

Abstract
Combination of passive targeting with active targeting is a promising approach to improve the therapeutic efficacy of nanotherapy. However, most reported polymeric systems have sizes above 100 nm, which limits effective extravasation into tumors that are poorly vascularized and have dense stroma. This will, in turn, limit the overall effectiveness of the subsequent uptake by tumor cells via active targeting. In this study, we combined the passive targeting via ultra-small-sized gemcitabine (GEM)-based nanoparticles (NPs) with the active targeting provided by folic acid (FA) conjugation for enhanced dual targeted delivery to tumor cells and tumor-associated macrophages (TAMs). We developed an FA-modified prodrug carrier based on GEM (PGEM) to load doxorubicin (DOX), for co-delivery of GEM and DOX to tumors. The co-delivery system showed small particle size of ∼10 nm in diameter. The ligand-free and FA-targeted micelles showed comparable drug loading efficiency and a sustained DOX release profile. The FA-conjugated micelles effectively increased DOX uptake in cultured KB cancer cells that express a high level of folate receptor (FR), but no obvious increase was observed in 4T1.2 breast cancer cells that have a low-level expression of FR. Interestingly, in vivo, systemic delivery of FA-PGEM/DOX led to enhanced accumulation of the NPs in tumor and drastic reduction of tumor growth in a murine 4T1.2 breast cancer model. Mechanistic study showed that 4T1.2 tumor grown in mice expressed a significantly higher level of FOLR2, which was selectively expressed on TAMs. Thus, targeting of TAM may also contribute to the improved in vivo targeted delivery and therapeutic efficacy.
KEY WORDS: Folic acid, Gemcitabine, Polymeric micelles, Breast cancer, Dual targeting, Tumor associated macrophages, Ultrasmall nanocarrier, Doxorubicin
Graphical abstract
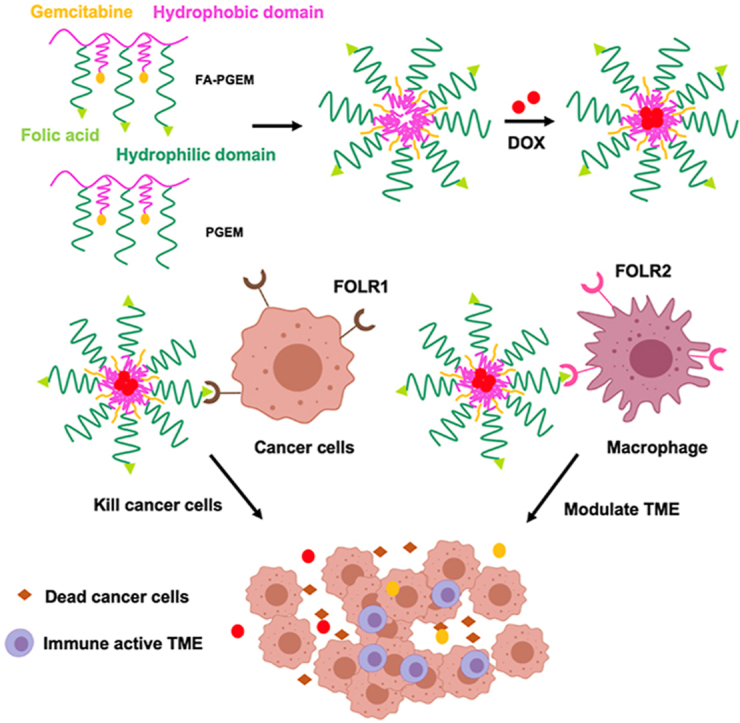
Folate receptors FOLR1 and FOLR2 are highly expressed on tumor cells and macrophages. FA-PGEM micellar carrier could enhance DOX delivery to both tumor cells and M2-like TAMs through folate-mediated active targeting effect.
1. Introduction
Due to the hyper-permeability of the tumor vasculature and the compromised lymphatic drainage, nanodelivery systems have been widely employed to selectively deliver therapeutics into solid tumors and minimize any off-target effect via the enhanced permeability and retention (EPR) effect. This passive targeting is dependent on both the tumor types and the physical properties of the drug delivery carrier. Some tumor types, such as pancreatic cancer and breast cancer, have very small vascular pore sizes (50–60 nm), which limits effective extravasation to NPs of smaller sizes1. However, many drug carriers that have been developed have relatively large size, often close to or larger than 100 nm. While these sizes are needed for high drug loading capacity, they restrict the effective extravasation of the NPs into tumor tissues2. Moreover, even if large NPs (>50 nm in diameter) are able to cross the tumor vessels, they might not be able to penetrate the dense extracellular matrix of the tumor interstitial space3. Although strategies are available to reduce the sizes of the carriers, these efforts also led to compromised drug loading efficiency. Thus, it is highly demanded to develop small sized carrier with excellent drug loading profiles.
On the other hand, the therapeutic efficacy of the passively targeted nanomedicine is not always satisfactory in clinical translation due to the heterogeneity of the EPR effect exhibited within a tumor, and inefficient tumor cell uptake4. Combination of passive targeting with active targeting has been demonstrated as a promising approach to further improve the therapeutic efficacy5. After the nanocarrier extravasates in the tumor tissue via EPR effect, the molecular ligand on the nanocarrier interacts with the receptor on the tumor cells and facilitates the cellular uptake via active targeting. In addition, active targeting only happens when the distance of ligand and receptor is less than 0.5 mm. Therefore, better EPR effect via small-sized nanocarrier is beneficial for the subsequent active targeting effect.
The folate receptor (FR) is a well-known tumor marker for active targeted delivery. The alpha isoform, FRα (or FOLR1) is overexpressed in most tumor cells6, 7, 8, 9, 10, 11. In contrast, FRβ (or FOLR2) is overexpressed in activated TAMs12, 13, 14. It has been reported that folic acid (FA) conjugated micelles and liposomes showed efficient targeted delivery into the tumor tissues. However, most studies were focused on the tumor types with high FRα expression in tumor cells, there are relatively few studies investigating FA-conjugated delivery systems for targeting FRβ positive TAMs. TAMs play an important role in tumor invasion, growth, angiogenesis, metastasis, and immunosuppression. Wei's group15 reported that FRβ overexpression in lung cancer TAMs was associated with poor prognosis. They developed a folate-modified lipoplex for targeting TAMs and demonstrated that FRβ positive TAMs are an attractive target for lung cancer therapy. However, FA-modified ultra-small polymeric nanocarrier has been rarely reported for combination of effective tumor penetration and active tumor targeting.
In this work, we combined the passive targeting via ultra-small sized GEM-based NPs with the active targeting provided by FA conjugation for enhanced dual targeting of both tumor cells and TAMs. GEM is a hydrophilic molecule that induces DNA damage to rapidly dividing cells. It is widely used clinically against a variety of solid tumors16. A number of studies showed that GEM synergistically improved the therapeutic activity with other anticancer agents such as cisplatin17, Taxol18, quercetin19, RAS inhibitors20, NLG91921, and aPDL122. However, the major potential problem in clinical translation of combination therapy is the difficulty in achieving effective codelivery of the different anticancer agents due to their different PK profiles and rapid clearance in vivo23. To resolve this issue, polymeric micelles are now extensively studied as nano-drug delivery systems. They are highly tunable amphiphilic polymers that can be conjugated or loaded with various hydrophilic and hydrophobic drugs24, 25, 26, 27. As a result, increased drug solubility and reduced uptake in normal tissues could be achieved, resulting in attenuation of toxicity that is commonly associated with conventional chemotherapy28,29. In our previous work, we found that coupling of GEM to PVD polymer (POEG-co-PVD) resulted in a drastic decrease in the particle size from 160 to 13 nm, leading to more efficient extravasation into tumors30. Surprisingly, this modification led to a further increase in drug loading capacity when serving as a prodrug carrier. Here, we further introduced FA as a ligand into this prodrug carrier to improve the targeting effect to tumor cells, and examined the opportunity of achieving a combination therapy through codelivery of GEM and DOX. The small sized polymeric nanocarrier is expected to enhance the penetration into the core of tumor tissues, which is beneficial for following active targeting effect. Interestingly, although FA targeting showed minimal benefit in cultured 4T1.2 tumor cells, systemic delivery of DOX via FA-PGEM led to a significant improvement in both tumor accumulation and overall therapeutic efficacy. Further studies showed that 4T1.2 tumor grown in mice expressed a significantly higher level of FR and that targeting of cancer cells and TAMs by this FA-conjugated nanotherapeutics synergistically led to the improved in vivo targeting.
