

Abstract
Despite the high morbidity and mortality rates associated with colorectal cancer (CRC), the aberrant genes and mechanisms driving CRC pathogenesis remain poorly understood. Chromosome instability (CIN), or ongoing changes in chromosome numbers, is a predominant form of genome instability associated with ~85% of CRCs, suggesting it may be a key mechanism driving CRC oncogenesis. CIN enables the acquisition of copy number alterations conferring selective growth, proliferation and survival advantages that promote cellular transformation. Despite these associations, the aberrant genes underlying CIN remain largely unknown. Candidate CIN gene FBXO7 encodes an F-box protein, a subunit of the SKP1-CUL1-FBOX (SCF) complex that confers substrate specificity to the complex and targets proteins for subsequent degradation by the 26S proteasome. Recently, the genes encoding the three core SCF complex members were identified as CIN genes; however, it is unknown whether F-box proteins exhibit similar integral roles in maintaining chromosome stability. Using short- small interfering RNA (siRNA) and long- (CRISPR/Cas9) term approaches, we show that reduced FBXO7 expression induces CIN in various colonic epithelial cell contexts, whereas FBXO7 knockout clones also exhibit hallmarks associated with cellular transformation, namely increased clonogenic and anchorage-independent growth. Collectively, these data demonstrate that FBXO7 is required to maintain genome stability identifying FBXO7 a novel CIN gene whose reduced expression may contribute to CRC development and progression.
Introduction
A stable genome is an essential characteristic of all eukaryotes that is fundamental to the maintenance and accurate partitioning of genetic material (i.e. chromosomes) into daughter cells. As such, genome stability is vital for normal cell function, physiology and species survival (1–3). Despite its indispensable nature, the extensive array of genes, proteins and cellular pathways regulating genome stability remain poorly understood; however, decades of focused research have provided tremendous insight into many key processes regulating chromosome stability including centrosome biology, chromosome dynamics, DNA replication and repair. As expected, aberrant expression of the genes encoding these pathways has pathogenic implications for many diseases such as cancer (4–7), and the regulation of these processes is now being explored for their clinical utility as diagnostic biomarkers, prognostic indicators or as drug targets that can be therapeutically leveraged for precision medicine purposes.
Colorectal cancer (CRC) is the third most diagnosed cancer in North America (United States and Canada only as comprehensive statistics are not available for Mexico), with ~175 000 new diagnoses and ~62 500 deaths attributed to the disease each year (8,9). Consequently, understanding the aberrant genes and pathways driving CRC development and progression is key to gain mechanistic insight that could lead to the development of novel therapies aimed at reducing current morbidity and mortality rates. Interestingly, ~85% of CRCs exhibit chromosome instability (CIN) (5,10), a common form of genome instability. CIN is defined as an increase in the rate at which whole chromosomes or large chromosomal fragments are gained or lost and is a driver of both genetic and cell-to-cell heterogeneity (5,10–12). Accordingly, CIN can confer selective growth, proliferation and survival advantages (10,11,13,14) associated with cellular transformation (7,15,16), tumor evolution (17), the acquisition of drug resistance (18) and poor patient outcomes (19,20). Thus, CIN is hypothesized to be an early etiologic event driving oncogenesis (4,6,7,13,17). Importantly, CIN is associated with ~85% of CRCs (10), suggesting it may be a key mechanism driving CRC oncogenesis. Despite the list of associations detailed above, the molecular determinants of CIN (i.e. aberrant genes, proteins and cellular pathways) remain poorly understood.
Recent evidence demonstrates that aberrant expression of key SKP1-CUL1-FBOX (SCF) complex members induces CIN (21–23). The SCF complex is comprised of three invariable core components (SKP1, S-phase kinase-associated protein 1; CUL1, Cullin 1; RBX1, Ring-Box 1), and one of 69 variable subunits called F-box proteins that confer substrate specificity to the SCF complex (24,25). The SCF complex is an E3 ubiquitin ligase whose principal function is to poly-ubiquitinate target proteins to effectively label them for proteolytic degradation by the 26S proteasome (24–26). As alluded to above, SKP1, CUL1 and RBX1 were recently identified as novel CIN genes, as their reduced expression induces CIN in CRC (21) and/or ovarian cancer contexts (22,23). Although emerging data demonstrate that the core SCF complex members harbor critical roles in maintaining genome stability in eukaryotic cell contexts, it remains unclear which, if any, of the F-box proteins also exhibit essential roles in maintaining genome and chromosome stability.
Using a complementary series of bioinformatics, genetic and single-cell quantitative imaging microscopy (QuantIM) approaches, we examined the impact altered FBXO7 expression has in CIN and CRC pathogenesis. FBXO7 was purposefully selected as a representative member of the F-box ‘other’ protein family for which no information is available specifically detailing its impact on genome instability, CIN or CRC pathogenesis. Accordingly, we sought to establish the clinical relevance of FBXO7 in CRC patient samples and determine the functional impact reduced FBXO7 expression has for chromosome stability in various colonic epithelial contexts. We first interrogated The Cancer Genome Atlas (TCGA) (27) and determined that FBXO7 copy number losses occur frequently in many solid cancer types. Most intriguingly, ~32% of CRCs exhibit copy number losses that correspond with significantly reduced messenger RNA (mRNA) expression levels relative to normal (diploid) cases. Moreover, copy number losses correlated with significantly worse outcomes relative to patients with normal copy numbers, suggesting that reduced FBXO7 expression may contribute to CRC development and/or progression. In agreement with this possibility, short-term FBXO7 silencing experiments performed in karyotypically stable malignant and non-malignant colonic epithelial cells corresponded with increases in CIN phenotypes, including changes in nuclear areas, micronucleus formation and aberrant chromosome numbers. To determine the long-term impact reduced FBXO7 expression has on CIN, heterozygous (FBXO7+/−) and homozygous (FBXO7−/−) clones were generated using CRISPR/Cas9 approaches. Clones were continually passaged and assessed at regular intervals over ~10 weeks with each clone exhibiting ongoing and dynamic CIN phenotypes relative to a control clone. Finally, we determined that reduced expression induced increases in cellular transformation phenotypes. Collectively, our findings show that FBXO7 is normally required to maintain genome stability and that reduced expression induces CIN and cellular transformation, which is consistent with reduced FBXO7 expression being a pathogenic contributor to CRC.
Results
Bioinformatic analyses prioritize FBXO7 as a strong candidate for subsequent study
As detailed above, CIN induces ongoing changes in chromosome complements (5), is a driver of genetic and cell-to-cell heterogeneity (10–12) and is arguably best understood in CRC, where it occurs in ~85% of cases (5). Despite this information, the causal origins of CIN in CRC are poorly understood. Recently, we determined that reduced expression of the three invariable core members of the SCF complex (SKP1, CUL1 and RBX1) induces CIN and has pathogenic implications for cancer development (21–23), but the specific impacts of the associated 69 F-box proteins remain unknown. To begin to explore the clinical relevance of the F-box proteins to ultimately prioritize one for functional analyses, a comprehensive bioinformatic analysis was performed in which both gene copy number losses and their associated impacts on patient outcomes were assessed in a TCGA cohort comprised of 526 CRC cases (27) using cBioPortal (28,29). First, gene copy number losses (deep and shallow deletions) were assessed (Supplementary Material, Fig. S1) and revealed that all 69 genes exhibit losses ranging from 0.6% (FBXL18) to ~67% (FBXO15), with shallow deletions being more prevalent than deep deletions, which are comparatively rare events (31 genes have 0 deep deletions [Supplementary Material, Table S1]). Although the 69 genes exhibit varying degrees of copy number losses, this does not mean that loss of each gene drives CRC pathogenesis and adversely impacts patient outcomes. Thus, to discern which gene-specific copy number losses correspond with worse clinical outcomes, three survival categories (progression-free, disease-specific and overall survival) were assessed for each gene. In general, copy number losses (deep and shallow deletions) correlated with significantly worse progression-free survival for 29 of 69 (42%) genes surveyed, whereas significantly worse disease-specific survival and overall survival were associated with 19 (28%) and 4 (6%) genes, respectively (Supplementary Material, Table S1). Remarkably, copy number losses of only four genes (FBXW12, FBXL2, FBXO7 and FBXO30) corresponded with significantly worse outcomes in all three outcome categories (progression-free, disease-specific and overall survival) and while FBXW12, FBXL2 and FBXO30 exhibit gene copy number losses of ~12–16%, FBXO7 is lost in ~32% of cases, identifying it as the most clinically relevant candidate to pursue in further bioinformatic and functional analyses.
