

Abstract
Corona Virus Disease‐19 (COVID‐19) is a pandemic disease mainly caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). It had spread from Wuhan, China, in late 2019 and spread over 222 countries and territories all over the world. Earlier, at the very beginning of COVID‐19 infection, there were no approved medicines or vaccines for combating this disease, which adversely affected a lot of individuals worldwide. Although frequent mutation leads to the generation of more deadly variants of SARS‐CoV‐2, researchers have developed several highly effective vaccines that were approved for emergency use by the World Health Organization (WHO), such as mRNA‐1273 by Moderna, BNT162b2 by Pfizer/BioNTech, Ad26.COV2.S by Janssen, AZD1222 by Oxford/AstraZeneca, Covishield by the Serum Institute of India, BBIBP‐CorV by Sinopharm, coronaVac by Sinovac, and Covaxin by Bharat Biotech, and the first US Food and Drug Administration‐approved antiviral drug Veklury (remdesivir) for the treatment of COVID‐19. Several waves of COVID‐19 have already occurred worldwide, and good‐quality vaccines and medicines should be available for ongoing as well as upcoming waves of the pandemic. Therefore, in silico studies have become an excellent tool for identifying possible ligands that could lead to the development of safer medicines or vaccines. Various phytoconstituents from plants and herbs with antiviral properties are studied further to obtain inhibitors of SARS‐CoV‐2. In silico screening of various molecular databases like PubChem, ZINC, Asinex Biol‐Design Library, and so on has been performed extensively for finding effective ligands against targets. Herein, in silico studies carried out by various researchers are summarized so that one can easily find the best molecule for further in vitro and in vivo studies.
Keywords: antiviral phytoconstituents, COVID‐19, in silico screening, molecular docking, SARS‐CoV‐2
In silico studies represent an excellent tool for identifying ligands that could lead to the development of safer medicines or vaccines. Various phytoconstituents with antiviral properties are being studied to obtain inhibitors of severe acute respiratory syndrome coronavirus 2. In silico screening of molecular databases like PubChem, ZINC, Asinex Biol‐Design Library, and so forth has been performed extensively to find effective ligands against various targets. This review summarizes the in silico studies carried out by many researchers to facilitate further in vitro and in vivo studies.
1. INTRODUCTION
In December 2019, a deadly viral infection started in the Wuhan province of China, where many people suffered from symptoms like pneumonia, flu, fever and headache. Because people from China travelled to other countries, this virus spread and became prevalent in almost 200 countries and territories within a short period of time and created havoc all over the world. Later, after considerable research, the root cause of this infection was found to be a virus that shows structural similarities to the SARS‐related coronavirus.[ 1 , 2 , 3 ] On 20 January 2020, the National Health Commission of China confirmed the outbreak of a viral disease named Corona Virus Disease‐19 (COVID‐19) that transmitted from human to human.[ 4 , 5 ] On 11 March 2020, COVID‐19 was declared a pandemic by the World Health Organization (WHO) because up to 11 March 2020, around 118,000 cases of this illness and 4291 deaths were reported worldwide. According to the WHO, to date, that is, 5 November 2021, 248,467,363 people had been infected, 5,027,183 confirmed cases of death have been reported and 7,027,377,238 people have been vaccinated.
The COVID‐19‐inducing virus is the seventh coronavirus that has been isolated from humans that causes severe infection.[ 6 ] Coronavirus that infects humans consists of a large family of viruses, that is, Alpha, Beta, Gamma and Delta coronaviruses. The COVID‐19 strain of the coronavirus is a new member of the Beta‐coronavirus that shows structural similarity to the SARS‐related coronavirus and bat coronavirus.[ 7 , 8 ] Under an electron microscope, severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) appears as a positive single‐stranded RNA virus with a crown‐like appearance having a genome size of approximately 30,000 bp.[ 7 , 9 ] SARS‐CoV‐2 possesses different types of glycoproteins, namely, spike glycoprotein (S), membrane protein (M), an envelope protein (E) and a nucleocapsid protein (N).[ 10 , 11 ] The S protein of SARS‐CoV‐2 is important for the entry of the virus into the host cell, while M and E proteins are important for viral assembly, transport and release of viral particles.[ 10 , 12 , 13 , 14 ] Inside the human body, SARS‐CoV‐2 uses transmembrane protease serine 2 (TMPRSS2), serine protease for S protein priming, and by using their S protein, it binds with the human receptor cells containing angiotensin‐converting enzyme 2 (ACE2).[ 15 ] At the boundary between its two domains, the S protein cleaves into S1 and S2. The S2 subunit is important for the fusion mechanism, while S1 enables binding with the receptor ACE2 as it contains a receptor‐binding domain (RBD).[ 16 ] After infection of cells by SARS‐CoV‐2, the RNA genome synthesizes two replicase polyproteins, that is, pp1a and pp1ab, which contain a replication/transcription complex, two proteases and several structural proteins.[ 17 ] In the case of other viruses such as HIV‐1 and hepatitis C virus, the protease plays an important role in maturation of the viral protein in host cells, due to which targeting of this main protease is very important for the development of antiviral drugs.[ 18 ] Similarly, in SARS‐CoV‐2, the main protease (Mpro) plays a significant role in cleavage of polyproteins into individual functional pieces, which is responsible for replication and transcription of new viruses. It is a homo‐dimeric protein that contains two protomers each and three domains, namely, Domain I, Domain II and Domain III, respectively. Among these, Domains I and II are made up of six antiparallel β‐barrels that encompass residues 8–110 and 102–184, respectively, whereas Domain III is made up of a cluster of α‐helices with residues from 201 to 303. Domain II is connected with Domain III with a long loop with residues from 185 to 200. There is a catalytic dyad Cys‐His in the cleft between Domains I and II that is believed to have active proteolytic activity.[ 19 , 20 ] For binding of any targeted drugs, there should be sufficient space in the binding or receptor site. In Mpro, the substrate‐binding site is located in the cleft between Domains I and II.[ 21 , 22 ] S1, S2, S3 and S4 are the subsites of the substrate‐binding site as shown in Figure 1. The amino acid substrates in Mpro are numbered ‐P4‐P3‐P2‐P1 and P1ʹ‐P2ʹ‐P3ʹ from N terminus to C terminus, and the cleavage site is present between P1 and P1ʹ, with glutamic residues present in the P1 position. Mpro performs a variety of important functions, such as maturing itself and other polyproteins, as well as cleaving two important polyproteins, pp1a and pp1ab, into 16 nonstructural proteins that are responsible for virus replication and maturation, as well as allowing viral particles to enter host cells.[ 20 ] Because of these important functions, Mpro is considered an effective target for anti‐coronavirus drugs.[ 23 , 24 , 25 ] In spite of many research and experimental reports, few of the drug moieties are considered to be active against Mpro, but they are in the clinical trial stage, whereas Veklury (remdesivir) is the first US Food and Drug Administration (FDA)‐approved antiviral drug for the treatment of COVID‐19. Recently, Pfizer developed an antiviral oral tablet formulation called Paxlovid, where nirmatrelvir acts as an active pharmaceutical ingredient, and it was approved by the US FDA. Paxlovid works by inhibiting the 3C‐like protease present in SARS‐CoV‐2. These new therapeutic agents will help in reducing mental, social, and economic stress and may improve quality of life.[ 26 ] However, the development of new drugs is very time consuming, and it should be cost‐effective. Therefore, computer‐aided drug design (CADD) can be used to test a large number of compounds in the lab and for screening of potent ligands or inhibitors that can target the majority of strains; this also helps to save cost, labour and time.[ 25 , 27 ]
Figure 1.
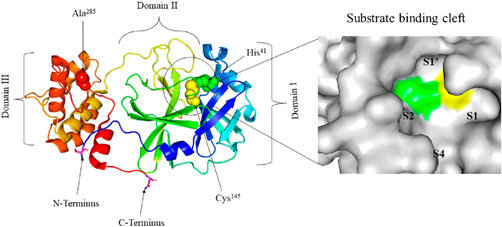
The crystal structure of the SARS‐CoV‐2 main protease (Mpro) is shown on the left, while a surface view of Mpro is shown on the right. Important domains and residues are shown in the crystal structure while four subsites of the substrate‐binding cleft S1, S2, S3 and S4 are shown on the right.[ 20 ] SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
2. TARGETING THE SARS‐COV‐2 MAIN PROTEASE MPRO
A Michael acceptor inhibitor, which is also known as the N3 inhibitor, was designed and developed using an in silico method with the potential to inhibit SARS‐CoV and MERS‐CoV.[ 28 , 29 , 30 ] Regarding SARS‐CoV‐2, N3 can fit into the substrate‐binding pocket of Mpro easily, which was identified by a molecular docking (MD) study, and N3 is quite effective against Mpro due to the capability of inhibiting Mpro in a time‐dependent manner.[ 31 ] Mpro of SARS‐CoV‐2 is a homodimer enzyme consisting of three domains and having a substrate‐binding site with a catalytic dyad of His41 and Cys145 that is located between Domains I and II, whereas Domain III plays a role in dimerization between protomers as shown in Figure 2. The inhibitors are designed to inhibit important catalytic function of this enzyme, which directly inhibits the virus from infecting further inside the host cells.[ 32 ]
Figure 2.
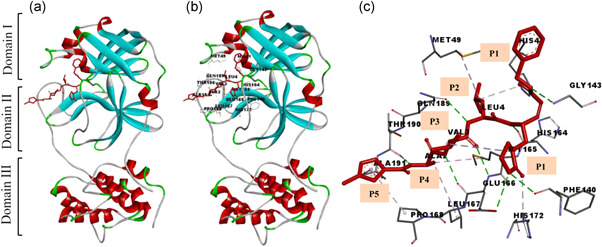
The main protease of SARS‐CoV‐2 contains the N3 inhibitor. (a) Schematic diagram of the dimeric Mpro‐N3 complex; (b) docking interaction of ligand N3 with protein (PDB: 6LU7); and (c) enlarged view of the binding interaction and subsites P1, P1ʹ, P2, P3, P4 and P5 of ligand N3. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
Havranek et al. carried out in silico studies to identify novel inhibitors as potential therapeutic targets against Mpro. Mpro is a 312 amino acid‐containing protein that is mainly involved in viral replication.[ 24 , 33 ] Since this protease is one of the potential targets for inhibition of COVID‐19,[ 34 ] the X‐ray crystal structure of this protease was used for docking purposes. Molecules 1–10 (Figure 3) have been used for docking against the SARS‐CoV‐2 main protease, PDB ID: 6LU7.[ 24 ]
Figure 3.
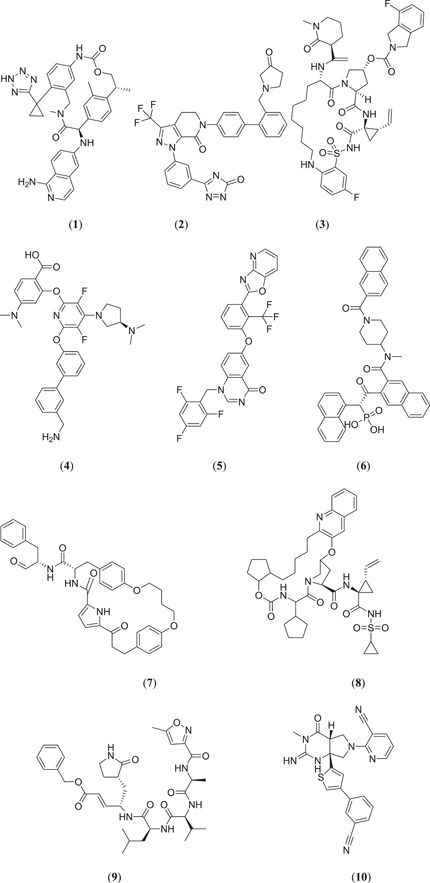
Structure of potential therapeutic agents that can inhibit SARS‐CoV‐2 Mpro
After docking and molecular dynamics simulation (MDS) studies, the binding affinities for the PDB and MD structures were analysed. Among them (Figure 3), compound (2R,15R)‐2‐[(1‐aminoisoquinolin‐6‐yl)amino]−4,15,17‐trimethyl‐7‐[1‐(2H‐tetrazol‐5‐yl)cyclopropyl]‐13‐oxa‐4,11‐diazatricyclo[14.2.2.16,10]henicosa‐1(18),6,8,10(21),16,19‐hexaene‐3,12‐dione (1) had a PDB structure and an MD structure that showed potent binding affinities of −10.6 and −10.0 kcal/mol, respectively. This molecule has macrocyclic tissue factor‐factor VIIa inhibitory activity, which can prevent blood coagulation after forming a complex with trypsin‐like serine protease factor Xa.[ 35 , 36 ] Du et al. reported that factor Xa is associated with the viral infectivity of SARS coronavirus by cleaving it into two functional units S1 and S2, which are further involved in enhancement of viral load.[ 37 ] Due to the presence of factor Xa in ACE2, the spike protein of SARS‐CoV‐2 can easily bind with ACE2 for viral entry. Therefore, macrocyclic tissue factor–factor VIIa inhibitor can be a useful target to inhibit SARS‐CoV‐2 replication because it inhibits factor Xa, which can further inhibit the entry of coronavirus into human cells.[ 24 ]
Khan et al. carried out a comparative molecular study to investigate potential inhibitors against Mpro. They chose 23 drug molecules and carried out a docking study using AutoDock Vina against Mpro (PDB ID: 6LU7), and identified 3 compounds (Figure 4) namely epirubicin (11), vapreotide (12) and saquinavir (13) with the best binding energies of −9.5, −9.1 and −9.5 kcal/mol, respectively.[ 38 ]
Figure 4.
