologies such as cancers, since causing the affection of the epigenome and transcriptional reprogramming. Here, we demonstrate that a loss-of-function mutation (LOF) of the major histone lysine methyltransferase SETDB1 possessing oncogenic activity in lung cancer cells leads to broad changes in the overall architecture and mechanical properties of the nucleus through genome-wide redistribution of heterochromatin, which perturbs chromatin spatial compartmentalization. Together with the enforced activation of the epithelial expression program, cytoskeleton remodeling, reduced proliferation rate and restricted cellular migration, this leads to the reversed oncogenic potential of lung adenocarcinoma cells. These results emphasize an essential role of chromatin architecture in the determination of oncogenic programs and illustrate a relationship between gene expression, epigenome, 3D genome and nuclear mechanics.
Graphical Abstract
Graphical Abstract.
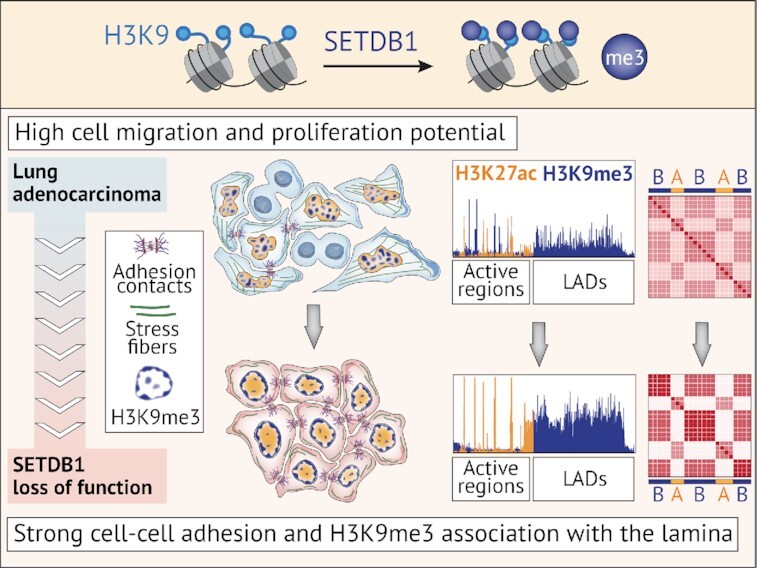
SETDB1 loss-of-function compromises oncogenic potential of lung adenocarcinoma cells decreasing migration and proliferation potential, and affecting 3D genome organization.
INTRODUCTION
The eukaryotic nucleus is separated from the cytoplasm by the nuclear envelope (NE) supported by a network of intermediate filament proteins referred to as lamins (1). Inside the nucleus, the genome is packaged and non-randomly organized to ensure efficient gene expression regulation. High-resolution C-techniques revealed that chromatin fibers fold into a hierarchical structure, including loops, topologically associated domains (TADs), and spatially segregated transcriptionally active (A) and repressive (B) compartments (2). The organized chromatin occupies specific positions relative to the nuclear periphery, with euchromatin shifting away from the nuclear periphery where heterochromatin is enriched (3). Perturbations in the higher-order chromatin organization and disruption of the peripheral heterochromatin pattern promote instability of the overall nuclear organization and are common for many pathologies, including cancer (4–7).
The methylation of histone 3 at lysine positions 9 (H3K9) and 27 (H3K27) is essential for the assembly of constitutive and facultative heterochromatin, respectively. H3K27 methylation is mediated by EZH (enhancer-of-zeste) lysine methyltransferases (KMTs) (8), which belong to the polycomb repressive complex 2 (PRC2), whereas H3K9 is mainly methylated by the KMTs of the SUV39 family composed of SUV39H1/2, G9A, GLP (9,10) and SETDB1 (11). Although in some cases these KMTs can substitute for each other, their functions are not redundant. In contrast to the SUV39H KMTs, which are key in the heterochromatin protein 1 (HP1)-dependent assembly of constitutive pericentromeric heterochromatin and heterochromatin tethering to the nuclear lamina (12), SETDB1 catalyzes mostly H3K9 di- and tri-methylation within the euchromatic regions (11) to repress gene expression and silence retrotransposons and other repetitive elements (13). SETDB1 is essential for the early embryo identity, pluripotency, self-renewal, and terminal differentiation of many progenitor cell types (14–17).
Imbalance in the functions of epigenetic modifiers can affect the global epigenetic status of the cells, causing widespread cascade consequences. The overexpression of SETDB1 is a common feature of different epithelial cancers (18–21). Pro-oncogenic properties of SETDB1 involve the promotion of cell cycle and cell proliferation, migration, and invasive potential of cancer cells (22,23). In addition to H3K9 methylation, SETDB1 might also impact the cancer phenotype by methylation of non-histone proteins, including the tumor suppressor p53 and the kinase AKT (21,24).
A decrease in SETDB1 levels remodels heterochromatin patterns (25,26) and affects the major characteristics of cancer progression, such as proliferation and migration, in various epithelial cancers (27,21,23,26). However, only certain issues have been addressed previously, which makes it difficult to draw a comprehensive model of the outcome of SETDB1 inactivation and gain insights into the underlying molecular mechanisms. Here, we analyzed the consequences of SETDB1 inactivation (LOF mutation) at multiple scales using A549 lung adenocarcinoma cell line as a model of epithelial migratory cancer cells with overexpressed SETDB1 (28). SETDB1 LOF triggers a widespread decrease of H3K9me3 at euchromatic regions with a substantial modulation of the gene expression program, resulting in reduced proliferation and migration of cancer cells. This was accompanied by an unanticipated increase of H3K9me3 in lamina-associated domains (LADs) and overall remodeling of the heterochromatin patterns at the nuclear periphery that led to the enforcement of chromatin compartment spatial segregation and acquisition of non-malignant structural and mechanical properties of the cell nucleus. These results demonstrate an important role of SETDB1 as an organizer of 2D and 3D chromatin architecture, the disbalance of which serves as a trigger for oncogenesis.
MATERIALS AND METHODS
Cell culture
A549 human male lung carcinoma cells (CCL-185) were obtained from ATCC and cultured in Dulbecco's modified Eagle's medium DMEM (Gibco; Cat#: 31966–021) supplemented with 10% fetal bovine serum (FBS; Gibco; Cat#: 10270-106) and 1% penicillin/streptomycin (Gibco; Cat#: 15140-122). Normal human bronchial epithelial (NHBE)-Bronchial Epi Cells for B-ALI from Lonza (CC-2540S) were cultured in PneumaCult-Ex Plus Medium (StemCell; Cat#: 05040) supplemented with 500 μl of Hydrocortisone (StemCell; Cat#: 07925). Cells were maintained at 37°C in the presence of 5% CO2 and were periodically screened for Mycoplasma contamination.
Mice
All animals were bred and maintained in specific pathogen-free facilities in accordance with FELASA and Animal Experimental Ethics Committee guidelines (University of Burgundy, France). This study was complied with all relevant ethical regulations for animal testing and research and received ethical approval from the Animal Experimental Ethics Committee (University of Burgundy, France). Animals had water ad libitum and were fed regular chow. Experiments were performed in 9- to 13-week-old extremely immunodeficient Nod/SCID-Gamma (NSG) mice (JAX stock #005557; Jackson Laboratory; USA) (29). Littermate animals from different cages were randomly assigned into experimental groups and were either co-housed or systematically exposed to other groups’ bedding to ensure equal exposure to common microbiota.
Cell lines establishment
To generate SETDB1 loss-of-function (SETDB1LOF) mutation in A549 cells, optimal sgRNA target sequences, closest to the genomic target sites, were designed using CRISPOR tool (30). The sgRNA were synthesized (Sigma-Aldrich) as oligonucleotide DNA sequences and cloned into pSpCas9(BB)-2A-GFP (PX458) (Addgene; Cat#: 48138) to construct two active CRISPR vectors; one construct targeted SETDB1 gene at exon 15 (SETDB1_sgRNA_ex15: 5′-ACAAACCGGTTGGTGCAACA-3′) and the other one targeted SETDB1 gene at exon 16 (SETDB1_sgRNA_ex16: 5′-GGCGGAGCTATGCTACCCGG-3′). Cloning was performed according to a protocol described by (31). For transfection, A549 cells were seeded into 10 cm dishes at 50–60% confluency and transfected with 10 μg of the appropriate sgRNA-containing PX458 plasmids, using jetOPTIMUS (Polyplus; Cat#: 117-01). The transfection was performed according to the manufacturer's recommended protocol, using a 1:1 ratio of jetOPTIMUS transfection reagent/DNA. At 48 h after transfection, single GFP-positive cells were isolated using FACSDiva version 6.1.3 into a 96-well plate. Genotyping PCRs were performed with Phusion High-Fidelity DNA Polymerase (NEB; Cat#: M0530S), using pair of primers located outside and inside of the targeted sequence. For further experiments, we selected a single A549 SETDB1 LOF clone (#32). Results of RNA-seq results were validated by RT-qPCR for several independent A549 SETDB1 LOF clones (Supplementary Figure S2F, G).
For the rescue experiments, full-length human SETDB1 cDNA was cloned into the NotI sites of the pcDNA3.1(+)-IRES-GFP (Addgene; Cat#: 51406). For transfection, A549 SETDB1LOF cells were seeded into 10 cm dishes at 50–60% confluency and transfected with 10 μg of the pcDNA3.1 (+)-SETDB1WT-IRES-GFP or pcDNA3.1(+)-IRES-GFP (control) plasmids, using jetOPTIMUS. The transfection was performed according to the manufacturer's recommended protocol, using a 1:1 ratio of jetOPTIMUS transfection reagent/DNA. At 48 h after transfection, selected reagent Neomycin (G418) (700 μg/ml; Gibco; Cat#: 10131-035) was added for 2 weeks, and stable cell lines were isolated.
Gene silencing by siRNA
siRNAs targeting human SUV39H1/H2 and negative control siRNAs (32) were from Sigma-Aldrich. Transfections were performed using Lipofectamine RNAiMax (Thermo Fisher; Cat#: 13778150) according to the manufacturer's instructions. Knockdown efficiency after 72 h of RNAi treatment was examined using RT-qPCR.
Immunofluorescence
Cells were seeded on glass coverslips at 60–70% confluence, fixed in 4% paraformaldehyde, incubated with 50 mM NH4Cl to quench formaldehyde, and permeabilized with 0.5% Triton X-100. Samples were subsequently incubated overnight with primary antibody against SETDB1 (Thermo Fisher; Cat#: MA5-15722), H3K9me3 (Abcam; Cat#: ab8898), H3K27me3 (Diagenode; Cat#: C15410069), Lamin A/C (Sigma-Aldrich; Cat#: SAB4200236), E-cadherin (Abcam; Cat#: ab40772), phalloidin-TRITC (Sigma-Aldrich; Cat#: P1951), paxillin (Millipore; Cat#: 05-417) overnight at 4°C, followed by incubation with appropriate fluorophore-conjugated secondary antibodies (Donkey F(ab’)2; Jackson ImmunoResearch Laboratories) for an additional 1 h. Cell nuclei were stained with DAPI (Life Technologies; Cat#: 62248). Coverslips were mounted with Vectashield mounting media (Clinisciences). Microscopy was performed using an inverted microscope Leica DMI-6000, using 40×, 63× or 100× immersion objectives. Images were taken with the HQ2 Coolsnap motorized by MetaMorph 7.10.2.240 software. All images were processed with ImageJ (Fiji) software.
Western blot
Cell lysis was carried out with RIPA lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS) supplemented with FPIC (Fast Protease Inhibitors cocktail; Sigma-Aldrich; Cat#: S8830-20TAB). The lysates were reduced in NuPAGE LDS Sample Buffer (Thermo Fisher; Cat#: NP0007) supplemented with NuPAGE Sample Reducing Agent (Thermo Fisher; Cat#: NP0009), separated by polyacrylamide gel in NuPAGE MOPS SDS Running Buffer (Thermo Fisher; Cat#: NP0001) and transferred into nitrocellulose membrane (Amersham; Cat#: 10600007). Membranes were blocked in 5% milk powder in PBST Buffer (1× PBS, 0.2% Tween 20) and incubated overnight at 4°C with the primary antibodies against SETDB1 (Abcam; Cat#: ab107225), H3 (Santa Cruz Biotechnology; Cat#: sc-8654), H3K9me3 (Abcam; Cat#: ab8898), H3K27me3 (Diagenode; Cat#: C15410069), Lamin A/C (Proteintech; Cat#: 10298-1-AP), Lamin B1 (Abcam; Cat#: ab16048), E-cadherin (BD Transduction Laboratory; Cat#: 610181), Paxillin (Millipore; Cat#: 05-417), β-actin (Sigma-Aldrich; Cat#: A5441). Membranes were incubated with the appropriate LI-COR IRDye secondary antibody (LI-COR Biosciences GmbH) and revealed by Odyssey Fc imaging system (LI-COR Biosciences GmbH). The images obtained from the slot blot assay were analyzed with the Image Studio Lite 5.2.5 software.
Cell proliferation
Cell proliferation analysis was performed with Cell Counting Kit-8 (WST-8/CCK8) (Abcam, Cat#: ab228554) according to the manufacturer's instructions. Cells were seeded in 96-well plates and cultured for 24, 48, 72 and 96 h, respectively. At indicated time points, 10 μl of CCK-8 reagent was added to the wells and incubated for another 1 h. The absorbance at OD490 was measured with the Gen5 microplate reader (BioTek).
Cell cycle analysis
Cells were seeded on 6-well plates at 60–70% confluence. After 48 h, cells were harvested and fixed with 70% cold ethanol overnight at 4°C. Samples were stained with 10 μg/ml propidium iodide (PI) staining solution from FITC Annexin V Apoptosis Detection Kit (BD Pharmingen; Cat#: 556547) and 0.2 mg/ml RNase A (Sigma-Aldrich; Cat#: R4642). Samples of at least 10 000 cells were acquired using a BD FACSCalibur flow cytometer. Subsequent analysis was done with FlowJo 10.7.1 software.
Mitotic index
Mitotic index was calculated as the number of positive cells stained with phospho-H3 (Ser10) (Millipore; Cat#: 05–598) in prometaphase, metaphase, anaphase, or telophase divided by the total number of nuclei stained with DAPI (Life Technologies; Cat#: 62248) × 100. Microscopy analysis was performed using an inverted microscope Leica DMI-6000, using 40× immersion objectives. Images were taken with the HQ2 Coolsnap motorized by MetaMorph 7.10.2.240 software. All images were processed with ImageJ (Fiji) software.
Apoptosis assay
Apoptosis detection was performed by using Annexin V-FITC Apoptosis Detection Kit (BD Pharmingen; Cat#: 556547) according to the manufacturer's instructions. Cells were seeded on six-well plates at 80–90% confluence. After 48 h, cells were harvested, washed twice in cold PBS and then resuspended in 100 μl of a 1× Binding Buffer (10 mM HEPES (pH 7.4), 150 mM NaCl, 2.5 mM CaCl2), stained with 5 μl of FITC Annexin V and 5 μl PI and incubated in the dark for 15 min at room temperature. Then, 400 μl binding buffer was added to the samples and at least 10 000 cells were acquired using a flow cytometer using a BD FACSCalibur flow cytometer. Subsequent analysis was done with FlowJo 10.7.1 software.