2. Materials and methods
2.1. Chemicals
Doxorubicin hydrochloride (DOX·HCl) was obtained from LC Laboratories (MA, USA)31. Vinylbenzyl chloride, potassium carbonate (K2CO3), azobisisobutyronitrile (AIBN), petroleum ether (PE), sodium hydroxide (NaOH), 2-hydroxyethyl methacrylate, di-tert-butyl decarbonate, 1,4-dioxane, dimethylformamide (DMF), tetrahydrofuran (THF), dimethyl sulfoxide (DMSO), N-hydroxy succinimide (NHS), N,N-diisopropylethylamine (DIPEA), triethylamine (TEA), 4-cyano-4-[(dodecylsulfanylthiocarbonyl) sulfanyl] pentanoic acid, poly(ethylene glycol, PEG2K), and poly(ethylene glycol) methyl ether methacrylate (OEGMA) were purchased from Fisher Scientific. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide HCl (EDC·HCL) and 1-hydroxybenzotriazole (HOBT) were purchased from GL Biochem (Shanghai, China).
2.2. Cell lines
Human oral epidermal carcinoma KB cell line was obtained from Dr. Larry Matherly at Ann Karmanos Cancer Center, Michigan, USA. Murine breast cancer cell line 4T1.2 was obtained from ATCC (Manassas, VA, USA). All the cell lines were routinely cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin at 37 °C in a humidified environment with 5% CO2. For in vitro studies, RPMI 1640 folate-free medium was used.
2.3. Animal tumor models
The female BALB/c mice (5–6 weeks) of 18–20 g were purchased from Charles River (Davis, CA, USA). The animal protocols were approved by Animal Use and Care Administrative Advisory Committee at the University of Pittsburgh. The mice were housed under pathogen free conditions and all experimental studies were completed according to the guidelines approved by the Ethics Committee of University of Pittsburgh.
2.4. Synthesis of FA-PGEM
The polymer PGEM was synthesized as per previous methods30. For conjugation of FA as a targeting ligand, NH2-PEG5K-NH2 was first reacted with FA. A relatively long PEG spacer (5 K instead of 2 K) was used to ensure that FA is accessible as a targeting ligand to FR overexpressing cancer cells. Briefly, FA (50 mg) was dissolved in 2 mL DMSO along with EDC·HCL (25 mg), DIPEA (30 μL) and NHS (15 mg). The mixture was stirred for 1 h in dark, followed by the addition of 400 mg NH2-PEG5K-NH2. After stirring overnight at room temperature, a mixture of chloroform/water (1:1) was added to the reaction mixture. The chloroform layer was collected and dried with anhydrous sodium sulfate. After precipitation in diethyl ether for two times, FA-PEG5K-NH2 was obtained. Then, FA-PEG5K-NH2 (50 mg) and GEM (40 mg) were linked to PVD polymer (30 mg) in the presence of EDC·HCL (100 mg), HOBT (60 mg) and DIPEA (100 μL). After reaction in 5 mL DMSO under stirring at 37 °C for 72 h, the reaction mixture was dialyzed against water for two days. The solution obtained was filtered, frozen at −80 °C and then freeze-dried overnight in a lyophilizer. The final product was dissolved in dichloromethane at a concentration of 26 mg/mL.
2.5. Preparation of DOX-free and DOX-loaded micelles
DOX·HCl was first treated with 3 mol equivalent of triethylamine in chloroform/methanal (1:1, v/v) to remove HCl. The PGEM and FA-PGEM polymers were mixed at different ratios to obtain a mixed polymer solution (1 mg/mL). To this, 20 μL of 5 mg/mL DOX was added and the final solution was air-dried to obtain a film (DOX/polymer:1/10, w/w). After removing all solvent in a vacuum pump, PBS was added to hydrate the film, forming DOX-loaded mixed micelles. Solutions were filtered through a syringe filter (pore size = 0.45 μm) to remove unloaded DOX. Blank micelles were prepared similarly without adding DOX.
2.6. Characterization of micelles
The mean hydrodynamic diameter, size distribution, and zeta potential of various formulations of micelles were evaluated by a Zetasizer. For the morphological analysis of blank and DOX-loaded micelles, low concentrations of samples were observed under transmission electron microscopy (TEM). To evaluate the drug loading efficiency and capacity, micelles were dissolved in DMSO to break the micelle structure and release DOX. The concentration of DOX loaded in the micelles was determined by UV Spectrometer (excitation 490 nm/emission 600 nm). The drug loading capacity (DLC) and efficiency (DLE) were calculated according to the following Eqs. (1), (2):
| (1) |
| (2) |
The critical micellar concentration (CMC) of FA-PGEM was determined using Nile red as a fluorescent probe. Twenty μL of Nile red (1.5 mg/mL) in dichloromethane each was added to twenty tubes and allowed to stay in a dark room overnight at room temperature to remove the solvent. Next, 2 mL of FA-PGEM micellar solution was added at logarithmic dilutions to each tube. Six hours later, the emission spectra of the solutions were measured (excitation wavelength of 550 nm and emission of 650 nm) and recorded. The absorbance values were plotted, and the CMC concentration was calculated at the intersection point of the two slopes.
2.7. DOX release kinetics
The PGEM/DOX and FA-PGEM/DOX micelle systems were tested for their in vitro release kinetics of DOX. For this experiment, 50 mL PBS (pH = 7.4) was used as the release medium and free DOX was employed as a control. In short, 250 μL of DOX-loaded micelles (5 mg DOX/mL) were sealed in dialysis bags (MWCO = 3.5 kDa). The dialysis bags were immersed in 50 mL PBS release medium in a beaker covered with Parafilm. The beakers were kept in an incubator shaker at 100 rpm and 37 °C32. At pre-determined time points, 1 mL of dialysate was collected and replaced with 1 mL fresh PBS. The concentrations of DOX in collected samples were measured by UV Spectrophotometer with the fluorescence detected at 580 nm. Error bars were obtained from triplicate samples.
2.8. RT-PCR analysis of FR expression
To evaluate the FRα (FOLR1) and FRβ (FOLR2) expression in 4T1.2 and KB cell lines, the cells were plated at high confluency in RPMI-1640 medium and cultured overnight. Their RNA was isolated, retrotranscribed and amplified using standard procedures. Oligonucleotide sequences for murine Folr1: (FP) 5′- GTGTCACAGGATTCAGGCCA-3′; (RP) 5′-TCGGGCTTCTTTGTCTCCAC-3′, Folr2: (FP) 5′-GTCACTTCATCCAAGACTCCTGC-3′; (RP) CACTGGTGACAGTCCTCTTTGC, and Gapdh: (FP) 5′-AGGTTGTCT CCTGCG ACTTCA-3′; (RP) 5′-TGGTCCAGGGTTTCTTACTCC-3′. Primers specific for human FOLR1: 5′-CTG GCTGGTGTTGGTAGAACAG-3′ (FP); 5′-AGGCCCCGAGGACAAGTT-3′ (RP); FOLR2: (FP) 5′-CATGTGCAGTGCCCAGGA-3′; (RP) 5′-CCAGGGACTGCATTGGTCAT-3′ and GAPDH: (FP) 5′-GCACCGTCAAGGCTGAGAAC-3′; (RP) 5′-TGGTGAAGACGCCAGTGGA-3′.