FBXO7 alterations are frequent in cancer and are associated with worse overall survival
Our previous work determined that the three core SCF complex members exhibit frequent copy number losses in cancer and that reduced expression corresponds with CIN (21–23). Unfortunately, little is known about the potential clinical impact aberrant FBXO7 expression has in cancer. To determine the prevalence and potential clinical impact FBXO7 copy number alternations may have in cancer, TCGA PanCancer Atlas data from 12 common cancer types were assessed for copy number status (Fig. 1A) (27–29). Overall, FBXO7 amplifications (≥2 additional copies) are rare (0% to 1%), whereas gains (one additional copy) occur in all 12 cancers and range from 0.6% to 22% in prostate and head and neck cancers, respectively. Similarly, deep (i.e. homozygous) deletions are rare (0% to 0.2%), whereas shallow (i.e. heterozygous) deletions are prevalent in all 12 cancer types and range from 8% to 71% in kidney and ovarian cancers, respectively. Of relevance to this study, FBXO7 copy number losses are more prevalent in CRC than copy number gains (Fig. 1A), with 0.2% (1/594 cases) and 32.5% (193 cases) of cases exhibiting deep and shallow deletions, respectively, relative to gains (4.4%; 26 cases) and amplifications (0.2%; 1 case). An overriding assumption of the copy number alteration data presented above is that they correspond with gene expression level changes. As shown in Figure 1B, a positive correlation exists between copy number changes and mRNA expression in CRC, with a significant difference observed between patients with shallow deletions and those with normal (diploid) copy numbers. Finally, health outcomes were specifically assessed for CRC and in general, patients with shallow deletions (i.e. low expression) fared worse than those with normal (diploid) copy numbers for progression-free, disease-specific and overall survival (Fig. 1C). Overall, these findings show that FBXO7 copy number alterations occur frequently in solid tumors with shallow deletions being the most prevalent, supporting a potential tumor suppressive role for FBXO7. This possibility is further bolstered by the health outcome data implicating reduced expression as an etiological factor in CRC, which warrants further investigation into the underlying mechanism of how reduced FBXO7 expression may contribute to oncogenesis.
Figure 1.

FBXO7 copy number alterations are frequent and associated with worse overall survival in cancer. (A) Bar graph presenting FBXO7 copy number alterations from TCGA PanCancer Atlas data for 12 common solid cancer types (total cases) (27–29). Note ~30% of CRC cases exhibit shallow deletions (i.e. heterozygous loss) of FBXO7. (B) Violin plots of TCGA PanCancer Atlas data for CRC identify positive linear relationships between FBXO7 copy number alterations and mRNA expression (27–29). Dotted and dashed lines within the violin plots identify 25th/75th and 50th percentiles, respectively. The various copy number categories (deep deletion; shallow deletion; diploid; gain; amplification) are indicated along the x-axis with the total number of cases indicated in brackets. Student’s t-test identifies a statistically significant difference in mean FBXO7 expression levels between samples harboring shallow deletions relative to normal diploid controls (****P-value <0.0001). (C) Kaplan–Meier curves reveal significantly worse progression-free (left), disease-specific (middle) and overall (right) survival for CRC patients harboring shallow FBXO7 deletions relative to those with normal (diploid) copy numbers (27–29). Case numbers are indicated within brackets.
Reduced FBXO7 expression induces increases in CIN-associated phenotypes in HCT116
The above data combined with those identifying SKP1, CUL1 and RBX1 as novel CIN genes (21–23) suggest that FBXO7 expression may normally be required to maintain chromosome stability. Accordingly, to determine whether reduced FBXO7 expression induces CIN, transient small interfering RNA (siRNA) based silencing was employed in HCT116 cells, a karyotypically stable CRC cell line regularly employed in CIN-based studies (21,30–33). However, we first evaluated the silencing efficiencies of four individual FBXO7 siRNA duplexes to identify the two most efficient (Fig. 2A; siFBXO7-2 and -4). These two duplexes, along with the siRNA pool (siFBXO7-P; see Materials and Methods) were utilized in all subsequent experiments and typically reduced FBXO7 abundance to <10% of endogenous levels.
Figure 2.

FBXO7 silencing corresponds with increases in CIN-associated phenotypes in HCT116 cells. (A) Semi-quantitative western blot showing reduced FBXO7 expression relative to siControl in HCT116 cells following transfection with four individual (siFBXO7-1, -2, -3, -4) and pooled (siFBXO7-P) siRNAs. FBXO7 expression is normalized to the α-tubulin loading control and is presented relative to siControl (set to 100%). SiFBXO7-2 and -4 are the two most efficient individual siRNA duplexes. (N = 2). (B) Low-resolution images of Hoechst-stained nuclei showing striking visual increases in nuclear areas and cell-to-cell heterogeneity following FBXO7 silencing relative to siControl. (C) Graph showing statistically significant increases (i.e. rightward shift) in cumulative nuclear area distribution frequencies following FBXO7 silencing relative to siControl (two-sample KS test; na, not applicable; ****P-value < 0.0001). (N = 3, n = 6). (D) High-resolution image of a Hoechst-stained nucleus and micronucleus (arrowhead). (E) Dot plot identifies significant increases in frequencies of micronuclei following FBXO7 silencing relative to siControl. The median values are indicated by red bars, whereas the fold increases in the median values are presented below the statistical information (Mann-Whitney (MW) test; **P-value < 0.01; N = 3, n = 6).
To begin to assess the impact reduced FBXO7 expression has in HCT116 cells, QuantIM was employed to identify changes in two CIN phenotypes, namely nuclear areas and micronucleus formation. Following FBXO7 silencing, visually striking increases in nuclear areas were readily apparent relative to siControl (Fig. 2B). Quantitative comparisons of the cumulative nuclear area frequency distributions revealed overall increases in nuclear area heterogeneity following silencing that were statistically significant relative to siControl (Fig. 2C; Supplementary Material, Table S2). Similarly, QuantIM identified significant increases in micronucleus formation (Fig. 2D) that were 3.4-, 8.4- and 6.0-fold greater than siControl in siFBXO7-2, -4 and -P, respectively (Fig. 2E; Supplementary Material, Table S3). Collectively, these data show that reduced FBXO7 expression corresponds with increases in CIN phenotypes in HCT116 cells, suggesting normal expression may be important for maintaining chromosome stability.
FBXO7 is required to maintain chromosome stability in HCT116 cells
To determine whether reduced FBXO7 expression adversely impacts chromosome stability by promoting chromosome gains and/or losses, mitotic chromosome spreads were generated (Fig. 3A) following FBXO7 silencing and quantitatively compared with siControl (HCT116 have a modal number of 45 chromosomes). In agreement with the nuclear area and micronucleus data, two-sample Kolmogorov–Smirnov (KS) tests identified statistically significant differences in the cumulative distribution frequencies following FBXO7 silencing relative to siControl (Fig. 3B; Supplementary Material, Table S4). More specifically, FBXO7 silencing induced significant increases (2.2- to 2.5-fold) in the frequency of aberrant mitotic spreads (n ≠ 45) relative to siControl (Fig. 3C; Supplementary Material, Table S5). Collectively, these chromosome data along with the nuclear area and micronucleus formation findings identify FBXO7 as a novel chromosome stability gene in HCT116 cells. They also show that reduced expression induces CIN, which is an aberrant phenotype associated with early etiological events in cancer including cellular transformation and oncogenesis (7,15,16).
Figure 3.

Reduced FBXO7 expression corresponds with increases in aberrant chromosome numbers in HCT116 cells. (A) Representative high-resolution images of DAPI-counterstained mitotic chromosome spreads displaying the modal number of 45 chromosomes (siControl), loss (<45), small-scale gain (46–59) and large-scale gain (≥60) in HCT116 cells. Chromosome numbers (N) within each spread are indicated in the top right corner. (B) Cumulative chromosome number distribution graph reveals significant differences following FBXO7 silencing relative to siControl (two-sample KS test; na, not applicable; *P-value ≤ 0.05; ***P-value < 0.001; ****P-value < 0.0001). (N = 3, n > 100 spreads/condition). (C) Dot plot presenting the frequencies of chromosome spreads with aberrant chromosome numbers following FBXO7 silencing. The fold increase relative to siControl is indicated at the top of each column with mean values identified by the red bar and the statistical analyses as indicated (Student’s t-test; *P-value ≤ 0.05; **P-value < 0.01; N = 3, n > 100 spreads/condition).
FBXO7 silencing induces CIN in non-malignant/non-transformed cells
Although the above findings support the possibility that reduced expression may have pathogenic implications for CRC, HCT116 cells are a transformed malignant cell line that cannot accurately inform upon the early etiological events driving cancer formation. Thus, to gain insight into CRC etiology, it was critical to perform analogous experiments with clinically appropriate models representing early disease states. In this regard, 1CT and its derivative cell lines, RPA and A1309, are karyotypically stable, non-malignant/non-transformed, male colonic epithelial cell lines (modal chromosome numbers = 46 [Supplementary Material, Fig. S2]). Although 1CT are immortalized with human telomerase reverse transcriptase (hTERT) and cyclin-dependent kinase 4 (CDK4), RPA and A1309 harbor additional genetic alterations frequently observed in precursor (precancerous) lesions including the expression of KRASG12V, along with reduced Tumor Protein P53 (TP53) and Adenomatous Polyposis Coli (APC) expression; A1309 cells also express a mutant form APC truncated at residue 1309 (34,35). Thus, 1CT, RPA and A1309 are ideal cellular models in which to assess the potential impact reduced FBXO7 expression has in early disease development.
To determine the impact reduced FBXO7 expression has in models of early disease development, analogous experiments to those detailed above were conducted in 1CT, RPA and A1309 cells. However, western blots were first performed that confirmed FBXO7 silencing reduced expression by >80% in all three cell line contexts (Fig. 4A). In agreement with the HCT116 data, nuclear area analyses revealed significant increases in cumulative nuclear area distribution frequencies for siFBXO7-4 and -P in 1CT, whereas all three silencing conditions were significant in both in RPA and A1309 cells (Fig. 4B; Supplementary Material, Table S6). Micronucleus formation analyses revealed trending (siFBXO7-4) or significant (siFBXO7-P) increases in 1CT, whereas RPA exhibited significant increases for siFBXO7-2 and -4 that were ~1.6-fold greater than siControl (Fig. 4C; Supplementary Material, Table S7). Similarly, silencing in A1309 corresponded with a 1.6- to 2.5-fold increase that was significant for siFBXO7-2 and siFBXO7-P (Fig. 4C; Supplementary Material, Table S7). Taken together, these findings demonstrate that reduced FBXO7 expression corresponds with increases in CIN phenotypes in early models of disease development.