Wound-healing and cell migration
Cells were seeded on a glass-bottom petri-dish (Fluorodish; Cat#: FD35-100) with the polydimethylsiloxane (PDMS, Sylgard 184; Dow Corning; Cat#: VAR0000625) coating for live microscopy. PDMS was prepared by mixing base and curing agent in a ratio of 1∶10. PDMS was homogeneously distributed on the surface of the dish to form a thin layer and then incubated at 80°C for 2 h. Some PDMS was poured over a square-shape mold, degassed, and cured at 80°C for 2 h to form square blocks. After curing, PDMS blocks were cut to form hollow 0.5 mm2 square-shape stencils. To allow cell adhesion and migration on the substrate, the Fluorodish surface was coated with 20 μg/ml of Fibronectin (Sigma-Aldrich; Cat#: F0556) diluted in PBS overnight at 4°C. Excess of Fibronectin washed away with PBS and the liquid solution was removed to allow the surface to dry. Once the surface was dried, a hollow square PDMS stencil was placed in the center of the petri dish. Cells were seeded inside the PDMS stencils. A549 control and SETDB1LOF cells were prepared at a 2/3 ratio due to their different proliferation rates, seeded in 2 separate dishes within concentrated drops of media, and incubated at 37°C for 45 min to allow cells to attach on the surface. The medium was then removed, to wash off unattached cells, and cells incubated with fresh medium overnight. After cells had reached confluence inside of the PDMS stencils (usually 24 h after seeding), samples were treated with mitosis-ceasing agent Mitomycin C (20 μg/ml, 1 h at 37°C; Sigma-Aldrich; Cat#: M4287) in order to block proliferation and record only active cell migration. PDMS stencils were then removed to allow cells to migrate away from their pool, and samples were transferred to a Biostation microscope (Nikon). To track cell movements, we used phase-contrast images and recorded multi-positional time-lapse movies. Ten minutes time step was applied between images, and movies were recorded over 16–25 h.
For cell–cell adhesion disruption experiments, we used plastic-bottom petri dishes (Corning; Cat#: 353001), suitable for live microscopy. Petri-dish were functionalized with 20 μg/ml of Fibronectin (Sigma-Aldrich; Cat#: F0556) diluted in PBS overnight at 4°C, and A549 SETDB1LOF cells were plated and treated as described above. EGTA was added to the sample media to disrupt Ca2+-dependent cell-cell adhesions at concentrations as mentioned in Supplementary Figure S1G.
Image processing was performed in the ImageJ (Fiji) program. Data analysis from processed files was performed using particle image velocimetry (PIV) from the MatPIV package (PIVlab v1.41) and implemented in the MATLAB program.
Xenograft studies in NSG mice
A549 control or SETDB1LOF cells (2 × 106) were suspended in 200 μl of mixture Dulbecco-modified phosphate buffered saline (DPBS, Sigma-Aldrich, Cat#: 59331C) and BD Matrigel Matrix (BD Biosciences; Cat#: 356234) in a ratio of 1∶1, and injected subcutaneously into the right flanks of NSG mice. Tumor growth was monitored every third day using digital calipers. Tumor size was calculated as: Tumor_volume = (length × width2)/2. Mice were euthanized at the end of the experiment, tumors were dissected and fixed in 10% formalin for subsequent immunohistochemical analysis.
Immunohistochemistry
Formalin-fixed paraffin-embedded (FFPE) tumor sections (5 μm) from A549 tumors were used for hematoxylin and eosin (H&E) staining and proliferation analysis with anti-Ki67 antibody (Leica Biosystems; Cat#: NCL-Ki-67p). Briefly, sections were deparaffinized, incubated overnight at 4°C with primary antibodies, washed, and incubated with secondary antibodies HRP Labelled Polymer Anti-Rabbit (Dako EnVision + System; Cat#: K4002) following the manufacturer's protocol. Slides were counterstained with hematoxylin counterstain and coverslips were mounted using non-aqueous mounting media. Images were scanned at 20× magnification using the Aperio Scanscope system.
RNA and quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was extracted from the cell lines or frozen tissue samples using RNeasy Mini Kit (Qiagen; Cat#: 74104). DNase treatment was performed using RNase-Free DNase Set (Qiagen; Cat#: 79254) to remove residual DNA. cDNA was prepared with a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher; Cat#: 4368814). Real-time quantitative PCR (RT-qPCR) was performed with GoTaq qPCR Master Mix (Promega; Cat#: A6002). Gene expression analysis was calculated following normalization to PPAI using the comparative Ct (cycle threshold) method. The primers used are provided in Supplementary Table S1.
RNA-seq
Total RNA was isolated using RNeasy Mini Kit (Qiagen; Cat#: 74104) followed by Turbo DNA-free Kit (Thermo Fisher; Cat#: AM1907). To construct libraries, 1 μg of high-quality total RNA (RIN > 9.9) was processed with Truseq Stranded Total RNA Library Prep Kit (Illumina; Cat#: 20020596) according to the manufacturer's instructions. Briefly, after removal of ribosomal RNA using Ribo-zero rRNA Kit (Illumina; Cat#: 20037135), confirmed by quality control on the Agilent 2100 Bioanalyzer, total RNA molecules were fragmented and reverse-transcribed using random primers. Replacement of dTTP by dUTP during the second strand synthesis permitted to achieve strand specificity. The addition of a single A base to the cDNA was followed by ligation of adapters. Libraries were quantified by qPCR using the KAPA Library Quantification Kit for Illumina Libraries (KapaBiosystems; Cat#: KR0405) and library profiles were assessed using the DNA High Sensitivity HS Kit (Agilent; Cat#: 5067-4626) on an Agilent Bioanalyzer 2100. Libraries were sequenced on an Illumina Nextseq 500 in a paired-end mode in three independent biological replicates at Platform GENOM’IC - Institute Cochin. Demultiplexing and quality of sequences were performed with the Aozan software v2.2.1.
Chromatin immunoprecipitation (ChIP)
Native chip (NChIP)
Around 1 × 107 cells were collected, washed with PBS and resuspended in 250 μl of ice-cold Douncing buffer (10 mM Tris–HCl (pH 7.5), 4 mM MgCl2, 1 mM CaCl2, 0.2% Triton X100), followed by homogenization with syringe with 26 gauge X1/2′ needles (BD Microlance; Cat#: BD 303800). To obtain mononucleosomes, cells were digested with 1.25 μl of micrococcal nuclease (Mnase) (0.5 U/μl; Sigma-Aldrich; Cat#: N3755-500UN) for 5 min at 37°C with 600 rpm. The reaction was stopped with 10 mM EDTA, pH 8. Nuclei were swollen to release chromatin after addition of Hypotonization buffer (0.2 mM EDTA (pH 8), FPIC (Fast Protease Inhibitors cocktail; Sigma-Aldrich; Cat#: S8830-20TAB), DTT). Chromatin corresponding to 8 μg of DNA was incubated with anti-H3K9me3 (Diagenode; Cat#: C15410193), anti-H3K27me3 (Diagenode; Cat#: C15410069), anti-H3K27ac (Diagenode; Cat#: C15410196) or anti-H3K4me3 (Diagenode; Cat#: C15410003) antibodies overnight at 4°C. The DNA–protein–antibody complexes were captured by DiaMag protein A-coated magnetic beads (Diagenode; Cat#: C03010020) by incubating at 4°C for 2 h. Magnetic beads were then washed with Low-Salt buffer (20 mM Tris–HCl (pH 8), 0.1% SDS, 1% Triton X100, 2 mM EDTA (pH 8), 150 mM NaCI and FPIC) and High-Salt buffer (20 mM Tris–HCl (pH 8), 0.1% SDS, 1% Triton X100, 2 mM EDTA (pH 8), 500 mM NaCI and FPIC). DNA was eluted with TE buffer (100 mM Tris–HCl (pH 8), 1% SDS, 1 mM EDTA (pH 8)) from the beads overnight, inputs corresponding to 1% of IP were treated as well, followed by incubation with 1 μl of RNase A (10 μg/ml; Sigma-Aldrich; Cat#: R4642) for 1 h. DNA was purified using MinElute PCR Purification Kit (Qiagen; Cat#: 28004).
Cross-linked ChIP (XChIP)
Around 1 × 107 cells were cross-linked directly in the culture plate with culture medium supplemented with 1% of formaldehyde (Sigma-Aldrich; Cat#: F8775) and 15 mM NaCl, 0.15 mM EDTA (pH 8), 0.075 mM EGTA and 0.015 mM HEPES (pH 8) during 10 min at RT. Formaldehyde was quenched with 0.125 M glycine and cells were washed in PBS. Fixed cells were prepared with buffers from Chromatin Shearing Optimization Kit, low SDS (Diagenode; Cat#: C01020013) for sonication for 5 min (30 s ON, 30 s OFF) (Bioruptor Diagenode), yielding genomic DNA fragments with a bulk size of 150–600 bp. 1% of chromatin extracts were taken aside for inputs. Chromatin corresponding to 8 μg of DNA was incubated with anti-CTCF (Active Motif; Cat#: 61311) antibody overnight at 4°C. The DNA-protein-antibody complexes were captured by DiaMag protein A-coated magnetic beads (Diagenode; Cat#: C03010020) by incubating at 4°C for 2 h. Magnetic beads were then washed with Low-Salt buffer (20 mM Tris–HCl (pH 8), 0.1% SDS, 1% Triton X100, 2 mM EDTA (pH 8), 150 mM NaCI and FPIC) and High-Salt buffer (20 mM Tris–HCl (pH 8), 0.1% SDS, 1% Triton X100, 2 mM EDTA (pH 8), 500 mM NaCI and FPIC). DNA was eluted with TE buffer (100 mM Tris–HCl (pH 8), 1% SDS, 1 mM EDTA (pH 8)) from the beads overnight, and then reverse cross-linked with 1 μl of RNase A (10 μg/ml; Sigma-Aldrich; Cat#: R4642), followed by Proteinase K digestion (5 μl of 20 mg/mL; Sigma-Aldrich; Cat#: P2308) during 2 h; inputs corresponding to 1% of IP were reverse cross-linked in the same conditions. DNA was purified using MinElute PCR Purification Kit (Qiagen; Cat#: 28004).
ChIP-seq
Libraries were prepared using the MicroPlex V2 library preparation Kit (Diagenode; Cat#: C05010012), following the manufacturer's instructions, 10 μl of DNA was used as starting material. After end repair of the double-stranded DNA templates, cleavable stem-loop adaptors were ligated, and then adaptor enriched DNA was amplified with 10 PCR cycles to add Illumina compatible indexes. The libraries were then purified with the Agencourt AMPure XP bead-based purification system. Final libraries were quantified with Qubit dsDNA HS assay (Thermo Fisher; Cat#: Q32851), and size distribution was monitored by capillary electrophoresis on Agilent 2100 Bioanalyzer using DNA High Sensitivity HS Kit. Libraries were then normalized to 10 pM and pooled before sequencing. Libraries were sequenced on an Illumina Nextseq 500 in a paired-end mode in two independent biological replicates at Platform GENOM’IC—Institute Cochin. Demultiplexing and quality of sequences were performed with the Aozan software v2.2.1.
Hi-C
Hi-C libraries were performed as described in (33). 10 million cells were cross-linked with fresh 1% formaldehyde (Sigma-Aldrich; Cat#: F8775) for 10 min at room temperature. Excess of formaldehyde was quenched with 125 mM glycine for 5 min. Cells were centrifuged (1000 × g, 10 min, 4°C), resuspended in 50 μl of 1 × PBS, snap-frozen in liquid nitrogen, and stored at -80°C. Defrosted cells were lysed in 1.5 ml of Isotonic buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.5% (v/v) NP-40 substitute, 1% (v/v) Triton-X100, 1× Protease Inhibitor Cocktail (Bimake; Cat#: B14001)) on ice for 15 min. Cells were centrifuged at 2500 × g for 5 min, resuspended in 100 μl of 1.1× DpnII buffer (NEB; Cat#: B0543S), and pelleted again. The pellet was resuspended in 200 μl of 0.3% SDS in 1.1× DpnII buffer and incubated at 37°C for 1 h. Then, 330 μl of 1.1× DpnII buffer and 53 μl of 20% Triton X-100 were added, and the suspension was incubated at 37°C for 1 h. Next, 600 U of DpnII enzyme (NEB; Cat#: R0543M) were added, and the chromatin was digested overnight (14–16 h) at 37°C with shaking (1400 rpm). On the following day, 200 U of DpnII enzyme were added, and the cells were incubated for an additional 2 h. DpnII was then inactivated by incubation at 65°C for 20 min. After DpnII inactivation, the nuclei were harvested for 10 min at 5000 × g, washed with 100 μl of 1× NEBuffer 2.1 (NEB; Cat#: B7202S), and resuspended in 50 μl of 1× NEBuffer 2.1. Cohesive DNA ends were biotinylated by the addition of 7.6 μl of the biotin fill-in mixture prepared in 1× NEBuffer 2.1 (0.025 mM dCTP (Thermo Fisher; Cat#: R0151), 0.025 mM dGTP (Thermo Fisher; Cat#: R0161), 0.025 mM dTTP (Thermo Fisher; Cat#: R0171), 0.025 mM biotin-14-dATP (Invitrogen; Cat#: 19524-016), and 0.8 U/μl Klenow enzyme (NEB; Cat#: M0210L). The samples were incubated at 37°C for 75 min with shaking (1400 rpm). Nuclei were centrifuged at 3000 × g for 5 min, resuspended in 100 μl of 1× T4 DNA ligase buffer (Thermo Fisher; Cat#: EL0011), and pelleted again. The pellet was resuspended in 300 μl of 1× T4 DNA ligase buffer, and 75 U of T4 DNA ligase (Thermo Fisher; Cat#: EL0011) were added. Chromatin fragments were ligated at 20°C for 6 h. The cross-links were reversed by overnight incubation at 65°C in the presence of Proteinase K (100 μg/ml; Sigma-Aldrich; Cat#: P2308). After cross-link reversal, the DNA was purified by single phenol-chloroform extraction followed by ethanol precipitation with 20 μg/ml glycogen (Thermo Scientific; Cat#: R0561) as the co-precipitator. After precipitation, the pellets were dissolved in 100 μl of 10 mM Tris–HCl (pH 8.0). To remove residual RNA, samples were treated with 50 μg of RNase A (Thermo Fisher; Cat#: R1253) for 45 min at 37°C. To remove residual salts and DTT, the DNA was additionally purified using Agencourt AMPure XP beads (Beckman Coulter; Cat#: A63881). Biotinylated nucleotides from the non-ligated DNA ends were removed by incubating the Hi-C libraries (2 μg) in the presence of 6 U of T4 DNA polymerase (NEB; Cat#: M0203L) in NEBuffer 2.1 supplied with 0.025 mM dATP (Thermo Fisher; Cat#: R0141) and 0.025 mM dGTP at 20°C for 4 h. Next, the DNA was purified using Agencourt AMPure XP beads. The DNA was then dissolved in 500 μl of sonication buffer (50 mM Tris–HCl (pH 8.0), 10 mM EDTA, 0.1% SDS) and sheared to a size of ∼100–500 bp using a VirSonic 100 (VerTis). The samples were concentrated (and simultaneously purified) using AMICON Ultra Centrifugal Filter Units (Millipore; Cat#: UFC503096) to a total volume of approximately 50 μl. The DNA ends were repaired by adding 62.5 μl MQ water, 14 μl of 10× T4 DNA ligase reaction buffer, 3.5 μl of 10 mM dNTP mix (Thermo Fisher; Cat#: R0191), 5 μl of 3 U/μl T4 DNA polymerase, 5 μl of 10 U/μl T4 polynucleotide kinase (NEB; Cat#: M0201L), 1 μl of 5 U/μl Klenow DNA polymerase, and then incubating at 25°C for 30 min. The DNA was purified with Agencourt AMPure XP beads and eluted with 50 μl of 10 mM Tris–HCl (pH 8.0). To perform an A-tailing reaction, the DNA samples were supplemented with 6 μl 10× NEBuffer 2.1, 1.2 μl of 10 mM dATP, 1 μl of MQ water, and 3.6 μl of 5 U/μl Klenow (exo-) (NEB; Cat#: M0212S). The reactions were carried out for 45 min at 37°C in a PCR machine, and the enzyme was then heat-inactivated by incubation at 65°C for 20 min. The DNA was purified using Agencourt AMPure XP beads and eluted with 100 μl of 10 mM Tris–HCl (pH 8.0). Biotin pulldown of the ligation junctions: 15 μl of MyOne Dynabeads Streptavidin C1 (Invitrogen; Cat#: 65001) beads washed with the TWB buffer (5 mM Tris–HCl (pH 8.0), 0.5 mM EDTA (pH 8.0), 1 M NaCl, 0.05% Tween-20), resuspended in 200 μl of 2× Binding buffer (10 mM Tris–HCl (pH 8.0), 1 mM EDTA, 2 M NaCl) and added to 200 μl of DNA. Biotin pulldown was performed for 30 min at 25°C with shaking. Next, beads with captured ligation junctions were washed once with TWB buffer, once with 1× T4 DNA ligase buffer, and then resuspended in 50 μl of adapter ligation mixture comprising 41.5 μl MQ water, 5 μl 10× T4 DNA ligase reaction buffer, 2.5 μl of Illumina TruSeq adapters, and 1 μl of 5 U/μl T4 DNA ligase. Adapter ligation was performed at 22°C for 2.5 h, and the beads were sequentially washed twice with 100 μl of TWB, once with 100 μl of CWB (10 mM Tris–HCl (pH 8.0) and 50 mM NaCl), and then resuspended in 25 μl of MQ water. Test PCR reactions containing 4 μl of the streptavidin-bound Hi-C library were performed to determine the optimal number of PCR cycles required to generate sufficient PCR products for sequencing. The PCR reactions were performed using KAPA High Fidelity DNA Polymerase (Kapa Biosystems; Cat#: 08201595001) and Illumina PE1.0 and PE2.0 PCR primers (10 pmol each). The temperature profile was 5 min at 98°C, followed by 6, 9, 12, 15 and 18 cycles of 20 s at 98°C, 15 s at 65°C and 20 s at 72°C. The PCR reactions were separated on a 2% agarose gel containing ethidium bromide, and the number of PCR cycles necessary to obtain a sufficient amount of DNA was determined based on the visual inspection of gels (typically 8–12 cycles). Four preparative PCR reactions were performed for each sample. The PCR mixtures were combined, and the products were purified with Agencourt AMPure XP beads. Libraries were sequenced on an Illumina Novaseq 6000 in a paired-end mode in two independent biological replicates at Genetico.
