

Abstract
Amino‐functionalized P(V) derivatives providing both N‐ and O‐donor modes have attracted interest owing to their potential to form interesting coordination assemblies with applications such as biological drugs. Novel coordination modes of two‐ and four‐dentate tris (pyridin‐2‐yl)phosphoric triamide OP[NH‐2Py]3 as ([Co(II){[O][NH‐2Py]P(O)[Ph]}2(DMF)2], 1) and ([Cu(II)Cl{[NH‐2Py]2P(O)[N‐2Py]}].DMF, 2) have been synthesized and structurally studied. The metal center environment is distorted octahedral for 1 and distorted square pyramidal for 2. The crystal structure of a new complex of Cu(II) with a Cu[N]4[Cl]2 environment ([Cu(II)Cl2(Pyrazole)4], 3) is also investigated. An evaluation of the inhibitory effect against the coronavirus (Main Protease [MPro] of SARS‐CoV‐2) was carried out by a molecular docking study and illustrates that these compounds have a good interaction tendency with CoV‐2, where 1 has the best binding affinity with the biological target comparable with other SARS‐CoV‐2 drugs. Moreover, theoretical QTAIM and natural bond orbital (NBO) calculations are used to evaluate the metal‐oxygen/‐nitrogen bonds suggesting that they are mainly electrostatic in nature with a slight covalent contribution. A molecular packing analysis using Hirshfeld surface (HS) analysis shows that N—H … O (in 1 and 2) and N—H … Cl (in 3) hydrogen bonds are the dominant interactions that contribute to the crystal packing cohesion. The semi‐empirical PIXEL method indicates that the electrostatic and repulsion energy components in the structures of 1 and 2 and the dispersion and electrostatic components in that of 3 are the major contributors to the total lattice energy.
Keywords: amino‐functionalized P(V) coordination compound, coronavirus inhibitor, density functional theory (DFT), molecular docking, PIXEL calculation
Novel coordination modes of two‐ and four‐dentate tris (pyridin‐2‐yl)phosphoric triamide OP[NH‐2Py]3 are structurally studied.
An evaluation of the inhibitory effect against the coronavirus for the studied compounds by the molecular docking simulation illustrates a good interaction tendency with CoV‐2 comparable with other SARS‐CoV‐2 drugs.
The molecular packing features are analyzed by Hirshfeld surface and semi‐empirical PIXEL analyses along with an evaluation of the metal‐oxygen/−nitrogen bonds by theoretical QTAIM and NBO calculations suggesting a mainly electrostatic nature with a slight covalent contribution.
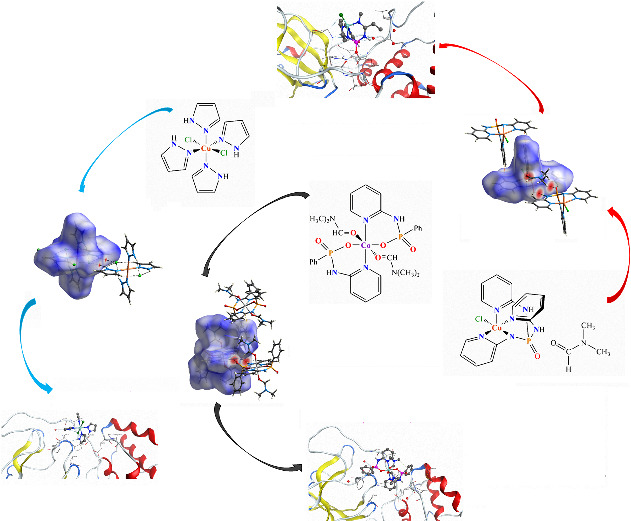
1. INTRODUCTION
Crystal engineering of supramolecular architectures is a multidisciplinary progress with focus on emerging technological applications, especially in the case of architectures with metal–ligand coordination bonds.[ 1 , 2 ] The design of such molecular architectures by the control of the various intermolecular interactions in the crystalline structure can provide favorable structures with applications in various fields.[ 3 , 4 ] Hence, the investigation of the structural features, molecular assemblies and the metal–ligand coordination bond strength has become of considerable interest in order to target desired material properties using a variety of metal complexes.[ 5 , 6 ]
Recently, novel various coordination assemblies featuring amino‐functionalized P(V) derivatives containing phosphorous imido bound (O═P―N) have received considerable attention.[ 7 , 8 , 9 ] Some of these phosphoramide compounds providing both N‐ and O‐donor functionalities can act as a class of certain flexible (half‐rigid) ligands. Among such ligands, pyridyl‐functionalized amino ligands bearing an (O)P[—NH‐Py] segment have an appropriate potential to construct exclusive examples of multi‐metallic architectures as metal–organic frameworks (MOFs) and porous coordination polymers.[ 10 , 11 , 12 ] From these amidophosphoryl‐based compounds interesting materials could be developed with technological applications in gas absorbers, dye‐sensitized solar cells and urease inhibitors.[ 13 , 14 , 15 ] Moreover, phosphoramide compounds and complexes synthesized from them have been tested as inhibitory factors such as anti‐cancer and anti‐viral agents, and prodrugs.[ 16 , 17 , 18 ] Recently, the anti‐coronavirus activity of some phosphoramides has been evaluated.[ 19 ] For this purpose, different strategies have been developed among which computational methods such as the molecular docking method are the fastest way to screen candidate drugs on their ligand‐protein interactions.[ 20 , 21 , 22 ]
For metal–ligand coordination compounds of PhP(O)[NH‐3,4Py]2 ligands (L 3,4) with both 3‐ and 4‐pyridinyl substitutions, a Cambridge Structural Database (CSD, version 5.40, with February 2019 update)[ 23 ] query gives 20 structures (18 for L 3 and 2 for L 4). These structures have N‐donor multi(2 or more)‐dentate L ligands coordinated to d‐block metal elements of Cu, Co, and Ni (with non‐coordinated P═O group) yielding polymers or MOFs. In the case of PhP(O)[NH‐2Py]2 (L 2) ligand bearing substitution 2‐pyridinyl, only one discrete chelate Cu (II) complex structure (RUYPUY)[ 24 ] with an N,O‐donor tridentate L 2 ligand (with coordinated P═O group) has been reported up to now. For metal complexes of P(O)[NH‐2,3,4Py]3 ligands (L′2,3,4), the CSD search yields 19 structures (4 for L′2 (Ag), 14 for L′3 (including different elements of Co, Ni, Cu, Zn, Cd, and Pd) and 1 for L′4 (Cu)) which construct polymers or MOFs. Recently, we reported the first discrete chelate complex of N,O‐donor tridentate L′2 ligand ([Co{(O)P[NH‐2Py]2[NH‐2PyH]}2]Cl3).[ 25 ]
In this article, in continuity of our previous work, we design and investigate the formation of novel coordination compounds using PhP(O)[NH‐2Py]2 and OP[NH‐2Py]3 ligands, which results in the following complexes: [Co(II){[O][NH‐2Py]P(O)[Ph]}2(DMF)2] (1) and [Cu(II)Cl{[NH‐2Py]2[N‐2Py]P(O)}].DMF (2). These complexes provide the first coordination compound with a [C]P(O)[N][O]―M segment building six‐membered chelate made of five different elements for 1 and the first P(O)[N]3‐based phosphoramide complex of copper with a Cu[N]4[Cl] environment in the case of 2. Moreover, they are the rare examples of discrete chelate phosphoramide complexes with non‐coordinated phosphoryl group. The structure of [Cu(II)Cl2(Pyrazole)4] (3) is also presented in this study for a comparison of the structural features and metal–ligand coordination bond strengths. The compounds are studied by single‐crystal X‐ray diffraction, FT‐IR spectroscopy and CHN elemental analysis. The X‐ray structural models are used to determine 3D Hirshfeld surfaces (HSs), 2D fingerprint plots, enrichment ratios (E) and interaction energies frameworks to evaluate the various intermolecular interactions in the crystal structures. From a theoretical point of view, density functional theory (DFT) and AIM calculations are performed to survey the coordination linkages and non‐covalent interactions in the studied structures. The PIXEL calculation method is also employed to analyze the lattice energies and intermolecular interaction energies in order to understand the packing principles in the studied complexes. A molecular docking study is performed to evaluate the inhibitory effect against the coronavirus (Main Protease [MPro] of SARS‐COV‐2).
2. EXPERIMENTAL
2.1. Materials and measurements
All chemicals materials and solvents were of analytical grade, obtained from commercial sources (Merck or Aldrich) and used without purification. Melting points were obtained using an Electrothermal IA‐9100 apparatus and are uncorrected. FT‐IR spectra were recorded on a Bruker ALPHA FT‐IR spectrometer with samples prepared as KBr pellets. Elemental analyses (C, H, and N) were performed using an Elementar Vario EL III elemental analyzer. Experiments of FESEM and EDX for the complexes 2 and 3 were performed by Zeiss Model Sigma 300‐HV which the obtained results can be seen in the supporting information.
2.2. Syntheses and crystallization
2.2.1. Preparation of [Co(II){[O][NH‐2Py]P(O)[Ph]}2(DMF)2] (1)
The phosphoramide ligand (O)P[Ph][NH‐2Py]2 was prepared according to a literature method of Srivastava et al.[ 24 ] FT‐ IR Data (KBr, , cm −1 ): 3113 (N—H), 3076, 2933, 2872, 1597, 1470, 1410, 1306, 1271, 1180 (P═O), 1155, 1122, 993, 962 (P—N), 810, 771, 756, 698, 644, 536, 496, 444. In order to obtain Co(II) complex, to a solution of (O)P[Ph][NH‐2Py]2 (0.64 mmol, 0.20 gm) in methanol (20 ml), a solution of CoCl2.6H2O (0.32 mmol, 0.04 gm) in methanol (15 ml) slowly was added, along with a few drops of DMF (N,N‐dimethylformamide) and the mixture was refluxed for 2 days. This reaction gave rise to the in situ generated ligand (O)P[O][Ph][NH‐2Py]− in the corresponding complex 1. Light blue single crystals were obtained from the reaction solution by slow evaporation of solvents at room temperature. Yield: 52% (112 mg); MP: 360°C; FT‐IR Data (KBr, , cm −1 ): 3130 (N—H), 3051, 2953, 2808, 1645, 1610, 1510, 1479, 1437, 1423, 1385, 1339, 1286, 1201 (P═O), 1159, 1136, 1105, 1072, 1001, 933 (P—N), 876, 789, 746, 698, 675, 634, 563, 546, 523, 424; Anal. Calcd. for C28H34CoN6O6P2: C, 46.26; H, 4.49; N, 12.57; found: C, 50.08; H, 5.10; N, 12.51.
2.2.2. Preparation of [Cu(II)Cl{[NH‐2Py]2[N‐2Py]P(O)}].DMF (2)
Tris (pyridin‐2‐yl)phosphoric triamide ligand (OP[NH‐2Py]3) was prepared according to a literature method of Tarahhomi et al.[ 25 ] For preparation of this complex, a solution of CuCl2.2H2O (0.61 mmol, 0.10 gm) in acetonitrile (15 ml) slowly was added to a solution of (O)P[NH‐2Py]3 (1.23 mmol, 0.40 gm) in methanol/DMF (20 ml) and the mixture was refluxed for 2 days. Green crystals of 2 suitable for X‐ray structural analysis were obtained by slow evaporation of solvents from the solution reaction at room temperature. Yield: 58% (177 mg); MP: 286–289°C; FT‐IR Data (KBr, , cm −1 ): 3186 (N—H), 3059 (N—H), 2908, 1663, 1605, 1508, 1477, 1460, 1321, 1277, 1190 (P═O), 1039, 997, 951, 928 (P—N), 793, 777, 663, 567, 519, 482, 424; Anal. Calcd. for C18H21ClCuN7O2P: C, 43.24; H, 3.94; N, 19.76; found: C, 43.47; H, 4.25; N, 19.71.
2.2.3. Preparation of [Cu(II)Cl2(Pyrazole)4] (3)
Light green single crystals of this complex were intrusively obtained from a reaction between a mixture of OPCl3 (1.00 mmol, 0.15 gm) and pyrazole (6.00 mmol, 0.41 gm) with CuCl2.2H2O (1.00 mmol, 0.17 gm) in methanol (20 ml) under reflux, followed by slow evaporation of solvents from the related solutions at room temperature. Yield: 46% (188 mg); MP: 229°C; FT‐IR Data (KBr, , cm −1 ): 3211 (N—H), 3113, 1512, 1477 (N‐C), 1404, 1360, 1350, 1269, 1254, 1169, 1130 (N‐C), 1074 (N‐N), 1049, 945, 910, 868, 781, 715, 615, 600; Anal. Calcd. for C12H16Cl2CuN8: C, 35.53; H, 3.65; N, 27.49; found: C, 35.43; H, 3.96; N, 27.55.
2.3. Crystal structure determination
For the structures 1 and 3, measurements were performed on a Gemini diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) and equipped with a Sapphire3 CCD detector. The data were corrected for absorption using redundant equivalent reflections with CrysAlisPro.[ 26 ] The charge‐flipping method implemented in the Superflip program[ 27 ] with standard algorithm parameters[ 28 ] was used for the structure solutions and the CRYSTALS program[ 29 ] was employed for the structure refinements. For structure 2, a single crystal X‐ray diffraction experiment was carried out at the XRD2 structural biology beamline, Syncrotrone Elettra Trieste. A wavelength λ of 0.70000 Å was selected using a dual crystal Si monochromator. The diffraction setup consisted of an Arinax MD2S high throughput diffractometer and a Pilatus 6M detector. The beam was defined using a 100‐μm aperture and further cleaned using a 200‐μm capillary, while sample cooling was performed using an open flow nitrogen cryostat at 100 K. A standard data collection was used, consisting of a 360° omega scan. Frame integration and scale and absorption corrections were done with XDS [ 30 ] and space group assignment was provided by Pointless from the CCP4 software suite,[ 31 ] as implemented in the XDS4 Elettra interface. The structure was solved using ShelxT and refined using ShelxL as implemented in the OLEX2 package.[ 32 ] For all structures, the H atoms were all located in a difference map, but those attached to carbon atoms were repositioned geometrically. The H atoms were initially refined with soft restraints on the bond lengths and angles to regularize their geometry (C―H and N―H and in the range 0.93–0.98 and 0.86–0.89 Å, respectively, O―H = 0.82 Å, and U iso[H] in the range 1.2–1.5 times U eq of the parent atom) after which the positions were refined with riding constraints.[ 33 ] PLATON [ 34 ] and Mercury [ 35 ] programs are utilized to draw the ORTEP and molecular packing diagrams.
