


Abstract
Histone H3.3 is an H3 variant which differs from the canonical H3.1/2 at four residues, including a serine residue at position 31 which is evolutionarily conserved. The H3.3 S31 residue is phosphorylated (H3.3 S31Ph) at heterochromatin regions including telomeres and pericentric repeats. However, the role of H3.3 S31Ph in these regions remains unknown. In this study, we find that H3.3 S31Ph regulates heterochromatin accessibility at telomeres during replication through regulation of H3K9/K36 histone demethylase KDM4B. In mouse embryonic stem (ES) cells, substitution of S31 with an alanine residue (H3.3 A31 –phosphorylation null mutant) results in increased KDM4B activity that removes H3K9me3 from telomeres. In contrast, substitution with a glutamic acid (H3.3 E31, mimics S31 phosphorylation) inhibits KDM4B, leading to increased H3K9me3 and DNA damage at telomeres. H3.3 E31 expression also increases damage at other heterochromatin regions including the pericentric heterochromatin and Y chromosome-specific satellite DNA repeats. We propose that H3.3 S31Ph regulation of KDM4B is required to control heterochromatin accessibility of repetitive DNA and preserve chromatin integrity.
INTRODUCTION
The eukaryotic genome is packaged into an ordered structure called chromatin, comprised of repeating units ∼147 bp of DNA wrapped around an octamer of four histones H2A, H2B, H3 and H4. The hydrophilic N-terminal histone tails extend from the nucleosome core and these tails are subjected to a diverse array of post-translational modifications (PTMs), which alter chromatin structure and dynamics. These PTMs are maintained through cell divisions, creating an epigenetic cellular memory which preserves gene expression patterns and cellular identity. In humans, the canonical histones, such as H3.1/H3.2, are synthesised only during S-phase and incorporated into chromatin in a DNA replication-dependent manner by the CAF1 histone chaperone complex (1–4). This coupling of DNA replication with histone assembly allows for preservation of chromatin states through cell divisions.
Unlike canonical histones, the histone variants are expressed and incorporated independent of DNA replication. Histone variants are used to replace canonical histones which are displaced outside of S-phase, such as during DNA repair and transcription. The highly conserved histone H3.3 is one such variant and H3.3 is important for maintaining epigenetic memory at both transcribed genes and repetitive heterochromatin. At transcribed genes, H3.3 is deposited by the HIRA (histone cell cycle regulator) chaperone complex, and this is important for maintaining transcriptional memory of active genes (5–7). In addition, H3.3 is also located at heterochromatin including telomeric DNA repeats, imprinted genes, retrotransposons and pericentric heterochromatin. At these sites, the H3.3 K9 residue is trimethylated (H3.3K9me3) which is required for the maintenance of the heterochromatic state (8–15).
H3.3 differs from canonical H3.1/2 at three residues (87–90) in the globular domain (AAIG in H3.3 versus SAVM in H3.1/2), and at residue 31 in the tail (S31 in H3.3 versus A31 in H3.1/2). The AAIG motif in the globular domain is important for interactions with H3.3-specific chaperone complexes and regulated deposition of H3.3 into chromatin. H3.3 deposition in euchromatin is dependent on HIRA while incorporation of H3.3 at telomeric DNA repeats, retrotransposons, pericentric heterochromatin and imprinted genes requires the DAXX (Fas death domain-associated protein) and ATRX (alpha-thalassemia/mental retardation, X-linked) histone chaperone complex (8–15). At these sites, H3.3 deposition is required for maintaining heterochromatin marks including H3K9me3, H4K20me3 and HP1α (also known as chromobox 5, CBX5) (7,9,14–17). In mouse ES cells, H3.3 is targeted for K9 trimethylation to establish a heterochromatic state enriched in H3.3K9me3 at the telomere (14,16), and this ATRX/H3.3 mediated chromatin assembly occurs within the PML (promyelocytic leukaemia) bodies (8,18–21). Despite being maintained as in a heterochromatic state, we have recently shown that an H3K9/K36 demethylase, KDM4B (lysine (K)-specific demethylase 4B), binds to and removes H3K9me3 (H3.3K9me3) at telomeres to increase chromatin accessibility and facilitate replication (22). Loss of KDM4B results in replication stress and DNA damage at telomeric heterochromatin. In addition to telomeres, H3.3 deposition by ATRX is also for silencing of endogenous retroviral elements (ERVs) including the intracisternal A-type particles (IAPs) and imprinted genes in mouse ES cells (15–17,23).
Several studies in flies, worms and plants have demonstrated a role for H3.3 in controlling gene expression, DNA repair, cell differentiation and embryonic development (24–31). In mouse, knockout of H3.3-encoding genes leads to infertility and embryo lethality (32–36), providing support for the importance of H3.3 in development and survival. In human, amino acid substitutions in H3.3 have been reported in several types of cancer, such as lysine to methionine at position 27 (H3.3-K27M) (37,38), glycine to arginine or valine at position 34 (H3.3-G34R/V), and mutations affecting their chaperones, such as DAXX/ATRX (37–42). These studies provide further evidence for the importance of H3.3 and its chaperones in chromatin-regulating pathways in vertebrates.
The serine 31 (S31) residue in H3.3 is conserved from yeast to mammals and can be phosphorylated (S31Ph). Phosphorylation of H3.3 S31 is driven by a network of mitotic kinases, including Checkpoint Kinase 1 (CHEK1) and Aurora Kinase B (43,44). In somatic cells, H3.3 S31Ph is enriched at pericentric satellite DNA repeats of metaphase chromosomes (45), while in pluripotent mouse embryonic stem (ES) cells, H3.3 S31Ph localises predominantly to telomeres (12). Upon differentiation of mouse ES cells, H3.3 S31Ph decreases at telomeres and increases at pericentric heterochromatin. H3.3 S31Ph is required for gastrulation in Xenopus, suggesting a role for this residue in controlling differentiation and cell fates (46). Recent studies have also highlighted the role of H3.3 S31Ph in driving gene transcription by promoting p300-dependent acetylation of H3.3 (47) and inhibiting recruitment of transcriptional co-repressor ZMYND11 (48). However, the role of H3.3 S31 at transcriptionally silent DNA repeat regions remains unknown. To address this, we have used H3.3 S31 mutant mouse ES models—H3.3 A31 (phospho-null) and H3.3 E31 (phospho-mimetic) mutants, to provide insight into the role of H3.3 S31Ph in chromatin homeostasis. Our results show that H3.3 S31Ph regulation of KDM4B histone demethylase is required for controlling heterochromatin accessibility and maintaining chromatin integrity at transcriptionally silent repetitive DNA regions including the telomeres, pericentric heterochromatin and Y chromosome satellite repeats.
MATERIALS AND METHODS
Cell culture
Mouse ES cells and derived cell lines including ATRX knockout, H3.3 S31, A31 and E31 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 12% heat-inactivated foetal calf serum, 103 units/ml leukaemia inhibitory factor (Merck), 0.1 mM β-mercaptoethanol, non-essential amino acids, l-glutamine and penicillin/streptomycin. Cells were maintained in 37°C incubator under 5% CO2.
Generation of H3.3 mutant clones
The H3.3 WT S31 and mutant (H3.3 A31 and H3.3 E31) cDNA fragments were commercially synthesised (Integrated DNA technologies). These fragments were cloned into pHL-EF1a-SphcCas9-iP-A (Addgene) at NcoI and EcoR1 sites. H3.3 S31, A31 and E31 expression constructs were linearised with FspI restriction enzyme prior to transfection into H3.3cKO (H3f3af/f H3f3bf/f) ES cells carrying all four conditional (f, for floxed) alleles of H3.3 genes. Single colonies expressing either H3.3 S31, A31 and E31 were selected by adding 2μg/ml of puromycin into cell culture media. The pCAGGS-Cre vector (Addgene) was transfected into H3.3cKO (H3f3af/f H3f3bf/f) ES cells expressing H3.3 S31, A31 and E31. Single colonies were cultured for an additional 4 weeks and successful deletion of endogenous H3f3a and H3f3b genes was assessed by Southern blot analyses. To validate the knockout of H3f3a genes, a 218 bp DNA probe that targets intron 3 was used and the fragment was amplified using H3f3a primers. The knockout of H3f3b genes was determined using a 377 bp DNA probe that targets the last exon and the fragment was amplified using H3f3b primers.
qPCR for expression analysis
RNA was extracted using Promega SV Total RNA Isolation Kit. cDNA was then synthesized using the High-Capacity cDNA Reverse Transcription Kit according to manufacturer's instructions (ThermoFisher Scientific). 20 ng of cDNA was combined with 0.5 μM of primers and FastStart DNA Master SYBR Green (Roche) in a 10 μl reaction and the expression levels of target genes were quantitated using the LightCycler® (Roche). As an internal control, primers specific for GAPDH were used in real-time polymerase chain reaction (PCR) analysis. The comparative cycle threshold (CT) method was used for data analyses and relative fold difference was expressed as 2−ΔΔCT. Primers for GAPDH, KDM4B, TERRA and H3.3 transcripts are shown in Supplementary Table S1.