Chromatin mobility
Cells were plated in Fluorodishes (Worldprecision; Cat#: FD35100) at the rate of 2 × 105 cells per dish the day before performing the live imaging. Thirty minutes before live imaging, the medium was replaced with 1 ml of fresh medium with two drops of NucBlue LiveProbe reagent (Thermo Fisher; Cat#: R37605). Live imaging was performed on a Leica DMI8 microscope, equipped with a CSU-X1 Yokogawa spinning disk module. The acquisition was realized with a 40× dry objective (N.A. 1.40) and collected by the Hamamatsu Orca flash 4.0 camera. The microscope was controlled by the Metamorph software v7.10.2.240 from Molecular Devices. Images were acquired in brightfield (one plane, exposure time 50 ms) with a 405 nm laser at 15% power (Z stack, seven planes centered on the middle plane of the nuclei, Z step of 0.5 μm, exposure time 150 ms) every 2 min for 1.5–2 h. Analysis was performed on the best Z plane chosen for each stage position, using a set of custom-written Fiji scripts. Briefly, the nuclei were segmented and registered for XY translation and rotation using the MultiStackReg plugin. The chromatin flow was then measured using the PIV plugin on the first 10 time points, restricted to the nuclei. The PIV magnitude value was obtained by averaging the values measured for the entire nucleus over the 10 time points.
Nuclear envelope fluctuations
Cells were transfected with the pEGFP-LAP2b (Euroscarf plasmid bank; Cat#: P30463) plasmid 24 h prior to imaging using jetOPTIMUS. The transfection was performed according to the manufacturer's recommended protocol, using a 1:1 ratio of jetOPTIMUS transfection reagent/DNA. Thirty minutes before live imaging, the medium was replaced with 1 ml of fresh medium with two drops of NucBlue LiveProbe reagent. Live imaging was performed on a Leica DMI8 microscope, equipped with CSU-X1 Yokogawa spinning disk module. The acquisition was realized with a 63× oil objective (N.A. 1.40) and collected by the Hamamatsu Orca flash 4.0 camera. The microscope was controlled by the Metamorph software v7.10.2.240 from Molecular Devices. Snapshots were first acquired in brightfield (one plane, exposure time 50 ms) with a 488 nm laser at 2% (one plane, exposure time 200 ms) and with a 405 nm laser at 15% power (one plane, exposure time 150 ms). Movies at high temporal resolution were then acquired with a 488 nm laser at 2% (exposure time 200 ms) at the rate of 1 frame every 250 ms for 700 timeframes (1.75 min). Analysis was performed with Fiji and Python with custom-written scripts. Briefly, the nuclei in the 488 nm channel were registered for XY translation and rotation using the StackReg plugin. For each nucleus, four lines were hand-drawn on the nuclear envelope (NE) and resliced over time to obtain 4 kymographs, one for each individual point in the NE. A custom-written Python script was used to determine the position of the NE at each timepoint of each kymograph. For each timepoint, a parabola was fitted on the pixel with the highest intensity and its 10 neighboring pixels. The maximum of the parabola was assigned as the NE position. For each kymograph, the average NE position was determined by averaging the successive NE positions. The square root of the Mean Square Displacement was calculated using the displacements calculated for each time point as the distance to the average NE position. Timepoints with displacements larger than twice the average displacement were interpreted as NE positioning errors and excluded from the analysis. The values per cell were obtained by averaging the values of the corresponding kymographs.
Immunofluorescence analysis
E-cadherin
Images were processed using the ‘rolling ball’ background subtraction method implemented in Fiji (rolling ball radius set to 400). Mean junctional E-cadherin intensity was calculated by measuring the average intensity inside fixed-width regions of interest (ROIs) placed around all visible cellular junctions in the field (∼20–50 junctions per image), excluding nodes. At least four fields were analyzed for each biological replicate. Images from three independent experiments were analyzed.
Paxillin
Images were processed using the ‘rolling ball’ background subtraction method implemented in Fiji (rolling ball radius set to 50). Normalized paxillin area was calculated by dividing the total paxillin-positive area by the number of nuclei in the field. To measure the total paxillin-positive area, the paxillin-stained images were thresholded above the intensity value, similar for all of the images in the batch, so that no cytosolic background was present. The nuclei were counted using the corresponding images with the DAPI-staining. The measurements were performed for at least four fields per biological replicate and the mean values for all of the fields for each replicate were retained. Image processing was looped using a custom-written macro.
F-actin
The Phalloidin (F-actin) distribution quantification was performed using the method adopted from (34). Images were processed using the ‘rolling ball’ background subtraction method implemented in Fiji (rolling ball radius set to 50), and the ROIs around all fully visible cells in the field were manually outlined. The mean Phalloidin intensity was measured inside of each ROI. Each of the cells was then treated as a separate image with the rest of the pixels outside of the ROI set to zero. Using the Fit Ellipse function, implemented in Fiji, the center of mass of each cell was detected and the line, passing through the center of mass and the frontier of the cell with the maximum Phalloidin intensity, was drawn. Using the macro, adapted from Zonderland et al. (34), the Phalloidin intensity profile along the line was obtained with the Fiji getProfile function, the line was trimmed on both sides by the first non-zero intensity value and then divided into 10 equal-size bins. The average intensity for all values inside each bin was calculated and the process was repeated for all of the cells in at least 4 fields for each biological replicate.
LaminA/C, H3K9me3 and H3K27me3 immunofluorescence
The images were subjected to background subtraction (rolling ball radius 50) and the ROIs around all fully visible nuclei were outlined. All ROIs were then analysed using the Clock Scan protocol (35) implemented as a Fiji Plugin. Briefly, the plugin sequentially collected multiple radial pixel-intensity profiles, scanned from the ROI center to the predetermined distance outside of the ROI border. The profiles were scaled according to the measured ROI radius, so that the distance from the center of the ROI to its border was represented in % of radius. The individual profiles were averaged into one integral radial pixel-intensity profile for each nucleus. The intensity determined outside of the ROI border (at a distance 100–120% of radius) was used for background correction.
The intensities were measured separately for the H3K9me3, H3K27me3, and Lamin A/C channels. The measurements were performed for 30–35 nuclei per biological replicate (92–104 nuclei per condition in total). All measured intensity values at each distance from the nucleus center were averaged and the mean intensity values (y) at each distance (x) were plotted to obtain the intensity distributions (у(x)). To minimize the effect of the difference in the nuclear shape on the intensity distribution functions were scaled: the radius was measured from the center of mass of the nuclei, and expressed in relative and not absolute values, allowing us to correct for the irregular shape of certain nuclei. The Kolmogorov–Smirnov test was used to compare control and A549 SETDB1LOF distribution functions.
Nuclear morphology assessment
Nuclear morphology was assessed using the images in the DAPI channel. Fully visible nuclei were outlined, and their area, perimeter, circularity and solidity were measured with Fiji. Circularity was defined as 4π × area/perimeter2, with a value of 1 indicating a perfect circle, and the values approaching 0 indicating a largely elongated shape. Solidity was defined as a ratio area/convex_area, with a value of 1 indicating a convex object, and values <1 indicating an invaginated object with an irregular boundary. To quantify the deviation of the nuclear shape from the ellipse, the irregularity index (defined as 1 – solidity) was used (36), with a value of 0 indicating a regular-shaped elliptical nuclei, and higher for nuclei with invaginated borders. The measurements were performed for 30–35 nuclei per biological replicate (92–104 nuclei per condition in total).
Immunohistochemistry analysis
The percentage of Ki-67 positive nuclei was determined using digital images of the tumor section by examining 8 fixed-area (500 000 mkm2) hot-spot fields. Hot spots were defined as areas in which Ki-67 staining was particularly higher relative to the adjacent areas. For each field the ratio of Ki-67-positive nuclei to the total number of nuclei was calculated. The fields contained 1500–2500 cells in total.
RNA-seq analysis
RNA-seq reads mapping and quantification
RNA-seq reads were mapped to the hg19 reference human genome assembly using STAR v2.6.1c (37) with GENCODE v33 (GRCh37) gene annotation (38). Read counts per gene were obtained using the ‘–quantMode’ parameter in STAR. Uniquely mapped reads with MAPQ > 30 were selected using SAMtools v1.5 (39). Reads overlapping with the hg19 blacklist regions (40) were discarded using bedtools v2.25.0 (41). The bigwig files with the CPM-normalized transcription signal in 50 bp bins were generated for merged replicates using bamCoverage function from deepTools v3.3.0 (42). Visualization of the signal profiles was created in the UCSC Genome Browser (43). Statistics of the RNA-seq data processing can be found in Supplementary Table S2A.
Differential gene expression
Differentially expressed genes were identified with edgeR v3.30.0 R package (44). Genes with low expression counts were filtered (filterByExp function), and the remaining genes were normalized using the trimmed mean of M values (TMM) method (45). To check the reproducibility between the replicates, a multidimensional scaling (MDS) plot of distances between the gene expression profiles was generated (46). Quasi-likelihood negative binomial generalized log-linear model (glmQLFit function) (47) and t-test relative to a fold change threshold of 1.5 (glmTreat function) (48) were used to calculate P-values per gene. The obtained P-values were adjusted with Benjamini–Hochberg correction for multiple testing (FDR). FDR threshold of 0.01 was used to define differentially expressed genes (Supplementary Table S3A).
Gene ontology analysis
Gene ontology (GO) analysis of differentially expressed genes was performed using GO term over-representation test from g:Profiler (49). The annotation of genes with GO terms from Ensembl (release 100) was utilized, and electronic GO annotations were removed prior to the analysis. FDR threshold of 0.05 was applied, and significant GO terms of size between 25 and 1500 were extracted (Supplementary Table S3B).
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was performed with GSEA v4.0.3 (50) in pre-ranked mode with 10 000 permutations (Supplementary Table S3C). Genes were pre-ranked by fold change derived from the differential expression analysis. Gene sets of size between 25 and 1500 from MSigDB v7.1 (51) were used.
Gene set variation analysis
Gene set variation analysis (GSVA) was performed using the GSVA v1.36.2 R package (52). Publicly available RNA-seq datasets of normal lung (N = 48) and NSCLC (N = 61) cell lines were used as a background. The datasets were downloaded from the GEO database (GEO accession numbers: GSE63900, GSE93526, GSE85447, GSE101993, GSE97036, GSE42006, GSE152446, GSE113185, GSE80386, GSE148729, GSE109821, GSE123769, GSE141666, GSE123631, GSE113493, GSE150809, GSE74866, GSE99015, GSE79051, GSE107637, GSE86337, GSE78531, GSE78628, GSE61955 and processed as described above (section ‘RNA-seq Reads Mapping and Quantification’) to obtain read counts per gene. Lung-specific gene set was extracted from the Human Protein Atlas v20.0 database (53), other gene sets were retrieved from the MSigDB v7.1 database. Ensembl gene identifiers were converted to Entrez using Biomart (54) with Ensembl genes (release 100). GSVA scores were estimated for log-transformed CPM gene counts for each sample (Supplementary Table S3D).
Repetitive elements expression analysis
RepEnrich2 (55) (https://github.com/nerettilab/RepEnrich2) was used to determine the expression levels of repetitive elements from the hg19 RepeatMasker annotation. First, RNA-seq reads were mapped to the hg19 reference human genome assembly using Bowtie v2.2.3 (56). Second, unique and multi-mapped reads were separated by a MAPQ cutoff of 30. Finally, fraction counts were calculated for each repetitive element. Differential expression analysis was performed with edgeR v3.30.0 R package. Library size was defined as Nreads_processed − Nreads_aligned_0_times, based on the log files from Bowtie2. Negative binomial generalized log-linear model (glmFit function) and likelihood ratio test (glmLRT function) (44) were used to calculate P-values per repetitive element. The obtained P-values were adjusted with Benjamini–Hochberg correction for multiple testing. FDR threshold of 0.05 was used to define differentially expressed repetitive elements.
ChIP-seq analysis
ChIP-seq reads mapping and normalization
ChIP-seq reads were mapped to the hg19 reference human genome assembly using Bowtie v2.2.3 with ‘–no-discordant’ and ‘–no-mixed’ options. Uniquely mapped reads with MAPQ >30 were selected using SAMtools v1.5 for further analysis. PCR duplicates were filtered out using the Picard v2.22 MarkDuplicates function. Reads overlapping with the hg19 blacklist regions were discarded using bedtools v2.25.0. The bigWig files with the ratio of RPKM-normalized ChIP-seq signal to the input in 50 bp bins were generated using deepTools v3.3.0 with pseudocount set to ‘0 1’ and smoothing window of three bins. Visualization of the signal profiles were created in the UCSC Genome Browser. Statistics of the ChIP-seq data processing can be found in Supplementary Table S2B.
Peak calling
Peak calling was performed with MACS2 v2.2.7.1 (57). For histone modifications (H3K4me3, H3K27ac, H3K9me3, H3K27me3) ChIP-seq, peaks were called both for replicates and pooled data. For H3K4me3 and H3K27ac, MACS2 was executed in a narrowPeak mode with tag size of 40 or 41 bp, mappable genome size set to ‘hs’, and q-value cutoff of 0.05. For H3K9me3 and H3K27me3, MACS2 was executed in a broadPeak mode with tag size of 40 or 41 bp, mappable genome size set to ‘hs’, and broad-cutoff of 0.1. The consensus peak list was obtained by overlapping the peaks, annotated for pooled replicates, with peaks from both replicates. Only the pooled peaks from canonical chromosomes that had an overlap of at least 50% with peaks from both replicates were retained. For CTCF ChIP-seq, peaks were called using the pooled data in a narrowPeak mode with tag size of 40 bp, mappable genome size set to ‘hs’, and P-value cutoff of 0.001. No additional peak calling filters were applied to the obtained CTCF peaks.
Super-enhancers annotation
To identify the super-enhancers, we used rank ordering of super-enhancers (ROSE) approach (58). For ROSE, H3K27ac peaks within 12.5 kb from one another were stitched into continuous enhancer clusters, within which the density of input-normalized H3K27ac ChIP-seq signal was calculated. These stitched regions were then ranked based on the total signal density, and plotted to geometrically define the cutoff. The cutoff was set at a point where a line with a slope of 1 was tangent to the curve, and regions above the curve were defined as super-enhancers (Supplementary Table S4A).
