

Abstract
Soluble epoxide hydrolase (sEH) is related to arachidonic acid cascade and is over-expressed in a variety of diseases, making sEH an attractive target for the treatment of pain as well as inflammatory-related diseases. A new series of memantyl urea derivatives as potent sEH inhibitors was obtained using our previous reported compound 4 as lead compound. A preferential modification of piperidinyl to 3-carbamoyl piperidinyl was identified for this series via structure-based rational drug design. Compound A20 exhibited moderate percentage plasma protein binding (88.6%) and better metabolic stability in vitro. After oral administration, the bioavailability of A20 was 28.6%. Acute toxicity test showed that A20 was well tolerated and there was no adverse event encountered at dose of 6.0 g/kg. Inhibitor A20 also displayed robust analgesic effect in vivo and dose-dependently attenuated neuropathic pain in rat model induced by spared nerve injury, which was better than gabapentin and sEH inhibitor (±)-EC-5026. In one word, the oral administration of A20 significantly alleviated pain and improved the health status of the rats, demonstrating that A20 was a promising candidate to be further evaluated for the treatment of neuropathic pain.
KEY WORDS: Soluble epoxide hydrolase, Analgesia, Synthesis, Neuropathic pain, Inhibitor
Graphical abstract
A series of memantyl urea-containing sEH inhibitors was created from an initial lead compound 4. Oral administration of A20 significantly alleviated pain and improved the health status of the rats.

1. Introduction
Neuropathic pain, an intractable pain from somatosensory nervous system damage or disease, which is triggered by pathogenic microorganisms, ischemia, traumatic stimulation, metabolic diseases, drug toxicity, etc., could cause anxiety, depression, even suicide that seriously affect the health and quality of patient's life1,2. Epidemiology statistics from January 1966 to December 2012 showed that approximately 6.9%–10% of the population worldwide suffered from neuropathic pain3. With the increasing trend of population aging, neuropathic pain will become a serious global health problem. Although the pathogenesis of neuropathic pain is still ambiguous, more and more evidence showed that proinflammatory and inflammatory factors play essential role in peripheral and central sensitization of neuropathic pain4,5.
After nerve cell injury, hyperactivated microglia could release a variety of proinflammatory cytokines, including monocyte chemotactic protein (MCP)-1, tumor necrosis factor (TNF)-α, macrophage inflammatory protein (MIP)-1α, interleukin (IL)-1β, IL-6, etc., which induce and maintain neuropathic pain6,7. Currently, drugs for the treatment of patients with neuropathic pain mainly include antiepileptic drugs (such as gabapentin), antidepressants, opioid analgesics, local anesthetics, and nonsteroidal antiinflammatory drugs (NSAIDs), etc.8. NSAIDs have poor therapeutic effect on neuropathic pain and display strong adverse events in gastrointestinal or cardiovascular system9. Because many patients with neuropathic pain are prescribed opioids to limit their symptoms, there are a lot of incidents with risk of opioid misuse, addiction, tolerance, respiratory depression, nausea and vomiting, etc.10. Furthermore, antiepileptic drugs also have some central nervous system (CNS) related adverse events, such as drowsiness, dizziness, which show poor therapeutic effect and patient compliance11.
Therefore, it is urgent to develop more effective therapeutic drugs for the management of neuropathic pain with novel mechanisms, strong analgesic effect and without addiction and CNS-related adverse events. Inflammatory cytokines—TNF-α, bradykinin, IL-1β, IL-6, etc.—serve as a basis in the process of neuropathic pain12. Therefore, eliminating the production of inflammatory cytokines may offer a new opportunity for the treatment of neuropathic pain13,14.
Arachidonic acid cascade derivatized CYP450 metabolic pathway could oxidize polyunsaturated fatty acid to corresponding epoxy-fatty acids (EpFAs)15. Endogenous mediators of EpFAs could act through various mechanisms to manage pain as well as inflammation, which was involved in activating peroxisome proliferators-activated receptors (PPARs), reducing endoplasmic reticulum (ER) stress, stabilizing mitochondrial dysfunction, and breaking or reversing endothelial cell dysfunction (ECD)16. Moreover, EpFAs could also reduce the activation of nuclear factor kappa-B (NF-κB) by three complementary cellular mechanisms, which is a core mechanism in the anti-inflammatory effect of EpFAs17. EpFAs could rapidly be hydrolyzed by soluble epoxide hydrolase (sEH) to corresponding pro-inflammatory diol products, leading to inactivation of endogenous active substances in vivo18. sEH inhibitors could stabilize EpFAs and provide a promising therapeutic strategy for pain and inflammatory-related diseases. As summarized in related reviews, this essential cellular mechanism underlies numerous diseases, such as metabolic disease, vascular disease, cytokine storm, etc., and more and more potential therapeutic targets are being gradually disclosed as the key role of ER stress in related diseases is proved through molecular biology technology16,19, 20, 21. Hence, pharmaceutical companies and research institutes involved in the Research & Development of sEH inhibitors have the “problems of the rich” in selecting a promising target and a practical indication from numerous possibilities21.
Currently, three sEH inhibitors are moved into the several clinical trials for the treatment of sEH-mediated diseases, as shown in Fig. 1. A great number of published articles revealed the efficacy of sEH inhibitors in regulating hypertension driven by the renin-angiotensin system (RAS)22,23. Therefore, AR-9281 (1) developed by Arete Therapeutics was selected as candidate drug to regulate hypertension, which was evaluated in phase II clinical trials24. Inflammation is provoked by various inflammatory leukocytes and cytokines thought to play crucial role in the development process of chronic obstructive pulmonary disease (COPD)25. sEH inhibitors could exert significant protective effects in rat model of COPD25. Therefore, GlaxoSmithKline (GSK) considered COPD as indication as well, and compound GSK-2256294 (2), developed by GSK, was evaluated in several phase I clinical trials for the treatment of COPD26. As it is known to all that neuropathic pain still doesn't meet medical demand in clinical practice; however, it is poorly remedied by existing medications. Inspired by the proof that sEH inhibitors could successfully treat severe equine laminitis21,27, EicOsis decided to move it from models into patients. In April 2020, the US Food and Drug Administration (FDA) granted EC-5026 by EicOsis with Fast Track Designation, as a non-addictive alternative to opioid analgesics, for the management of neuropathic pain (NCT04228302), which created favorable conditions for accelerating its clinical development and launch21,28.
Figure 1.

Chemical structures of sEH inhibitors in clinic trials.
Herein, we introduce the discovery and chemical optimization of lead compound 4 based on our previous study29. Through chemical modification of lead compound, we identified racemate molecule that demonstrating strong inhibition of recombinant human sEH (HsEH) in vitro assay. A detailed structure–activity relationship (SAR) investigation improved the potency of the single configuration molecule by nearly 4 times, which demonstrated significantly analgesic effect with dose-dependence in spared nerve injury model. Our work supports the development of an orally administered sEH inhibitor for the treatment of neuropathic pain.
2. Results and discussion
2.1. Design strategy
We were devoted to discovering more potent sEH inhibitors containing memantinyl urea and terminal piperidinamide moiety, which were further structurally optimized from previously reported compound29. We visualized those new compounds might demonstrate low melting point, and good water solubility with favorable pharmacokinetic (PK) properties. To facilitate the design of new sEH inhibitors, we further analyzed the binding modes of lead compound 4 with sEH protein (PDB ID: 3WKE) through Discovery Studio 2016 software, as disclosed in Fig. 2. Binding surface results displayed that piperidinyl moiety of compound 4 was exposed to solvent cavity, which offered several opportunities to increase potency of inhibitors as there were several extra amino acid residues situated in the solvent cavity of sEH. In addition, substitution at piperidinyl moiety provided an opportunity to introduce polar functional groups, which was contributed to modulating the physiochemical properties of the compounds. Therefore, memantyl urea and benzamide groups were retained, carboxyl was introduced to piperidinyl moiety at C3 and C4 position, which was further derivatized with amines or alcohol leading to target compounds (Scheme 1).
Figure 2.