2.9. Cellular cytotoxicity assay
4T1.2 and KB cells were plated in a 96-well plate and cultured overnight at 37 °C with 5% CO2. Next, the cells were treated with various concentrations of DOX, PGEM, PGEM/DOX, FA-PGEM, FA-PGEM/DOX and FA-PGEM/DOX in the presence of free folate (1 mmol/L, 1:100, v/v) for 30 min. The drug-containing medium was then replaced with fresh medium and cells were cultured for another 24 h. The media from each well was replaced with 100 μL MTT in PBS (10%) and cells were incubated for additional 4 h. Finally, the MTT solution was replaced by 100 μL of filtered DMSO to dissolve the formalin crystals, giving rise to purple-colored solution. The absorbance of each well was recorded using a UV spectrophotometer and the cellular viability (%) was calculated according to the following Eq. (3):
| (3) |
2.10. Cellular uptake study
KB or 4T1.2 cells were seeded into each well of 6-well plates (3 × 105 cells/well) and grown overnight at 37 °C and 5% CO2. The medium was replaced by fresh folate-free RPMI 1640 medium containing free DOX, PGEM/DOX, FA-PGEM/DOX, and FA-PGEM/DOX in the presence of 1 mmol/L free folic acid. Each formulation was freshly prepared at an equivalent DOX concentration of 6 μg/mL. The cells were incubated for 30 min at 37 °C, and then washed three times with cold PBS followed by fixation with 10% paraformaldehyde for 10 min on ice. Cells were then washed with cold PBS thrice. Next, the nuclei were stained with DAPI for 5 min and cells were again washed three times with cold PBS. Finally, the intracellular uptake of DOX in various formulations was observed under a fluorescence microscope.
For further quantification of the DOX uptake, KB and 4T1.2 cells were seeded in a 6-well plate at a high density (1 × 106 cells/well) and incubated overnight at 37 °C and 5% CO2. Cells were then similarly treated with various formulations as described above. Thirty min later, the medium was removed, and cells were washed three times with 1 mL cold PBS. The cells were harvested with 0.5 mL trypsin treatment and cell pellets were collected after centrifugation. Cells were then washed with 1 mL PBS three times to remove traces of cell culture media. Next, the cells were fixed with 10% paraformaldehyde for 10 min. Cells were then washed with 1 mL PBS twice and the cell pellets were suspended in 200 μL PBS and transferred to the flow cytometry tubes for the flow cytometry analysis with MACS Quant Analyzer. A total of 20,000 events were collected for each sample. The DOX in samples was excited with an argon laser (480 nm), and fluorescence was detected at 570 nm.
2.11. In vivo biodistribution of DOX
Female BALB/c mice were inoculated with 2 × 105 4T1.2 cells/mouse s.c. and the tumor sizes were observed daily. After the tumors reached ∼200 mm3 in sizes, the mice were randomly assigned to three different treatment groups (n = 3) and received a single i.v. dose of free DOX, PGEM/DOX, and FA-PGEM/DOX at a DOX dosage of 10 mg/kg. Twenty-four hours post administration, the mice were sacrificed, and their tumors and other major organs were harvested. Frozen sections of 10 μm thickness were prepared using CryoStar NX50 and observed for DOX signals under a fluorescence microscope.
The in vivo distribution profiles of different formulations were further examined with near infrared imaging (NIRI). Groups of three 4T1.2 tumor (∼200 mm3)-bearing mice received i.v. injection of free 1,10-dioctadecyl-3,3,30,30-tetramethylindotricarbocyanine Iodide (DiR), PGEM/DIR, and FA-PGEM/DIR, respectively, with the polymer to DIR ratio at 20:1 (w/w) for all formulations. At different times post injection, the mice were subjected to whole-body NIR using an IVIS Imaging system. Mice were then euthanized, and their tumors and major organs were excised and subjected to ex-vivo imaging.
2.12. In vivo antitumor efficacy study
Thirty female BALB/c mice were inoculated with 4T1.2 cells (2 × 105 cells/mouse) and the tumor sizes were observed daily. Once the tumor reached a volume of ∼50 mm3, the mice were randomly divided into five groups and received three tail vein injections of saline, DOX, PGEM, FA-PGEM, and FA-PGEM/DOX. The DOX dosage was kept at 5 mg/kg. The tumor volume, measured with a digital calliper, was recorded using Eq. (4):
| (4) |
where L and W are length and width of each tumor respectively. To compare the various groups, relative tumor volume (RTV) was calculated, using Eq. (5):
| (5) |
Additionally, the body weights of all mice were recorded to monitor toxicity. The tumor growth inhibition rate (IR) was assessed by Eq. (6):
| (6) |
At the completion of therapy study, tumor tissues were collected, fixed with 4% (v/v) paraformaldehyde in PBS (pH 7.4), embedded in paraffin, and sectioned into 4 μm slices. Each section was processed for hematoxylin and eosin (H&E) staining, and Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.
Serum samples were collected at the end of the therapy study and subjected to analysis of the levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) enzyme activity, two biomarkers for liver toxicity.
2.13. Folate-mediated targeting of FOLR2 in M2-like TAMs
To test the potential targeting of FOLR2 on macrophages by our FA-PGEM, we evaluated the expression of FOLR2 on M1 and M2 macrophages. Macrophages were isolated from the spleen of naive mice (5–6 weeks old) following a published protocol33. Macrophages were separated from other mononuclear cells and lymphocytes based on the adherence property of macrophages. To induce an M1 polarization, the macrophages were treated with LPS (100 ng/mL) + IFN-γ (20 ng/mL). To induce M2 polarization, the macrophages were treated with IL-4 (250 ng/mL) and IL-13 (250 ng/mL) for 18 h33. The mRNA expression levels of several classic markers for each phenotype were examined by RT-PCR with the following primers34: iNOS (M1 marker): (FP) 5′-CGAAACGCTTCACTTCCAA-3′, (RP) 5′-TGAGCCTATATTGCTGTGGCT-3′; Arginase 1 (M2 marker): (FP) 5′-AACACGGCAGTGGCTTTAACC-3′, (RP) 5′-GGTTTTCATGTGGCGCATTC-3′; Interleukin 1 beta (Il1b, M1 marker): (FP) 5′-GCAACTGTTCCTGAACTCAACT-3′, (RP) 5′-ATCTTTTGGGGTCCGTCAACT-3′; CD206 (M2 marker): (FP) 5′-CTCTGTTCAGCTATTGGACGC-3′, (RP) 5′-CGGAATTTCTGGGATTCAGCTTC-3′.
The mRNA expression levels of Folr2 in M2 cells were also examined with the following primers: (FP) 5′-GTCACTTCATCCAAGACTCCTGC-3′; (RP) 5′-CACTGGTGACAGTCCTCTTTGC-3′.
Cellular uptake of DOX by M0, M1 and M2 cells following treatments with various DOX formulations was similarly performed as described in an earlier section. In addition, the numbers of tumor-infiltrating M2 macrophages were examined by flow cytometry following various treatments. Tumor-bearing mice received different treatments on Days 6, 9 and 12, respectively. Single cell suspensions were then prepared and multi-parameter staining was used to identify the M2 macrophage (CD45+, CD11b+, F4/80+, and CD206+).