Differential enrichment analysis
DiffBind v2.16.0 (59) was utilized to analyze the differential enrichment of the H3K9me3 and H3K27ac ChIP-seq peaks, and differentially regulated super-enhancers. To analyze heterochromatin domains and continuous enhancer clusters, peaks within 12.5 kb from one another for H3K9me3 and H3K27ac data were stitched together (60). Stitched ChIP-seq peaks and annotated super-enhancers were pooled together for control and A549 SETDB1LOF conditions and used for differential enrichment analysis. DiffBind pipeline was executed with default parameters. FDR threshold of 0.01 and absolute log2FC of 1 were used to define differentially regulated regions (Supplementary Table S4B–D).
Differential enrichment of CTCF was determined based on the fold change of the ChIP-seq signal density at the pooled peaks level. Absolute fold change threshold of 1.5 was used to define differentially regulated CTCF peaks (Supplementary Table S4E).
Genomic location of differential H3K27ac and CTCF peaks was assessed using ChIPseeker v1.24.0 (61) with GENCODE v33 (GRCh37) gene annotation.
Lamina-associated domains
For the pile-ups generation, genomic regions of the constitutive (62), HeLa (63,64), IMR90 (65–67), TIG-3 (68) and NHDF (69,64) lamina-associated domains (LADs) were used. Overlaps of differentially regulated H3K9me3 domains with conserved LADs and inter-LADs were calculated with intersect function from bedtools v2.25.0. Jaccard similarity coefficients between differentially regulated H3K9me3 domains and LADs were calculated with jaccard function from bedtools v2.25.0.
Pile-ups generation
Pile-ups of the ChIP-seq signals were calculated using the computeMatrix function from deepTools v3.3.0. Pile-ups at the genes were generated in scale-regions mode with parameters ‘–beforeRegionStartLength’ 3000, ‘–regionBodyLength’ 5000, ‘–afterRegionStartLength’ 3000, ‘–missingDataAsZero’, and ‘–skipZeros’. Pile-ups at the H3K9me3 domains and LADs were generated in scale-regions mode with parameters ‘–beforeRegionStartLength’ 500000, ‘–regionBodyLength’ 1000000, ‘–afterRegionStartLength’ 500000, ‘-bs’ 10000, ‘–unscaled5prime’ 10000, ‘–unscaled3prime’ 10000, ‘–missingDataAsZero’, and ‘–skipZeros’. Pile-ups of the H3K27ac and CTCF peaks were generated in reference-point mode with parameters ‘–referencePoint’ center, ‘-a’ 10000 (‘-a’ 2500 for CTCF), ‘-b’ 10000 (‘-b’ 2500 for CTCF), ‘–missingDataAsZero’, and ‘–skipZeros’. Pile-ups were visualized using plotHeatmap function from deepTools v3.3.0.
Association between enhancers and gene expression
To assess the degree of association between differentially regulated enhancers and gene expression changes, Genomic Regions Enrichment of Annotations Tool (GREAT) v4.0.4 (70) was utilized. GREAT was run in basal extension mode excluding regions 5 kb upstream and 1 kb downstream of TSS, and searching for H3K27ac peaks up to distal 500 kb. Genes associated with differential H3K27ac peaks were extracted and analyzed with g:Profiler to calculate GO terms enrichment, as described above for the differentially expressed genes (section ‘Gene Ontology Analysis’, Supplementary Table S4F, G). To associate enhancer-linked genes with gene expression, fgsea v1.14.0 R package (71) was used. It was run in pre-ranked mode with 10000 permutations, and parameter scoreType set to ‘pos’ and ‘neg’ for genes linked with increased and decreased H3K27ac peaks respectively.
Motifs search
HOMER v4.11 (72) was used to identify the motifs present in the ATAC-seq peaks within the annotated super-enhancers. ATAC-seq peaks for the A549 cell line from the ENCODE Project Consortium were used (ENCODE accession numbers ENCSR032RGS and ENCSR220ASC). Peak coordinates were converted from hg38 to hg19 reference human genome assembly with UCSC liftOver tool. HOMER motif discovery algorithm (findMotifsGenome.pl) was executed with default parameters, and known motifs with P-values <0.01 were used for the analysis. The obtained motifs were filtered for transcription factor (TFs) motifs only using the Human Transcription Factor Database v1.01 (73) and converted to the orthologous human gene names if necessary. In case more than one motif for TF was present, the motif with higher fold enrichment was retained.
Hi-C analysis
Hi-C reads mapping and filtering
Hi-C reads were mapped to the reference human genome hg19 assembly using Bowtie v2.2.3 with the iterative mapping procedure implemented in hiclib Python package (74). The mapping was performed in the ‘–very-sensitive’ mode. Minimal read size was set to 25 bp, and the read was extended by 5 bp during the iterative mapping procedure, until a maximal read length was reached. Non-uniquely mapped reads, ‘same fragment’ and ‘dangling end’ reads, PCR duplicates, reads from restriction fragments shorter than 100 bp and longer than 100 kb, and reads from the top 0.5% of restriction fragments with the greatest number of reads were discarded. Read pairs were then aggregated into genomic bins to produce contact matrices. Low coverage bins removal and iterative correction of the matrices were performed using balance function from cooler v0.8.5 (75). Statistics of the Hi-C data processing can be found in Supplementary Table S2C.
Annotation of chromatin compartments
A/B-compartments were annotated using cooltools v0.3.2 call-compartments function for 100 kb resolution contact matrices. The orientation of the eigenvectors (PC1) was selected such that it correlates positively with GC content. Consequently, B compartment bins were assigned with negative eigenvector values, and A compartment bins were assigned with positive. To validate the compartment annotation, histone modification ChIP-seq signals from the ENCODE Project Consortium were used (ENCODE accession numbers ENCSR000AUL, ENCSR000AUN, ENCSR000AUI, ENCSR000ATP, ENCSR000AUK, ENCSR000AVI, ENCSR000ASH, ENCSR000ASV, and ENCSR000AUM). ChIP-seq signal fold change was calculated for each compartment, and was defined as the mean signal in the compartment divided by the mean signal across the whole genome.
Identification of compartment switches
Compartment switches were identified using the genome-wide eigenvector difference (ΔPC1) between A549 SETDB1LOF and control conditions. Only genomic bins where both conditions had annotated A/B compartment state were considered. Genomic bins with ΔPC1 difference between conditions >1.645 standard deviations from the mean ΔPC1 were annotated as compartment switches. The annotated compartment switches were then classified into Stable A, Stable B, A to B, and B to A compartments based on the sign of the eigenvector difference (Supplementary Table S5). To correlate ΔPC1 and ChIP-seq profiles or RNA-seq data, the fold changes of ChIP-seq or RNA-seq data were averaged across 100 kb bins.
Saddle plots
Saddle plots were generated using cooltools v0.3.2 compute-saddle function using 100 kb observed-over-expected cis and trans matrices, respectively. The expected contact matrices were obtained using cooltools v0.3.2 compute-expected function with default parameters. To exclude the extreme eigenvector values from the analysis, top and bottom 2.5 percentiles of the genome-wide eigenvector were clipped. The obtained saddle plots were zoomed into 25 equally sized bins by quantiles.
Compartment strength
Compartment strength was calculated from the compartmentalization saddle plots using two metrics as described in (76) and (77) (Supplementary Figure S5G). AA stands for the average contact enrichment within top 20% of the genome-wide eigenvector values, BB stands for the average contact enrichment within bottom 20% of the genome-wide eigenvector values, and AB/BA stands for the average contact depletion between AA and BB.
Domains detection
Topologically associating domains (TADs) were annotated using the Armatus (78) algorithm implementation from the lavaburst Python package (77) for 20 kb resolution contact matrices. In this algorithm, the average size and the number of TADs were controlled by the scaling parameter γ. To find the optimal γ for TAD partition, the domains at γ values from 0 to 2.5 were called. Median TAD size, number of TADs and genome coverage with TADs were then calculated for each annotation and plotted as curves. Based on these curves, γControl = 0.46 and γSETDB1-KO = 0.52 were selected. TADs smaller than 60 kb were dropped out due to their poor resolution. The remaining domains were subjected to manual refinement in order to increase the annotation quality.
Domain analysis
Insulation score was calculated using the insulation score algorithm (79) implemented in cooltools v0.3.2 diamond-insulation function for 20 kb resolution contact matrices with window size of 360 kb. The average TAD and average TAD boundary were calculated using coolpup.py v0.9.5 (80) from 20 and 5 kb observed-over-expected contact matrices respectively. The expected contact matrices were obtained using cooltools v0.3.2 compute-expected function with ‘–ignore-diags’ set to 0. For the average TAD pile-up, coolpup.py was run with options ‘–local’, ‘–rescale’, and ‘–rescale_size’ set to 99 pixels. For the average TAD boundary, coolpup.py was run with options ‘–local’ and ‘–pad’ set to 200 kb.
P(s) curves
P(s) curves were computed using hiclib and the range between 20 kb and 100 Mb was extracted.
Statistical tests
Statistical analyses were performed using Graphpad Prism v8.4.1 software, MATLAB, R and Python. Statistical significance was determined by the specific tests indicated in the corresponding figure legends.
RESULTS
SETDB1 LOF reduces proliferation and migration velocity of A549 lung adenocarcinoma cells
To impair the H3K9 methylation function of SETDB1 in A549 lung adenocarcinoma cells, we performed CRISPR/Cas9-induced deletion in the catalytic SET domain of SETDB1 (Figure 1A, B and Supplementary Figure S1A). The A549 cell line with a stable LOF mutation of SETDB1 (SETDB1LOF) did not show an increase in a fraction of apoptotic cells (Supplementary Figure S1B). However, SETDB1LOF cells displayed a reduced mitotic index (Supplementary Figure S1C) without significant changes in the duration of mitotic phases (Supplementary Figure S1C); a decreased proliferation rate (Figure 1C), and an increased G1/S ratio (55.7/31% in SETDB1LOFvs. 49.8/34.6% in the control cells; Supplementary Figure S1D).
Figure 1.

SETDB1 loss-of-function reduces A549 lung adenocarcinoma cell proliferation and migration velocity. (A) Overall experimental strategy design. (B) Upper: diagram of the strategy used to generate SETDB1 loss-of-function mutation (SETDB1LOF) in A549 cells by CRISPR/Cas9-mediated deletion in the catalytic SET domain (15th and 16th exons). Lower: RNA-seq data (CPM-normalized coverage) for SETDB1 mRNA levels in control (cyan) and SETDB1LOF (pink) A549 cells. Deleted region where the designed guide RNAs (gRNAs) target the 15th (forward gRNA) and the 16th (reverse gRNA) exons of the SETDB1 gene (see Methods) is highlighted. (C) Cell proliferation curve for control (cyan) and SETDB1LOF (pink) A549 cells at the indicated time points (t, hours) (N ≥ 3 independent experiments; mean ± SEM of eight wells; **P-value < 0.01; ***P-value < 0.001, Mann–Whitney U-test). (D) Wound-healing assay. Left: representative cell velocity measured by wound-healing assay for control (cyan) and SETDB1LOF (pink) A549 cells. Right: average cell velocity for control (cyan) and SETDB1LOF (pink) A549 cells. Each point represents a migration rate in one location (N = 3 independent experiments, on average of 32 measurements for the control and 35 measurements for SETDB1LOF cells; mean ± SEM; ****P-value < 0.0001, t-test). (E) Representative immunofluorescence images of control and SETDB1LOF A549 cells stained with phalloidin-TRITC (F-actin), anti-Paxillin or anti-E-cadherin antibodies. DNA was labeled with DAPI. Scale bar, 10 μm. (F) Quantification of F-actin cellular signal. Left: representation of F-actin cellular distribution stained with phalloidin-TRITC in control and SETDB1LOF A549 cells; each point represents the mean of the F-actin signal intensity in one of the 10 bins along the cellular axis (N = 3 independent experiments; mean ± SEM, 92–104 of cells analyzed; *P-value < 0.05; **P-value < 0.01, Mann–Whitney U-test). Right: quantification of overall F-actin intensity (mean ± SEM; ns, not significant, Mann–Whitney U-test). A corresponding representative image is presented in (E). (G) Quantification of Paxillin and E-cadherin signals. Left: quantification of Paxillin area normalized to the number of nuclei. Right: quantification of overall E-cadherin intensity at cellular junctions. Corresponding representative images are presented in (E) (N ≥ 3 independent experiments; mean ± SEM; *P-value < 0.05; ***P-value < 0.001, Mann–Whitney U-test). (H) Volume of tumors in NSG mice injected with control (N = 8 mice, cyan) or SETDB1LOF A549 (N = 7 mice, pink) A549 cells at the indicated post-injection time in days (****P-value < 0.0001, two-way ANOVA test). (I) Percentage of Ki-67-positive nuclei in control (cyan) or SETDB1LOF (pink) A549 tumor sections (mean ± SEM; ***P-value < 0.001, Mann–Whitney U-test). Corresponding representative images are presented in (J). (J) Representative images of control and SETDB1LOF A549 tumor sections stained with hematoxylin and eosin (H&E) or with an anti-Ki-67 antibody. Scale bar, 1 mm and 50 μm.
We next asked whether SETDB1 LOF affects cell migration using a live imaging wound-healing assay. To avoid bias by cell proliferation rates, cells were pre-treated with a mitosis-blocking agent Mitomycin C (as described in Materials and Methods). SETDB1LOF cells showed a reduced overall displacement (Figure 1D) and cell detachment (Supplementary Movie S1), displaying collective migration, whereas some control cells in the leading front exhibited elongated shapes (Supplementary Figure S1E and Movie S1), often detached from the cohort (Supplementary Movie S1), and demonstrated lamellipodia-like migration (Supplementary Figure S1E, Movie S1 and Movie S2). Furthermore, F-actin stained with phalloidin-TRITC formed thinner and poorly oriented stress fibers, as compared to control cells (Figure 1E, F and Supplementary Figure S1F). The focal adhesion complex, which ensures the correct communication between the cell and extracellular matrix during migration (81), was also affected in SETDB1LOF cells, as highlighted by a markedly increased number of focal adhesion points revealed by paxillin immunostaining (Figure 1E, G and Supplementary Figure S1F). Moreover, SETDB1 LOF promoted the formation of strong E-cadherin-based cell-cell junctions (Figure 1E, G and Supplementary Figure S1F). To discriminate the effects of cytoskeleton remodeling and altered cell-cell adhesion in the suppression of cell migration, we impaired cell-cell junctions by chelation of divalent ions with EGTA (82) and measured the cellular migration velocity. In the presence of 2.5 mM EGTA in the growth media, the SETDB1LOF cells did not show significant changes in the average cellular velocity (Supplementary Figure S1G). A progressive decrease in cell velocity was observed at 3 mM EGTA due to the disruption of cell-cell contacts and detachment of cells from the surface. Thus, we conclude that SETDB1 LOF-mediated migration suppression is primarily determined by cytoskeleton remodeling, in particular by F-actin stress fiber reorganization.
To check whether SETDB1 LOF affected cancer cell proliferation in vivo, we performed a xenograft assay by subcutaneous injection of control and SETDB1LOF cells into immunocompromised Nod-SCID-Gamma (NSG) mice. Tumor growth of SETDB1LOF cells was significantly delayed compared to the control cells (Figure 1H). Hematoxylin and Eosin (H&E) staining revealed a decreased cell density in SETDB1LOF xenograft tumors compared to control (Figure 1J). Apoptosis or necrosis was not microscopically observed upon SETDB1 LOF (Figure 1J). The cell proliferation antigen Ki-67 immunostaining of xenografts (Figure 1I, J) showed a significantly lower number of Ki-67-positive cells in SETDB1LOF xenografts as compared to the control, indicating that SETDB1 LOF reduced tumor aggressiveness.
Collectively, these data suggest that SETDB1 LOF slows the proliferation and migration of lung adenocarcinoma cells. In agreement with these and previous results (83,84), we found that lower SETDB1 mRNA expression was associated with higher survival of NSCLC patients, as revealed by Kaplan–Meier analysis of publicly available data (85) (Supplementary Figure S1H).