Immunofluorescence analyses and fluorescence in situ hybridization (FISH)
Cells were treated with 100 ng/ml Colcemid (Gibco) for 1 h at 37°C to enrich for mitotic cells. Cells were harvested and resuspended in a hypotonic solution of 0.075 M KCl before being cytospun onto glass slides. Cells were rinsed with KCM buffer (120 mM KCl, 20 mM NaCl, 10 mM Tris–HCl at pH 7.2, 0.5 mM ethylenediaminetetraacetic acid (EDTA) 0.1% [vol/vol] Triton X-100 and protease inhibitor), followed by incubations in a KCM extraction buffer containing 0.5% Triton X-100, and a KCM blocking buffer containing 1% BSA (wt/vol). They were then incubated with the relevant primary and secondary antibodies for 1 h at 37°C. After each round of antibody incubation, slides were washed thrice in KCM buffer. Slides were then fixed in KCM with 4% formaldehyde and mounted in mounting medium (Vectashield). Images were collected using a fluorescence microscope linked to a CCD camera system.
For FISH analyses following immunofluorescence analyses, slides were dehydrated in a 80%, 95% and 100% ethanol series and air-dried. FISH was performed by hybridization overnight at 37°C with either telomeric TTAGGG (PNA Bio), Y chromosome satellite repeat (i.e. Yq mega-satellite repeat) (49) and mouse major satellite (44,50) DNA probes in 10 mM NaHPO4, pH 7.4, 10 mM NaCl, 20 mM Tris, pH 7.5 and 50% formamide. Slides were washed in 2× SSC at room temperature followed by two rounds of washes at 50°C in 0.5× SSC and one round at 50°C in 0.1× SSC. Slides were then mounted in mounting medium (Vectashield). The 1.8kb Yq mega-satellite repeat DNA probe (49) was amplified using PCR primers Yq satellite Yr_f and Yr_r, and cloned into pGEMT-Easy (Promega).
Immunofluorescence and FISH images were collected using a fluorescence microscope linked to a CCD camera system and signal intensities were processed using the Zen software 2011 according to the manufacturer's instruction (Carl Zeiss Microscopy).
qPCR analysis of telomere length
Genomic DNA was prepared from mouse ES cell lines for real time quantitative PCR analysis of telomere length (51,52) using the LightCycler and analysed with LightCycler® 480 Software. The average telomere length was measured by quantifying telomeric DNA relative to a single copy gene. 36B4 was used as the single copy reference gene. 2 ng of genomic DNA, 300 nM primers, and DNA SYBR Green PCR Master mix (Roche) were used for the qPCR reactions. For telomere DNA amplification, the PCR cycling parameters used were 95°C for 10 min, 30 cycles of 95°C for 15 s and a 56°C anneal-extension step for 1 min. For 36B4 DNA amplification, the PCR cycling parameters used were 95°C for 10 min, 35 cycles of 95°C for 15 s, 52°C annealing for 20 s and 72°C extension, for 30 s. A standard curve was set up for each primer set using wildtype genomic DNA template over a range of 0.8–100 ng. Relative telomere length was represented by the value of telomere divided by the value of the 36B4 gene.
Immunoprecipitation
H3.3 S31, A31 and E31 expressing ES cells were transfected with FLAG-tagged KDM4B (53,54) using Lipofectamine 2000 transfection Reagent (Thermo Scientific). After 24 h, transfected cells were lysed in cold high salt RIPA buffer (50 mM Tris–HCl pH 7.5, 1 mM EDTA, 250 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.05% SDS and protease inhibitors) and sonicated before centrifugation. The supernatant was collected and adjusted with low-salt RIPA buffer (50 mM Tris–HCl pH 7.5, 1 mM EDTA, 90 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.05% SDS and protease inhibitors). The lysate was assessed for protein concentration using the Pierce™ BCA Protein Assay Kit. Equal amount of protein lysates were subjected to immunoprecipitation using anti- FLAG-antibody (Sigma Aldrich, F1804) overnight. The immunoprecipitated proteins were captured using protein G conjugated magnetic beads. The beads were washed with RIPA buffer (50 mM Tris–HCl pH 7.5, 1 mM EDTA, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.05% SDS and protease inhibitors). Immunoprecipitated proteins were eluted by boiling for 5 min in 2× SDS PAGE sample buffer containing β-mercaptoethanol. The eluates were subjected to SDS/PAGE gel electrophoresis and immunoblotting with H3.3 and FLAG antibodies, respectively. To assess the binding between H3.3 and KDM4B, relative intensities of input, immunoprecipitated H3.3 (H3.3 Ip) and Flag-KDM4B (KDM4B Ip) were quantified using Image Lab Software (Biorad Laboratories) and ratios of H3.3 Ip/KDM4B Ip calculated. The changes in H3.3/KDM4B binding affinity in A31 and E31 mutant cell lines were calculated by normalising their H3.3 Ip/KDM4B Ip ratios to the H3.3 Ip/KDM4B Ip ratio of WT H3.3 S31 cells.
Histone demethylase assay
250 ng of H3.3 K36me3, H3.3 S31PhK36me3, H3.3 A31K36me3 or H3.3 E31K36me3 peptides (China peptides) were incubated with 50 nM KDM4B recombinant protein (Active Motif) in reaction buffer (50μl) containing 50 mM HEPES pH 7.5, 0.02% Triton X-100, 100 μM 2OG, 100 μM ascorbate, 50 μM (NH4)2Fe(SO4)2·6(H2O), 1 mM TCEP for 2 h at room temperature. Samples were co-spotted onto an MTP anchorChip 800/384 TF MALDI target plate with Matrix solution of 10 mg/ml a-cyano-4-hydroxycinnamic acid (Laser BioLabs, Sophia-Antipolis, France) in 50% acetonitrile 0.1% TFA. Samples were analysed on a Bruker Daltonics (Bremen, Germany) ULTRAFLEX MALDI TOF/TOF in reflector mode with an m/z range of 1200 to 3500 Da, using Smartbeam parameter set 4, and detector gain 2.5× for 1000 shots. The data was processed using Flexanalysis (Bruker, Version 3.4, build 50). The spectra were externally calibrated against a 4700 peptide mix standard including angiotensin (1296 m/z), Glu-Fibrinopeptide B (1570m/z), ACTH (2093 m/z), ACTH (2465 m/z) and ACTH (3657 m/z), which were spotted on adjacent calibration wells.
Chromatin immunoprecipitation (ChIP)
Cells were crosslinked with either 2 mM ethylene glycol bis (succinimidyl succinate) (EGS, Pierce) for 45 min followed by a 1% formaldehyde for 20 min or 1% formaldehyde for 10 min before quenching with 250 mM glycine. Chromatin was released by sequential lysis with cells lysis buffer (10 mM Tris pH 8, 10 mM NaCl and 0.2% NP40) and nuclear lysis buffer (50 mM Tris pH 8, 10 mM EDTA and 1% SDS). Chromatin was sheared to ∼300 bp with 20 rounds of sonication (30 s on and 30 s off) on a Bioruptor (Diagenode) and a portion was kept as input. Chromatin was incubated overnight at 4 °C with 5 μg antibodies against either H3K9me3 (Abcam, ab8898), H3K36me3 (Abcam, ab9050), H3 (Abcam, ab1791), ATRX (Santa Cruz, SC15408), KDM4B (Abcam, ab191434) or H3.3 S31Ph (Active Motif) before immunoprecipitation with Protein A agarose beads (Sigma-Aldrich, 05015979001) and sequentially washing with low salt (20 mM Tris pH 8, 2 mM EDTA, 50 mM NaCl, 1% Triton X-100, 0.1% SDS), high salt (20 mM Tris pH 8, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.01% SDS), LiCl buffers (10 mM Tris pH 8, 1 mM EDTA, 0.25 M LiCl, 1% NP40, 1% sodium deoxycholate) and TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8). Chromatin was eluted with elution buffer (1% SDS, 100 mM NaHCO3), incubated with Proteinase K and de-crosslinked overnight at 65°C before phenol chloroform extraction and ethanol precipitation. Purified ChIP DNA was used as a template for qPCR with appropriate primers.