2.14. Re-analysis of single-cell RNA-seq data
Several publicly available scRNA-Seq data were analyzed including breast invasive carcinoma (BRCA, GSE11472 and GSE143423), non-small cell lung cancer (NSCLC, GSE117570, GSE127465, and GSE143423), ovarian cancer (OV, GSE118828), pancreatic cancer (PAAD, CRA001160 and GSE111672), and skin cutaneous melanoma (SKCM, GSE72056). Expression of FOLR1 and FOLR2 was first examined in tumor cells and tumor-infiltrating immune cells. The expression in several subpopulations of immune cells was then further examined.
2.15. Statistical analysis
Data was processed by GraphPad Prism 7. All values are reported as mean ± SEM (standard error of mean). In all statistical analyses, the significance level was set at a probability of P < 0.05. All results were analyzed by Student's t-test for two groups, and One-way ANOVA for multiple groups.
3. Results and discussion
3.1. Synthesis and characterization of PGEM and FA-PGEM polymers
The synthesis route of PGEM polymer was shown in Scheme 1A. First, the vinyl benzyl monomer (VD monomer) was synthesized with a disulfide linkage as previously reported30. Then, through RAFT co-polymerization of VD monomer and OEG950 monomer, POEG-co-PVD polymer was obtained. This polymer was then conjugated with GEM using the EDC/HOBt coupling reaction. GEM loading capacity was determined by HPLC‒UV analysis via the alkaline hydrolysis method. The GEM loading in the POEG-co-PVDGEM (PGEM) polymeric carrier was determined to be ∼7% (w/w). FA-PGEM was similarly synthesized as shown in Scheme 1B30. FA-PEG was firstly obtained by the reaction of FA with NH2-PEG5K-NH2. Then, PVD polymer was obtained by polymerization of VD monomer. After conjugation with FA-PEG and GEM, FA-PGEM was obtained. The structure of the polymer was characterized by 1H NMR spectrum, and the numbers of GEM and FA units per polymer molecule were determined to be 5 and 1, respectively (Supporting Information Fig. S1).
Scheme 1.

Synthetic schemes for PGEM and FA-GEM polymers. (A) The polymer backbone POEG-co-PVD was synthesized via RAFT polymerization, followed by post conjugation with gemcitabine via EDC/HOBT coupling reaction. (B) The polymer backbone PVD was synthesized via RAFT polymerization, followed by post conjugation with gemcitabine and folic acid-modified PEG.
3.2. Preparation of FA-targeted PGEM micelles
In our previous work, we found that coupling of GEM to PVD polymer backbone resulted in a drastic decrease in the particle size from 160 to 13 nm30. This modification led to a further increase in DLC when serving as a prodrug carrier. Thus, we first compared the DOX loading capacity of PGEM nanocarrier with that of larger NPs made from PVD polymer backbone. We found that PVD polymer was able to load DOX with a DLC of as high as 13.7% (Supporting Information Table S1). In comparison, PGEM showed a higher DOX loading capacity of 25.1%.
Here, we prepared the FA-targeted PGEM micelles by introducing FA ligand into the ultra-small PGEM prodrug carrier to improve the targeting effect to tumor cells. As a first step to optimize the ligand density on the micellar carrier, the two polymers FA-PGEM and PGEM were mixed at various ratios to obtain mixed micelles with different percentages of FA-conjugated PGEM (Scheme 2). All the micellar formulations readily formed small sized particles with an average hydrodynamic diameter of ∼10 nm (as measured by DLS, Table 1). The zeta potentials for all the formulations were slightly negative, suggesting that at physiological pH, individual micelle units would be repelled from each other, which helps to prevent aggregation. Importantly, conjugation of FA did not significantly alter the size of PGEM micelles. For the subsequent studies, a targeted delivery system with a 5% density of FA-PGEM was used.
Scheme 2.

(A) Self-assembly of DOX-loaded FA-PGEM micelles. (B) FA-PGEM micelles for dual targeting of both tumor cells and M2-like TAMs. Folate receptors FOLR1 and FOLR2 are highly expressed on tumor cells and macrophages, respectively. FA-PGEM micellar carrier could enhance DOX delivery to both tumor cells and M2-like TAMs through folate-mediated active targeting effect.
Table 1.
Characterizations of blank and DOX-loaded PGEM and FA-GEM micelles.
| FA%a | Micelles | Size (d) (nm)b | Zeta potential (mV)b | DLE (%)c | DLC (%)d |
|---|---|---|---|---|---|
| 0% | PGEM | 11.24 ± 0.24 | ‒0.32 | 86.31 | 7.84 |
| PGEM/DOX | 8.8 ± 0.96 | ‒1.14 | |||
| 1% | FA-PGEM | 11 ± 0.33 | ‒3 | 80.13 | 7.28 |
| FA-PGEM/DOX | 10.28 ± 0.98 | ‒1.36 | |||
| 5% | FA-PGEM | 11.08 ± 0.13 | ‒3.96 | 82.43 | 7.49 |
| FA-PGEM/DOX | 9.17 ± 1.04 | ‒2.29 | |||
| 10% | FA-PGEM | 12.07 ± 1.01 | ‒0.85 | 95.6 | 8.69 |
| FA-PGEM/DOX | 10.17 ± 0.86 | ‒1.45 | |||
| 20% | FA-PGEM | 9.96 ± 1.7 | ‒1.54 | 96 | 8.72 |
| FA-PGEM/DOX | 11.17 ± 0.53 | ‒1.91 |
Percentage of FA-PGEM mixed with PGEM.
Measured by dynamic light scattering particle sizer.
DLE = Drug loading efficiency as measured by UV spectrophotometer.
DLE = Drug loading capacity of micelle calculated using DLC.
DOX could be readily loaded into the micelles with a DLC of 7%–9% (Table 1). Incorporation of DOX into these micelles did not have any impact on the size or zeta potential. Micelles were stable in PBS for up to two weeks at 4 °C without significant changes in sizes.
The morphology of blank and DOX-loaded FA-PGEM was visualized by TEM. Homogenously distributed micelles of spherical shape and uniform size were observed (Fig. 1A and B).
Figure 1.

In vitro biophysical characterizations of PGEM and FA-PGEM micelles. (A) TEM images of blank and DOX-loaded PGEM and FA-PGEM micelles (scale bar, 100 nm). (B) DLS analysis of micelles (carrier:drug = 10:1, w/w). (C) CMC measurement of FA-PGEM micelles by using nile red as a fluorescent probe. (D) DOX release profiles of DOX-loaded PGEM and FA-PGEM micelles with free DOX as the control. PBS was used as the release medium. Data are presented as means ± SEM (n = 3).
The CMC of the micelles was established using Nile red as a fluorescence probe and was calculated to be 0.037 mg/mL (Fig. 1C). The low value indicates that it could provide a good stability upon dilution in bloodstream post intravenous injection.
3.3. DOX release kinetics from micelles
The release profile of DOX-loaded micelles was tested in PBS (pH 7.4). As expected, free DOX easily diffused through the dialysis bags and, within 12 h, 80% DOX was found in the release medium. For our micelles, less than 43% DOX was released from micelles after 24 h (Fig. 1D). This slow release of DOX from the DOX-loaded formulations can be attributed to the strong π–π stacking, and hydrophobic interactions between the PGEM carrier and DOX.
3.4. FR expression in tumor cells and immune cells
The mRNA expression levels of FRα and FRβ in 4T1.2 and KB cell lines were evaluated via real-time RT-PCR. For both isoforms, KB cell line showed a significantly higher expression (Fig. 2), concurring with results from other research groups35. In addition, higher mRNA levels of FOLR1 (FRα) were observed in KB cell lines (Fig. 2A) compared to FOLR2 (FRβ, Fig. 2B).