Figure 4.
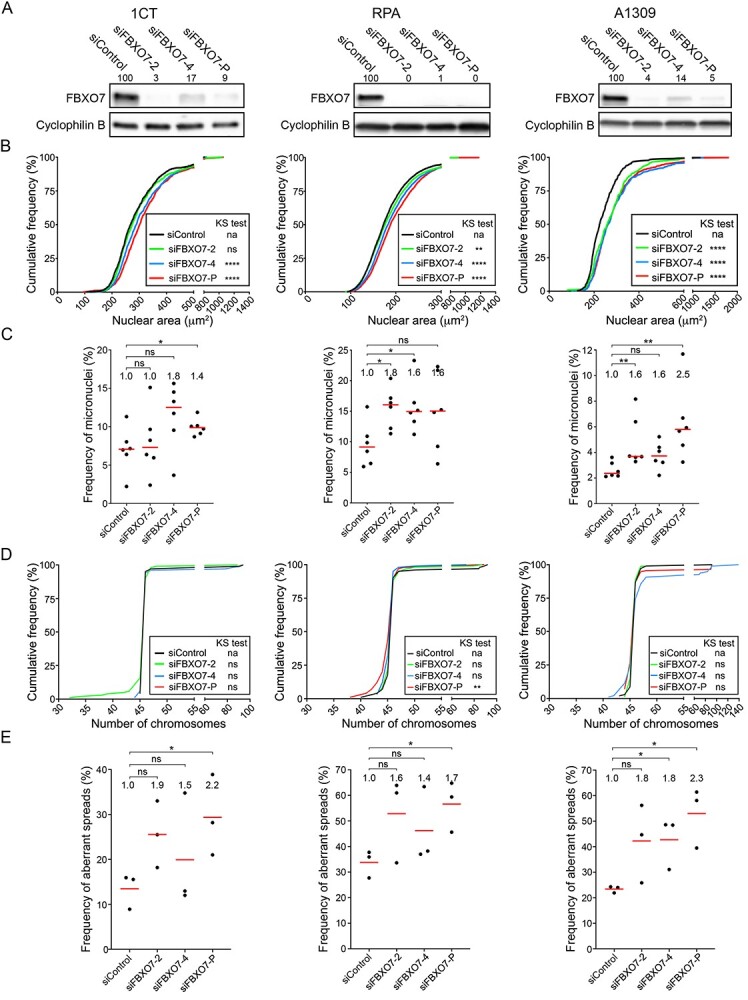
FBXO7 silencing in 1CT, RPA and A1309 cells corresponds with increases in CIN-associated phenotypes. (A) Semi-quantitative western blots showing reduced FBXO7 expression following silencing in 1CT (left), RPA (middle) and A1309 (right) cells. Cells were treated with individual (siFBXO7-2, -4) and pooled (siFBXO7-P) siRNA duplexes. FBXO7 expression is normalized to the corresponding loading control (Cyclophilin B) and is presented relative to siControl. (B) Nuclear area frequency distributions following FBXO7 silencing relative to siControl in 1CT (left), RPA (middle) and A1309 (right) cells. Two-sample KS tests reveal significant or trending increases (i.e. rightward shifts) in distributions relative to siControl (na, not applicable; ns, not significant; [P-value > 0.05]; **P-value < 0.01; ****P-value < 0.0001). (C) Dot plots identify trending and significant increases in micronucleus formation following FBXO7 silencing relative to siControl. Red lines identify the median of six replicate cells, with statistical significance as determined by MW tests (ns, not significant; *P-value ≤ 0.05; **P-value < 0.01; N = 3; n = 6). The fold increase in the median frequency of micronuclei relative to siControl is presented at the top of each column. (D) Representative graphs presenting the cumulative chromosome number distribution frequencies following FBXO7 silencing in 1CT, RPA and A1309 cells. Two-sample KS tests reveal that the siFBXO7-P condition induces a significant change in distributions within the RPA and A1309 cells (ns, not significant [P-value > 0.05]; **P-value < 0.01; ****P-value < 0.0001). (E) Dot plots presenting the frequency of abnormal mitotic chromosome spreads (N ≠ 46 chromosomes) following FBXO7 silencing relative to siControl. Red bars identify mean values with the mean fold increase in aberrant spreads relative to siControl presented at the top of each column. Student’s t-tests reveal significant increases relative to siControl as indicated (ns not significant [P-value > 0.05], *P-value ≤ 0.05; N = 3; n > 100 chromosome spreads/condition).
To establish whether reduced FBXO7 expression corresponds with changes in chromosome numbers, mitotic chromosome spreads were assessed following silencing in 1CT, RPA and A1309 cells. Although Figure 4D shows that the cumulative chromosome number distribution frequencies exhibit visual changes within the tail regions of the curves relative to siControl (particularly for RPA and A1309), the distributions were only deemed significant for the siFBXO7-P condition within RPA cells (Supplementary Material, Table S8); however, it should be noted that two-sample KS tests are insensitive to changes within tail regions (36). Nevertheless, when the frequency of aberrant spreads (n ≠ 46) is compared for each biological replicate (Fig. 4E; Supplementary Material, Table S9), a trending and sometimes significant (siFBXO7-4, siFBXO7-P) increase in aberrant chromosome numbers occurred within each cell line. Overall, the mean fold increase in the frequency of aberrant chromosome numbers ranges from 1.4- to 2.3-fold for all FBXO7 silencing conditions in all three cell lines, but the greatest fold increases generally occurred within A1309. Taken together, these findings identify FBXO7 as a novel chromosome stability gene in non-malignant/non-transformed colonic epithelial cells and show that the most pronounced aberrant phenotypes occur within A1309 cells. These data strengthen the possibility that reduced FBXO7 expression may be an early etiological event in CRC pathogenesis.
Heterozygous and homozygous loss of FBXO7 corresponds with dynamic CIN phenotypes in cellular models of early disease development
Having identified copy number losses as the most common FBXO7 alteration in CRC patients, we modeled this alteration and assessed the temporal impact that constitutive reduction in FBXO7 expression has on CIN in long-term (~10 weeks) experiments. Although the siRNA-based approaches reduce FBXO7 expression to levels below those expected within CRC patient samples harboring shallow deletions (i.e. heterozygous loss), they do not completely eliminate protein expression as is expected in samples harboring deep deletions (i.e. homozygous deletion). Thus, we sought to generate heterozygous knockout cells to model the most prevalent clinical CRC context and homozygous knockout cells to not only model the rare clinical context but to also assess complete loss-of-function. Using CRISPR/Cas9 approaches, two heterozygous (FBXO7+/−1 and FBXO7+/−2) and two homozygous (FBXO7−/−A and FBXO7−/−B) clones, along with a non-targeting sgRNA clone (NT-Control) were generated in A1309 cells. A1309 were purposefully selected as they are a non-transformed, early model of disease development that produced the most pronounced CIN phenotypes following FBXO7 silencing (Fig. 4). Western blots determined FBXO7 expression was reduced to ~40% of endogenous levels within FBXO7+/−1 and FBXO7+/−2 and was absent in both FBXO7−/− clones (Supplementary Material, Fig. S3A). DNA sequencing determined that FBXO7+/−1 and FBXO7+/−2 harbor a single base pair (bp) insertion and deletion, respectively, whereas FBXO7−/−A harbors a 2 bp deletion in allele 1 and a 1 bp deletion in allele two and FBXO7−/−B possesses a 28 bp deletion in allele 1 and 2 bp deletion in allele 2 (Supplementary Material, Fig. S3B). The allele-specific edit(s) in each clone introduce a premature stop codon (Supplementary Material, Fig. S3C) predicted to induce nonsense-mediated mRNA decay that is supported by the lack of protein products corresponding to the estimated molecular weights.
To determine the long-term impact heterozygous and homozygous loss has on CIN, the FBXO7+/−, FBXO7−/− and NT-Control clones were continually passaged and assessed for changes in nuclear areas, micronucleus formation and chromosome numbers every four passages (p; approximately every 2 weeks) for ~10 weeks. Overall, CIN was prevalent and dynamic within all FBXO7 clones relative to NT-Control (Fig. 5). More specifically, FBXO7+/−1 exhibited significant increases (i.e. rightward shifts) in cumulative nuclear area distribution frequencies at p0, p4 and p12, but ultimately exhibited smaller distributions (i.e. leftward shifts) at p16 and p20 compared with NT-Control, whereas FBXO7+/−2 exhibited similar trends, but the changes were more pronounced (Fig. 5A; Supplementary Material, Table S10). With respect to the homozygous knockout clones, FBXO7−/−A and FBXO7−/−B exhibited opposing and significant cumulative nuclear area distribution frequencies as nuclear areas were smaller in FBXO7−/−A and larger in FBXO7−/−B relative to NT-Control at p0 but vice versa at p4. Interestingly, their distinct evolutionary trajectories appear to begin converging at p8, as they did not exhibit significant differences relative to NT-Control at p8 but began to exhibit statistically significant changes at p12 (FBXO7−/−A) and from p16 to p20 (FBXO7−/−A and FBXO7−/−B) with largely overlapping distributions observed at p16 and p20.
Figure 5.