2.4. Theoretical methods
2.4.1. DFT and PIXEL procedures
Quantum mechanical calculations based on DFT were carried out by using the GAUSSIAN09 package.[ 36 ] The B3LYP functional,[ 37 ] the LANL2DZ basis set for Cu and Co and the 6‐311++G(d,p) basis set for other atoms were used in all calculations.[ 38 ] Natural bond orbital (NBO) analysis[ 39 ] was applied to study the electronic aspects of the studied structures. The topological properties at the bond critical points (BCPs) were analyzed by the quantum theory of atoms in molecules (QTAIM)[ 40 ] procedure, using the AIMALL program.[ 41 ] The PIXEL calculations were performed by using the PIXEL program[ 42 ] using a density determined at the B3LYP/6‐31G(d,p) level. The PIXEL results were analyzed using Shishkin's energy vector models[ 43 , 44 ] and processed by using the processPIXEL program[ 45 ] and visualized using Mercury.[ 35 ]
2.4.2. Molecular docking procedure
The protein–ligand interactions between the complexes 1–3 and the target protein 6M03 (Protein Data Bank identifier), which is related to Main Protease (MPro) of SARS‐CoV‐2 were determined by molecular docking with Autodock Vina.[ 46 ] Water molecules, heteroatoms and ligands of the structure of 6M03 were removed using the Chimera and Discovery Studio software[ 47 ] and hydrogen atoms were placed automatically in the structure of this stripped version of 6M03. A grid box was set up with dimensions of 100, 100, and 126 Å in the x, y, and z directions, respectively, and a voxel width of 0.9 Å. For a better insight in the studied biological activity of the complexes 1–3, a molecular dynamics study of the 1–3 with the modified target protein 6M03 was performed on a time scale of 100 nanoseconds with the GROMACS software[ 48 ] and then the molecular docking procedure was again carried out in the active sites.
2.5. HS analysis
HS analysis[ 49 , 50 ] along with the graphical maps of three‐dimensional (3D) HSs and the associated two‐dimensional (2D) fingerprint plots (FPs) were generated by using the software package Crystal Explorer 17.5.[ 51 ] The Crystallographic Information File (CIF) was used as the input file to this software. In the HS plotted over d norm, the red and blue surfaces indicate contacts with distances shorter (close contact) and longer (distinct contact) than the sum of van der Waals radii, respectively, and the white color indicates distances equal to the sum of the van der Waals radii.[ 52 ] The shape‐index property of the surface is a tool to visualize the C―H … π and π … π stacking by the presence of the red π‐hole for former and the adjacent red and blue triangles for later.[ 53 ] Moreover, the enrichment ratio (E) is derived from the HS analysis to mark the high and low propensity to form contacts in crystals as an ratio larger (E > 1, called as favored) or lower (E < 1, dis‐favored) than unity, respectively.[ 54 ]
3. RESULTS AND DISCUSSION
3.1. Description of the structures
Details of crystal data, data collection and structure refinement are summarized in Table 1. Selected bond lengths and angles are listed in Table S1. The studied structures and formation reactions of 1–3 are shown in Schemes 1 and 2.
TABLE 1.
Crystal data and structure refinement for compounds 1–3
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| CCDC number | 2034829 | 2035415 | 2035414 |
| Chemical formula | C28H34CoN6O6P2 | C15H14ClCuN6OP.C3H7NO | C12H16Cl2CuN8 |
| M r (gr mol−1) | 671.48 | 497.38 | 406.78 |
| Temperature (K) | 293 | 100 | 293 |
| Crystal system, Space group | Triclinic, P | Monoclinic, P21/n | Monoclinic, C2/c |
| a, b, c (Å) | 9.3128 (4), 10.1772 (4), 16.7278 (7) | 14.207 (1), 9.416 (1), 16.743 (1) | 13.6437 (5), 9.1806 (2), 14.7232 (5) |
| α, β, γ (°) | 79.116 (4), 86.539 (4), 80.850 (4) | 90, 112.665 (8), 90 | 90, 116.779 (5), 90 |
| V (Å3) | 1536.35 (12) | 2066.8 (3) | 1646.40 (11) |
| Z | 2 | 4 | 4 |
| Radiation type | Mo Kα | Synchrotron, λ = 0.700 Å | Mo Kα |
| (mm−1) | 0.71 | 1.24 | 1.66 |
| F(000) | 698 | 1020 | 828 |
| Crystal size (mm) | 0.25 × 0.10 × 0.05 | 0.05 × 0.03 × 0.02 | 0.25 × 0.15 × 0.12 |
| Crystal color/habit | Light blue/Stick | Green/Prism | Light green/Prism |
| Diffractometer | Xcalibur, Sapphire3, Gemini | Huber 4‐circles Kappa Goniometer, Pilatus 6 M detector | Xcalibur, Sapphire3, Gemini |
| Theta range for data collection (°) | 2.1 to 29.1 | 2.5 to 29.7 | 2.8 to 29.0 |
| Absorption correction | Multi‐scan/CrysAlis PRO | Multi‐scan/DENZO/SCALEPACK | Multi‐scan/CrysAlis PRO |
| T min, T max | 0.959, 1.000 | 0.95, 0.98 | 0.915, 1.000 |
| No. of measured, independent and observed [I > 2.0σ(I)] reflections | 22,430, 7244, 6104 | 37,959, 5971, 5786 | 5905, 1943, 1780 |
| R int | 0.037 | 0.018 | 0.027 |
| (sin /λ)max (Å−1) | 0.684 | 0.708 | 0.682 |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.052, 0.135, 0.95 | 0.021, 0.060, 0.99 | 0.030, 0.076, 0.97 |
| No. of reflections | 7243 | 5971 | 1942 |
| No. of parameters | 391 | 280 | 114 |
| No. of restraints | 0 | 8 | 8 |
| H‐atom treatment | H‐atom parameters constrained | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement |
| Δρ max, Δρ min (e.Å−3) | 0.51, −0.69 | 0.45, −0.46 | 0.25, −0.45 |
SCHEME 1.
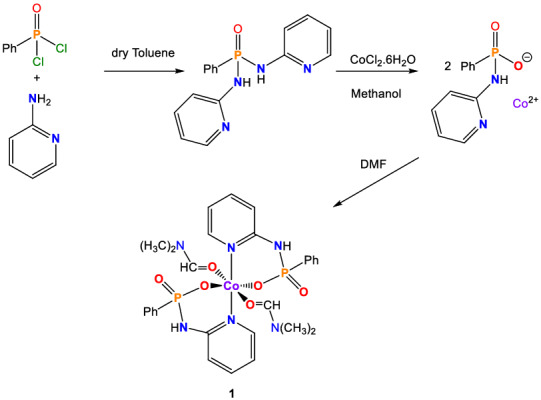
Structure and formation reaction of 1 reflecting the metal‐assisted P–N bond hydrolysis
SCHEME 2.
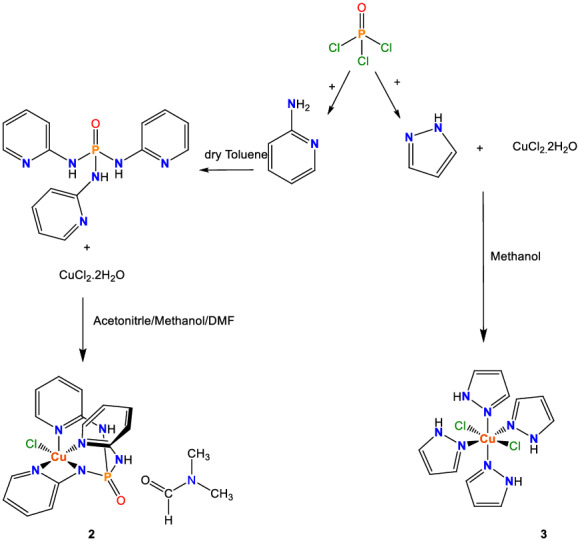
Structures and formation reactions of 2 and 3
3.1.1. Crystal structure of 1
The reaction of the ligand (O)P[Ph][NH‐2Py]2 with a solution of CoCl2.6H2O (in methanol along with a few drops of DMF) give the light blue crystals of complex 1, [Co(II){[O][NH‐2Py]P(O)[Ph]}2(DMF)2] derived from in situ generated pyridyl‐functionalized amidophosphate Ligand (O)P[O][Ph][NH‐2Py]−. The in situ formation of the amidophosphate ligand may be attributed to the low Lewis acidic nature of the Co (II) salt which can cause a metal‐assisted P–N bond hydrolysis at some point in the reaction (Scheme 1). Such metal‐assisted P–N bond hydrolysis has also been observed in some similar phosphoramide complexes, such as Zn (II) and Cu (II) complexes derived from in situ generated pyridyl‐functionalized bis (amido)phosphate Ligands.[ 10 , 13 ] It is noted that only one distorted octahedral Cu (II) complex of (O)P[Ph][NH‐2Py]2 ligand with an Cu(N)4(O)2 environment (RUYPUY: [Cu(II){(O)P[NH‐2Py]2[Ph]}2](NO3) has been reported up to now.[ 24 ]
The complex 1 (Figure 1) crystallizes in the triclinic space group P with Z′ = 0.5 and an asymmetric unit consisting of 2 × 0.5 [Co(II){[O][NH‐2Py]P(O)[Ph]}2(DMF)2]. For each symmetrically independent complex, the Co (II) cation is found in a hexa‐coordinate environment Co(N)2(O)4 adopting a distorted octahedral geometry featuring two pyridine nitrogens (NPy) and two O− contacts derived from the chelating N,O‐coordinations of the two amidophosphate ligands and two carbonyl oxygen atoms coordinations from two DMF molecules (Figure 1). Indeed, in this structure, each phosphoramide molecule acts as a bidentate N,O‐donor ligand providing two metal binding sites around the central cation Co (II) besides two other metal binding sites created by two DMF molecules led to the formation of the first discrete chelate phosphoramide complex with two bidentate [C]P(O)[N][O]‐based ligands.
FIGURE 1.
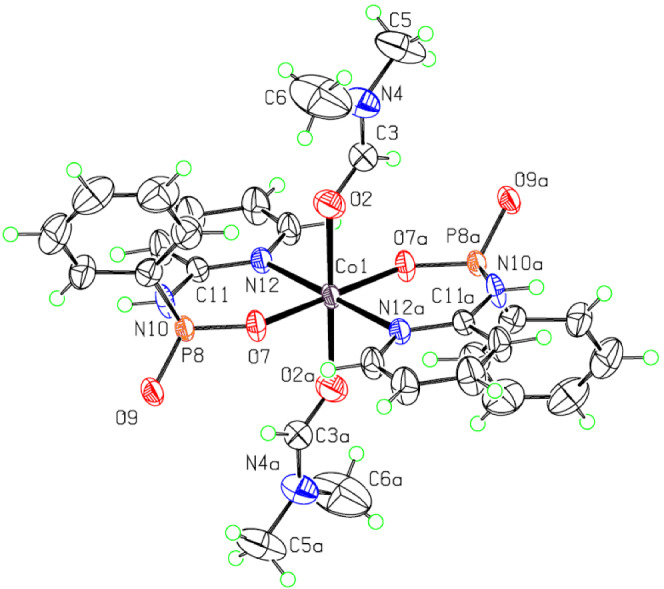
Displacement ellipsoid plot (50% probability level) and the atom numbering scheme for the molecule Co1 of 1. H atoms are drawn as circles of arbitrary radii and carbon atoms of aromatic rings are not numbered for more clarity
In each independent complex, the bonds around Co (II) center are the same in pairs due to the presence of an inversion center of symmetry. The Co–N and Co–O (of O═P) distances are 2.219 (2) (for Co1)/2.185 (2) (for Co23) Å and 1.993 (2)/2.023 (2) Å, respectively, and that of Co–ODMF measures 2.172 (2)/2.161 (2) Å. The trans N–Co–N and O–Co–O angles are about 180.0° and those for cis N–Co–O and O–Co–O angles vary between 86.29 (10) and 93.71 (10)° and 89.92 (9) to 90.33 (9)°, respectively, confirming a disordered geometry around the central Co (II) cation in both symmetrically independent complexes. Moreover, the P–N bonds are the same in each independent complex due to the presence of above‐mentioned symmetry at 1.679 (3) Å (for Co1)/1.689 (3) Å (for Co23) which are slightly longer than those in the free ligand (with CSD refcode: RUYQEJ[ 24 ]; P–N = 1.652 (1) and 1.657 (1) Å). The uncoordinated P═O bond lengths (1.486 (2) Å for both molecules Co1 and Co23) are also slightly longer than that in the free ligand (1.484 (1) Å).