Global transcriptional changes induced by SETDB1 LOF correlate with malignant phenotype attenuation
To obtain insights into the molecular mechanisms behind the aforementioned SETDB1 LOF-induced cellular phenotype, we performed differential gene expression analysis of RNA-seq data and identified 2848 differentially expressed genes (DEGs), with 1887 up- and 961 down-regulated ones (Figure 2A and Supplementary Figure S2A). Analysis of transcriptomic data revealed the activation of 179 genes encoding KRüppel-associated box (KRAB)-containing zinc finger transcription factors, or KRAB-ZNFs (Supplementary Figure S2B). These proteins function as a DNA-binding recognition scaffold for the silencing of endogenous retroviruses through heterochromatin establishment by SETDB1 in association with and its cofactor KRAB-associated protein 1 (KAP1) (13,86). We thus assumed that SETDB1 LOF results in massive genome-wide deregulation of ERVs. Indeed, analysis of RNA-seq data with RepEnrich2 (55) tool revealed changes in the expression level for all main classes of transposable elements (TEs) (Supplementary Figure S2C). Among 46 types of TEs analyzed, four ERV-TEs, Alu (belonging to SINE TEs), and L1-LINE TEs were mostly overrepresented in up-regulated TEs in SETDB1LOF cells, which is in agreement with the known role of SETDB1 in the repression of these TEs (87). Another group of genes that was massively de-repressed upon SETDB1 LOF was the clustered Protocadherin (PCDH) genes (Supplementary Figure S2B) involved in the regulation of cell-cell adhesions (88), cell death, and proliferation (89), and are known to be regulated by SETDB1 (90,91).
Figure 2.

Global transcriptional changes induced by SETDB1 LOF correlate with malignant phenotype attenuation (A) Volcano plot showing significant SETDB1 LOF-induced changes in gene expression in A549 cells (N = 3 independent experiments; FDR < 0.01, t-test relative to a threshold). Up-regulated genes (red): 1887 (log2(fold change) > 0). Down-regulated genes (blue): 961 (log2(fold change) < 0). (B) Gene ontology terms over-represented in the differentially expressed genes in SETDB1LOFvs. control A549 cells. (C) Gene set enrichment analysis in SETDB1LOFvs. control A549 cells. Normalized enrichment score (NES) and FDR are indicated. (D) Gene set variation analysis scores for each gene set in the normal lung (N = 48), NSCLC (N = 61), control (cyan), and SETDB1LOF (pink) A549 cells. Dashed vertical lines correspond to the median, 25th and 75th percentiles. (E) mRNA expression of selected mesenchymal and epithelial markers, and genes related to the WNT/β-catenin signaling pathway in control (cyan) and SETDB1LOF (pink) A549 cells. RT-qPCR data were normalized to the PPIA mRNA expression values in control A549 cells that were set as 100% (N ≥ 3 independent experiments; mean ± SEM; *P-value < 0.05, Mann–Whitney U-test). (F) Effect of rescued expression of SETDB1 on mRNA expression of selected mesenchymal and epithelial markers, and genes related to the WNT/β-catenin signaling pathway in A549 SETDB1LOF transfected with pcDNA3.1(+)-IRES-GFP or pcDNA3.1(+)-SETDB1WT-IRES-GFP. RT-qPCR data were normalized to the PPIA mRNA expression values in A549 SETDB1LOF transfected with pcDNA3.1(+)-IRES-GFP that were set as 100% (N = 3 independent experiments; mean ± SEM; *P-value < 0.05, Mann–Whitney U-test). (G) mRNA expression of selected mesenchymal and epithelial markers, and genes related to the WNT/β-catenin signaling pathway in control (N = 8 mice, cyan) and SETDB1LOF A549 xenografts (N = 7 mice, pink) (see Figure 1H). RT-qPCR data were normalized to the PPIA expression values in control A549 xenografts that were set as 100% (mean ± SEM; ns, not significant, ***P-value < 0.001, Mann–Whitney U-test).
Gene ontology (GO) enrichment analysis revealed that DEGs upon SETDB1 LOF were significantly enriched in terms of cell adhesion, migration, proliferation, and epithelium and mesenchyme development (Figure 2B). Analysis of previously published RNA-seq data from SETDB1 knockout cells (cervical adenocarcinoma cell line HeLa (25); mouse postnatal forebrain neurons (91); retinal pigment epithelial cells RPE1 (92)) showed similar GO terms, such as cell adhesion, proliferation, migration, canonical WNT/β-catenin signaling pathway, and cell death regulation (Supplementary Figure S2D), thus demonstrating that SETDB1 LOF-mediated effects on the transcription program are similar in non-related cancer and normal cell types.
To determine the gene signature transcriptional changes, we applied gene set enrichment analysis (GSEA). We found that in SETDB1LOF cells, genes controlling cell-cell adhesion were significantly up-regulated (Figure 2C); whereas NSCLC-associated signatures were down-regulated (Figure 2C and Supplementary Figure S2E), such as WNT/β-catenin (93), mTORC1 (94), and Hedgehog (95) signaling pathways were down-regulated (Figure 2C and Supplementary Figure S2E); MYC targets and proteins providing the G2M checkpoint breakthrough, including effector cyclin-dependent kinases (96) were also down-regulated (Supplementary Figure S2E). Together, these data suggest that SETDB1 LOF attenuates the NSCLC-like phenotype of A549 cells.
To assess the overall trend of gene expression program changes in SETDB1LOF cells, we re-analyzed public transcriptomic data for 48 normal lung and 61 NSCLC cell lines datasets and compared the expression levels of several relevant gene sets using gene set variation analysis (GSVA). The apparent feature of SETDB1LOF cells was a correlation with normal epithelial cell lines across several gene sets, including MYC-targets, cell-cell adhesion, G2M-checkpoint, DNA replication, cell proliferation, embryonic stem cell (ESC)-specific genes, and genes associated with poor survival of NSCLC patients (Figure 2D).
We confirmed RNA-seq data by RT-qPCR and found a reduction in mRNA levels of mesenchymal genes and genes related to WNT/β-catenin signaling (MYC, CCND1, FN1, SERPINE1) and an increase in mRNA levels of epithelial genes (CDH1, CDH3, PCDHA6, PCDHB6, PCDHGA4) (Figure 2E). We checked the reproducibility of the observed effects in several SETDB1LOF clones (Supplementary Figure S2F, S2G). Different SETDB1 LOF clones exhibited the same trend, as compared to the controls (Supplementary Figure S2G), and this trend was reversed after the rescue of SETDB1 expression in SETDB1LOF cells (Figure 2F and Supplementary Figure S2F). Finally, we confirmed similar trends for mRNA expression of selected genes on the xenograft tumors from the NSG mice (Figure 2G). Therefore, we concluded that SETDB1LOF cells (re)acquired features of a normal epithelial phenotype underpinned by the gene expression program changes.
Global transcriptional changes induced by SETDB1 LOF are linked to genome-wide modulation of enhancer and super-enhancer activities
To elucidate the mechanism of SETDB1 LOF-induced transcriptional changes, we examined the epigenetic status of potential regulatory elements by ChIP-seq assay for H3K27ac and H3K4me3 (Supplementary Figure S3A), which mark active enhancers and promoters, respectively. Increased and decreased H3K27ac peaks were predominantly located in intronic and intergenic regions, suggesting a possible association with enhancers (Figure 3A, B). As expected, an increase of H3K27ac levels was accompanied by the up-regulation of associated genes, and vice versa (Figure 3C). Moreover, genes associated with increased H3K27ac regions contributed to the GO terms associated with the epithelial phenotype, such as cell adhesion and cell-cell junction organization (Figure 3D), whereas genes associated with down-regulated H3K27ac regions were involved in mesenchymal development and epithelial-to-mesenchymal (EMT) transition, cell migration, stress fiber assembly, and other terms, typical for cancer progression (Figure 3D).
Figure 3.

Global transcriptional changes induced by SETDB1 LOF are linked to genome-wide modulation of enhancer activity (A) Pile-ups of the H3K27ac enrichment at the differentially enriched H3K27ac peaks in control and SETDB1LOF A549 cells. (B) Genomic locations of the differentially enriched H3K27ac peaks (**P-value < 0.01; ****P-value < 0.0001, chi-squared test). (C) Gene expression changes associated with increased and decreased H3K27ac enrichments revealed by gene set enrichment analysis in SETDB1LOF versus control A549 cells. Enrichment score (ES) and P-values are indicated. (D) Gene ontology terms overrepresented in genes associated with differentially enriched H3K27ac peaks. (E) Volcano plot showing differential (FDR < 0.01, absolute log2(fold change) > 1, Wald test) super-enhancers (SEs) in control and SETDB1LOF A549 cells (N = 2 independent experiments). Increased SEs (red): 104; decreased SEs (blue): 26. Genes associated with SEs linked to NSCLC progression are indicated.
Since super-enhancers (SEs) are responsible for cell identity (58,60) and play an important role in oncogenesis (97), we asked whether SETDB1 LOF affected SEs genome-wide (i.e. those with an altered level of H3K27ac). First, to distinguish SEs from typical enhancers, we used the rank ordering of SE (ROSE) approach (58) and annotated 832 and 883 SEs in the control and SETDB1LOF cells (Supplementary Figure S3B and C). Next, we assessed the H3K27ac level at SEs and identified 130 differentially regulated SEs in SETDB1LOF as compared to the control cells (Figure 3E). Among 104 SEs up-regulated in SETDB1LOF cells, we found SEs associated with ZNF331, TOX3, PCDH9, PTPRK and LIMA1 genes involved in the suppression of EMT, cell proliferation, cell migration, invasion and metastasis (98–102) (Figure 3E and Supplementary Figure S3D). Among the 26 SEs down-regulated in SETDB1LOF, we found those associated with MICAL2, RSPO3, and HJURP genes involved in cancer progression (103–105) (Figure 3E and Supplementary Figure S3D). Finally, we applied HOMER (72) to identify transcription factor (TF) binding motifs enriched in ATAC-seq peaks within differentially regulated SEs (ENCODE ATAC-seq for A549 cell line, as described in Materials and Methods) (Supplementary Figure S3E). We found that motifs of TFs involved in the regulation of proliferation, migration, and invasion of lung cancer cells, including FOSL1, ATF3, BATF, AP-1, SIX4 and NFE2L2 were enriched in differential SEs (Supplementary Figure S3E), denoting a potential role of these factors in the observed phenotype. Together, these data suggest that SETDB1 LOF-induced changes in the transcriptional pattern of A549 cells are mediated by the modulation of enhancer and SE activities.
SETDB1 LOF induces H3K9me3 and H3K27me3 redistribution within the nuclear space and along the genome
To investigate how SETDB1 LOF affects heterochromatin profiles, we first assayed the total abundance of H3K9me3 and H3K27me3 marks by western blot analysis. A moderate, but significant decrease of both marks was observed in SETDB1LOF cells (on average, by 25% for H3K9me3 and by 10% for H3K27me3; Supplementary Figure S4A). Immunofluorescence imaging showed an accumulation of H3K9me3 at the nuclear periphery in SETDB1LOF nuclei (Figure 4A). To quantitatively estimate the H3K9me3 shifting, we calculated the distribution of the immunofluorescence signal intensity along the nuclear radius. In control cells, H3K9me3 signal monotonically radially increased (Figure 4B), whereas, in SETDB1LOF cells, the distribution of H3K9me3 had a pronounced peak at the nuclear edge, co-localizing with the nuclear lamina as revealed by anti-Lamin-A/C staining (Figure 4A, B and Supplementary Figure S4B, C). Notably, in normal human bronchial epithelial (NHBE) cells, H3K9me3 distribution demonstrated a pattern similar to that in SETDB1LOF cells, with a well-defined peak at the nuclear lamina (Supplementary Figure S4B, C). Concerning the H3K27me3 pattern, we observed an opposite and less pronounced trend, with a slight shifting of H3K27me3 signal intensity from the nuclear lamina towards the nuclear interior in SETDB1LOF cells, without detectable changes in the curve shape (Figure 4A, B).
Figure 4.

SETDB1 LOF induces H3K9me3 and H3K27me3 redistribution within the nuclear space and along the genome (A) Representative immunofluorescence images of control and SETDB1LOF A549 cells stained with anti-H3K9me3, anti-Lamin A/C, and anti-H3K27me3 antibodies. DNA was labeled with DAPI. Scale bar, 10 μm. (B) Nuclear distribution of H3K9me3, Lamin A/C, and H3K27me3 in control (cyan) and SETDB1LOF (pink) A549 cells. Intensities are plotted as a function of distance from the nuclear center (N = 3 independent experiments; mean intensity ± SEM; 92–104 of cells analyzed; ***P-value < 0.001, Kolmogorov–Smirnov test). The region with maximum Lamin A/C intensity is indicated with a grey dashed line. (C) Pile-ups of the H3K9me3 and H3K27me3 enrichments at the differentially expressed genes in control and SETDB1LOF A549 cells. Up- and down-regulated genes are shown in red and blue, respectively. (D) Pie chart of the genome-wide dynamics of H3K9me3-enriched regions in SETDB1LOF versus control A549 cells. The percentages indicate the fraction of changed H3K9me3 peaks from the total peaks number. S – the total length of changed H3K9me3 regions. (E) Correlation between H3K27me3 and H3K9me3 enrichment in 100-kb genomic bins in control and SETDB1LOF A549 cells. Spearman's correlation coefficients are indicated for bins with increased and decreased H3K9me3 levels separately. (F) Pile-ups of the H3K9me3 and H3K27me3 enrichment at increased H3K9me3 regions in control and SETDB1LOF A549 cells. (G) Pie charts of the overlap between increased, decreased, and stable H3K9me3 enriched regions with constitutive LADs from (62).
Next, to characterize genome-wide the SETDB1 LOF-driven changes in the distributions of constitutive and facultative heterochromatin, we profiled H3K9me3 and H3K27me3 marks using ChIP-seq. In SETDB1LOF cells we observed dramatic changes in H3K9me3 and H3K27me3 peak positions (Supplementary Figure S4D) concomitant to an overall alteration in levels of these marks across large segments of the genome, as compared to the control cells (Supplementary Figure S4F). In particular, H3K9me3 levels were substantially reduced along all genes, including the down-regulated ones (Figure 4C and Supplementary Figure S4E). In contrast, H3K27me3 levels decreased and increased at up-regulated and down-regulated genes, respectively (Figure 4C and Supplementary Figure S4E). Hence, we suggest that the up-regulation of gene expression upon SETDB1 LOF is associated with the loss of both constitutive and facultative heterochromatin, whereas transcription repression is not linked to the H3K9me3 (Supplementary Figure S4G), but appears to be mediated by other mechanisms, including Polycomb repressive (106).
Surprisingly, the overall decrease in H3K9me3 levels evidenced by western blot in SETDB1LOF cells (Supplementary Figure S4A) was accompanied by an increase in H3K9me3 levels at specific genomic regions (Figure 4D, F and Supplementary Figure S4F), with a simultaneous decrease in H3K27me3 levels (Figure 4E, F and Supplementary Figure S4F), as revealed by differential enrichment analysis. Visual inspection of the ChIP-seq profiles in the genome browser revealed that these regions are typically gene-poor, heterochromatin rich (Supplementary Figure S4F), and relatively long (up to 7.4 Mb in length, 202 kb on average, Supplementary Figure S4H). The majority of regions with increased H3K9me3 in SETDB1LOF overlapped with constitutive LADs (cLADs) from Lenain et al. (2017) (62) that are conserved in several non-related cell types, whereas regions with decreased H3K9me3 coincided with inter-LADs (Figure 4G and Supplementary Figure S4I, K). These ChIP-seq results were validated by ChIP-qPCR for several genes, confirming a significant increase in H3K9me3 occupancy in cLAD regions in SETDB1LOF cells (Supplementary Figure S4J). The predominant location of regions with increased H3K9me3 (Supplementary Figure S4K) and decreased H3K27me3 (Supplementary Figure S4L) levels within LADs was also confirmed by the comparison with LAD positions in fibroblast cell lines: IMR90, TIG-3, and NHDF. Moreover, analysis of published H3K9me3 ChIP-seq data in HeLa and RPE1 cells after SETDB1 knockout (25,107,92) showed an increase in H3K9me3 occupancy at constitutive LADs, as compared to the corresponding control cells (Supplementary Figure S4M), confirming that the observed increase in H3K9me3 upon SETDB1 LOF is not an artifact and not cell-type specific.