CIN-associated phenotypes are dynamic in FBXO7+/− and FBXO7−/− clones. (A) Cumulative nuclear area distribution frequencies reveal dynamic and significant changes in nuclear area distributions in FBXO7+/− and FBXO7−/− clones over ~10 weeks (p0 to p20) relative to NT-Control (two-sample KS test; *P-value ≤ 0.05; ***P-value < 0.001; ****P-value < 0.0001; N = 1, n ≥ 600 cells/condition/timepoint). (B) Dot plots presenting the frequency of micronuclei in FBXO7+/− and FBXO7−/− clones from p0 to p20 relative to NT-Control. MW tests identify significant increases at p4, p8, p12 and p20 (ns, not significant [P-value > 0.05]; *P-value ≤ 0.05; **P-value < 0.01; N = 1, n = 6). (C) Bar graphs presenting the frequencies of chromosome losses (< 46; white), small-scale gains (47–59; gray) and large-scale gains (≥ 60; black) in FBXO7+/− and FBXO7−/− clones over time. The fold increases in total aberrant chromosome numbers relative to the corresponding NT-Control are presented above each bar (N = 1, n > 100 spreads/condition/timepoint).
Next, micronucleus formation was compared between FBXO7 and NT-Control clones. As shown in Figure 5B, there was a general trend towards increases in micronucleus formation in FBXO7+/−1 and FBXO7+/−2 relative to NT-Control at p0 (Supplementary Material, Table S11). At remaining timepoints, FBXO7+/−1 exhibited decreases in micronucleus frequencies, except for p12 and p20 where increases occurred. Contrastingly, FBXO7+/−2 generally exhibited increases in micronucleus formation that were deemed significant at all timepoints except for p0 and p16. On the other hand, FBXO7−/−A and FBXO7−/−B exhibited ongoing dynamics relative to NT-Control. Initially, these increases were subtle at p0 and became more pronounced at p8 and p12, but ultimately decreased relative to NT-Control at p16 and p20. Collectively, these dynamics in distributions and micronucleus formation agree with the ongoing changes in cell-to-cell heterogeneity associated with the CIN phenotype.
To determine whether dynamic changes in chromosome numbers correspond with the heterogeneous CIN phenotypes observed above, mitotic chromosome spreads were assessed that identified dynamic changes in chromosome numbers between timepoints (Fig. 5C). More specifically, the FBXO7+/− clones exhibited 3.3- and 4.5-fold increases in the frequency of aberrant chromosome numbers that were maximal at p0 (FBXO7+/−1, ~95% aberrant spreads; FBXO7+/−2, ~70% aberrant spreads) relative to NT-Control (21% aberrant spreads), which decreased over the 10 weeks, with FBXO7+/−1 and FBXO7+/−2 exhibiting 1.8- and 1.4-fold increases at p20. The FBXO7−/− clones also exhibited distinct and dynamic increases relative to NT-Control from p0 to p20; however, they were typically less pronounced than those of the FBXO7+/− clones. For example, FBXO7−/−A exhibited increases as large as 2.6-fold (p12) and as small as 1.4-fold (p8) relative to NT-Control, whereas FBXO7−/−B displayed increases up to 2.7-fold (p12) and as little as 1.3-fold (p20). Overall, it is noteworthy that the FBXO7+/− clones often produced the strongest aberrant phenotypes within the three CIN assays conducted. Taken together, these data demonstrate that FBXO7 copy number losses, including both heterozygous and homozygous models, induce dynamic temporal changes in CIN phenotypes and agree with an early etiological role in CRC development.
Heterozygous and homozygous loss of FBXO7 induces cellular transformation
CIN is an enabling hallmark of cancer (37) frequently associated with cellular transformation (7,15). Accordingly, we sought to determine whether the FBXO7 clones exhibit key phenotypes indicative of cellular transformation, including changes in proliferation, clonogenic potential and anchorage-independent growth. Overall, no overt changes in cellular proliferation were detected within the clones (Fig. 6A), as the doubling times for the FBXO7+/− clones were 17.6 h and 20.7 h relative to NT-control (17.5 h), whereas the FBXO7−/− clones were 16.8 h and 18.8 h. Next, clonogenic potential was assessed using 2D colony formation assays. On the basis of the number and sizes of colonies, all FBXO7+/− and FBXO7−/− clones displayed increased clonogenic capacity relative to NT-Control (Fig. 6B–D). More specifically, FBXO7+/−1 and FBXO7+/−2 exhibited increases in mean colony numbers (4.8- and 14.3-fold, respectively) and median colony sizes (0.16 mm2 and 0.13 mm2, respectively) relative to NT-Control (0.03 mm2). Interestingly, FBXO7−/−A and FBXO7−/−B also exhibited enhanced clonogenic potential, albeit milder than their heterozygous counterparts, as mean fold increases in colony numbers were 4.0 (FBXO7−/−A) and 2.8 (FBXO7−/−B), whereas the median nuclear areas were 0.05 mm2 and 0.09 mm2, respectively. Finally, 3D colony formation assays were performed to assess anchorage-independent growth (Fig. 6E–G) that revealed increases in the mean colony numbers (2.0-fold; 118 mean colonies) and median colony sizes (0.015 mm2) for FBXO7+/−1 relative to NT-Control (60.5 mean colonies; median size = 0.012 mm2), whereas FBXO7+/−2 produced an average of 3.5 colonies with a smaller median size (0.010 mm2). Similarly, FBXO7−/−A generated an overall increase in mean colony number (~3.5-fold; 210.5 median colonies) and median size (median = 0.014 mm2), whereas FBXO7−/−B exhibited reduced colony formation capacity, with a mean of 21 colonies and a median size of 0.011 mm2. Collectively, these data show that both heterozygous and homozygous loss of FBXO7 can induce cellular transformation, which further supports the possibility that reduced FBXO7 is an early etiological event in CRC.
Figure 6.

Reduced FBXO7 expression induces cellular transformation phenotypes. (A) Graph presenting the growth curves from FBXO7+/− and FBXO7−/− clones over 7 days. Cell numbers were normalized to the mean cell number on Day 1 and dots show the mean cell number ± standard deviation (SD). (N = 2, n = 6). (B) Representative low-resolution images of 2D colony formation 2 weeks post-seeding in duplicate wells. Cells were stained with crystal violet for visualization. (C) Bar graph presenting the mean (±SD) number of colonies (≥100 μm in diameter), with the fold increase relative to NT-Control presented above each bar. (D) Dot plot showing increases in colony sizes in FBXO7+/− and FBXO7−/− clones compared with the NT-Control. Fold increases in median values are indicated at the top of each column, whereas the red bars identify median values. (N = 2, n = 2). (E) Representative low-resolution images of 3D colony formation 4 weeks post-seeding in duplicate wells (top and middle rows) in agarose. Cells were fixed and stained with crystal violet for visualization. Black bounding boxes highlight magnified regions (bottom row). (F) Bar graph presenting the mean (±SD) number of colonies. The fold increases in mean colony number relative to NT-Control are presented above each bar. (G) Dot plot presenting increases in colony sizes in FBXO7+/− and FBXO7−/− clones compared with NT-Control. Fold increases in median sizes are indicated at the top of each column, whereas the red bars identify median values. (N = 2, n = 2).
Discussion
In this study, we employed three complementary QuantIM approaches in both transient (siRNA) and stable (CRISPR/Cas9) genetic models to demonstrate that normal FBXO7 expression is required to maintain chromosome stability. Thus, these findings identify FBXO7 as a novel CIN gene in both malignant and non-malignant/non-transformed colonic epithelial cellular contexts. Next, we determined that both heterozygous and homozygous loss of FBXO7 is associated with increases in key indicators of cellular transformation, namely clonogenic potential and anchorage-independent growth. These findings, coupled with the observations that FBXO7 is frequently lost in ~30% of CRCs and that reduced expression corresponds with significantly worse progression-free, disease-specific and overall survival, strongly support the possibility that reduced FBXO7 expression is an early etiologic event contributing to the development of CRC.
CIN is defined as an increase in the rate at which chromosomes are gained and/or lost and is a driver of both cellular and genetic heterogeneity (5,10–12). As such, it is typically expected that nuclear areas and chromosome enumeration should identify corresponding increases (or decreases) relative to controls. Interestingly, the siRNA-based experiments identified increases in cumulative nuclear area distributions following FBXO7 silencing, whereas chromosome enumeration revealed both gains and losses in chromosome numbers. Similarly, the FBXO7 clones also exhibited analogous trends where the nuclear area analyses did not necessarily correspond with the findings from the chromosome enumeration assays. These inverse relationships may be explained by biological phenomena and/or inherent technical differences existing between the various assays employed. For example, large-scale chromosome gains are better tolerated than large-scale chromosome losses, as the latter are predicted to adversely impact cell viability and effectively deplete those cells from a given experimental condition/population. Indeed, large-scale chromosome losses in cervical cancer and glioblastoma cell lines are less compatible with viability (38). Moreover, changes in nuclear areas are most often associated with large-scale changes in DNA (i.e. ploidy) content (14,39–41), whereas the impact small-scale chromosome gains and/or losses (e.g. ≤3 chromosomes) have on nuclear areas is unclear, especially when considering the variable sizes of human chromosomes [e.g. chromosome 1 (~250 million bp) versus chromosome 21 (~48 million bp)]. As such, cells harboring only small-scale changes involving a few chromosomes will likely go undetected in nuclear area analyses but would be identified following chromosome enumeration. Lastly, the populations analyzed at the experimental endpoint of nuclear area and chromosome spread analyses differ due to inherent differences between the assays. Nuclear area analyses are conducted on asynchronous interphase populations, whereas mitotic chromosome spreads only analyze cells capable of entering mitosis and will exclude cells undergoing established mechanisms leading to increased nuclear areas including endoreduplication (extra rounds of DNA replication independent of cytokinesis). It should also be noted that chromosome gains and losses appear to be largely random, and therefore, certain growth advantages (or disadvantages) will be associated with the specific chromosomes gained or lost within a given cell. In this regard, early alterations conferring selective advantages will be maintained and propagated within a given population, whereas those conferring growth disadvantages will be eliminated from the population. Accordingly, the independent assays performed in this study (i.e. nuclear area and chromosome enumeration assays) are expected to be impacted by the early (random) selective events occurring within a given cellular population and therefore are not expected to produce identical outcomes in all instances. Thus, the difference in the random early selective events is the underlying impetus of why multiple complementary CIN assays must be employed to determine the reproducibility of CIN phenotypes (i.e. consistently induces CIN) irrespective of the specific trends (i.e. gains versus losses) observed.