ChIP sequencing
H3.3 S31Ph ChIP sequencing was performed on mitotic mouse ES cell population. Cells were synchronised at G2/M by treatment with 9 μM of RO-3306 (Sigma-Aldrich) for 12 hours and released into culture medium containing 50ng/ml nocodazole following three 1x PBS washes. Mitotic synchronised cells were harvested 11 hours after the release, fixed with formaldehyde, and subjected to ChIP with an antibody against H3.3 S31Ph (Active Motif) using the ChIP protocol described above. For ChIP sequencing, DNA sample concentrations were determined by Qubit (ThermoFisher Scientific) and 20 ng of DNA was used as starting material. ChIP libraries were prepared with Nugen Ovation Ultralow System V2 (Nugen protocol M01379v1, 2014) with 10 cycles of amplification. Libraries quality was assessed by Qubit, Bioanalyzer (Agilent) and qPCR, and a single equimolar pool was made based on size adjusted qPCR quantitation. Following denaturation, 12 pM of library pools were used for cBot hybridisation and cluster generation (Illumina Protocol 15006165 v02 Feb 2016), and samples were sequenced on an Illumina HiSeq 1500 rapid mode (50 bp SR sequencing, Illumina Protocol 15035788 Rev D, April 2014). Fastq files of H3.3 S31Ph ChIP-seq and matched input samples were aligned to a repeat database with Repeat Enrichment Estimator v1.0 (55). In brief, a repeat assembly file was generated using the Repbase database and reads were aligned to this library and counted. Further details are available in reference (55). The H3.3 S31Ph ChIP sequencing dataset is available on the Gene Expression Omnibus (GEO) database.
siRNA depletion experiments and real-time PCR analyses
Four sets of siRNA oligonucleotides specific for mouse KDM4B (siKdm4b) were commercially synthesised (GenePharma). 40 nM of each siRNA oligonucleotides set was transfected into cells with Lipofectamine 2000 according to the manufacturer's instructions (Thermosciences). As controls, medium GC-content scramble siRNA oligonucleotides (siControl) (GenePharma) were included in the experiments. The lipid-siRNA complex, prepared at a Lipofectamine:siRNA ratio of 2:1, was added into mammalian cells containing cell culture media lacking penicillin and streptomycin. The cells were harvested and examined after 48h of transfection. qPCR was used to measure KDM4B expression levels after siRNA transfection.
Supplementary Information contains information on primer sequences (Supplementary Table S1), antibodies (Supplementary Table S2), peptide sequences (Supplementary Table S3) and siRNA sequences (Supplementary Table S4).
RESULTS
Generation of H3.3 S31, A31 and E31 expressing mouse ES cell lines
In mouse ES cells, H3.3 S31Ph is enriched at telomeres on mitotic chromosomes and increases at pericentric heterochromatin upon the induction of differentiation, suggesting a role for H3.3 S31Ph in regulating heterochromatin silencing (12). In line with this, ChIP-seq analysis of mitotic mouse ES cells showed that H3.3 S31Ph is present at the telomere and also at other DNA repeats including the IAP/ERV retrotransposons (15,23) (Figure 1A). These findings further support the link between H3.3 S31Ph and heterochromatin silencing. To investigate the role of H3.3 S31Ph at DNA repeats, we employed a two-step strategy to obtain mouse ES cells expressing only either the wild-type (WT) H3.3 S31, mutant H3.3 A31 (serine to alanine) or mutant H3.3 E31 (serine to glutamic acid) protein. The A31 substitution mimics H3.1/2 and is non-phosphorylable, (phospho-null) while the glutamic acid E31 substitution is a structural mimic of a phosphorylated serine. In mouse, histone H3.3 is encoded by two unlinked genes, H3f3a and H3f3b. We previously established a Cre/loxP conditional allelic replacement system targeted to H3f3a and H3f3b in mice (24,33). We bred a 129S1/SvImJ mouse strain which was homozygous for all four conditional (f, for floxed) alleles, i.e. H3f3af/f, H3f3bf/f, and derived XY euploid embryonic stem (ES) cells (hereafter termed H3.3 cKO) (33). The H3.3 S31, H3.3 A31 and H3.3 E31 expression cassettes were stably transfected into H3.3cKO cells. The H3f3a and H3f3b genes were ablated upon exposure to Cre recombinase and replaced with YFP and CFP reporter genes (Supplementary Figure S1A, B). Southern blot analysis on WT ES cells, H3.3cKO (H3f3af/f H3f3bf/f), H3.3 S31, A31 and E31 clones yielded fragment sizes which were consistent with successful deletion of H3.3 alleles (Supplementary Figure S1A, C). Cells with H3f3a–/– H3f3b+/+ and H3f3a+/+ H3f3b–/– H3.3 genotypes were included as controls. Western blot analysis with an antibody against YFP/CFP proteins confirmed the removal of endogenous H3.3 (Supplementary Figure S1D). Expression of H3.3 S31, A31 and E31 was verified by H3.3 S31Ph staining and qPCR analyses (Figure 1 B, C and Supplementary Figure S1E). The S31A or S31E substitution did not result in a global change in H3.3 protein levels or chromatin states as shown by western blot analyses of mutant H3.3 A31 and H3.3 E31 cells (Supplementary Figure S1F–L). The anti-H3.3 antibody used here has been previously been validated using H3.3 knockout mouse ES cell lines (14) (Supplementary Figure S1M).
Figure 1.

Substitution of H3.3 S31 with A31 affects ATRX binding and heterochromatin formation at telomeres in mouse ES cells. (A) DNA repeats which are enriched for H3.3 S31Ph relative to input. Bars represent ChIP-sequencing reads of H3.3 S31Ph in mitotic cells normalised to total read count, with reads from input sequencing as a control. Repeats with minimum 100 mapped reads and two-fold over enriched over input are shown. (B) Immunofluorescence analysis on H3.3 S31 conditional (H3f3 f/fH3f3 f/f), S31 (wildtype), A31 (phospho-null) and E31 (phospho-mimetic) mouse ES cell lines with an antibody against H3.3 S31Ph (green), followed by FISH analysis using a DNA probe against telomeric TTAGGG repeats (red). H3.3 S31Ph is enriched at telomeres (arrows) in H3f3 f/fH3f3 f/f and H3.3 S31 cells but not in H3.3 A31 and E31 cells. Scale bar represents 4 μm. (C) H3.3 S31, A31 and E31 transcript levels were determined by quantitative RT-PCR analyses using specific primers against H3.3 S31, A31 and E31 cDNA, where the last 2 nucleotides of the 5′ forward primers recognise the first 2 nucleotides of H3.3 S31, A31 and E31 codons, respectively. The same 3′ reverse primer was used. The relative expression levels of H3.3 S31, A31 and E31 are normalised to GAPDH expression levels (mean ± SEM, n = 3). (D) Immunostaining of ATRX (green), followed by Tel-FISH (Telomere-fluorescence in situ hybridisation) analysis with a DNA probe against telomeric TTAGGG repeats (red). Clear localisation of ATRX at telomeres (shown by arrows) was detected in wildtype (WT), H3.3 S31 and E31 mouse ES cell lines, but the co-localised foci were significantly reduced in H3.3 A31 cells. Scale bar represents 4 μm. (E) ChIP-qPCR analyses (mean ± SEM, n = 3) showing relative enrichment levels of ATRX/Input, H3.3/Input, H3K9me3/Input and total H3/Input at telomeres. P-values calculated using Student's t-test (** P< 0.05; * P< 0.1; ns, non-significant;). In H3.3 E31 cells, both ATRX (P = 0.0695) and H3K9me3 (P= 0.013) levels are increased at telomeres, but H3.3 A31 cells showed decreases in ATRX (P= 0.097) and H3K9me3 (P= 0.121) levels.
H3.3 S31Ph regulates ATRX binding and heterochromatin formation at the telomere
We and others have reported that ATRX deposits H3.3 to form a heterochromatin structure enriched with H3K9me3 and HP1α at the telomere in mouse ES cells (7,9,12,13), and this ATRX-mediated heterochromatin assembly occurs within the PML bodies (8). H3.3 S31 is phosphorylated during mitosis in mammalian cells (12,45). This PTM is enriched at heterochromatic DNA repeats including the telomere in pluripotent mouse ES cells (12) and pericentric heterochromatin of metaphase chromosomes (45) (Figure 1B). We sought to determine if H3.3 S31 and its phosphorylated form is required for H3.3 deposition by ATRX at telomeres. Immunofluorescence analysis showed a reduced association of ATRX with telomeres in H3.3 A31 phospho-null cells (Figure 1D). In line with this, ChIP-qPCR analysis also showed a reduced ATRX binding at the telomere in H3.3 A31 cells but ATRX level at the telomere was increased in H3.3 E31 phospho-mimetic cells (Figure 1E). Considering that ATRX regulates heterochromatin formation, we also assessed the level of H3K9me3 at telomeres (Figure 1E). H3.3 E31 cells showed an increase in H3K9me3 level at the telomere, while H3.3 A31 cells showed a decreasing trend (Figure 1E). In addition, we noted an increase in H3 level at the telomere in H3.3 E31 cells, indicating increased nucleosome occupancy (Figure 1E). These findings indicate that ATRX/H3K9me3 maintenance at the telomere was upregulated in H3.3 E31 cells. Considering that ATRX mediated heterochromatin assembly at telomeres occurs within the PML bodies (8,18–20), we also assessed the impact of H3.3 S31 substitution on the localisation of telomeres to PML bodies. Strikingly, telomeres localisation with PML bodies increased in H3.3 E31 cells, but reduced in H3.3 A31 cells compared to H3.3. S31 cells (Figure 2A, B). In addition, H3.3 A31 cells displayed smaller PML body foci, comparable to those in ATRX knockout cells (Figure 2A). Based on these findings, we propose that H3.3 S31 and its phosphorylated form play a role in preserving telomeric ATRX/H3K9me3 heterochromatin formation within the PML bodies.
Figure 2.