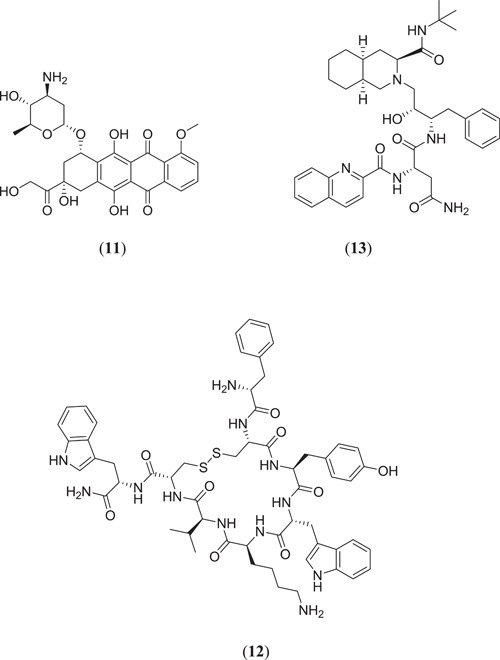
Structures of epirubicin, vapreotide and saquinavir
Gaudencio and Pereira[ 26 , 39 ] used a CADD that consisted of a ligand and a structure‐based method to find out the lead molecule against SARS‐CoV‐2 main protease from a marine natural product (MNP). They used the QSAR model to screen the huge database CheMBL and successfully selected 20 molecules, which were further subjected to a MD study against Mpro (PDB ID: 6LU7) using AutoDock Vina (version 1.1).[ 40 ] Before the docking study, intrinsic ligand N3 and water molecules from 6LU7 were removed using AutoDock Tools. After the docking study, it was found that 5 important natural marine products (Figure 5) (4aR,6aR,12aS,12bS)‐9‐(3,4‐dimethoxyphenyl)‐4a,12a‐dihydroxy‐4,4,6a,12b‐tetramethyl‐4a,5,6,6a,12,12a‐hexahydrobenzo[f]pyrano[4,3‐b]chromene‐1,11(4H,12bH)‐dione (14), (7aR,12aS,13aS)‐3,3,14,14‐tetramethyl‐11,12,13,13a,14,15‐hexahydro‐7a,12a‐(epiminomethano)indolizino[6,7‐h]pyrano[3,2‐a]carbazole‐7,8,16(3H,10H)‐trione (15), (2R,3S,4S,5S,6R)‐2‐(hydroxymethyl)‐6‐{[(3S,4S,4aR,6aS,12bS,12cS)‐4,12b,12c‐trimethyl‐4‐(4‐methylpent‐3‐en‐1‐yl)‐1,2,3,4,4a,5,6,6a,7,12,12b,12c‐dodecahydrobenzo[6]indeno[1,2‐b]indol‐3‐yl]oxy}tetrahydro‐2H‐pyran‐3,4,5‐triol (16), [4‐(6‐bromo‐1H‐indol‐3‐yl)‐1H‐imidazol‐2‐yl](1H‐indol‐2‐yl)methanone (17) and (6‐bromo‐1H‐indol‐2‐yl)[4‐(6‐bromo‐1H‐indol‐3‐yl)‐1H‐imidazol‐2‐yl]methanone (18) showed the best lead‐like properties with a good range of binding energies. Therefore, these molecules from marine origin can be further studied experimentally to identify a potential lead against the deadliest SARS‐CoV‐2 main protease.[ 39 ]
Figure 5.
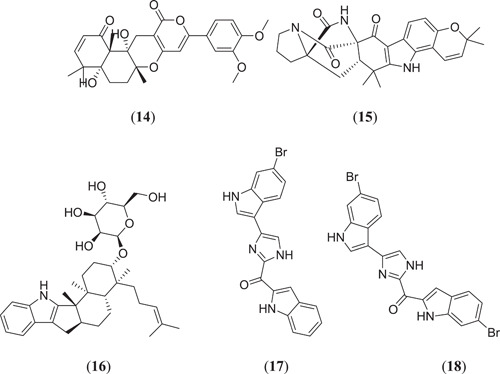
Structures of phytoconstituents obtained from marine natural products
In SARS‐CoV‐2, two kinds of proteins are present: structural and nonstructural proteins; nonstructural proteins are involved in different viral functions such as formation of a replicase and transcriptase complex, whereas structural proteins like membrane, spike, envelope and nucleocapsid proteins are involved in the attachment, fusion and release mechanism of viral contents.[ 26 ] Alazmi et al. carried out in silico virtual screening, characterization and docking and MD studies against SARS‐CoV‐2 proteins, in which they used 4 important proteins that were found to be very virulent for COVID‐19 disease. These proteins are nonstructural proteins 4 (NSP4) associated with viral replication, RNA‐dependent RNA polymerase (RdRp) associated with viral replication and transcription mechanism, uridylate‐specific endoribonuclease (NendoU/nsps15) involved in cleaving of bond and, finally, human ACE2, which was associated with viral entry into the human body.[ 41 , 42 ] Primary screening from the natural compound library (containing around 100,000 compounds) was performed to select the top 5 best docked ligands. The ligands used in docking studies are natural compounds (Figure 6), namely baicalin (19), kaempferol (20), limonin (21), nimbolide (22) and quercetin (23). The toxicities of ligand were checked using the ProTox‐II toxicity study model.[ 43 ] All these ligands were docked using AutoDock 4.2 against three target proteins, which were downloaded from the RCSB Protein Data Bank (PDB), that is, nsp15 (PDB ID: 6VWW), RdRp (PDB ID:6YYT) and human ACE2 (PDB ID: 6M1D Chain B), respectively.
Figure 6.
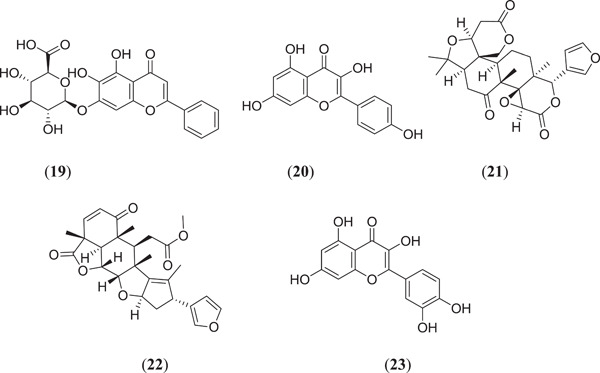
Structures of naturally obtained Mpro inhibitors
From the docking results, it was found that baicalin showed the best docking results against NSP4, NSP15 and RdRp, with binding energies of −6.8 ± 0.78, −7.4 ± 0.52 and −8.7 ± 0.35 kcal/mol, respectively, whereas limonin showed a good binding score of −11.0 ± 0.18 kcal/mol against ACE2 target proteins. The possible key interactions involved in the binding of baicalin with target proteins are as follows: NSP4: H bond with Ser496; NSP15: H bond with Thr341, Lys290, Ser294, Leu346 and Tyr343; and RdRp: H bond with Asp167, Ser798, Asp621, Tyr622, Lys801 and Lys624.[ 42 ]
India has been a global hub for many herbal and medicinal plants.[ 44 ] Calendula officinalis is one such important medicinal plant, which is also known as marigold, that has various important phytochemicals and bioactive compounds, due to which it is widely used as an antiseptic, antiulcer, antibacterial, wound healing and antigenotoxic agent.[ 45 , 46 , 47 ] Das et al. carried out in silico studies to identify potential inhibitors of SARS‐CoV‐2 main protease from flavonoid‐based phytoconstituents of C. officinalis. They obtained the crystal structure of SARS‐CoV‐2 main protease (Mpro) from PDB ID: 6LU7 and prepared the protein using Discovery Studio Visualizer (DSV) 2017 and important phytochemicals of C. officinalis that act as ligands.
Docking of all the possible phytoconstituents was carried out using AutoDock Vina against PBD ID: 6LU7. Among the 15 compounds, the top 3 compounds (Figure 7) rutin (24), isorhamnetin‐3‐β‐D (25) and calendoflaside (26), with the best binding energies of −8.8, −8.7 and −8.5 kcal/mol, respectively, were screened for further MDS to determine their conformational stability or flexibility and dynamic properties.[ 48 ] Aghee et al. investigated novel SARS‐CoV‐2 main protease inhibitors by in silico studies such as pharmacophore modelling, virtual screening, MD and MDS. After docking of a large database against PDB ID: 6LU7, CDocker energy was considered the selection criterion. Most of the compounds failed to show drug‐like properties because of Lipinski rule violation, but the compound ethyl 3‐hydroxy‐6‐methyl‐4‐{3‐methyl‐2‐[3‐methyl‐2‐(3‐methylbutanamido)butanamido]butanamido}heptanoate (27) (Figure 8) showed a good binding energy of −74.01 kcal/mol and drug‐like properties; hence, it can be further studied to establish its possible use against SARS‐CoV‐2.[ 49 ]
Figure 7.
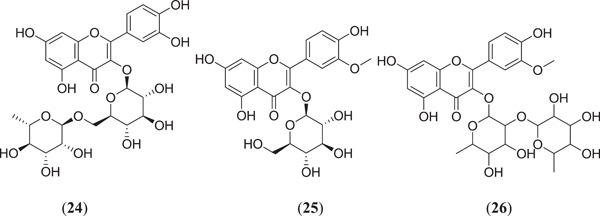
Structures of flavonoids obtained from Calendula officinalis
Figure 8.
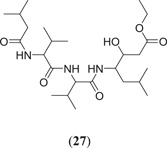
In silico screening of natural compounds against SARS‐CoV‐2. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
Joshi et al. performed in silico studies on 318 phytochemicals from 11 different plants by taking the main protease Mpro and ACE2 as the targets. Molecular docking was performed, and the top three compounds (Figure 9) quercetin‐3‐glucoronide‐7‐glucoside (28), quercetin‐3‐vicianoside (29) and absinthin (30) were found to have better binding energies of −7.9, −11.3; −8.3, −11.0 and −8.2, −11.8 kcal/mol, respectively, with both the targets Mpro and ACE2.[ 50 ] Krupanidhi et al. investigated the possible SARS‐CoV‐2 protease inhibitor from the phytochemicals present in Tinospora cordifolia, where five compounds were found to show good binding affinity towards the active site of the main protease (3CLpro); among them (Figure 9), tinosponone (31) was found to be the best compound against the target, with the best binding energy of −7.7 kcal/mol.[ 51 ] Borquaye et al. carried out in silico studies on important alkaloids obtained from Cryptolepis sanguinolenta, and MD was performed against Mpro and RdRp; 13 alkaloids were found to have a strong binding affinity towards the target, and among them, compounds (Figure 9) cryptospirolepine (32), cryptomisrine (33), bis‐cryptolepine (34) and cryptoquindoline (35) were found to show better binding energies of −10.00, −9.80; −10.60, −9.80; −8.80, −8.90 and −9.50, −8.75 kcal/mol against Mpro and RdRp, respectively.[ 52 ]
Figure 9.
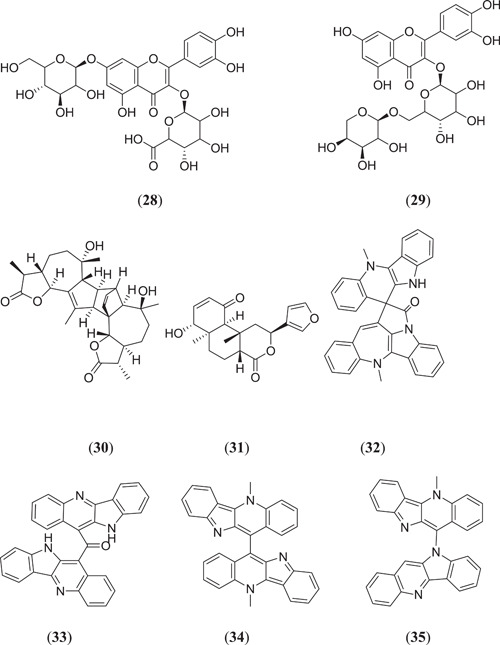
Natural phytoconstituents as Mpro inhibitors
Lokhande et al. investigated possible Mpro inhibitors from bioflavonoids containing Rhus succedanea using MD and MDS studies, where compounds amentoflavone (36) and agastiflavone (37) (Figure 10) interacted strongly with the catalytic site of Mpro, with binding energies of −27.04 and −25.87 kcal/mol, respectively.[ 53 ] Similarly, the diflavone compounds (Figure 10), amentoflavone (36), bilobetin (38) and ginkgetin (39), from the plant Torreya nucifera, and polyphenols from green tea such as epigallocatechin (40), (–)‐epicatechin gallate (41) and gallocatechin‐3‐gallate (42) were also found to act as Mpro inhibitors, with binding energies −9.2, −9.1, −9.0, −7.6, −8.2 and −9.0 kcal/mol, respectively.[ 54 ] Holanda et al. carried out in silico studies on Mpro inhibitors from 1,2,3‐triazole‐phthalimide derivatives, where the seven best compounds were subjected to a MD study; among them, one compound 2‐[1‐(1‐{[1‐(pyridin‐4‐yl)‐1H‐1,2,3‐triazol‐4‐yl]methyl}‐1H‐benzo[d]imidazol‐2‐yl)ethyl]isoindoline‐1,3‐dione (43) showed a promising docking result, with a binding energy of −10.26 kcal/mol, as compared to the binding energy of −7.0 kcal/mol of the N3 ligand.[ 55 ] Tripathi et al. carried out docking and molecular simulation studies of bioactive molecules from Withania somnifera against Mpro, in which 40 compounds were screened for Mpro‐inhibiting activity; 4 compounds showed the best binding energy after an MDS study. Withanoside V (44) showed better stability and binding affinity to the target protein; hence, it could act as a possible Mpro inhibitor.[ 56 ] Kumar et al. screened several metabolites obtained from natural sources, and their in silico study was carried out using MD and MDS; they found that ursolic acid (45), carvacrol (46) and oleanolic acid (47) showed binding energies of −4.0, −0.0 and −5.9 kcal/mol, respectively.[ 57 ]
Figure 10.
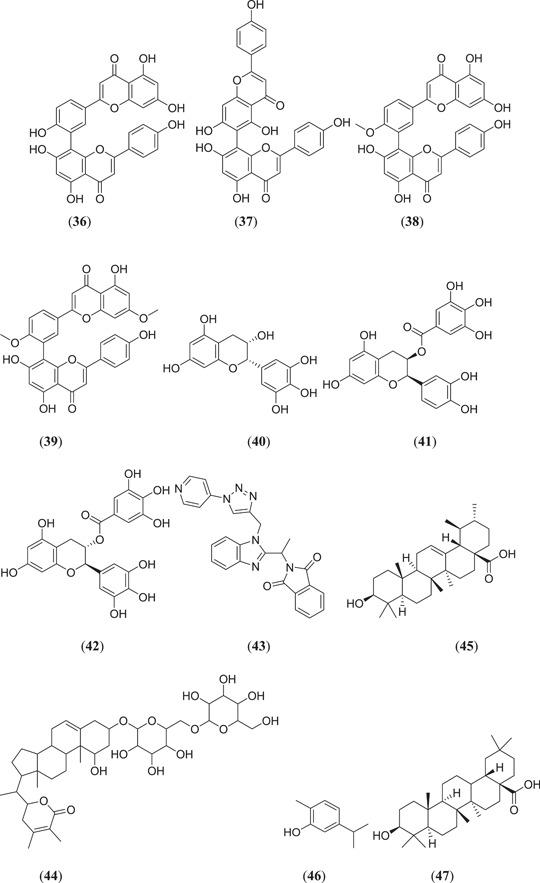
Diflavone metabolites from natural sources and steroidal compounds as Mpro inhibitors
Ghosh et al. screened some polyphenols from Broussonetia papyrifera and tested them for Mpro‐inhibiting activity; six phenolic compounds (Figure 11), namely, papyriflavonol A (48), broussochalcone A (49), 3ʹ‐(3‐methylbut‐2‐enyl)‐3ʹ,4ʹ,7ʹ‐trihydroxyflavone (50), broussoflavan A (51), kazinol J (52) and kazinol F (53), showed good binding energies in the range of −7.6 to −8.2 kcal/mol.[ 58 ] Ibrahim et al. screened 18 anti‐COVID‐19 drug candidates and performed an in silico study; they found that compound (3R,3aS,6aR)‐hexahydrofuro[2,3‐b]furan‐3‐yl ((2S,3R)‐4‐{2‐[(1‐cyclopentylpiperidin‐4‐yl)amino]‐N‐isobutylbenzo[d]thiazole‐6‐sulfonamido}‐3‐hydroxy‐1‐phenylbutan‐2‐yl)carbamate (54) showed good binding energy and good stability tested by a MDS.[ 59 ] Choudhury investigated the active phytoconstituents of the Indian medicinal herb T. cordifolia (Giloy) against the Mpro protein, where (Figure 11) berberine (55) showed better binding affinity with (−7.3 kcal/mol).[ 60 ] Rout et al. carried out an in silico study on spice molecules to determine the inhibitory activity against SARS‐CoV‐2 RBD Spro and Mpro, where (Figure 11) piperine (56) was found to have good binding activity in the range of −6.4 and −7.3 kcal/mol against both the target proteins.[ 61 ] Aanouz et al. investigated Moroccan medicinal plants as Mpro inhibitors using in silico studies, where, among 67 natural compounds, 3 compounds (Figure 11), namely, β‐eudesmol (57), digitoxigenin (58) and crocin (59), showed satisfactory results as Mpro inhibitors, with binding energies of −7.1, −7.2 and −8.2 kcal/mol, respectively.[ 62 ]
Figure 11.
