


Abstract
Vibrio cholerae biofilm formation/maintenance is controlled by myriad factors; chief among these are the regulator VpsR and cyclic di-guanosine monophosphate (c-di-GMP). VpsR has strong sequence similarity to enhancer binding proteins (EBPs) that activate RNA polymerase containing sigma factor σ54. However, we have previously shown that transcription from promoters within the biofilm biogenesis/maintenance pathways uses VpsR, c-di-GMP and RNA polymerase containing the primary sigma factor (σ70). Previous work suggested that phosphorylation of VpsR at a highly conserved aspartate, which is phosphorylated in other EBPs, might also contribute to activation. Using the biofilm biogenesis promoter PvpsL, we show that in the presence of c-di-GMP, either wild type or the phospho-mimic VpsR D59E activates PvpsL transcription, while the phospho-defective D59A variant does not. Furthermore, when c-di-GMP levels are low, acetyl phosphate (Ac∼P) is required for significant VpsR activity in vivo and in vitro. Although these findings argue that VpsR phosphorylation is needed for activation, we show that VpsR is not phosphorylated or acetylated by Ac∼P and either sodium phosphate or potassium phosphate, which are not phosphate donors, fully substitutes for Ac∼P. We conclude that VpsR is an unusual regulator that senses phosphate directly, rather than through phosphorylation, to aid in the decision to form/maintain biofilm.
INTRODUCTION
Rapid detection and adaptation to fluctuating environmental changes are essential for bacterial survival and proliferation. Microorganisms sense a multitude of extracellular signals, including physical and chemical parameters such as temperature, pH, osmolarity, autoinducers, ions and host cell contact. Mechanisms that can transmit those signals then lead to changes in gene expression.
In bacteria, a major signaling pathway is the two-component signal transduction system (TCS), comprised of a sensor kinase (SK) and a soluble cytoplasmic response regulator (RR) (1,2). After an environmental signal is perceived by the sensory domain or a signaling partner of the SK, phospho-transmission or a phospho-relay results in the phosphorylation of an aspartic acid residue within the RR. This, in turn, induces an RR conformational change, which alters protein–protein and/or protein–DNA properties and in many cases leads to changes in gene expression. As a phosphorylated aspartic acid is typically labile (3,4), phosphorylation can be lost either passively or by a specific phosphatase, returning the RR to an inactive, nonphosphorylated state. Substitution of the aspartic acid with an alanine typically results in an inactive, phospho-defective variant, while a D→E substitution yields a constitutively active, phospho-mimic protein (5–7).
Vibrio cholerae is a Gram-negative, motile bacterial pathogen that causes the gastrointestinal disease cholera. In areas where cholera is endemic, disease occurrence parallels seasonal pattern and climate changes (8), while in regions where cholera is nonendemic, the introduction of the pathogenic bacteria together with poor sanitation leads to rapid bacterial dissemination via the fecal–oral route (9). Despite causing illness in human hosts, V. cholerae is a natural aquatic inhabitant and must survive in myriad environmental niches, including both tropical and temperate waters (10–12). With dozens of predicted SKs and putative RRs (13), V. cholerae employs TCSs as a major tool to sense and respond to these environmental conditions, helping it to survive stressors such as changes in temperature and salinity (14).
Regulation of biofilm formation is vital to V. cholerae since it facilitates survival in the aquatic environment and transmission to the human host (15). A putative V. cholerae RR, VpsR, is the master regulator of biofilm formation (16–21). Of the dozens of RRs in V. cholerae, 11 established or potential RRs have been shown to also be involved in the positive or negative regulation of biofilm formation. For example, VpsT, LuxO and VxrB have been shown to activate biofilm formation, while PhoB, VarA, VieA, VarR, VC1348 and VCA0210 repress formation (13,17,18,21–29).
VpsR was originally classified as an enhancer binding protein (EBP) based on its amino acid homology within three distinct EBP domains: an N-terminal receiver (REC) region with a conserved aspartic residue for phosphorylation; a central ATPase associated with diverse cellular activities (AAA+) domain that can interact with the alternate sigma factor, σ54, of RNA polymerase (RNAP); and a C-terminal helix–turn–helix, DNA-binding domain (30,31). However, it is now clear that VpsR is an atypical EBP. Both its REC and AAA+ domains contain substitutions at highly conserved residues needed for interaction with σ54 and for ATPase activity (32), and VpsR activates transcription by RNAP containing the primary sigma factor (σ70 in Escherichia coli), not σ54 (17,21). Furthermore, a recent crystal structure of a truncated VpsR, containing the REC–AAA+ domains and bound to a nonhydrolyzable ATP analogue, indicates a novel ATP binding pocket that is distinct from AAA+ binding (33). Interestingly, while both the full-length and the truncated VpsR exhibit ATPase and GTPase activity under some conditions (33), we did not detect ATP hydrolysis under conditions that are used for in vitro transcription (17). Thus, the role of the ATPase is unclear.
Despite these differences from typical EBPs, the VpsR REC domain retains the highly conserved aspartic acid residue (D59), suggesting that the protein might be phosphorylated (Supplementary Figure S1). This idea has been bolstered by previous studies, which demonstrated that the V. cholerae VpsR D59A variant cannot form biofilms, while the D59E variant produces biofilms at levels similar to or greater than wild type (WT) (34,35). However, the VpsR REC–AAA+ structure demonstrates that other residues needed for transfer of a phosphate or stabilization of a phosphorylated aspartate appear to be absent (33).
Besides VpsR, the second messenger cyclic di-guanosine monophosphate (c-di-GMP) is also needed to regulate biofilm formation in V. cholerae in response to environmental signals (16,18,36,37). c-di-GMP, which is generated from two GTPs by a diguanylate cyclase (DGC), is degraded into GMP or pGpG by a phosphodiesterase (38). Increasing levels of c-di-GMP drive the transition from a motile planktonic state to a sedentary biofilm lifestyle (39). For some regulators, it is known that c-di-GMP is needed for RR dimerization or DNA binding, indicating that c-di-GMP functions to change the conformational state of the protein or to improve its affinity for DNA (32,37,40–55).
Using an in vitro transcription system, we have previously shown that VpsR and c-di-GMP together directly activate transcription from promoters present within the biofilm biogenesis/maintenance operons: PvpsL, PvpsU, PrbmF and PrbmA in the presence of σ70-RNAP (17,21). Surprisingly, at PvpsL, c-di-GMP does not function through stimulation of VpsR dimerization or by improving DNA binding. Instead, the presence of c-di-GMP changes the protein–DNA contacts within the PvpsL/VpsR/RNAP complex, resulting in the generation of the transcriptionally active, ‘open’ complex. These results were obtained using a purified VpsR that had been heterologously produced in E. coli, and transcriptional activation was observed without treatment with an SK or acetyl phosphate (Ac∼P), a chemical phosphate donor that is known to phosphorylate some RRs in vitro and in vivo. Thus, the phosphorylation status of VpsR has remained unknown, a possible SK responsible for phosphorylating VpsR has not been identified and the role of phosphorylation, if any, in VpsR transcriptional activation has not been determined.
Here, we have asked whether VpsR is directly phosphorylated and what factors besides c-di-GMP can affect its activity, using the phospho-mimic variant D59E, the phospho-defective variant D59A and a purified denatured/renatured VpsR (VpsRren). We find that in vitro VpsRren preincubated either with Ac∼P, which is a phosphate (Pi) donor, or with sodium phosphate or potassium phosphate, which are not able to phosphorylate VpsR, activates transcription from PvpsL when concentrations of c-di-GMP are lower. However, as the concentration of c-di-GMP increases, the need for Pi diminishes. We also observe that the presence of Ac∼P is needed for PvpsL activation in vivo when the level of c-di-GMP is low. Our results argue that the activity of VpsR is affected by the presence of either Ac∼P or Pi, rather than by protein phosphorylation, and that either of these molecules together with c-di-GMP directly modulates the ability of VpsR to activate biofilm formation.
MATERIALS AND METHODS
DNA
pMLH06 contains the vpsL promoter from −97 to +213 cloned into the EcoRI and HindIII restriction enzyme sites of pRLG770 (56). pMLH10 contains the vpsL promoter from −97 to +213 cloned into the SpeI and BamHI restriction sites of pBBRlux (22). pBH625, containing the vpsL-lux fusion, and pMLH17, containing WT vpsR, were constructed as previously described (17,28,57). pMLH18 and pMLH19 contain vpsR D59A and D59E, respectively, and were constructed from pMLH17 using QuikChange (Agilent). Primer sequences are available upon request. pMLH11 is a pET28b(+) derivative (Novagen) that contains vpsR with an N-terminal his6-tag, cloned as previously described (17). pMLH14 and pMLH15, which contain the vpsR alleles encoding VpsR D59A and VpsR D59E, respectively, were constructed from pMLH11 using QuikChange (Agilent). pCMW75, which contains an active Vibrio harveyi DGC, qrgB, was used for generating high levels of c-di-GMP in the strains used for the gene reporter assays, while pCMW98 encodes qrgB with a mutation in the catalytic active site of the enzyme to function as a negative control (36). Fragments containing PvpsL used for electrophoretic mobility shift assays (EMSAs) and DNase I footprinting were obtained as PCR products as previously described (17), using primers that annealed from positions −97 to +113 relative to the transcription start site (TSS) of vpsL.
Strains and growth conditions
Escherichia coli ElectroMAX DH10B (Invitrogen) and E. coli DH5α (New England Biolabs) were used for cloning, and BL21 (DE3) (58) and Rosetta2 (DE3)/pLysS (New England Biolabs) were used for protein synthesis. The V. cholerae strains CW2034 (ΔvpsL), WN310 (ΔvpsL ΔvpsR), BP52 (ΔvpsR), NK001 (vpsR D59A), NK002 (vpsR D59E) and NK007 (ΔackA1/2 Δpta) were derived from the El Tor biotype strain C6707str2 (59). Strains carrying mutations in vpsR were constructed using the pKAS32 suicide vector as described previously (60).
For the lux fusion assays, strains were grown in Luria–Bertani broth (1% tryptone, 0.5% yeast extract, 1% NaCl at pH 7.5) with kanamycin (50 μg/ml), chloramphenicol (5 μg/ml), 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG, final concentration) or 0.2% arabinose as needed. In Figure 1, overnight strains of E. coli containing the vpsL-lux reporter, the IPTG-inducible QrgB or QrgB* expression vector, and an arabinose-inducible expression vector for WT VpsR, VpsR D59A or VpsR D59E were diluted 1:100 in 150 μl in a solid white 96-well plate (36). Luminescence was measured after 12 h of incubation at 35°C with shaking. In Figure 7, overnight strains of V. cholerae strains WT or ΔackA1/2 Δpta encoding the QrgB or QrgB* expression vector and the vpsL-lux reporter were diluted 1:100 in 150 μl in a solid white 96-well plate. Luminescence was measured after 7.5 h of incubation at 35°C with shaking. In Figure 10, cultures of V. cholerae ΔvpsL (strain CW2034) encoding the vpsL-lux reporter and pEVS141 (vector control) or the QrgB expression plasmid were grown with shaking at 35°C to an OD600 of 1.0. Four hundred microliters of this culture was pelleted in a microcentrifuge and washed with 500 μl of phosphate-free MOPS minimal media supplemented with 0.5% glycerol and 25 mM l-asparagine, l-arginine, l-glutamate and l-serine and trace metals without trinitriloacetic acid as previously described (61). The washed cultures were resuspended in MOPS minimal media with the same concentrations of kanamycin, chloramphenicol and 0.1 mM IPTG with 0 or 50 μM KH2PO4 as described (62). One hundred fifty microliters of this culture were distributed to the wells of a solid white 96-well plate and incubated without shaking for 3 h at 35°C before measuring luciferase and OD600. Luminescence was read and measured on an Envision multilabel counter (PerkinElmer) and reported in relative luminescent units (R.L.U., counts min-1 mL-1/OD600 unit) as previously described (17). Statistical analysis and replication are reported in the figure legends.