Docked pose of compound 4 in blue bound to sEH (PDB ID: 3WKE).
Scheme 1.

Design strategy based on lead compound 4.
In vitro biological activity screening showed that substituents at C3 position of piperidinyl moiety demonstrated better inhibitory effect than C4 position. Amongst, compound with carbamoyl displayed strong inhibitory potency on sEH. Considering that lead compound 4 was easily metabolized in vitro and in vivo; therefore, F and Cl atoms were respectively introduced to central phenyl group to improve the metabolic stability (Scheme 1). Meanwhile, these compounds are featured with para-disubstituted with strong molecular symmetry. The introduction of halogen atoms at phenyl group could reduce structural symmetry, which was beneficial to reduce melting point and improve solubility. The introduction of halogen atoms afforded the most potency compounds with good physical properties. Substituents at C3 position of piperidinyl moiety include a chiral carbon atom, biological activity assay screening revealed that S-configurational molecule was a more potent sEH inhibitor than R-configuration by nearly 2 times. According to above results, compound with S-configuration and F substituent was obtained, which demonstrated strong inhibitory potency on sEH (Scheme 1).
In consideration of chemical character of piperidinyl as well as significant inhibitory effects of derivatives in vitro, piperidinyl was respectively replaced with 4-aminopiperidinyl and piperazinyl, then further acylated with different acids to obtain target compounds B1–B8 and C1–C8, respectively. Referring to the compound A20, we replaced 4-aminopiperidinyl with (S)-3-aminopiperidinyl, and introduced F atom at phenyl group, leading to compound B9, as shown in Scheme 1.
2.2. Chemistry
The strategy adopted for the synthesis of compound A1 (Scheme 2) started with commercially available para-nitrobenzoyl chloride, which underwent acylation with piperidine-4-carboxylic acid to provide compound 6. Reduction of 6 using 5% Pd-C at H2 atmosphere under mildly condition provided 7. Subsequent acylation of the amino group present in 7 with phenyl chloroformate, and resulting key intermediate 8 was nucleophilic substituted with memantine to give A1. Esterification of the carboxyl of A1 with EtOH in the presence of SOCl2 provided compound A2. Chlorination of A1 with SOCl2 in THF cleanly afforded the acyl chloride intermediate (not shown). Subsequent acylation (ammonia, THF, 0 °C) yield compound A3. Commercially available amines were then coupled to the compound A1 with the assistance of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI) and 1-hydroxybenzotriazole (HOBt) in DCM solution to give A4−A7. Compounds A8−A14 were obtained in similar route starting from commercially available piperidine-3-carboxylic acid, as shown in Scheme 3.
Scheme 2.

Synthetic route to compounds A1−A7. Reactions and conditions: i) piperidine-4-carboxylic acid, K2CO3, THF/H2O; (ii) H2 (g), 5% Pd-C, EtOH, 60 °C, 12 h; (iii) K2CO3, PhOCOCl, THF, 0 °C→ rt; iv) memantine, Et3N, THF, 75 °C, 8 h; v) SOCl2, EtOH, 0 °C→reflux, 2 h; vi) (1) SOCl2, DMF, THF; (2) NH3 (g), THF; vii) amines, EDCI, HOBt, Et3N, DCM, rt.
Scheme 3.

Synthetic route to compounds A8−A14. Reactions and conditions: i) piperidine-3-carboxylic acid, K2CO3, THF/H2O; (ii) H2 (g), 5% Pd-C, EtOH, 60 °C, 12 h; (iii) K2CO3, PhOCOCl, THF, 0 °C→rt; iv) memantine, Et3N, THF, 75 °C, 8 h; v) SOCl2, EtOH, 0 °C→reflux, 2 h; vi) (1) SOCl2, DMF, THF; (2) NH3 (g), THF; vii) amines, EDCI, HOBt, Et3N, DCM, rt.
Halogen substituted compounds A15 and A16 were synthesized as exhibited in Scheme 4. Chlorination of 3-fluoro-4-nitrobenzoic acid 12 and 3-chloro-4-nitrobenzoic acid 13 with SOCl2 in THF cleanly afforded the corresponding acyl chloride intermediate (not shown). Subsequent acylation with ethyl piperidine-3-carboxylate respectively yield compounds 14 and 15. It is noteworthy that reduction of intermediates 14 and 15 containing halogen atom using 5% Pd-C at H2 atmosphere condition didn't provide corresponding target compounds, but leading to side dehalogenation reaction. Therefore, anilines 16 and 17 were obtained in good to excellent yields via reduction of nitro intermediates 14 and 15 in the presence of Fe/NH4Cl under standard conditions. Moreover, phenyl chloroformate has been firstly utilized for urea formation, which was similar with Scheme 2, Scheme 3. However, corresponding intermediates containing halogen atom couldn't react with memantine. Hence, we replaced phenyl chloroformate with triphosgene (BTC) to obtain urea compounds. Amino groups in 16 and 17 were transformed to the corresponding isocyanate (not shown) in the presence of BTC, and then reacted with memantine to respectively give compounds 18 and 19, which were hydrolyzed under basic conditions to affording the desired acid intermediates. Chlorination of the acids 20 and 21 using SOCl2, followed by coupling with ammonia, generated the corresponding target compounds A15 and A16.
Scheme 4.

Synthetic route to compounds A15−A17. Reactions and conditions: i) (1) SOCl2, DMF, THF, 0 °C→rt; (2) ethyl piperidine-3-carboxylate, Et3N,THF, 0 °C→rt; ii) Fe, NH4Cl, EtOH/H2O, 70 °C; iii) (1) BTC, Et3N, DCM, 0 °C→reflux; (2) memantine, Et3N, DCM, 0 °C→rt; iv) (1) NaOH, H2O, EtOH; (2) Conc. HCl; v) (1) SOCl2, DMF, THF, 0 °C→rt; (2) NH3 (g), THF, 0→rt; vi) SOCl2, MeOH, 0 °C→reflux, 2 h; vii) 50% (w/w) NH2OH solution, 1 mol/L NaOH, MeOH, 0 °C.
The preparation of compound A17 started with the previously described A1 (Scheme 4). Esterification of the carboxyl of A1 with MeOH in the presence of SOCl2 provided 22. Subsequent aminolysis of the methyl ester in hydroxylamine aqueous afforded A17.
The synthesis of chiral compounds A18 and A19 (Scheme 5) was accomplished via acylation of commercially available (R)-ethyl piperidine-3-carboxylate and (S)-ethyl piperidine-3-carboxylate with 4-nitrobenzoyl chloride to give 23 and 24. Hydrogenation reduction of the nitro group in compounds 23 and 24 yielded the amino intermediates. Subsequent acylation of the amino group present in 25 and 26 with phenyl chloroformate, and resulting key intermediates 27 and 28 were nucleophilic substituted with memantine to respectively give 29 and 30, followed by hydrolysis of ester under basic condition afforded 31 and 32. Chlorination of the acids 31 and 32 using SOCl2, followed by coupling with ammonia, generated the corresponding target compounds A18 and A19.
Scheme 5.