The enrichment in H3K9me3 at LADs upon SETDB1 LOF could be established by other H3K9-specific methyltransferases, SUV39H1, which co-exists with SETDB1 in the common complex(es) (108), and/or its homolog SUV39H2, associated with H3K9me3 establishment in lamina-associated chromatin (109). Indeed, we found that the knockdown of both SUV39H1 and SUV39H2 in SETDB1LOF cells abolished the H3K9me3 enrichment at cLADs at several tested loci (Supplementary Figure S4N).
Collectively, these results demonstrate that SETDB1 LOF triggers a widespread decrease in H3K9me3 at euchromatic regions, accompanied by a SUV39H1/H2-mediated increase in H3K9me3 within LADs that is coupled with a slight decrease in H3K27me3.
SETDB1 LOF leads to H3K9me3 redistribution between A/B compartments and affects both large-scale and local chromatin topology
To test whether the SETDB1 LOF-induced global heterochromatin re-distribution along the genome and in the nuclear space affected the 3D genome organization, we performed in situ Hi-C and generated contact maps at a resolution of up to 20 kb. The dependence of relative contact probability on the genomic distance between pairs of loci was essentially the same in SETDB1LOF and control cells (Supplementary Figure S5A). Thus, SETDB1 LOF did not affect the general properties of the 3D genome. At the same time, visual inspection of heatmaps revealed that SETDB1 LOF substantially disturbed contact profiles at a subset of genome loci, as manifested by changes in the ‘plaid pattern’ (110), reflecting genome compartmentalization changes (Figure 5A). To analyze the chromatin contact profile systematically, we annotated A (active) and B (inactive) chromatin compartments using principal component analysis (PCA) (111,110) (Figure 5A) and used publicly available profiles of histone marks to validate compartment segmentation (Supplementary Figure S5B). The first principal component (PC1) was highly correlated between SETDB1LOF and control cells (Pearson's r = 0.89; Figure 5B and Supplementary Figure S5C), and the proportion of A and B compartments was similar in the two cell lines (Supplementary Figure S5D), suggesting that the overall compartmentalization was preserved upon SETDB1 LOF. Nonetheless, we found that 5.5% (N = 1454) and 3.8% (N = 1009) of 100-kb genomic bins switched from B to A and from A to B compartment, respectively (Figure 5B, C).
Figure 5.

SETDB1 LOF leads to H3K9me3 redistribution between A/B compartments and affects both large-scale and local chromatin topology (A) Hi-C interaction matrices at 100-kb resolution showing examples of chromatin compartment switches in control and SETDB1LOF A549 cells for Chr. 4: 73.0–115.0 Mb (left panel) and Chr. 12: 102.0–133.0 Mb (right panel). Switching regions are highlighted with grey boxes and black arrows. Eigenvector (PC1 track) for compartments A (red) and B (blue) is shown. Lower panels: ChIP-seq profiles (RPKM- and input-normalized ratio) for H3K9me3 and H3K4me3 in control and SETDB1LOF A549 cells are shown according to the UCSC genome browser. (B) Scatter plot of PC1 values at 100-kb resolution for the control and SETDB1LOF A549 cells. S – total length of the regions that switched compartment state (absolute z-score of ΔPC1 > 1.645) from B to A (red) or A to B (blue). (C) Pie chart showing the dynamics of compartment bins in SETDB1LOFvs. control A549 cells. N – number of 100-kb bins. (D) Distribution of RPKM-normalized H3K4me3 and H3K27ac ChIP-seq signals within stable A and B compartment bins and in switched from B to A, or A to B compartment bins in control (cyan) and SETDB1LOF (pink) A549 cell lines (ns, not significant; **P-value < 0.01; ****P-value < 0.0001, Mann–Whitney U-test). (E) Distribution of RPKM-normalized H3K27me3 and H3K9me3 ChIP-seq signals within stable A and B compartment bins and in switched from B to A, or A to B compartment bins in control (cyan) and SETDB1LOF (pink) A549 cell lines (**P-value < 0.01, ****P-value < 0.0001, Mann–Whitney U-test). (F) Left: saddle plots of contact enrichments in cis and in trans between 100-kb genomic bins belonging to A and B compartments in control and SETDB1LOF A549 cells. Numbers represent the compartment strength values calculated as described in (76). Right: subtraction of A549 SETDB1LOF from control saddle plots for cis and trans contacts. (G) Hi-C interaction matrices at 20- (top) and 5-kb (bottom) resolutions showing the PCDH gene cluster on Chr. 5: 135.5 – 145.0 Mb (left panels) and ZNF genes on Chr. 19: 50.0–58.0 Mb (right panels). Strengthened TAD boundaries in the SETDB1LOF as compared to control A549 cells are highlighted with black arrows. Eigenvector (PC1 track) for compartments A and B is shown in red and blue, respectively. Genes, RNA-seq profile (CPM-normalized coverage), ChIP-seq profiles (RPKM- and input-normalized ratio) for H3K9me3, H3K27me3, H3K4me3, H3K27ac, and CTCF in control and SETDB1LOF A549 cells are shown according to the UCSC genome browser.
To further analyze these compartment changes, we divided all 100-kb genomic bins into four groups according to their compartment state (Figure 5C): bins belonging to A and to B compartments in both cell lines (‘stable A’ and ‘stable B’, respectively), and bins that switched their compartment state in SETDB1LOF (‘A-to-B’ and ‘B-to-A’). As expected, the H3K4me3 and H3K27ac histone marks and transcription were substantially enriched within ‘stable A’ bins, as compared to ‘stable B’ bins; the decrease and increase of these marks correlated with the ‘A-to-B’ and ‘B-to-A’ switched bins, respectively (Figure 5D and Supplementary Figure S5F). The changes in the H3K27me3 mark in the switched bins mirrored those of the active chromatin (Figure 5E and Supplementary Figure S5E), probably due to colocalization of H3K4me3 and H3K27me3 within the so-called bivalent domains (112). However, we observed a remarkably distinct trend for the H3K9me3 mark. In control cells, in contrast to the active marks displaying high differences between compartments, H3K9me3 abundance was only 1.4-fold enriched in the B as compared to the A compartment (Figure 5E). In SETDB1LOF cells, H3K9me3 increased in ‘stable B’ and in ‘A-to-B’ bins and decreased in the two other bin groups, resulting in a 4.2-fold overall enrichment of H3K9me3 in the B vs. A compartment. This finding agrees with the above-described increase of this mark in LADs (Figure 4G), which mostly reside in the B compartment. As a result, we observed a gain of spatial contacts within the B compartment in cis and a decrease of inter-compartment interactions in SETDB1LOF cells, suggesting an increased density of the B compartment folding and further segregation of compartments within the nuclear space (Figure 5F). Consequently, the compartment strength was augmented upon SETDB1 LOF (Supplementary Figure S5G), in agreement with previously published Hi-C data from mouse postnatal forebrain neurons after SETDB1 knockout (91) (Supplementary Figure S5H).
Notably, the aforementioned changes in compartmentalization upon SETDB1 LOF displayed a remarkably stronger correlation with differential enrichment in H3K4me3 (Spearman's ρ = 0.5), H3K27ac (Spearman's ρ = 0.41), and H3K9me3 (Spearman's ρ = –0.33) marks, as compared to changes in transcription level (Spearman's ρ = 0.24; Supplementary Figure S5I, J). Next, we asked whether SETDB1 LOF impacted the organization of TADs, and found that TAD boundary positions were largely unaffected in SETDB1LOF cells (Supplementary Figure S5K). A minor increase of TAD boundary insulation (Supplementary Figure S5L, M) and no changes in intra-TAD interactions (Supplementary Figure S5N) were detected. In parallel, we observed substantial alterations in the TAD profiles within several loci. One such example is the PCDH locus, containing 53 genes within three clusters: PCDH-alpha (PCDHA), PCDH-beta (PCDHB) and PCDH-gamma (PCDHG) (Figure 5G). In A549 control cells and many other cancer cells, the major part of the PCDH locus is transcriptionally repressed and marked with H3K9me3 (89,113,114). SETDB1 LOF resulted in a global reactivation of the PCDH locus (Supplementary Figure S2B), with a concomitant decrease in H3K9me3 and an increase in active histone marks (Figure 5G). This change was coupled with the partition of the entire PCDH locus into several contact domains, the boundaries of which demarcated multi-gene clusters from each other (Figure 5G). Interestingly, the up-regulation of the PCDH locus was correlated with the emergence of numerous CTCF ChIP-seq peaks within the locus, and the majority of them did not coincide with the positions of new TAD boundaries in SETDB1LOF cells (Figure 5G). An increase in CTCF binding strength (Supplementary Figure S5O) and in the number of CTCF-occupied sites (including promoters (Supplementary Figure S5R) and TAD boundaries (Supplementary Figure S5S)) without an increase in the total CTCF protein level (Supplementary Figure S5O, P, Q) turned out to be a general trend in SETDB1 LOF condition. This is in agreement with published data, demonstrating that SETDB1 is required for shielding the genome from excess CTCF binding (91).
Another example of TAD profile changes upon SETDB1 LOF is a ZNF locus on chromosome 19, which contains numerous KRAB-ZNF genes, including the tumor suppressor ZNF331 (115) (Figure 5G). Here, the entire region, located in a large virtually unstructured TAD in the control cells, was partitioned in SETDB1LOF cells into several TADs with well-defined boundaries. Similar to the PCDH locus, this region lost H3K9me3 methylation (Figure 5G) and was transcriptionally up-regulated (Supplementary Figure S2B) upon SETDB1 LOF. Thus, local changes in TAD profiles upon SETDB1 LOF occur within specific multi-gene loci, where alterations in H3K9me3 and active epigenetic marks coincide with large-scale perturbations of the transcription profile.
SETDB1 LOF changes A549 cell nuclei shape and mechanical properties
To understand how changes in the epigenome and 3D genome organization upon SETDB1 LOF contribute to suppression of the malignant phenotype of A549 cells, we studied characteristics of nuclei and chromatin, which potentially reflect mechanical properties and were previously shown to influence cell migration (116). Examination under an optical microscope revealed the differences in the nuclear shape between the control and SETDB1LOF cells (Figures 4A, 6A and Supplementary Figure S4B), manifested by more invaginations and protrusions in the control cells. We assessed the overall nuclear shape by quantification of the nuclear irregularity index (36) and found that SETDB1LOF cells have an average nuclear irregularity index of 0.029, which perfectly matches that of normal primary NHBE cells (Figure 6A, B). In contrast, A549 control cells have an abnormal nuclear morphology, with a significantly higher irregularity index (0.038; Figure 6A, B). Correspondingly, the circularity index and the average nucleus area (Figure 6B) of SETDB1LOF cells were more similar to NHBE, rather than to the control A549.
Figure 6.

SETDB1 LOF changes the shape and mechanical properties of the A549 cell nuclei (A) Representative DAPI-stained nuclei of control (cyan) and SETDB1LOF A549 (pink), and NHBE (grey) cells showing different nuclear morphology. Numbers denote the nuclear irregularity index of the corresponding nucleus. Bar, 10 μm. (B) Quantification of irregularity index (left panel), circularity index (middle panel), and nucleus area (right panel) for control (cyan), SETDB1LOF A549 (pink), and NHBE cells (grey). Each point represents the measurement for individual nuclei, horizontal lines correspond to median, error bars represent the interquartile range (ns, not significant; ****P-value < 0.0001, Kruskall-Wallis test with Dunn's correction for multiple comparisons). (C) Quantification of NE fluctuations for control (cyan) and SETDB1LOF (pink) A549, and NHBE cells (grey). Each point represents the average for each individual cell on 2 to 4 kymographs of the square root of the NE mean square displacement relative to its mean position; horizontal lines correspond to the median, error bars represent the interquartile range (ns, not significant; ****P-value < 0.0001, one-way ANOVA–Kruskal–Wallis test). (D) Quantification of DNA displacement for control (cyan) and SETDB1LOF (pink) A549, and NHBE nuclei (grey). Each point represents the average for each individual cell on 10 timeframes of the displacement between two consecutive frames; horizontal lines correspond to the median, error bars represent the interquartile range (ns, not significant; ****P-value < 0.0001, one-way ANOVA- Kruskal–Wallis test). (E) Representative particle image velocimetry (PIV) of DNA within nuclei in control and SETDB1LOF A549, and NHBE cells showing the overall flow of DNA displacement. Scale bar, 5 μm. (F) Graphical representation of the SETDB1 LOF-induced effects in lung adenocarcinoma cells.
The nuclear shape is primarily determined by the nuclear envelope's (NE) mechanical properties, consisting of the nuclear membrane and nuclear lamina (117). Therefore, we assayed the NE dynamics in vivo by measuring the NE fluctuations constrained by the cytoskeleton from the cytoplasm on one side, and chromatin and lamins from the nucleoplasm, on the other side. Surprisingly, we found that the NE fluctuation magnitude was the same between control and SETDB1LOF cells, and, on average, two-fold higher than in the normal NHBE cells (Figure 6C). Another factor influencing the overall mechanical properties of the nucleus is the state of the bulk chromatin, particularly the constitutive heterochromatin positioned near the nuclear lamina (118). Thus, to follow the spatiotemporal dynamics of chromatin, we measured the chromatin movement by in vivo imaging of Hoechst 33342-stained nuclei. Particle image velocimetry (PIV) analysis showed that the average chromatin displacement significantly decreased from 3.044 pixels displacement/time frame in the control cells to 2.659 in SETDB1LOF cells, which was similar to that in NHBE cells (Figure 6D). Moreover, the observed decrease of chromatin mobility was more prominent at the nuclear periphery (Figure 6E). Together with the above-described H3K9me3-marked heterochromatin redistribution towards the nuclear periphery and strengthening of the B compartments, these results show an increase in the density of tightly compacted chromatin at the periphery upon SETDB1 LOF. This increase could make nuclei more robust, preventing changes in their shape and restricting cell mobility.
DISCUSSION
Here, we sought to disambiguate the multiple roles of SETDB1 in cancer and their consequences on various scales, which fuel the oncogenic process. To this end, we have studied a plethora of effects triggered by SETDB1 loss of function in NSCLC cells, ranging from gene expression, and epigenome analyses, the 3D genome organization, and the mechanical properties of chromatin and nuclei, to xenograft tumor assays.
SETDB1 target loci are predominantly located within euchromatin gene-rich chromosome segments, where SETDB1 is involved in gene repression and silencing of repetitive elements (13,119). We suppose that the decrease of H3K9me3 level at these loci is the first outcome of SETDB1 LOF, preceding and then directly or indirectly promoting all the other observed effects (proposed model is described in Figure 6F). Notably, it has been previously shown that H3K9me2 is also decreased in SETDB1 deficient cells at euchromatic regions (120). Writing and erasing epigenetic enzyme complexes for both activating and repressive histone modifications typically compete within these genomic regions in determining their epigenetic state (121,122). Our observations suggest that SETDB1 LOF shifts the equilibrium to the benefit of histone lysine acetyltransferases in hundreds of loci genome-wide. This shift results in a drastic increase of H3K27 acetylation responsible for an apparent activation of numerous super-enhancers, and an overall transcriptional reprogramming leading to massive up-regulation of gene repertoire underpinning the reacquired normal epithelial phenotype of SETDB1LOF cells. These changes, together with increased focal adhesion and stress fiber reorganization, restrict cell migration.