It was also noted that the CIN phenotypes observed within the immortalized cellular contexts (i.e. 1CT; RPA; A1309) are typically milder than those observed within the malignant HCT116 cells. Although the precise mechanisms accounting for these differences remain unknown, there are various biological possibilities that may explain these differences. First, HCT116 and 1CT (and its derivative lines) represent two genetically distinct contexts. More specifically, HCT116 is a malignant CRC cell line with a MutL Homolog 1 (MLH1) deficiency that underlies a DNA mismatch repair defect (42). As a result, these cells continually accumulate small indels (typically 1–3 bp in size) and/or mutations throughout their genome that will impact additional genes (e.g. oncogenes, tumor suppressor, DNA repair) that may synergize with FBXO7 silencing to produce more pronounced phenotypes. On the other hand, 1CT, RPA and A1309 are all non-malignant cell lines (see Materials and Methods for descriptions) that are DNA mismatch repair proficient and therefore do not exhibit the spectrum of mutations contained within HCT116. Moreover, others have shown that re-expression of TERT enhances DNA repair and genome stability, albeit typically at telomeres (43), potentially rendering 1CT, RPA and A1309 cells more resistant to CIN. Accordingly, it is conceivable that HCT116 may be genetically predisposed to develop CIN following reduced FBXO7 expression. Beyond these genetic differences, inherent differences in population doubling times may also impact the magnitude of the various phenotypes assessed. For example, HCT116 have a population doubling time of ~20 h, whereas 1CT, RPA and A1309 have doubling times of ~24 h. Accordingly, over the 4-day time course of the transient silencing experiments, HCT116 are expected to undergo ~4.8 population doublings, whereas 1CT, RPA and A1309 will only undergo ~4 doublings. Thus, the extra population doubling associated with the HCT116 cells may afford an additional opportunity for CIN to occur, particularly if it occurs during mitosis. Collectively, although the above information may account for the stronger phenotypes observed within the HCT116 cells, the underlying biological and technical mechanisms accounting for these differences remain to be fully elucidated.
Although reduced FBXO7 expression induced reproducible CIN phenotypes within each model employed, heterogeneous responses occurred between the various conditions (siRNA and CRISPR/Cas9) and contexts (HCT116, 1CT, RPA or A1309). As indicated above, these heterogeneous responses are expected as CIN drives cellular and genetic heterogeneity, which produces ongoing changes in gene copy numbers (gene dosage) that differentially impact the various pathways promoting CIN (44,45). Thus, the heterogeneous nature of CIN may also account for the different evolutionary trajectories observed among the FBXO7 clones and the more extreme phenotypes observed within the FBXO7+/− clones. However, additional mechanisms may also account for the specific differences observed between clones. Conceptually, the FBXO7+/− clones still retain a capacity to selectively target protein substrates for proteolytic degradation via the 26S proteasome, albeit at reduced rates relative to the diploid (FBXO7+/+) condition, whereas the FBXO7−/− clones lose all substrate targeting capacity. Thus, it is conceivable that the reduced substrate targeting associated with the FBXO7+/− clones will slowly promote substrate accumulation that may not be immediately perceived by the cell versus the rapid accumulation of substrates occurring within the FBXO7−/− clones. As such, the backup mechanisms compensating for complete FBXO7 loss (i.e. functional compensation by other F-box proteins and/or suppressor mutations) will not be activated and enable higher levels of CIN. Indeed, there are numerous comprehensive reviews summarizing the status of F-box proteins and the pathways they regulate including lists of overlapping protein substrates targeted by multiple F-box proteins (46,47). As such, it is possible that one of the remaining 68 F-box proteins may functionally substitute for FBXO7 within the FBXO7−/− clones to restore substrate targeting more effectively, thereby minimizing CIN. However, this possibility is speculative and future studies are required to fully identify the spectrum of substrates and pathways regulated by FBXO7 before it can be determined whether functional compensation occurs. Alternatively, the acquisition of suppressor mutations or a mutation in an unlinked gene that serves to functionally suppress the CIN phenotypes normally associated with complete FBXO7 loss may have occurred early in the generation of the FBXO7−/− clones. Indeed, it is well established that suppressor mutations occur most frequently in response to selective pressures and has been observed in many model systems (e.g. yeast, flies, mice and human cell lines) as a mechanism to compensate for loss-of-function genotypes (48–50). Although this remains a possibility, it is unlikely that a suppressor mutation occurred within the same gene in both FBXO7−/− clones. Nevertheless, this could be explored through transcriptomic analyses involving all FBXO7 clones, which could identify the transcripts and/or pathways that compensate for the complete absence of FBXO7 within the FBXO7−/− clones relative to the FBXO7+/− and NT-control clones.
FBXO7 has classically been considered an oncogene as overexpression occurs in a variety of cancer patient samples (51–54). For example, Laman et al. (53) performed an immunohistochemical evaluation and determined that FBXO7 was overexpressed in CRC patient samples (n = 40) relative to normal colonic samples (n = 4) supporting a potential oncogenic role. However, more recent studies have begun to challenge this singular notion by suggesting FBXO7 may harbor oncogene- or tumor suppressor-like activities depending on the cellular and/or tissue-specific contexts in which it is studied. Moreover, FBXO7 overexpression corresponds with enhanced cellular Inhibitor of Apoptosis Protein 1 (cIAP1) degradation (51) and stabilization of p105 (55) that are associated with both over-activation of the anti-apoptotic Nuclear Factor Kappa B (NF-kB) signaling pathway and increased cellular proliferation, which are characteristics of many CRCs (56). Contrastingly, FBXO7 also encodes tumor suppressive activities by promoting hepatoma upregulated protein (HURP) degradation, a putative hepatocellular carcinoma oncogene that supports TP53 activity (54,57) and is associated with negative regulation of the NF-kB signaling pathway (58,59). Importantly, the bioinformatic analyses conducted in this study show that FBXO7 copy number losses occur more frequently than gains across a large range of cancer types, including CRC, which agrees with a putative tumor suppressor role. Thus, it appears that its role in cancer pathogenesis is likely tied to the functional roles of the substrates that it normally targets for proteolytic degradation.
The overall impact reduced FBXO7 expression has in CRC is only beginning to be realized and its role in the SCF complex is a promising mechanism warranting further investigation. Indeed, the current study builds upon previous work that identified each of the three core SCF complex members, namely SKP1, CUL1 and RBX1 as novel CIN genes in CRC and ovarian cancer contexts (21–23). Recent work has begun to identify the potential mechanisms by which reduced SKP1, CUL1 and RBX1 expression induces CIN. Intriguingly, diminished expression of the core SCF complex members induced increases in Cyclin E1 (CCNE1) abundance (21–23) and aberrant CENPA (centromere protein A) localization, which are associated with chromosome segregation errors and CIN (21,60). Importantly, as Cyclin E1 is not an established target of the SCFFBXO7 complex, it is highly likely that alternative mechanisms account for the CIN observed in cells with reduced FBXO7 expression. Unfortunately, very few SCFFBXO7 targets have been identified to date; however, the cell cycle regulatory protein HURP is one established target. Coincidentally, HURP is frequently overexpressed in many cancer types including hepatocellular, breast and bladder cancers (27) and its overexpression is associated with induction and aneuploidy and genome instability revealing a potential mechanistic link between reduced FBXO7 expression, a lack of HURP turnover and CIN, although this remains to be formally tested. In addition, Delgado-Camprub et al. (61) demonstrated that an FBXO7 deficiency in human dopaminergic neuroblastoma cells is associated with impaired mitochondrial metabolism and elevated intracellular levels of reactive oxygen species. Interestingly, oxidative stress is associated with CIN (62) and coincidentally occurs in cells with increased Cyclin E1 abundance (16). Thus, although these potential relationships are not CRC specific, they do present promising areas for further investigations aimed at identifying additional SCFFBXO7 substrates to ultimately discover the underlying mechanism(s) giving rise to CIN in cells harboring reduced FBXO7 expression. Accordingly, these previous findings coupled with those of the current study implicate aberrant SCF function and reduced FBXO7 expression as early etiological events contributing to the development of CRC. As such, CRCs harboring FBXO7 copy number losses may be ideal conditions to therapeutically exploit using synthetic lethal paradigms [reviewed in Thompson et al. (63)].