Synthetic route to compounds A18 and A19. Reactions and conditions: i) R- or S-ethyl piperidine-3-carboxylate, Et3N, THF, 0 °C→rt; ii) H2 (g), 5% Pd-C, EtOH, 60 °C, 12 h; (iii) K2CO3, PhOCOCl, THF, 0 °C→rt; iv) memantine, Et3N, THF, 75 °C, 8 h; v) (1) NaOH, H2O, EtOH; (2) Conc. HCl; vi) (1) SOCl2, DMF, THF; (2) NH3 (g), THF.
The synthesis of compounds A20 and A21 (Scheme 6) was accomplished starting from 3-fluoro-4-nitrobenzoic acid. Chlorination of 3-fluoro-4-nitrobenzoic acid 12 with SOCl2 in THF cleanly afforded the corresponding acyl chloride intermediate (not shown). Subsequent acylation with (S)-ethyl piperidine-3-carboxylate or (R)-ethyl piperidine-3-carboxylate yield compounds 33 and 34. Anilines 35 and 36 were prepared in excellent yield via reduction of nitro compounds 33 and 34 in the presence of Fe/NH4Cl under standard conditions. Amino group in 35 and 36 was transformed to the corresponding isocyanate (not shown) in the presence of BTC, and then reacted with memantine to give compounds 37 and 38, which after hydrolysis under basic conditions gave the desired intermediate acids 39 and 40. Chlorination of the acids 39 and 40 using SOCl2, followed by coupling with ammonia, generated the desired compounds A20 and A21.
Scheme 6.

Synthetic route to compound A20 and A21. Reactions and conditions: i) (1) SOCl2, DMF, THF, 0 °C→60 °C; (2) (S)-ethyl piperidine-3-carboxylate or (R)-ethyl piperidine-3-carboxylate, Et3N, THF, 0 °C→rt; ii) Fe, NH4Cl, EtOH/H2O, 70 °C; iii) (1) BTC, Et3N, DCM, 0 °C→reflux; (2) memantine, Et3N, DCM, 0 °C→rt; iv) (1) NaOH, H2O, EtOH; (2) Conc. HCl; v) (1) SOCl2, DMF, THF, 0 °C→rt; (2) NH3 (g), THF, 0 °C→rt.
The preparation of compound B1 (Scheme 7) started with para-nitrobenzoyl chloride, which underwent acylation with tert-butyl piperidin-4-ylcarbamate to provide compound 41. Reduction of 41 using 5% Pd-C at H2 atmosphere under mildly condition provided 42. Subsequent acylation of the amino group present in 42 with phenyl chloroformate, and resulting key intermediate 43 was nucleophilic substituted with memantine to give 44. Removal of the Boc group in 44 with diluted trifluoroacetic acid (TFA) furnished the amine B1 quantitatively. Subsequent acylation of the amino group present in B1 with commercially available methanesulfonyl chloride, acetyl chloride and propionyl chloride to give corresponding compounds B2–B4. Then, commercially available acids (not shown) were then coupled to the compound B1 with the assistance of EDCI and HOBt in DCM solution to give B5–B7 and 45. Treatment of 45 with TFA deprotected the Boc group to give compound B8.
Scheme 7.

Synthetic route to compounds B1–B8. Reactions and conditions: (i) tert-butyl piperidin-4-yl-carbamate, Et3N, THF, 0 °C→rt, 6 h; (ii) H2 (g), 5% Pd-C, EtOH, 60 °C, 12 h; (iii) K2CO3, PhOCOCl, THF, 0 °C→rt, 7 h; (iv) memantine, Et3N, THF, 75 °C, 8 h; v) TFA, DCM, 0 °C→rt, 2 h; vi) (a) corresponding acyl chlorides, Et3N, DCM; or (b) corresponding acids, EDCI, HOBt, Et3N, DCM, rt.
The synthesis of compound B9 (Scheme 8) was accomplished using 3-fluoro-4-nitrobenzoic acid as starting material. Chlorination of 3-fluoro-4-nitrobenzoic acid 12 with SOCl2 in THF cleanly afforded the corresponding acyl chloride intermediate (not shown). Subsequent acylation with tert-butyl (S)-piperidin-3-ylcarbamate yield compound 46. Aniline 47 was prepared in excellent yield via reduction of nitro compound 46 in the presence of Fe/NH4Cl under standard conditions. Amino group in 47 was transformed to isocyanate (not shown) in the presence of BTC, and then reacted with memantine to give compound 48. Removal of the Boc group in 48 with diluted TFA furnished the amine 49 quantitatively. Subsequent acylation of the amino group present in 49 with commercially available methanesulfonyl chloride to give compound B9.
Scheme 8.

Synthetic route to compound B9. Reactions and conditions: i) (1) SOCl2, DMF, THF, 0 → 60 °C; (2) tert-butyl (S)-piperidin-3-yl-carbamate, Et3N, THF, 0 °C→rt; ii) Fe, NH4Cl, EtOH/H2O, 70 °C; iii) (1) BTC, Et3N, DCM, 0 °C→reflux; (2) memantine, Et3N, DCM, 0 °C→rt; iv) TFA, DCM, 0 °C→rt; vi) MsCl, Et3N, DCM, 0 °C→rt.
The preparation of compound C1 (Scheme 9) started with 4-nitrobenzoyl chloride, which underwent acylation with tert-butyl piperazine-1-carboxylate to provide compound 50. Reduction of 50 using 5% Pd-C at H2 atmosphere under mildly condition provided 51. Subsequent acylation of the amino group present in 51 with phenyl chloroformate, and resulting key intermediate 52 was nucleophilic substituted with memantine to give 53. Removal of the Boc protecting group in 53 with diluted TFA furnished the amine C1 quantitatively. Subsequent acylation of the amino group present in C1 with commercially available methanesulfonyl chloride, acetyl chloride and propionyl chloride to give corresponding compounds C2–C4. Then, commercially available acids (not shown) were then coupled to the compound C1 with the assistance of EDCI and HOBt in DCM solution to give C5–C7 and 54. Treatment of 54 with TFA deprotected the Boc group to give compound C8.
Scheme 9.

Synthetic route to compounds C1–C8. Reactions and conditions: (i) tert-butyl piperazine-1-carboxylate, Et3N, THF, 0 °C→rt, 6 h; (ii) H2 (g), 5% Pd-C, EtOH, 60 °C, 12 h; (iii) K2CO3, PhOCOCl, THF, 0 °C→rt, 7 h; (iv) memantine, Et3N, THF, 75 °C, 8 h; v) TFA, DCM, 0 °C→rt, 2 h; vi) (a) corresponding acyl chlorides, Et3N, DCM; or (b) corresponding acids, EDCI, HOBt, Et3N, DCM, rt.
2.3. Molecular docking
We used computer aided drug design to simulate possible binding modes of compounds A20 and B9 in the active pocket of sEH protein (PDB ID: 3WKE) through Discovery Studio 2016 software, as depicted in Fig. 3. Compounds A20, B9 and t-AUCB adopt the same conformation to bind to the sEH active pocket (Fig. 3A). The urea group forms hydrogen bonds with catalytic triad constituted with backbones of residues Asp335, Tyr383, and Tyr466 (Fig. 3B and C). The F atom of the phenyl group makes halogen bond with Phe267. The phenyl group of A20 and B9 not only binds in a hydrophobic cavity but also makes key π−cation interaction with residues of His524. Carbamoyl moiety of A10 and methanesulfonamide group of B9 extend to the solvent exposure area, which form hydrogen bond with His524, respectively. In addition, methanesulfonamide group of B9 makes extra hydrogen bond with Trp525. These may explain why compounds A10 and B9 could enhance inhibitory potency comparing to lead compound 4. The methyl groups of the memantyl moiety extend to a hydrophobic pocket formed by the backbones of residues Trp336 and Met339. These possibly binding modes suggest that the piperidinamide tail group may be contributing strongly to the binding affinity for the sEH protein.
Figure 3.