Figure 2.

FOLR1 (A) and FOLR2 (B) mRNA expression levels in 4T1.2 and KB cells analyzed by quantitative real-time PCR. Relative mRNA levels were determined by the ΔΔCt method using GAPDH for internal cross-normalization. Data are presented as means ± SEM (n = 3). ∗∗∗∗P < 0.0001.
We have also analyzed the expression profiles of FOLR1 and FOLR2 in various cancer types including breast cancer (BRCA), non-small cell lung cancer (NSCLC), ovarian cancer (OV), pancreatic cancer (PAAD), and skin cancer (SKCM) from publicly available datasets (Supporting Information Fig. S2). FOLR1 was mainly expressed in tumor cells (Fig. S2A) while FOLR2 was largely expressed in tumor-infiltrating immune cells (Fig. S2B). Furthermore, among tumor-infiltrating immune cells, TAMs showed the highest levels of FOLR2 expression (Fig. S2C).
3.5. Cellular uptake study
To study the uptake of DOX into KB and 4T1.2 cells in vitro, the cultured cells were treated with different micellar formulations and their DOX uptake was assessed using a fluorescence microscope. While the nuclei stained with DAPI appear blue, DOX inherently emits a red fluorescence and this signal can be readily visualized by the fluorescence microscopy. Fig. 3A and B shows the fluorescence images of the cells 30 min post treatment. Free DOX was rapidly taken up by the KB cells as shown by the strong fluorescence signals inside KB cells (Fig. 3A). Incorporation of DOX into the ligand-free PGEM resulted in a significant decrease in the uptake of DOX (Fig. 3A). However, coupling with FA led to a significant recovery in the uptake of DOX, the fluorescence signals in FA-PGEM/DOX-treated KB cells were even higher than those in free DOX-treated cells (Fig. 3A). Upon addition of free FA to the media at a concentration of 1 mmol/L, a loss of fluorescence intensity was observed indicating an inhibition in the uptake of DOX (Fig. 3A). This competition between free FA and the FA-conjugated micelles supports the notion that the uptake is mediated via FR that is abundantly expressed on the surface of KB cancer cells.
Figure 3.

Cellular uptake of various DOX formulations. Fluorescence microscopic images of KB (A) and 4T1.2 (B) cells treated with various formulations, in the presence or absence of free folate at 37 °C for 30 min. DOX concentration was kept at 6 μL/mL and nuclei were stained with DAPI (scale bar = 50 μm); DOX uptake in KB (C) and 4T1.2 (D) cells was quantified by flow cytometry (MFI = mean fluorescence intensity, n = 3). ∗∗∗P < 0.001 ∗∗∗∗P < 0.0001.
The uptake of DOX into the KB cells was further quantified using flow cytometry as shown in Fig. 3C. The quantitative data are consistent with the imaging data from microscopic study. Fig. 3B and D shows the results of uptake study in 4T1.2 cells. Free DOX was efficiently taken up by 4T1.2 cells. Similar to what was observed in KB cells, incorporation of DOX into PGEM micelles led to a significant decrease in DOX uptake. However, no recovery in DOX uptake was seen in 4T1.2 cells (Fig. 3D) following conjugation with FA. Interestingly, addition of FA further decreased the uptake of FA-PGEM/DOX while it had no effect on the uptake of PGEM/DOX (data not shown), suggesting that the two formulations were taken up by 4T1.2 cells via different mechanisms despite their comparable efficiencies in cellular uptake.
3.6. In vitro cytotoxicity
Fig. 4A shows the cytotoxicity of free DOX and various micellar DOX formulations in KB cells (see Table 2). Free DOX exhibited cytotoxicity towards KB cells in a concentration-dependent manner. PGEM carrier alone also showed a low level of cytotoxicity and conjugation with FA led to a slight increase in cytotoxicity. Incorporation of DOX into ligand-free micelles (PGEM) resulted in a significant decrease in cytotoxicity. This is likely due to the fact DOX was effectively taken up by cells through a mechanism(s) that was more efficient compared to that for the uptake of PGEM/DOX. Nonetheless, the cytotoxicity of PGEM/DOX was significantly improved following conjugation with folate. Importantly, the enhanced cytotoxicity of FA-PGEM/DOX was drastically blocked by an excess amount of free folate, suggesting that the cytotoxicity of FA-PGEM/DOX towards KB cells is dependent on FR-mediated intracellular delivery of DOX and GEM.
Figure 4.

In vitro cytotoxicity of different formulations against (A) KB and (B) 4T1.2 cells. Cells were treated with DOX, PGEM, PGEM/DOX, FA-PGEM, FA-PGEM/DOX, and FA-PGEM/DOX in the presence of free folate for 30 min, followed by incubation in drug-free medium for another 24 h. Cytotoxicity was analyzed by MTT assay. Shown in right panels are the cell viabilities of KB and 4T1.2 cells after various treatments at an equivalent DOX dose of 20 μg/mL. Data are presented as means ± SEM (n = 3) ∗∗P < 0.01.
Table 2.
IC50 of various formulations in KB and 4T1.2 cells.
| IC50 (μg/mL) | DOX | PGEM | PGEM/DOX | FA-PGEM | FA-PGEM/DOX | FA-PGEM/Dox/Free FA |
|---|---|---|---|---|---|---|
| KB cells | 3.913 | 21.59 | 15.05 | 20.66 | 5.220 | 184.8 |
| 4T1.2 cells | 5.876 | 24.81 | 11.98 | 23.82 | 10.25 | 201.5 |
Fig. 4B shows the cytotoxicity of various formulations in 4T1.2 cells. Comparable levels of cytotoxicity were observed among free DOX, PGEM/DOX and FA-PGEM/DOX. Lack of difference between PGEM/DOX and FA-PGEM/DOX suggests minimal benefit of folate targeting in 4T1.2 cells, which was different from what was seen in KB cells (Fig. 4A). This is consistent with the earlier data that 4T1.2 cells expressed a low level of FR (Fig. 2) and that introduction of FA into PGEM resulted in minimal improvement in cellular uptake (Fig. 3B and D).
3.7. Biodistribution studies
The above data show that DOX can be effectively loaded to both PGEM and FA-PGEM and delivered to cultured tumor cells. However, active targeting mediated by FA was only demonstrated in KB cells but not 4T1.2 cells. We went on to further investigate the biodistribution of different formulations in tumors and other major organs in 4T1.2 tumor-bearing mice. DiR was used as a fluorescent probe in this study. In brief, free DiR, PGEM/DiR and FA-PGEM/DiR were injected i.v. at a DiR dosage of 0.1 mg/kg. Twenty-four hours post-injection, the whole body-imaging of tumor-bearing mice was obtained. Major organs from each group were then excised for ex vivo imaging. As shown in Fig. 5A‒D, free DiR was largely eliminated from the mice and little signals were seen in tumors and other normal organs and tissues except liver, spleen and lung. Delivery of DiR via PGEM led to obvious accumulation in tumor tissues (Fig. 5A and C). This is likely attributed to the extended circulation time of PEG-conjugated micelles and EPR effect. However, substantial amounts of signals were also observed in liver and spleen (Fig. 5B and D). This is likely due to the nonspecific uptake of NPs by RES. Introduction of FA as a targeting ligand led to further increases in the DiR signals in tumor tissues. Importantly, concomitant decreases in accumulations in liver and spleen were also observed in FA-targeted group. Similar results were seen at two other time points (12 and 36 h post injection, Supporting Information Fig. S3). In addition, the signals in FA-PGEM-treated tumors remained at peak level even at 36 h after the injection (Fig. S3).