An analysis of the H‐bond network indicates that the uncoordinated phosphoryl oxygen atoms are involved in the N―H … O hydrogen bonding (H‐bonding) interactions with protons of the pyridyl amino moieties building the eight‐membered motif rings. For each symmetrically independent complex, the net effect of these H‐bonding interactions connecting the dependent neighboring complex molecules along the b axis gives rise to a one‐dimensional linear arrangement. Thus, two symmetry‐independent 1D chains are formed. The packing diagram of this structure shows the formation of a 2D assembly parallel to the (50‐4) plane (Figure 2) in which the 1D chains are connected to each other via weak C—H … C and C—H … π (Table 2) interactions. Moreover, the above‐mentioned 2D network of 1 is stabilized via the participation of pyridine rings to some H … H interactions (Table 2) leading to the formation of a three‐dimensional (3D) supramolecular in this structure. Some intramolecular interactions between the CH group of aminopyridine rings of one amidophosphate ligand with the oxygen phosphate atom of another amidophosphate ligand (C13—H131 … O7 and C35—H351 … O29, Table 2) are also observed.
FIGURE 2.
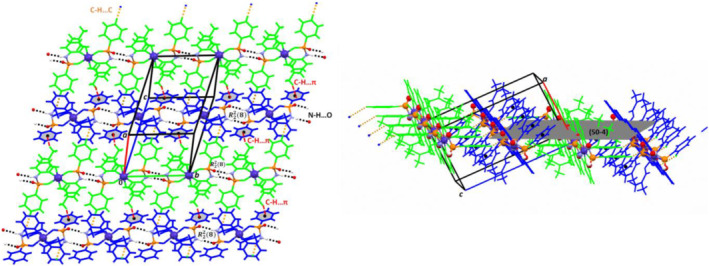
Views of two‐dimensional supramolecular structure of 1 formed parallel to the (50‐4) plane. Two symmetry‐independent 1D chains constructed along the b axis through the N—H … O (black dashed lines, Table 2) classical hydrogen bonds building the motif rings are connected together by the C—H … C (orange dashed lines, Table 2) and C—H … π (red dashed lines, Table 2) interactions. Different colors have been shown for symmetrically independent molecules. The “P(O)NH” segments involving the N—H … O classical hydrogen bonds are depicted as “ball and stick” and cobalt metal centers are shown as violet colored big balls
TABLE 2.
Hydrogen bond and intermolecular interaction geometries (Å, °) for compounds 1–3
| D—H···A | D—H | H···A | D···A* | ∠D—H···A |
|---|---|---|---|---|
| 1 | ||||
| N10—H101···O9i | 0.85 | 1.98 | 2.825 (6) | 172 |
| N32—H321···O31ii | 0.88 | 2.00 | 2.879 (6) | 179 |
| C20—H201···C33iii | 0.93 | 2.79 | 3.620 (6) | 149 |
| C16—H161···O9i | 0.92 | 2.65 | 3.370 (4) | 136 |
| C13—H131···O7iii | 0.93 | 2.26 | 2.932 (6) | 129 |
| C35—H351···O29iv | 0.93 | 2.32 | 2.959 (6) | 126 |
| C27—H273···π v | ― | 2.919 | ― | 149 |
| H351 … H361 | ― | ― | 2.319 | ― |
| H371 … H381 | ― | ― | 2.325 | ― |
| 2 | ||||
| N5—H51···O14i | 0.84 (1) | 1.94 (1) | 2.771 (2) | 177 (1) |
| N15—H151···O26 | 0.85 (1) | 1.99 (1) | 2.819 (2) | 165 (1) |
| C21—H211···O26 | 0.94 | 2.51 | 3.235 (2) | 134 |
| C22—H221···O14i | 0.93 | 2.60 | 3.321 (2) | 135 |
| C10—H101···Cl2ii | 0.94 | 2.86 | 3.509 (1) | 128 |
| C29—H291···N7iii | 0.95 | 2.69 | 3.481 (2) | 140 |
| C20—H201···C10iv | 0.95 | 2.78 | 3.636 (2) | 151 |
| C27—H271···C19v | 0.94 | 2.71 | 3.623 (2) | 164 |
| C25—H251···C22vi | 0.93 | 2.85 | 3.327 (1) | 114 |
| C30—H303···π i | ― | 2.974 | ― | 126 |
| π… π iv | ― | ― | 3.7837 (8) | ― |
| 3 | ||||
| N9—H91···Cl2i | 0.86 (2) | 2.83 (2) | 3.476 (2) | 133 (2) |
| C12—H121···Cl2ii | 0.92 | 2.942 | 3.638 (2) | 133.3 |
| C5—H51···π iii | ― | 2.814 | ― | 135 |
| C5—H51···π iii | ― | 3.401 | ― | 126 |
| π···π iv | ― | ― | 3.860 (2) | ― |
Note: Symmetry transformations used to generate equivalent atoms for 1: (i) −x, −y + 1, −z; (ii) –x + 1, −y + 3, −z + 1; (iii) −x, −y + 2, −z; (iv) –x + 1, −y + 2, −z + 1; (v) x, y, z + 1; for 2: (i) –x + 1, −y + 1, −z + 1; (ii) –x + 2, −y, −z + 1; (iii) x, y + 1, z; (iv) –x + 2, −y + 1, −z + 1; (v) x + , −y + , z + ; (vi) −x + , y − , −z + ; for 3: (i) x – , y + , z; (ii) –x + 1, y, −z + ; (iii) x – , y ‐ , z; (iv) –x + 1, −y + 1, −z + 1.*For π … π interactions d(D…A) = Cg … Cg and for C–H … π interactions d(H….A) = H … Cg and ∠D—H···A = ∠D—H···Cg, where Cg = centroid of the aromatic phenyl (for 1), pyridine (for 2) or pyrazole (for 3) rings.
3.1.2. Crystal structure of 2
The molecular structure of 2 is displayed in Figure 3. The complex 2 crystallizes in the monoclinic space group P21/n. The asymmetric unit consists of one molecular complex ([Cu(II)Cl{[NH‐2Py]2[N‐2Py]P(O)}], in which the charge balance is restored by one anionic phosphoramide ligand and one Cl− anion, and one DMF molecule. Indeed, in coordinated phosphoramide ligand [NH‐2Py]2[N‐2Py]P(O), one of NH units of 2‐aminopyridine moieties connected to phosphoryl group is disprotonated and coordinated to the metallic center and gives a anionic form (L −) of phosphoramide ligand. In this complex, the Cu (II) cation is found in a distorted five‐coordinate Cu(N)4(Cl) square‐pyramidal geometry with three Npyridyl contacts and one NP‐N contact, derived from the chelating four‐dentate N,N,N,N‐coordination of one phosphoramide ligand with uncoordinated phoshporyl group, and one Cl− coordinating anion. Based on a CSD query,[ 23 ] the complex 2 is the first example of a discrete chelate phosphoramide in which phosphoramide compound acts as a flexible four‐dentate ligand with uncoordinated phoshporyl group. Moreover, such structure of a phosphoramide complex with a penta‐coordinate environment M(N)4(Cl) has not been reported up to now. It should be noted that only some Ag(I) multi‐nuclear complexes of (O)P[NH‐2Py]3 ligand (CSD refcodes: VEFVAF, VEFVEJ, VEFVEJ01, and VEFVIN) have been reported up to now.[ 11 ]
FIGURE 3.
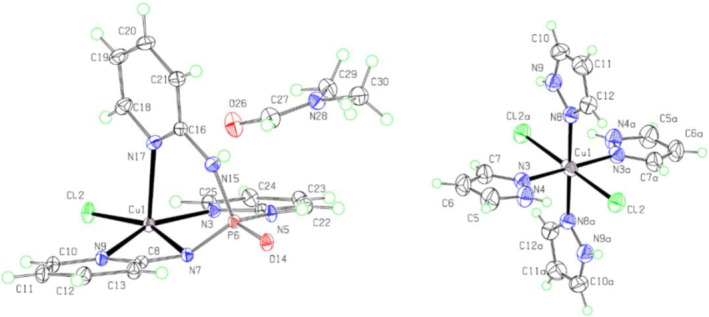
The molecular structures in the crystal of 2 (left) and 3 (right) with atom labeling. Displacement ellipsoids are drawn at the 50% probability level. H atoms are drawn as circles of arbitrary radii
In the structure of 2, the Cu–N distances range from 2.0391 (9) to 2.3371(9) Å, and of the Cu–Cl distance is 2.2363 (3) Å. The N–Cu–N and N–Cu–Cl angles in the square plane range from 66.03 (3) to 100.73 (3)° for the cis‐forms and are 158.68 (3) and 159.93 (3)° for the trans‐forms. The Naxial–Cu–N angles range from 90.34(3) to 97.93(3)° and is 102.15 (3)° for the Naxial–Cu–Cl angle. These angles values suggest a distorted square‐pyramidal geometry around the Cu (II) center, where the axial position is occupied by one pyridyl nitrogen atom whereas the equatorial square plane is defined by the other coordinated nitrogen atoms along with the Cl− anion. In this distorted geometry, the copper is not in the plane and is displaced slightly toward the axial pyridyl nitrogen by 0.202 Å. Moreover, the P–NH bonds (1.665 (1) and 1.671 (1) Å) are slightly longer than those in the free ligand (with CSD refcodes: LAFNAI and LAFNAI01; P–N = from 1.639 (2) to 1.653 (2) Å), whereas the P–N− bond (1.6061 (9) Å) is shorter. The uncoordinated P═O bond length (1.4861 (8) Å) is slightly longer than those in the free ligand (1.471 (2) and 1.473 (2) Å).
In the crystal structure of 2, the molecules are packed in a four‐connected motif composed of two phosphoramide complexes and two DMF molecules by the N—H … O classical hydrogen bond interactions (Table 2, Figure 4). In this packing, the uncoordinated phosphoryl group takes part in an N—H … O═P interaction connecting two complex molecules as a dimeric H‐bonded pattern building the motif ring. Two DMF molecules are adjoined to this dimeric pattern via the N—H … O═C hydrogen bonds between the NH units of phosphoramide complexes and the carbonyl groups of DMF molecules. Moreover, the crystal lattice of 2 exhibits weak C―H … O═P, C―H … Cl, C―H … N, C―H … π and π … π intermolecular interactions (Table 2) that stabilize the crystal packing by connecting the quoted H‐bonded four‐connected motifs together forming a 3D network (Figure 4). In this packing feature, the C―H … π interaction is formed between the methyl group of the DMF molecule with the centroid Cg of the pyridine ring. An additional π … π interaction is found between the centroids Cg of the pyridine rings.
FIGURE 4.
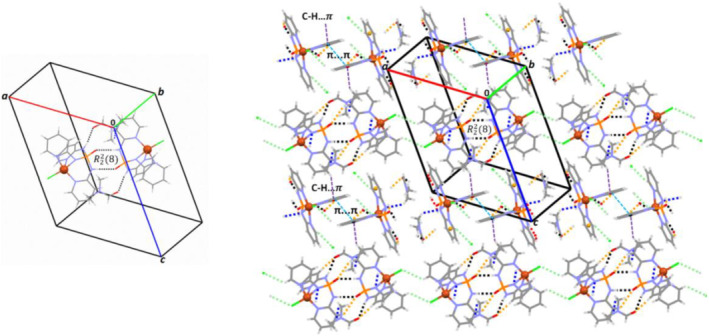
Left: Presentation of classical hydrogen bond interactions (N―H … O, black dashed lines, Table 2) involved in four‐membered hydrogen‐bonded motif of 2; Right: A view of the 3D network formed by various intermolecular interactions in 2 (C―H … O (orange dashed lines), C―H … Cl (light green), C―H … N (dark blue), C―H … π (purple) and π … π (light blue), Table 2). Copper centers are shown as big balls
3.1.3. Crystal structure of 3
This structure crystallizes in the monoclinic space group C2/c. A view of the structure of 3 is shown in Figure 3. The asymmetric unit is composed of one‐half of the [Cu(II)Cl2(Pyrazole)4] complex. The Cu (II) atom is hexa‐coordinated by four pyrazole molecules and two Cl− anions adopting a distorted octahedral geometry. This distorted geometry is confirmed by the different Cu–N (2.0032 (14) and 2.0207 (15) Å) and Cu–Cl (2.8372 (5) Å) distances and the bond angles around Cu (II) center with the values 88.49 (6)° and 91.51 (6)° for cis N–Cu–N angles and from 87.80 (5)° to 92.20 (5)° for cis N–Cu–Cl and about 180.0° for trans N–Cu–N and Cl–Cu–Cl angles (Table S1).
The H‐bond‐assisted supramolecular assembly by C—H … Cl and N—H … Cl interactions in 3 leads to a 3D network. A view of the 2 × 2 × 2 unit cell packing diagram is shown in Figure 5. The presence of pyrazole rings in this network results in π interactions (C—H … π and π … π, Table 2) stabilizing further the 3D assembly.
FIGURE 5.
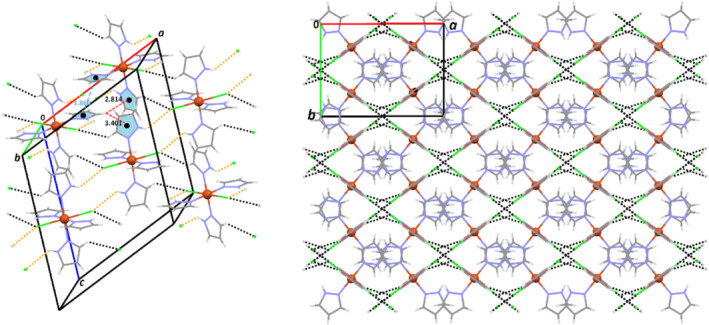
Left: A selected molecule of 3 and its hydrogen bond interaction connectivities with neighboring molecules as dashed lines (black: C—H … Cl and orang: N—H … Cl, Table 2) has been shown. The C—H … π and π … π interactions (Table 2) between pyrazole rings are displayed as red and blue dash lines, respectively, in this view; Right: the 2 × 2 × 2 unit cell packing diagram of 3 viewed along the c axis has been presented showing the 3D network formed by the quoted intermolecular interactions (black dashed lines)
3.2. Theoretical results and discussion
The molecular structures 1–3 were fully optimized by DFT method at the SDD (6–311++G(d,p)/LANL2DZ) basis set. Input files were constructed from the X‐ray crystal structures for the chemical calculations. The selected optimized geometrical parameters of complexes 1–3 are summarized in Table S2. From the results collected in Table S2, the optimized parameters for all compounds support the experimental data from X‐ray crystallography results for all compounds. The highest differences between the experimental (X‐ray crystallography) and calculated parameters for bond lengths and angles, respectively, are found about 0.2 Å (for Co―O) in 1 and about 5° (for N–Cu–N) in 2. NBO and QTAIM analyses were performed for the fully optimized structures in order to evaluate the nature and strength of the metal‐oxygen/‐nitrogen/‐chlorine bonds.