Molecular simulation of compounds A20 and B9 bound to sEH (PDB ID: 3WKE). (A) showed docked poses of compounds A20 (purple), B9 (blue) and t-AUCB (green) in sEH binding pocket. (B) indicated sEH bound to compound A20 in purple. (C) displayed sEH bound to compound B9 in blue. Hydrogen bond was showed as dash in yellow.
2.4. Structure–activity relationships of series derivatives
The memantyl urea moiety has been shown to be promising with respect to the sEH inhibitory effect of compounds. Cell-free recombinant HsEH IC50 assays based on biochemical method were performed on series compounds30, and the results of these experiments were compared against the cell-free IC50 of lead compound 4. These data were provided in Table 1, Table 2, Table 3.
Table 1.
Exploration of pipecolic acid derivatives SAR.
| Compd. | Structure | Subst. | IC50 (nmol/L)a | cLogPb | LLEc |
|---|---|---|---|---|---|
| A1 | X = H, R1 = OH | 4 | >1000 | 3.017 | – |
| A2 | X = H, R1 = OEt | 4 | 0.44 ± 0.26 | 3.591 | 5.762 |
| A3 | X = H, R1 = NH2 | 4 | 0.99 ± 0.18 | 2.390 | 6.616 |
| A4 | X = H, R1 = NHMe | 4 | 0.74 ± 0.23 | 2.595 | 6.538 |
| A5 | X = H, R1 = NMe2 | 4 | 0.75 ± 0.54 | 2.801 | 6.321 |
| A6 | X = H, R1 = NHCH(CH2)2 | 4 | 1.14 ± 0.77 | 3.085 | 5.859 |
| A7 | X = H, R1 = NHOMe | 4 | 0.92 ± 0.25 | 2.441 | 6.596 |
| A8 | X = H, R1 = OH | 3 | 0.42 ± 0.36 | 3.152 | 6.222 |
| A9 | X = H, R1 = OEt | 3 | 0.21 ± 0.19 | 3.726 | 5.942 |
| A10 | X = H, R1 = NH2 | 3 | 0.25 ± 0.11 | 2.525 | 7.076 |
| A11 | X = H, R1 = NHMe | 3 | 0.45 ± 0.39 | 2.730 | 6.614 |
| A12 | X = H, R1 = NMe2 | 3 | 0.37 ± 0.21 | 2.936 | 6.499 |
| A13 | X = H, R1 = NHCH(CH2)2 | 3 | 0.36 ± 0.23 | 3.220 | 6.219 |
| A14 | X = H, R1 = NHOMe | 3 | 0.34 ± 0.22 | 2.576 | 6.887 |
| A15 | X = F, R1 = NH2 | 3 | 0.15 ± 0.13 | 2.730 | 7.094 |
| A16 | X = Cl, R1 = NH2 | 3 | 0.18 ± 0.11 | 3.189 | 6.561 |
| A17 | X = H, R1 = NHOH | 3 | 0.39 ± 0.29 | 2.537 | 6.868 |
| A18 | X = H, R1 = NH2 | 3(R) | 0.31 ± 0.30 | 2.525 | 7.683 |
| A19 | X = H, R1 = NH2 | 3(S) | 0.12 ± 0.10 | 2.525 | 6.988 |
| A20 | X = F, R1 = NH2 | 3(S) | 0.06 ± 0.05 | 2.730 | 7.487 |
| A21 | X = F, R1 = NH2 | 3(R) | 0.08 ± 0.03 | 2.730 | 7.367 |
| 4 | – | – | 0.40 ± 0.25 | 3.832 | 5.566 |
| (±)-EC-5026 | – | – | 0.09 ± 0.02 | 4.227 | 5.825 |
| AR-9281 | – | – | 13.8 | 1.300 | 6.570 |
The cell-free IC50 values are recorded on HsEH protein based on biochemical method. The IC50 values were expressed as mean ± standard error of mean (SEM).
Discovery Studio 2016 software was used to predict clogP values.
Ligand lipophilicity efficiency (LLE) was calculated by following equation: LLE = pIC50‒clogP.
Table 2.
Exploration of aminopiperidine derivatives SAR.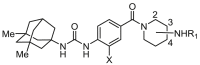
| Compd. | Structure | Subst. | IC50 (nmol/L)a | cLogPb | LLEc |
|---|---|---|---|---|---|
| B1 | X = H, R1 = H | 4 | 2.14 ± 1.19 | 2.337 | 6.332 |
| B2 | X = H, R1 = Ms | 4 | 0.79 ± 0.30 | 1.923 | 7.178 |
| B3 | X = H, R1 = Ac | 4 | 1.36 ± 0.98 | 2.132 | 6.734 |
| B4 | X = H, R1 = 
|
4 | 0.60 ± 0.37 | 2.799 | 6.420 |
| B5 | X = H, R1 = 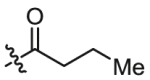
|
4 | 0.50 ± 0.39 | 3.255 | 6.043 |
| B6 | X = H, R1 = 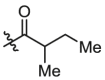
|
4 | 0.45 ± 0.21 | 3.718 | 5.629 |
| B7 | X = H, R1 = 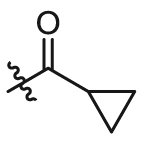
|
4 | 0.60 ± 0.44 | 2.890 | 6.330 |
| B8 | X = H, R1 = 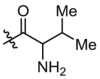
|
4 | 0.45 ± 0.40 | 2.498 | 6.846 |
| B9 | X = F, R1 = Ms | 3 | 0.11 ± 0.03 | 2.593 | 7.347 |
| 4 | – | – | 0.40 ± 0.25 | 3.830 | 5.570 |
| (±)-EC-5026 | – | – | 0.09 ± 0.02 | 4.227 | 5.825 |
| AR-9281 | – | – | 13.8 | 1.300 | 6.570 |
The cell-free IC50 values are recorded on HsEH protein based on biochemical method. The IC50 values were expressed as mean ± standard error of mean (SEM).
Discovery Studio 2016 software was used to predict clogP values.
Ligand lipophilicity efficiency (LLE) was calculated by following equation: LLE = pIC50‒clogP.
Table 3.
Exploration of piperazine derivatives SAR.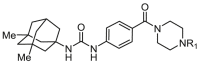
| Compd. | R1 | IC50 (nmol/L)a | cLogPb | LLEc |
|---|---|---|---|---|
| C1 | H | >1000 | 2.058 | – |
| C2 | Ms | 0.73 ± 0.41 | 1.870 | 7.269 |
| C3 | Ac | 1.21 ± 1.16 | 2.08 | 6.839 |
| C4 | |
1.09 ± 0.68 | 2.747 | 6.215 |
| C5 | |
0.89 ± 0.22 | 3.203 | 5.846 |
| C6 | |
0.64 ± 0.39 | 3.665 | 5.531 |
| C7 | |
0.94 ± 0.37 | 2.838 | 6.188 |
| C8 | |
2.66 ± 1.90 | 2.445 | 6.131 |
| 4 | – | 0.40 ± 0.25 | 3.830 | 5.570 |
| (±)-EC-5026 | – | 0.09 ± 0.02 | 4.227 | 5.825 |
| AR-9281 | – | 13.8 | 1.300 | 6.570 |
The cell-free IC50 values are recorded on HsEH protein based on biochemical method. The IC50 values were expressed as mean ± standard error of mean (SEM).
Discovery Studio 2016 software was used to predict clogP values.
Ligand lipophilicity efficiency (LLE) was calculated by following equation: LLE = pIC50‒cLogP.
According to the activities shown in Table 1, it appears that substituent at C3 position was beneficial and the terminal amide or ester moiety was fundamental for activity retention. Inhibitory potency on sEH of compounds substituted at C3 position (A8−A14) was slightly better than substituents at C4 position (A1−A7). Amongst, compound A1 substituted with 4-COOH group displayed lowest inhibitory potency, with IC50 value > 1000 nmol/L. Replacing carboxyl with amide or ester resulted in improving potency significantly. The best results were achieved with the ethyl ester-containing compound A9, showing IC50 value of 0.21 nmol/L. Among the different primary amide (‒NH2 A10), secondary amides (‒NHMe A11, cyclopropylamine A13, NHOMe A14) and tertiary amide (‒NMe2 A12) examined, the best results were obtained with the carbamoyl-containing compound A10, showing IC50 value of 0.25 nmol/L. Compound containing hydroxamic acid group (A17) displayed similar potency with carboxyl compound (A8) with IC50 values of 0.39 and 0.42 nmol/L. Although compound A9 was more potency than A10 in the inhibition of sEH in vitro, A9 was more easily metabolized by hydrolysis than A10 according to chemical stability. In addition, compound A10 possessed higher ligand lipophilicity efficiency (LLE)31. Accordingly, we selected compound A10 as the template for further optimization.