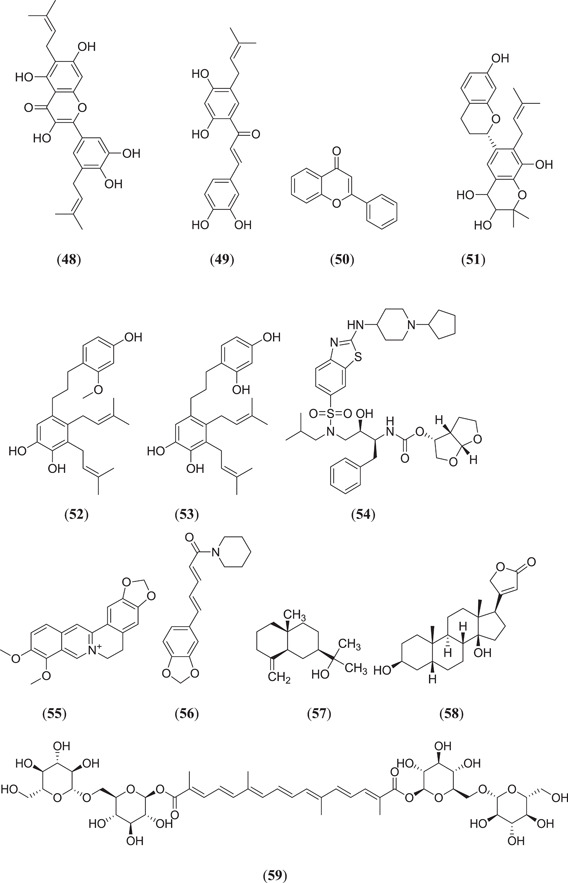
Polyphenolic anti‐COVID drugs and natural phytoconstituents from Moroccan medicinal plants
Keretsu et al. investigated some previously used antiviral drug moieties and carried out in silico MD and dynamic simulation studies to identify potential inhibitors of Mpro, in which three compounds (Figure 12), namely, aclarubicin (60), TMC‐310911 (61) and faldaprevir (62), showed potential Mpro‐inhibiting property.[ 63 ] The ZINC database has become a useful tool for in silico studies. Kumar et al. screened a large database of molecules and selected two molecules, 5‐(furan‐2‐yl)‐N‐{1‐[2‐(piperidin‐1‐yl)ethyl]‐1H‐benzo[d]imidazol‐2‐yl}‐7‐(trifluoromethyl)pyrazolo[1,5‐a]pyrimidine‐3‐carboxamide (63) and (4‐benzylpiperazin‐1‐yl)[5‐(thiophen‐2‐yl)−7‐(trifluoromethyl)‐[1,2,4]triazolo[1,5‐a]pyrimidin‐2‐yl]methanone (64), for MD and dynamic simulation studies, and both the molecules showed comparable binding affinity as the cocrystal ligand N3 of the Mpro protein.[ 64 ] Shree et al. carried out an in silico study on active phytoconstituents from three medicinal plants, namely W. somnifera, T. cordifolia and Ocimum sanctum, from which compounds were docked against Mpro (PDB ID: 6LU7), and it was found that withanoside V (44) (Figure 10) (10.32 kcal/mol) and somniferine (65) (9.62 kcal/mol) from W. somnifera, tinocordiside (66) (8.10 kcal/mol) from T. cordifolia and vicenin (67), isorientin 4‐O‐p‐hydroxy‐benzoate (68) and ursolic acid (45) (Figure 10) from O. sanctum showed good binding energies of 8.97, 8.55 and 8.52 kcal/mol, respectively.[ 65 ] Bioactive molecules from plants and herbs can be excellent inhibitors of COVID‐19, with very negligible risk of toxicity.[ 66 ] Tallei et al. carried out research on bioactive molecules, where compounds (Figure 12) hesperidin (69), nabiximols (70), pectolinarin (71) and rhoifolin (72) were found to act as potential Mpro inhibitors as compared to chloroquine and hydroxychloroquine sulphate.[ 67 ]
Figure 12.
Some antiviral and natural phytoconstituents as Mpro inhibitors
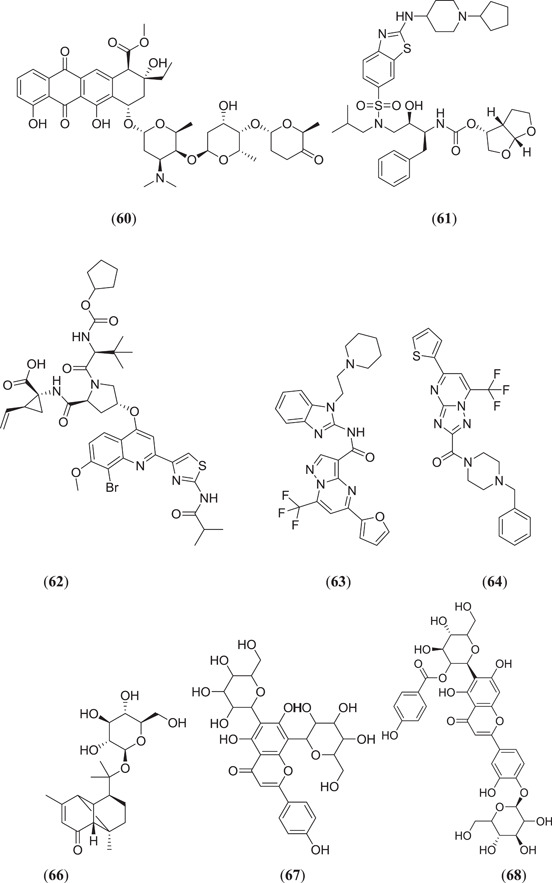
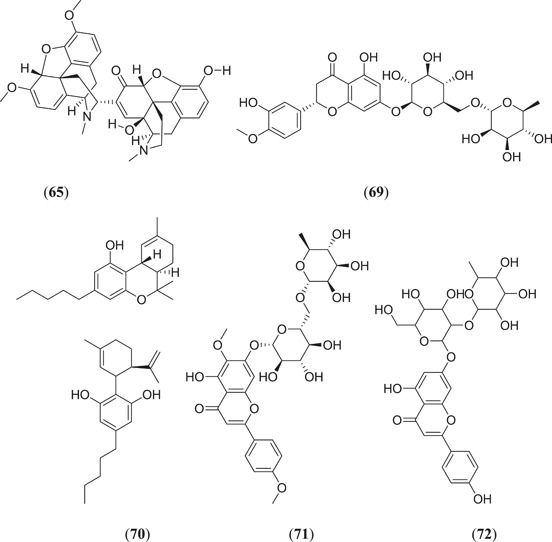
Various ligands from the ZINC database were used for in silico studies to identify new hit molecules against the target protein. Razzaghi‐Asl et al. investigated various ligands against Mpro, where compound (Figure 13) (R)‐1‐[5‐((1S,6S)‐bicyclo[4.1.0]heptane‐7‐carboxamido)pyridin‐2‐yl]piperidine‐3‐carboxamide (73) showed good binding affinity as well as good thermodynamic stability after binding with the target protein.[ 68 ] Andrographolides (Figure 13) (74) obtained from Andrographis peninculata were subjected to various in silico studies such as target analysis, toxicity study and MD against Mpro, and showed good binding affinity, with a binding energy of −3.094 kcal/mol.[ 69 ] In silico studies on various phytochemicals can lead to the discovery of new Mpro inhibitors. Twelve important phytochemicals were subjected to various in silico studies, of which compounds (Figure 13) glycyrrhizin (75), tryptanthrine (76), rhein (77) and berberine (55) (Figure 11) showed significant binding affinity, with binding energies of −8.1, −8.2, −8.9 and −8.1 kcal/mol, respectively, and all these molecules also showed good drug‐like properties.[ 70 ] From in silico studies of 20 compounds, four metabolites namely amentoflavone (36) (Figures 10 and 13), guggulsterone (78), puerarin (79) and piperine (56) (Figure 11) showed strong binding affinity with Mpro of SARS‐CoV‐2, and further molecular dynamic studies confirmed their thermodynamic stability.[ 71 ] Prasanth et al. investigated 48 phytoconstituents from Cinnamon, and MD was performed; compounds (Figure 13) tenuifolin (80) and pavetannin C1 (81) showed good binding energies of −8.8 and −11.1 kcal/mol, respectively.[ 72 ]
Figure 13.
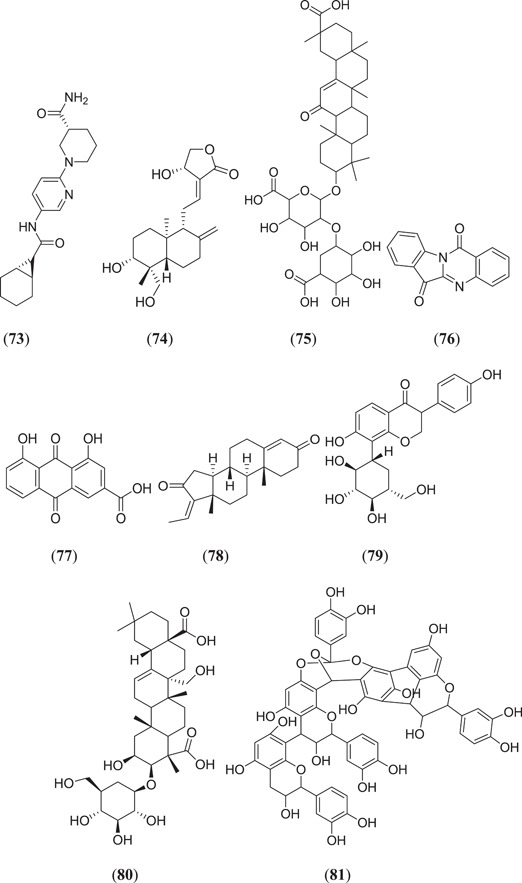
Structures obtained from the ZINC database and natural phytoconstituents
Jin et al. carried out in silico screening studies on 10,000 compounds to identify Mpro inhibitors; six compounds (Figure 14) ebselen (82), disulfiram (83), tideglusib (84), carmofur (85), shikonin (86) and PX‐12 (87) showed drug‐like properties and good binding affinity with the target protein.[ 31 ] Screening of the Asinex Biol‐Design Library was performed by Kanhed and his team to identify Mpro inhibitors; 20 compounds (Figure 14) from four classes, namely, disubstituted pyrazoles 4‐[5‐(3‐ethoxy‐4‐hydroxyphenyl)‐1‐(2‐hydroxyethyl)‐1H‐pyrazol‐3‐yl]benzene‐1,2‐diol (88), 4‐[5‐(3‐ethoxy‐4‐hydroxyphenyl)‐1‐(2‐hydroxyethyl)‐1H‐pyrazol‐3‐yl]benzene‐1,3‐diol (89), 4‐[5‐(4‐hydroxy‐3‐methoxyphenyl)‐1‐(2‐hydroxyethyl)‐1H‐pyrazol‐3‐yl]‐2,6‐dimethoxyphenol (90), 2‐[5‐(4‐hydroxy‐3‐methoxyphenyl)‐1‐(2‐hydroxyethyl)‐1H‐pyrazol‐3‐yl]benzene‐1,4‐diol (91), 4‐(2‐amino‐2‐oxoethoxy)‐3‐[5‐(2,3‐dimethoxyphenyl)‐1‐phenethyl‐1H‐pyrazol‐3‐yl]phenyl hydrogen carbonate (92), cyclic amide containing compound 4‐ethyl‐N‐[1‐(4‐fluoro‐2‐phenoxyphenoxy)propan‐2‐yl]‐6‐isobutyl‐5,8,12‐trioxo‐3,4,5,6,7,8,9,10,11,12‐decahydro‐2H‐benzo[m][1,4,7,11]oxatriazacyclotetradecine‐10‐carboxamide (93), 3‐benzyl‐6‐(hydroxymethyl)‐N‐[2‐(4‐methoxyphenoxy)ethyl]‐5,8,12‐trioxo‐3,4,5,6,7,8,9,10,11,12‐decahydro‐2H‐benzo[m][1,4,7,11]oxatriazacyclotetradecine‐10‐carboxamide (94), 6‐benzyl‐N‐{2‐[2‐(3‐chlorophenoxy)‐4‐fluorophenoxy]ethyl}‐4‐ethyl‐5,8,12‐trioxo‐3,4,5,6,7,8,9,10,11,12‐decahydro‐2H‐benzo[m][1,4,7,11]oxatriazacyclotetradecine‐10‐carboxamide (95), 7‐(4‐hydroxybenzyl)‐6,9,14‐trioxo‐N‐[2‐(pyridin‐2‐yl)ethyl]‐3,4,5,6,7,8,9,10,11,12,13,14‐dodecahydro‐2H‐benzo[o][1,5,8,13]oxatriazacyclohexadecine‐12‐carboxamide (96), 3‐benzyl‐N‐[2‐(4‐fluorophenoxy)ethyl]‐6‐(hydroxymethyl)‐5,8,12‐trioxo‐3,4,5,6,7,8,9,10,11,12‐decahydro‐2H‐benzo[m][1,4,7,11]oxatriazacyclotetradecine‐10‐carboxamide (97), 3‐benzyl‐6‐isopropyl‐5,8,12‐trioxo‐N‐(3‐phenylpropyl)−3,4,5,6,7,8,9,10,11,12‐decahydro‐2H‐benzo[m][1,4,7,11]oxatriazacyclotetradecine‐10‐carboxamide (98), 6‐benzyl‐N‐{1‐[2‐(3‐chlorophenoxy)‐4‐fluorophenoxy]propan‐2‐yl}‐4‐ethyl‐5,8,12‐trioxo‐3,4,5,6,7,8,9,10,11,12‐decahydro‐2H‐benzo[m][1,4,7,11]oxatriazacyclotetradecine‐10‐carboxamide (99), 6‐benzyl‐N‐{1‐[2‐(3‐chlorophenoxy)‐6‐fluorophenoxy]propan‐2‐yl}−4‐ethyl‐5,8,12‐trioxo‐3,4,5,6,7,8,9,10,11,12‐decahydro‐2H‐benzo[m][1,4,7,11]oxatriazacyclotetradecine‐10‐carboxamide (100), 3‐benzyl‐N‐[3‐(4‐ethoxy‐3‐methoxyphenyl)propyl]‐6‐(hydroxymethyl)‐5,8,12‐trioxo‐3,4,5,6,7,8,9,10,11,12‐decahydro‐2H‐benzo[m][1,4,7,11]oxatriazacyclotetradecine‐10‐carboxamide (101), pyrrolidine‐based N‐(2‐fluoro‐1‐{2‐[6‐(3‐fluorobenzyl)pyrazin‐2‐yl]pyrrolidin‐1‐yl}‐1‐oxo‐3‐phenylpropan‐2‐yl)acetamide (102), 1‐[4‐(aminomethyl)benzoyl]‐N‐(4‐fluorobenzyl)‐4‐(4‐fluorophenyl)‐3‐hydroxypyrrolidine‐2‐carboxamide (103), 3‐(2‐aminoethoxy)‐N‐{[3‐hydroxy‐1‐(1,6‐naphthyridin‐4‐yl)pyrrolidin‐3‐yl]methyl}benzamide (104), 1‐[3‐(2‐aminoethoxy)benzoyl]‐N‐benzhydryl‐4‐(4‐fluorophenyl)‐3‐hydroxypyrrolidine‐2‐carboxamide (105) and miscellaneous types 1‐[3‐(5‐{[6‐hydroxy‐4‐(2‐methoxyacetyl)‐6‐[(p‐tolyloxy)methyl]‐1,4‐diazepan‐1‐yl]methyl}‐2‐methoxyphenoxy)propyl]azepan‐2‐one (106), 4‐(6‐methoxynicotinoyl)‐1‐propyl‐3‐[2‐(pyridin‐2‐yl)benzyl]piperazin‐2‐one (107), were found to show Mpro‐inhibiting activity after docking against the target protein.