Surprisingly, we have also found that SETDB1 LOF is followed by a SUV39H1/H2-dependent increase of H3K9me3 levels within LADs. A previous study (123) showed that alpha-satellite RNA transcripts contributed to the stable association of SUV39H1 with constitutive heterochromatin. Recently, we have shown that SETDB1 cooperates with MPP8 to silence satellite DNA repeats (124). Thus, activation of alpha-satellite RNA transcripts in SETDB1 LOF cells might contribute to the stabilization of SUV39H1 at LADs. Since H3K9me3 is an anchor for chromatin attachment to the lamina through the recruitment of HP1-beta interacting with the Lamin B receptor (125–127), the increase in H3K9me3 levels could be responsible for the observed deposition of H3K9me3-marked chromatin at the nuclear periphery. H3K9me3 occupancy positively correlates with chromatin compaction driven by the oligomerization of H3K9me3 binding HP1 (128–130) and the formation of liquid condensates (131,132). Moreover, attachment to the lamina per se is sufficient for chromatin compaction (133). These facts could explain the increased density of chromatin packaging in the B compartment, which is enriched in LADs, upon SETDB1 LOF, and tighter H3K9me3-driven association of LADs with the nuclear lamina, as revealed by immunofluorescence analysis. Thus, in SETDB1LOF cells, tightly packed H3K9me3-marked chromatin forms a clearly visible layer adjacent to the nuclear lamina, similarly to non-cancer cells. The highly compact state and low mobility of these chromatin regions imply that they could serve as a tough carcass for the NE. The presence of this layer is associated with a more spherical shape of the NE, although it does not affect its fluctuations. Since the ability of nuclei to change their shape is an essential property of migrating cancer cells, suppression of this ability upon SETDB1 LOF, ensured by the tightly packed heterochromatin located at the lamina, may contribute to the reduction of cell migration, and, along with the decreased proliferation rate, restrict their oncogenic potential.
Taken together, our findings demonstrate that the loss of H3K9 methylation activity of SETDB1 reshapes the epigenetic profiles within both active and inactive chromatin. Ultimately, this causes perturbations in the spatial genome organization, affecting both the gene expression patterns and mechanical properties of the cell nuclei. Collectively, these changes result in the suppression of the lung adenocarcinoma cell's malignant phenotype.
DATA AVAILABILITY
Raw and processed data for Hi-C, RNA-seq, and ChIP-seq are available in the GEO repository under the accession number GSE168233. All custom analysis scripts, macro and data that support the conclusions are available from the authors on request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Ekaterina Boyarchuk (UMR7216, Université de Paris, Paris, France) for technical help and Sonja Boland (Institut Jacques Monod, Paris, France) for providing the NHBE cell line, advice, and help with their culture. We thank Natalia Klimenko (Institute of Gene Biology, Russian Academy of Science, Moscow, Russia) for advice on the ChIP-seq data analysis. We also thank Mathieu Maurin (U932, Institute Curie, INSERM, Paris, France) for help with image analysis.
Author contributions: V.V.Z. and S.A. conceived the project; V.V.Z. engineered and cultured cell lines, performed functional and validation studies, and immunofluorescence imaging; V.V.Z. and L.D.-M. prepared ChIP-seq and RNA-seq libraries; S.V.U. prepared Hi-C libraries; M.D.M. performed computational analysis of NGS and publicly available data; V. J. engineered constructs for the rescue experiments; A.G. and R.-M.M. performed in vitro migration assays; B.U. and O.D. performed in vivo studies; A.W. and M.P. performed mechanical studies; A.K. and Y.S.V. performed the analysis of immunofluorescence images; V.V.Z., S.V.U., S.V.R., Y.S.V., M.D.M. and S.A wrote the manuscript with input from all authors.
Contributor Information
Vlada V Zakharova, Epigenetics and Cell Fate (EDC) department, UMR7216, CNRS, Université Paris Cité, F-75013 Paris, France.
Mikhail D Magnitov, Institute of Gene Biology, Russian Academy of Science, Moscow 119334, Russia.
Laurence Del Maestro, Epigenetics and Cell Fate (EDC) department, UMR7216, CNRS, Université Paris Cité, F-75013 Paris, France.
Sergey V Ulianov, Institute of Gene Biology, Russian Academy of Science, Moscow 119334, Russia; Faculty of Biology, M.V. Lomonosov Moscow State University, Moscow 119991, Russia.
Alexandros Glentis, Institute Jacques Monod, CNRS, Université Paris Cité, F-75013 Paris, France.
Burhan Uyanik, INSERM UMR1231, LipSTIC, University of Burgundy Franche-Comté F-21000, Dijon, France.
Alice Williart, Institut Curie and Institut Pierre Gilles de Gennes, PSL Research University, CNRS, UMR 144, 75248 Paris, France.
Anna Karpukhina, UMR9018, CNRS, Université Paris-Sud Paris-Saclay, Institut Gustave Roussy; 94805 Villejuif, France; Koltzov Institute of Developmental Biology, 119334 Moscow, Russia.
Oleg Demidov, INSERM UMR1231, LipSTIC, University of Burgundy Franche-Comté F-21000, Dijon, France; Institute of Cytology, RAS, 194064 St. Petersburg, Russia; NTU Sirius, 354340 Sochi, Russia.
Veronique Joliot, Epigenetics and Cell Fate (EDC) department, UMR7216, CNRS, Université Paris Cité, F-75013 Paris, France.
Yegor S Vassetzky, UMR9018, CNRS, Université Paris-Sud Paris-Saclay, Institut Gustave Roussy; 94805 Villejuif, France; Koltzov Institute of Developmental Biology, 119334 Moscow, Russia.
René-Marc Mège, Institute Jacques Monod, CNRS, Université Paris Cité, F-75013 Paris, France.
Matthieu Piel, Institut Curie and Institut Pierre Gilles de Gennes, PSL Research University, CNRS, UMR 144, 75248 Paris, France.
Sergey V Razin, Institute of Gene Biology, Russian Academy of Science, Moscow 119334, Russia; Faculty of Biology, M.V. Lomonosov Moscow State University, Moscow 119991, Russia.
Slimane Ait-Si-Ali, Epigenetics and Cell Fate (EDC) department, UMR7216, CNRS, Université Paris Cité, F-75013 Paris, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Work in Ait-Si-Ali's laboratory was supported by the Fondation pour la Recherche Medicale (FRM) « Equipe FRM » [DEQ20160334922]; Association Française contre les Myopathies Telethon (AFM-Telethon) [22480]; Institut National du Cancer (INCa) [2012-1-PL BIO-03-CNRS B-1]; Agence Nationale de la Recherche (ANR) « MuSiC » [ANR-17-CE12-0010-01]; work in Razin's laboratory was supported by the Russian Science Foundation (RSF) [21-64-00001]; Interdisciplinary Scientific and Educational School of Moscow University ‘Molecular Technologies of the Living Systems and Synthetic Biology’; work in Demidov's laboratory was supported by the ARC Foundation for cancer research [R20066MM]; Hi-C experiments were performed using the equipment of IGB RAS facilities, supported by the Ministry of Science and Higher Education of the Russian Federation and facilities of the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, IGB RAS. Funding for open access charge: FRM, AFM-Telethon, ANR, RSF, INCa.
Conflict of interest statement. None declared.
REFERENCES
- 1. Gruenbaum Y., Foisner R.. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015; 84:131–164. [DOI] [PubMed] [Google Scholar]
- 2. Rowley M.J., Corces V.G.. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018; 19:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Steensel B., Belmont A.S.. Lamina-Associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell. 2017; 169:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carone D.M., Lawrence J.B.. Heterochromatin instability in cancer: from the barr body to satellites and the nuclear periphery. Semin. Cancer Biol. 2013; 23:99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zink D., Fischer A.H., Nickerson J.A.. Nuclear structure in cancer cells. Nat. Rev. Cancer. 2004; 4:677–687. [DOI] [PubMed] [Google Scholar]
- 6. Valton A.-L., Dekker J.. TAD disruption as oncogenic driver. Curr. Opin. Genet. Dev. 2016; 36:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ibrahim D.M., Mundlos S.. Three-dimensional chromatin in disease: what holds us together and what drives us apart?. Curr. Opin. Cell Biol. 2020; 64:1–9. [DOI] [PubMed] [Google Scholar]
- 8. Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R.S., Zhang Y.. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002; 298:1039–1043. [DOI] [PubMed] [Google Scholar]
- 9. Lehnertz B., Ueda Y., Derijck A.A.H.A., Braunschweig U., Perez-Burgos L., Kubicek S., Chen T., Li E., Jenuwein T., Peters A.H.F.M.. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003; 13:1192–1200. [DOI] [PubMed] [Google Scholar]
- 10. Shinkai Y., Tachibana M.. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011; 25:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schultz D.C., Ayyanathan K., Negorev D., Maul G.G., Rauscher F.J.. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002; 16:919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peters A.H., O’Carroll D., Scherthan H., Mechtler K., Sauer S., Schöfer C., Weipoltshammer K., Pagani M., Lachner M., Kohlmaier A.et al.. Loss of the suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001; 107:323–337. [DOI] [PubMed] [Google Scholar]
- 13. Matsui T., Leung D., Miyashita H., Maksakova I.A., Miyachi H., Kimura H., Tachibana M., Lorincz M.C., Shinkai Y.. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. 2010; 464:927–931. [DOI] [PubMed] [Google Scholar]
- 14. Song Y.J., Choi J.H., Lee H.. Setdb1 is required for myogenic differentiation of C2C12 myoblast cells via maintenance of MyoD expression. Mol. Cells. 2015; 38:362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beyer S., Pontis J., Schirwis E., Battisti V., Rudolf A., Le Grand F., Ait-Si-Ali S.. Canonical wnt signalling regulates nuclear export of setdb1 during skeletal muscle terminal differentiation. Cell Discov. 2016; 2:16037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tan S.-L., Nishi M., Ohtsuka T., Matsui T., Takemoto K., Kamio-Miura A., Aburatani H., Shinkai Y., Kageyama R.. Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Dev. Camb. Engl. 2012; 139:3806–3816. [DOI] [PubMed] [Google Scholar]
- 17. Bilodeau S., Kagey M.H., Frampton G.M., Rahl P.B., Young R.A.. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 2009; 23:2484–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodriguez-Paredes M., Martinez de Paz A., Simó-Riudalbas L., Sayols S., Moutinho C., Moran S., Villanueva A., Vázquez-Cedeira M., Lazo P.A., Carneiro F.et al.. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene. 2014; 33:2807–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Batham J., Lim P.S., Rao S.. SETDB-1: a potential epigenetic regulator in breast cancer metastasis. Cancers. 2019; 11:E1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ceol C.J., Houvras Y., Jane-Valbuena J., Bilodeau S., Orlando D.A., Battisti V., Fritsch L., Lin W.M., Hollmann T.J., Ferré F.et al.. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011; 471:513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fei Q., Shang K., Zhang J., Chuai S., Kong D., Zhou T., Fu S., Liang Y., Li C., Chen Z.et al.. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat. Commun. 2015; 6:8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Y., Huang J., Li Q., Chen K., Liang Y., Zhan Z., Ye F., Ni W., Chen L., Ding Y.. Histone methyltransferase SETDB1 promotes cells proliferation and migration by interacting withTiam1 in hepatocellular carcinoma. BMC Cancer. 2018; 18:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun Q.-Y., Ding L.-W., Xiao J.-F., Chien W., Lim S.-L., Hattori N., Goodglick L., Chia D., Mah V., Alavi M.et al.. SETDB1 accelerates tumorigenesis by regulating WNT signaling pathway. J. Pathol. 2015; 235:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo J., Dai X., Laurent B., Zheng N., Gan W., Zhang J., Guo A., Yuan M., Liu P., Asara J.M.et al.. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat. Cell Biol. 2019; 21:226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Timms R.T., Tchasovnikarova I.A., Antrobus R., Dougan G., Lehner P.J.. ATF7IP-mediated stabilization of the histone methyltransferase SETDB1 is essential for heterochromatin formation by the HUSH complex. Cell Rep. 2016; 17:653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Griffin G.K., Wu J., Iracheta-Vellve A., Patti J.C., Hsu J., Davis T., Dele-Oni D., Du P.P., Halawi A.G., Ishizuka J.J.et al.. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature. 2021; 595:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cao N., Yu Y., Zhu H., Chen M., Chen P., Zhuo M., Mao Y., Li L., Zhao Q., Wu M.et al.. SETDB1 promotes the progression of colorectal cancer via epigenetically silencing p21 expression. Cell Death. Dis. 2020; 11:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Watanabe H., Soejima K., Yasuda H., Kawada I., Nakachi I., Yoda S., Naoki K., Ishizaka A.. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008; 8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shultz L.D., Lyons B.L., Burzenski L.M., Gott B., Chen X., Chaleff S., Kotb M., Gillies S.D., King M., Mangada J.et al.. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. Baltim. Md. 2005; 1950; 174:6477–6489. [DOI] [PubMed] [Google Scholar]
- 30. Concordet J.-P., Haeussler M.. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018; 46:W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ait-Si-Ali S., Guasconi V., Fritsch L., Yahi H., Sekhri R., Naguibneva I., Robin P., Cabon F., Polesskaya A., Harel-Bellan A.. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J. 2004; 23:605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ulianov S.V., Khrameeva E.E., Gavrilov A.A., Flyamer I.M., Kos P., Mikhaleva E.A., Penin A.A., Logacheva M.D., Imakaev M.V., Chertovich A.et al.. Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res. 2016; 26:70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zonderland J., Wieringa P., Moroni L.. A quantitative method to analyse F-actin distribution in cells. MethodsX. 2019; 6:2562–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dobretsov M., Petkau G., Hayar A., Petkau E.. Clock scan protocol for image analysis: ImageJ plugins. J. Vis. Exp. JoVE. 2017; 124:55819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stephens A.D., Liu P.Z., Banigan E.J., Almassalha L.M., Backman V., Adam S.A., Goldman R.D., Marko J.F.. Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol. Biol. Cell. 2018; 29:220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S.et al.. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.1000 Genome Project Data Processing Subgroup . The sequence alignment/map format and SAMtools. Bioinforma. Oxf. Engl. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Amemiya H.M., Kundaje A., Boyle A.P.. The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 2019; 9:9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinforma. Oxf. Engl. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Navarro Gonzalez J., Zweig A.S., Speir M.L., Schmelter D., Rosenbloom K.R., Raney B.J., Powell C.C., Nassar L.R., Maulding N.D., Lee C.M.et al.. The UCSC genome browser database: 2021 update. Nucleic Acids Res. 2021; 49:D1046–D1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McCarthy D.J., Chen Y., Smyth G.K.. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012; 40:4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Robinson M.D., Oshlack A.. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010; 11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lund S.P., Nettleton D., McCarthy D.J., Smyth G.K.. Detecting differential expression in RNA-sequence data using quasi-likelihood with shrunken dispersion estimates. Stat. Appl. Genet. Mol. Biol. 2012; 11: 10.1515/1544-6115.1826. [DOI] [PubMed] [Google Scholar]
- 48. McCarthy D.J., Smyth G.K.. Testing significance relative to a fold-change threshold is a TREAT. Bioinforma. Oxf. Engl. 2009; 25:765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J.. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019; 47:W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P.. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015; 1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hänzelmann S., Castelo R., Guinney J.. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013; 14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A.et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 54. Durinck S., Spellman P.T., Birney E., Huber W.. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009; 4:1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Criscione S.W., Zhang Y., Thompson W., Sedivy J.M., Neretti N.. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics. 2014; 15:583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Langmead B., Salzberg S.L.. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of chip-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A.. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013; 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ross-Innes C.S., Stark R., Teschendorff A.E., Holmes K.A., Ali H.R., Dunning M.J., Brown G.D., Gojis O., Ellis I.O., Green A.R.et al.. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012; 481:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A.. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yu G., Wang L.-G., He Q.-Y.. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinforma. Oxf. Engl. 2015; 31:2382–2383. [DOI] [PubMed] [Google Scholar]