Next, we further modified the phenyl linker to improve potency of compounds. Excitingly, replacing the H atom at X with F and Cl resulted in compounds A15 and A16, which displayed about 2.5- and 2-fold potency improvements comparing to A10 with IC50 values of 0.15 and 0.18 nmol/L. In addition, substitution with halogen atom might prevent the metabolism of the phenyl group, and improve physical properties, e.g., melting point and solubility. Then, exploration on the chirality center of C3 position in A10 would be expected to further improve the potency. The two enantiomers A18 (R) and A19 (S) were respectively synthesized to determine the chiral preference for sEH inhibition. It was observed that the inhibitory potency of (S) steric configuration (A19) was 2-fold stronger than (R) enantiomer (A18) with IC50 values of 0.12 and 0.31 nmol/L, respectively. Taken together, data showed that (S)-carbamoyl substituent at C3 position of piperidinyl moiety and F substituent at phenyl linker (A20) was the optimal combination, not only providing strong inhibitory potency against the HsEH (IC50 = 0.06 nmol/L) but also demonstrating better metabolic stability and high LLE. Similar potency improvement (A21) was observed at enantiomer of A20, which was slightly weaker than A20 against HsEH, with IC50 values of 0.08 and 0.06 nmol/L.
Next, we performed structural modifications by changing the piperidinyl moiety of the lead compound 4. As shown in Table 2, replacing piperidinyl moiety (4) with 4-aminopiperidinyl (B1) resulted in decreasing inhibitory potency with IC50 value of 2.14 nmol/L. The incorporation of amides tail at the 4-aminopiperidinyl moiety resulted in compounds (B2–B8) with quite potent biochemical activity against HsEH. Among them, compound B3, bearing the acetyl group yielded the lowest potency with IC50 value of 1.36 nmol/L. Furthermore, methanesulfonamide group increased both potency and LLE, which might improve solubility and give more favorable physicochemical properties. Taken together, replacing 4-aminopiperidinyl with (S)-3-aminopiperidinyl and introducing of F atom at phenyl linker resulted in more potent inhibitor B9 with IC50 value of 0.11 nmol/L, which displayed higher LLE (7.347) than lead compound 4 (5.570).
As shown in Table 3, replacing piperidinyl moiety (4) with piperazinyl moiety (C1) resulted in significantly decreasing inhibitory potency with IC50 value > 1000 nmol/L. The incorporation of amides tail at the NH group of piperazinyl moiety resulted in compounds (C2–C8) with high inhibitory potency against HsEH. Among them, compound C2, bearing the methanesulfonamide group yielded the best potency with IC50 value of 0.73 nmol/L and increased favorable LLE (7.269). These data concluded that piperazinyl derivatives demonstrated weaker potency than compound 4, and other biological screenings weren't further evaluated in vitro and in vivo. Therefore, considering the potency and drug like properties, compounds A10 and A20 were selected as candidate compounds for further evaluated in vivo.
2.5. Microsomal stability
To determine the microsomal stability, preliminary microsomal stability studies (see Table 4) were carried out for the compounds A10 and A20. Both compounds displayed better metabolic stability than lead compound 4 in vitro. Compound A10 could remain in human and rat liver microsomes, with half-life (t1/2) of 69 min and 84 min. Excitingly, A20 displayed best microsomal stability for both human and rat species than compound A10 and lead compound 4, with t1/2 of 174 and 120 min, respectively. The results showed that F atom substituent could increase metabolic stability. Based on melting point and bioavailability, the F atom substitution (A20) further reduced the melting point and improved LLE, and A20 indicated most promise as it displayed the highest LLE.
Table 4.
Half-lifes of compounds A10 and A20 in human and rat microsomal buffer and melting point.
| Compd. | Human t1/2 (min) | Rat t1/2 (min) | Melting point | LLE |
|---|---|---|---|---|
| A10 | 69 | 84 | 196−198 °C | 7.076 |
| A20 | 174 | 120 | 165−166 °C | 7.487 |
| 4 | 25 | 31 | 204−205 °C | 6.570 |
2.6. Pharmacokinetic study in vivo
Considering to their good inhibitory potency, as well as suitable metabolic stability in vitro, the pharmacokinetics profile of A10 and A20 were further evaluated. After oral (po) and intravenous (iv) administration at a single dose, blood levels were analyzed to 12 or 24 h and pharmacokinetic parameters were shown in Table 5. The compounds A10 and A20 had a very similar PK profile. The maximum concentration of A20 (1.00 nmol/L) was reached on 0.63 h after oral administration; the area under the curve (AUC0–24 h) was 5.57 nmol h/L; A20 had a plasma t1/2 of 5.18 h. The maximum concentration of A20 (7.11 nmol/L) was reached on 4.8 min after iv administration; the area under the curve (AUC0–12 h) was 3.90 nmol h/L; A20 had a plasma t1/2 of 6.25 h. The bioavailability of A20 was 28.6%. However, the maximum concentration of A10 (0.12 nmol/L) was reached on 2.10 h after oral administration, and bioavailability of A10 was 3.1%, as shown in Table 5. It's worth noting that compound A10 exhibited high apparent volume of distribution (Vd) and clearance (CL), which demonstrating that A10 could been widely distributed in the surrounding tissues or bound to plasma proteins as well as quickly metabolized.
Table 5.
Pharmacokinetics parameters of A10 and A20 in rats after oral and intravenous administrationa.
| Parameter | A10 (n = 6) |
A20 (n = 6) |
||
|---|---|---|---|---|
| iv (10 mg/kg) | po (10 mg/kg) | iv (10 mg/kg) | po (50 mg/kg) | |
| Tmax (h) | 0.08 | 2.10 | 0.08 | 0.633 |
| Cmax (nmol/L) | 30.11 | 0.12 | 7.11 | 1.00 |
| t1/2 (h) | 2.66 | 2.93 | 6.25 | 5.18 |
| CL (mL/h) | 749.73 | 33580.26 | 1213.60 | 4070.93 |
| Vz (L) | 3.48 | 224.16 | 3.683 | 79.146 |
| AUC0‒t (nmol·h/L) | 10.14 | 0.31 | 3.90 | 5.57 |
| AUC0‒∞ (nmol·h/L) | 10.20 | 0.32 | 3.96 | 8.02 |
| F (%) | 3.1 | 28.6 | ||
After administration, blood samples were collected at different time (A10: 0, 0.083, 0.17, 0.5, 1, 2, 4, 6, 8, 12 h; A20: 0, 0.083, 0.17, 0.33, 0.5, 0.75, 1, 2, 4, 6, 8, 12, 24 h). Compounds formulated in 0.5% CMC-Na solution was administered orally at the indicated doses. The combination of compounds in a ratio of 8% ethanol absolute, 4% Tween-80 and 88% normal saline was chosen for the intravenous injection formulation.
The plasma protein binding rate (%PPB) of SD rat was measured through classic equilibrium dialysis devices. The assay disclosed that A20 showed moderate %PPB (88.6%). Because the therapeutic effect of drug was contributed to the concentration of free drug, this moderate %PPB (88.6%) could be beneficial to analgesic effect in vivo.
2.7. Safety of compound A20
To further evaluate the potential toxicological effect and adverse events, acute toxicity in Institute of Cancer Research (ICR) mice was carried out. The compound A20 was administered via po at a single dose of 6.0 g/kg. Body weight, and behavioral status was continuously recorded for 14 days. According to body weight and apparent behavior problems, no adverse events were observed, indicating that A20 was well tolerated and had no toxicity at dose of 6.0 g/kg (Fig. 4).
Figure 4.