[ 73 ] Vijayakumar et al. carried out in silico studies on flavonoids, where chalcone derivative (E)‐3‐(1H‐indol‐3‐yl)‐1‐(naphthalen‐2‐yl)prop‐2‐en‐1‐one (108) and quercetin (23) (Figure 6) showed the best binding affinity with Mpro, with binding energies of −10.4 and −9.2 kcal/mol, respectively.[ 74 ] A structure‐based study revealed that various phytochemicals including polyphenols and alkaloids have Mpro‐ and spike protein‐inhibiting activity.[ 75 , 76 , 77 ] Anisotine (Figure 14) (109) is an important alkaloid from the plant Justicia adhatoda that showed a good binding energy of −7.9 kcal/mol after docking against Mpro.[ 54 ] Ibrahim et al. docked 2017 flavone molecules against Mpro, and of these, two compounds (Figure 14) 3‐[((1R,2R,3S,4R,5S)‐2,3‐dihydroxy‐4‐{[(1R,2R,3R,4R,5S)‐2,3,4‐trihydroxy‐5‐(hydroxymethyl)cyclohexyl]oxy}‐5‐{[((2R,3R,4S,5R,6S)‐3,4,5‐trihydroxy‐6‐methyltetrahydro‐2H‐pyran‐2‐yl)oxy]methyl}cyclohexyl)oxy]‐2‐(3,4‐dihydroxyphenyl)‐5,7‐dihydroxy‐4H‐chromen‐4‐one (110) and (1R,2S,3R,4R,6R)‐6‐{[2‐(3,4‐dihydroxyphenyl)‐5,7‐dihydroxy‐4‐oxo‐4H‐chromen‐3‐yl]oxy}‐2,3dihydroxy‐4‐{[((2R,3R,4R,5R,6S)‐3,4,5‐trihydroxy‐6‐methyltetrahydro‐2H‐pyran‐2‐yl)oxy]methyl}cyclohexyl‐3,4,5‐trihydroxybenzoate (111) showed considerable thermodynamic stability and good binding affinity of −10.7 and −9.5 kcal/mol, respectively.[ 78 ] Umar et al. studied some active compounds obtained from Azadirachta indica, Mangifera indica and Moringa oleifera, after docking of selected compounds against Mpro, and found that mangiferin (Figure 14) (112) showed the highest binding affinity towards target protein, with a binding energy of −8.4 kcal/mol.[ 79 ] Swargiary et al. investigated the binding affinity of various phytoconstituents with 3‐chymoytrpsin‐like (3CLpro; PDB ID: 6M2N) and papain‐like protease (PLpro; PDB ID: 7JN2); out of 32 compounds only two compounds, amentoflavone (36) (Figure 10) and gallocatechin gallate (113), showed the best binding affinity towards the target protein.[ 80 ]
Figure 14.
Disubstituted pyrazole, cyclic amide, pyrrolidine‐based, and some flavonoids and alkaloids as Mpro inhibitors
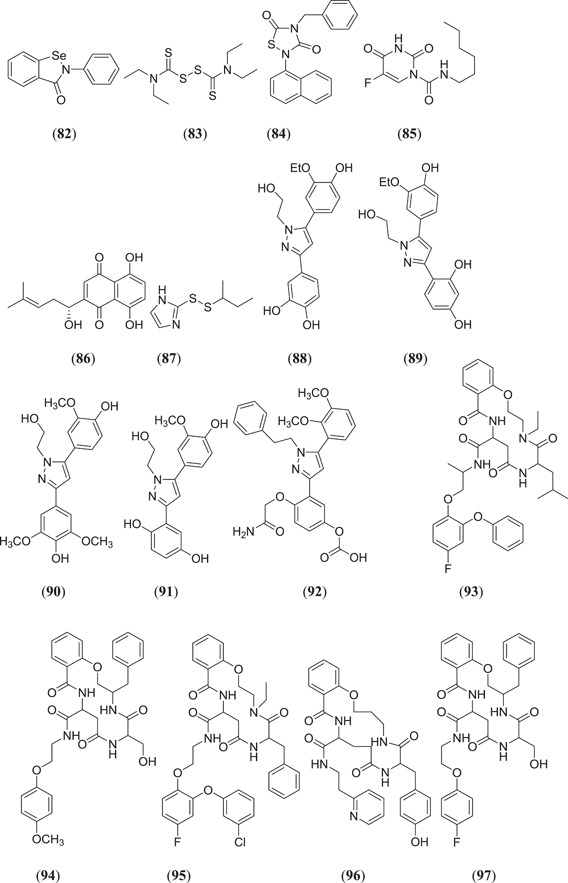
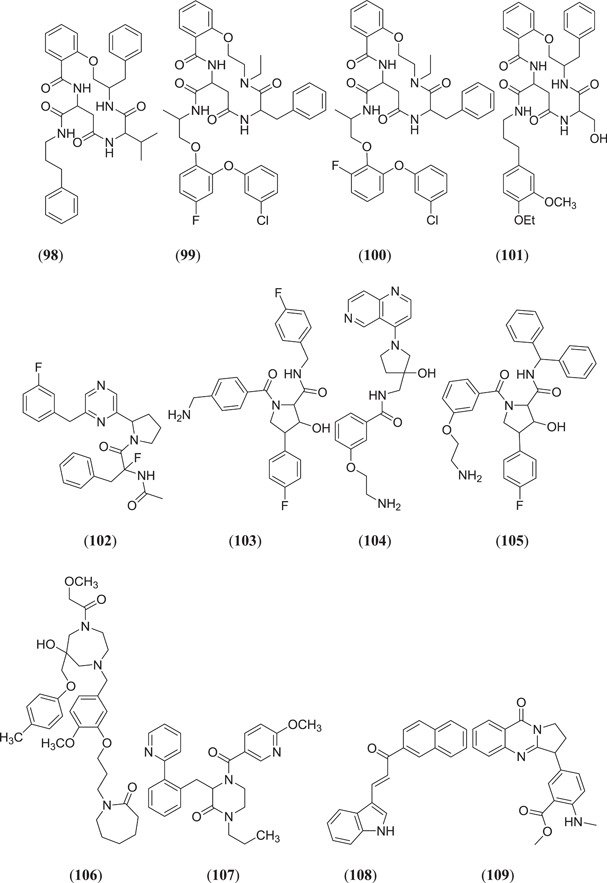
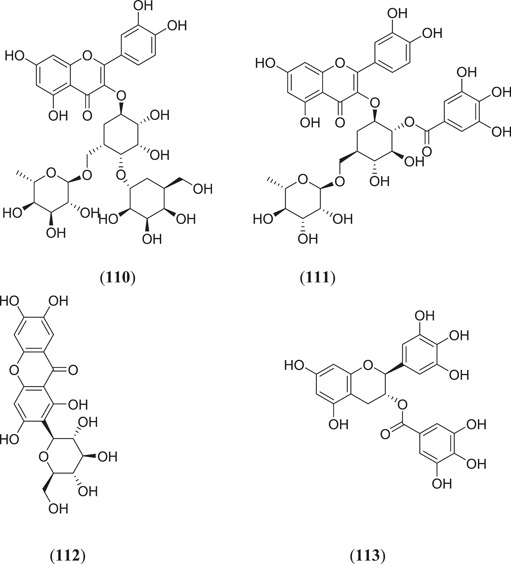
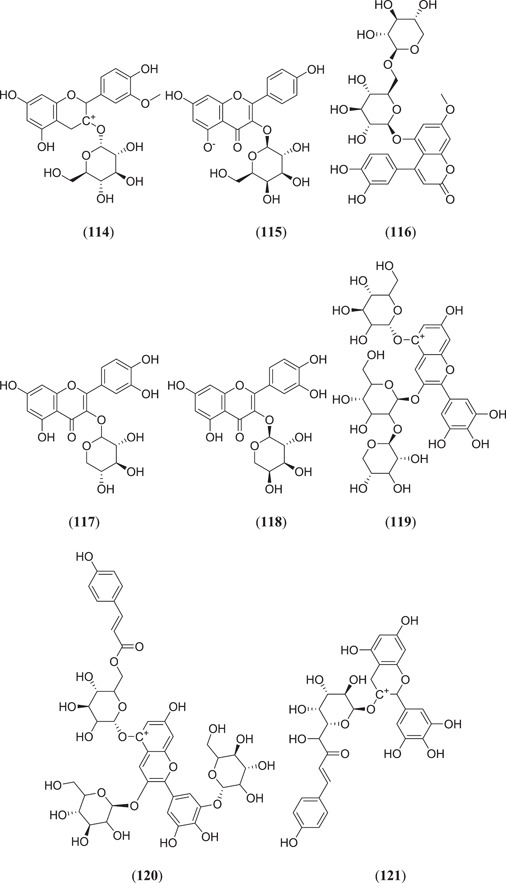
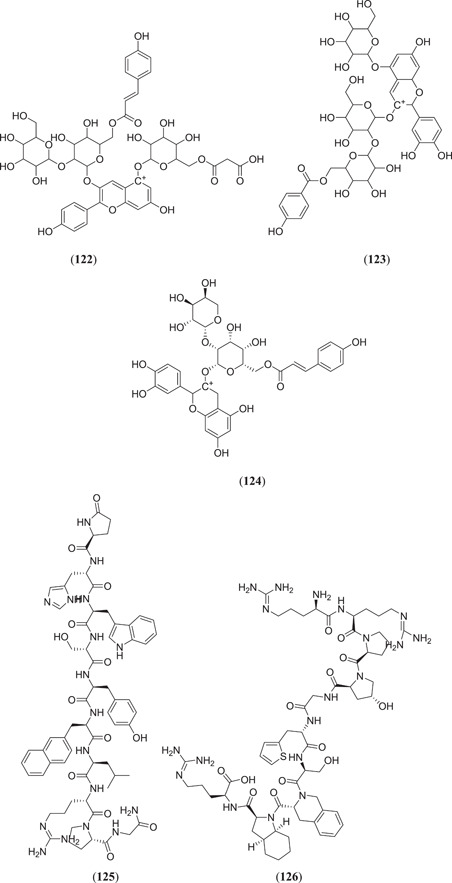
Majumder and Mandal screened the Sigma‐Aldrich plant profiler chemical library by in silico docking against the main protease PDB ID: 6LU7 to identify Mpro inhibitors; of these (Figure 15), compounds that showed the best binding energy are peonidin 3‐O‐glucoside (114), kaempferol 3‐O‐β‐rutinoside (115), 4‐(3,4‐dihydroxyphenyl)‐7‐methoxy‐5‐[(6‐O‐β‐d‐xylopyranosyl‐β‐d‐glucopyranosyl)oxy]−2H−1‐benzopyran‐2‐one (116), quercetin‐3‐d‐xyloside (117) and quercetin 3‐O‐α‐l‐arabinopyranoside (118), and their thermodynamic stability was confirmed by performing a MDS study.[ 81 ] Ateeq Ahmed, in his study, screened more than 51 phytoconstituents of Juniperus procera Hochst against Mpro, of which rutin (24) (Figure 7) showed the highest interaction score of −9.00 kcal/mol against the selected target.[ 82 ] Das and his team investigated 33 molecules from natural compounds, antifungal, antiviral, antiprotozoal and antinematodes against Mpro; among them, rutin (24) showed the highest inhibitory activity, with a ∆G value of −9.55 kcal/mol.[ 83 ] Fakhar et al. screened anthocyanin‐derived compounds against Mpro and they found the six best compounds (Figure 15): 3‐{((2S,5S)‐4,5‐dihydroxy‐6‐(hydroxymethyl)‐3‐[((2S,3R,5R)‐3,4,5‐trihydroxytetrahydro‐2H‐pyran‐2‐yl)oxy]tetrahydro‐2H‐pyran‐2‐yl)oxy}‐7‐hydroxy‐5‐{[(2S,4S,5S)‐3,4,5‐trihydroxy‐6‐(hydroxymethyl)tetrahydro‐2H‐pyran‐2‐yl]oxy}‐2‐(3,4,5‐trihydroxyphenyl)‐5H‐chromen‐5‐ylium (119), 2‐(3,4‐dihydroxy‐5‐{[(2S,4S,5S)‐3,4,5‐trihydroxy‐6‐(hydroxymethyl)tetrahydro‐2H‐pyran‐2‐yl]oxy}phenyl)‐7‐hydroxy‐5‐{[(2S,4S,5S)‐3,4,5‐trihydroxy‐6‐({[(E)‐3‐(4‐hydroxyphenyl)acryloyl]oxy}methyl)tetrahydro‐2H‐pyran‐2‐yl]oxy}‐3{[(2S,4S,5S)‐3,4,5‐trihydroxy‐6‐(hydroxymethyl)tetrahydro‐2H‐pyran‐2‐yl]oxy}‐5H‐chromen‐5‐ylium (120), 5,7‐dihydroxy‐3‐({(2S,3R,4S,5R,6S)‐3,4,5‐trihydroxy‐6‐[(E)‐1‐hydroxy‐4‐(4‐hydroxyphenyl)‐2‐oxobut‐3‐en‐1‐yl]tetrahydro‐2H‐pyran‐2‐yl}oxy)‐2‐(3,4,5‐trihydroxyphenyl)chroman‐3‐ylium (121), (E)‐5‐({6‐[(2‐carboxyacetoxy)methyl]‐3,4,5‐trihydroxytetrahydro‐2H‐pyran‐2‐yl}oxy)‐3‐{[4,5‐dihydroxy‐6‐({[3‐(4‐hydroxyphenyl)acryloyl]oxy}methyl)‐3‐{[3,4,5‐trihydroxy‐6‐(hydroxymethyl)tetrahydro‐2H‐pyran‐2‐yl}oxy)tetrahydro‐2H‐pyran‐2‐yl]oxy}‐7‐hydroxy‐2‐(4‐hydroxyphenyl)‐5H‐chromen‐5‐ylium (122), 3‐({4,5‐dihydroxy‐6‐(hydroxymethyl)‐3‐[(3,4,5‐trihydroxy‐6‐{[(4‐hydroxybenzoyl)oxy]methyl}tetrahydro‐2H‐pyran‐2‐yl)oxy]tetrahydro‐2H‐pyran‐2‐yl}oxy)‐2‐(3,4‐dihydroxyphenyl)‐7‐hydroxy‐5‐{[3,4,5‐trihydroxy‐6‐(hydroxymethyl)tetrahydro‐2H‐pyran‐2‐yl]oxy}‐3,8a‐dihydro‐2H‐chromen‐3‐ylium (123) and 3‐({(2S,3R,4R,5S,6S)‐4,5‐dihydroxy‐6‐({[(E)‐3‐(4‐hydroxyphenyl)acryloyl]oxy}methyl)‐3‐[((2R,3R,4S,5S)‐3,4,5‐trihydroxytetrahydro‐2H‐pyran‐2‐yl)oxy]tetrahydro‐2H‐pyran‐2‐yl}oxy)‐2‐(3,4‐dihydroxyphenyl)‐5,7‐dihydroxychroman‐3‐ylium (124), with binding energy ranging from −12.37 to −9.58 kcal/mol.[ 84 ] Chatterjee et al. screened the top five hit compounds against SARS‐CoV‐2 Mpro and found that nafarelin (125) and icatibant (126) showed the best binding energies of −712.94 and −851.74 kJ/mol, respectively.[ 85 ]
Figure 15.