Figure 1.

Mutation of VpsR residue D59 to an alanine impairs VpsR activity at PvpsL in vivo and in vitro. (A) Expression of PvpsL-lux was determined in E. coli containing WT vpsR, vpsR D59A or vpsR D59E expressed from an arabinose-inducible promoter on a plasmid under unaltered or high c-di-GMP conditions via IPTG induction of QrgB* (white bars) or QrgB (black bars), respectively. Error bars indicate standard deviation of three independent cultures and significance was determined using a two-way ANOVA with a Šídák correction (ns, not significant; **P < 0.01). (B) Gel showing single round of in vitro transcription from the plasmid template PvpsL with E. coli σ70-RNAP and WT VpsR, VpsR D59A or VpsR D59E with or without 12.5 μM c-di-GMP. (C) Quantification of values in panel (B) with standard deviations relative to transcription by RNAP alone (lane 1) is shown. Values are as follows: lane 2: 1.09, 0.59; lane 3: 0.76, 0.75, 1.17; lane 4: 5.72, 5.81, 4.35; lane 5: 1.66, 1.46, 1.02; lane 6: 7.92, 4.24, 5.81, 5.77 (one-tailed t-test for two independent means for lanes 4–6 relative to lane 3: ns, not significant; **P < 0.01).
Figure 7.

Pathway for the biosynthesis of Ac∼P and vpsL-lux gene reporter assays in V. cholerae WT and ΔackA1/2Δpta. (A) Ac∼P is generated by the enzyme Pta and is degraded by the two enzymes AckA1 and AckA2 in V. cholerae. (B) Expression from pBH625 (vpsL-lux) is shown for either V. cholerae WT (black) or ΔackA1/2 Δpta (gray) containing pBH625 (vpsL-lux) and with overexpression of either the DGC QrgB or QrgB* to generate high or unaltered intracellular c-di-GMP, respectively. Error bars indicate standard deviation of six independent cultures and significance was determined using a multiple unpaired t-test with a two-stage Benjamini, Krieger and Yekutieli correction (**P < 0.01).
Figure 10.
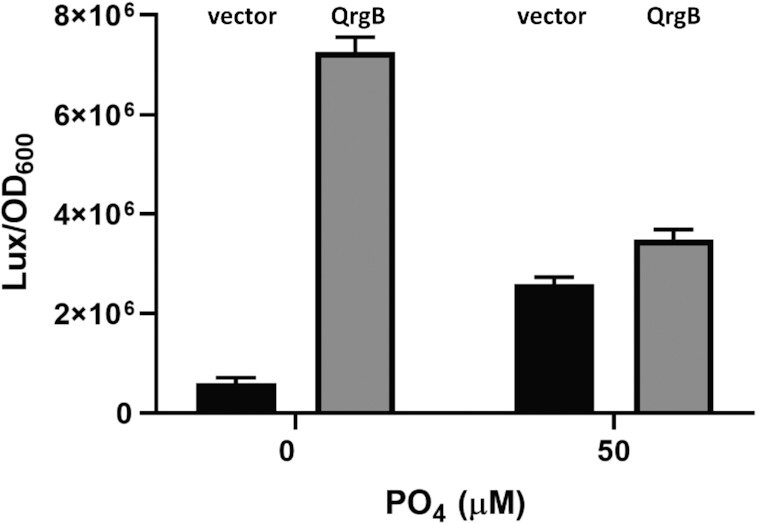
Phosphate enhances vpsL expression in vivo at lower concentrations of c-di-GMP. The vpsL-lux transcriptional fusion was measured after 3 h of incubation in MOPS + NRES minimal media supplemented with 0 or 50 μM added PO4 (black bars, unaltered c-di-GMP, vector; gray bars, high c-di-GMP, QrgB). The mean and standard deviation of three independent replicates are plotted, and all comparisons exhibit a statistically significant difference with P < 0.01 using a two-way ANOVA followed by a Šídák correction.
Proteins
Escherichia coli RNAP core was purchased from Epicenter Technologies and New England Biolabs. Escherichia coli σ70 was purified as previously described (63). WT VpsR, and the D59A and D59E protein variants (all His6-tagged) were expressed and purified from Rosetta2 (DE3)/pLysS (Novagen) containing pMLH11, pMLH14 or pMLH15, respectively, as previously described (17).
VpsRren was expressed and purified as previously described for WT VpsR (17) with the following modifications. After centrifugation of the induced culture at 13 000 × g for 10 min, the cell pellet was resuspended in 20 ml Binding Buffer [20 mM Tris–HCl (pH 7.5), 500 mM NaCl, 5 mM imidazole, 1 mM phenylmethylsulfonyl fluoride], sonicated, centrifuged at 17 500 × g for 20 min and resuspended again in Binding Buffer. We estimate from protein gels that this pellet contains >90% of the VpsR protein; the soluble fraction, which was used to isolate WT VpsR, contains ∼10% or less. After centrifugation at 17 500 × g for 15 min, the pellet was resuspended in 8 ml Binding Buffer containing 6 M urea (Binding Buffer/6 M urea), rocked gently for 1 h and centrifuged at 17 500 × g for 15 min. The supernatant was transferred to a new centrifuge bottle, centrifuged as before and the supernatant filtered through a 0.8 micron syringe filter. After pre-equilibration of Ni-NTA His-binding resin (Qiagen) with Binding Buffer/6 M urea, 2 ml of the supernatant was mixed with 2 ml of the resin slurry and gently rocked for at least 1 h, and then loaded into a 10-ml disposable column (Bio-Rad). The resin was washed with 12 ml Binding Buffer/6 M urea followed by 12 ml Wash Buffer [20 mM Tris–HCl (pH 7.5), 500 mM NaCl, 10 mM imidazole, 6 M urea]. VpsR was eluted twice with 3 ml Elute Buffer [20 mM Tris–HCl (pH 7.5), 500 mM NaCl, 6 M urea, 60 mM imidazole]. The eluted protein was renatured by sequential dialysis in the following buffers: 1× Reconstitution Buffer (RB) [50 mM Tris–HCl (pH 7.5), 1 mM ethylenediaminetetraacetic acid (EDTA), 0.1% Triton X-100, 20% glycerol, 1 mM DTT] containing 6 M urea, three times; 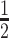 × RB containing 6 M urea;
× RB containing 6 M urea; 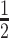 × RB containing 3 M urea; 1× RB containing 3 M urea;
× RB containing 3 M urea; 1× RB containing 3 M urea; 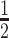 × RB containing 3 M urea;
× RB containing 3 M urea; 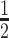 × RB; 1× RB, three times; and the indicated VpsR Storage Buffer four times, either Tris/low-salt buffer [20 mM Tris–HCl (pH 7.5), 1 mM EDTA, 10 mM NaCl, 20% glycerol, 0.1 mM DTT] or Pi/high-salt buffer [20 mM sodium phosphate (pH 7.8), 250 mM NaCl, 20% glycerol, 7 mM β-mercaptoethanol]. Dialyzed protein was stored at −80°C. Protein concentrations were determined by comparison with known amounts of RNAP core after SDS-PAGE and gel staining with Colloidal Blue (Invitrogen).
× RB; 1× RB, three times; and the indicated VpsR Storage Buffer four times, either Tris/low-salt buffer [20 mM Tris–HCl (pH 7.5), 1 mM EDTA, 10 mM NaCl, 20% glycerol, 0.1 mM DTT] or Pi/high-salt buffer [20 mM sodium phosphate (pH 7.8), 250 mM NaCl, 20% glycerol, 7 mM β-mercaptoethanol]. Dialyzed protein was stored at −80°C. Protein concentrations were determined by comparison with known amounts of RNAP core after SDS-PAGE and gel staining with Colloidal Blue (Invitrogen).
For in vitro treatment of VpsR proteins with Ac∼P, 200 mM Ac∼P was prepared by freshly dissolving lithium potassium acetyl phosphate (Sigma) in 20 mM Tris–Cl (pH 7.5). Proteins, present in the indicated protein storage buffer, were then treated at room temperature for 30 min in the presence of 20 mM Ac∼P and 2 mM Tris–Cl (pH 7.5) or in the presence of 2 mM Tris–Cl (pH 7.5) as the control.
Electrophoretic mobility shift assays
Protein–DNA complexes were formed as described previously (17), in which 5 nM of 32P-labeled DNA was incubated at 37°C for 10 min with the following, as indicated: VpsR (final concentration from 0.2 to 2 μM); 0.16 μM reconstituted RNAP (σ:core ratio of 2.5:1); and 50 μM c-di-GMP at 37°C for 10 min in 1× transcription buffer [40 mM Tris–acetate (pH 7.9), 150 mM potassium glutamate, 4 mM magnesium acetate, 0.1 mM EDTA (pH 7.0), 0.01 mM DTT, 100 μg/ml BSA]. For VpsR/DNA complexes, poly[dI-dC] (1 μg) was added and the final binding volume of 20 μl also contained 14.5 μl of VpsR in the indicated storage buffer, either Tris/low-salt buffer or Pi/high-salt buffer. The VpsR–DNA complexes were loaded onto 5% (w/v), nondenaturing, polyacrylamide gels already running at 100 V in 1× Tris/borate/EDTA (TBE) buffer and electrophoresed for 1.5 h. VpsR/RNAP/DNA transcription complexes were challenged by the addition of heparin (500 μg) for 15 s immediately before electrophoresis on 4% (w/v) nondenaturing, polyacrylamide gels already running at 100 V in 1× TBE buffer. Samples were electrophoresed for 3 h at 380 V/h. After autoradiography, films were scanned using a Powerlook 2100XL densitometer and amounts quantified using Quantity One software (Bio-Rad). Kd(app) values were calculated by determining the VpsR concentration needed to shift 50% of the free DNA. This was determined from the loss of free DNA and not from the retarded species.