The effect of the compound A20 on the body weight of ICR mice. A20 formulated in 0.5% CMC-Na solution was administered orally at the dose of 6.0 g/kg.
In addition, patch clamp technique was performed to detected inhibitory effect of A20 on human ether-a-go-go related gene (hERG) potassium channels in HEK293 cell lines. Compound A20 had low hERG activity at 3 μmol/L and 30 μmol/L with inhibitory rates as 0.46% and 41.97%, respectively, which indicating that A20 couldn't lead to potential adverse effects on heart.
2.8. Pharmacology study of A20in vivo
Following successful structural optimization of 4, the behavior of A20 was further investigated in SD rat with neuropathic pain model induced by spared nerve injury (SNI). The rats were randomly divided into gabapentin (60 mg/kg/day), (±)-EC-5026 (3 mg/kg/day) and A20 groups, which was further included 1, 3, and 9 mg/kg/day groups. After oral administration with a 12 h dosing interval (BID), mechanical hyperalgesia by von Frey test in rats subjected to evaluate the alleviation of pain on 1, 2, 4, 8, 16, 20 day throughout the study, which was presented as increasing of mechanical paw withdrawal threshold (PWT)32.
Treatment with A20 was well-tolerated at three doses in SD rats, as shown in Fig. 5. On first day, compound A20 demonstrated significantly analgesic effect at the 3 and 9 mg/kg/day dose levels, both groups were superior to (±)-EC-5026 and gabapentin. At the lowest dose (1 mg/kg/day) tested in this study, treatment with A20 didn't exhibit significantly analgesic effect. On Day 4, maximum analgesic effect of A20 at 9 mg/kg/day dose level was achieved. Although the antinociceptive effect of A20 was weaker than gabapentin before 16 days, the efficacy of gabapentin gradually reduced. Remarkably, on Day 20, antinociceptive effect of A20 at the 3 and 9 mg/kg/day dose levels were significantly stronger than (±)-EC-5026 and gabapentin. Comparing to the model group, A20 showed a time- and dose-dependent alleviation of pain and was well tolerated. Treatment with A20 at dose of 9 mg/kg/day showed best analgesic effect; furthermore, analgesic effect of A20 at 3 mg/kg/day dose of 3 mg/kg/day was also stronger than (±)-EC-5026 and gabapentin. To our delight, the molar dose of A20 was only 2% of gabapentin.
Figure 5.