H3.3 S31Ph regulates telomeric localisation to PML bodies and KDM4B activity in mouse ES cells. (A) Immunostaining of PML (red) and TERF1 (green; marker for telomere) showing co-localisation of PML and TERF1 (shown by arrows) in wildtype (WT), H3.3 S31, E31 and Atrx–/ymouse ES cell lines, but the co-localised signal was greatly reduced in H3.3 A31 and Atrx–/y mouse ES cell lines. Scale bars: 4 μm. (B) Percentages (mean ± SEM, n = 3, 16 nuclei were analysed in each experiment) of co-localized PML/TERF1 foci in wildtype (WT), H3.3 S31, A31, E31 and Atrx–/y mouse ES cell lines are shown. (C) Protein immunoprecipitation with an anti-Flag antibody in H3.3 S31, A31 and E31 mouse ES cell lines expressing Flag-tagged KDM4B, followed by western blot analysis with anti-Flag and H3.3 antibodies, respectively. The changes in H3.3/KDM4B binding affinities were presented as the relative ratios of H3.3 and KDM4B immunoprecipitated (H3.3 Ip/KDM4B Ip) in WT, A31 and E31 cell lines, respectively. (D) ChIP-qPCR analyses (mean ± SEM, n = 3) of KDM4B in H3.3 S31, A31 and E31 mouse ES cell lines, showing increased KDM4B binding (KDM4B/Input) at the telomere. (E) Mass spectrometry analysis of in vitro KDM4B histone demethylase assays. KDM4B recombinant protein was incubated with either H3.3 S31, A31, E31 or S31Ph K36me3 peptides. Dashed lines indicate expected masses for K36 me0, me1, me2 and me3. (B, D) P-values calculated using Student's t-test (** P< 0.05; * P< 0.1; ns, non-significant).
H3.3 S31Ph inhibits KDM4B binding and function
As the H3.3 S31 substitutions affect ATRX binding, H3K9me3 heterochromatin and association with PML bodies, we speculated that this residue might interfere with a H3K9me3 demethylase. We recently showed that the dual histone H3K9/K36 demethylase KDM4B binds the telomeres and regulates chromatin accessibility by antagonising H3K9me3/HP1α levels (22). While ATRX forms a heterochromatin structure (14–16), KDM4B (56,57) removes methyl groups from H3K9me3 at telomeres during replication (22). The removal of H3K9me3 relieves heterochromatin to facilitate accessibility to DNA polymerase during replication. We also showed that H3K9me3/HP1α containing heterochromatin is required for the association of telomeres with PML bodies, and overexpression of KDM4B disrupts PML body formation. We investigated if the S31 substitutions would affect the association and activity of KDM4B at the telomere. We immunoprecipitated KDM4B in H3.3 S31, A31 and E31 cells expressing FLAG-tagged KDM4B (53,54), and immuno-blotted for H3.3 binding. We found that FLAG-KDM4B pulled down higher levels of H3.3 A31 compared to WT H3.3 S31, while the opposite was true for H3.3 E31 (Figure 2C). This demonstrates that the S31A substitution increases binding preference for KDM4B while H3.3 E31 inhibits KDM4B association. In line with this, we also found increased association between KDM4B with telomeres in H3.3 A31 cells by ChIP-qPCR (Figure 2D). These findings indicate that the A31 substitution leads to a greater binding of the KDM4B demethylase that removes H3K9me3 at telomeres.
Given that KDM4B binds preferentially to the phospho-null H3.3 A31, we investigated if H3.3 S31Ph may inhibit activity of KDM4B. To investigate this, we performed in vitro demethylation assays with recombinant KDM4B and the WT S31, A31 and E31 K36me3 peptides, and analysed the results by mass spectrometry (Figure 2E). KDM4B demethylated K36me3 in the WT H3.3 S31 and A31 peptides with equal efficiency. In contrast, both the H3.3 S31Ph and H3.3 E31 peptides substantially inhibited the demethylation of K36me3 by KDM4B (Figure 2E). These results support a model where H3.3 S31Ph and H3.3 E31 phospho-mimic inhibit the demethylase activity of KDM4B. We have recently shown that KDM4B binds to and removes H3K9me3 at telomeres to increase chromatin accessibility and facilitate replication (22). Since KDM4B is a dual H3K9/K36 demethylase, an increased interaction between KDM4B and H3.3 A31 phospho-null mutant (Figure 2C) would remove H3K9me3 at telomeres in H3.3 A31 cells (Figure 1E). Conversely, the increased levels of H3K9me3 and ATRX at telomeres in H3.3 E31 mutants could be induced by the inhibition of KDM4B by H3.3 E31 phospho-mimic (Figure 1D, E). If the loss of H3K9me3 at telomeres in H3.3 A31 is driven by increased interactions with KDM4B, then depletion of KDM4B should rescue heterochromatin in these cells. To investigate this, we performed RNAi knockdown of KDM4B in H3.3 A31 cells (Supplementary Figure S2A). Compared to WT H3.3 S31 cells subjected to scramble siRNA control knockdown, depletion of KDM4B led to an increase in the level of H3K9me3 at telomeres in H3.3 A31 cells, but not in H3.3 E31 cells (Figure 3A, B). The depletion of KDM4B also increased association of telomeres with HP1α (Supplementary Figure S2B) and PML bodies (Figure 3C, D), indicating a restored formation of a heterochromatic structure at telomeres. These data are consistent with a role for H3.3S31Ph in ATRX-mediated heterochromatin modification at telomeres, through regulation of KDM4B binding and activity.
Figure 3.
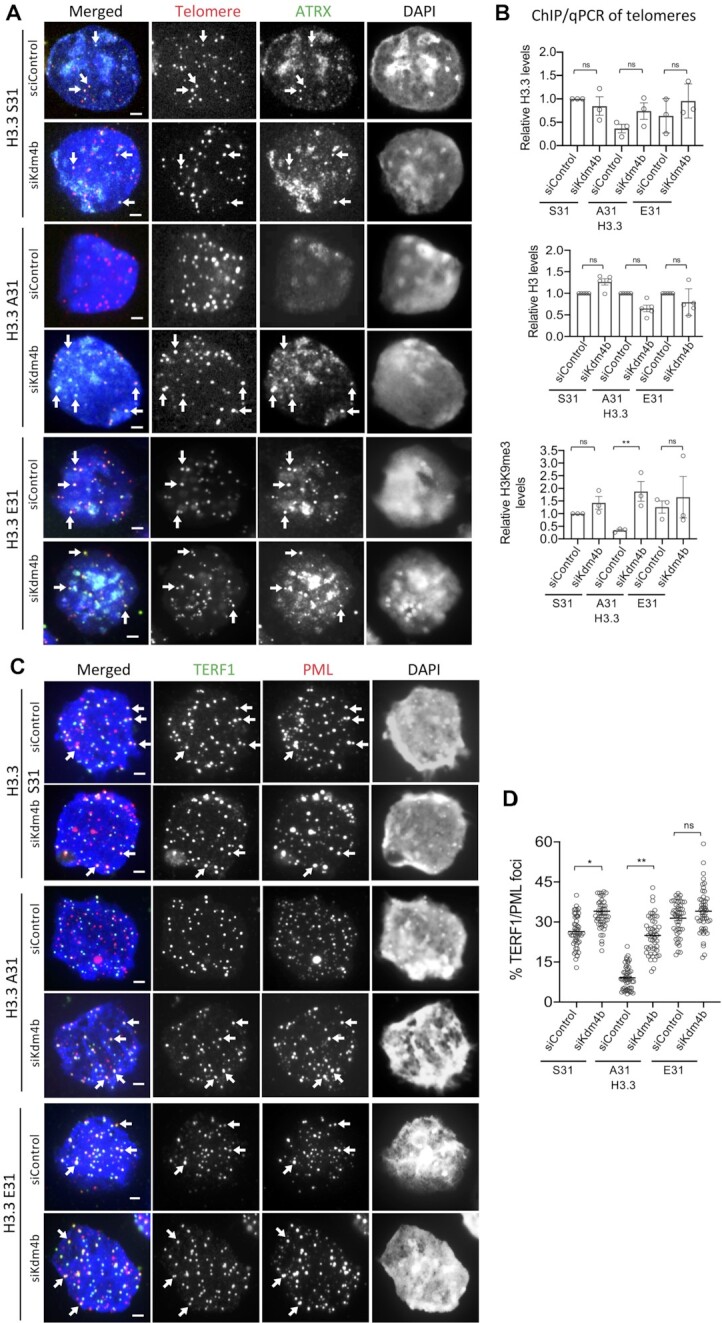
KDM4B depletion restores ATRX binding, heterochromatin formation and PML localisation at telomeres in H3.3 A31 mouse ES cells. (A) Immunostaining of ATRX (green), followed by Tel-FISH analysis with a DNA probe against telomeric TTAGGG repeats (red) in H3.3 S31, A31 and E31 mouse ES cell lines. Compared to control siRNA (siControl), Kdm4b siRNA (siKdm4b) mediated knockdown led to restored ATRX binding at telomeres (shown by arrows). Scale bars: 4 μm. (B) ChIP-qPCR analyses showing relative levels of H3.3, H3K9me3 and H3 at telomeres (mean ± SEM, n = 3) in H3.3 S31 and A31 mouse ES cell lines subjected to control siRNA (siControl) and Kdm4b siRNA (siKdm4b) knockdown, compared to WT H3.3 S31 cells. (C) Immunostaining of TERF1 (green; marker for telomere) and PML (red) showing increased co-localisation of PML and TERF1 (shown by arrows) in H3.3 A31 cells, following the knockdown of KDM4B expression. Scale bars: 4 μm. (D) Percentages (mean ± SEM, n = 4, 24 nuclei were analysed in each experiment) of co-lcalized PML/TERF1 foci in H3.3 S31 and A31 mouse ES cell lines with and without knockdown of KDM4B (siKdm4b). B, D P-values calculated using Student's t-test (** P< 0.05; * P< 0.1; ns, non-significant).