Figure 5.

Tissue biodistribution of various formulations in 4T1.2 tumor-bearing mice. Whole body near-infrared images (A) and ex vivo fluorescence imaging (B) of tumors and other major organs 24 h following treatment with free DiR, PGEM/DiR and FA-PGEM/DiR, respectively. (C and D) Quantitative analysis (fluorescence intensity) of the data shown in panels A and B, respectively. (E) DiR accumulation in 4T1.2 tumor sections at 24 h after injection of free DiR, PGEM/DiR, and FA-PGEM/DiR, respectively. (F) DOX accumulation in 4T1.2 tumor sections at 24 h following i.v. administration of free DOX, PGEM/DOX and FA-PGEM/DOX, respectively (magnification, 40×; Scale bar = 50 μm; DOX dose, 5 mg/kg).
Consistent with the data of whole body and ex vivo imaging, widespread DiR signals were observed in the sections of tumors treated with PGEM/DiR (Fig. 5E). This is likely attributed to the efficient tumor extravasation and subsequent deep penetration of PGEM micelles due to their ultrasmall sizes. Similarly, decoration of PGEM micelles with folate led to further increases in the amount of DiR signals in tumor sections. Similar results were obtained when we examined the DOX fluorescence in the sections of tumors 24 h following treatment with DOX, PGEM/DOX and FA-PGEM/DOX (Fig. 5F).
3.8. In vivo antitumor efficacy
We continued our evaluation of in vivo antitumor activity of various DOX formulations in 4T1.2, an aggressive murine breast cancer model. Tumor volumes were measured to evaluate the efficacy of different groups and plotted in a growth curve as relative tumor volume (tumor volume at a given time point/initial tumor volume before 1st injection, Fig. 6A). Free DOX-treated mice showed significantly smaller tumors as compared to saline group (P = 0.02). Interestingly, compared to DOX, the blank carrier PGEM alone showed a higher therapeutic efficacy, which can be attributed to the EPR effect of the NPs. Conjugating FA to the carrier further improved the anti-tumor efficacy. Among all treatment groups, FA-PGEM/DOX showed the best antitumor activity with an IR of 85 ± 5.5% (Fig. 6A and B).
Figure 6.

In vivo evaluation of anti-tumor efficacy of various formulations. (A) Relative tumor growth curves of various formulations in 4T1.2 tumor model followed every three days for 16 days. DOX dose, 5 mg/kg. Polymer to drug ratio, 20:1 (mg/mg); (B) Inhibition rates of various treatments at the completion of study (Day 16); (C) Representative photomicrographs of H&E staining (upper) and TUNEL fluorescence staining (bottom) of tumor sections after various treatments (scale bar = 50 μm). The results are presented as means ± SEM (n = 5). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
H&E-stained tumor sections in the saline-treated group showed typical high density of tumor cells with large nuclei. However, the tumor sections showed bulk necrosis in the groups of FA-PGEM and FA-PGEM/DOX (Fig. 6C). Next, we analyzed the levels of apoptosis in various groups by TUNEL assay. As expected, tumors treated with FA-PGEM or FA-PGEM/DOX showed higher levels of fluorescence intensity, indicating more apoptotic cells. The superior therapeutic efficacy of FA-PGEM/DOX is likely attributed to the effective accumulation at tumor tissue due to the small sizes of the NPs, and the FA-mediated active targeting. The combination/synergistic effect between DOX and GEM shall play an important role.
3.9. Safety evaluations
No noticeable changes in mouse appearance and movement or significant body weight changes were observed throughout the in vivo study in any treatment group (Fig. 7A). In addition, no significant changes were found in the serum levels of ALT and AST, suggesting minimal impact on liver function (Fig. 7B). At the end of the study, mice were sacrificed and their major organs including the heart, lung, liver, spleen and kidney were harvested and fixed in paraffin. H&E staining of the paraffin-fixed tissues showed no obvious changes in histology, suggesting that our PGEM-based micelle system was well tolerated at the dose used (Fig. 7C).
Figure 7.

Safety/toxicity evaluations of various treatments. (A) Body weights of mice monitored every three days after various treatments; (B) Liver function assays after various treatments. Results were expressed as the mean ± SEM (n = 5); (C) Representative photomicrographs of H&E staining of heart, lung, liver, spleen and kidney (Magnification 40×, scale bar = 50 μm).
3.10. FRβ targeting in M2 macrophages
The above results clearly demonstrate the improved tumor-targeting of folate-decorated PGEM/DOX in 4T1.2 tumor-bearing mice despite that the cultured 4T1.2 cells showed low expression levels of both FOLR1 and FOLR2 with little, if any, benefits of FA-mediated active targeting in uptake and cytotoxicity. These data suggest that there might be other cell types in the tumor microenvironment susceptible to active targeting by FA-PGEM nanocarrier. Interestingly, 4T1.2 tumor tissues expressed significantly higher mRNA levels of FOLR2 compared to cultured 4T1.2 cells (Fig. 8A). TAMs, a subpopulation of macrophages present in the tumor microenvironment, have been reported to be associated with angiogenesis, tumor growth and metastasis, and immunosuppression36, 37, 38. M2 cells were also reported to overexpress FOLR2 (FRβ)39. We then went to examine if M2 macrophages could be targeted by our FR-targeted nanocarrier to elucidate a potential role of M2-targeting in the overall in vivo targeting efficiency of our nanocarrier. Macrophages were isolated from mouse spleen and polarized towards M2 phenotype using IL 4 and 13. A dominant M2 phenotype was confirmed by reduced expression of iNOS and significantly induced Arg1 and CD206 expression. We also confirmed an increase in FOLR2 expression in these polarized cells (Fig. 8B). The M1 polarization was induced by LPS and IFN-γ and was confirmed by increased expression of IL-1b (Supporting Information Fig. S4).
Figure 8.

FR-mediated targeting of FA-PGEM NPs to M2 cells in vitro and in vivo. (A) FOLR2 mRNA expression levels in cultured 4T1.2 cells and 4T1.2 tumor tissues; (B) mRNA expression levels of iNOS, Arginase, CD206, and FOLR2 in M2 polarized cells as compared to M0 cells; (C) Fluorescence microscopic images of M2 cells 30 min following treatment with DOX and DOX-loaded PGEM and FA-GEM micelle, in the presence or absence of free folate at 37 °C. DOX concentration was maintained at 6 μL/mL and nuclei were stained with DAPI (Magnification 40×, scale bar = 50 μm). (D) Quantitative analysis (mean fluorescence intensity) of the data in panel C. (E) Flow cytometric analysis of DOX uptake by M0, M1 or M2 cells 30 min following treatment with various DOX formulations. (F) Flow cytometric analysis of the percentage of M2 macrophages in the tumor tissues treated with various formulations (3 dosages). Results are expressed as the mean ± SEM (n = 3); ∗P < 0.05 ∗∗P < 0.01.
The cellular uptake of DOX was then analyzed on the M2 cells. Cells were treated with free DOX, PGEM/DOX, FA-PGEM/DOX, and FA-PGEM/DOX in the presence of 1 mmol/L free folic acid for 30 min. The DOX concentration was maintained at 6 μg/mL. As shown in Fig. 8C and D, strong fluorescence signals were found to be associated with M2 cells treated with FA-PGEM/DOX, much higher than those in cells treated with PGEM/DOX. In addition, free FA significantly decreased the uptake of DOX in M2 cells. These data suggest that upon extravasation of FA-PGEM/DOX into the tumor tissues through EPR effect, the FA-mediated active targeting of both tumor cells and M2 TAMs contributed synergistically to the enhanced accumulation at the tumor site and improved antitumor activity.