In vitro transcriptions and primer extensions
When indicated, VpsR or VpsRren was pretreated with 20 mM Ac∼P as described earlier and collected on ice. Transcription reactions were assembled on ice with 0.02 pmol of supercoiled template, 0–3.0 pmol of VpsR, 0–250 μM of c-di-GMP (as indicated), reconstituted RNAP (0.2 pmol of σ70 plus 0.05 pmol of core) and 1× transcription buffer (see earlier) in a total volume of 5 μl. After incubation at 37°C for 10 min, a solution (1 μl) containing ribodeoxynucleoside triphosphates (1.43 mM ATP, GTP, CTP and 35 μM [α-32P] UTP at ∼5 × 104 dpm/pmol) with or without 500 ng heparin was added to initiate a single or multiple rounds of transcription, respectively. Reactions were incubated for 10 min at 37°C and subsequently collected on dry ice with the addition of 15 μl formamide load solution [1 mM EDTA (pH 7), 1% bromophenol blue, 1% xylene cyanol FF in deionized formamide] or 25 μl of urea loading solution (1× TBE, 7 M urea, 1% bromophenol blue, 1% xylene cyanol FF). Aliquots were electrophoresed on 4% (w/v) polyacrylamide and 7 M urea denaturing gels in 0.5× TBE buffer. After electrophoresis, gels were exposed to X-ray films, films were scanned and radioactivity was quantified as described earlier. Primer extensions were performed as described (17).
DNase I footprinting
Solutions were assembled as described earlier for EMSAs. DNA (0.04 μM) was incubated with the following as indicated: 1.4 μM VpsR; 50 μM c-di-GMP; and/or 0.16 μM of reconstituted RNAP (σ:core ratio of 2.5:1). After an incubation of 10 min at 37°C, heparin was added to complexes with RNAP for 15 s to ensure binding specificity prior to addition of a 1.5 μl solution containing 0.3 U of DNase I. No additional competitor was added to complexes containing VpsR alone and DNA +/− c-di-GMP since these reactions already contained 1 μg of poly(dI-dC). After an additional 30 s of incubation at 37°C, samples were immediately loaded onto 4% (w/v) nondenaturing, polyacrylamide gels already running at 100 V in 1× TBE buffer. Samples were electrophoresed for 3 h at 380 V/h. After autoradiography, the protein/DNA complexes were excised, and the DNA was extracted. This challenge/gel isolation technique, which has been used previously in other systems (64), was important to ensure that we were observing the footprint of the stable and specific protein complex of interest rather than nonspecific or unstable complexes. The extracted DNA was then electrophoresed on denaturing gels as described (64).
Potassium permanganate footprinting
For potassium permanganate (KMnO4) footprinting, reactions were assembled as described earlier for DNase I footprinting (17). Briefly, after addition of 500 ng of heparin to ensure transcription complex stability, a final concentration of 2.5 mM KMnO4 was added prior to incubation at 37°C for 2.5 min. Reactions were quenched with 5 μl of 14 M β-mercaptoethanol and further processed as described (64).
Labeling proteins with 32P Ac∼P
High specific activity Ac∼32P was generated as described (65,66) by incubating [γ-32P] ATP (13 μl, 6000 Ci/mmol, PerkinElmer), acetate kinase (AckA) [Sigma; 3 μl of 1 unit/μl, freshly dissolved in cold 100 mM triethanolamine (pH 7.6)] and 5× AKP buffer [3.6 μl; 125 mM Tris–Cl (pH 7.6), 300 mM potassium acetate, 50 mM MgCl2] for 30 min at room temperature. The resulting Ac∼32P was purified by passage through a 30-kDa cutoff membrane (Amicon Ultra, Millipore). Protein (50 pmol, 4–10 μl) was then treated for 30 min at room temperature with 2.5 μl of either this Ac∼32P or a lower specific activity Ac∼32P obtained by the addition of the nonradioactive 200 mM Ac∼P solution (described earlier) to give a final concentration of 13 mM during protein incubation. c-di-GMP was included as indicated. Following incubation, proteins were collected on ice. Aliquots were mixed with SDS glycine load solution (Bio-Rad) containing β-mercaptoethanol but were not boiled before electrophoresis on 12% Tris–glycine SDS gels (Invitrogen). After electrophoresis, gels were exposed to X-ray films and films were scanned as described earlier. When indicated, aliquots were also used for in vitro transcription reactions.
Mass spectrophotometric analysis of VpsRren
Intact protein mass of each sample was measured on an X500B Q-TOF (Sciex) mass spectrometer coupled to an Exion UHPLC. VpsRren solutions were acidified with 1% formic acid immediately before injection onto an Aeris 3.6 widepore XB-C8 column. Data were analyzed using Explorer and mass reconstruction was performed in Bio Tool Kit both within the SCIEX OS software.
RESULTS
The activities of the VpsR phospho-variants D59A and D59E suggest that D59 may require phosphorylation for activity
Although VpsR has considerable sequence conservation with the EBP family of RRs, it lacks both a GAFTGA motif and the typical DE residues within the Walker B motif that are signature features of the EBP interaction with σ54 (30). Furthermore, our previous work has indicated that at the biofilm biogenesis/maintenance promoters PvpsL, PvpsU, PrbmF and PrbmA, VpsR functions with σ70-RNAP rather than with σ54-RNAP (17,21). However, VpsR does have the typical site of phosphorylation for an EBP, a highly conserved aspartic acid (residue D59 within VpsR) located within the N-terminal receiver domain (Supplementary Figure S1). Several studies have found that the substitution of a conserved aspartic acid with an alanine (a phospho-defective mutation) typically renders an RR inactive, while an aspartic to glutamic substitution (a phospho-mimic mutation) generates a constitutive phenotype (5–7). This same result has been observed for VpsR in biofilm formation. Both WT VpsR and the D59E variant form robust biofilms, while biofilm production is poor in the presence of the D59A variant (34,35). These results have suggested that D59 might be phosphorylated even though no cognate SK for VpsR has been identified.
To investigate whether VpsR phosphorylation might be involved in transcription activation at PvpsL, we compared the activities of WT VpsR, VpsR D59A and VpsR D59E in transcriptional lux fusion assays in E. coli and in in vitro transcription assays. We found that expression of a D59A vpsR mutation from a plasmid in E. coli significantly impairs activation of PvpsL in response to increased c-di-GMP driven by IPTG induction of the V. harveyi DGC QrgB, which generates physiological levels of c-di-GMP (67) (Figure 1A). Alternatively, the vpsR D59E mutant displayed elevated transcription compared to WT from PvpsL in vivo both in the presence and in the absence of c-di-GMP, but importantly it remained responsive to c-di-GMP (Figure 1A). For the in vitro work, we purified the phospho-variants using the same procedure used for WT VpsR. Similarly, in vitro transcription of PvpsL was reduced by VpsR D59A, but not by VpsR D59E (Figure 1B and C). Previously, we determined the TSS for WT VpsR in vivo and in vitro (17). We confirmed that the start sites for the in vitro and in vivo RNA produced in the presence of VpsR D59E are identical (Supplementary Figure S2A and B, respectively).
Purification of a VpsR that becomes active in transcription after pretreatment with Ac∼P
The results with the phospho-variants were consistent with the idea that phosphorylation of VpsR D59 is needed for activity. However, other findings were confounding. In our previous in vitro studies (17,21) and the experiments described so far, we used purified VpsR (referred to here as WT VpsR) that was heterologously produced in E. coli from a plasmid construct. Consequently, no V. cholerae SK was present to phosphorylate WT VpsR during its production. Thus, if VpsR were phosphorylated, this would have had to occur through crosstalk with an E. coli SK or from Ac∼P present in the cell. However, in either case, we would have expected that D59 would lose this phosphorylation during purification since a phosphorylated aspartic acid is typically labile (3,4). With other purified RRs, pretreatment with Ac∼P in vitro is a common method to ensure phosphorylation of the RR after purification (68). However, our purified WT VpsR was active in the absence of Ac∼P, and pretreatment with Ac∼P did not increase the level of transcription (Figure 2A). In addition, treatment with Ac∼P did not change the migration of WT VpsR in Phos-tag gels (data not shown), a technique routinely used to assay RR phosphorylation (64,69–73). Taken together, these results seemed inconsistent with the activities of the phospho-variants.
Figure 2.

Effect of Ac∼P on WT VpsR (A) and VpsRren (B) in activated transcription from PvpsL. Multiple (A) or single (B) rounds of in vitro transcriptions with the plasmid template PvpsL, E. coli σ70-RNAP, WT VpsR or VpsRren pretreated with Ac∼P (or control buffer), and c-di-GMP as indicated. For the graph in panel (A), values and standard deviations represent the average of three or more experiments and values for lanes 2–5 are relative to lane 1 (transcription in the absence of WT VpsR, Ac∼P and c-di-GMP). For lanes 2–5, the values are as follows: lane 2, 0.98, 1.28, 1.04, 0.84; lane 3: 3.07, 3.84, 2.92, 3.02; lane 4: 1.11, 1.22, 0.94, 0.85; lane 5: 1.85, 3.67, 2.87, 2.78. Results of one-tailed t-test for two independent means are shown for lanes 3–5 relative to lane 2 (ns, not significant; **P < 0.01). For the graph in panel (B), values in lanes 1–6 represent the average of two experiments; values and standard deviations in lanes 7–12 represent averages of four or more experiments. For lanes 2–6, values are relative to lane 1 (transcription in the absence of VpsRren, Ac∼P and c-di-GMP); for lanes 8–12, values are relative to lane 7 (transcription in the absence of Ac∼P and c-di-GMP). Values are as follows: lane 8: 2.6, 1.6, 2.2, 1.6, 3.5; lane 9: 7.8, 10, 6.4, 9.6, 8.1; lane 10: 1.1, 1.0, 1.0, 0.90, 1.3; lane 11: 3.6, 6.3, 5.5, 10, 9.1; lane 12: 6.0, 8.0, 8.4, 8.4, 11. Results of one-tailed t-test for two independent means are shown for lane 8 relative to lane 11 (**P < 0.01).
We hypothesized that if WT VpsR was phosphorylated, the phosphorylation might be stabilized by a particular protein conformation or protein pocket, as was described for the response regulator DrrA of Thermotoga maritima (74). If so, denaturation/renaturation of WT VpsR might result in a dephosphorylated protein. Previous VpsR purification methods (17,37,55), including ours, have involved purifying the protein from the soluble lysate even though most of the VpsR protein remains within an insoluble pellet. Consequently, we purified VpsR from the insoluble pellet by denaturation in buffer containing 6 M urea and then slowly renatured the protein, resulting in the protein referred to here as VpsRren. As will become relevant, it should be noted that the final storage buffer for VpsRren (Tris/low-salt buffer) differed from that for WT VpsR and the phospho-variants (Pi/high-salt buffer) (see the ‘Materials and Methods’ section for details).
Unlike WT VpsR, VpsRren required a pretreatment with Ac∼P for significant activity when c-di-GMP concentrations were lower (25 μM) (Figure 2B, lane 8 versus lane 11). The level of activation achieved with Ac∼P pretreatment (lane 11) was similar to what we had observed using WT VpsR either with or without Ac∼P (17). However, when the c-di-GMP concentration was increased 10-fold (to 250 μM), VpsRren induced transcription of vpsL in vitro to high amounts with or without Ac∼P (Figure 2B, lane 9 versus lane 12). Thus, denaturation/renaturation to generate VpsRren now resulted in a protein that requires treatment with Ac∼P for high activity when c-di-GMP levels are lower.