H3.3 S31Ph regulates chromatin integrity at heterochromatin in the genome
We have previously shown that KDM4B controls accessibility at telomeric heterochromatin during replication and that loss of KDM4B leads to increased H3K9me3, replication stress and DNA damage at telomeres (22). As KDM4B function is regulated by phosphorylation of H3.3 S31, we investigated if the H3.3 E31 substitution, which inhibited KDM4B activity, also led to increased damage and loss of telomere integrity. Indeed, we found that H3.3 E31 expression led to a substantial increase in γH2AX level at telomeres (Figure 4A-B), liken to the increase in damage at the telomere in Kdm4b knockout mouse ES cells (22). The H3.3 A31 mutant which promotes KDM4B activity, also showed an increase in telomeric DNA damage as assessed by γH2AX staining, but to a lesser extent (Figure 4A-B). These findings were verified by ChIP-qPCR of γH2AX in WT and H3.3 mutant lines (Figure 4C). We also investigated if the changes in telomeric chromatin state (Figure 1E) and integrity (Figure 4A-C) affect the maintenance of telomere length and transcript level in H3.3 A31 and E31 cells (Supplementary Figure S3A, B). A decrease in telomere length was detected in H3.3 E31 cells, however, no significant change was found in H3.3 A31 cells (Supplementary Figure S3A, B). As for telomeric transcription, both H3.3 A31 and E31 cells showed a reduction in TERRA transcription (Supplementary Figure S3B).
Figure 4.

Increase in γH2AX level at telomeres in H3.3 E31 mouse ES cells. (A) Immunostaining of TERF1 (red; marker for telomere) and γH2AX (green) showing increased γH2AX at telomeres (indicated by arrows) in both interphase and mitotic H3.3 E31 cells. Y chromosomes are indicated by arrowheads. Scale bars: 4 μm. (B) Percentages (mean ± SEM, n = 3, 16 nuclei were analysed in each experiment) of co-localized TERF1/γH2AX foci (TIF) are shown, and increased TIFs are found in H3.3 E31 and Kdm4b–/– cells. (C) ChIP-qPCR analyses (mean ± SEM, n = 3) of γH2AX at telomeres and Foxp3 (a control gene) in H3.3 S31, A31 and E31 mouse ES cell lines. (B, C) P-values calculated using Student's t-test (** P< 0.05; * P< 0.1; ns, non-significant).
In addition to telomeres, we also observed a high level of γH2AX on what appears to be the Y chromosome in H3.3 E31 cells (Figure 4A, arrowhead). The Y chromosome is highly repetitive in sequence and contains large regions of H3K9me3 heterochromatin especially on its q arm (e.g. Yq mega-satellite DNA repeat) (49). It is therefore possible that the H3.3 E31 phospho-mimic mutation can interfere with KDM4B and induce damage at other heterochromatin regions in the genome, including the Yq mega-satellite repeats. To investigate this, we first determined if H3.3 S31Ph was present on Yq mega-satellite repeats by immunofluorescence analysis in WT ES cells. We stained WT cells with an antibody against H3.3 S31Ph, followed by FISH using a Yq mega-satellite DNA repeat probe (49) and detected clear H3.3 S31Ph staining across the long arm of the Y chromosome (Figure 5A, B). We then investigated if E31 phospho-mimic mutant increased DNA damage on the Y chromosome. Immunofluorescence analysis was performed with an antibody against γH2AX, followed by FISH using the Yq mega-satellite repeat DNA probe (49). The H3.3 E31 cells showed substantially increased DNA damage signals on the Y chromosome relative to the WT and H3.3 A31 cells (Figure 5C, D). These findings support the idea that H3.3 S31Ph also regulates chromatin accessibility at other heterochromatin regions in the genome including Y chromosome satellite repeats, in addition to the telomeres.
Figure 5.
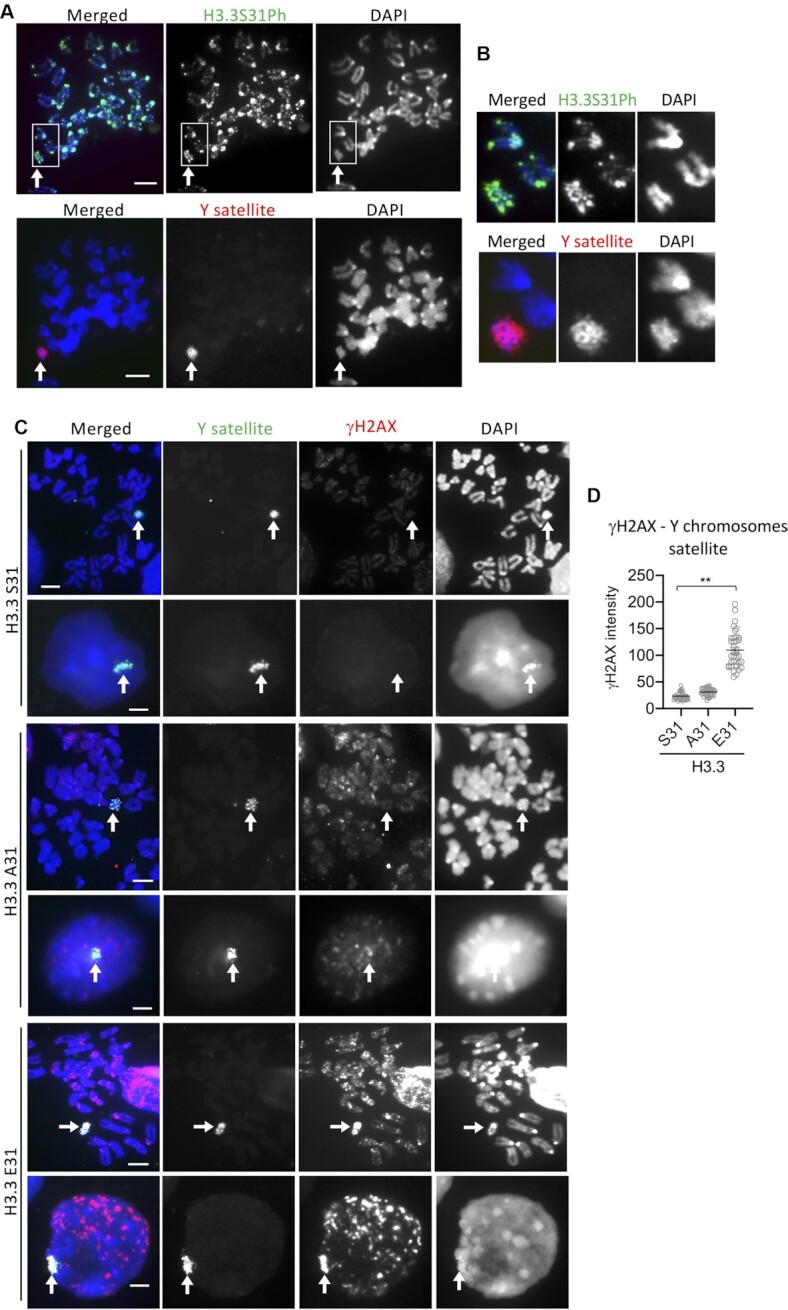
Increase in γH2AX level at Y chromosome satellite repeats in H3.3 E31 mouse ES cells. (A, B) Immunostaining of H3.3 S31Ph (green) in wildtype (WT) mouse ES cells (top panel), followed by FISH analysis with a DNA probe against Yq megasatellite repeats (red, bottom panel). (C, D) Immunostaining of γH2AX (red; marker for damage), followed by FISH analysis with a DNA probe against Yq megasatellite repeats (green) in interphase and metaphase H3.3 S31, A31 and E31 mouse ES cell lines (C). Increased γH2AX was also detected on Y chromosome in H3.3 E31 cells (arrows). Scale bars: 4 μm. The relative intensities (mean ± SEM, n = 3, 16 nuclei were analysed in each experiment) of γH2AX signals on Y chromosomes is shown in (D). P-values calculated using Student's T-test (** P< 0.05).
We have previously shown that H3.3 S31Ph is enriched at pericentric heterochromatin of metaphase chromosomes in mouse ES cells that have been subjected to cellular differentiation (12). It is possible that H3.3 S31Ph is also important for regulating H3K9me3 heterochromatin accessibility to prevent DNA damage at these repeats. To investigate this, we assessed the effect of the H3.3 E31 substitution on pericentric heterochromatin in mouse ES cells subjected to cellular differentiation by the withdrawal of leukemia inhibitory factor and the addition of retinoic acid to the culture media (12,45). As expected, H3.3 S31 WT ES cells showed increased H3.3 S31Ph signal at pericentric heterochromatin 3 days after the induction of differentiation (Figure 6A), indicating that this modification is important at pericentric heterochromatin in differentiated cells. In line with this, we detected an increase in DNA damage, as assessed by γH2AX intensities at pericentric heterochromatin in H3.3 E31 cells, but not in H3.3 A31 cells (Figure 6B, C). These findings support the idea that H3.3 S31Ph also regulates chromatin accessibility at pericentric heterochromatin. Collectively, these results suggest that H3.3 S31Ph is required for DNA integrity at repetitive regions including the telomeres, pericentric heterochromatin and Y chromosome satellite DNA repeats.