Structures from the Sigma‐Aldrich plant library, phytoconstituents from Juniperus procera and already existing antiviral, antiprotozoal agents as Mpro inhibitors
3. TARGETING THE SARS‐COV‐2 SPIKE PROTEIN (PDB ID: 6M0J)
It is known that the human ACE2 receptors are attacked by spike proteins of SARS‐CoV‐2.[ 86 , 87 , 88 ] The SARS‐CoV‐2 spike glycoproteins are homo‐trimeric in nature, contain the S1 and S2 subunits in each of the spike monomers, which play a role in binding to cell receptors, lead to fusion between the viral membrane and host cell membrane, followed by successive entry of the virus, where cellular protease and serine protease TMPRSS2 promote cleavage of these two subunits. Cryo‐electron microscopy studies revealed that RBD carries out dissociation between the S1 subunit and ACE2, whereas the S2 subunit transforms from a metastable prefusion state to a more stable postfusion state, which is very much essential for membrane fusion.[ 89 , 90 ] The RBD of SARS‐CoV‐2 with twisted pentameric β‐sheets contains subunits β1, β2, β3, β4 and β7 with loops and connecting helices that form the main core. Β5 and β6 are short subunits that are present in between β4 and β7. The β‐sheets of RBD are stabilized by various pairs of cysteine residues; among them, three important pairs are Cys379‐Cys432, Cys336‐Cys361 and Cys391‐Cys525 and the remaining pair Cys480‐Cys488 promotes connection of the loop in the distal end of the receptor‐binding motif (RBM) of RBD as shown in Figure 16.[ 91 ]
Figure 16.
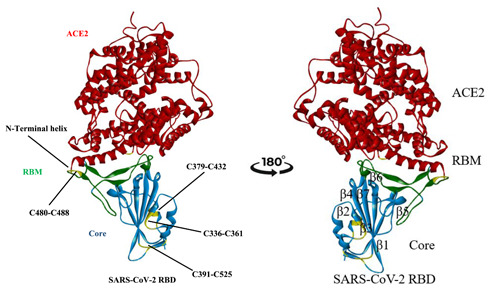
Crystal structure of the SARS‐CoV‐2 spike receptor‐binding domain (RBD) bound with ACE2 (PDB ID: 6M0J). Red represents ACE2, cyan represents the SARS‐CoV‐2 RBD core and green represents RBM. Yellow disulphide linkages in the SARS‐CoV‐2 RBD are shown as sticks. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
The potential drug target for the spike protein can be of three types such as host protease inhibition, fusion inhibition and neutralizing antibodies. Under host protease inhibition, TPC2, TMPRSS2 and cathepsin inhibitors can be used, under fusion inhibition, peptides that can target the HR1 domain can be one option and viracept can be used, which is an HIV‐protease inhibitor, and finally, by using potential neutralizing antibodies, the binding tendency of the virus with various host cell targets such as RBD, S1, S2 and the N‐terminus can be inhibited.[ 92 ]
For an in silico model of identification of drugs, virtual screening and pharmacophore‐based modelling play the most important roles.[ 93 ] Shehroz et al. used a CADD to identify the lead against spike protein of SARS‐CoV‐2. Screening of molecules with molecular weight <1.2 kDa was performed using the Cambridge Structural Database, the ZINC database, the Drug Bank and the TIMBAL database.[ 94 ] Molecules with protein–ligand interaction potential and those that followed the Lipinski rule were selected as leads against the desires pharmacophore.[ 95 , 96 ] Out of 1327 hits, 10 compounds that showed the best fit scores above >50 were selected and docking was performed using AutoDock Vina against RBD of the S protein. SARS‐CoV‐2 and all these eight compounds (Figure 17) N‐ethyl‐2,3‐difluoro‐N‐[(3R,4R)‐3‐hydroxy‐1‐(thiophene‐2‐carbonyl)piperidin‐4‐yl]benzamide (127), 1‐[4‐(hexylamino)‐2‐(pyridin‐3‐yl)‐5,6‐dihydropyrido[3,4‐d]pyrimidin‐7(8H)‐yl]ethanone (128), 6‐(1H‐imidazol‐1‐yl)‐N‐[1‐(3,4,5‐trifluorophenyl)ethyl]pyrimidin‐4‐amine (129), N‐[3‐(3‐{1‐[3‐(4‐fluorophenyl)−1,2,4‐oxadiazol‐5‐yl]ethyl}ureido)‐4‐methyl phenyl]acetamide (130), 1‐{4‐[(3‐phenylpropyl)amino]‐2‐(pyridin‐3‐yl)‐5,6‐dihydropyrido[3,4‐d]pyrimidin‐7(8H)‐yl}ethanone (131), 1‐[2‐(4‐methyl‐6,7‐dihydro‐5H‐cyclopenta[d]pyrimidin‐2‐yl)ethyl]‐3‐[3‐(oxazol‐5‐yl)phenyl]urea (132), 1‐[4‐methyl‐3‐(2‐oxopyrrolidin‐1‐yl)phenyl]‐3‐[(tetrahydro‐2H‐pyran‐4‐yl)methyl]urea (133) and 1‐(isoxazol‐3‐ylmethyl)‐3‐[4‐methyl‐3‐(2‐oxopyrrolidin‐1‐yl)phenyl]urea (134) showed good binding affinity towards the selected target.[ 94 ]
Figure 17.
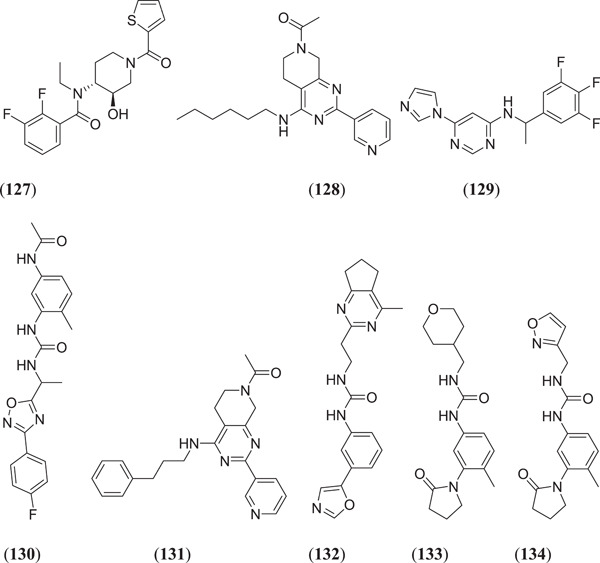
Structures obtained from the Cambridge Structural Database, the ZINC database, Drug Bank and the TIMBAL database for use against the SARS‐CoV‐2 spike protein. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
Chikhale et al. investigated the phytoconstituents present in W. somnifera (Indian Ginseng) for SARS‐CoV‐2 host entry and spike protein inhibition; compound (Figure 18) quercetin glucoside (135) showed the best binding affinity against target proteins.[ 97 ] Teli et al. conducted an in silico study of natural compounds obtained from various plant species; compounds (Figure 18) solanine (136) and rutin (24) (Figure 7) showed both Mpro and spike glycoprotein RBD receptor antagonistic properties.[ 98 ] Patel et al. investigated phytochemicals from Curcuma longa, and docking was carried out against spike protein (PDB ID: 6M0J) using AutoDock 4.2; it was found that bis‐demethoxycurcumin (137) showed a binding energy of −10.01 kcal/mol with the target protein.[ 99 ] Tallei et al. carried out in silico research on various bioactive molecules, and the MD results showed that hesperidin (69), nabiximols (70), pectolinarin (71), epigallocatechin (40) (Figure 10) and rhoifolin (72) (Figure 12) can act as possible SARS‐CoV‐2 spike protein inhibitors.[ 67 , 100 ] Vijayakumar and his team studied various flavonoids using an in silico approach, where postdocked analysis found that quercetin (23) (Figure 6) binds with the target protein with a binding affinity of −7.8 kcal/mol, and it can act as a competitive inhibitor against spike protein.[ 74 ] Propolis is a kind of supplement that is present in natural compounds and mostly used in Indonesia.[ 101 ] Various natural components of propolis were screened and docked against spike protein subunit S2, and it was found that four flavonoids, namely (Figure 18), neoblavaisoflavone (138), methylphiopogonone (139), 3ʹ‐methoxydaidzin (140) and genistein (141), showed good binding energies of −8.3, −8.1, −8.2 and −8.3 kcal/mol, respectively, as compared to pravastatin (−7.3 kcal/mol).[ 102 ] Panja et al. studied the mutation in the spike protein's genomic sequences, and five calcium chelating drugs were docked against this spike protein; of these, compounds (Figure 18) penicillamine (142) and dimercaprol (143) showed good inhibitory activity against Furin and ACE2 of the spike protein.[ 103 ] Pandey et al. investigated several phytochemicals for SARS‐CoV‐2 spike proteins through a MD study, and they found three compounds, namely, fisetin (144), quercetin (23) and kaempferol (20), with promising binding affinity against the target protein; MD was performed to confirm their thermodynamic stability.[ 104 ] Wen et al. investigated and prepared a library of 8820 compounds after MD and found a single compound, trichostatin A (145), with significant Mpro inhibition activity.[ 105 ]
Figure 18.
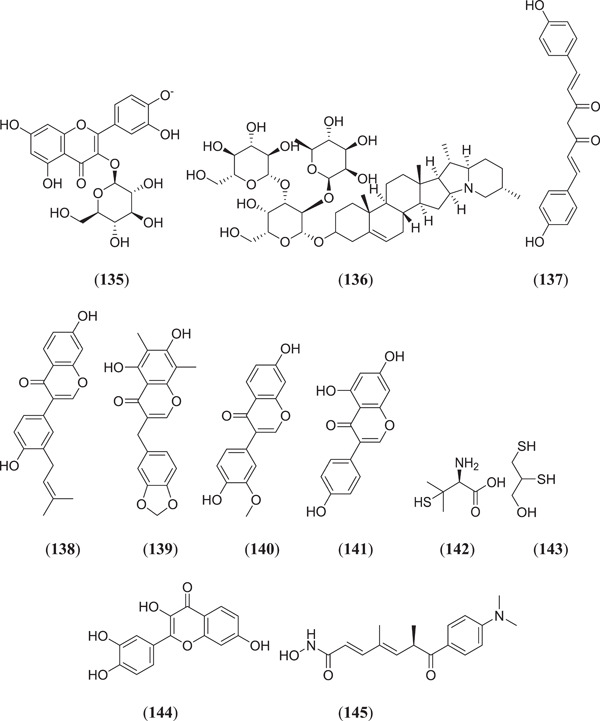
Various flavonoids as SARS‐CoV‐2 spike protein inhibitors. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
4. TARGETING SARS‐CoV‐2 RNA‐DEPENDENT RNA POLYMERASE (RdRp)
RdRp, which is commonly known as nsp12, is involved in the mechanism of replication and transcription of SARS‐CoV‐2 viral proteins.[ 106 ] Polyproteins 1a and 1ab from open reading frames 1a (ORF1a) and ORF1ab cleaved to form the RdRp core along with conjugation of core nsp7/nsp8.[ 107 ] The structure of the protein resembles a curled right‐hand grip divided into three subdomains, the finger domain (amino acid residues 398–581, 628–919), the palm domain (582–627, 688–815) and the thumb domain (816–919), as shown in Figure 19.[ 41 , 109 ] For additional stability of RdRp, Zn ions are also required. One of the Zn ions attached with four amino acid residues (His295, Cys301, Cys306 and Cys310) of the N terminus, whereas the second Zn ion attached with the finger domain region consisting of amino acid residues Cys487, His642, Cys645 and 646.[ 107 ]
Figure 19.
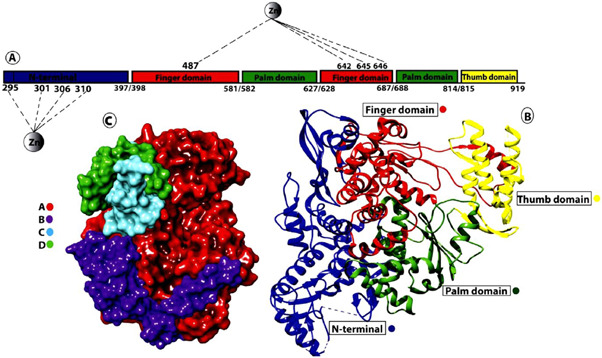
(a) Linear structure of RdRp of SARS‐Cov‐2 showing the N‐terminus and other functional domains. Attachment of the Zn ion with various amino acid residues. (b) Crystal structure of RdRp along with important domains. (c) Surface view of RdRp along with its cofactors.[ 108 ] RdRp, RNA‐dependent RNA polymerase; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
Ogunyemi et al. performed an in silico study to identify SARS‐CoV‐2 RdRp inhibitors from alkaloid‐ and flavonoid‐containing African plants, where 226 bioactive compounds were used for docking against RdRp (PDB ID: 6M71) by using remdesivir and sovosivir as reference molecules, and of these, three alkaloidal drugs, namely, tinosponone (Figure 9) (31), (Figure 20) 10ʹ‐hydroxyusambaresine (146) and strychnopentamine (147), and two flavonoid compounds, namely usararotenoid A (148) and 12α‐epi‐milletosin (149), showed the highest binding energies of −10.6, −10.1, −9.4, −8.4 and −8.8 kcal/mol against the target protein, respectively.[ 110 ] Vijayakumar et al. studied various flavonoids using an in silico approach, and postdocked analysis showed that cyanidin (150) binds with the target protein with a binding affinity of −7.7 kcal/mol, which confers the ability to hinder its replication mechanism.[ 74 ] Elfiky et al. investigated the RdRp‐inhibiting potential of various FDA‐approved antiviral drugs along with some new molecules using an in silico approach; two derivatives of compound IDX‐184, namely (3,5‐dihydroxyphenyl)oxidanyl (151) and (3‐hydroxyphenyl)oxidanyl (152), showed promising results in terms of binding energy along with good thermodynamic stability.[ 111 ] Singh et al. screened different polyphenols against RdRp and found that (–)‐epigallocatechin gallate (153), theaflavin‐3ˊ‐O‐gallate (154), theaflavin‐3ˊ‐gallate (155) and theaflavin‐3,3ˊ‐digallate (156) showed the best inhibiting activity on RdRp, with binding energies of −7.3, −9.3, −9.6 and −9.9 kcal/mol, respectively.[ 112 ]
Figure 20.