Materials and Methods
Copy number alterations, reduced expression and patient outcome analyses
Publicly available genomic (i.e. gene copy number) and mRNA expression data were extracted using cBioPortal (28,29) from TCGA PanCancer Atlas datasets (27) for 12 common cancer types (breast, cervical, CRC, glioblastoma, head and neck, kidney, liver, lung, ovarian, pancreatic, prostate and uterine) and analyzed using online tools available through cBioPortal (bioinformatic workflow presented in Supplementary Material, Fig. S1). Briefly, user defined onco-query commands (HOMDEL; HETLOSS; GAIN; AMP) were performed in cBioPortal to identify FBXO7 copy number alterations as detailed previously (22,23). Putative copy number alterations reported in cBioPortal are estimates identified from patient data using Genomic Identification of Significant Targets in Cancer (GISTIC) (64), where patient-specific thresholds were used to reveal genomic regions harboring homozygous (deep) or heterozygous (shallow) deletions, gains, amplifications or normal (diploid) copy numbers as detailed elsewhere (64,65). Thresholds are determined on a case-by-case basis but are based on the minimum median arm-level deletion/amplification copy number identified for a given sample. FBXO7 mRNA expression data associated with deep and shallow deletions, diploid, gains and amplifications were imported into Prism v9 (GraphPad) and shallow deletions and normal (diploid) cases were statistically compared by Student’s t-test. Patient outcomes including progression-free, disease-specific and overall survival were extracted from TCGA data (27) and stratified on the basis of FBXO7 shallow deletions or normal (diploid) copy number. Data were imported into Prism where survival curves were generated and statistically compared using log-rank tests with a P-value ≤0.05 considered significant.
Cell lines and culture
The human (male) malignant CRC cell line HCT116 (modal chromosome number = 45) was purchased from American Type Culture Collection (Rockville, MD), whereas the three (male) non-malignant colonic epithelial cell lines, 1CT and two derivative lines (RPA and A1309) were generously provided by Dr Jerry Shay (University of Texas Southwestern Medical Center, TX) (34,35). 1CT, RPA and A1309 (modal chromosome numbers = 46; Supplementary Material, Fig. S2A) are non-transformed lines immortalized with hTERT and CDK4; RPA also expresses mutant KRASG12V and has reduced TP53 and APC expression, whereas A1309 is an RPA derivative that also expresses a mutant APC protein truncated at residue 1309 (Supplementary Material, Fig. S2B) (34,35,66). HCT116 cells were cultured in McCoy’s medium (HyClone) supplemented with 10% fetal bovine serum (Sigma-Aldrich), whereas 1CT and derivatives were grown in Dulbecco’s Modified Eagle Medium with high glucose/medium 199 (HyClone) supplemented with cosmic calf serum (HyClone). All cell lines were authenticated on the basis of protein expression and karyotypic analyses (Supplementary Material, Fig. S2). HCT116 cells were grown in a humidified incubator at 37°C with 5% CO2, whereas 1CT and derivatives were grown in low-oxygen chambers filled with 2% O2, 7% CO2 and 91% N2 at 37°C.
FBXO7 silencing and western blot
FBXO7 silencing was conducted by transfecting ON-TARGETplus siRNA duplexes (GE Dharmacon) into cells using RNAiMAX (Life Technologies). Four individual siRNA duplexes (siFBXO7-1, -2, -3, -4) targeting unique regions within the FBXO7 coding sequence or a pool (siFBXO7-P) comprised of equimolar amounts of each individual siRNAs and a non-targeting control (siControl) were employed. The two most efficient siRNA duplexes (siFBXO7-2 and -4) were initially identified, and along with the siFBXO7-P were employed in all subsequent experiments. Silencing efficiencies were assessed by western blot 4 days post-transfection as detailed previously (67) with the antibodies and dilutions indicated in Supplementary Material, Table S12. Relative protein expression levels were determined using semi-quantitative image analysis in which FBXO7 band intensities were first normalized to the corresponding loading control (α-tubulin or cyclophilin B) and are presented relative to siControl (siRNA) or NT-Control (CRISPR/Cas9).
QuantIM
QuantIM approaches were employed to assess CIN-associated phenotypes including changes in cumulative nuclear area frequency distributions and micronucleus formation as detailed elsewhere (14,68). Briefly, cells were seeded in 96-well plates, silenced in sextuplet and permitted to grow for 4 days, following which cells were fixed (4% paraformaldehyde), stained (Hoechst 33342) and imaged using a Cytation 3 Cell Imaging Multi-Mode Reader (BioTek) equipped with a 20× objective (0.45 numerical aperture). Nine non-overlapping images were collected per well with experiments performed in triplicate. Nuclear areas and micronucleus formation were automatically quantified using Gen5 (BioTek) image acquisition and analysis software as detailed previously (14,68). All quantitative data were imported into Prism where descriptive statistics and non-parametric tests were performed including two-sample KS tests comparing cumulative nuclear area distribution frequencies to report D-statistics (the maximum deviation between distribution curves) and Mann–Whitney tests assessing differences in micronucleus formation. All graphs were generated in Prism, whereas figures were assembled in Photoshop 2021 (Adobe).
Mitotic chromosome spread generation and enumeration
Mitotic chromosome spreads were generated as detailed elsewhere (67,69). A minimum of 100 spreads per condition were enumerated, with all experiments performed in triplicate, except for the long-term temporal analyses that were performed once at each timepoint for each CRISPR/Cas9 clone. Two-sample KS tests were employed to identify statistically significant differences in cumulative chromosome number distribution frequencies relative to siControl or NT-Control.
CRISPR/Cas9 approach to generate FBXO7 knockout A1309 clones
FBXO7 knockout clones were generated using a two-step CRISPR/Cas9 approach in A1309 cells with FBXO7-targeting and non-targeting control synthetic guide RNAs (sgRNAs) according to the manufacturer (Sigma, Aldrich) and as detailed previously (22). Briefly, A1309 cells were transduced with lentivirus particles containing two independent FBXO7 sgRNAs or a NT-Control that co-expressed blue fluorescent protein (BFP). Cells were sorted (BFP+) by fluorescence-activated cell sorting (FACS), with successfully transduced populations transiently transfected with a Cas9 and green fluorescent protein (GFP) expression plasmid. FACS was used to recover BFP+/GFP+ cells, with individual clones isolated through subsequent serial dilutions. Putative FBXO7 knockout clones were identified by western blot (reduced FBXO7 abundance), with DNA sequencing analysis used to identify the allele-specific edits (Genome Quebec, Montreal, Canada).
Proliferation assay
Proliferation rates were determined by enumerating nuclei (cells) every 24 h for 7 days. Cells (700/well) were seeded in sextuplet in one 96-well plate/timepoint, and every 24 h cells were fixed, stained (Hoechst) and imaged in a 4 × 4 matrix of non-overlapping images with a Cytation 3 imager (20× lens) as detailed above. To account for potential differences in initial seeding densities, cell numbers were normalized to the mean cell number of the corresponding condition for Day 1. Proliferation curves were generated by graphing the mean normalized nuclear counts on a logarithmic scale as a function of time. Cell doubling times were calculated using the following formula:
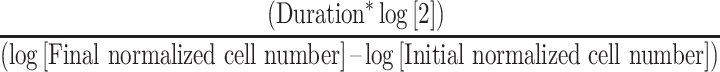 |
[[DmEquation1]]
Experiments were performed in duplicate.
Clonogenic assay
Clonogenic growth was assessed by seeding FBXO7+/−, FBXO7−/− and NT-Control clones at low density (350 cells/well) into six-well plates. Cells were grown for 14 days, at which point they were fixed (4% paraformaldehyde), stained (0.005% crystal violet) and scanned using a HP Officejet 4620 series scanner. Images were imported into ImageJ for processing and analysis in which colony numbers and sizes were automatically determined. Colonies were operationally defined as those with a diameter ≥ 100 μm in size. Experiments were performed twice.
Soft-agar colony formation assay
Colony formation assays were performed as described previously (67,70). Cells were seeded (20 000 cells/well) in 0.4% agar into a six-well plate containing a base layer of 0.6% agar, with HCT116 cells serving as a positive control (67). Cells were supplemented with media that was replaced every week for 4 weeks, following which cells were fixed (4% paraformaldehyde), stained (0.005% crystal violet) and imaged with a Cytation 3 (BioTek) equipped with a 4× objective. Gen5 software was employed to enumerate and measure colonies, with colonies being operationally defined as those with a diameter ≥ 100 μm in size (e.g. minimum 50 cells/colony). Experiments were performed twice.
Supplementary Material
Acknowledgements
We acknowledge that the CancerCare Manitoba Research Institute is located on original lands of Anishinaabeg, Cree, Oji-Cree, Dakota and Dene peoples, and on the homeland of the Métis Nation. We respect the Treaties that were made on these territories and acknowledge the harms and mistakes of the past. We dedicate ourselves to move forward in partnership with Indigenous communities in a spirit of reconciliation and collaboration. We thank members of the McManus laboratory for constructive criticisms during the writing of this review. We also thank Dr Jerry Shay for providing the 1CT and derivative cell lines and acknowledge the strong support of the CancerCare Manitoba Research Institute and the CancerCare Manitoba Foundation. Conflict of Interest statement. None declared.
Contributor Information
Michaela C L Palmer, CancerCare Manitoba Research Institute, CancerCare Manitoba, Winnipeg, MB R3E 0V9, Canada; Department of Biochemistry & Medical Genetics, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB R3E 0J9, Canada.
Nicole M Neudorf, CancerCare Manitoba Research Institute, CancerCare Manitoba, Winnipeg, MB R3E 0V9, Canada; Department of Biochemistry & Medical Genetics, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB R3E 0J9, Canada.
Ally C Farrell, CancerCare Manitoba Research Institute, CancerCare Manitoba, Winnipeg, MB R3E 0V9, Canada; Department of Biochemistry & Medical Genetics, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB R3E 0J9, Canada.