We also compared the cellular uptake of free DOX, PGEM/DOX, FA-PGEM/DOX by M0, M1 and M2 cells (Fig. 8E). Compared to M1 and M2 cells, free DOX showed higher levels of uptake by M0 cells. Both PGEM/DOX and FA-PGEM/DOX showed low levels of uptake by M1 cells. In contrast, FA-PGEM/DOX showed significantly more uptake by M0 and M2 compared to PGEM/DOX. The low-level uptake of free DOX by M2 cells suggest that M2 cells might be rendered resistant to cytotoxic effect of DOX. On the other hand, incorporation of DOX into FA-PGEM NPs may enhance the selective cytotoxicity towards M2 cells while sparing M1 cells. We then went to examine changes in the numbers of M2 cells in tumor tissues following treatments with various DOX formulations. It was interesting to note that the numbers of M2 cells in tumors were significantly increased following treatment with either PGEM or PGEM/DOX (Fig. 8F). The underlying mechanism is unclear at present. However, it has been reported that tumor cells may respond to chemotherapy by increasing production of various types of cytokines or chemokines40, which can promote M2 polarization and/or recruitment of M2 macrophages. Nonetheless, decoration of either PGEM or PGEM/DOX with FA led to significant reduction of the numbers of infiltrated M2 macrophages (Fig. 8F), demonstrating the potential of FR-targeted NPs in selectively eliminating M2 macrophages.
4. Conclusions
In summary, we modified a novel GEM-based pro-drug micellar carrier by conjugating FA as an active targeting ligand for the co-delivery of DOX and GEM. Micelles were ultrasmall (∼10 nm), and FA modification did not affect the size of the carrier. The FA-conjugated micelles significantly enhanced DOX uptake in KB cells in vitro, leading to increased cellular cytotoxicity. Such benefit of targeting was not observed in 4T1.2 in vitro. However, results from in vivo NIR imaging and therapeutic study revealed that introduction of FA to PGEM/DOX micelles led to enhanced accumulation in tumor tissues and significantly improved anti-tumor activity in a 4T1.2 cancer model. Mechanistically the improved performance of FA-decorated PGEM may be attributed to, at least partially, to a synergistic targeting of both tumor cells and TAMs.
Acknowledgments
This work was supported by National Institute of Health grants R01CA174305, R01CA219399, R01CA223788 (Song Li, USA), R21CA249649 (Jingjing Sun, USA) and a grant from Shear Family Foundation (Song Li, USA).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.09.024.
Contributor Information
Jingjing Sun, Email: jis84@pitt.edu.
Song Li, Email: sol4@pitt.edu.
Author contributions
Song Li and Jingjing Sun designed the research. Pearl Moharil and Zhuoya Wan carried out the experiments and performed data analysis. Apurva Pardeshi, Jiang Li, Haozhe Huang, Zhangyi Luo, Sanjay Rathod, Ziqian Zhang, Yuang Chen, Bei Zhang and Jingjing Sun participated part of the experiments. Christian A Fernandez provided experimental drugs and quality control. Pearl Moharil, Jingjing Sun, and Zhuoya Wan wrote the manuscript. Song Li and Jingjing Sun revised the manuscript. All of the authors have read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Chauhan V.P., Jain R.K. Strategies for advancing cancer nanomedicine. Nat Mater. 2013;12:958–962. doi: 10.1038/nmat3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cabral H., Matsumoto Y., Mizuno K., Chen Q., Murakami M., Kimura M., et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 3.Pluen A., Boucher Y., Ramanujan S., McKee T.D., Gohongi T., di Tomaso E., et al. Role of tumor–host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc Natl Acad Sci U S A. 2001;98:4628–4633. doi: 10.1073/pnas.081626898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park J., Choi Y., Chang H., Um W., Ryu J.H., Kwon I.C. Alliance with EPR effect: combined strategies to improve the EPR effect in the tumor microenvironment. Theranostics. 2019;9:8073. doi: 10.7150/thno.37198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hrkach J., Von Hoff D., Ali M.M., Andrianova E., Auer J., Campbell T., et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4 doi: 10.1126/scitranslmed.3003651. 128ra39. [DOI] [PubMed] [Google Scholar]
- 6.Ao L.J., Wang B., Liu P., Huang L., Yue C.X., Gao D.Y., et al. A folate-integrated magnetic polymer micelle for MRI and dual targeted drug delivery. Nanoscale. 2014;6:10710–10716. doi: 10.1039/c4nr02484b. [DOI] [PubMed] [Google Scholar]
- 7.Chen H.M., Ahn R.,V.J., Thompson D.H., O'Halloran T.V. Folate-mediated intracellular drug delivery increases the anticancer efficacy of nanoparticulate formulation of arsenic trioxide. Mol Cancer Ther. 2009;8:1955–1963. doi: 10.1158/1535-7163.MCT-09-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Q., Zheng J.W., Yuan X.Z., Wang J.F., Zhang L.J. Folic acid grafted and tertiary amino based pH-responsive pentablock polymeric micelles for targeting anticancer drug delivery. Mater Sci Eng C. 2018;82:1–9. doi: 10.1016/j.msec.2017.08.026. [DOI] [PubMed] [Google Scholar]
- 9.Lee E.S., Na K., Bae Y.H. Polymeric micelle for tumor pH and folate-mediated targeting. J Control Release. 2003;91:103–113. doi: 10.1016/s0168-3659(03)00239-6. [DOI] [PubMed] [Google Scholar]
- 10.Gao Z.G., Tian L., Hu J., Park I.S., Bae Y.H. Prevention of metastasis in a 4T1 murine breast cancer model by doxorubicin carried by folate conjugated pH sensitive polymeric micelles. J Control Release. 2011;152:84–89. doi: 10.1016/j.jconrel.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hami Z., Amini M., Ghazi-Khansari M., Rezayat S.M., Gilani K. Doxorubicin-conjugated PLA-PEG-folate based polymeric micelle for tumor-targeted delivery: synthesis and in vitro evaluation. J Pharm Sci. 2014;22:30. doi: 10.1186/2008-2231-22-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Low P.S., Kularatne S.A. Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol. 2009;13:256–262. doi: 10.1016/j.cbpa.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Kurahara H., Takao S., Kuwahata T., Nagai T., Ding Q., Maeda K., et al. Clinical significance of folate receptor β-expressing tumor-associated macrophages in pancreatic cancer. Ann Surg Oncol. 2012;19:2264–2271. doi: 10.1245/s10434-012-2263-0. [DOI] [PubMed] [Google Scholar]
- 14.Varghese B., Vlashi E., Xia W., Ayala Lopez W., Paulos C.M., Reddy J., et al. Folate receptor-β in activated macrophages: ligand binding and receptor recycling kinetics. Mol Pharm. 2014;11:3609–3616. doi: 10.1021/mp500348e. [DOI] [PubMed] [Google Scholar]
- 15.Tie Y., Zheng H., He Z.Y., Yang J.Y., Shao B., Liu L., et al. Targeting folate receptor β positive tumor-associated macrophages in lung cancer with a folate-modified liposomal complex. Signal Transduct Target Ther. 2020;5:1–15. doi: 10.1038/s41392-020-0115-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pauwels B., Korst A.E., Pattyn G.G., Lambrechts H.A., Vermeulen K., Lenjou M., Pooter C.M., et al. Cell cycle effect of gemcitabine and its role in the radiosensitizing mechanism in vitro. Int J Radiat Biol. 2003;57:1075–1083. doi: 10.1016/s0360-3016(03)01443-3. [DOI] [PubMed] [Google Scholar]