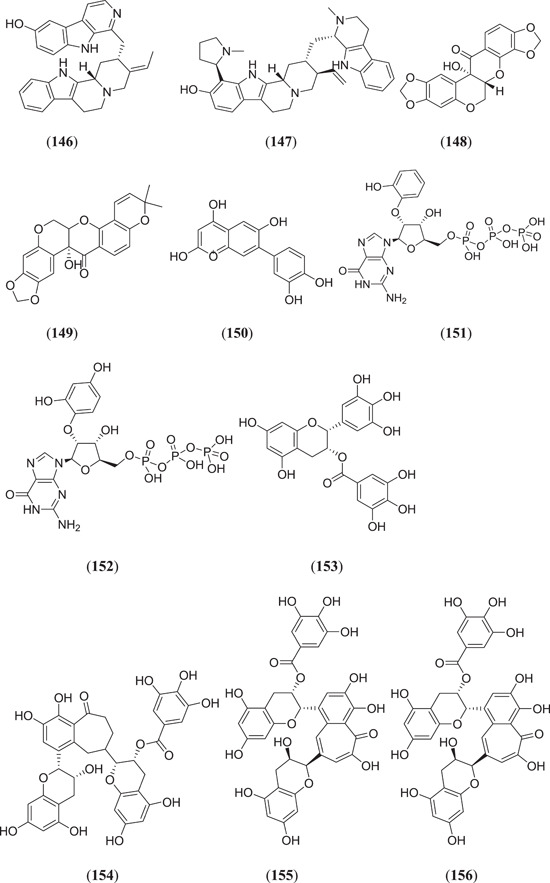
In silico screening to identify SARS‐CoV‐2 RdRp inhibitors from alkaloids and flavonoids. RdRp, RNA‐dependent RNA polymerase; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
5. TARGETING ACE2
ACE2 is a membrane protein receptor that is mainly expressed in the lungs, kidneys, heart and intestine.[ 113 ] It serves as a main portal for the entry of SARS‐CoV‐2 into the host cell; therefore, inhibition of ACE2 has become a potential novel target to minimize infections by preventing the entry of SARS‐CoV‐2.
Abdelli et al. carried out an in silico study of active phytoconstituents from Ammoides verticillata found in western Algeria, where compound (Figure 21) isothymol (157) showed a good binding affinity of −5.7853 kcal/mol as compared to the cocrystal ligand β‐d‐mannose of ACE2.[ 114 ] Gyebi et al. investigated some drug‐like alkaloids as potential inhibitors for SARS‐CoV‐2 cell entry, that is, inhibitors for spike glycoprotein, human ACE2 and TMPRSS2 protein, which are involved in the mechanism of entry of this virus. They found that compounds cryptospirolepine (32) (Figures 9), 10‐hydroxyusambarensine (146) (Figure 20) and cryptoquinoline (35) (Figure 9) showed the best binding affinity with the target protein.[ 76 ] Shakya et al. studied a set of phytoconstituents from Morus alba Linn. for their ability to inhibit TMPRSS2. Docking of a series of compounds was performed by Glide, and five molecules (1R,2S,3R,4R,6R)‐4‐{[((1R,2R,3R,4S,5R)‐2,4‐dihydroxy‐3,5‐dimethylcyclohexyl)oxy]methyl}‐6‐phenethoxycyclohexane‐1,2,3‐triol (158), 8‐((1E,5E)‐2,6‐dimethylocta‐1,5‐dien‐1‐yl)‐5,7‐dihydroxy‐2‐(4‐hydroxyphenyl)‐4H‐chromen‐4‐one (159), 2‐(2,4‐dihydroxyphenyl)‐5‐hydroxy‐8,8‐dimethyl‐3‐(3‐methylbut‐2‐en‐1‐yl)pyrano[2,3‐f]chromen‐4(8H)‐one (160), 3,5,7‐trihydroxy‐2‐(4‐hydroxyphenyl)‐4H‐chromen‐4‐one (161) and (2S,8R)−2‐(2,4‐dihydroxyphenyl)−5‐hydroxy‐8‐methyl‐8‐(4‐methylpent‐3‐en‐1‐yl)−2,3‐dihydropyrano[2,3‐f]chromen‐4(8H)‐one (162) were found to have good binding affinities of −9.547, −7.980, −7.888, −7.807 and −7.426 kcal/mol, respectively; they all showed good thermodynamic stability against target proteins.[ 115 ] Computational study revealed that flavonoids curcumin (163) and catechin (164) showed good binding affinity towards ACE2, with binding energies of −7.8 and −8.9 kcal/mol, respectively.[ 116 ] Horne and Vohl studied the effect of the polyphenolic compound resveratrol (165) and ACE2; after an in silico study it was found that the selected compounds showed good binding affinity with the target protein.[ 117 ]
Figure 21.
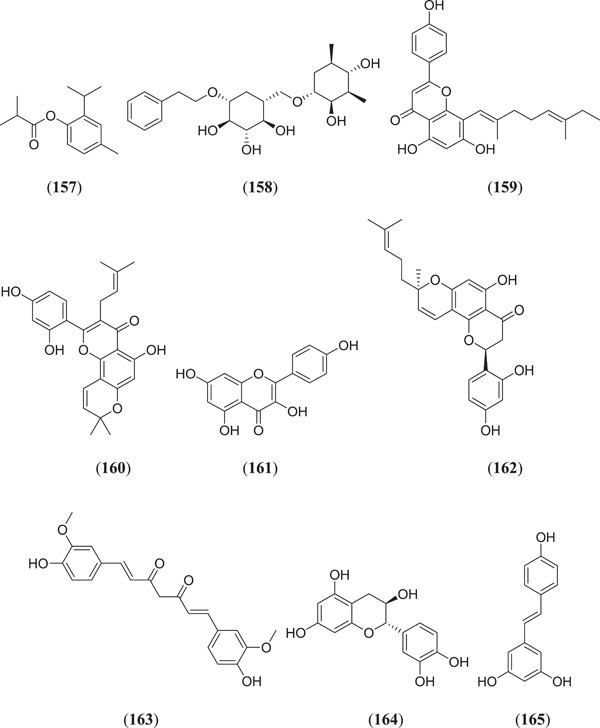
Structures of some phytoconstituents from Ammoides verticillate, Morus alba and alkaloidal and flavonoid drugs as ACE2 inhibitors. ACE2, angiotensin‐converting enzyme 2
6. TARGETING MULTIPLE LIGANDS
To prevent COVID‐19, researchers are making efforts to identify multitargeting ligands. It will be useful to develop inhibitors that target the entry of virus, virus multiplication as well as the RBD at the same time by using a single ligand. Shawan et al. investigated 43 flavonoid compounds against Mpro/3CLpro, Plpro and ACE2, and two potent flavonoid compounds, namely (Figure 22), luteolin (166) and abyssinone II (167), showed the highest binding affinity to all the targets.[ 118 ] Mrid et al. investigated several Moroccan medicinal plants that are known to have antiviral properties. Selected molecules were docked against three targets namely Mpro, PLpro and spike protein of PDB ID: 6LU7, 6W9C and 6VYB, respectively; it was found that compounds kaempferol (20), quercetin (23) (Figure 6), hesperidin (69) (Figure 12), luteolin (166), procynidin B1 (168), aloe emodin (169), betulinic acid (170) and β‐sitosterol (171) showed significant binding energy against all the targets.[ 119 ]
Figure 22.
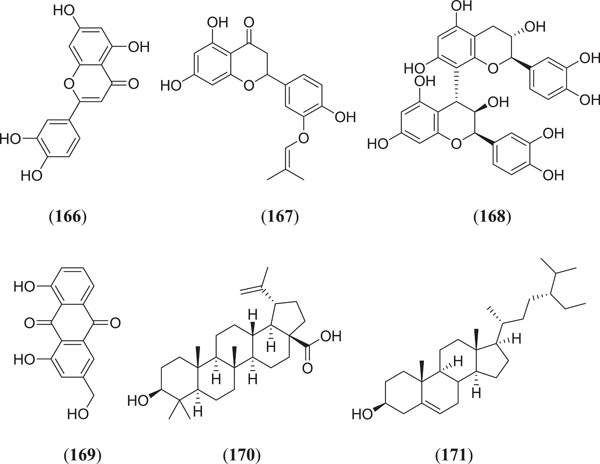
Some flavonoids as multitarget ligands inhibiting SARS‐CoV‐2. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
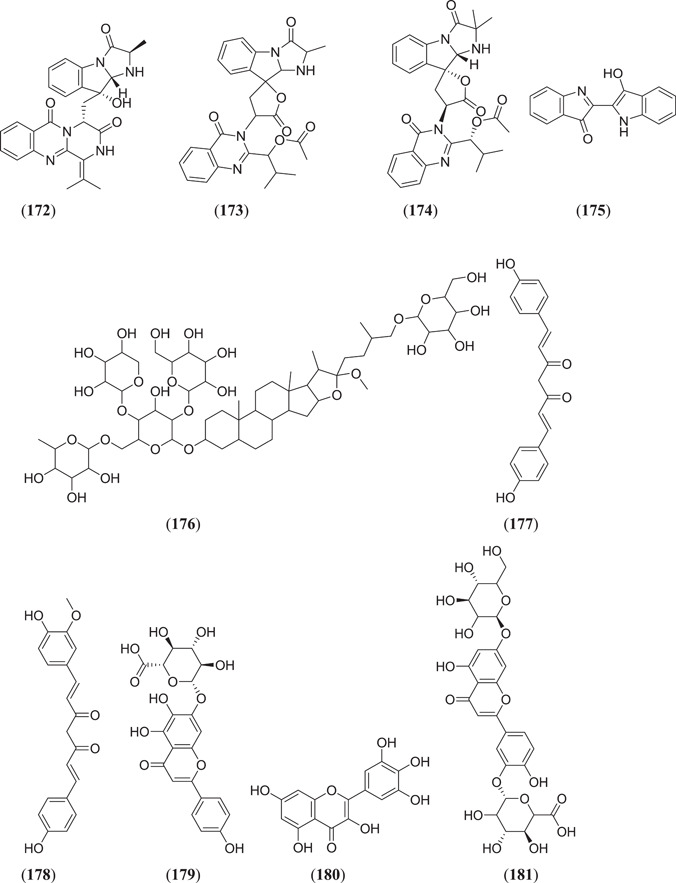
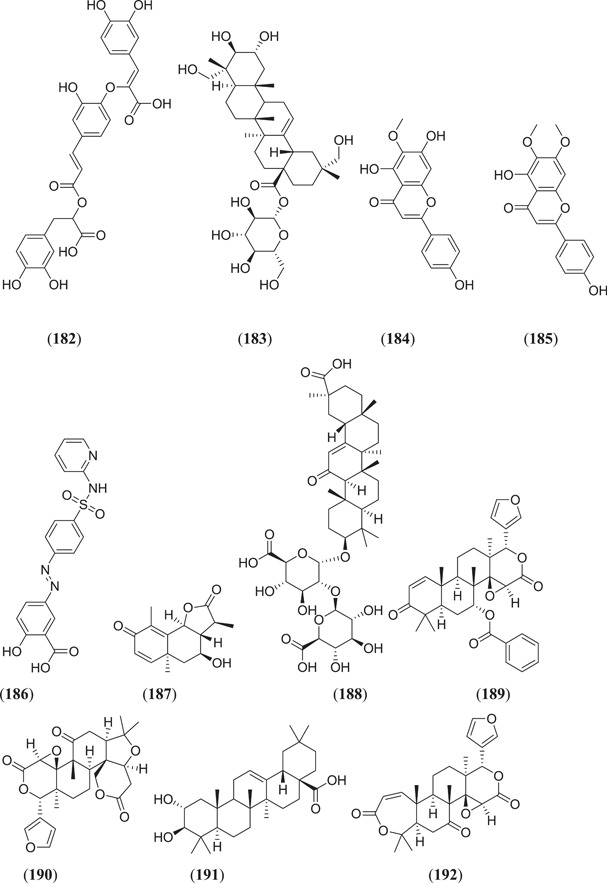
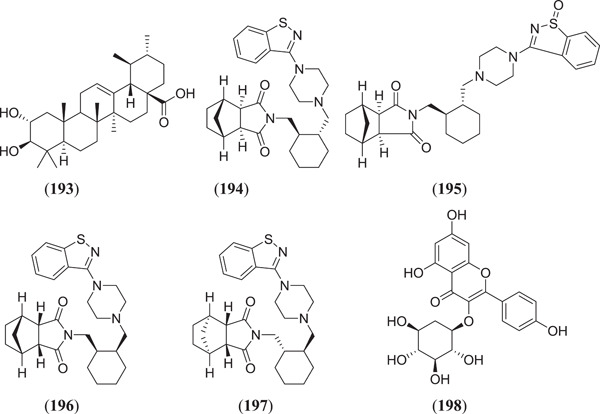
Esraa et al. screened seventy‐one quinoline and quinazoline alkaloids against three potential targets of SARS‐COV‐2, that is Mpro (PDB ID: 6LU7), spike glycoprotein (PDB ID: 6LZG) and human ACE2 (PDB ID: 1R42); among these, three compounds (Figure 23) norquinadoline A (172), deoxynortryptoquivaline (173) and deoxytryptoquivaline (174) showed the highest binding affinity.[ 120 ] Rameshwar et al. investigated 12 natural compounds against SARS spike glycoprotein human ACE2 complexes, among which indigo blue (175) showed the highest affinity with a docking score of −11.2 kcal/mol against both the selected targets.[ 121 ] Chikhale et al. screened 32 different phytoconstituents of Asparagus racemosus Wild. against spike protein and NSP15 endoribonuclease of SARS‐CoV‐2. In their study, they found that asparoside‐C (176) showed the highest binding energies of −62.61 and −55.19 kcal/mol with spike protein (6M0J) and NSP15 endoribonuclease (6W0J), respectively.[ 97 ] Sharma et al. selected eight plant‐derived compounds and two reported drugs of SARS‐CoV‐2 and screened them against Mpro (3CLpro) and endoribonuclease (NSP15) of SARS‐CoV‐2. They found that (Figure 23) bisdemethoxycurcumin (177), demethoxycurcumin (178), quercetin (23) (Figure 6), scutellarin (179) and myricetin (180) showed the best binding affinities of −7.3, −7.02, −6.58, −7.13 and −6.15 kcal/mol, respectively, against Mpro and −6.56, −7.51, −6.49, −6.97 and −6.52 kcal/mol, respectively, against endoribonuclease protein.[ 122 ] Prasanth et al. screened 12 phytoconstituents from Melissa officinalis against Mpro (6LU7) and spike protein (6LZG) of SARS‐CoV‐2. From their experiment, they found that among 12 phytoconstituents, three compounds (Figure 23) luteolin‐7‐glucoside‐3ʹ‐glucuronide (181), melitric acid‐A (182) and quadranoside‐III (183) showed the best binding affinities of −8.5, −8.2 and −8.6 kcal/mol against Mpro and −10.1, −10 and −9.2 kcal/mol against spike protein, respectively.[ 123 ] Omar Sekiou et al. screened different natural compounds against Mpro (6LU7) and angiotensin‐converting enzyme (1r42) of SARS‐CoV‐2 and performed a comparison with hydroxyl‐chloroquine. In their study, they found that quercetin (23), (Figure 23) hispidulin (184), cirsimaritin (185), sulfasalazine (186), curcumin (163) (Figure 21) and artemisin (187) showed better inhibiting activity than hydroxyl‐chloroquine against selected targets. The binding affinities of quercetin, hispidulin, cirsimaritin, sulfasalazine, curcumin and artemisin on Mpro were −7.5, −7.3, −7.2, −7.2, −6.8 and −6.8 kcal/mol, respectively, and the binding affinities of hispidulin, cirsimaritin, curcumin and artemisin on angiotensin‐converting enzyme were −7.8, −7.6, −7.2 and −7.2 kcal/mol, respectively.[ 124 ] Vardhan et al. screened 154 phytochemicals from limonoids and triterpenoids against five protein targets of SARS‐CoV‐2, which are 3CLpro (main protease), PLpro (papain‐like protease), SGp‐RBD (spike glycoprotein‐receptor‐binding domain), ACE2 and RdRp (RNA‐dependent RNA polymerase). From the screening, it was found that (Figure 23) glycyrrhizic acid (188), 7‐deacetyl‐7‐benzoylgedunin (189), limonin (190), maslinic acid (191), obacunone (192), corosolic acid (193) and ursolic acid (45) (Figure 10) showed the best result against selected targets.[ 125 ] Thurakkal et al. screened 76 organosulphur compounds against five protein targets of SARS‐CoV‐2 that is Mpro (main protease), PLpro (papain‐like protease), Spro, helicase and RdRp (RNA‐dependent RNA polymerase). They found that lurasidone (194) and its derivatives that is lurasidone sulphoxide (195), lurasidone endo (196) and lurasidone exo (197) showed the best binding affinity against selected targets.[ 126 ] Adejoro et al. screened 198 compounds from five selected medicinal plants that is Ageratum conyzoides, Phyllanthus amarus, Androgrphis paniculata (Burn. F.), A. indica and Momordica charantia against Mpro (6LU7) and spike glycoprotein (6LZG and 6VXX) of SARS‐CoV‐2. They found that of 198 compounds, astragalin (198) showed the best binding energies of −8.5, −8.0 and −7.6 kcal/mol for 6LU7, 6LZG and 6VXX, respectively, against selected targets.[ 127 ]
Figure 23.