Figure 6.

Increase in γH2AX level at pericentric heterochromatin in differentiated H3.3 E31 mouse ES cells. (A) Immunostaining of H3.3 S31Ph (green), followed by FISH analysis with a DNA probe against mouse major satellite (red). H3.3 S31Ph is enriched at the pericentric heterochromatin (arrows) in differentiated mouse ES cells. Scale bars: 4 μm. (B) Immunostaining of γH2AX (red; marker for damage), followed by FISH analysis with a DNA probe against mouse major satellite (green). Increased γH2AX was also detected on pericentric heterochromatin in H3.3 E31 cells (arrows). Scale bars: 4 μm. (C) Percentages (mean ± SEM, n = 3, 16 nucleic were analysed in each experiment) of co-localized pericentric heterochromatin (major satellite)/γH2AX foci (TIF) are shown, and increased percentage of γH2AX-enriched foci are found in H3.3 E31 cells. P-values calculated using Student's t-test (** P< 0.05).
DISCUSSION
Histone H3.3 is a key player in diverse processes including gene regulation, cell differentiation and embryonic development. The presence of H3.3 at actively transcribed genes is essential for preserving transcriptional memory (5). At transcriptionally silent regions, H3.3 is deposited by the ATRX/DAXX complex and this is required for maintaining heterochromatin (14–16). The phosphorylatable serine 31 residue distinguishes H3.3 from the H3.1/2 canonical histones. The S31(Ph) residue is highly conserved in eukaryotes, suggesting that this residue is an important epigenetic regulator of chromatin organisation. Recent studies have linked H3.3 S31Ph to active transcription (47,48,58), however, the role of H3.3 S31 at transcriptionally silenced repetitive DNA regions remains unknown. Here we provide evidence for an unrecognized role of H3.3 S31Ph as a phosphorylation switch that regulates the function of KDM4B demethylase at telomeres. The unphosphorylated form of H3.3 S31 binds KDM4B and allows H3K9me3 demethylation, while phosphorylated H3.3 S31 inhibits KDM4B activity and preserves H3K9me3 at telomeric heterochromatin. Accordingly, H3.3 A31 phospho-null cells show increased KDM4B binding at telomeres, resulting in reductions of H3K9me3, ATRX and HP1α and reduced association of telomeres with PML bodies. Additionally, depletion of KDM4B in H3.3 A31 cells restores ATRX, H3K9me3 and HP1α levels at telomeres, and enhanced telomere localization at PML bodies. These observations agree with our recent finding that H3K9me3/HP1α containing heterochromatin is required for the association of telomeres with PML bodies, and that overexpression of KDM4B disrupts PML body formation (22). PML bodies are phase-separated liquid compartments which form through multivalent interactions between proteins and nucleic acids. HP1α is a strong driver of phase separation (59,60) and it binds the H3K9me3 residue which is demethylated by KDM4B (61). In H3.3 A31 cells, the missing phosphorylation switch could account for an uncontrolled demethylation by KDM4B, resulting in the loss of H3K9me3 and inability to nucleate PML bodies at telomeres.
H3.3 S31Ph has recently been proposed to act either by attracting or repulsing protein factors (46). Specifically, H3.3 S31Ph has been shown to regulate binding of histone acetyltransferase P300 and repressor ZMYND11 (a factor that recognises H3K36me3 and involved in intron retention that recognises H3K36me3) (47,48,58). Here we provide evidence that H3.3 S31Ph acts as a phosphorylation switch that regulates the recruitment of KDM4B demethylase to telomeres. Black et al. demonstrated that a related H3K9me3 demethylase, KDM4A, controls chromatin accessibility of a late-replicating heterochromatic satellite region by antagonising H3K9me3/HP1α (56,62), and over-expression of KDM4A advances S-phase entry (62). Our findings support the idea that KDM4B regulates heterochromatin accessibility at telomeres during replication (22). Although telomeres are enriched in heterochromatin, KDM4B binds telomeres in S-phase to promote accessibility during replication. Depletion of KDM4B results in increased H3K9me3 and HP1α at telomeres, and this in turn leads to increased replication stress and damage at telomeres (22). Consistent with this idea, the damage at telomere in H3.3 E31 cells may be caused by the inability of KDM4B to bind and demethylate H3K9me3 during replication. As a consequence, the high level of H3K9me3 could act as a barrier to DNA polymerase during replication, thereby inducing damage at the telomere and compromising the ability to maintain telomere length. Consistent with this idea, the gain in H3K9me3 heterochromatin at the telomere could account for the reduction in TERRA in H3.3 E31 cells. However, a lower level of telomeric transcript/TERRA was also found in H3.3 A31 cells, although these cells showed loss of H3K9me3 heterochromatin. This could be driven by the formation of a secondary DNA structure that blocks TERRA transcription. Supporting this, the presence of ATRX at telomeres has been shown to play a central role in suppressing deleterious DNA secondary structures including r-loops that form at transcribed telomeric repeats, (63). Further investigation is required to unravel the impact of H3.3 S31 and its phosphorylated form in suppressing r-loops and regulating telomeric transcription.
H3.3 E31 cells also showed increased damage at other H3.3 S31Ph heterochromatin regions in addition to the telomeres, including Y chromosome satellite repeats and pericentric heterochromatin. Although KDM4B has not been reported to control chromatin accessibility at these regions, overexpression of KDM4A H3K9me3 demethylase has been found to antagonize HP1α recruitment to pericentric heterochromatin (56). Based on our findings, we propose that H3.3 S31Ph acts as a phosphorylation switch which regulates H3K9me3 heterochromatin maintenance and accessibility at repetitive DNA regions including the telomeres, pericentric heterochromatin and Y chromosome satellite repeat (Figure 7). It has been well established that ATRX/DAXX mediated H3.3 deposition promotes H3K9me3 heterochromatin formation, however, KDM4B counteracts this by removing H3K9me3. H3.3 S31 and its phosphorylation play key roles in regulating the balance between H3K9me3 heterochromatin maintenance and KDM4B function at repetitive DNA regions. At these sites, the absence of H3.3 phosphorylation in S phase allows the recruitment of KDM4B to reduce H3K9me3 and to facilitate replication at heterochromatin. Following the completion of replication, S31 is phosphorylated, preventing removal of H3K9me3 by KDM4B and thereby maintaining a heterochromatic state from mitosis until the next G1/S phase. Our results yield mechanistic insights into how ATRX, H3.3 S31Ph and KDM4B act together to preserve heterochromatin. Notably, H3.3 S31Ph signal is substantially increased across the chromosome arms in ATRX-mutated cancers that utilise the ALT (Alternative Lengthening of telomeres) telomere maintenance pathway (43). Further work is essential to determine if H3.3 S31Ph regulates KDM4B activity and heterochromatin maintenance in these cancers. Altogether, we show that H3.3 S31Ph acts as a switch that regulates protein binding, providing insight into the function of H3.3 in regulating genome integrity.
Figure 7.
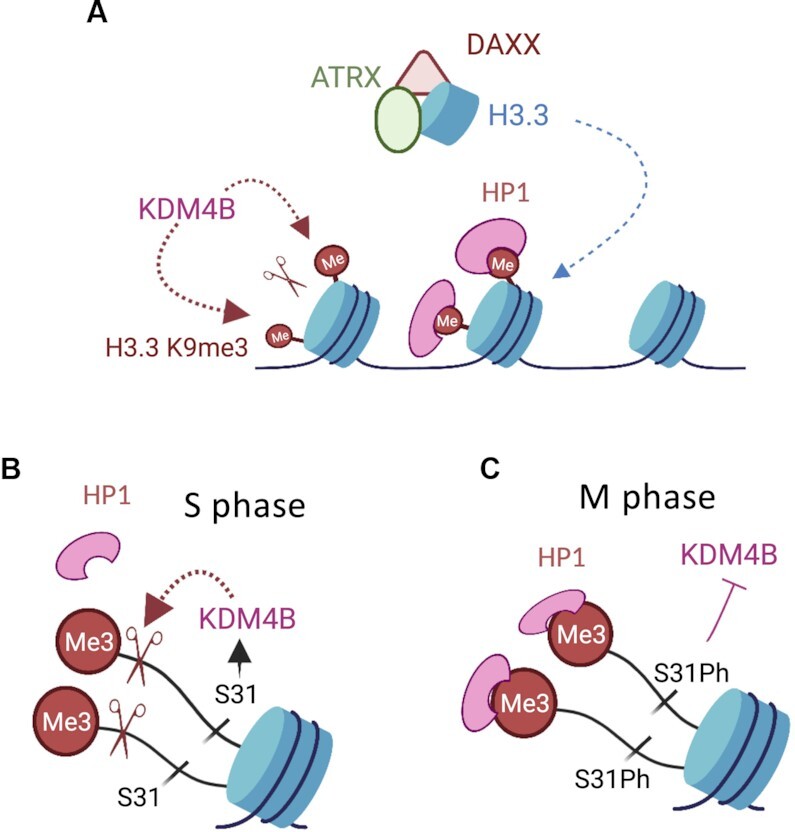
H3.3 S31Ph acts as a phosphorylation switch which regulates KDM4B binding and heterochromatin accessibility at the telomeres. (A) ATRX and KDM4B act to maintain telomeric heterochromatin integrity. ATRX complexes with DAXX to deposit H3.3 at telomeres to form H3K9me3 heterochromatin, enabling binding of HP1α via its interaction with H3K9me3. Despite being enriched with H3K9me3, telomeres are also enriched with KDM4B that demethylates H3K9me3. We propose that ATRX and KDM4B act to maintain the level of H3K9me3 at the telomere, and this is achieved via H3.3 S31Ph switch (B) During replication, the absence of H3.3 phosphorylation in S phase allows KDM4B to bind and demethylate H3K9me3 to promote heterochromatin accessibility at telomeres. (C) After replication and as cells progress into M phase, H3.3 S31 is phosphorylated, preventing removal of H3K9me3 by KDM4B and thereby maintaining a heterochromatic state from mitosis until the next G1/S phase. Figure made in BioRender.com.