3.2.1. Charge density analysis
The electron density and its Laplacian (ρ(r) and ∇2 ρ(r)), and the energetic parameters at the BCP obtained using the QTAIM theory can be used to evaluate the strength and characteristic of a bond. The energetic parameters include the terms V(r), G(r), and H(r), that is, the potential, kinetic, and total energy densities, respectively. Large values of ρ(r) along with values of ∇2 ρ(r) < 0, H(r) < 0 and > 2 are calculated for shared interactions (covalent bonds). In contrast, small ρ(r) values with ∇2 ρ(r) > 0, H(r) > 0 and < 1 refer to closed‐shell, non‐covalent interactions such as ionic, hydrogen bond and van der waals (vdw) interactions.[ 55 , 56 ] We employ here the QTAIM method as a more appropriate index to characterize better the metal‐oxygen/nitrogen contacts in the studied complexes. Molecular graphs and distribution of BCPs for the structures 1–3 are presented in Figure S1 and the AIM parameters are collected in Table 3.
TABLE 3.
Distances (Å), topological properties ρ(r) and ∇2 ρ(r) (a.u.), and energetic parameters (G(r), V(r) and H(r) in a.u.) for BCPs of the metal‐oxygen/nitrogen contacts in the complexes 1–3
| Compound | Distance | ρ(r)/∇2 ρ(r) |
|
G(r) | H(r) | |
|---|---|---|---|---|---|---|
| 1 | ||||||
| Co … OPT | 1.927 | 0.079/0.549 | 0.149 | 0.143 | −0.006 | |
| Co … ODMF | 2.332 | 0.031/0.188 | 0.038 | 0.043 | 0.004 | |
| Co … NPy | 2.087 | 0.065/0.364 | 0.101 | 0.096 | −0.005 | |
| 2 | ||||||
| Cu … NPy | 2.163[ a ] | 0.079, 0.075, 0.037/0.386, 0.361, 0.144 | 0.111, 0.104, 0.039 | 0.103, 0.097, 0.038 | −0.007, −0.007, −0.002 | |
| Cu … N | 2.055 | 0.076/0.334 | 0.101 | 0.092 | −0.009 | |
| Cu … Cl | 2.323 | 0.059/0.199 | 0.058 | 0.054 | −0.004 | |
| 3 | ||||||
| Cu … N | 2.029 | 0.077/0.397 | 0.112 | 0.106 | −0.007 | |
| Cu … Cl | 2.874 | 0.023/0.043 | 0.019 | 0.015 | −0.004 | |
Average values are reported.
For all metal‐oxygen/nitrogen (M—O/N) contacts in the three studied complexes 1–3, exception for Co … ODMF contact in 1, the values of ρ(r) at the BCPs are in the range 0.065 to 0.079 a.u., in where the positive values are found for the electron density Laplacian (∇2 ρ(r) > 0) showing a depletion of electron density in the atomic basin (Table 3). These data suggest a closed‐shell (non‐covalent) nature for the metal‐oxygen/nitrogen contacts. However, small negative values for the total energy density (H(r) < 0 in the range from −0.002 to −0.009 a.u.) and with magnitude of 1 < < 2 for the ratio of potential and kinetic energy densities are found which may suggest a rather strong bond with mainly ionic character and a partial covalent nature for these contacts. Similar results are observed for the metal‐chlorine contacts with the ρ(r)/∇2 ρ(r) values of 0.059/0.199 and 0.023/0.043 a.u..
Moreover, the close values for both M—OPT and M—N bonds justify the close competition of negative oxygen (of phosphoryl group) and neutral nitrogen (of pyridine rings) atoms to coordinate to metal center, and the ability of such pyridyl‐functionalized amidophosphoryl ligands to provide the multiple metal binding sites presenting the multi‐dentate N,O‐donor ligands. In structure 1, a comparison of M—O/N bonds shows higher ρ(r)/∇2 ρ(r) values (Table 3) at the Co—OPT BCP than those at the Co—NPy BCP, although the and H(r) terms include almost similar values for the these contacts. This can be attributed the better capability of the P—O group relative to the pyridyl nitrogen to connect to the metal center in a complexation process. Interestingly, in the structure 2 with a non‐coordinated P═O group, the Cu—NPy BCP has higher ρ(r)/∇2 ρ(r) values than those at the Co—NPy BCP in 1, whereas these ρ(r)/∇2 ρ(r) values are almost the same as those at the Co—OPT BCP of 1. In the case of the Co … ODMF contact in 1, the terms ∇2 ρ(r) > 0, H(r) > 0 and < 1 confirm a complete non‐covalent character, where the smaller values of ρ(r) and ∇2 ρ(r) for this contact relative to the ones in other M—O/N contacts also support the weaker strength for the Co … ODMF contact.
3.2.2. NBO analysis
The electronic and energetic parameters such as natural charge, bond order (BO) and electron delocalization energy (E 2) calculated by NBO analysis[ 57 ] are presented in Table 4 and used to evaluate the M—O/N contacts in the studied complexes 1–3.
TABLE 4.
Results of NBO analysis (natural charge (Q), bond order (BO) and delocalization energy (E 2)) determined by DFT method at the B3LYP/SDD level for 1–3
| Bond | BO | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| M―OPT | 0.420 | ― | ― |
| M―ODMF | 0.187 | ― | ― |
| M―N[ a ] | 0.376 |
0.297/0.281/0.284(NN‐Py) 0.187(Nax) |
0.298 |
| M―Cl | ― |
0.582 ― |
0.229 |
| Atom/Q | 1 | 2 | 3 |
|---|---|---|---|
| M (metal) | 0.810 | 0.814 | 0.851 |
| OPT | −1.039 | — | — |
| OMDF | −0.653 | — | — |
| N[ a ] | −0.524 |
−0.576/−0.554/−0.585(Naxial) −1.044(NN‐Py [ b ]) |
−0.356 |
| Cl | — | −0.553 | −0.724 |
| Donor–acceptor interactions | |||
|---|---|---|---|
| Compound | Donor | Acceptor | E 2(kcal/mol) |
| 1 | LP OPT | LP[ a ] Co | 80.83 |
| LP ODMF | LP[ a ] Co | 28.60 | |
| LP N | LP[ a ] Co | 49.94 | |
| 2 | |||
| LP Neq | LP[ a ] Cu | 40.53/41.06(NN‐Py)/36.77 | |
| LP Nax | LP[ a ] Cu | 23.47 | |
| LP Cl | LP[ a ] Cu | 87.21 | |
| 3 | |||
| LP N | LP[ a ] Cu | 41.66/41.72 | |
| LP Cl | LP[ a ] Cu | 29.01/29.03 | |
N is nitrogen atom of pyridine (for 1 and 2) or pyrazole (for 3) ring coordinated to metal.
NN‐Py is negative nitrogen (N−) of (O)P(N−—Py) segment of PT ligand coordinated to metal for 2.
As is seen in Table 4, the higher values of BO for M—O/N bonds are observed for complex 1 suggesting slightly stronger contacts in this complex compared to 2 and 3 which is in agreement with the QTAIM results. A comparison of BO of M—OPT and M—NPy bonds in 1 shows a higher BO for M—OPT in line with the better capability of the P—O group compared to the pyridyl nitrogen to coordinate to the metal center. The more electronegative nitrogen atoms with higher negative natural charges attached to the metal center are found for 1 and 2 (with the almost same positive charge for metal) compared to 3. However, the positive charge of Cu atom in 3 (0.851) is larger than the positive charge of Co in 1 (0.810) and Cu in 2 (0.814).
The values of the electron delocalization energies (E 2, kcal mol−1) of LP O/N → LP* M in the studied complexes presented in Table 4 also display a similar trend, where the higher values of E 2 are obtained for 1 compared to those in 2 and 3. Indeed, when an oxygen/nitrogen atom of the amidophosphoryl ligand is attached to metal center, a transfer of the electron density from the donor orbital of oxygen/nitrogen lone pair LP O/N into the acceptor orbital of lone pair antibonding of metal LP* M occurs. Such a calculation of second‐order perturbation energies for electronic delocalization can be a suitable criterion to estimate of the strengths of the M—O/N bonds. Therefore, the higher values of these energies (E 2) confirm the slightly stronger M—O/N bonds in complex 1 compared to those in complexes 2 and 3. In this way, the six‐coordinated complex 1 (produced by the amidophosphate ligand) are stabilized at energies around 81/50 kcal mol−1 versus the stabilization of the five‐coordinate complex 2 (from phosphoric triamide ligand) at energies about 41/24 kcal mol−1 and the six‐coordinate complex 3 (without phosphoramide ligand) by about 42 kcal mol−1. However, it should be noted that the M—O/N bond strengths are also influenced by different coordination modes and metal centers.
In the case of the Co … ODMF contact in 1, the lower values of the natural charges, bond order (BO) and electron delocalization energy (E 2) compared to other M—O/N bonds in the studied complexes confirm a considerable weaker strength for this bond.
3.2.3. PIXEL analysis
The lattice energies and intermolecular interaction energies are analyzed by help of the PIXEL calculation method[ 58 ] in order to understand the packing principles in complexes 1–3. This method reveals the inherent character of an intermolecular interaction by using the partition of the cohesive energy between two molecules into coulombic (E coul), polarization (E pol), dispersion (E disp), and repulsion (E rep) terms based on the quantum chemical electron density.[ 42 ] The PIXEL calculations can be used to rank the strengths of supramolecular interactions, and also to identify intermolecular interactions that are associated with repulsion, but nevertheless recognized as binding, that is, “antagonist units”, or interactions that are characterized by negligible attractive or repulsive forces, that is, “neutral units.”[ 59 ]
The lattice energies of the studied complexes 1–3 calculated by the PIXEL method are presented in Table 5. Energy partitioning shows that the crystal package of 1 has the lowest negative energy (−300.2 kJ mol−1) indicating a higher stability for this complex compared to 2 and 3. As discussed above, similar results have been obtained by DFT calculations showing that complex 1 has slightly stronger contacts and higher values of E 2 of LP O/N → LP* M compared to 2 and 3. The lattice energy absolute values slightly reduce in the case of 3 and the lower value is found for 1. This result may be attributed to the polymeric cohesions of 1 constructed by the classical N–H … O═P hydrogen bond interaction, while the crystal packing of 2 is restricted to a four‐membered hydrogen‐bonded motif of classical N–H … O hydrogen bond interactions and in the case of 3, the crystal packing is devoid of any classical interaction. Interestingly, the 3D cohesion of 3 constructed by the weak N/C–H … Cl interactions with a higher lattice energy displays a higher stability than 2 as was mentioned before. For all complexes, the major contributions toward the lattice stabilization come from the coulombic and dispersion components. The remaining contribution is provided by the polarization energy.
TABLE 5.
Lattice energies of compounds 1–3 partitioned into different energy components
| Compound | E coul (kJ mol−1) | E pol (kJ mol−1) | E disp (kJ mol−1) | E rep (kJ mol−1) | E tot (kJ mol−1) |
|---|---|---|---|---|---|
| 1 | −188.4 | −76.8 | −281.7 | 246.6 | −300.2 |
| 2 | −102.6 | −42.3 | −135.9 | 135.3 | −145.4 |
| 3 | −101.5 | −40.0 | −213.6 | 128.7 | −226.4 |
In order to further evaluate the packing modes, the different intermolecular interactions in 1–3 along with their interaction energies partitioned into the various energy components are tabuled in Table 6. The highest interaction energy is found for the linked molecules by the hydrogen bond interactions formed by the favorite donor and acceptor units N–H and P═O, i.e. the classical hydrogen bond N–H … O═P in 1 and 2. In the case of the structure 3 which does not have a phosphoryl group, the non‐classical hydrogen bond N–H … Cl constructed via the favorite donor unit N–H shows the highest interaction energy. These molecular connections in 1 and 2 are primarily stabilized by electrostatic and repulsion components found as well in analogous compounds,[ 60 , 61 ] while in the case of 3, dispersion and electrostatic components dominate. Interestingly, in the case of weak non‐classical interactions for all three complexes, the calculated interaction energies show the preference of the dispersion component compared to the electrostatic one.
TABLE 6.