Quinoline and quinazoline alkaloids against three potential targets of SARS‐CoV‐2. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
7. TARGETING THE NSPS9
Junior et al. investigated lapachol derivatives for nsps9 of COVID‐19 inhibitory activity through an in silico study where (Figure 24) lapachol VI (199) and lapachol IX (200) demonstrate good binding energies and stable dynamics with the target protein.[ 128 ]
Figure 24.
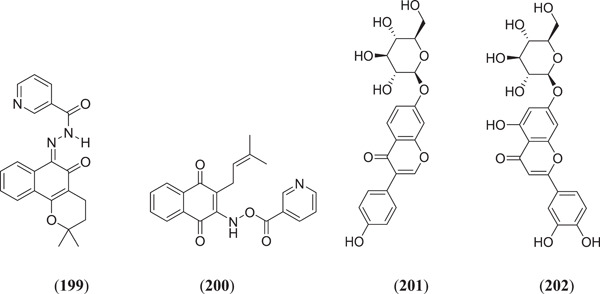
Lapachol derivatives for nsps9 of COVID‐19 inhibitory activity. COVID‐19, Corona Virus Disease‐19
8. OTHERS
Chandra et al., from their studies, found four natural antiviral compounds, that is, amentoflavone (36) (Figure 10), (Figure 24) daidzin (201), luteoloside (202) and baicalin (19) (Figure 6), to show strong inhibiting activity against methyltransferase of SARS‐CoV‐2, with binding energies of −9.3, −8.3, −8.7 and −8.3 kcal/mol, respectively.[ 129 ] Rolta et al. investigated 100 phytoconstituents of Rheum emodi against three different active sites of the RNA binding domain of nucleocapsid phosphoprotein of SARS‐CoV‐2. They found that three compounds, that is, aloe‐emodin (169) (Figure 22), anthrarufin (203) and alizarin (204) (Figure 25), showed the best binding energies of −8.508, −8.456 and −8.441 kcal/mol on active site A, −6.433, −6.345 and −6.598 kcal/mol on active site B and −8.508, −8.538 and −8.841 kcal/mol on active site C, respectively.[ 130,131 ] Ibrahim Khalifa et al. examined 19 hydrolysable tannins against 3CLpro of SARS‐CoV‐2, and they found that of these (Figure 25), pedunculagin (205), tercatain (206) and castalin (207) showed the best binding affinities of −18.58, −23.11 and −14.04 kcal/mol, respectively, on the selected target.[ 132 ]
Figure 25.
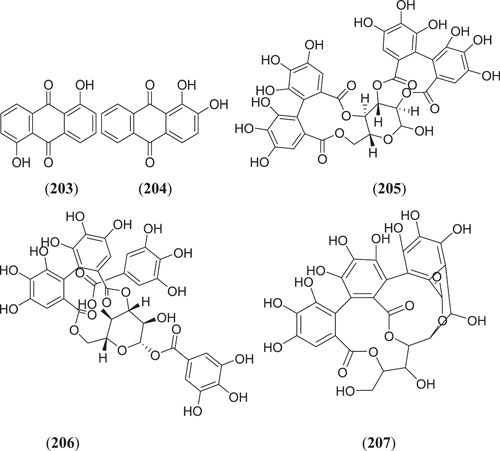
Natural antiviral compounds showing strong inhibiting activity against methyltransferase of SARS‐CoV‐2
9. CONCLUSION
COVID‐19 is a viral disease that mainly affects the respiratory system of humans. It is caused by SARS‐CoV‐2. After considerable research and analyses, a few vaccines have been developed that have been approved by the WHO, such as mRNA‐1273 by Moderna, BNT162b2 by Pfizer/BioNTech, Ad26.COV2.S by Janssen, AZD1222 by Oxford/AstraZeneca, Covishield by the Serum Institute of India, BBIBP‐CorV by Sinopharm and coronaVac by Sinovac. Worldwide, people are receiving their scheduled vaccines as a protective measure against SARS‐CoV‐2. Moreover, Veklury (remdesivir) became the first US FDA‐approved antiviral drug for the treatment of COVID‐19‐infected patients older than 12 years of age. In December 2021, the US FDA also granted permission for emergency use of nirmatrelvir in combination with ritonavir for COVID‐19 treatment. Although vaccines have been developed, development of proper antiviral drugs for the treatment of SARS‐CoV‐2 still remains a challenge as the virus is prone to rapid mutations. For this purpose, in silico studies have become a useful tool for developing new drug molecules against various targets of SARS‐CoV‐2 before in vitro and in vivo testing of these molecules. Along with phytoconstituent screening, databases like PubChem, ZINC, etc. are also being screened to identify SARS‐CoV‐2 inhibitors. There might be many potent molecules that can act as possible Mpro inhibitors, namely, ebselen, berberine, nabiximols, hesperidin, pectolinarin, rhoifolin and epigallocatechin. Methylene blue, resveratrol, curcumin and catechin show good potential in inhibiting the spike protein of SARS‐CoV‐2. Luteolin, abyssinone II, procynidin B1 and indigo blue showed the highest binding against all the target proteins of SARS‐CoV‐2 such as Mpro, spike protein and ACE2. Crytospirolepine, 10ʹ‐hydroxyusambaresine, strychnopentamine, usararotenoid A and 12α‐epi‐milletosin showed good binding affinity towards inhibition of RdRp of SARS‐CoV‐2. Here, the best molecules that were found through in silico studies were compiled and they need to be studied further for effective drug development.
CONFLICT OF INTERESTS
The authors declare no conflict of interest.
ACKNOWLEDGEMENT
The financial support from the All India Council for Technical Education (AICTE), New Delhi, India is gratefully acknowledged (8‐21/FDC/RPS (NER)/POLICY‐1/2020‐21).
Ali F., Alom S., Shakya A., Ghosh S. K., Singh U. P., Bhat H. R.. Implication of in silico studies in the search for novel inhibitors against SARS‐CoV‐2. Arch. Pharm. 2022;355:e2100360. 10.1002/ardp.202100360
REFERENCES
- 1. Bogoch I. I., Watts A., Thomas‐Bachli A., Huber C., Kraemer M. U. G., Khan K., J. Travel. Med. 2020, 27, taaa011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paules C. I., Marston H. D., Fauci A. S., JAMA, J. Am. Med. Assoc. 2020, 323, 707. [DOI] [PubMed] [Google Scholar]
- 3. Tang X., Wu C., Li X., Song Y., Yao X., Wu X., Duan Y., Zhang H., Wang Y., Qian Z., Cui J., Lu J., Natl. Sci. Rev. 2020, 7, 1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. C., Sohrabi , Z., Alsafi , N., O'Neill , M., Khan , A., Kerwan , A., Al‐Jabir , C., Iosifidis , R., Agha , Int. J. Surg. 2020, 76, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B., Lancet 2020, 395, 497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Munster V. J., Koopmans M., van Doremalen N., van Riel D., de Wit E., N. Engl. J. Med. 2020, 382, 692. [DOI] [PubMed] [Google Scholar]
- 7. Fehr A. R., Perlman S., Coronaviruses Methods Protocols, Springer, New York: 2015, p. 1. [Google Scholar]
- 8. Fang L., Annu. Rev. Virol. 2016, 3, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shehroz M., Zaheer T., Hussain T., Heliyon 2020, 6, e05278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Belouzard S., Millet J. K., Licitra B. N., Whittaker G. R., Viruses 2012, 4, 1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boopathi S., Poma A. B., Kolandaivel P., J. Biomol. Struct. Dyn. 2021, 39, 3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ruch T. R., Machamer C. E., Viruses 2012, 4, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Siu Y. L., Teoh K. T., Lo J., Chan C. M., Kien F., Escriou N., Tsao S. W., Nicholls J. M., Altmeyer R., Peiris J. S. M., Bruzzone R., Nal B., J. Virol. 2008, 82, 11318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bosch B. J., van der Zee R., de Haan C. A. M., Rottier P. J. M., J. Virol. 2003, 77, 8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hoffmann M., Kleine‐Weber H., Krüger N., Müller M., Drosten C., Pöhlmann S., BioRxiv 2020. 10.1101/2020.01.31.929042 [DOI] [Google Scholar]
- 16. Walls A. C., Park Y. J., Tortorici M. A., Wall A., McGuire A. T., Veesler D., Cell 2020, 181, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ziebuhr J., Siddell S. G., J. Virol. 1999, 73, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lv Z., Chu Y., Wang Y., Palliat. Care 2015, 7, 95. [Google Scholar]
- 19. Xue X., Yang H., Shen W., Zhao Q., Li J., Yang K., Chen C., Jin Y., Bartlam M., Rao Z., J. Mol. Biol. 2007, 366, 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mengist H. M., Dilnessa T., Jin T., Front. Chem. 2021, 9, 622898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qamar M. T., Alqahtani S. M., Alamri M. A., Chen L.‐L., J. Pharm. Anal. 2020, 10, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R., Science 2020, 368, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thiel V., Ivanov K. A., Putics Á., Hertzig T., Schelle B., Bayer S., Weißbrich B., Snijder E. J., Rabenau H., Doerr H. W., Gorbalenya A. E., Ziebuhr J., J. Gen. Virol. 2003, 84, 2305. [DOI] [PubMed] [Google Scholar]
- 24. Havranek B., Islam S. M., J. Biomol. Struct. Dyn. 2021, 39, 4304. 10.1080/07391102.2020.1776158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen Y., Wang G., Ouyang L., Signal Transduct. Target. Ther. 2020, 5, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gaudêncio S. P., Pereira F., Mar. Drugs 2020, 18, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chtita S., Belhassan A., Aouidate A., Belaidi S., Bouachrine M., Lakhlifi T., Comb. Chem. High Throughput Screen. 2021, 24, 441. [DOI] [PubMed] [Google Scholar]
- 28. Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K. Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z., PLoS Biol. 2005, 3, e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xue X., Yu H., Yang H., Xue F., Wu Z., Shen W., Li J., Zhou Z., Ding Y., Zhao Q., Zhang X. C., Liao M., Bartlam M., Rao Z., J. Virol. 2008, 82, 2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang F., Chen C., Tan W., Yang K., Yang H., Sci. Rep. 2016, 61, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L. W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H., Nature 2020, 582, 289. [DOI] [PubMed] [Google Scholar]
- 32. Amporndanai K., Meng X., Shang W., Jin Z., Rogers M., Zhao Y., Rao Z., Liu Z. J., Yang H., Zhang L., O'Neill P. M., Samar Hasnain S., Nat. Commun. 2021, 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu X., Zhang B., Jin Z., Yang H., Rao Z., PDB Release. 2020, 119, 17. [Google Scholar]
- 34. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E., Nucleic Acids Res. 2000, 28, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Al‐Horani R. A., Desai U. R., Expert Opin. Ther. Pat. 2016, 26, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. (Uladzimir) Ladziata V., Glunz P. W., Zou Y., Zhang X., Jiang W., Jacutin‐Porte S., Cheney D. L., Wei A., Luettgen J. M., Harper T. M., Wong P. C., Seiffert D., Wexler R. R., Priestley E. S., Bioorg. Med. Chem. Lett. 2016, 26, 5051. [DOI] [PubMed] [Google Scholar]
- 37. Du L., Kao R. Y., Zhou Y., He Y., Zhao G., Wong C., Jiang S., Yuen K. Y., Jin D. Y., Zheng B. J., Biochem. Biophys. Res. Commun. 2007, 359, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Khan M. A., Mahmud S., Alam A. S. M. R. U., Rahman M. E., Ahmed F., Rahmatullah M., J. Biomol. Struct. Dyn. 2021, 39, 6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Trott O., Olson A. J., J. Comput. Chem. 2009, 31, NA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Prajapat M., Sarma P., Shekhar N., Avti P., Sinha S., Kaur H., Kumar S., Bhattacharyya A., Kumar H., Bansal S., Medhi B., Indian J. Pharmacol. 2020, 52, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L., Ge J., Zheng L., Zhang Y., Wang H., Zhu Y., Zhu C., Hu T., Hua T., Zhang B., Yang X., Li J., Yang H., Liu Z., Xu W., Guddat L. W., Wang Q., Lou Z., Rao Z., Science 2020, 368, 779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alazmi M., Motwalli O., J. Biomol. Struct. Dyn. 2021, 39, 6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Banerjee P., Eckert A. O., Schrey A. K., Preissner R., Nucleic Acids Res. 2018, 46, W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pandey M. M., Rastogi S., Rawat A. K. S., Altern. Med. 2013, 2013, 376327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muley B. P., Khadabadi S. S., Banarase N. B., J. Pharmacol. Res. 2009, 8, 455. [Google Scholar]
- 46. C. G., Sindhu , Int. J. Res. Ayurveda Pharm. 2010, 1, 131. [Google Scholar]
- 47. Ashwlayan V. D., Kumar A., Verma M., Garg V. K., Gupta S., Pharm. Pharmacol. Int. J. 2018, 6, 149. [Google Scholar]
- 48. Das P., Majumder R., Mandal M., Basak P., J. Biomol. Struct. Dyn. 2021, 39, 6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aghaee E., Ghodrati M., Ghasemi J. B., Inform. Med. Unlocked 2021, 23, 100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Joshi T., Joshi T., Sharma P., Mathpal S., Pundir H., Bhatt A., Chandra S., Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4529. [DOI] [PubMed] [Google Scholar]