WT VpsR, VpsR D59E and VpsRren pretreated with Ac∼P form similar open complexes with RNAP
Given that VpsRren underwent a denaturation procedure and that both VpsRren in the absence of Ac∼P and the phospho-defective VpsR D59A were impaired in transcription, we characterized VpsRren and the phospho-variants further and compared their activities to those of the previously purified WT VpsR. To determine DNA-binding activity, we performed EMSAs using 32P-labeled PvpsL DNA (Figure 3). As observed before with WT VpsR (17), we observed multiple retarded species. Consequently, Kd(app) values were determined from the loss of free DNA rather than from the shifted DNA bands. WT VpsR, D59A and D59E all demonstrated similar binding affinity in the presence or absence of c-di-GMP (averages between 1 and 2 μM), indicating that neither the D59A or D59E mutation nor the presence of c-di-GMP significantly affected DNA-binding activity relative to WT VpsR (Figure 3A–C). In addition, the DNase I footprints obtained with any of these proteins and PvpsL DNA were similar in the presence or absence of c-di-GMP (Figure 4A and B, lanes 2–7), suggesting that the protein–DNA contacts were also similar.
Figure 3.

DNA binding to PvpsL measured by EMSAs. Representative gels showing the retardation of the 32P-labeled DNA harboring −97 to +113 of PvpsL with increasing amounts of the indicated VpsR (concentrations of 0, 0.2, 0.4, 0.8 and 2 μM) either in the absence or in the presence of 50 μM c-di-GMP, as indicated. Black arrows indicate retarded complexes, while gray arrow indicates free DNA. Apparent DNA-binding dissociation constants (Kd(app)), determined from at least three replicate EMSAs, were calculated as the concentration of VpsR needed for the loss of 50% of the free DNA. WT VpsR and the variants (A–C) were present in Pi/high-salt buffer, while VpsRren (D and E) was present in the Tris/low-salt buffer. Consequently, the determined Kd(app) values determined in panels (A–C) are not comparable to those obtained in panels (D) and (E).
Figure 4.

DNase I footprinting of PvpsL complexes containing WT VpsR, VpsR D59A and VpsR D59E on nontemplate (A) or template (B) DNA. GA corresponds to G + A ladder. VpsR, c-di-GMP and RNAP are present as indicated. To the right of each gel image, a schematic indicates the −10 and −35 regions and the +1 TSS. The VpsR binding site is indicated as a dashed line. DNase I protection regions and hypersensitivity sites seen with the activated complex of RNAP, VpsR, c-di-GMP and DNA are depicted as black rectangles and horizontal arrows, respectively. In each panel, images are from the same gel; the white line indicates where intervening lanes were removed.
The binding affinity of VpsRren for the DNA was also unchanged with/without c-di-GMP or with/without pretreatment with Ac∼P (Figure 3D and E). These results indicated that the presence of c-di-GMP or Ac∼P did not significantly affect the DNA binding affinity of VpsRren. While in this analysis the estimated Kd(app) for VpsRren (Figure 3D and E) was ∼10-fold higher than that of WT VpsR (Figure 3A), we attribute this difference to the different buffer conditions present in these binding analyses. WT VpsR and VpsRren were present in different buffers and >70% of the buffer in the EMSA was from the protein itself. Consequently, the determined Kd(app) values for WT VpsR and VpsRren are not comparable.
To determine whether the transcription complexes formed with the various VpsR proteins differed from those characterized with WT VpsR, we first used KMnO4 footprints, which identify single-stranded thymines within the open transcription bubble surrounding the TSS (75–77). These analyses indicated that in the presence of c-di-GMP, RNAP generates similar open transcription bubbles in three cases: WT VpsR in the absence of Ac∼P (Figure 5A and B, lane 2) (17), VpsR D59E in the absence of Ac∼P (Figure 5A and B, lane 6) and VpsRren pretreated with Ac∼P (Figure 5C, lane 6). As expected from the transcription results, VpsRren without Ac∼P pretreatment (Figure 5C, lane 4) and VpsR D59A (Figure 5A and B, lane 4) are impaired in open complex formation with RNAP.
Figure 5.

KMnO4 footprinting to determine the transcription bubble in transcription complexes made with WT VpsR, VpsR D59A, VpsR D59E and VpsRren. (A) Reactive thymines within the transcription bubble are observed at positions −6 and −7 on nontemplate DNA. (B, C) Reactive thymines within the transcription bubble (indicated by vertical line) are observed at positions −11, −4, −3, −2, −1, +1 and +2 on template DNA. RNAP, c-di-GMP, VpsR, Ac∼P and KMnO4 were present as indicated. GA corresponds to G + A ladder. In each panel, images are from the same gel; the white line indicates where intervening lanes were removed.
We performed DNase I footprints with VpsRren either with or without pretreatment with Ac∼P and +/− c-di-GMP or with the phospho-variants VpsR D59E and D59A +/− c-di-GMP to observe whether there were differences among the protein–DNA contacts with these various proteins. We had previously reported the DNase I footprints of RNAP alone, RNAP/c-di-GMP, RNAP/WT VpsR and RNAP/c-di-GMP/WT VpsR at PvpsL (17). We repeated these footprints to compare with the complexes formed with the phospho-variants or formed with VpsRren pretreated with or without Ac∼P. As observed before (17), DNase I footprints at PvpsL obtained with the basal transcription complexes formed with RNAP alone (Figure 4A and B, lane 9; Figure 6A and B, lane 2), RNAP/c-di-GMP (Figure 4A and B, lane 10) or RNAP/WT VpsR (Figure 4A and B, lane 11) were similar and consistent with the formation of a weak transcription complex. Likewise, when using VpsRren pretreated without Ac∼P (Figure 6A and B, lanes 3 and 4), VpsRren pretreated with Ac∼P but lacking c-di-GMP (Figure 6A and B, lane 5) or D59A in the presence c-di-GMP (Figure 4A and B, lane 13), we also only observed evidence of a weak complex when assayed by DNase I footprinting.
Figure 6.

DNase I footprinting of PvpsL transcription complexes containing WT VpsR or VpsRren on nontemplate (A) or template (B) DNA. GA corresponds to G + A ladder. Reactions contained VpsR WT, VpsRren or VpsRren pretreated with Ac∼P, 50 μM c-di-GMP and/or RNAP as indicated. To the right of each gel image, a schematic indicates the −10 and −35 regions and the +1 TSS. The VpsR binding site is indicated as a dashed black line. DNase I protection regions and hypersensitive sites seen with the activated complex containing RNAP, DNA, c-di-GMP, and either VpsRren pretreated with Ac∼P or WT VpsR are depicted as rectangles and horizontal arrows, respectively.
In contrast, as we have reported previously (17), the complex of RNAP/WT VpsR/c-di-GMP at PvpsL generated clear footprints, consistent with a transcriptionally open complex, in which the DNA is protected from −58 to +30 (Figure 4A and B, lane 12; Figure 6A and B, lane 7). This footprint includes both the RNAP contacts and the VpsR binding site from −31 to −52 on the nontemplate strand and from −34 to −53 on the template strand. A distinct feature of the transcription complex is an alteration in the protein–DNA pattern at the VpsR binding site (17). In this regard, notice the increased protection of positions −34, −35 and −38 on the nontemplate strand seen in the presence of c-di-GMP and RNAP (Figure 4A, lane 12; Figure 6A, lane 7), but not with VpsR alone (Figure 4A, lane 5).
Similar protection patterns were also observed for the c-di-GMP activated complexes using either VpsR D59E (Figure 4A and B, lane 14) or VpsRren that had been pretreated with Ac∼P (Figure 6A and B, lane 6). These results together with the transcription and EMSA results indicated that VpsR D59E and VpsRren pretreated with Ac∼P are fully functional proteins whose c-di-GMP-dependent transcription complexes are similar to those formed with WT VpsR. However, unexpectedly, in the absence of Ac∼P, the DNA/VpsRren/RNAP/c-di-GMP complex did yield enhanced cleavages at positions −54 and −53 on the nontemplate strand (Figure 6A and B, lane 4) that were also seen with the activated complexes. The reason for these specific cleavages is currently not clear.
The presence of Ac∼P stimulates VpsR activation in vivo
To investigate whether the results observed with VpsRrenin vitro were physiologically relevant, we quantified expression from PvpsL using a luciferase reporter assay in V. cholerae strains that have different concentrations of Ac∼P and c-di-GMP. We modulated the level of Ac∼P by using strains with mutations in the phosphotransacetylase (Pta)–AckA pathway (Figure 7A), in which Pta converts acetyl-CoA and Pi to Ac∼P and CoA while AckA converts Ac∼P to acetate, generating ATP from ADP in the process. However, the AckA reaction is reversible, generating Ac∼P from acetate. Thus, in the absence of both Pta and AckA, very little Ac∼P is present (78,79). However, unlike E. coli, V. cholerae contains two ackA genes. One gene, ackA1, is in an operon together with pta, while the second ackA gene, ackA2, is present on the second chromosome. Previous studies have shown that the two ackA genes encode functionally redundant proteins; thus, both proteins must be deleted to probe the role of Ac∼P (80,81).
To investigate whether the level of Ac∼P in vivo affects transcription from PvpsL, we performed transcriptional lux fusion assays in WT V. cholerae and ΔackA1/2 Δpta strains (Figure 7B). We found that under normal c-di-GMP concentrations, vpsL-lux activity was induced 9.75-fold in the WT versus the ΔackA1/2 Δpta strain, implicating Ac∼P in the generation of active VpsR at PvpsL in vivo (Figure 7B, QrgB*). However, in the increased c-di-GMP state generated by QrgB induction, which generates intracellular c-di-GMP concentrations that are still physiologically relevant (67), the difference in vpsL-lux expression in the WT versus ΔackA1/2 Δpta strain was only 2.5-fold (Figure 7B, QrgB). This finding was consistent with our in vitro transcriptions showing that the presence of Ac∼P significantly enhances VpsR activation of PvpsL when levels of c-di-GMP are lower and suggested that our studies with the VpsRren protein recapitulated a process observed in vivo. However, we cannot eliminate the possibility that a low level of phosphorylated VpsR is present in vivo in the absence of Ac∼P.
Given the results with VpsRren and the phospho-variants, we considered the possibility that pretreatment with Ac∼P phosphorylates VpsRren, perhaps at residue D59. In this scenario, our WT VpsR did not need pretreatment with Ac∼P in vitro because WT VpsR was phosphorylated by Ac∼P in vivo and this phosphorylation was maintained during the original purification process. However, phosphorylation of VpsR was lost in the denaturation/renaturation procedure used for VpsRren, making pretreatment with Ac∼P in vitro a necessary step before VpsR could significantly stimulate transcription when c-di-GMP was low.