sEH inhibitors attenuate neuropathic pain in SD rat model induced by spared nerve injury. Data are expressed as mean ± standard error of mean (n = 6); ∗P < 0.05, ∗∗P < 0.01, vs. Treatment/Model; #P < 0.05, vs. Treatment/Model; $P < 0.05, $$P < 0.01, vs. Treatment/Model; &P < 0.05, vs. Treatment/Model; @P < 0.05, vs. Treatment/Model; ▲P < 0.05, vs. Treatment/Model. A20 formulated in 0.5% CMC-Na solution was administered orally at the indicated doses.
On Day 20, the time–effect curve of A20 after last administration was measured, as shown in Fig. 6. At the doses of 3 and 9 mg/kg/day, treatment with A20 alleviated neuropathic pain for up to 6 h after the last dose in the SNI model, which was significantly longer than (±)-EC-5026 and gabapentin. In addition, A20 demonstrated faster onset after administration than both positive controls.
Figure 6.

sEH inhibitors attenuate neuropathic pain in rat model induced by spared nerve injury. Data are expressed as mean ± standard error of mean (n = 6).
3. Conclusions
In summary, we described the identification a new series of memantyl urea derivatives as potent sEH inhibitors, which were derived from an initial lead compound 4 through structure-based rational drug design. Different sites in the lead molecule were systematically optimized, leading to the identification of A20 as a highly potent sEH inhibitor, which was suitable for evaluating analgesic effect in preclinical neuropathic pain model in vivo. The improvement of 1–2 unit LLE was achieved via lead modification of piperidinyl to S-3-carbamoyl piperidinyl and by the incorporation of F atom at the phenyl group. These events culminated with the discovery of promising molecule A20. Oral dosing of A20 at 3 mg/kg/day in the SD rat with neuropathic pain symptom induced by SNI showed a robust increasing of the PWT level, and analgesic effect of A20 was stronger than (±)-EC-5026 and gabapentin. Delightfully, the molar dose of A20 is only 2% of gabapentin. According to the promising results obtained with compound A20, more research around memantyl urea‒containing sEH inhibitors for the treatment of neuropathic pain is currently ongoing.
4. Experimental
4.1. General information
All reagents and starting materials were purchased from commercial suppliers, and used without further purification. Nuclear magnetic resonance (NMR) spectra were obtained on an Ascent 400 MHz spectrometer (1H frequency of 400 MHz; 13C frequency of 100 MHz). Chemical shifts (δ) are reported in parts per million (ppm) relative to Me4Si as internal standard. Melting points were determined with an X-4 melting point devices. High-resolution mass spectrometry (HRMS) analysis was performed on an Agilent Technologies 6530 Accurate-Mass Q-TOF MS instrument using electrospray ionization (ESI) as ionization source. Details of compound synthesis and characterization were provided in Supporting Information. The purity analysis of target compounds was carried out by high-performance liquid chromatograph (HPLC).
4.2. Biological activity assays in vitro
Compounds were screened for their ability to inhibit HsEH by monitoring the hydrolase of (3-phenyloxiranyl)acetic acid cyano(6-methoxynaphthalen-2-yl)methyl ester (PHOME) to corresponding 6-methoxy-2-naphthaldehyde29. The fluorescent assay was performed on purified recombinant HsEH protein. After addition of PHOME solution ([S]final = 50 μmol/L), the mixture consisted of HsEH protein ([C]final = 6.4 nmol/L) and inhibitor in 25 mmol/L Tris-HCl buffer (pH = 7.4, containing 0.1 mg/mL of BSA) were incubated at 37 °C for 10 min. Fluorescence intensity was read on a SpectraMax M2 instrument at Ex/Em = 330/465 nm. IC50 values were measured by regression analysis using IBM SPSS Statistics 20 software.
4.3. Microsomal stability
Hepatic microsomes were commercially obtained from Research Institute for Liver Diseases Co., Ltd. (Shanghai, China). The detailed experimental procedure followed the procedures reported of our previously work29. The incubation mixture was composed of 490 μL of microsomal protein in 100 mmol/L phosphate buffered saline buffer (pH = 7.4) and 10 μL of A20 (3.7 mg/mL). The content of human and SD rat's hepatic microsomal protein was 0.4 and 0.53 mg/mL, respectively. NADPH was dissolved in phosphate buffered saline buffer to give 1 mmol/L NADPH solution, which was added to above mixture. After that, the mixture was incubated at 37 °C on 0, 5, 10, 20, 30 and 60 min. Then, the reaction was finished by cool MeCN (500 μL). The resulting mixture was vortexed for 1 min, and EtOAc (400 μL) was added into the mixture (200 μL). The mixture was further vortexed for 1 min, and centrifuged at 3000 rpm for 5 min. After that, supernatant (400 μL) was concentrated with Termovap Sample Concentrator. Then, the residue was reconstituted with 150 μL MeCN:H2O (7:3, v/v), the resulting mixture was vortexed for 1 min, and centrifuged at 10,000 rpm for 5 min. After that, supernatant was respectively analyzed by HPLC. The concentrations of A20 were measured by injecting 20 μL of sample onto a reverse-phase WondaSil C18 Superb column (150 mm × 4.6 mm, 5 μm) at a flow rate of 1 mL/min under the mobile phase conditions: MeCNH2O = 70:30 (v/v). Analytical UV detection was recorded at 210 and 230 nm.
4.4. Pharmacokinetics study in vivo
SD rats were used in this study (12 females, 12 males, and 8 weeks old), and purchased from Anhui Medical University (Anhui, China) (License No.: SCXK (Wan) 2017-001). All animal procedures were consisted with the Guiding Principles for Research Involving Animals of Anhui University of Chinese Medicine and all animal experiments were approved by the Animal Ethics Committee of Anhui University of Chinese Medicine (Anhui, China). The SD rats were raised at sterile conditions (18–22 °C, humidity 35%–65%, and ventilation) under daily cycles of light/darkness with a 12 h interval. They went through a period of 3 days of acclimatization before experimental procedure. 24 SD rats were assigned to 4 groups randomly (6 females and 6 males for A10; 6 females and 6 males for A20). Compounds formulated in 0.5% CMC-Na solution was administered orally at the indicated doses. The combination of compounds in a ratio of 8% ethanol absolute, 4% Tween-80 and 88% normal saline was chosen for the intravenous injection formulation. The SD rats were administered A10 (10 mg/kg) via iv or po routes. The SD rats were administered compound A20 via iv or po routes with different doses of 10 and 50 mg/kg. SD rats were weighed to calculate the required volume before each administration. Blood samples were obtained at indicated time points (A10: 0, 0.083, 0.17, 0.5, 1, 2, 4, 6, 8, 12 h; A20: 0, 0.083, 0.17, 0.33, 0.5, 0.75, 1, 2, 4, 6, 8, 12, 24 h). Blood sample was taken from the orbit vein at indicated time points, and collected on eppendorf containing of anticoagulant EDTA-K2, and centrifuged (4 °C) at 1100 rpm for 5 min. After that, each plasma sample (50 μL) was mixed with tadalafil (50 μL) in MeCN (20 ng/mL) and MeCN (300 μL), resulting mixture was further centrifuged at 13,000 rpm for 10 min. Then, supernatant (50 μL) was reconstituted with MeCN (200 μL), and resulting concentration of compounds were respectively analyzed by TSQ Altis Triple Quadrupole LC/MS instrument (Thermo Scientific). All the pharmacokinetic parameters were obtained using Phoenix WinNonlin software.
4.5. Percentage plasma protein binding (%PPB)
The detailed experimental procedure followed the procedures reported of our previously work29. The experimental assay was calibrated using warfarin of known %PPB: rat plasma (99.5%).
4.6. Efficacy of A20in vivo
SD rats were used in this study (males, and 8 weeks old, 180–220 g), and purchased from Jinan Pengyue Experimental Animal Breeding Co., Ltd. (License No.: SCXK (Lu) 20190003). All animal experiments were carried out under the guidelines of the Yantai University Committee for Use and Care of Animals. The animals were raised at sterile conditions (18–22 °C, humidity 35%–65%, and ventilation) under daily cycles of light/darkness with a 12 h interval. They went through a period of 3 days of acclimatization before surgical procedure. The A20 was suspended in 0.5% carboxymethylcellulose sodium aqueous. All test items were pre-prepared before one day and stored at 4 °C.
Surgical procedure for SD rats with neuropathic pain model induced by spared nerve injury (SNI) was prepared according reported method33, 34, 35. The SNI could produce sensory symptoms, such as hypersensitivity to mechanical and thermal stimuli, which develop immediately after 3 days. After successful modeling, rats were randomly divided into vehicle, (±)-EC-5026, gabapentin and test groups (n = 6). After oral administration with a 12 h dosing interval (BID), mechanical hyperalgesia by von Frey test in rats subjected to evaluate the alleviation of pain throughout the study. On Days 1, 2, 4, 8, 16, 20, PWT of A20 group was tested. The data was calculated via Eq. (1):
| (1) |
Statistical analysis was obtained by GraphPad Prism 8.0.2. Data are expressed as mean ± standard error of mean.
Acknowledgments