Tooba Razi, CancerCare Manitoba Research Institute, CancerCare Manitoba, Winnipeg, MB R3E 0V9, Canada; Department of Biochemistry & Medical Genetics, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB R3E 0J9, Canada.
Zelda Lichtensztejn, CancerCare Manitoba Research Institute, CancerCare Manitoba, Winnipeg, MB R3E 0V9, Canada; Department of Biochemistry & Medical Genetics, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB R3E 0J9, Canada.
Kirk J McManus, CancerCare Manitoba Research Institute, CancerCare Manitoba, Winnipeg, MB R3E 0V9, Canada; Department of Biochemistry & Medical Genetics, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB R3E 0J9, Canada.
Funding
Research in the McManus laboratory was generously supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) Canadian Graduate Scholarship (MCLP), a Research Manitoba/CancerCare Manitoba MSc Studentship (NMN), a Graduate Enhancement of Tri-Agency Stipends Award (ACF), an NSERC Discovery Grant (KJM; RGPIN: 2018-05007) and a CancerCare Manitoba Foundation Operating Grant (KJM).
References
- 1. Dion-Cote, A.M. and Barbash, D.A. (2017) Beyond speciation genes: an overview of genome stability in evolution and speciation. Curr. Opin. Genet. Dev., 47, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang, J. and Lindahl, T. (2016) Maintenance of genome stability. Genomics Proteomics Bioinformatics, 14, 119–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nicholson, J.M. and Cimini, D. (2015) Link between aneuploidy and chromosome instability. Int. Rev. Cell Mol. Biol., 315, 299–317. [DOI] [PubMed] [Google Scholar]
- 4. Sieber, O.M., Heinimann, K. and Tomlinson, I.P.M. (2003) Genomic instability - the engine of tumorigenesis? Nat. Rev. Cancer, 3, 701–708. [DOI] [PubMed] [Google Scholar]
- 5. Lengauer, C., Kinzler, K.W. and Vogelstein, B. (1997) Genetic instability in colorectal cancers. Nature, 386, 623–627. [DOI] [PubMed] [Google Scholar]
- 6. Vishwakarma, R. and McManus, K.J. (2020) Chromosome instability; implications in cancer development, progression, and clinical outcomes. Cancers (Basel), 12, 824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nowak, M.A., Komarova, N.L., Sengupta, A., Jallepalli, P.V., Shih Ie, M., Vogelstein, B. and Lengauer, C. (2002) The role of chromosomal instability in tumor initiation. Proc. Natl. Acad. Sci. U. S. A., 99, 16226–16231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brenner, D.R., Weir, H.K., Demers, A.A., Ellison, L.F., Louzado, C., Shaw, A., Turner, D., Woods, R.R., Smith, L.M. and Canadian Cancer Statistics Advisory, C (2020) Projected estimates of cancer in Canada in 2020. CMAJ, 192, E199–E205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Siegel, R.L., Miller, K.D., Fuchs, H.E. and Jemal, A. (2021) Cancer statistics, 2021. CA Cancer J. Clin., 71, 7–33. [DOI] [PubMed] [Google Scholar]
- 10. Geigl, J.B., Obenauf, A.C., Schwarzbraun, T. and Speicher, M.R. (2008) Defining 'chromosomal instability'. Trends Genet., 24, 64–69. [DOI] [PubMed] [Google Scholar]
- 11. Lepage, C.C., Morden, C.R., Palmer, M.C.L., Nachtigal, M.W. and McManus, K.J. (2019) Detecting chromosome instability in cancer: approaches to resolve cell-to-cell heterogeneity. Cancers (Basel), 11, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Orr, B. and Compton, D.A. (2013) A double-edged sword: how oncogenes and tumor suppressor genes can contribute to chromosomal instability. Front. Oncol., 3, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Markowitz, S.D. and Bertagnolli, M.M. (2009) Molecular origins of cancer: molecular basis of colorectal cancer. N. Engl. J. Med., 361, 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thompson, L.L. and McManus, K.J. (2015) A novel multiplexed, image-based approach to detect phenotypes that underlie chromosome instability in human cells. PLoS One, 10, e0123200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miura, M., Miura, Y., Padilla-Nash, H.M., Molinolo, A.A., Fu, B., Patel, V., Seo, B.M., Sonoyama, W., Zheng, J.J., Baker, C.C.et al. (2006) Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation. Stem Cells, 24, 1095–1103. [DOI] [PubMed] [Google Scholar]
- 16. Aziz, K., Limzerwala, J.F., Sturmlechner, I., Hurley, E., Zhang, C., Jeganathan, K.B., Nelson, G., Bronk, S., Fierro Velasco, R.O., vanDeursen, E.J.et al. (2019) Ccne1 overexpression causes chromosome instability in liver cells and liver tumor development in mice. Gastroenterology, 157, 210–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heng, H.H., Steven, W.B., Stevens, J.B., Horne, S.D., Liu, G., Abdallah, B.Y., Ye, K.J. and Ye, C.J. (2013) Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev., 32, 325–340. [DOI] [PubMed] [Google Scholar]
- 18. Lee, A.J., Endesfelder, D., Rowan, A.J., Walther, A., Birkbak, N.J., Futreal, P.A., Downward, J., Szallasi, Z., Tomlinson, I.P., Howell, M.et al. (2011) Chromosomal instability confers intrinsic multidrug resistance. Cancer Res., 71, 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carter, S.L., Eklund, A.C., Kohane, I.S., Harris, L.N. and Szallasi, Z. (2006) A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet., 38, 1043–1048. [DOI] [PubMed] [Google Scholar]
- 20. Sotillo, R., Schvartzman, J.M., Socci, N.D. and Benezra, R. (2010) Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature, 464, 436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thompson, L.L., Baergen, A.K., Lichtensztejn, Z. and McManus, K.J. (2020) Reduced SKP1 expression induces chromosome instability through aberrant cyclin E1 protein turnover. Cancers (Basel), 12, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lepage, C.C., Palmer, M.C.L., Farrell, A.C., Neudorf, N.M., Lichtensztejn, Z., Nachtigal, M.W. and McManus, K.J. (2021) Reduced SKP1 and CUL1 expression underlies increases in cyclin E1 and chromosome instability in cellular precursors of high-grade serous ovarian cancer. Br. J. Cancer, 124, 1699–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bungsy, M., Palmer, M.C.L., Jeusset, L.M., Neudorf, N.M., Lichtensztejn, Z., Nachtigal, M.W. and McManus, K.J. (2021) Reduced RBX1 expression induces chromosome instability and promotes cellular transformation in high-grade serous ovarian cancer precursor cells. Cancer Lett., 500, 194–207. [DOI] [PubMed] [Google Scholar]
- 24. Willems, A.R., Schwab, M. and Tyers, M. (2004) A hitchhiker's guide to the cullin ubiquitin ligases: SCF and its kin. Biochim. Biophys. Acta, 1695, 133–170. [DOI] [PubMed] [Google Scholar]
- 25. Jackson, P.K. and Eldridge, A.G. (2002) The SCF ubiquitin ligase: an extended look. Mol. Cell, 9, 923–925. [DOI] [PubMed] [Google Scholar]
- 26. Lee, E.K. and Diehl, J.A. (2014) SCFs in the new millennium. Oncogene, 33, 2011–2018. [DOI] [PubMed] [Google Scholar]
- 27. Hoadley, K.A., Yau, C., Hinoue, T., Wolf, D.M., Lazar, A.J., Drill, E., Shen, R., Taylor, A.M., Cherniack, A.D., Thorsson, V.et al. (2018) Cell-of-origin patterns dominate the molecular classification of 10,000 Tumors from 33 types of cancer. Cell, 173(291–304), e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cerami, E., Gao, J., Dogrusoz, U., Gross, B.E., Sumer, S.O., Aksoy, B.A., Jacobsen, A., Byrne, C.J., Heuer, M.L., Larsson, E.et al. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov, 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao, J., Aksoy, B.A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S.O., Sun, Y., Jacobsen, A., Sinha, R., Larsson, E.et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal., 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Asbaghi, Y., Thompson, L.L., Lichtensztejn, Z. and McManus, K.J. (2017) KIF11 silencing and inhibition induces chromosome instability that may contribute to cancer. Genes Chromosomes Cancer, 56, 668–680. [DOI] [PubMed] [Google Scholar]
- 31. Baergen, A.K., Jeusset, L.M., Lichtensztejn, Z. and McManus, K.J. (2019) Diminished Condensin gene expression drives chromosome instability that may contribute to colorectal cancer pathogenesis. Cancers (Basel), 11, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leylek, T.R., Jeusset, L.M., Lichtensztejn, Z. and McManus, K.J. (2020) Reduced expression of genes regulating cohesion induces chromosome instability that may promote cancer and impact patient outcomes. Sci. Rep., 10, 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barber, T.D., McManus, K.J., Yuen, K.W.Y., Reis, M., Parmigiani, G., Shen, D., Barrett, I., Nouhi, Y., Spencer, F., Markowitz, S.et al. (2008) Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. U. S. A., 105, 3443–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roig, A.I., Eskiocak, U., Hight, S.K., Kim, S.B., Delgado, O., Souza, R.F., Spechler, S.J., Wright, W.E. and Shay, J.W. (2010) Immortalized epithelial cells derived from human colon biopsies express stem cell markers and differentiate in vitro. Gastroenterology, 138(1012–1021), e1011–e1015. [DOI] [PubMed] [Google Scholar]