To test this hypothesis, we asked whether VpsRren could be labeled with 32P after treatment with Ac∼32P. As a control we used the B. pertussis RR BvgA, which is known to be phosphorylated by Ac∼P at an aspartic residue (70). We first used a low concentration (0.41 μM) of Ac∼32P with a high specific activity (1.7 × 107 dpm/pmol) and conditions that had been reported previously for observing phosphorylation of the Borrelia burgdorferi RR Rrp2 (65). Labeling of the 23 kDa BvgA was observed under these conditions (Figure 8A, lanes 1 and 4), but no labeling of VpsRren was detected without (lane 2) or with (lane 3) the addition of 50 μM c-di-GMP. We then used a much higher concentration of Ac∼32P (13.3 mM at 400 dpm/pmol) and conditions that we had used previously to pretreat VpsRren with Ac∼P before transcription. As before, labeling of BvgA was detected (Figure 8A, lane 7). However, again no labeling of VpsRren was observed in the absence or presence of 50 μM c-di-GMP (Figure 8A, lanes 5 and 6, respectively).
Figure 8.
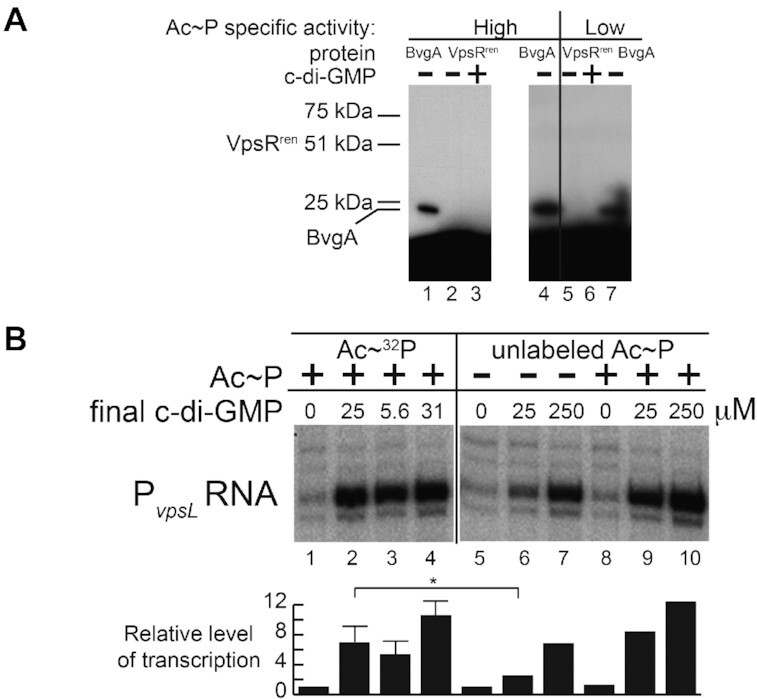
Incubation of VpsRren with Ac∼32P does not label the protein even though it is active in transcription. (A) SDS protein gels (one of two experiments) showing the products after treatment of BvgA or VpsRren with Ac∼32P (high specific activity: 0.41 μM at ∼1.7 × 107 dpm/pmol, lanes 1–4; low specific activity: 13.3 mM at ∼400 dpm/pmol, lanes 5–7) and, as indicated, 50 μM c-di-GMP. The positions of standard proteins of 25 and 75 kDa and of BvgA (23 kDa) are indicated. No labeling of VpsRren is detected, but the expected position of the protein is indicated. (B) Representative gel (of four experiments) showing the products of in vitro transcription reactions obtained using plasmid template PvpsL, RNAP and VpsRren pretreated with Ac∼32P (13.3 mM at ∼400 dpm/pmol, lanes 1–4), VpsRren without Ac∼P (lanes 5–7) or VpsRren pretreated with 20 mM unlabeled Ac∼P (+, lanes 8–10), and the indicated concentration of c-di-GMP in the transcription reaction. The same VpsRren shown in (A) lane 5 was used for (B) lanes 1 and 2; the same VpsRren shown in (A) lane 6 was used for (B) lanes 3 and 4. The transcription experiment in panel (B) was performed twice, with repeats of the transcription reactions in lanes 1–4 done a total of four times; lanes 5–10 were done in duplicate. For lanes 2–4, values are relative to lane 1; for lanes 6–10, values are relative to lane 5. Values are as follows: lane 2: 9.56, 5.08, 8.67, 4.27; lane 3: 7.53, 5.62, 5.81, 2.36; lane 4: 10.3, 9.20, 13.9, 8.83; lane 6: 3.08,1.92; lane 7: 7.65, 5.99; lane 8: 1.55, 0.93; lane 9: 8.90, 7.80; lane 10: 15.0, 9.74. Results of one-tailed t-test for two independent means are shown for lane 2 relative to lane 6 (*P < 0.05).
To confirm that VpsRren was activated by the above treatment with Ac∼32P even though we could not detect phosphorylation, aliquots from the reactions in Figure 8A, lanes 5 and 6, were used directly for in vitro transcription reactions. As seen in Figure 8B, VpsRren in these aliquots behaved as expected. Lack of c-di-GMP resulted in basal transcription (lane 1 versus normal condition in lane 5). However, when 25 μM c-di-GMP was present in the transcription reaction, a high level of activated transcription was observed whether VpsR had been pretreated with the Ac∼32P (the aliquot from Figure 8A, lane 5) or had been pretreated with unlabeled Ac∼P (compare lanes 2 and 9). Pretreatment with Ac∼32P in the presence of 25 μM c-di-GMP (resulting in a final transcription concentration of 5.6 μM c-di-GMP, lane 3) or pretreatment with Ac∼32P in the presence of 25 μM c-di-GMP followed by the addition of 25 μM c-di-GMP in the transcription reaction (resulting in a final c-di-GMP concentration of 31 μM, lane 4) also yielded an enhanced level of transcription as that seen under our normal transcription condition (lane 9). Taken together, these experiments indicate that VpsRren is activated by pretreatment with Ac∼P, but it is not phosphorylated.
Ac∼P does not acetylate VpsRren in vitro
In addition to phosphorylation, Ac∼P is also capable of transferring an acetyl group to a protein (82,83). Protein acetylation by Ac∼P has been documented by mass spectrometry analysis, which reveals an increase of 42 Da for each acetyl moiety (84).
We applied this analysis to VpsRren, which had been stored in the Tris/low-salt buffer and treated with no Ac∼P, with Ac∼P alone or with Ac∼P in the presence of c-di-GMP, or to VpsRren, which been stored in the Pi/high-salt buffer previously used for WT VpsR. In each case, the same molecular weight (50 649 Da) was observed (Supplementary Figure S3), indicating that there are no detectable moieties added to any of these proteins. This weight is lower than that calculated for His-tagged VpsR (50 806 Da) but is consistent with cleavage of the N-terminal Met and Gly and oxidation of two Met sidechains, which can be an artifact of protein handling. Thus, we conclude that none of these proteins are acetylated. Furthermore, this analysis eliminated the remote possibility that our inability to detect phosphorylation of VpsRren by Ac∼32P was because the protein was already stably phosphorylated, even after denaturation/renaturation.
The presence of Pi rather than phosphorylation stimulates VpsR activation
The inability to detect phosphorylation or acetylation of VpsRren after treatment with Ac∼P suggested that Ac∼P stimulation of VpsRrenin vitro (Figure 2B) and VpsR in vivo (Figure 7B) arises without direct phosphorylation or acetylation. Consequently, we considered other ways that Ac∼P might stimulate VpsRren activity.
As noted previously, a major difference between VpsRren and WT VpsR was their storage buffers: WT VpsR was stored in a Pi/high-salt storage buffer (containing 20 mM sodium phosphate, pH 7.8; 250 mM NaCl), while VpsRren was stored in a Tris/low-salt buffer (containing 20 mM Tris, pH 7.5; 10 mM NaCl). Although the added transcription buffer (containing 40 mM Tris–acetate, pH 7.9, and 150 mM potassium glutamate) comprised the bulk of salt and buffer in the transcription reaction, we considered the possibility that the sodium phosphate present in the WT VpsR storage buffer was stimulatory, eliminating the need to pretreat WT VpsR with Ac∼P.
To explore this idea, we investigated how pretreatment of VpsRren with 20 mM sodium phosphate affected transcription compared to pretreatment with 20 mM Ac∼P. We found that the presence of either form of phosphate, Ac∼P or Pi, stimulated VpsRren similarly at low c-di-GMP concentrations (Figure 9). Importantly, high phosphate alone was unable to induce transcription, showing that c-di-GMP is required for VpsR to activate RNAP. As another test, we repurified VpsRren using the denaturation/renaturation procedure, but this time we stored the protein in the Pi/high-salt storage buffer. The protein was now similarly active in the presence of 25 μM c-di-GMP with or without pretreatment with Ac∼P (Supplementary Figure S4). Finally, we eliminated the possibility that Na+ ion, rather than Pi, was the stimulatory agent by comparing the activation of VpsRren after pretreatment with either 20 mM NaCl versus 20 mM sodium phosphate or 20 mM KCl versus 20 mM potassium phosphate (Supplementary Figure S5). In this case, pretreatment with either sodium phosphate or potassium phosphate was stimulatory, while pretreatment with 20 mM NaCl or KCl was not. Since neither sodium phosphate nor potassium phosphate is a phosphate donor, we conclude that the presence of Pi, rather than phosphorylation, is sufficient to stimulate VpsRren activity. This suggests that Ac∼P functions simply through the presence of the phosphate rather than by its ability to donate the phosphate to VpsR.
Figure 9.

The presence of either 20 mM Ac∼P (A) or 20 mM NaPO4 (B) in the pretreatment step of VpsRren renders it more active. Graphs (top) show the fold activation for transcription obtained using the plasmid template PvpsL and VpsRren in the Tris/low-salt storage buffer pretreated, as indicated, in the absence of Ac∼P or NaPO4 (A, B), in the presence of 20 mM Ac∼P (A) or in the presence of 20 mM sodium phosphate, pH 7.8 (B) versus the indicated final concentration of c-di-GMP present during transcription. Graphs show all the values obtained from two experiments. Lines were drawn through the averages of the points. Underneath are sections of representative gels showing the in vitro transcription products from PvspL and from the control promoter on the plasmid for RNAI.
To test whether phosphate enhances vpsL expression at low c-di-GMP concentrations in vivo, we assessed the expression of the vpsL-lux transcriptional fusion in a ΔvpsL V. cholerae strain in MOPS minimal media supplemented with low and high Pi as previously described (62). vpsL-lux was assessed at unaltered concentrations of c-di-GMP (i.e. the vector control) or a strain with elevated c-di-GMP generated by overexpressing the DGC QrgB from a plasmid. Based on our in vitro results demonstrating that phosphate enhances vpsL expression at low c-di-GMP, we predicted that the fold induction of vpsL expression at these two c-di-GMP conditions would decrease with increasing phosphate. Indeed, we observed that at low Pi, vpsL was induced 12-fold by increasing c-di-GMP, while only a 1.2-fold induction was observed at high phosphate concentrations (Figure 10). At high c-di-GMP, vpsL-lux expression decreased from low to high Pi for unknown reasons, but we attribute this difference to the many regulatory factors that impact vspL expression (13,17,18,21–29). Importantly, vpsL-lux expression increased from low to high phosphate, supporting our in vitro results that demonstrate phosphate enhances VpsR activation of vpsL at lower concentrations of c-di-GMP.