- 62. Lenain C., de Graaf C.A., Pagie L., Visser N.L., de Haas M., de Vries S.S., Peric-Hupkes D., van Steensel B., Peeper D.S.. Massive reshaping of genome-nuclear lamina interactions during oncogene-induced senescence. Genome Res. 2017; 27:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lund E.G., Duband-Goulet I., Oldenburg A., Buendia B., Collas P.. Distinct features of lamin A-interacting chromatin domains mapped by chip-sequencing from sonicated or micrococcal nuclease-digested chromatin. Nucl. Austin Tex. 2015; 6:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Paulsen J., Sekelja M., Oldenburg A.R., Barateau A., Briand N., Delbarre E., Shah A., Sørensen A.L., Vigouroux C., Buendia B.et al.. Chrom3D: three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biol. 2017; 18:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sadaie M., Salama R., Carroll T., Tomimatsu K., Chandra T., Young A.R.J., Narita M., Pérez-Mancera P.A., Bennett D.C., Chong H.et al.. Redistribution of the lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2013; 27:1800–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shah P.P., Donahue G., Otte G.L., Capell B.C., Nelson D.M., Cao K., Aggarwala V., Cruickshanks H.A., Rai T.S., McBryan T.et al.. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013; 27:1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dou Z., Xu C., Donahue G., Shimi T., Pan J.-A., Zhu J., Ivanov A., Capell B.C., Drake A.M., Shah P.P.et al.. Autophagy mediates degradation of nuclear lamina. Nature. 2015; 527:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guelen L., Pagie L., Brasset E., Meuleman W., Faza M.B., Talhout W., Eussen B.H., de Klein A., Wessels L., de Laat W.et al.. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008; 453:948–951. [DOI] [PubMed] [Google Scholar]
- 69. Lund E., Oldenburg A.R., Collas P.. Enriched domain detector: a program for detection of wide genomic enrichment domains robust against local variations. Nucleic Acids Res. 2014; 42:e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010; 28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A.. Fast gene set enrichment analysis. 2021; bioRxiv doi:01 February 2021, preprint: not peer reviewed 10.1101/060012. [DOI]
- 72. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 172:650–665. [DOI] [PubMed] [Google Scholar]
- 74. Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., Dekker J., Mirny L.A.. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods. 2012; 9:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Abdennur N., Mirny L.A.. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinforma. Oxf. Engl. 2020; 36:311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nora E.P., Goloborodko A., Valton A.-L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G.. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017; 169:930–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schwarzer W., Abdennur N., Goloborodko A., Pekowska A., Fudenberg G., Loe-Mie Y., Fonseca N.A., Huber W., Haering C.H., Mirny L.et al.. Two independent modes of chromatin organization revealed by cohesin removal. Nature. 2017; 551:51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Filippova D., Patro R., Duggal G., Kingsford C.. Identification of alternative topological domains in chromatin. Algorithms Mol. Biol. AMB. 2014; 9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Crane E., Bian Q., McCord R.P., Lajoie B.R., Wheeler B.S., Ralston E.J., Uzawa S., Dekker J., Meyer B.J.. Condensin-driven remodelling of x chromosome topology during dosage compensation. Nature. 2015; 523:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Flyamer I.M., Illingworth R.S., Bickmore W.A.. Coolpup.py: versatile pile-up analysis of Hi-C data. Bioinforma. Oxf. Engl. 2020; 36:2980–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tojkander S., Gateva G., Lappalainen P.. Actin stress fibers–assembly, dynamics and biological roles. J. Cell Sci. 2012; 125:1855–1864. [DOI] [PubMed] [Google Scholar]
- 82. Rothen-Rutishauser B., Riesen F.K., Braun A., Günthert M., Wunderli-Allenspach H.. Dynamics of tight and adherens junctions under EGTA treatment. J. Membr. Biol. 2002; 188:151–162. [DOI] [PubMed] [Google Scholar]
- 83. Inoue Y., Matsuura S., Kurabe N., Kahyo T., Mori H., Kawase A., Karayama M., Inui N., Funai K., Shinmura K.et al.. Clinicopathological and survival analysis of japanese patients with resected non-small-cell lung cancer harboring NKX2-1, SETDB1, MET, HER2, SOX2, FGFR1, or PIK3CA gene amplification. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer. 2015; 10:1590–1600. [DOI] [PubMed] [Google Scholar]
- 84. Lafuente-Sanchis A., Zúñiga Á., Galbis J.M., Cremades A., Estors M., Martínez-Hernández N.J., Carretero J.. Prognostic value of ERCC1, RRM1, BRCA1 and SETDB1 in early stage of non-small cell lung cancer. Clin. Transl. Oncol. 2016; 18:798–804. [DOI] [PubMed] [Google Scholar]
- 85. Győrffy B., Surowiak P., Budczies J., Lánczky A.. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013; 8:e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rowe H.M., Jakobsson J., Mesnard D., Rougemont J., Reynard S., Aktas T., Maillard P.V., Layard-Liesching H., Verp S., Marquis J.et al.. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature. 2010; 463:237–240. [DOI] [PubMed] [Google Scholar]
- 87. Fukuda K., Shinkai Y.. SETDB1-Mediated silencing of retroelements. Viruses. 2020; 12:E596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schreiner D., Weiner J.A.. Combinatorial homophilic interaction between gamma-protocadherin multimers greatly expands the molecular diversity of cell adhesion. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:14893–14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dallosso A.R., Øster B., Greenhough A., Thorsen K., Curry T.J., Owen C., Hancock A.L., Szemes M., Paraskeva C., Frank M.et al.. Long-range epigenetic silencing of chromosome 5q31 protocadherins is involved in early and late stages of colorectal tumorigenesis through modulation of oncogenic pathways. Oncogene. 2012; 31:4409–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhu Y., Sun D., Jakovcevski M., Jiang Y.. Epigenetic mechanism of SETDB1 in brain: implications for neuropsychiatric disorders. Transl. Psychiatry. 2020; 10:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jiang Y., Loh Y.-H.E., Rajarajan P., Hirayama T., Liao W., Kassim B.S., Javidfar B., Hartley B.J., Kleofas L., Park R.B.et al.. The methyltransferase SETDB1 regulates a large neuron-specific topological chromatin domain. Nat. Genet. 2017; 49:1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sun Z., Chadwick B.P.. Loss of SETDB1 decompacts the inactive x chromosome in part through reactivation of an enhancer in the IL1RAPL1 gene. Epigenetics Chromatin. 2018; 11:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Akiri G., Cherian M.M., Vijayakumar S., Liu G., Bafico A., Aaronson S.A.. Wnt pathway aberrations including autocrine wnt activation occur at high frequency in human non-small-cell lung carcinoma. Oncogene. 2009; 28:2163–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gately K., Al-Alao B., Dhillon T., null Mauri F., Cuffe S., Seckl M., O’Byrne K.. Overexpression of the mammalian target of rapamycin (mTOR) and angioinvasion are poor prognostic factors in early stage NSCLC: a verification study. Lung Cancer Amst. Neth. 2012; 75:217–222. [DOI] [PubMed] [Google Scholar]
- 95. Yuan Z., Goetz J.A., Singh S., Ogden S.K., Petty W.J., Black C.C., Memoli V.A., Dmitrovsky E., Robbins D.J.. Frequent requirement of hedgehog signaling in non-small cell lung carcinoma. Oncogene. 2007; 26:1046–1055. [DOI] [PubMed] [Google Scholar]
- 96. Imielinski M., Berger A.H., Hammerman P.S., Hernandez B., Pugh T.J., Hodis E., Cho J., Suh J., Capelletti M., Sivachenko A.et al.. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012; 150:1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhang X., Choi P.S., Francis J.M., Imielinski M., Watanabe H., Cherniack A.D., Meyerson M.. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet. 2016; 48:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wang Y., He T., Herman J.G., Linghu E., Yang Y., Fuks F., Zhou F., Song L., Guo M.. Methylation of ZNF331 is an independent prognostic marker of colorectal cancer and promotes colorectal cancer growth. Clin. Epigenetics. 2017; 9:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chen Y., Xiang H., Zhang Y., Wang J., Yu G.. Loss of PCDH9 is associated with the differentiation of tumor cells and metastasis and predicts poor survival in gastric cancer. Clin. Exp. Metastasis. 2015; 32:417–428. [DOI] [PubMed] [Google Scholar]
- 100. Xu X., Li D., Liu J., Ma Z., Huang H., Min L., Dai L., Dong S.. Downregulation of PTPRK promotes cell proliferation and metastasis of NSCLC by enhancing STAT3 activation. Anal. Cell. Pathol. Amst. 2019; 2019:4265040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ohashi T., Idogawa M., Sasaki Y., Tokino T.. p53 mediates the suppression of cancer cell invasion by inducing LIMA1/EPLIN. Cancer Lett. 2017; 390:58–66. [DOI] [PubMed] [Google Scholar]
- 102. Sanders A.J., Martin T.A., Ye L., Mason M.D., Jiang W.G.. EPLIN is a negative regulator of prostate cancer growth and invasion. J. Urol. 2011; 186:295–301. [DOI] [PubMed] [Google Scholar]
- 103. Zhou W., Liu Y., Gao Y., Cheng Y., Chang R., Li X., Zhou Y., Wang S., Liang L., Duan C.et al.. MICAL2 is a novel nucleocytoplasmic shuttling protein promoting cancer invasion and growth of lung adenocarcinoma. Cancer Lett. 2020; 483:75–86. [DOI] [PubMed] [Google Scholar]
- 104. Gong X., Yi J., Carmon K.S., Crumbley C.A., Xiong W., Thomas A., Fan X., Guo S., An Z., Chang J.T.et al.. Aberrant RSPO3-LGR4 signaling in Keap1-deficient lung adenocarcinomas promotes tumor aggressiveness. Oncogene. 2015; 34:4692–4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wei Y., Ouyang G.-L., Yao W.-X., Zhu Y.-J., Li X., Huang L.-X., Yang X.-W., Jiang W.-J.. Knockdown of HJURP inhibits non-small cell lung cancer cell proliferation, migration, and invasion by repressing Wnt/β-catenin signaling. Eur. Rev. Med. Pharmacol. Sci. 2019; 23:3847–3856. [DOI] [PubMed] [Google Scholar]
- 106. Comet I., Riising E.M., Leblanc B., Helin K.. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer. 2016; 16:803–810. [DOI] [PubMed] [Google Scholar]
- 107. Tchasovnikarova I.A., Timms R.T., Matheson N.J., Wals K., Antrobus R., Göttgens B., Dougan G., Dawson M.A., Lehner P.J.. GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science. 2015; 348:1481–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Fritsch L., Robin P., Mathieu J.R.R., Souidi M., Hinaux H., Rougeulle C., Harel-Bellan A., Ameyar-Zazoua M., Ait-Si-Ali S.. A subset of the histone H3 lysine 9 methyltransferases suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol. Cell. 2010; 37:46–56. [DOI] [PubMed] [Google Scholar]
- 109. Bian Q., Khanna N., Alvikas J., Belmont A.S.. β-Globin cis-elements determine differential nuclear targeting through epigenetic modifications. J. Cell Biol. 2013; 203:767–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O.et al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Belaghzal H., Dekker J., Gibcus J.H.. Hi-C 2.0: an optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods San Diego Calif. 2017; 123:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Barski A., Cuddapah S., Cui K., Roh T.-Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K.. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823–837. [DOI] [PubMed] [Google Scholar]
- 113. Novak P., Jensen T., Oshiro M.M., Watts G.S., Kim C.J., Futscher B.W.. Agglomerative epigenetic aberrations are a common event in human breast cancer. Cancer Res. 2008; 68:8616–8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wang K.-H., Lin C.-J., Liu C.-J., Liu D.-W., Huang R.-L., Ding D.-C., Weng C.-F., Chu T.-Y.. Global methylation silencing of clustered proto-cadherin genes in cervical cancer: serving as diagnostic markers comparable to HPV. Cancer Med. 2015; 4:43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Yu J., Liang Q.Y., Wang J., Cheng Y., Wang S., Poon T.C.W., Go M.Y.Y., Tao Q., Chang Z., Sung J.J.Y.. Zinc-finger protein 331, a novel putative tumor suppressor, suppresses growth and invasiveness of gastric cancer. Oncogene. 2013; 32:307–317. [DOI] [PubMed] [Google Scholar]
- 116. Denais C., Lammerding J.. Nuclear mechanics in cancer. Adv. Exp. Med. Biol. 2014; 773:435–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Polychronidou M., Grobhans J.. Determining nuclear shape: the role of farnesylated nuclear membrane proteins. Nucl. Austin Tex. 2011; 2:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Stephens A.D., Banigan E.J., Marko J.F.. Chromatin's physical properties shape the nucleus and its functions. Curr. Opin. Cell Biol. 2019; 58:76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Liu S., Brind’Amour J., Karimi M.M., Shirane K., Bogutz A., Lefebvre L., Sasaki H., Shinkai Y., Lorincz M.C.. Setdb1 is required for germline development and silencing of H3K9me3-marked endogenous retroviruses in primordial germ cells. Genes Dev. 2014; 28:2041–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Fukuda K., Shimura C., Miura H., Tanigawa A., Suzuki T., Dohmae N., Hiratani I., Shinkai Y.. Regulation of mammalian 3D genome organization and histone H3K9 dimethylation by H3K9 methyltransferases. Commun. Biol. 2021; 4:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hyun K., Jeon J., Park K., Kim J.. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017; 49:e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Dawson M.A. The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science. 2017; 355:1147–1152. [DOI] [PubMed] [Google Scholar]
- 123. Johnson W.L., Yewdell W.T., Bell J.C., McNulty S.M., Duda Z., O’Neill R.J., Sullivan B.A., Straight A.F.. RNA-dependent stabilization of SUV39H1 at constitutive heterochromatin. Elife. 2017; 6:e25299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Cruz-Tapias P., Robin P., Pontis J., Maestro L.D., Ait-Si-Ali S.. The H3K9 methylation writer SETDB1 and its reader MPP8 cooperate to silence satellite DNA repeats in mouse embryonic stem cells. Genes. 2019; 10:E750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Polioudaki H., Kourmouli N., Drosou V., Bakou A., Theodoropoulos P.A., Singh P.B., Giannakouros T., Georgatos S.D.. Histones H3/H4 form a tight complex with the inner nuclear membrane protein LBR and heterochromatin protein 1. EMBO Rep. 2001; 2:920–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Poleshko A., Mansfield K.M., Burlingame C.C., Andrake M.D., Shah N.R., Katz R.A.. The human protein PRR14 tethers heterochromatin to the nuclear lamina during interphase and mitotic exit. Cell Rep. 2013; 5:292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lachner M., O’Carroll D., Rea S., Mechtler K., Jenuwein T.. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001; 410:116–120. [DOI] [PubMed] [Google Scholar]
- 128. Hiragami-Hamada K., Soeroes S., Nikolov M., Wilkins B., Kreuz S., Chen C., De La, Rosa-Velázquez I.A., Zenn H.M., Kost N.et al.. Dynamic and flexible H3K9me3 bridging via HP1β dimerization establishes a plastic state of condensed chromatin. Nat. Commun. 2016; 7:11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Canzio D., Chang E.Y., Shankar S., Kuchenbecker K.M., Simon M.D., Madhani H.D., Narlikar G.J., Al-Sady B.. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol. Cell. 2011; 41:67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Yamamoto K., Sonoda M.. Self-interaction of heterochromatin protein 1 is required for direct binding to histone methyltransferase, SUV39H1. Biochem. Biophys. Res. Commun. 2003; 301:287–292. [DOI] [PubMed] [Google Scholar]
- 131. Strickfaden H., Tolsma T.O., Sharma A., Underhill D.A., Hansen J.C., Hendzel M.J.. Condensed chromatin behaves like a solid on the mesoscale in vitro and in living cells. Cell. 2020; 183:1772–1784. [DOI] [PubMed] [Google Scholar]
- 132. Larson A.G., Elnatan D., Keenen M.M., Trnka M.J., Johnston J.B., Burlingame A.L., Agard D.A., Redding S., Narlikar G.J.. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature. 2017; 547:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ulianov S.V., Doronin S.A., Khrameeva E.E., Kos P.I., Luzhin A.V., Starikov S.S., Galitsyna A.A., Nenasheva V.V., Ilyin A.A., Flyamer I.M.et al.. Nuclear lamina integrity is required for proper spatial organization of chromatin in Drosophila. Nat. Commun. 2019; 10:1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed data for Hi-C, RNA-seq, and ChIP-seq are available in the GEO repository under the accession number GSE168233. All custom analysis scripts, macro and data that support the conclusions are available from the authors on request.