DATA AVAILABILITY
Data that support the findings of this study are available from the corresponding author on reasonable request. H3.3 S31Ph ChIP sequencing dataset is available on the Gene Expression Omnibus (GEO) database under the accession number GSE181692.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by Worldwide Cancer Research, Brain Tumour Charity UK and NHMRC Australia. High-throughput sequencing was performed at the MHTP Medical Genomics Facility. This research was supported by use of the Nectar Research Cloud, a collaborative Australian research platform supported by the NCRIS-funded Australian Research Data Commons (ARDC).
Author contributions: M.U., B.V., F-L.C., L.H., A.G., D.S., P.T., J.R.M., H.P.V. and L.H.W. performed the experiments and analysed the data. P.K and P.C. provided materials and contributed to discussion. M.U., L.H., A.G., D.S., J.R.M, P.P.D, P.K., P.C., H.P.V. and L.H.W. wrote and edited the manuscript. M.U. and B.V. contributed equally to this work.
Contributor Information
Maheshi Udugama, Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Clayton, Victoria 3800, Australia.
Benjamin Vinod, Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Clayton, Victoria 3800, Australia.
F Lyn Chan, Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Clayton, Victoria 3800, Australia.
Linda Hii, Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Clayton, Victoria 3800, Australia.
Andrew Garvie, Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Clayton, Victoria 3800, Australia.
Philippe Collas, Department of Molecular Medicine, Institute of Basic Medical Sciences, Faculty of Medicine, University of Oslo, 0317 Oslo, Norway; Department of Immunology and Transfusion Medicine, Oslo University Hospital, 0424 Oslo, Norway.
Paul Kalitsis, Victorian Clinical Genetics Service, Murdoch Children's Research Institute and Department of Paediatrics, University of Melbourne, Royal Children's Hospital, Parkville, Victoria 3052, Australia.
David Steer, Biomedical Proteomics Facility, Monash University, Wellington Road, Clayton, Victoria 3800, Australia.
Partha P Das, Department of Anatomy and Developmental Biology, Biomedicine Discovery Institute, Monash University, Clayton, Victoria 3800, Australia.
Pratibha Tripathi, Department of Anatomy and Developmental Biology, Biomedicine Discovery Institute, Monash University, Clayton, Victoria 3800, Australia.
Jeffrey R Mann, Department of Anatomy and Developmental Biology, Biomedicine Discovery Institute, Monash University, Clayton, Victoria 3800, Australia.
Hsiao P J Voon, Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Clayton, Victoria 3800, Australia.
Lee H Wong, Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Clayton, Victoria 3800, Australia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Worldwide Cancer Research and Brain Tumour Charity UK. Funding for open access charge: Monash University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Tagami H., Ray-Gallet D., Almouzni G., Nakatani Y.. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004; 116:51–61. [DOI] [PubMed] [Google Scholar]
- 2. Ray-Gallet D., Woolfe A., Vassias I., Pellentz C., Lacoste N., Puri A., Schultz D.C., Pchelintsev N.A., Adams P.D., Jansen L.E.et al.. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell. 2011; 44:928–941. [DOI] [PubMed] [Google Scholar]
- 3. Groth A., Corpet A., Cook A.J., Roche D., Bartek J., Lukas J., Almouzni G.. Regulation of replication fork progression through histone supply and demand. Science. 2007; 318:1928–1931. [DOI] [PubMed] [Google Scholar]
- 4. Smith S., Stillman B.. Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell. 1989; 58:15–25. [DOI] [PubMed] [Google Scholar]
- 5. Chen P., Zhao J., Wang Y., Wang M., Long H., Liang D., Huang L., Wen Z., Li W., Li X.et al.. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev. 2013; 27:2109–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ahmad K., Henikoff S.. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell. 2002; 9:1191–1200. [DOI] [PubMed] [Google Scholar]
- 7. Goldberg A.D., Banaszynski L.A., Noh K.M., Lewis P.W., Elsaesser S.J., Stadler S., Dewell S., Law M., Guo X., Li X.et al.. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010; 140:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang F.T., McGhie J.D., Chan F.L., Tang M.C., Anderson M.A., Mann J.R., Andy Choo K.H., Wong L.H. PML bodies provide an important platform for the maintenance of telomeric chromatin integrity in embryonic stem cells. Nucleic Acids Res. 2013; 41:4447–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Drane P., Ouararhni K., Depaux A., Shuaib M., Hamiche A.. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010; 24:1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Law M.J., Lower K.M., Voon H.P., Hughes J.R., Garrick D., Viprakasit V., Mitson M., De Gobbi M., Marra M., Morris A.et al.. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell. 2010; 143:367–378. [DOI] [PubMed] [Google Scholar]
- 11. Lewis P.W., Elsaesser S.J., Noh K.M., Stadler S.C., Allis C.D.. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:14075–14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wong L.H., Ren H., Williams E., McGhie J., Ahn S., Sim M., Tam A., Earle E., Anderson M.A., Mann J.et al.. Histone H3.3 incorporation provides a unique and functionally essential telomeric chromatin in embryonic stem cells. Genome Res. 2009; 19:404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wong L.H., McGhie J.D., Sim M., Anderson M.A., Ahn S., Hannan R.D., George A.J., Morgan K.A., Mann J.R., Choo K.H.. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010; 20:351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Udugama M., Chang F.T., Chan F.L., Tang M.C., Pickett H.A., McGhie J.D., Mayne L., Collas P., Mann J.R., Wong L.H.. Histone variant H3.3 provides the heterochromatic H3 lysine 9 tri-methylation mark at telomeres. Nucleic Acids Res. 2015; 43:10227–10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elsasser S.J., Noh K.M., Diaz N., Allis C.D., Banaszynski L.A.. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature. 2015; 522:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Voon H.P., Hughes J.R., Rode C., De La, Rosa-Velazquez I.A., Jenuwein T., Feil R., Higgs D.R., Gibbons R.J.. ATRX plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep. 2015; 11:405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Voon H.P., Wong L.H.. New players in heterochromatin silencing: histone variant H3.3 and the ATRX/DAXX chaperone. Nucleic Acids Res. 2016; 44:1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Delbarre E., Ivanauskiene K., Kuntziger T., Collas P.. DAXX-dependent supply of soluble (H3.3-H4) dimers to PML bodies pending deposition into chromatin. Genome Res. 2013; 23:440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Delbarre E., Ivanauskiene K., Spirkoski J., Shah A., Vekterud K., Moskaug J.O., Boe S.O., Wong L.H., Kuntziger T., Collas P.. PML protein organizes heterochromatin domains where it regulates histone H3.3 deposition by ATRX/DAXX. Genome Res. 2017; 27:913–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ivanauskiene K., Delbarre E., McGhie J.D., Kuntziger T., Wong L.H., Collas P.. The PML-associated protein DEK regulates the balance of H3.3 loading on chromatin and is important for telomere integrity. Genome Res. 2014; 24:1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Voon H.P.J., Collas P., Wong L.H.. Compromised telomeric heterochromatin promotes ALTernative lengthening of telomeres. Trends Cancer. 2016; 2:114–116. [DOI] [PubMed] [Google Scholar]
- 22. Udugama M., Hii L., Garvie A., Cervini M., Vinod B., Chan F.L., Das P.P., Mann J.R., Collas P., Voon H.P.J.et al.. Mutations inhibiting KDM4B drive ALT activation in ATRX-mutated glioblastomas. Nat. Commun. 2021; 12:2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sadic D., Schmidt K., Groh S., Kondofersky I., Ellwart J., Fuchs C., Theis F.J., Schotta G.. Atrx promotes heterochromatin formation at retrotransposons. EMBO Rep. 2015; 16:836–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Skene P.J., Henikoff S.. Histone variants in pluripotency and disease. Development. 2013; 140:2513–2524. [DOI] [PubMed] [Google Scholar]
- 25. Hodl M., Basler K.. Transcription in the absence of histone H3.3. Curr. Biol. 2009; 19:1221–1226. [DOI] [PubMed] [Google Scholar]