DISCUSSION
Bacteria need to quickly adapt to a variety of different environments, and TCSs represent a widespread mechanism for this adaptation. However, despite the extensive biochemical characterizations and identifications of hundreds of TCSs, research undercovering how regulators sense signals and then work to activate transcription is still emerging.
In the case of the V. cholerae EBP-like activator and putative RR VpsR, it is well established that the level of c-di-GMP provides a crucial signal in the decision to form biofilm. However, it has been unclear whether other signals feed into this decision and particularly whether phosphorylation is involved. VpsR contains a predicted site for phosphorylation, a highly conserved aspartic acid D59 (Supplementary Figure S1). This conserved site is known to be phosphorylated in EBPs that are similar to VpsR. Furthermore, biofilm formation by V. cholerae, which is activated by VpsR, is impaired by the phospho-defective substitution D59A, but is similar to WT with the phospho-mimic D59E (34,35). These results have strongly suggested that phosphorylation of D59 is needed for VpsR activity and biofilm biogenesis. However, the recent crystal structure of truncated VpsR containing the REC–AAA+ domains suggests that the region surrounding D59 differs from that observed in other REC domains of EBPs, with an absence of residues needed to facilitate and stabilize the phosphorylated Asp (33). In addition, there is no cognate SK upstream of vpsR, and so this phosphorylation, if present, would have to arise either through crosstalk from a noncognate SK or from the phosphate donor Ac∼P, which is present in the cell.
Previously, we established an in vitro transcription system and demonstrated that in the presence of c-di-GMP and σ70-RNAP, VpsR directly activates the major promoters of the V. cholerae biofilm biogenesis/maintenance genes (17,21). The WT VpsR used in this system was purified after production in E. coli from a plasmid-encoded vpsR, and we demonstrated that it is transcriptionally active in vitro without the presence of a kinase or pretreatment with Ac∼P, despite the usual lability of a phosphorylated aspartic acid residue. Furthermore, the addition of Ac∼P does not increase WT VpsR activity (Figure 2A). These results are unusual for a phosphorylated RR. This led us to further investigate the possibility of VpsR phosphorylation by testing the activities of the phospho-variants D59A and D59E at PvpsL in vivo and the in vitro properties of these phospho-variants as well as VpsRren, a VpsR that was purified by a protocol involving denaturation/renaturation.
Despite the fact that the phospho-mimic D59E is active while the phospho-defective D59A mutation seriously impairs VpsR activity and that under certain conditions VpsRren needs Ac∼P for full activity in vitro, we show that VpsR does not require phosphorylation to induce transcription. Phosphorylation of VpsRren by Ac∼32P is not detected despite the protein being fully active under the labeling conditions (Figure 8), and mass spectrometry analyses eliminate the possibility that VpsR is acetylated or that purified VpsR maintains a stably conjugated phosphate (Supplementary Figure S3). Finally, either sodium phosphate or potassium phosphate, which are not Pi donors, can fully substitute for the stimulatory effect of Ac∼P (Figure 9, Supplementary Figure S5). This dependence of VpsRren on Pi does not arise simply because the denaturation/renaturation process generates an impaired protein since VpsRren is active when the level of c-di-GMP is high (Figure 2B).
Importantly, the stimulation of VpsR by phosphate is biologically relevant. We show that VpsR activation of PvpsL is dependent on Ac∼P when c-di-GMP levels are low in vivo (Figure 7), similar to what we observe in vitro. Thus, VpsR is a unique regulator that senses Pi directly without needing to be phosphorylated by an SK or by a chemical Pi donor. Although signaling through phosphorylation is a well-established mechanism, to our knowledge this is the first evidence of a putative RR that directly responds to phosphate.
Our results are consistent with previous studies indicating that there is a relationship between Pi levels and biofilm formation in V. cholerae. Under limiting Pi conditions, such as the marine environment, the RR PhoB decreases biofilm formation by decreasing the expression of VpsR (24). PhoB belongs to the PhoR/B TCS in which the inner membrane SK, PhoR, responds to periplasmic orthophosphate and, after autophosphorylation, transfers a phosphate to PhoB, which then subsequently activates the Pho regulon. The Pho regulon includes genes important for bacterial adaptation and survival in low Pi conditions. Our data suggest that another level of regulation arises through the direct sensing of Pi levels by VpsR. Furthermore, a pst (phosphate-specific transport system) mutation decreases biofilm formation and restores motility in V. cholerae (23), further suggesting a connection between phosphate levels and the transition between sessile and motile lifestyles. We observed an increase of vpsL expression from low to high phosphate at unaltered concentrations of c-di-GMP, consistent with these findings (Figure 10). However, at high c-di-GMP concentrations, vpsL expression decreased with increasing phosphate via an unknown regulatory mechanism that remains to be elucidated. It is important to note that the levels of c-di-GMP that we are using are biologically relevant. When grown in rich complex media, intracellular concentrations of c-di-GMP range from 10 μM at low cell density when biofilms are induced to 0.5 μM at high cell density when biofilms are repressed (36); in other conditions, the concentration of c-di-GMP can be even higher (85,86). Finally, it has been shown that the two Pi uptake systems and PhoB are upregulated within V. cholerae biofilms and are needed for full infectivity in the infant mouse model (23,62,87). These previous results and our findings demonstrate an intricate connection between phosphate and c-di-GMP to modulate V. cholerae biofilm formation.
Other bacteria also appear to exhibit this phenomenon. In Pseudomonas aeruginosa, low Pi levels help promote hyperswarming via rhlR expression by PhoB upregulation (88), while in P. fluorescens, bacteria grown in a Pi limited environment lose their ability to form biofilms due to Pho regulon activation (89). Such regulation could suggest that a lack of phosphate is a key signal for nutrient depletion, stimulating bacteria to disperse and colonize more nutrient-rich environments. Whether the influence of Pi in these systems also involves direct sensing is yet to be determined.
Overall, our results indicate that while VpsR either alone or in the presence of c-di-GMP binds DNA similarly, only an activated VpsR with c-di-GMP generates the active transcription complex with RNAP at the biofilm biogenesis/maintenance promoters (17,32) (Figure 11). However, the presence of Pi increases the level of VpsR activation at lower intracellular concentrations of c-di-GMP. Consequently, the direct sensing of Pi concentration by VpsR can be a factor in the formation or maintenance of biofilms, tipping the balance toward either the motile or sessile lifestyle. Because the affinity of VpsRren for the DNA is similar with or without pretreatment with Ac∼P and +/− c-di-GMP (Figure 3D and E), it seems unlikely that Pi aids in the interaction of VpsR alone with the DNA. However, the VpsR/DNA interaction within the activated transcription complex with RNAP and c-di-GMP differs from that of VpsR alone (Figure 4) (17); consequently, Pi might aid in the transition to the active transcription complex. Given that the presence of Pi becomes unnecessary as the concentration of c-di-GMP increases, a simple mechanism could be that the presence of Pi increases the binding affinity of VpsR for c-di-GMP. However, our attempts to demonstrate this directly have been inconclusive, suggesting that a more complicated mechanism may be at work.
Figure 11.
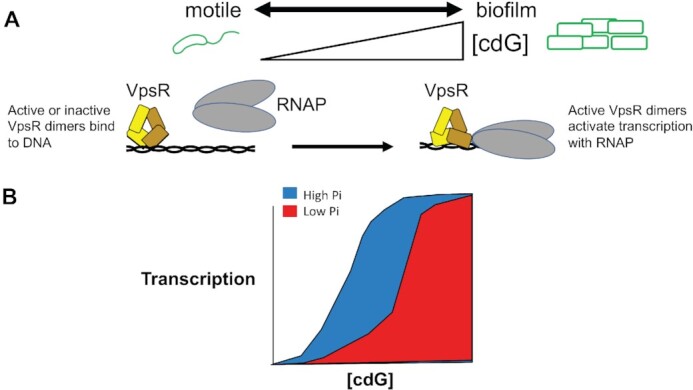
Model for the action of phosphate (Pi) and the concentration of c-di-GMP (cdG) on activation of PvpsL and biofilm formation. (A) The level of cdG controls the transition of V. cholerae from the motile to the biofilm lifestyle. Although VpsR dimers bind the DNA in the absence or presence of cdG, only the VpsR dimers formed in the presence of cdG are transcriptionally active with RNAP at PvpsL. Formation of this complex results in a different interaction of VpsR with the DNA as observed by the DNase I footprints (indicated by the change of the VpsR dimer conformation). (B) While cdG is required for active VpsR formation, higher levels of Pi tip the balance toward more active VpsR, which then increases transcription. Thus, the level of Pi also participates in the decision between the motile and biofilm lifestyles.
It appears then that even though the VpsR residue D59 is important and can be substituted with a glutamic acid, it is not phosphorylated. The recently reported VpsR REC–AAA+ structure predicts that the negatively charged D59 is 3.2 Å from the positively charged arginine R86 (33). Consequently, it may be that this electrostatic interaction is needed, perhaps for protein structure or stability, and that it can be replaced by a negatively charged glutamic acid, but not by an alanine. Our work provides a cautionary tale against assuming that the activities of phospho-variants and/or the need for Ac∼P under certain conditions automatically represent the presence of a phosphorylated aspartic acid. Further work will be needed to dissect the contribution of this conserved residue.
DATA AVAILABILITY
All original autoradiographs are available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Melissa Arroyo-Mendoza, Abraham Correa-Medina, Kyung Moon, Jennifer Patterson-West, Bokyung Son, Elisa Kleinfeld and members of the Waters laboratory for helpful discussions and comments on the manuscript and Bonnie Bassler for early support of this research.
Notes
Present address: Meng-Lun Hsieh, Department of Surgery, University of Texas Southwestern Center, Dallas, TX, USA.
Contributor Information
Meng-Lun Hsieh, Gene Expression and Regulation Section, Laboratory of Biochemistry and Genetics, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, USA; Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI 48823, USA.
Niklas Kiel, Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, MI 48824, USA.
Lisa M Miller Jenkins, Collaborative Protein Technology Resource, Laboratory of Cell Biology, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Wai-Leung Ng, Department of Molecular Biology and Microbiology, Tufts University School of Medicine, Boston, MA 02111, USA.
Leslie Knipling, Gene Expression and Regulation Section, Laboratory of Biochemistry and Genetics, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Christopher M Waters, Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, MI 48824, USA.
Deborah M Hinton, Gene Expression and Regulation Section, Laboratory of Biochemistry and Genetics, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases [to M.-L.H., L.K. and D.M.H.]; National Institutes of Health [F30GM123632 to M.-L.H.; R01GM109259, R01AI158433 and R35GM139537 to C.M.W.; R01AI21337 to W.-L.N.]; Michigan State University DO/PhD Program [to M.-L.H.]; National Science Foundation [MCB1253684 to C.M.W.]; Intramural Research Program of the National Cancer Institute [to L.M.M.J.]; Heinrich-Heine University Düsseldorf [to N.K.]. Funding for open access charge: Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases.