- 17.Maase H.V., Andersen L., Crino L., Weinknecht S., Dogliotti L. Weekly gemcitabine and cisplatin combination therapy in patients with transitional cell carcinoma of the urothelium: a phase II clinical trial. Ann Oncol. 1999;10:1461–1465. doi: 10.1023/a:1008331111654. [DOI] [PubMed] [Google Scholar]
- 18.Matsumura N., Nakamura Y., Kohjimoto Y., Nishizawa S., Kikkawa K., Iba A., et al. Overexpression of ribonucleotide reductase subunit M1 protein predicts shorter survival in metastatic bladder cancer patients treated with gemcitabine-containing combination chemotherapy. Int J Urol. 2017;24:230–235. doi: 10.1111/iju.13274. [DOI] [PubMed] [Google Scholar]
- 19.Serri C., Quagliariello V., Iaffaioli R.V., Fusco S., Botti G., Mayol L., et al. Combination therapy for the treatment of pancreatic cancer through hyaluronic acid-decorated nanoparticles loaded with quercetin and gemcitabine: a preliminary in vitro study. J Cell Physiol. 2019;234:4959–4969. doi: 10.1002/jcp.27297. [DOI] [PubMed] [Google Scholar]
- 20.Sun J.Z., Blaskovich M.A., Knowles D., Qian Y.M., Ohkanda J., Bailey A.D., et al. Antitumor efficacy of a novel class of non-thiol-containing peptidomimetic inhibitors of farnesyltransferase and geranylgeranyltransferase I. Cancer Res. 1999;59:4919–4926. [PubMed] [Google Scholar]
- 21.Sun J.J., Wan Z.Y., Xu J.N., Luo Z.Y., Ren P.F., Zhang B., et al. Tumor size-dependent abscopal effect of polydopamine-coated all-in-one nanoparticles for immunochemo-photothermal therapy of early-and late-stage metastatic cancer. Biomaterials. 2021;269:120629. doi: 10.1016/j.biomaterials.2020.120629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang C., Wang J.Q., Zhang X.D., Yu S.J., Wen D., Hu Q., et al. In situ formed reactive oxygen species-responsive scaffold with gemcitabine and checkpoint inhibitor for combination therapy. Sci Transl Med. 2018;10:3682. doi: 10.1126/scitranslmed.aan3682. [DOI] [PubMed] [Google Scholar]
- 23.Sun J.J., Sun L.Y., Li J.C., Xu J.N., Wan Z.Y., Ouyang Z.B., Liang L., et al. A multi-functional polymeric carrier for simultaneous positron emission tomography imaging and combination therapy. Acta Biomater. 2018;75:312–322. doi: 10.1016/j.actbio.2018.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Batrakova E.V.B.T., Vetro J.A., Kabanov A.V. Polymer micelles as drug carriers. Adv Drug Deliv Rev. 2006:57–93. [Google Scholar]
- 25.Sun J.J., Chen Y.C., Huang Y.X., Fu X.F., Zhang X.L., Zhao W.C., Wei Y., et al. A prodrug micellar carrier assembled from polymers with pendant farnesyl thiosalicylic acid moieties for improved delivery of paclitaxel. Acta Biomater. 2016;43:282–291. doi: 10.1016/j.actbio.2016.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pillai J.J., Thulasudasan A.K., Anto R.J., Devika N.C., Ashwanikumara N., Kumar G.S. Curcumin entrapped folic acid conjugated PLGA–PEG nanoparticles exhibit enhanced anticancer activity by site specific delivery. RSC Adv. 2015;5:25518–25524. [Google Scholar]
- 27.Wan Z.Y., Zheng R.H., Moharil P., Liu Y.Z., Chen J., Sun R.Z., et al. Polymeric micelles in cancer immunotherapy. Molecules. 2021;26:1220. doi: 10.3390/molecules26051220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biswas S., Kumari P., Lakhani P.M., Ghosh B. Recent advances in polymeric micelles for anti-cancer drug delivery. Eur J Pharm Sci. 2016;83:184–202. doi: 10.1016/j.ejps.2015.12.031. [DOI] [PubMed] [Google Scholar]
- 29.KwonGS, Croy S.R. Polymeric micelles for drug delivery. Curr Pharm Des. 2006;12:4669–4684. doi: 10.2174/138161206779026245. [DOI] [PubMed] [Google Scholar]
- 30.Sun J.J., Chen Y.C., Xu J.N., Song X.P., Wan Z.Y., Du Y.Q., et al. High loading of hydrophobic and hydrophilic agents via small immunostimulatory carrier for enhanced tumor penetration and combinational therapy. Theranostics. 2020;10:1136. doi: 10.7150/thno.38287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y.H., Sun J.J., Huang Y.X., Chen Y.C., Li J., Liang L., et al. Metformin-conjugated micellar system with intratumoral pH responsive de-shielding for co-delivery of doxorubicin and nucleic acid. Biochem Pharmacol. 2021:114453. doi: 10.1016/j.bcp.2021.114453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wan Z.Y., Sun J.J., Xu J.N., Moharil P., Chen J., Xu J., et al. Dual functional immunostimulatory polymeric prodrug carrier with pendent indoximod for enhanced cancer immunochemotherapy. Acta Biomater. 2019;90:300–313. doi: 10.1016/j.actbio.2019.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mia S., Warnecke A., Zhang X.M., Malmstrom V., Harris R.A. An optimized protocol for human M2 macrophages using M-CSF and IL-4/IL-10/TGF-beta yields a dominant immunosuppressive phenotype. Scand J Immunol. 2014;79:305–314. doi: 10.1111/sji.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis M.J., Tsang T.M., Qiu Y., Dayrit J.K., Freij J.B., Huffnagle G.B., et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. mBio. 2013;4 doi: 10.1128/mBio.00264-13. 00264-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heijden J.W., Oerlemans R., Dijkmans B.A., Qi H., Laken C.J., Lems W.F., et al. Folate receptor β as a potential delivery route for novel folate antagonists to macrophages in the synovial tissue of rheumatoid arthritis patients. Arthritis Rheumatol. 2009;60:12–21. doi: 10.1002/art.24219. [DOI] [PubMed] [Google Scholar]
- 36.Hu J.M., Liu K., Liu J.H., Jiang X.L., Zou H., Pang L.J., Wang X.L., Liu C.X., et al. CD163 as a marker of M2 macrophage, contribute to predict aggressiveness and prognosis of Kazakh esophageal squamous cell carcinoma. Oncotarget. 2017;8:21526–21538. doi: 10.18632/oncotarget.15630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen J.Y., Hu Y.W., Putt K.S., Singhal S., Han H.Y., Visscher D.W., Murphy L.M., et al. Assessment of folate receptor alpha and beta expression in selection of lung and pancreatic cancer patients for receptor targeted therapies. Oncotarget. 2018;9:4485–4495. doi: 10.18632/oncotarget.23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song X., Wan Z.Y., Chen T.J., Fu Y., Jiang K.J., Yi X., et al. Development of a multi-target peptide for potentiating chemotherapy by modulating tumor microenvironment. Biomaterials. 2016;108:44–56. doi: 10.1016/j.biomaterials.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Feng Y., Shen J., Streaker E.D., Lockwood M., Zhu Z., Low P.S., et al. A folate receptor beta-specific human monoclonal antibody recognizes activated macrophage of rheumatoid patients and mediates antibody-dependent cell-mediated cytotoxicity. Arthritis Res. 2011;13:59. doi: 10.1186/ar3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y.C., Sun J.J., Huang Y.X., Lu B.F., Li S. Improved cancer immunochemotherapy via optimal co-delivery of chemotherapeutic and immunomodulatory agents. Mol Pharm. 2018;15:5162–5173. doi: 10.1021/acs.molpharmaceut.8b00717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.