List of intermolecular interaction energies (kJ mol−1) for short contacts of 1–3
| Molecular Pairs | Interactions | Symmetry Code | X … A (Å) | D─X … A (°) | Centroid … Centroid Distance (Å) | E coul | E pol | E disp | E rep | E tot |
|---|---|---|---|---|---|---|---|---|---|---|
| Compound 1 | ||||||||||
| A … A | N10—H101···O9 | x, y – 1, z | 2.825 | 172 | 10.177 | −129.9 | −54.0 | −73.8 | 139.9 | −117.8 |
| A … A | C16—H161···O9 | x – 1, y, z | 3.370 | 136 | 9.313 | −18.7 | −10.4 | −48.6 | 32.1 | −45.6 |
| A … B | C27—H273···π | x, y, z – 1 | 2.919 | 149 | 9.324 | −22.0 | −11.1 | −73.8 | 40.9 | −66.0 |
| A … B | C20—H201···C33 | x – 1, y, z – 1 | 3.620 | 149 | 9.815 | −18.2 | −8.1 | −53.8 | 28.8 | −51.3 |
| B … B | N32—H321···O31 | x, y – 1, z | 2.879 | 179 | 10.177 | −117.3 | −47.0 | −66.7 | 113.5 | −117.5 |
| B … B | H351···H361 | x – 1, y, z | 2.319 | ― | 9.313 | −23.4 | −14.5 | −47.8 | 33.9 | −51.9 |
| Compound 2 | ||||||||||
| A … A | N5—H51···O14 | –x + 1, −y + 1, −z + 1 | 2.771 | 177 | 8.167 | −134.9 | −56.1 | −39.1 | 132.9 | −97.2 |
| A … A | C10—H101···Cl2 | –x + 2, −y, −z + 1 | 3.509 | 128 | 9.328 | −26.7 | −12.0 | −30.3 | 29.4 | −39.7 |
| A … A | C20—H201···C10 | –x + 2, −y + 1, −z + 1 | 3.636 | 151 | 7.625 | −15.0 | −6.8 | −47.9 | 34.0 | −35.7 |
| A … A | C25—H251···C22 | –x + , y – , −z + | 3.327 | 114 | 8.683 | −14.4 | −8.6 | −39.9 | 29.4 | −33.4 |
| A … A | C18···C20 (π … π) | x, y – 1, z | 3.784 | ― | 9.416 | −5.1 | −2.3 | −9.4 | 2.2 | −14.6 |
| A … B | N15—H151···O26 | x, y, z | 2.819 | 165 | 6.482 | −47.0 | −20.8 | −22.4 | 53.3 | −36.9 |
| A … B | C21—H211···O26 | –x + 1, −y + 1, −z + 1 | 3.235 | 134 | 5.658 | −13.1 | −4.0 | −25.7 | 16.2 | −26.7 |
| A … B | C29—H291···N7 | x, y – 1, z | 3.481 | 140 | 5.248 | −13.6 | −5.2 | −27.4 | 21.6 | −24.6 |
| A … B | C27—H271···C19 | x + , −y + , z + | 3.623 | 164 | 5.922 | −8.4 | −4.2 | −22.3 | 19.4 | −15.5 |
| A … B | C30—H303···π | –x + , y – , −z + | 2.974 | 126 | 8.449 | −8.0 | −3.4 | −7.2 | 5.6 | −13.0 |
| Compound 3 | ||||||||||
| A … A | N9—H91···Cl2 | x – , y + , z | 3.476 | 133 | 8.222 | −38.2 | −16.3 | −28.4 | 22.8 | −60.1 |
| A … A | C12—H121···Cl2 | x – , −y + , z – | 3.638 | 133 | 7.448 | −13.6 | −13.6 | −49.4 | 29.4 | −47.3 |
| A … A | C5—H51···π | x – , y – , z | 2.814 | 135 | 8.222 | −9.7 | −6.3 | −49.0 | 31.7 | −33.3 |
| A … A | C5—H51···π | x, y − 1, z | 3.401 | 126 | 9.181 | −18.5 | −7.2 | −19.9 | 13.8 | −31.8 |
| A … A | π···π | x, −y + 2, z – | 3.860 | ― | 8.675 | −10.3 | −4.6 | −30.6 | 15.5 | −30.0 |
Moreover, a visual inspection of the results is given in Figures 6, S2, and S3 using energy‐vector diagrams.[ 43 , 44 ] The energy‐vector diagrams indicate that for 1 and 2, the formation of the framework along the b (for 1) and a (for 2) axis is mostly directed by the component of electrostatic nature, due to the stabilization of the adjacent complexes linked as the N―H … O═P hydrogen bonded chains (for 1)/dimers (for 2). In this direction, the dispersion and polarization components have lower contributions. As is seen in Figure S3, for 3, in the different directions, the dispersion component provides the more contribution in the energy frameworks compared to the other components and the electrostatic takes the second status, where the formation of the 3D framework in this structure is provided by the weak N/C―H … Cl interactions. Finally, from the collected results of the PIXEL calculations, a dominant electrostatic nature is assigned to the classical hydrogen bond interactions N–H … O in 1 and 2, while the non‐classical N/C―H … Cl hydrogen bond interactions in 3 reveal both electrostatic and dispersion components.
FIGURE 6.
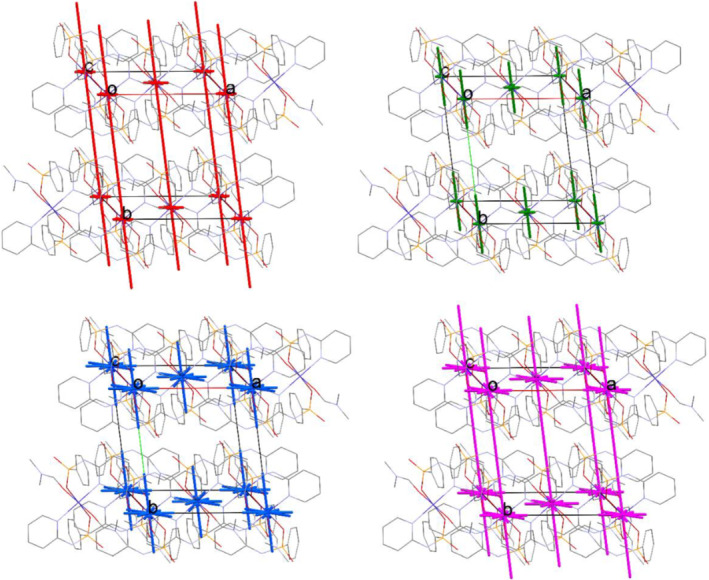
Energy‐vector diagrams representing the results of PIXEL calculations for 1 showing different components electrostatic (red wireframe), polarization (green), dispersion (blue), and total (purple)
3.3. Molecular docking study
In order to evaluate the inhibitory potential of 1–3 by their binding affinities targeting MPro (6M03) which is one of important proteins involved in the mechanism of action of SARS‐CoV‐2, the best position of the ligand showing the highest negative binding affinity with receptor was selected and analyzed. The binding energies of complexes 1–3 are given in Table 7. The binding affinities for all three complexes are negative; so 1–3 can all be used as inhibitor of MPro of SARS‐COV‐2. Complex 1 displays the largest negative binding affinity (−6.1 kcal mol−1), slightly better than those of complexes 2 and 3 (−5.8 and −4.7 kcal mol−1, respectively), where these values are related to the results before the MD simulation.
TABLE 7.
Resulted parameters (kcal mol−1) from interaction between ligand and main protease of Autodock Vina for the studied complexes 1–3 before and after molecular dynamics (MD)
| Compound | Affinity before MD | Affinity after MD |
|---|---|---|
| 1 | −6.1 | −8.0 |
| 2 | −5.8 | −6.4 |
| 3 | −4.7 | −5.3 |
Figures 7, S4, and S5 display the various interactions of the studied compounds 1–3, respectively, with selected biological targets of the 6M03 protein. The organic and aromatic segments of 1 interact with the lipophilic amino acid residues of Pro A108, Pro A241, Met A235, Pro A132, and Phe A134, and its polar segments interact with the polar amino acid residues of Glu A240, Thr A198, Tyr A239, Thr A196, Gly A195, and Asn A133. In the case of complexes 2 (Figure S4) and 3 (Figure S5), similar interactions are observed between the organic segments of complexes with lipophilic amino acid residues (Leu A286, Leu A287 for 2 and Phe A134, Pro A108, Pro A132, Pro A241 for 3) and between polar amino acid residues (Thr A199, Asp A289, Arg A131, Glu A290, Thr A198, Asp A197, and Lys A137 for 2 and Asn A133, Thr A198, Glu A240, Thr A196, and Gly A195 for 3) and polar segments of complex with opposite charge. In general, the results illustrate that the studied complexes are well correlated with the active sites of amino acid residue of 6M03 protein via various π‐type, polar and van der Waals interactions and thus, these complexes can be suggested as the suitable inhibitors to disturb the function of 6M03 protein. Indeed, by disrupting the function of this protein which is of spike (S) proteins, the keys that the coronavirus uses to enter host cells, these complexes can be suggested to inhibit SARS‐COV‐2.
FIGURE 7.
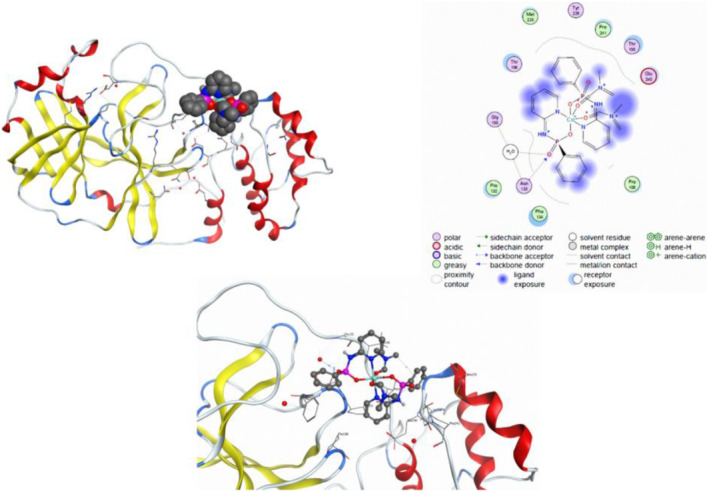
Up left: The best poses of the 3D complex 1 in the binding pocket of 6M03. The 2D (up right) and 3D (down) ligand maps of the complex 1 with amino acid sites inside of the active pocket of 6M03. These results are related to the molecular docking before molecular dynamic calculation
In order to a more exact analysis of the best docking modes according to the ligand‐protein interactions, molecular dynamic (MD) simulation are carried out, where such simulation describe the more stable complexes with the higher negative binding affinity compared to ones before MD. Indeed, the optimized ligand‐protein interactions by MD give an improved affinity of all complexes 1–3 with 6M03 (Figures 8, S6, and S7 and Table 7) and the results are more reliable. Complex 1 is still displaying the highest negative binding affinity (−8.0 kcal mol−1) compared to 2 and 3. This value is very similar to those of introduced coronavirus inhibitors such as Remdesivir (−7.8 kcal mol−1)[ 21 ] or Epigallocatechine Gallate (EGCG) or KDH (−7.4 kcal mol−1),[ 22 ] showing that 1 exhibits a very good inhibitory activity against coronavirus.
FIGURE 8.
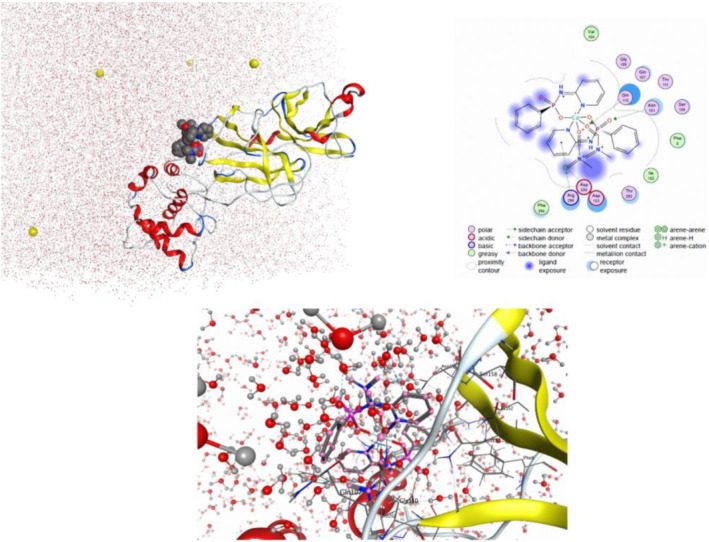
Up left: The best poses of the 3D complex 1 in the binding pocket of 6M03. The 2D (up right) and 3D (down) ligand maps of the complex 1 with amino acid sites inside of the active pocket of 6M03. These results are related to the molecular docking after molecular dynamics calculation
The results after MD simulation have been displayed in Figures 8, S6, and S7. Rather similar interactions between 1–3 and 6M03 are observed before and after MD, except for small differences in the amino acid residues. Indeed, after MD the organic and aromatic segments of complex 1 have good interactions with lipophilic amino acid residues of Phe A294, Ile A152, Phe A8, and Val A104, and the polar segments interact with polar amino acid residues of Arg A298, Asp A295, Asp A153, Thr A292, Ser A158, Asn A151, Thr A111, Gln A107, Gln A110, and Gly A109. In the case of complexes 2 (Figure S7) and 3 (Figure S8), suitable interactions are found between organic segments of complexes with lipophilic amino acid residues (Met A276, Leu A272, Leu A271, Leu A268, Leu A287, and Leu A286 for 2 and Pro A108, Leu A167, and Leu A27 for 3) and between polar amino acid residues (Tyr A237, Gly A275, and Tyr A239 for 2 and Glu A166, Cys A145, His A164, His A41, Thr A45, Cys A44, Ser A46, and Gln A189 for 3) and polar segments of complexes with opposite charge.
3.4. HS analysis and enrichment ratio (E XY)
The relative contributions of intermolecular interactions to the corresponding HS areas and the enrichment ratios (E XY) are listed in Table 8. For each complex, the HSs mapped over the d norm and shape index properties obtained using a standard (high) surface resolution along with full 2D fingerprint plots are shown in Figures 9 and S8–S10, respectively, for the molecule Co1 of 1, and the molecule Co23 of 1, 2, and 3. Figures S7–S10 give the 2D decomposed FPs. It is should be noted that for 2, the HS has been generated on the [Cu(II)Cl{[NH‐2Py]2[N‐2Py]P(O)}] molecule of this structure. Moreover, in the following discussion the label of the metal atoms are used to introduce the symmetrically independent molecules of a structure.
TABLE 8.