- 35. Zhang, L., Kim, S., Jia, G., Buhmeida, A., Dallol, A., Wright, W.E., Fornace, A.J., Al-Qahtani, M. and Shay, J.W. (2015) Exome sequencing of normal and isogenic transformed human colonic epithelial cells (HCECs) reveals novel genes potentially involved in the early stages of colorectal tumorigenesis. BMC Genomics, 16(Suppl 1), S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mason, D.M. and Schuenemeyer, J.H. (1983) A modified Kolmogorov-Smirnov test sensitive to tail alternatives. Ann. Stat., 11, 933–946. [Google Scholar]
- 37. Hanahan, D. and Weinberg, R.A. (2011) Hallmarks of cancer: the next generation. Cell, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 38. Kops, G.J., Foltz Dr Fau Cleveland, D.W. and Cleveland, D.W. (2004) Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. U. S. A., 101, 8699–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nielsen, K., Petersen, S.E. and Orntoft, T. (1989) A comparison between stereological estimates of mean nuclear volume and DNA flow cytometry in bladder tumours. APMIS, 97, 949–956. [DOI] [PubMed] [Google Scholar]
- 40. Petersen, I., Kotb, W.F., Friedrich, K.H., Schluns, K., Bocking, A. and Dietel, M. (2009) Core classification of lung cancer: correlating nuclear size and mitoses with ploidy and clinicopathological parameters. Lung Cancer, 65, 312–318. [DOI] [PubMed] [Google Scholar]
- 41. Zeimet, A.G., Fiegl, H., Goebel, G., Kopp, F., Allasia, C., Reimer, D., Steppan, I., Mueller-Holzner, E., Ehrlich, M. and Marth, C. (2011) DNA ploidy, nuclear size, proliferation index and DNA-hypomethylation in ovarian cancer. Gynecol. Oncol., 121, 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kolodner, R.D., Hall, N.R., Lipford, J., Kane, M.F., Morrison, P.T., Finan, P.J., Burn, J., Chapman, P., Earabino, C., Merchant, E.et al. (1995) Structure of the human MLH1 locus and analysis of a large hereditary nonpolyposis colorectal carcinoma kindred for mlh1 mutations. Cancer Res., 55, 242–248. [PubMed] [Google Scholar]
- 43. Sharma, G.G., Gupta, A., Wang, H., Scherthan, H., Dhar, S., Gandhi, V., Iliakis, G., Shay, J.W., Young, C.S. and Pandita, T.K. (2003) hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene, 22, 131–146. [DOI] [PubMed] [Google Scholar]
- 44. Potapova, T. and Gorbsky, G.J. (2017) The consequences of chromosome segregation errors in mitosis and meiosis. Biology (Basel), 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rancati, G., Pavelka, N., Fleharty, B., Noll, A., Trimble, R., Walton, K., Perera, A., Staehling-Hampton, K., Seidel, C.W. and Li, R. (2008) Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell, 135, 879–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Randle, S.J. and Laman, H. (2016) F-box protein interactions with the hallmark pathways in cancer. Semin. Cancer Biol., 36, 3–17. [DOI] [PubMed] [Google Scholar]
- 47. Tekcham, D.S., Chen, D., Liu, Y., Ling, T., Zhang, Y., Chen, H., Wang, W., Otkur, W., Qi, H., Xia, T.et al. (2020) F-box proteins and cancer: an update from functional and regulatory mechanism to therapeutic clinical prospects. Theranostics, 10, 4150–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ye, C.J., Sharpe, Z. and Heng, H.H. (2020) Origins and consequences of chromosomal instability: from cellular adaptation to genome chaos-mediated system survival. Genes (Basel), 11, 1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Buglo, E., Sarmiento, E., Martuscelli, N.B., Sant, D.W., Danzi, M.C., Abrams, A.J., Dallman, J.E. and Züchner, S. (2020) Genetic compensation in a stable slc25a46 mutant zebrafish: a case for using F0 CRISPR mutagenesis to study phenotypes caused by inherited disease. PLoS One, 15, e0230566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. El-Brolosy, M.A. and Stainier, D.Y.R. (2017) Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet., 13, e1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chang, Y.F., Cheng, C.M., Chang, L.K., Jong, Y.J. and Yuo, C.Y. (2006) The F-box protein Fbxo7 interacts with human inhibitor of apoptosis protein cIAP1 and promotes cIAP1 ubiquitination. Biochem. Biophys. Res. Commun., 342, 1022–1026. [DOI] [PubMed] [Google Scholar]
- 52. Kang, J. and Chung, K.C. (2015) The F-box protein FBXO7 positively regulates bone morphogenetic protein-mediated signaling through Lys-63-specific ubiquitination of neurotrophin receptor-interacting MAGE (NRAGE). Cell. Mol. Life Sci., 72, 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Laman, H., M, F.J., Ye, H., Henderson, S., Galinanes-Garcia, L., Hara, E., Knowles, P., McDonald, N. and Boshoff, C. (2005) Transforming activity of Fbxo7 is mediated specifically through regulation of cyclin D/cdk6. EMBO J., 24, 3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nelson, D.E., Randle, S.J. and Laman, H. (2013) Beyond ubiquitination: the atypical functions of Fbxo7 and other F-box proteins. Open Biol., 3, 130131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Udasin, R.G., Gottfried, Y., Fabre, B., Bercovich, B., Ziv, T. and Ciechanover, A. (2021) The p105 NF-ĸB precursor is a pseudo substrate of the ubiquitin ligase FBXO7, and its binding to the ligase stabilizes it and results in stimulated cell proliferation. Biochem. Biophys. Res. Commun., 558, 224–230. [DOI] [PubMed] [Google Scholar]
- 56. Dolcet, X., Llobet, D., Pallares, J. and Matias-Guiu, X. (2005) NF-kB in development and progression of human cancer. Virchows Arch., 446, 475–482. [DOI] [PubMed] [Google Scholar]
- 57. Hsu, J.M., Lee, Y.C., Yu, C.T. and Huang, C.Y. (2004) Fbx7 functions in the SCF complex regulating Cdk1-cyclin B-phosphorylated hepatoma up-regulated protein (HURP) proteolysis by a proline-rich region. J. Biol. Chem., 279, 32592–32602. [DOI] [PubMed] [Google Scholar]
- 58. Spagnol, V., Oliveira, C.A.B., Randle, S.J., Passos, P.M.S., Correia, C., Simaroli, N.B., Oliveira, J.S., Mevissen, T.E.T., Medeiros, A.C., Gomes, M.D.et al. (2021) The E3 ubiquitin ligase SCF(Fbxo7) mediates proteasomal degradation of UXT isoform 2 (UXT-V2) to inhibit the NF-κB signaling pathway. Biochim. Biophys. Acta Gen. Subj., 1865, 129754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kuiken, H.J., A, E.D., Laman, H., Bernards, R., Beijersbergen, R.L. and Dirac, A.M. (2012) Identification of F-box only protein 7 as a negative regulator of NF-kappaB signalling. J. Cell. Mol. Med., 16, 2140–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takada, M., Zhang, W., Suzuki, A., Kuroda, T.S., Yu, Z., Inuzuka, H., Gao, D., Wan, L., Zhuang, M., Hu, L.et al. (2017) FBW7 loss promotes chromosomal instability and tumorigenesis via cyclin E1/CDK2-mediated phosphorylation of CENP-A. Cancer Res., 77, 4881–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Delgado-Camprubi, M., Esteras, N., Soutar, M.P., Plun-Favreau, H. and Abramov, A.Y. (2017) Deficiency of Parkinson's disease-related gene Fbxo7 is associated with impaired mitochondrial metabolism by PARP activation. Cell Death Differ., 24, 2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Limoli, C.L. and Giedzinski, E. (2003) Induction of chromosomal instability by chronic oxidative stress. Neoplasia, 5, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Thompson, L.L., Jeusset, L.M., Lepage, C.C. and McManus, K.J. (2017) Evolving therapeutic strategies to exploit chromosome instability in cancer. Cancers (Basel), 9, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Beroukhim, R., Getz, G., Nghiemphu, L., Barretina, J., Hsueh, T., Linhart, D., Vivanco, I., Lee, J.C., Huang, J.H., Alexander, S.et al. (2007) Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc. Natl. Acad. Sci. U. S. A., 104, 20007–20012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cancer Genome Atlas Research, N (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang, L., Theodoropoulos, P.C., Eskiocak, U., Wang, W., Moon, Y.A., Posner, B., Williams, N.S., Wright, W.E., Kim, S.B., Nijhawan, D.et al. (2016) Selective targeting of mutant adenomatous polyposis coli (APC) in colorectal cancer. Sci. Transl. Med., 8, 361ra140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sajesh, B.V., Bailey, M., Lichtensztejn, Z., Hieter, P. and McManus, K.J. (2013) Synthetic lethal targeting of superoxide dismutase 1 selectively kills RAD54B-deficient colorectal cancer cells. Genetics, 195, 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lepage, C.C., Thompson, L.L., Larson, B. and McManus, K.J. (2020) An automated, single cell quantitative imaging microscopy approach to assess micronucleus formation, genotoxicity and chromosome instability. Cell, 9, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sajesh, B.V., Lichtensztejn, Z. and McManus, K.J. (2013) Sister chromatid cohesion defects are associated with chromosome instability in Hodgkin lymphoma cells. BMC Cancer, 13, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Borowicz, S., Van Scoyk, M., Avasarala, S., Karuppusamy Rathinam, M.K., Tauler, J., Bikkavilli, R.K. and Winn, R.A. (2014) The soft agar colony formation assay. J. Vis. Exp., 92, e51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.