- 26. Sakai A., Schwartz B.E., Goldstein S., Ahmad K.. Transcriptional and developmental functions of the H3.3 histone variant in drosophila. Curr. Biol. 2009; 19:1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Delaney K., Mailler J., Wenda J.M., Gabus C., Steiner F.A.. Differential expression of histone H3.3 genes and their role in modulating temperature stress response in caenorhabditis elegans. Genetics. 2018; 209:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bonnefoy E., Orsi G.A., Couble P., Loppin B.. The essential role of drosophila HIRA for de novo assembly of paternal chromatin at fertilization. PLoS Genet. 2007; 3:1991–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Orsi G.A., Algazeery A., Meyer R.E., Capri M., Sapey-Triomphe L.M., Horard B., Gruffat H., Couble P., Ait-Ahmed O., Loppin B.. Drosophila yemanuclein and HIRA cooperate for de novo assembly of H3.3-containing nucleosomes in the male pronucleus. PLoS Genet. 2013; 9:e1003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frey A., Listovsky T., Guilbaud G., Sarkies P., Sale J.E.. Histone H3.3 is required to maintain replication fork progression after UV damage. Curr. Biol. 2014; 24:2195–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Romanello M., Schiavone D., Frey A., Sale J.E.. Histone H3.3 promotes IgV gene diversification by enhancing formation of AID-accessible single-stranded DNA. EMBO J. 2016; 35:1452–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang M.C., Jacobs S.A., Mattiske D.M., Soh Y.M., Graham A.N., Tran A., Lim S.L., Hudson D.F., Kalitsis P., O’Bryan M.K.et al.. Contribution of the two genes encoding histone variant h3.3 to viability and fertility in mice. PLoS Genet. 2015; 11:e1004964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tang M.C., Jacobs S.A., Wong L.H., Mann J.R.. Conditional allelic replacement applied to genes encoding the histone variant H3.3 in the mouse. Genesis. 2013; 51:142–146. [DOI] [PubMed] [Google Scholar]
- 34. Loppin B., Bonnefoy E., Anselme C., Laurencon A., Karr T.L., Couble P.. The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature. 2005; 437:1386–1390. [DOI] [PubMed] [Google Scholar]
- 35. Couldrey C., Carlton M.B., Nolan P.M., Colledge W.H., Evans M.J.. A retroviral gene trap insertion into the histone 3.3A gene causes partial neonatal lethality, stunted growth, neuromuscular deficits and male sub-fertility in transgenic mice. Hum. Mol. Genet. 1999; 8:2489–2495. [DOI] [PubMed] [Google Scholar]
- 36. Bush K.M., Yuen B.T., Barrilleaux B.L., Riggs J.W., O’Geen H., Cotterman R.F., Knoepfler P.S.. Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin. 2013; 6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu G., Broniscer A., McEachron T.A., Lu C., Paugh B.S., Becksfort J., Qu C., Ding L., Huether R., Parker M.et al.. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012; 44:251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sturm D., Witt H., Hovestadt V., Khuong-Quang D.A., Jones D.T., Konermann C., Pfaff E., Tonjes M., Sill M., Bender S.et al.. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012; 22:425–437. [DOI] [PubMed] [Google Scholar]
- 39. Schwartzentruber J., Korshunov A., Liu X.Y., Jones D.T., Pfaff E., Jacob K., Sturm D., Fontebasso A.M., Quang D.A., Tonjes M.et al.. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012; 482:226–231. [DOI] [PubMed] [Google Scholar]
- 40. Heaphy C.M., de Wilde R.F., Jiao Y., Klein A.P., Edil B.H., Shi C., Bettegowda C., Rodriguez F.J., Eberhart C.G., Hebbar S.et al.. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011; 333:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiao Y., Shi C., Edil B.H., de Wilde R.F., Klimstra D.S., Maitra A., Schulick R.D., Tang L.H., Wolfgang C.L., Choti M.A.et al.. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011; 331:1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fontebasso A.M., Papillon-Cavanagh S., Schwartzentruber J., Nikbakht H., Gerges N., Fiset P.O., Bechet D., Faury D., De Jay N., Ramkissoon L.A.et al.. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014; 46:462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang F.T., Chan F.L., JD R.M., Udugama M., Mayne L., Collas P., Mann J.R., Wong L.H.. CHK1-driven histone H3.3 serine 31 phosphorylation is important for chromatin maintenance and cell survival in human ALT cancer cells. Nucleic Acids Res. 2015; 43:2603–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chan F.L., Vinod B., Novy K., Schittenhelm R.B., Huang C., Udugama M., Nunez-Iglesias J., Lin J.I., Hii L., Chan J.et al.. Aurora kinase b, a novel regulator of TERF1 binding and telomeric integrity. Nucleic Acids Res. 2017; 45:12340–12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hake S.B., Garcia B.A., Kauer M., Baker S.P., Shabanowitz J., Hunt D.F., Allis C.D.. Serine 31 phosphorylation of histone variant H3.3 is specific to regions bordering centromeres in metaphase chromosomes. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:6344–6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sitbon D., Boyarchuk E., Dingli F., Loew D., Almouzni G.. Histone variant H3.3 residue S31 is essential for xenopus gastrulation regardless of the deposition pathway. Nat. Commun. 2020; 11:1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martire S., Gogate A.A., Whitmill A., Tafessu A., Nguyen J., Teng Y.C., Tastemel M., Banaszynski L.A.. Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat. Genet. 2019; 51:941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Armache A., Yang S., Martinez de Paz A., Robbins L.E., Durmaz C., Cheong J.Q., Ravishankar A., Daman A.W., Ahimovic D.J., Klevorn T.et al.. Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature. 2020; 583:852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Soh Y.Q., Alfoldi J., Pyntikova T., Brown L.G., Graves T., Minx P.J., Fulton R.S., Kremitzki C., Koutseva N., Mueller J.L.et al.. Sequencing the mouse y chromosome reveals convergent gene acquisition and amplification on both sex chromosomes. Cell. 2014; 159:800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wong L.H., Brettingham-Moore K.H., Chan L., Quach J.M., Anderson M.A., Northrop E.L., Hannan R., Saffery R., Shaw M.L., Williams E.et al.. Centromere RNA is a key component for the assembly of nucleoproteins at the nucleolus and centromere. Genome Res. 2007; 17:1146–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cawthon R.M. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002; 30:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Callicott R.J., Womack J.E.. Real-time PCR assay for measurement of mouse telomeres. Comp. Med. 2006; 56:17–22. [PubMed] [Google Scholar]
- 53. Das P.P., Shao Z., Beyaz S., Apostolou E., Pinello L., De Los Angeles A., O’Brien K., Atsma J.M., Fujiwara Y., Nguyen M.et al.. Distinct and combinatorial functions of jmjd2b/kdm4b and jmjd2c/kdm4c in mouse embryonic stem cell identity. Mol. Cell. 2014; 53:32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Voon H.P.J., Udugama M., Lin W., Hii L., Law R.H.P., Steer D.L., Das P.P., Mann J.R., Wong L.H.. Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat. Commun. 2018; 9:3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Day D.S., Luquette L.J., Park P.J., Kharchenko P.V.. Estimating enrichment of repetitive elements from high-throughput sequence data. Genome Biol. 2010; 11:R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Klose R.J., Yamane K., Bae Y., Zhang D., Erdjument-Bromage H., Tempst P., Wong J., Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006; 442:312–316. [DOI] [PubMed] [Google Scholar]
- 57. Berry W.L., Janknecht R.. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013; 73:2936–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wen H., Li Y., Xi Y., Jiang S., Stratton S., Peng D., Tanaka K., Ren Y., Xia Z., Wu J.et al.. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014; 508:263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Larson A.G., Elnatan D., Keenen M.M., Trnka M.J., Johnston J.B., Burlingame A.L., Agard D.A., Redding S., Narlikar G.J.. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature. 2017; 547:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Strom A.R., Emelyanov A.V., Mir M., Fyodorov D.V., Darzacq X., Karpen G.H.. Phase separation drives heterochromatin domain formation. Nature. 2017; 547:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bannister A.J., Zegerman P., Partridge J.F., Miska E.A., Thomas J.O., Allshire R.C., Kouzarides T.. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001; 410:120–124. [DOI] [PubMed] [Google Scholar]
- 62. Black J.C., Allen A., Van Rechem C., Forbes E., Longworth M., Tschop K., Rinehart C., Quiton J., Walsh R., Smallwood A.et al.. Conserved antagonism between JMJD2A/KDM4A and HP1gamma during cell cycle progression. Mol. Cell. 2010; 40:736–748. [DOI] [PubMed] [Google Scholar]
- 63. Nguyen D.T., Voon H.P.J., Xella B., Scott C., Clynes D., Babbs C., Ayyub H., Kerry J., Sharpe J.A., Sloane-Stanley J.A.et al.. The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 2017; 18:914–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data that support the findings of this study are available from the corresponding author on reasonable request. H3.3 S31Ph ChIP sequencing dataset is available on the Gene Expression Omnibus (GEO) database under the accession number GSE181692.