Main contributions of intermolecular contacts, surface contacts (S X), random contacts (R XX/R XY) and enrichment ratios (E XX/E XY) for 1–3 are presented
| Contacts | Co1 of 1 | Co23 of 1 | 2 | 3 | S X | H | C | N | O | Cl | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H … H | 49.8 | 56.2 | 42.8 | 52.7 | Co1 of 1 | 74.65 | 16.35 | 2.05 | 6.45 | ― | |||
| H … C/C … H | 32.7 | 27.0 | 21.3 | 21.5 | Co23 of 1 | 77.95 | 13.50 | 1.75 | 6.50 | ― | |||
| H … O/O … H | 12.9 | 13.0 | 12.3 | ― | 2 | 68.95 | 13.35 | 4.20 | 6.15 | 5.15 | |||
| H … N/N … H | 4.1 | 3.5 | 8.4 | 8.4 | 3 | 74.10 | 13.75 | 5.50 | ― | 6.45 | |||
| H … Cl/Cl … H | ― | ― | 10.3 | 12.9 | |||||||||
| C … N/N … C | ― | ― | ― | 2.6 | |||||||||
| C … C | ― | ― | 2.7 | 1.7 |
| Atoms | H | C | N | O | H | C | N | O | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Co1 of 1 | R XX/R XY | E XX/E XY | Co1 of 1 | ||||||||||
| H | 55.73 | 24.41 | 3.06 | 9.63 | H | 0.89 | 1.34 | 1.34 | 1.34 | ||||
| C | 24.41 | ― | ― | ― | C | 1.34 | ― | ― | ― | ||||
| N | 3.06 | ― | ― | ― | N | 1.34 | ― | ― | ― | ||||
| O | 9.63 | ― | ― | ― | O | 1.34 | ― | ― | ― |
| Co23 of 1 | R XX/R XY | E XX/E XY | Co23 of 1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H | 60.76 | 21.05 | 2.73 | 10.13 | H | 0.92 | 1.28 | 1.28 | 1.28 | ||||
| C | 21.05 | ― | ― | ― | C | 1.28 | ― | ― | ― | ||||
| N | 2.73 | ― | ― | ― | N | 1.28 | ― | ― | ― | ||||
| O | 10.13 | ― | ― | ― | O | 1.28 | ― | ― | ― |
| Atoms | H | C | N | O | Cl | H | C | N | O | Cl | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | R XX/R XY | E XX/E XY | 2 | ||||||||||
| H | 47.54 | 18.41 | 5.79 | 8.48 | 7.10 | H | 0.90 | 1.16 | 1.45 | 1.45 | 1.45 | ||
| C | 18.41 | 1.78 | ― | ― | ― | C | 1.16 | 1.52 | ― | ― | ― | ||
| N | 5.79 | ― | ― | ― | ― | N | 1.45 | ― | ― | ― | ― | ||
| O | 8.48 | ― | ― | ― | ― | O | 1.45 | ― | ― | ― | ― | ||
| Cl | 7.10 | ― | ― | ― | ― | Cl | 1.45 | ― | ― | ― | ― |
| Atoms | H | C | N | Cl | H | C | N | Cl | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | R XX/R XY | E XX/E XY | 3 | ||||||||||
| H | 54.91 | 20.38 | 8.15 | 9.56 | H | 0.96 | 1.05 | 1.03 | 1.35 | ||||
| C | 20.38 | 1.89 | 1.51 | ― | C | 1.05 | 0.90 | 1.72 | ― | ||||
| N | 8.15 | 1.51 | ― | ― | N | 1.03 | 1.72 | ― | ― | ||||
| Cl | 9.56 | ― | ― | ― | Cl | 1.35 | ― | ― | ― |
FIGURE 9.
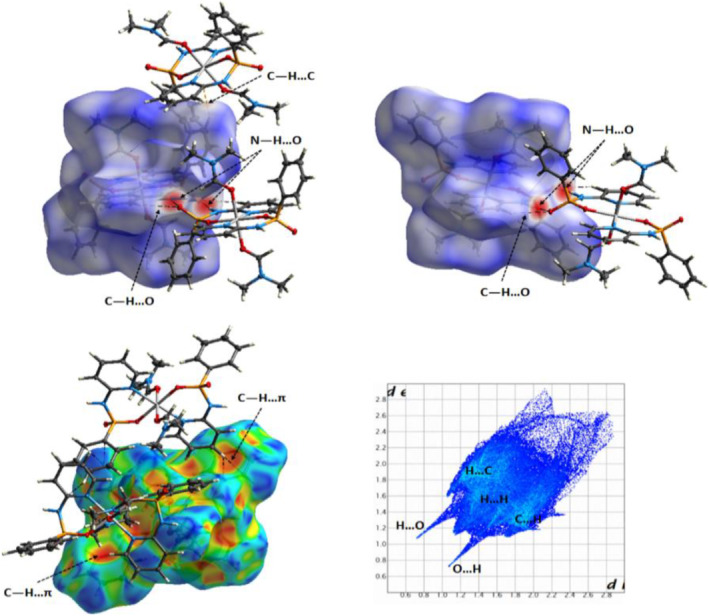
HS mapped for the molecule Co1 of 1 with the d norm (up) and shape index (left down) properties showing main intermolecular contacts (N―H … O, C―H … O, C―H … C and C―H … π, Table 2) between the selected molecule inside the surface and its neighboring molecules. Full 2D fingerprint plot marking the main close contacts are represented on right down of figure. For decomposed FPs of various intermolecular contacts refer to supporting information
3.4.1. HSs, FPs, and E XY of the complex 1
From the d norm HSs of the molecule Co1 of 1 in Figure 9, the red regions on the surface depict H … O/O … H contacts associated to classical N―H … O hydrogen bond interactions between the uncoordinated NH and P═O units of phosphoramide ligands. The areas delineated into the C―H … C and C―H … O hydrogen bonds appear as very small red spots. The C―H … π interactions between the pyridine rings and CH groups are recognized by patterns of red π‐holes on the shape index surface displayed in Figure 9. The whole fingerprint region represented as a combination of d e and d i along with the marked main intermolecular interactions is also displayed in Figure 9. The H … H contacts have the largest contribution (49.8%) to the HS and are shown as the nearly blue sharp regions on the diagonal focused around 1.1 Å ˂ d e, d i ˂ 2.5 Å. The H … O/O … H (12.9% contribution) interactions appear as blue spikes in the top and bottom parts of the left side of the plot with minimum values of d i + d e ≈ 1.8 Å which is less than the sum of the van der Waals radii of hydrogen and oxygen atoms (≈ 2.7 Å). The H … C/C … H (32.7% contribution) are shown as wings and the H … N/N … H (4.1% contributions) interactions are located in the upper left and lower right sides of the plot.
For molecule Co23 of 1, similar to molecule Co1, the major finding refers to the H … O/O … H contacts arising mainly from the classical N―H … O hydrogen bond interactions (Figure S8). Very small red areas on HS are related to H…H and C―H … C interactions. Further, the pattern of the red π‐hole on the shape index surface displayed in Figure S8, above the pyridine ring areas, shows the C―H … π interaction as a result of interaction between the pyridine ring and CH3 group of coordinated DMF. From the full FP in lower right part of Figure S8, the H … H contacts make the largest contribution (56.2%, in the range 1.0 Å ˂ d e, d i ˂ 2.4 Å) to the HS and the H⋯O/O⋯H (13.0%) interactions are mainly displayed as two sharp spikes providing the minimum values of d i + d e ≈ 1.8 Å which are less than sum of the van der Waals radius of hydrogen and oxygen atoms (Figure S12). The H … C/C … H and H … N/N … H contacts are represented as points in the top left part (d e > d i, H … C and H … N) and bottom right part (d e < d i, C … H and N … H) of the FP comprising 27.0% and 3.5% of the surface, respectively.
Finally, for both molecules Co1 and Co23 of this structure, the favored interactions recognized by the enrichment ratios larger than unity (Table 8) are found for those of the H … O/O … H, H … C/C … H and H … N/N … H type, where the molecules relatively rich in oxygen, carbon and nitrogen on the corresponding HSs (S O,C,N > 2.00). In this way, the classical hydrogen bond interactions N–H … O from type H … O/O … H contacts are known as favored interactions. The H … H contacts are introduced as disfavored with enrichment ratios (E HH) lower than unity for both molecules, whereas these compounds have a large number of H atoms on their surface (S H large) and comprise the significant contributions of the corresponding surface areas.
3.4.2. HSs, FPs, and E XY of the complex 2
As it is seen from Figure S9, the deep red areas on the HS plotted over d norm appearing near O atoms of phosphoryl (of complex molecule) or carbonyl (of DMF molecule) groups, and hydrogen atoms of N―H units display their roles as the respective acceptors and donors in the classical N―H⋯O(═P/C) hydrogen bond interactions. Some little red spots on the white regions of d norm surface are also observed resulting from the C―H … O, C―H … C, C―H … Cl or C―H … N hydrogen bond interactions. The shape index of the HS is utilized as a tool to visualize the C―H … π and π⋯π interactions by the presence of the patterns of the red π‐hole (for the former) or adjacent red and blue triangles (for the latter) on the shape index surface. Figure S9 clearly shows that both patterns are found on the shape index surface of 2 suggesting the presence of C―H … π and π⋯π interactions in this complex.
According to the FP of 2 in Figure S9 and its information in Table 8, the H … H interactions are found as the most important interactions contributing 42.8% to the overall HS. These contacts are reflected in Figure S13 as widely scattered points in the center of the plot with a spike at d e = d i and minimum values of d i + d e ≈ 2.2 Å which are almost equal to twice the hydrogen van der Waals radius (≈2.2 Å). The second main contribution to the intermolecular interactions arises from H … C/C … H contacts, comprising 21.3% of the total HS area (Table 8), and are shown in the FP as scattered points in the top left (d e > d i, H … C) and lower right part (d e < d i, C … H). Another important interaction is the H⋯O/O … H contact that comprises 12.3% of the total HS area (Table 8) being displayed as pair of spikes with minimum values of d i + d e ≈ 1.8 Å (Figure S13) which is less than the sum of the van der Waals radius of hydrogen and oxygen atoms (≈2.60 Å). A close inspection of other intermolecular contacts in this structure reveals lower proportions of H … N/N … H contacts (8.4%) and H⋯Cl/Cl … H (10.3%) contacts (Table 2 and Figure S13). Moreover, the structure of 2 is described by C … C contacts, comprising a negligible proportion of 2.7% of the total HS area representing on the FP as the characteristic pattern on the diagonal at d e = d i (in the range 1.6 to 2.2 Å for d e and 1.6 to 2.3 Å for d i, Figure S13). These contacts can be introduced as an evidence for π⋯π stacking interactions as are observed on the shape index of the HS.
In this structure, more contacts (H⋯C/C … H, H⋯N/N … H, H⋯O/O … H, H⋯Cl/Cl … H, and C … C) are favored since their enrichment ratios E are larger than unity (Table 8). Therefore, the classical N–H … O hydrogen bond interactions of type H … O/O … H contacts are identified as favored. The most enriched contact is explored for C … C which can be explained by the formation of the π⋯π stacking interactions between aminopyridien rings in this structure. Contrarily, the H … H contact is disfavored (E HH = 0.90), but this contact generates a majority (42.8%) of the interaction surface.
3.4.3. HSs, FPs, and E XY of the complex 3
The main contact for the complex 3 can be seen as little bright red areas on the d norm HS and is indicative of an N―H⋯Cl hydrogen bond interaction (Figure S10). The other significant intermolecular interaction presented as very little red spot on white area of the d norm HS is related to the C―H⋯Cl hydrogen bond interaction. Apart from hydrogen bond interactions, the π‐effect interactions of C―H⋯π and π … π are seen on the shape index surface as red π‐hole and adjacent red/blue triangles patterns, respectively.
Figure S10 depicts the full FP marking the main close contacts and shows H … H contacts as the major term of the scattered points on the plot comprising 52.7% of the total HS. These contacts appear on the diagonal plot at d e = d i with the minimum values of d i + d e ≈ 2.4 Å which is larger than 2 × the hydrogen van der Waals radius (≈2.2 Å). The H … C/C … H contacts comprise 21.5% of the total HS and represent two wings in the region of (1.7 Å, 2.5 Å). The H … Cl/Cl … H interactions appear as a pair of almost symmetrical spikes on the plot with d i + d e ≈ 2.8 Å (Figure S14) which is slightly less than sum of the hydrogen and chlorine van der Waals radius (≈2.85 Å). The C … N/N … C and C … C contacts, comprising an insignificant proportion of 2.6% and 1.7% of the total HS area, respectively, represent on the FP as the characteristic patterns on the diagonal at d e = d i (in the range 1.7 to 2.1 Å for former and 1.8 to 2.2 Å for later, Figure S14). A survey of other intermolecular contacts in this structure discloses a proportion of H … N/N … H (8.4%) contacts (Table 2 and Figure S14).
The list of enrichment ratios in Table 8 marks the H … Cl/Cl … H (E HCl = 1.35) and C … N/N … C (E CN = 1.72) contacts which turn out to be fully favored in the crystal packing, whereas those of type H … C/C … H (E HCl = 1.05) and H … N/N … H (E CN = 1.03) are slightly enriched. In this way, in the absence of classical hydrogen bond interactions, the importance of these relatively weak interactions in the molecular assembly of such structure is confirmed. The H … H and C … C contacts are characterized as approximately non‐enriched (disfavored) interactions, as they display the enrichment ratios less than unity, E HH = 0.96 and E CC = 0.90. However, in the case of H … H, these contacts cover the most significant contribution to the total HS (52.7%) and the surface contact S H is about 74.