This work was funded by the Liaoning Revitalization Talents Program (XLYC1908031, China), Basic Research Project of Department of Education of Liaoning Province-natural sciences (2020LJC02, China), Major Basic Research Project of Natural Science Foundation of Shandong Province (ZR2018ZC1056, China), and partial support was provided by the NIH-NIEHS RIVER Award (R35 ES030443-01, USA), the NIEHS Superfund Research Program (P42 ES004699, USA).
Author contributions
Fangyu Du: conceptualization, methodology, formal analysis, writing‒original draft, writing‒review & editing, visualization; Ruolin Cao: visualization, software; Chen Lu: software, investigation; Yajie Shi: resources, investigation; Jianwen Sun: resources, formal analysis; Yang Fu: investigation, validation; Bruce D. Hammock: resources, funding acquisition; Zhonghui: formal analysis, software; Zhongbo Liu: formal analysis, writing‒review & editing, funding acquisition, supervision; Guoliang Chen: writing‒review & editing, project administration, funding acquisition, supervision. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2021.09.018.
Contributor Information
Zhongbo Liu, Email: 546265581@qq.com.
Guoliang Chen, Email: chenguoliang@syphu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Treede R.D., Jensen T.S., Campbell J.N., Cruccu G., Dostrovsky J.O., Griffin J.W., et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2008;70:1630–1635. doi: 10.1212/01.wnl.0000282763.29778.59. [DOI] [PubMed] [Google Scholar]
- 2.Bouhassira D. Neuropathic pain: definition, assessment and epidemiology. Rev Neurol (Paris) 2019;175:16–25. doi: 10.1016/j.neurol.2018.09.016. [DOI] [PubMed] [Google Scholar]
- 3.van Hecke O., Austin S.K., Khan R.A., Smith B.H., Torrance N. Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain. 2014;155:654–662. doi: 10.1016/j.pain.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 4.St John S.E. Advances in understanding nociception and neuropathic pain. J Neurol. 2018;265:231–238. doi: 10.1007/s00415-017-8641-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baron R., Binder A., Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010;9:807–819. doi: 10.1016/S1474-4422(10)70143-5. [DOI] [PubMed] [Google Scholar]
- 6.Ji R.R., Nackley A., Huh Y., Terrando N., Maixner W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology. 2018;129:343–366. doi: 10.1097/ALN.0000000000002130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fiore N.T., Austin P.J. Are the emergence of affective disturbances in neuropathic pain states contingent on supraspinal neuroinflammation?. Brain Behav Immun. 2016;56:397–411. doi: 10.1016/j.bbi.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 8.Song J., Yu H., Liu Y. Current status of treatment and drug discovery for neuropathic pain. Acta Pharm Sin. 2021;56:679–688. [Google Scholar]
- 9.Shinozaki T., Yamada T., Nonaka T., Yamamoto T. Acetaminophen and non-steroidal anti-inflammatory drugs interact with morphine and tramadol analgesia for the treatment of neuropathic pain in rats. J Anesth. 2015;29:386–395. doi: 10.1007/s00540-014-1953-0. [DOI] [PubMed] [Google Scholar]
- 10.Martinez-Navarro M., Maldonado R., Banos J.E. Why mu-opioid agonists have less analgesic efficacy in neuropathic pain. Eur J Pain. 2019;23:435–454. doi: 10.1002/ejp.1328. [DOI] [PubMed] [Google Scholar]
- 11.Altiparmak B., Cil H., Celebi N. Effect of melatonin on the daytime sleepiness side-effect of gabapentin in adults patients with neuropathic pain. Rev Bras Anestesiol. 2019;69:137–143. doi: 10.1016/j.bjane.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner K., Yang J., Inceoglu B., Hammock B.D. Soluble epoxide hydrolase inhibition is antinociceptive in a mouse model of diabetic neuropathy. J Pain. 2014;15:907–914. doi: 10.1016/j.jpain.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macone A., Otis J.A.D. Neuropathic pain. Semin Neurol. 2018;38:644–653. doi: 10.1055/s-0038-1673679. [DOI] [PubMed] [Google Scholar]
- 14.Gopalsamy B., Farouk A.A.O., Tengku Mohamad T.A.S., Sulaiman M.R., Perimal E.K. Antiallodynic and antihyperalgesic activities of zerumbone via the suppression of IL-1β, IL-6, and TNF-α in a mouse model of neuropathic pain. J Pain Res. 2017;10:2605–2619. doi: 10.2147/JPR.S143024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaspera R., Totah R.A. Epoxyeicosatrienoic acids: formation, metabolism and potential role in tissue physiology and pathology. Expet Opin Drug Metabol Toxicol. 2009;5:757–771. doi: 10.1517/17425250902932923. [DOI] [PubMed] [Google Scholar]
- 16.Wagner K.M., Gomes A., Mcreynolds C.B., Hammock B.D. Soluble epoxide hydrolase regulation of lipid mediators limits pain. Neurotherapeutics. 2020;17:900–916. doi: 10.1007/s13311-020-00916-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morisseau C., Hammock B.D. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol. 2013;53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morin C., Sirois M., Echavé V., Albadine R., Rousseau E. 17,18-Epoxyeicosatetraenoic acid targets PPARγ and p38 mitogen-activated protein kinase to mediate its anti-inflammatory effects in the lung: role of soluble epoxide hydrolase. Am J Resp Cell Mol. 2010;43:564–575. doi: 10.1165/rcmb.2009-0155OC. [DOI] [PubMed] [Google Scholar]
- 19.Wagner K.M., McReynolds C.B., Schmidt W.K., Hammock B.D. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol Ther. 2017;180:62–76. doi: 10.1016/j.pharmthera.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wagner K.M., Atone J., Hammock B.D. Soluble epoxide hydrolase inhibitor mediated analgesia lacks tolerance in rat models. Brain Res. 2020;1728:146573. doi: 10.1016/j.brainres.2019.146573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammock B.D., McReynolds C.B., Wagner K., Buckpitt A., Cortes-Puch I., Croston G., et al. Movement to the clinic of soluble epoxide hydrolase inhibitor EC5026 as an analgesic for neuropathic pain and for use as a nonaddictive opioid alternative. J Med Chem. 2021;64:1856–1872. doi: 10.1021/acs.jmedchem.0c01886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiamvimonvat N., Ho C.M., Tsai H.J., Hammock B.D. The soluble epoxide hydrolase as a pharmaceutical target for hypertension. J Cardiovasc Pharmacol. 2007;50:225–237. doi: 10.1097/FJC.0b013e3181506445. [DOI] [PubMed] [Google Scholar]
- 23.Imig J.D., Zhao X., Capdevila J.H., Morisseau C., Hammock B.D. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 24.Chen D., Whitcomb R., MacIntyre E., Tran V., Do Z.N., Sabry J., et al. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single-and multiple-dose studies in healthy human subjects. J Clin Pharmacol. 2012;52:319–328. doi: 10.1177/0091270010397049. [DOI] [PubMed] [Google Scholar]
- 25.Wang L., Yang J., Guo L., Uyeminami D., Dong H., Hammock B.D., et al. Use of a soluble epoxide hydrolase inhibitor in smoke-induced chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2012;46:614–622. doi: 10.1165/rcmb.2011-0359OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lazaar A.L., Yang L., Boardley R.L., Goyal N.S., Robertson J., Baldwin S.J., et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br J Clin Pharmacol. 2016;81:971–979. doi: 10.1111/bcp.12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mao J., Gold M.S., Backonja M.M. Combination drug therapy for chronic pain: a call for more clinical studies. J Pain. 2011;12:157–166. doi: 10.1016/j.jpain.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee K.S.S., Ng J.C., Yang J., Hwang S.H., Morisseau C., Wagner K., et al. Preparation and evaluation of soluble epoxide hydrolase inhibitors with improved physical properties and potencies for treating diabetic neuropathic pain. Bioorg Med Chem. 2020;28:115735. doi: 10.1016/j.bmc.2020.115735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du F., Sun W., Morisseau C., Hammock B.D., Bao X., Liu Q., et al. Discovery of memantinyl urea derivatives as potent soluble epoxide hydrolase inhibitors against lipopolysaccharide-induced sepsis. Eur J Med Chem. 2021;223:113678. doi: 10.1016/j.ejmech.2021.113678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolf N.M., Morisseau C., Jones P.D., Hock B., Hammock B.D. Development of a high-throughput screen for soluble epoxide hydrolase inhibition. Anal Biochem. 2006;355:71–80. doi: 10.1016/j.ab.2006.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leeson P., Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 32.Chaplan S.R., Bach F.W., Pogrel J.W., Chung J.M., Yaksh T.L. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 33.Guida F., De Gregorio D., Palazzo E., Ricciardi F., Boccella S., Belardo C., et al. Behavioral, biochemical and electrophysiological changes in spared nerve injury model of neuropathic pain. Int J Mol Sci. 2020;21:3396. doi: 10.3390/ijms21093396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boccella S., Guida F., Palazzo E., Marabese I., de Novellis V., Maione S., et al. Spared nerve injury as a long-lasting model of neuropathic pain. Methods Mol Biol. 2018;1727:373–378. doi: 10.1007/978-1-4939-7571-6_28. [DOI] [PubMed] [Google Scholar]
- 35.Jin X., Luo A., Zhang G. Comparison of the establishment and afficacy of three neuropathic pain models. J Clin Anesth. 2005;21:338–340. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.