Conflict of interest statement. None declared.
REFERENCES
- 1. Ulrich L.E., Zhulin I.B.. The MiST2 database: a comprehensive genomics resource on microbial signal transduction. Nucleic Acids Res. 2010; 38:D401–D407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nguyen M.P., Yoon J.M., Cho M.H., Lee S.W.. Prokaryotic 2-component systems and the OmpR/PhoB superfamily. Can. J. Microbiol. 2015; 61:799–810. [DOI] [PubMed] [Google Scholar]
- 3. Hess J.F., Oosawa K., Kaplan N., Simon M.I.. Phosphorylation of three proteins in the signaling pathway of bacterial chemotaxis. Cell. 1988; 53:79–87. [DOI] [PubMed] [Google Scholar]
- 4. Zapf J., Madhusudan M., Grimshaw C.E., Hoch J.A., Varughese K.I., Whiteley J.M.. A source of response regulator autophosphatase activity: the critical role of a residue adjacent to the Spo0F autophosphorylation active site. Biochemistry. 1998; 37:7725–7732. [DOI] [PubMed] [Google Scholar]
- 5. Correa N.E., Lauriano C.M., McGee R., Klose K.E.. Phosphorylation of the flagellar regulatory protein FlrC is necessary for Vibrio cholerae motility and enhanced colonization. Mol. Microbiol. 2000; 35:743–755. [DOI] [PubMed] [Google Scholar]
- 6. Freeman J.A., Bassler B.L.. A genetic analysis of the function of LuxO, a two-component response regulator involved in quorum sensing in Vibrioharveyi. Mol. Microbiol. 1999; 31:665–677. [DOI] [PubMed] [Google Scholar]
- 7. Klose K.E., Weiss D.S., Kustu S.. Glutamate at the site of phosphorylation of nitrogen-regulatory protein NTRC mimics aspartyl-phosphate and activates the protein. J. Mol. Biol. 1993; 232:67–78. [DOI] [PubMed] [Google Scholar]
- 8. Alam M., Islam A., Bhuiyan N.A., Rahim N., Hossain A., Khan G.Y., Ahmed D., Watanabe H., Izumiya H., Faruque A.S.et al.. Clonal transmission, dual peak, and off-season cholera in Bangladesh. Infect. Ecol. Epidemiol. 2011; 1: 10.3402/iee.v1i0.7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morris J.G. Jr Cholera—modern pandemic disease of ancient lineage. Emerg. Infect. Dis. 2011; 17:2099–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Magny G.C., Mozumder P.K., Grim C.J., Hasan N.A., Naser M.N., Alam M., Sack R.B., Huq A., Colwell R.R.. Role of zooplankton diversity in Vibrio cholerae population dynamics and in the incidence of cholera in the Bangladesh Sundarbans. Appl. Environ. Microbiol. 2011; 77:6125–6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vezzulli L., Pezzati E., Moreno M., Fabiano M., Pane L., Pruzzo C., VibrioSea Consortium. Benthic ecology of Vibrio spp. and pathogenic Vibrio species in a coastal Mediterranean environment (La Spezia Gulf, Italy). Microb. Ecol. 2009; 58:808–818. [DOI] [PubMed] [Google Scholar]
- 12. Collin B., Rehnstam-Holm A.S.. Occurrence and potential pathogenesis of Vibrio cholerae, Vibrio parahaemolyticus and Vibrio vulnificus on the South Coast of Sweden. FEMS Microbiol. Ecol. 2011; 78:306–313. [DOI] [PubMed] [Google Scholar]
- 13. Teschler J.K., Cheng A.T., Yildiz F.H.. The two-component signal transduction system VxrAB positively regulates Vibrio cholerae biofilm formation. J. Bacteriol. 2017; 199:267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Conner J.G., Teschler J.K., Jones C.J., Yildiz F.H.. Staying alive: Vibrio cholerae’s cycle of environmental survival, transmission, and dissemination. Microbiol. Spectr. 2016; 4:4.2.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Silva A.J., Benitez J.A.. Vibrio cholerae biofilms and cholera pathogenesis. PLoS Negl. Trop. Dis. 2016; 10:e0004330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beyhan S., Bilecen K., Salama S.R., Casper-Lindley C., Yildiz F.H.. Regulation of rugosity and biofilm formation in Vibrio cholerae: comparison of VpsT and VpsR regulons and epistasis analysis of vpsT, vpsR, and hapR. J. Bacteriol. 2007; 189:388–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hsieh M.L., Hinton D.M., Waters C.M.. VpsR and cyclic di-GMP together drive transcription initiation to activate biofilm formation in Vibrio cholerae. Nucleic Acids Res. 2018; 46:8876–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yildiz F.H., Dolganov N.A., Schoolnik G.K.. VpsR, a member of the response regulators of the two-component regulatory systems, is required for expression of vps biosynthesis genes and EPS(ETr)-associated phenotypes in Vibrio cholerae O1 El Tor. J. Bacteriol. 2001; 183:1716–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Teschler J.K., Zamorano-Sanchez D., Utada A.S., Warner C.J., Wong G.C., Linington R.G., Yildiz F.H.. Living in the matrix: assembly and control of Vibriocholerae biofilms. Nat. Rev. Microbiol. 2015; 13:255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yildiz F.H., Schoolnik G.K.. Vibrio cholerae O1 El Tor: identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. Proc. Natl Acad. Sci. U.S.A. 1999; 96:4028–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsieh M.L., Waters C.M., Hinton D.M.. VpsR directly activates transcription of multiple biofilm genes in Vibrio cholerae. J. Bacteriol. 2020; 202:e00234-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hammer B.K., Bassler B.L.. Regulatory small RNAs circumvent the conventional quorum sensing pathway in pandemic Vibrio cholerae. Proc. Natl Acad. Sci. U.S.A. 2007; 104:11145–11149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pratt J.T., McDonough E., Camilli A.. PhoB regulates motility, biofilms, and cyclic di-GMP in Vibrio cholerae. J. Bacteriol. 2009; 191:6632–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sultan S.Z., Silva A.J., Benitez J.A.. The PhoB regulatory system modulates biofilm formation and stress response in El Tor biotype Vibrio cholerae. FEMS Microbiol. Lett. 2010; 302:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Casper-Lindley C., Yildiz F.H.. VpsT is a transcriptional regulator required for expression of vps biosynthesis genes and the development of rugose colonial morphology in Vibrio cholerae O1 El Tor. J. Bacteriol. 2004; 186:1574–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tischler A.D., Camilli A.. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol. Microbiol. 2004; 53:857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bilecen K., Fong J.C., Cheng A., Jones C.J., Zamorano-Sanchez D., Yildiz F.H.. Polymyxin B resistance and biofilm formation in Vibrio cholerae are controlled by the response regulator CarR. Infect. Immun. 2015; 83:1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lenz D.H., Miller M.B., Zhu J., Kulkarni R.V., Bassler B.L.. CsrA and three redundant small RNAs regulate quorum sensing in Vibrio cholerae. Mol. Microbiol. 2005; 58:1186–1202. [DOI] [PubMed] [Google Scholar]
- 29. McKee R.W., Kariisa A., Mudrak B., Whitaker C., Tamayo R.. A systematic analysis of the in vitro and in vivo functions of the HD-GYP domain proteins of Vibrio cholerae. BMC Microbiol. 2014; 14:272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rappas M., Bose D., Zhang X.. Bacterial enhancer-binding proteins: unlocking sigma54-dependent gene transcription. Curr. Opin. Struct. Biol. 2007; 17:110–116. [DOI] [PubMed] [Google Scholar]
- 31. Gao F., Danson A.E., Ye F., Jovanovic M., Buck M., Zhang X.. Bacterial enhancer binding proteins—AAA+ proteins in transcription activation. Biomolecules. 2020; 10:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hsieh M.-L., Hinton D.M., Waters C.M.. Chou S.-H., Guiliani N., Lee V.T., Römling U.. Cyclic di-GMP regulation of gene expression. Microbial Cyclic Di-Nucleotide Signaling. 2020; Cham: Springer International Publishing; 379–394. [Google Scholar]
- 33. Chakrabortty T., Roy Chowdhury S., Ghosh B., Sen U.. Crystal structure of VpsR revealed novel dimeric architecture and c-di-GMP binding site: mechanistic implications in oligomerization, ATPase activity and DNA binding. J. Mol. Biol. 2022; 434:167354. [DOI] [PubMed] [Google Scholar]
- 34. Lauriano C.M., Ghosh C., Correa N.E., Klose K.E.. The sodium-driven flagellar motor controls exopolysaccharide expression in Vibrio cholerae. J. Bacteriol. 2004; 186:4864–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu D.C., Zamorano-Sanchez D., Pagliai F.A., Park J.H., Floyd K.A., Lee C.K., Kitts G., Rose C.B., Bilotta E.M., Wong G.C.L.et al.. Reciprocal c-di-GMP signaling: incomplete flagellum biogenesis triggers c-di-GMP signaling pathways that promote biofilm formation. PLoS Genet. 2020; 16:e1008703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Waters C.M., Lu W., Rabinowitz J.D., Bassler B.L.. Quorum sensing controls biofilm formation in Vibrio cholerae through modulation of cyclic di-GMP levels and repression of vpsT. J. Bacteriol. 2008; 190:2527–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Srivastava D., Harris R.C., Waters C.M.. Integration of cyclic di-GMP and quorum sensing in the control of vpsT and aphA in Vibrio cholerae. J. Bacteriol. 2011; 193:6331–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krasteva P.V., Giglio K.M., Sondermann H.. Sensing the messenger: the diverse ways that bacteria signal through c-di-GMP. Protein Sci. 2012; 21:929–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Romling U., Galperin M.Y., Gomelsky M.. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol. Mol. Biol. Rev. 2013; 77:1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baraquet C., Harwood C.S.. FleQ DNA binding consensus sequence revealed by studies of Fleq-dependent regulation of biofilm gene expression in Pseudomonas aeruginosa. J. Bacteriol. 2015; 198:178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baraquet C., Harwood C.S.. Cyclic diguanosine monophosphate represses bacterial flagella synthesis by interacting with the Walker A motif of the enhancer-binding protein FleQ. Proc. Natl Acad. Sci. U.S.A. 2013; 110:18478–18483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baraquet C., Murakami K., Parsek M.R., Harwood C.S.. The FleQ protein from Pseudomonas aeruginosa functions as both a repressor and an activator to control gene expression from the pel operon promoter in response to c-di-GMP. Nucleic Acids Res. 2012; 40:7207–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matsuyama B.Y., Krasteva P.V., Baraquet C., Harwood C.S., Sondermann H., Navarro M.V.. Mechanistic insights into c-di-GMP-dependent control of the biofilm regulator FleQ from Pseudomonas aeruginosa. Proc. Natl Acad. Sci. U.S.A. 2016; 113:E209–E218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schumacher M.A., Zeng W., Findlay K.C., Buttner M.J., Brennan R.G., Tschowri N.. The Streptomyces master regulator BldD binds c-di-GMP sequentially to create a functional BldD2–(c-di-GMP)4 complex. Nucleic Acids Res. 2017; 45:6923–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schumacher M.A., Zeng W.. Structures of the activator of K. pneumonia biofilm formation, MrkH, indicates PilZ domains involved in c-di-GMP and DNA binding. Proc. Natl Acad. Sci. U.S.A. 2016; 113:10067–10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wilksch J.J., Yang J., Clements A., Gabbe J.L., Short K.R., Cao H., Cavaliere R., James C.E., Whitchurch C.B., Schembri M.A.et al.. MrkH, a novel c-di-GMP-dependent transcriptional activator, controls Klebsiella pneumoniae biofilm formation by regulating type 3 fimbriae expression. PLoS Pathog. 2011; 7:e1002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li W., He Z.G.. LtmA, a novel cyclic di-GMP-responsive activator, broadly regulates the expression of lipid transport and metabolism genes in Mycobacterium smegmatis. Nucleic Acids Res. 2012; 40:11292–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chambers J.R., Liao J., Schurr M.J., Sauer K.. BrlR from Pseudomonas aeruginosa is a c-di-GMP-responsive transcription factor. Mol. Microbiol. 2014; 92:471–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liao J., Schurr M.J., Sauer K.. The MerR-like regulator BrlR confers biofilm tolerance by activating multidrug efflux pumps in Pseudomonas aeruginosa biofilms. J. Bacteriol. 2013; 195:3352–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Raju H., Sharma R.. Crystal structure of BrlR with c-di-GMP. Biochem. Biophys. Res. Commun. 2017; 490:260–264. [DOI] [PubMed] [Google Scholar]