Finally, HS analysis for 1–3 shows that the classical hydrogen bonds N―H … O are dominant interactions in the structures 1 and 2 evidenced as obvious red areas on the HS and two sharp spikes on the FP, whereas, in the structure 3, only some little red spots and two short spikes, respectively, on the HS and FP related to the weak N―H … Cl interactions are found. The presence of some π‐effect interactions (C―H … π and/or π … π) in the structures 1–3 is confirmed by the related characteristic patterns on the shape index surfaces. For all structures, the contribution of H … H and H … C/C … H contacts is larger than other contacts and then, H … O/O … H in the case of 1 and 2, and H … Cl/Cl … H for 3 are in the next position, where the H … O/O … H contacts are not found in structure 3. The H … C/C … H and H … N/N … H contacts are favored in all structures 1–3 since the enrichment ratios E HC and E HN are larger than unity (Table 8). Other favored contacts are of the H … O/O … H type for 1 and 2, and of the H … Cl/Cl … H type for 2 and 3. This may be explained by the fact that the N―H … O (in the crystal structures 1 and 2) and N―H … Cl (in 3) dominant interactions are covered by these favored contacts. In contrast, the H … H contacts are disfavored in 1 and 2 with E HH ≤ 0.92, although these contacts have the largest contribution to the interaction surfaces (at least 42%). The structure 3 shows an slightly increased propensity to form such contacts (E HH = 0.96), where a higher amount of H … H contacts of the total HS area is found in 3 compared to those in 1 and 2 (Table 8). Eventually, a visual inspection of FPs reveals that points at the largest d e and d i values on the full FPs are more diffused in the case of 1 (0.7 Å < d i, d e < 2.9 Å) and 2 (0.7 Å < d i, d e < 2.6 Å) compared to those in 3 (1.1 Å < d i, d e < 2.5 Å). This is explained by the presence of classical hydrogen bond interactions in the crystal packing of 1 and 2 versus the absence of such classical interactions in 3.
3.5. Comparison of results with some other metal complexes
To gain a better evaluation of the obtained outcomes, the results collected in this work are compared with those of a well‐known family of metal complexes, namely, metal‐Salen Schiff base complexes. Metal complexes derived from Salen Schiff base type ligands providing the inner N2O2 coordination modes are an important class of coordination compounds. These such complexes have been recently received attention due to their catalytic, magnetic, electric, luminescence and fluorescence properties and also, for their biomedical applications such as anticancer, antibacterial, antimicrobial and antiviral.[ 62 , 63 , 64 , 65 , 66 ]
In the first comparison, the AIM and NBO analyses results are considered. The charge density analysis for 1–3 shows the values of ρ(r) (around +0.073 a.u.) and ∇2 ρ(r) > 0 (around +0.36 and +0.55 a.u.), H(r) < 0 (around −0.005 a.u.), and 1 < < 2 for the M—O and M—N contacts suggesting a rather strong bond with mainly ionic character and a partial covalent nature for these contacts. It is also obtained a slightly stronger Co—OPT bond compared to the Co—NPy bond in 1. Usman et al. reported the similar results of the positive values of ρ(r) (around +0.12 a.u.) and ∇2 ρ(r) (+0.54 to +0.60 a.u) with the negative values of H(r) < 0 (around −0.04 a.u.), and also, 1 < < 2 for the Zn—O and Zn—N contacts of the Zn(II) Salen complex.[ 67 ] Tetravalent f‐element complexes of the type [M (salen)2] (M = Ce, Th, Pa, U, Np, Pu) investigated by Kloditz et al. illustrate the values about +0.09 a.u. for ρ(r), about +0.33 a.u. for ∇2 ρ(r) and around −0.02 a.u. for H(r) with 1 < < 2.[ 68 ] In these literatures, authors suggest the typical signature of ionic bonding with a degree of covalency for the M—O and M—N contacts. Moreover, in both our work and referenced literatures, it is suggested that O‐donor atoms are stronger and more covalent than bonds to the N‐donor atoms in the phosphoramide or Salen ligand. The bond order results are also confirmed a stronger M—O contact than M—N in the metal–ligand (phosphoramide or Salen) coordination.
In the case of the molecular docking study on the complexes 1–3 studied in here, the negative binding affinity of −8.0, −6.4, and −5.3 kcal mol−1, respectively, for the ligand‐protein (6M03) interactions are obtained as discussed in above. In comparison, a molecular docking simulation study on some Salen complexes against SARS‐CoV‐2 (6Y84) shows the binding affinity around −7.5 kcal mol−1.[ 66 ] This result is almost similar to those obtained in this work, especially for the complex 1, which can suggest a suitable inhibitory ability against coronavirus for some of amino‐functionalized P(V) coordination compounds competing with other compounds under investigation as anti‐CoV‐2 such as Salen metal complexes.
Form HS analysis, the H … H and H … C/C … H contacts and then, H … O/O … H and H … N/N … H (for 1 and 2) or H … Cl/Cl … H and H … N/N … H (for 3) are contacts with the most contribution related to the total HS. As it is seen, the H … O/O … H contact dominate the H … N/N … H for the metal‐phosphramide complexes 1 and 2, whereas in the Salen complexes,[ 63 , 69 ] the H … N/N … H is maximum than the H … O/O … H. This can be attributed to the uncoordinated phosphoryl oxygen atoms in 1 and 2 which are involved in the N―H … O hydrogen bonding, where in the phosphoramide complexes with the coordinated phosphoryl oxygen atoms,[ 25 ] similarly the H … N/N … H input is larger than the H … O/O … H.
Finally, with considering the above‐mentioned similarities, a novel feature of phosphoramide complexes can be investigated where such compounds with suitable amino substituents operating as N,O‐donor ligands can be compared with well‐know Salen complexes. Indeed, a bias toward focusing on similarities between these two classes of metal complexes can be a turning point to study and develop the desired and emerging applications of phosphoramide complexes.
4. CONCLUSION
This study presents the crystal structures of novel coordination compounds of pyridyl‐functionalized amidophosphoryl ligands OP[Ph][NH‐2Py]2 and OP[NH‐2Py]3 as two discrete chelate phosphoramide complexes with environments Co(O)4(N)2 (1) and Co(N)4(Cl) (2) along with a six‐coordinate Cu (II) complex of pyrazole with environment Co(N)4(Cl)2 (3). The crystal packing investigations done for these structures indicate that the origin of the difference in packing mode between 1 and 2 on the one hand and 3 on the other hand stems from the presence (1 and 2) or absence (3) of classical hydrogen bond interactions. QTAIM and NBO calculations suggest that the M—O and M—N bonds are principally electrostatic in nature with a slightly covalent character. To gain additional insight into the molecular packing features, a HS analysis was done on complexes 1–3 showing that H … C/C … H and H … N/N … H interactions are contributing to the lattice cohesion for 1–3, H … O/O … H for 1 and 2 and H … Cl/Cl … H for 2 and 3 besides some π‐effect interactions (C―H … π and/or π … π). The PIXEL calculations reveal that the most stabilizing intermolecular interaction energies define a 1D classical N–H … O═P framework in 1 and a N–H … O four‐membered motif in 2 with the major contribution of the electrostatic and repulsion components, compared to a 3D non‐classical N/C–H … Cl framework in 3 with the more dominance of the electrostatic and dispersion energy terms. Moreover, a biological study based on the molecular docking method shows a good potential of inhibiting Mpro of SARS‐CoV‐2 for especially complex 1.
AUTHOR CONTRIBUTIONS
Mobina Najarianzadeh: Data curation; investigation; methodology. Atekeh Tarahhomi: Formal analysis; investigation; project administration; supervision. Samaneh Pishgo: Data curation. Arie van der Lee: Data curation; formal analysis.
CONFLICT OF INTEREST
Authors declare no potential conflict of interest.
Supporting information
TABLE S1. Selected bond lengths (Å) and angles (°) for compounds 1–3.
Table S2. Selected experimental and theoretical bond lengths (Å) and angles (°) for compounds 1–3.
Figure S1. Molecular graphs and distribution of bond critical points (BCPs) for 1 (left), 2 (middle) and 3 (right). BCPs and bond paths connecting BCPs are represented as red spheres and black lines and dashed lines, respectively.
Figure S2. Energy‐vector diagrams representing the results of PIXEL calculations for 2 showing different components electrostatic (red wireframe), polarization (green), dispersion (blue) and total (purple).
Figure S3. Energy‐vector diagrams representing the results of PIXEL calculations for 3 showing different components electrostatic (red wireframe), polarization (green), dispersion (blue) and total (purple).
Figure S4. Up left: The best poses of the 3D complex 2 in the binding pocket of 6 M03; The 2D (up right) and 3D (down) ligand maps of the complex 2 with amino acid sites inside of the active pocket of 6 M03. These results are related to the molecular docking before molecular dynamics calculation.
Figure S5. Up left: The best poses of the 3D complex 3 in the binding pocket of 6 M03; The 2D (up right) and 3D (down) ligand maps of the complex 3 with amino acid sites inside of the active pocket of 6 M03. These results are related to the molecular docking before molecular dynamics calculation.
Figure S6. Up left: The best poses of the 3D complex 2 in the binding pocket of 6 M03; The 2D (up right) and 3D (down) ligand maps of the complex 2 with amino acid sites inside of the active pocket of 6 M03. These results are related to the molecular docking after molecular dynamics calculation.
Figure S7. Up left: The best poses of the 3D complex 3 in the binding pocket of 6 M03; The 2D (up right) and 3D (down) ligand maps of the complex 3 with amino acid sites inside of the active pocket of 6 M03. These results are related to the molecular docking after molecular dynamics calculation.
Figure S8. HS mapped for the molecules Co23 of 1 with the d norm (up) and shape index (left down) properties showing main intermolecular contacts (N―H … O, C―H … C, H … H and C―H … π, Table 3) between the selected molecule inside the surface and its neighboring molecules. Full 2D fingerprint plot marking the main close contacts are represented on right down of figure. For decomposed FPs of various intermolecular contacts refer to supporting information.
Figure S9. HS mapped for the complex 2 with the d norm (up) and shape index (left and middle down) properties showing main intermolecular contacts (N―H … O, C―H … O, C―H … C, C―H … Cl, C―H … N, C―H … π and π … π, Table 2) between the selected molecule inside the surface and its neighboring molecules. Full 2D fingerprint plot marking the main close contacts are represented on right down of figure. For decomposed FPs of various intermolecular contacts refer to supporting information.
Figure S10. HS mapped for the complex 3 with the d norm (left up) and shape index (right up and left down) properties showing main intermolecular contacts (N―H … Cl, C―H … Cl, C―H … π and π … π, Table 2) between the selected molecule inside the surface and its neighboring molecules. Full 2D fingerprint plot marking the main close contacts are represented on right down of figure. For decomposed FPs of various intermolecular contacts refer to supporting information.
Figure S11. Decomposed FPs of various intermolecular contacts H … H, H … C/C … H, H … O/O … H and H … N/N … H for the molecule Co1 of 1.
Figure S12. Decomposed FPs of various intermolecular contacts H … H, H … C/C … H, H … O/O … H and H … N/N … H for the molecule Co23 of 1.
Figure S13. Decomposed FPs of various intermolecular contacts H … H, H … C/C … H, H … O/O … H, H … Cl/Cl … H, H … N/N … H and C … C for the complex 2.
Figure S14. Decomposed FPs of various intermolecular contacts H … H, H … C/C … H, H … Cl/Cl … H, H … N/N … H, C … N/N … C, C … C for the complex 3.
ACKNOWLEDGMENTS
Support of this investigation by Semnan University is gratefully acknowledged. Moreover, we sincerely thank Mr. Mohammadreza Aallaei, Imam Hossein University, Tehran, Iran, for his assistance with the molecular docking calculations. Maurizio Polentarutti is thanked for collecting X‐ray data at the XRD1 beamline of the Elettra synchrotron (Trieste, Italy) for compound 2 during the beamtime allocated for project 20200264.
Najarianzadeh M., Tarahhomi A., Pishgo S., van der Lee A., Appl Organomet Chem 2022, 36(5), e6636. 10.1002/aoc.6636
Contributor Information
Atekeh Tarahhomi, Email: tarahhomi.at@semnan.ac.ir.
Arie van der Lee, Email: arie.van-der-lee@umontpellier.fr.
DATA AVAILABILITY STATEMENT
Data openly available in a public repository that issues datasets with DOIs.
REFERENCES
- 1. Moulton B., Zaworotko M. J., Chem. Rev. 2001, 101, 1629. [DOI] [PubMed] [Google Scholar]
- 2. Desiraju G. R., Angew. Chem., Int. Ed. 2007, 46, 8342. [DOI] [PubMed] [Google Scholar]
- 3. Schmidt G. M. J., Pure Appl. Chem. 1971, 27, 647. [Google Scholar]
- 4. Monro S., Colón K. L., Yin H., Roque J. III, Konda P., Gujar S., Thummel R. P., Lilge L., Cameron C. G., McFarland S. A., Chem. Rev. 2019, 119, 797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Janiak C., Dalton Trans. 2003, 2781. [Google Scholar]
- 6. Barceló‐Oliver M., Baquero B. A., Bauzá A., García‐Raso Á., Vich R., Mata I., Molins E., Terrón À., Frontera A., Dalton Trans. 2013, 42, 7631. [DOI] [PubMed] [Google Scholar]
- 7. Bortoluzzi M., Castro J., Gobbo A., Ferraro V., Pietrobon L., Dalton Trans. 2020, 49, 7525. [DOI] [PubMed] [Google Scholar]
- 8. Gupta R., Paithankar H., Chugh J., Boomishankar R., Inorg. Chem. 2021, 60, 10468. [DOI] [PubMed] [Google Scholar]
- 9. Nikpour S., Dorosti N., Afshar F., Kubicki M., Appl. Organomet. Chem. 2020, 34, e5724. [Google Scholar]
- 10. Gupta A. K., Srivastava A. K., Mahawar I. K., Boomishankar R., Cryst. Growth des. 2014, 14, 1701. [Google Scholar]
- 11. Gupta A. K., Steiner A., Boomishankar R., Dalton Trans. 2012, 41, 9753. [DOI] [PubMed] [Google Scholar]
- 12. Li N., Jiang F., Chen L., Li X., Chen Q., Hong M., Chem. Commun. 2011, 47, 2327. [DOI] [PubMed] [Google Scholar]
- 13. Gupta A. K., Nagarkar S. S., Boomishankar R., Dalton Trans. 2013, 42, 10964. [DOI] [PubMed] [Google Scholar]
- 14. Shariatinia Z., Tuba R. S., J. sol. Energy Eng. 2015, 137, 011006. [Google Scholar]
- 15. Domínguez M. J., Sanmartín C., Font M., Palop J. A., San Francisco S., Urrutia O., Houdusse F., García‐Mina J. M., J. Agric. Food Chem. 2008, 56, 3721. [DOI] [PubMed] [Google Scholar]
- 16. McGuigan C., Thiery J.‐C., Daverio F., Jiang W. G., Davies G., Mason M., Bioorg. Med. Chem. 2005, 13, 3219. [DOI] [PubMed] [Google Scholar]
- 17. Romanowska J., Sobkowski M., Szymańska‐Michalak A., Kozodziej K., Dąbrowska A., Lipniacki A., Piasek A., Pietrusiewicz Z. M., Figlerowicz M., Guranowski A., Boryski J., Stawiński J., Kraszewski A., J. Med. Chem. 2011, 54, 6482. [DOI] [PubMed] [Google Scholar]
- 18. Davey M. S., Malde R.i., Mykura R. C., Baker A. T., Taher T. E., Le Duff C. S., Willcox B. E., Mehellou Y., J. Med. Chem. 2018, 61, 2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gholivand K., Mohammadpanah F., Pooyan M., Roohzadeh R., J. Mol. Struct. 2022, 1248, 131481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu D., Shao C., Guan W., Su Z., Sun J., J. Inorg. Biochem. 2007, 101, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deshpande R. R., Tiwari A. P., Nyayanit N., Modak M., Eur. J. Pharmacol. 2020, 886, 173430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Sagaama A., Brandan S. A., Ben Issa T., Issaoui N., Heliyon 2020, 6, e04640. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Aallaei M., Molaakbari E., Mostafav P., Salarizadeh N., Eshaghi Maleksah R., Afzali D., J. Mol. Struct. 2022, 1253, 132301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Groom C. R., Bruno I. J., Lightfoot M. P., Ward S. C., Acta Crystallogr. Sect. B 2016, 72, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Srivastava A. K., Divya P., Praveenkumar B., Boomishankar R., Chem. Mater. 2015, 27, 5222. [Google Scholar]
- 25. Sebghati M., Tarahhomi A., Bozorgvar M. S., Dumitrescu D. G., van der Lee A., RSC Adv. 2021, 11, 8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Agilent , CrysAlis PRO, Version 1.171.38.43c, Agilent Technologies, Yarnton, Oxfordshire, England: 2011. [Google Scholar]
- 27. Palatinus L., Chapuis G., J. Appl. Crystallogr. 2007, 40, 786. [Google Scholar]
- 28. van der Lee A., J. Appl. Crystallogr. 2013, 46, 1306. [Google Scholar]
- 29. Betteridge P. W., Carruthers J. R., Cooper R. I., Prout K., Watkin D. J., J. Appl. Crystallogr. 2003, 36, 1487. [Google Scholar]
- 30. Kabsch W., XDS. Acta Crystallogr. Sect. D 2010, 66, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Acta Crystallogr. Sect. D 2011, 67, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dolomanov O. V., Bourhis L. J., Gildea R. J., Howard J. A. K., Puschmann H., J. Appl. Crystallogr. 2009, 42, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cooper R. I., Thompson A. L., Watkin D. J., J. Appl. Crystallogr. 2010, 43, 1100. [Google Scholar]
- 34. Spek A. L., Acta Crystallogr. Sect. D 2009, 65, 148.19171970 [Google Scholar]
- 35. Macrae C. F., Bruno I. J., Chisholm J. A., Edgington P. R., McCabe P., Pidcock E., Rodriguez‐Monge L., Taylor R., van de Streek J., Wood P. A., J. Appl. Crystallogr. 2008, 41, 466. [Google Scholar]
- 36. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J. A. Jr., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A. G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al‐Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., Pople J. A., Gaussian 09 (Linux version). 2009. [Google Scholar]
- 37. Lee C., Yang W., Parr R. G., Phys. Rev. 1998, B37, 785. [DOI] [PubMed] [Google Scholar]
- 38.a) Jensen F., WIREs Comput. Mol. Sci. 2013, 3, 273; [Google Scholar]; b) Dunning T. H. Jr., Hay P. J., “In Methods of Electronic Structure Theory”, Chapter 1, Springer, Boston, MA: 1977. 1; [Google Scholar]; c) Lewars E. G., Computational Chemistry: Introduction to the Theory and Applications of Molecular and Quantum Mechanics, 1st ed., Springer; 2011. [Google Scholar]
- 39. Reed A. E., Curtiss L. A., Weinhold F., Chem. Rev. 1998, 88, 899. [Google Scholar]
- 40. Bader R. F. W., In Atoms in Molecules, a Quantum Theory, Oxford University Press, New York, NY: 1990. [Google Scholar]
- 41. Todd A., Keith T. K., Gristmill Software, Overland Park KS, AIMAll, USA: 2013. [Google Scholar]
- 42. Dunitz J. D., Gavezzotti A., Chem. Soc. Rev. 2009, 38, 2622. [DOI] [PubMed] [Google Scholar]
- 43. Shishkin O. V., Dyakonenko V. V., Maleev A. V., Schollmeyer D., Vysotsky M. O., CrystEngComm 2011, 13, 800. [Google Scholar]
- 44. Shishkin O. V., Dyakonenko V. V., Maleev A. V., CrystEngComm 2012, 14, 1795. [Google Scholar]
- 45. Bond A. D., J. Appl. Crystallogr. 2014, 47, 1777. [Google Scholar]
- 46. Trott O., Olson A. J., J. Comput. Chem. 2010, 31, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.a) Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., J. Comput. Chem. 2004, 25, 1605; [DOI] [PubMed] [Google Scholar]; b) Discovery Studio Visualizer, v20.1.0.19259; 2009. Available online: https://discover.3ds.com/discovery-studio-visualizer (accessed on October 2020). [Google Scholar]
- 48. Berendsen H. J. C., van der Spoel D., van Drunen R., Comput. Phys. Commun. 1995, 91, 43. [Google Scholar]
- 49. Spackman M. A., McKinnon J. J., CrystEngComm 2002, 4, 378. [Google Scholar]
- 50. Spackman M. A., Jayatilaka D., CrystEngComm 2009, 11, 19. [Google Scholar]
- 51. Turner M. J., McKinnon J. J., Wolff S. K., Grimwood D. J., Spackman P. R., Jayatilaka D., Spackman M. A., CrystalExplorer 17.5, University of Western Australia, Australia: 2017. [Google Scholar]
- 52. McKinnon J. J., Jayatilaka D., Spackman M. A., Chem. Commun. 2007, 3814. [DOI] [PubMed] [Google Scholar]
- 53. McKinnon J. J., Spackman M. A., Mitchell A. S., Acta Crystallogr. Sect. B 2004, 60, 627. [DOI] [PubMed] [Google Scholar]
- 54. Jelsch C., Ejsmont K., Huder L., IUCrJ 2014, 1, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bader R. F. W., Monatsh. Chem. 2005, 136, 819. [Google Scholar]
- 56.a) Arnold W. D., Oldfield E., J. Am. Chem. Soc. 2000, 122, 12835; [Google Scholar]; b) Cremer D., Kraca E., Croat. Chem. Acta 1984, 57, 1259. [Google Scholar]
- 57. Weinhold F., J. Comput. Chem. 2012, 33, 2363. [DOI] [PubMed] [Google Scholar]
- 58. Gavezzotti A., J. Phys. Chem. B. 2002, 106, 4145. [Google Scholar]
- 59. Corpinot M. K., Bučar D. ‐K., Cryst. Growth des. 2019, 19, 1426. [Google Scholar]
- 60. Tarahhomi A., der Lee A., Monatsh. Chem. 2018, 149, 1759. [Google Scholar]
- 61. Bozorgvar M. S., Tarahhomi A., van der Lee A., J. Mol. Struct. 2021, 1223, 128942. [Google Scholar]
- 62. Majumdar D., Agrawal Y., Thomas R., Ullah Z., Santra M. K., Das S., Pal T. K., Bankura K., Mishra D., Appl. Organomet. Chem. 2020, 34, e5269. [Google Scholar]
- 63. Majumdar D., Kumar Pal T., Sakib S. A., Das S., Bankura K., Mishra D., Inorg. Chem. Commun. 2021, 128, 108609. [Google Scholar]
- 64. Majumdar D., Das S., Thomas R., Ullah Z., Sreejith S. S., Das D., Shukla P., Bankura K., Mishra D., Inorganica Chim. Acta 2019, 492, 221. [Google Scholar]
- 65. Poladian Q., Şahin O., Karakurt T., İlhan‐Ceylan B., Kurt Y., Polyhedron 2021, 201, 115164. [Google Scholar]
- 66. Oruma U. S., Ukoha P. O., Uzoewulu C. P., Ndefo J. C., Ugwuoke S. C., Ukwueze N. N., Eze T. E., Ekowo L. C., Eze F. U., Chinaegbomkpa U. V., Okafor S. N., Ezeorah C. J., Mol. Ther. 2021, 26, 4379. [Google Scholar]
- 67. Usman M., Ahmad Khan R., Alsalme A., Alharbi W., Alharbi K. H., Jaafar M. H., Khanjer M.h. A., Tabassum S., Crystals 2020, 10, 259. [Google Scholar]
- 68. Kloditz R., Radoske T., Schmidt M., Heine T., Stumpf T., Patzschke M., Inorg. Chem. 2021, 60, 2514. [DOI] [PubMed] [Google Scholar]
- 69. Bhattacharyya A., Bauzá A., Sproules S., Natrajan L. S., Frontera A., Chattopadhyay S., Polyhedron 2017, 137, 332. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1. Selected bond lengths (Å) and angles (°) for compounds 1–3.
Table S2. Selected experimental and theoretical bond lengths (Å) and angles (°) for compounds 1–3.
Figure S1. Molecular graphs and distribution of bond critical points (BCPs) for 1 (left), 2 (middle) and 3 (right). BCPs and bond paths connecting BCPs are represented as red spheres and black lines and dashed lines, respectively.
Figure S2. Energy‐vector diagrams representing the results of PIXEL calculations for 2 showing different components electrostatic (red wireframe), polarization (green), dispersion (blue) and total (purple).
Figure S3. Energy‐vector diagrams representing the results of PIXEL calculations for 3 showing different components electrostatic (red wireframe), polarization (green), dispersion (blue) and total (purple).
Figure S4. Up left: The best poses of the 3D complex 2 in the binding pocket of 6 M03; The 2D (up right) and 3D (down) ligand maps of the complex 2 with amino acid sites inside of the active pocket of 6 M03. These results are related to the molecular docking before molecular dynamics calculation.
Figure S5. Up left: The best poses of the 3D complex 3 in the binding pocket of 6 M03; The 2D (up right) and 3D (down) ligand maps of the complex 3 with amino acid sites inside of the active pocket of 6 M03. These results are related to the molecular docking before molecular dynamics calculation.
Figure S6. Up left: The best poses of the 3D complex 2 in the binding pocket of 6 M03; The 2D (up right) and 3D (down) ligand maps of the complex 2 with amino acid sites inside of the active pocket of 6 M03. These results are related to the molecular docking after molecular dynamics calculation.
Figure S7. Up left: The best poses of the 3D complex 3 in the binding pocket of 6 M03; The 2D (up right) and 3D (down) ligand maps of the complex 3 with amino acid sites inside of the active pocket of 6 M03. These results are related to the molecular docking after molecular dynamics calculation.
Figure S8. HS mapped for the molecules Co23 of 1 with the d norm (up) and shape index (left down) properties showing main intermolecular contacts (N―H … O, C―H … C, H … H and C―H … π, Table 3) between the selected molecule inside the surface and its neighboring molecules. Full 2D fingerprint plot marking the main close contacts are represented on right down of figure. For decomposed FPs of various intermolecular contacts refer to supporting information.
Figure S9. HS mapped for the complex 2 with the d norm (up) and shape index (left and middle down) properties showing main intermolecular contacts (N―H … O, C―H … O, C―H … C, C―H … Cl, C―H … N, C―H … π and π … π, Table 2) between the selected molecule inside the surface and its neighboring molecules. Full 2D fingerprint plot marking the main close contacts are represented on right down of figure. For decomposed FPs of various intermolecular contacts refer to supporting information.
Figure S10. HS mapped for the complex 3 with the d norm (left up) and shape index (right up and left down) properties showing main intermolecular contacts (N―H … Cl, C―H … Cl, C―H … π and π … π, Table 2) between the selected molecule inside the surface and its neighboring molecules. Full 2D fingerprint plot marking the main close contacts are represented on right down of figure. For decomposed FPs of various intermolecular contacts refer to supporting information.
Figure S11. Decomposed FPs of various intermolecular contacts H … H, H … C/C … H, H … O/O … H and H … N/N … H for the molecule Co1 of 1.
Figure S12. Decomposed FPs of various intermolecular contacts H … H, H … C/C … H, H … O/O … H and H … N/N … H for the molecule Co23 of 1.
Figure S13. Decomposed FPs of various intermolecular contacts H … H, H … C/C … H, H … O/O … H, H … Cl/Cl … H, H … N/N … H and C … C for the complex 2.
Figure S14. Decomposed FPs of various intermolecular contacts H … H, H … C/C … H, H … Cl/Cl … H, H … N/N … H, C … N/N … C, C … C for the complex 3.
Data Availability Statement
Data openly available in a public repository that issues datasets with DOIs.