- 51. Wang F., He Q., Yin J., Xu S., Hu W., Gu L.. BrlR from Pseudomonas aeruginosa is a receptor for both cyclic di-GMP and pyocyanin. Nat. Commun. 2018; 9:2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fazli M., McCarthy Y., Givskov M., Ryan R.P., Tolker-Nielsen T.. The exopolysaccharide gene cluster Bcam1330–Bcam1341 is involved in Burkholderia cenocepacia biofilm formation, and its expression is regulated by c-di-GMP and Bcam1349. MicrobiologyOpen. 2013; 2:105–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fazli M., O’Connell A., Nilsson M., Niehaus K., Dow J.M., Givskov M., Ryan R.P., Tolker-Nielsen T.. The CRP/FNR family protein Bcam1349 is a c-di-GMP effector that regulates biofilm formation in the respiratory pathogen Burkholderia cenocepacia. Mol. Microbiol. 2011; 82:327–341. [DOI] [PubMed] [Google Scholar]
- 54. Krasteva P.V., Fong J.C., Shikuma N.J., Beyhan S., Navarro M.V., Yildiz F.H., Sondermann H.. Vibrio cholerae VpsT regulates matrix production and motility by directly sensing cyclic di-GMP. Science. 2010; 327:866–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zamorano-Sanchez D., Fong J.C., Kilic S., Erill I., Yildiz F.H.. Identification and characterization of VpsR and VpsT binding sites in Vibrio cholerae. J. Bacteriol. 2015; 197:1221–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ross W., Thompson J.F., Newlands J.T., Gourse R.L.. E. coli Fis protein activates ribosomal RNA transcription in vitro and in vivo. EMBO J. 1990; 9:3733–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hammer B.K., Bassler B.L.. Distinct sensory pathways in Vibrio cholerae El Tor and classical biotypes modulate cyclic dimeric GMP levels to control biofilm formation. J. Bacteriol. 2009; 191:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Studier F.W., Rosenberg A.H., Dunn J.J., Dubendorff J.W.. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990; 185:60–89. [DOI] [PubMed] [Google Scholar]
- 59. Thelin K.H., Taylor R.K.. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect. Immun. 1996; 64:2853–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Skorupski K., Taylor R.K.. Positive selection vectors for allelic exchange. Gene. 1996; 169:47–52. [DOI] [PubMed] [Google Scholar]
- 61. Tamayo R., Schild S., Pratt J.T., Camilli A.. Role of cyclic di-GMP during El Tor biotype Vibrio cholerae infection: characterization of the in vivo-induced cyclic di-GMP phosphodiesterase CdpA. Infect. Immun. 2008; 76:1617–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mudrak B., Tamayo R.. The Vibrio cholerae Pst2 phosphate transport system is upregulated in biofilms and contributes to biofilm-induced hyperinfectivity. Infect. Immun. 2012; 80:1794–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hsieh M.L., James T.D., Knipling L., Waddell M.B., White S., Hinton D.M.. Architecture of the bacteriophage T4 activator MotA/promoter DNA interaction during sigma appropriation. J. Biol. Chem. 2013; 288:27607–27618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Boulanger A., Moon K., Decker K.B., Chen Q., Knipling L., Stibitz S., Hinton D.M.. Bordetella pertussis fim3 gene regulation by BvgA: phosphorylation controls the formation of inactive vs. active transcription complexes. Proc. Natl Acad. Sci. U.S.A. 2015; 112:E526–E535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xu H., Caimano M.J., Lin T., He M., Radolf J.D., Norris S.J., Gherardini F., Wolfe A.J., Yang X.F.. Role of acetyl-phosphate in activation of the Rrp2–RpoN–RpoS pathway in Borrelia burgdorferi. PLoS Pathog. 2010; 6:e1001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Quon K.C., Marczynski G.T., Shapiro L.. Cell cycle control by an essential bacterial two-component signal transduction protein. Cell. 1996; 84:83–93. [DOI] [PubMed] [Google Scholar]
- 67. Pursley B.R., Fernandez N.L., Severin G.B., Waters C.M.. The Vc2 cyclic di-GMP-dependent riboswitch of Vibrio cholerae regulates expression of an upstream putative small RNA by controlling RNA stability. J. Bacteriol. 2019; 201:e00293-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wolfe A.J. The acetate switch. Microbiol. Mol. Biol. Rev. 2005; 69:12–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Barbieri C.M., Stock A.M.. Universally applicable methods for monitoring response regulator aspartate phosphorylation both in vitro and in vivo using Phos-tag-based reagents. Anal. Biochem. 2008; 376:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Boulanger A., Chen Q., Hinton D.M., Stibitz S.. In vivo phosphorylation dynamics of the Bordetella pertussis virulence-controlling response regulator BvgA. Mol. Microbiol. 2013; 88:156–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kinoshita E., Kinoshita-Kikuta E.. Improved Phos-tag SDS-PAGE under neutral pH conditions for advanced protein phosphorylation profiling. Proteomics. 2011; 11:319–323. [DOI] [PubMed] [Google Scholar]
- 72. Wayne K.J., Li S., Kazmierczak K.M., Tsui H.C., Winkler M.E.. Involvement of WalK (VicK) phosphatase activity in setting WalR (VicR) response regulator phosphorylation level and limiting cross-talk in Streptococcus pneumoniae D39 cells. Mol. Microbiol. 2012; 86:645–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Horstmann N., Saldana M., Sahasrabhojane P., Yao H., Su X., Thompson E., Koller A., Shelburne S.A. 3rd. Dual-site phosphorylation of the control of virulence regulator impacts group a streptococcal global gene expression and pathogenesis. PLoS Pathog. 2014; 10:e1004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Goudreau P.N., Lee P.J., Stock A.M.. Stabilization of the phospho-aspartyl residue in a two-component signal transduction system in Thermotoga maritima. Biochemistry. 1998; 37:14575–14584. [DOI] [PubMed] [Google Scholar]
- 75. Decker K.B., Hinton D.M.. Transcription regulation at the core: similarities among bacterial, archaeal, and eukaryotic RNA polymerases. Annu. Rev. Microbiol. 2013; 67:113–139. [DOI] [PubMed] [Google Scholar]
- 76. Feklistov A., Darst S.A.. Structural basis for promoter-10 element recognition by the bacterial RNA polymerase sigma subunit. Cell. 2011; 147:1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang Y., Feng Y., Chatterjee S., Tuske S., Ho M.X., Arnold E., Ebright R.H.. Structural basis of transcription initiation. Science. 2012; 338:1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Weinert B.T., Iesmantavicius V., Wagner S.A., Scholz C., Gummesson B., Beli P., Nystrom T., Choudhary C.. Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol. Cell. 2013; 51:265–272. [DOI] [PubMed] [Google Scholar]
- 79. Kuhn M.L., Zemaitaitis B., Hu L.I., Sahu A., Sorensen D., Minasov G., Lima B.P., Scholle M., Mrksich M., Anderson W.F.et al.. Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS One. 2014; 9:e94816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Muzhingi I., Prado C., Sylla M., Diehl F.F., Nguyen D.K., Servos M.M., Flores Ramos S., Purdy A.E.. Modulation of CrbS-dependent activation of the acetate switch in Vibrio cholerae. J. Bacteriol. 2018; 200:e00380-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liimatta K., Flaherty E., Ro G., Nguyen D.K., Prado C., Purdy A.E.. A putative acetylation system in Vibrio cholerae modulates virulence in arthropod hosts. Appl. Environ. Microbiol. 2018; 84:e01113-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wolfe A.J. Physiologically relevant small phosphodonors link metabolism to signal transduction. Curr. Opin. Microbiol. 2010; 13:204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Singh K.K., Athira P.J., Bhardwaj N., Singh D.P., Watson U., Saini D.K.. Acetylation of response regulator protein MtrA in M. tuberculosis regulates its repressor activity. Front. Microbiol. 2020; 11:516315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zee B.M., Garcia B.A.. Validation of protein acetylation by mass spectrometry. Methods Mol. Biol. 2013; 981:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Koestler B.J., Waters C.M.. Exploring environmental control of cyclic di-GMP signaling in Vibrio cholerae by using the ex vivo lysate cyclic di-GMP assay (TELCA). Appl. Environ. Microbiol. 2013; 79:5233–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Massie J.P., Reynolds E.L., Koestler B.J., Cong J.P., Agostoni M., Waters C.M.. Quantification of high-specificity cyclic diguanylate signaling. Proc. Natl Acad. Sci. U.S.A. 2012; 109:12746–12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Merrell D.S., Hava D.L., Camilli A.. Identification of novel factors involved in colonization and acid tolerance of Vibrio cholerae. Mol. Microbiol. 2002; 43:1471–1491. [DOI] [PubMed] [Google Scholar]
- 88. Blus-Kadosh I., Zilka A., Yerushalmi G., Banin E.. The effect of pstS and phoB on quorum sensing and swarming motility in Pseudomonas aeruginosa. PLoS One. 2013; 8:e74444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Monds R.D., Newell P.D., Gross R.H., O’Toole G.A. Phosphate-dependent modulation of c-di-GMP levels regulates Pseudomonas fluorescens Pf0-1 biofilm formation by controlling secretion of the adhesin LapA. Mol. Microbiol. 2007; 63:656–679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original autoradiographs are available upon request.