- 51. Krupanidhi S., Abraham Peele K., Venkateswarulu T. C., Ayyagari V. S., Nazneen Bobby M., John Babu D., Venkata Narayana A., Aishwarya G., J. Biomol. Struct. Dyn. 2021, 39, 5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Borquaye L. S., Gasu E. N., Ampomah G. B., Kyei L. K., Amarh M. A., Mensah C. N., Nartey D., Commodore M., Adomako A. K., Acheampong P., Mensah J. O., Mormor D. B., Aboagye C. I., Biomed. Res. Int. 2020, 2020, 5324560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lokhande K., Nawani N., Venkateswara S. K., Pawar S., J. Biomol. Struct. Dyn. 2020, 0, 1. 10.1080/07391102.2020.1858165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ghosh R., Chakraborty A., Biswas A., Chowdhuri S., J. Mol. Struct. 2021, 1229, 129489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Holanda V. N., de A. Lima E. M., da Silva W. V., Maia R. T., de L. Medeiros R., Ghosh A., de M. Lima V. L., de de Figueiredo R. C. B. Q., J. Biomol. Struct. Dyn. 2021, 1. 10.1080/07391102.2020.1871073 [DOI] [Google Scholar]
- 56. Tripathi M. K., Singh P., Sharma S., Singh T. P., Ethayathulla A. S., Kaur P., J. Biomol. Struct. Dyn. 2021, 39, 5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kumar A., Choudhir G., Shukla S. K., Sharma M., Tyagi P., Bhushan A., Rathore M., J. Biomol. Struct. Dyn. 2021, 39, 3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ghosh R., Chakraborty A., Biswas A., Chowdhuri S., J. Biomol. Struct. Dyn. 2021, 39, 6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ibrahim M. A. A., Abdelrahman A. H. M., Allemailem K. S., Almatroudi A., Moustafa M. F., Hegazy M. E. F., Protein J. 2021, 40, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chowdhury P., J. Biomol. Struct. Dyn. 2021, 39, 6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rout J., Swain B. C., Tripathy U., J. Biomol. Struct. Dyn. 2022, 40, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aanouz I., Belhassan A., El‐Khatabi K., Lakhlifi T., El‐ldrissi M., Bouachrine M., J. Biomol. Struct. Dyn. 2021, 39, 2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Keretsu S., Bhujbal S. P., Cho S. J., Sci. Rep. 2020, 10, 17716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kumar D., Kumari K., Vishvakarma V. K., Jayaraj A., Kumar D., Ramappa V. K., Patel R., Kumar V., Dass S. K., Chandra R., Singh P., J. Biomol. Struct. Dyn. 2021, 39, 4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shree P., Mishra P., Selvaraj C., Singh S. K., Chaube R., Garg N., Tripathi Y. B., J. Biomol. Struct. Dyn. 2022, 40, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vellingiri B., Jayaramayya K., Iyer M., Narayanasamy A., Govindasamy V., Giridharan B., Ganesan S., Venugopal A., Venkatesan D., Ganesan H., Rajagopalan K., Rahman P. K. S. M., Cho S. G., Kumar N. S., Subramaniam M. D., Sci. Total Environ. 2020, 725, 138277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tallei T. E. Tumilaar S. G. Niode N. J. Fatimawali, Kepel B. J. Idroes R. Effendi Y. Sakib S. A. Bin Emran T. Scientifica 2020, 2020, 6307457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Razzaghi‐Asl N., Ebadi A., Shahabipour S., Gholamin D., J. Biomol. Struct. Dyn. 2021, 39, 6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Enmozhi S. K., Raja K., Sebastine I., Joseph J., J. Biomol. Struct. Dyn. 2021, 39, 3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Narkhede R. R., Pise A. V., Cheke R. S., Shinde S. D., Nat. Prod. Bioprospect. 2020, 10, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mishra A., Pathak Y., Choudhir G., Kumar A., Mishra S. K. & Tripathi V. Mishra A., Pathak Y., Kumar A., Mishra S. K., Tripathi V., Asian Pac. J. Trop. Biomed. 2021, 11, 155. [Google Scholar]
- 72. Prasanth D. S. N. B. K., Murahari M., Chandramohan V., Panda S. P., Atmakuri L. R., Guntupalli C., J. Biomol. Struct. Dyn. 2021, 39, 4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kanhed A. M., Patel D. V., Teli D. M., Patel N. R., Chhabria M. T., Yadav M. R., Mol. Divers. 2021, 25, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vijayakumar B. G., Ramesh D., Joji A., Jayachandra Prakasan J., Kannan T., Eur. J. Pharmacol. 2020, 886, 173448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bahadur Gurung A., Ajmal Ali M., Lee J., Abul Farah M., Mashay Al‐Anazi K., J. King Saud Univ. Sci. 2020, 32, 2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gyebi G. A., Adegunloye A. P., Ibrahim I. M., Ogunyemi O. M., Afolabi S. O., Ogunro O. B., J. Biomol. Struct. Dyn. 2020, 0, 1. 10.1080/07391102.2020.1835726 [DOI] [Google Scholar]
- 77. Joshi R. S., Jagdale S. S., Bansode S. B., Shankar S. S., Tellis M. B., Pandya V. K., Chugh A., Giri A. P., Kulkarni M. J., J. Biomol. Struct. Dyn. 2021, 39, 3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ibrahim M. A. A., Mohamed E. A. R., Abdelrahman A. H. M., Allemailem K. S., Moustafa M. F., Shawky A. M., Mahzari A., Hakami A. R., Abdeljawaad K. A. A., Atia M. A. M., J. Mol. Graphics Modell. 2021, 105, 107904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Umar H. I., Josiah S. S., Saliu T. P., Jimoh T. O., Ajayi A., Danjuma J. B., J. Taibah Univ. Med. Sci. 2021, 16, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Swargiary A., Mahmud S., Saleh M. A., J. Biomol. Struct. Dyn. 2020, 0, 1. 10.1080/07391102.2020.1835729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Majumder R., Mandal M., J. Biomol. Struct. Dyn. 2022, 40, 696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Al‐Zahrani A. A., Nat. Prod. Commun. 2020, 15, 1934578X2095395. [Google Scholar]
- 83. Das S., Sarmah S., Lyndem S., Singha Roy A., J. Biomol. Struct. Dyn. 2021, 39, 3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fakhar Z., Faramarzi B., Pacifico S., Faramarzi S., J. Biomol. Struct. Dyn. 2021, 39, 6171. [DOI] [PubMed] [Google Scholar]
- 85. Chatterjee S., Maity A., Chowdhury S., Islam M. A., Muttinini R. K., Sen D., J. Biomol. Struct. Dyn. 2021, 39, 5290. [DOI] [PubMed] [Google Scholar]
- 86. South A. M., Tomlinson L., Edmonston D., Hiremath S., Sparks M. A., Nat. Rev. Nephrol. 2020, 16, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K. Y., Wang Q., Zhou H., Yan J., Qi J., Cell 2020, 181, 894.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhang H., Penninger J. M., Li Y., Zhong N., Slutsky A. S., Intensive Care Med. 2020, 46, 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gui M., Song W., Zhou H., Xu J., Chen S., Xiang Y., Wang X., Cell Res. 2017, 27, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Song W., Gui M., Wang X., Xiang Y., PLoS Pathog. 2018, 14, e1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X., Nature 2020, 581, 215. [DOI] [PubMed] [Google Scholar]
- 92. Huang Y., Yang C., Xu X., Xu W., Liu S., Acta Pharmacol. Sin. 2020, 41, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yang J., Zhang Y., Curr. Protoc. Bioinf. 2015, 52, 5.8.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shehroz M., Zaheer T., Hussain T., Heliyon 2020, 6, e05278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. C. R., Groom , F. H., Allen , Angew. Chem Int. Ed. 2014, 53, 662. [DOI] [PubMed] [Google Scholar]
- 96. Kapetanovic I. M., Chem.‐Biol. Interact. 2008, 171, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chikhale R. V., Gurav S. S., Patil R. B., Sinha S. K., Prasad S. K., Shakya A., Shrivastava S. K., Gurav N. S., Prasad R. S., J. Biomol. Struct. Dyn. 2021, 39, 4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Teli D. M., Shah M. B., Chhabria M. T., Front. Mol. Biosci. 2021, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Patel A., Rajendran M., Shah A., Patel H., Pakala S. B., Karyala P., J. Biomol. Struct. Dyn. 2021, 0, 1. 10.1080/07391102.2020.1868338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Basu A., Sarkar A., Maulik U., Sci. Rep. 2020, 10, 17699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Braakhuis A., Nutrients 2019, 11, 2705. [Google Scholar]
- 102. Harisna A. H., Nurdiansyah R., Syaifie P. H., Nugroho D. W., Saputro K. E., Firdayani, Prakoso C. D., Rochman N. T., Maulana N. N., Noviyanto A., Mardliyati E., Biochem. Biophys. Rep. 2021, 26, 100969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sundar Panja A., Sarkar A. 2020. 10.20944/preprints 202004.0282.v1
- 104. Pandey P., Rane J. S., Chatterjee A., Kumar A., Khan R., Prakash A., Ray S., J. Biomol. Struct. Dyn. 2021, 39, 6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wen L., Tang K., Chik K. K.‐H., Chan C. C.‐Y., Tsang J. O.‐L., Liang R., Cao J., Huang Y., Luo C., Cai J.‐P., Ye Z.‐W., Yin F., Chu H., Jin D.‐Y., Yuen K.‐Y., Yuan S., Chan J. F.‐W., Int. J. Biol. Sci. 2021, 17, 1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Tan Y. W., Fung T. S., Shen H., Huang M., Liu D. X., Virology 2018, 513, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kirchdoerfer R. N., Ward A. B., Nat. Commun. 2019, 10, 2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Khalifa I., Zhu W., Mohammed H. H. H., Dutta K., Li C., J. Food Biochem. 2020, 44, e13432. 10.1111/jfbc.13432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mcdonald S. M., Wiley Interdiscip. Rev.: RNA 2013, 4, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ahmad J., Ikram S., Ahmad F., Rehman I. U., Mushtaq M., Heliyon 2020, 6, e04502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Elfiky A. A., J. Biomol. Struct. Dyn. 2021, 39, 3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Singh S., Sk M. F., Sonawane A., Kar P., Sadhukhan S., J. Biomol. Struct. Dyn. 2021, 39, 6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., Donovan M., Woolf B., Robison K., Jeyaseelan R., Breitbart R. E., Acton S., Circ. Res. 2000, 87, E1. [DOI] [PubMed] [Google Scholar]
- 114. Abdelli I., Hassani F., Bekkel Brikci S., Ghalem S., J. Biomol. Struct. Dyn. 2021, 39, 3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Shakya A., Chikhale R. V., Bhat H. R., Alasmary F. A., Almutairi T. M., Ghosh S. K., Alhajri H. M., Alissa S. A., Nagar S., Islam M. A., Mol. Divers. 2021. 10.1007/s11030-021-10209-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Jena A., Kanungo N., Nayak V., Chainy G. B. N., Dandapat J., Sci. Rep. 2021, 11, 2043 [DOI] [PMC free article] [PubMed]
- 117. Horne J. R., Vohl M. C., Am. J. Physiol.: Endocrinol. Metab. 2020, 318, E830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Shawan M. M. A. K., Halder S. K., Hasan M. A., Bull. Natl. Res. Cent. 2021, 45, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ben Mrid R., Bouchmaa N., Kabach I., Sobeh M., Zyad A., Nhiri M., Yasri A. 2020, 1. 10.21203/rs.3.rs-38104/v1 [DOI]
- 120. Ismail E. M. O. A., Shantier S. W., Mohammed M. S., Musa H. H., Osman W., Mothana R. A., J. Chem. 2021, 2021, 3613268. 10.1155/2021/3613268 [DOI] [Google Scholar]
- 121. Cheke R. S., Narkhede R. R., Shinde S. D., Ambhore J. P., Jain P. G., Biointerface Res. Appl. Chem. 2020, 11, 10628. [Google Scholar]
- 122. Sharma A., Goyal S., Yadav A. K., Kumar P., Gupta L., J. Biomol. Struct. Dyn. 2022, 40, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Prasanth D. S. N. B. K., Murahari M., Chandramohan V., Bhavya G., Lakshmana Rao A., Panda S. P., Rao G. S. N. K., Chakravarthi G., Teja N., Suguna Rani P., Ashu G., Purnadurganjali C., Akhil P., Vedita Bhavani G., Jaswitha T., Mol. Simul. 2021, 47, 457. [Google Scholar]
- 124. Omar S., Bouziane I., Bouslama Z., Djemel A., ChemRxiv 2020, 2. 10.26434/chemrxiv.12181404.v1 [DOI]
- 125. Vardhan S., Sahoo S. K., Comput. Biol. Med. 2020, 124, 103936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Thurakkal L., Singh S., Roy R., Kar P., Sadhukhan S., Porel M., Chem. Phys. Lett. 2021, 763, 138193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Adejoro I. A., Babatunde D. D., Tolufashe G. F., J. Taibah Univ. Sci. 2020, 14, 1563. [Google Scholar]
- 128. Junior N. N., Santos I. A., Meireles B. A., Nicolau M. S. P., Lapa I. R., Aguiar R. S., Jardim A. C. G., José D. P., J. Biomol. Struct. Dyn. 2021, 1. 10.1080/07391102.2021.1875050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chandra A., Chaudhary M., Qamar I., Singh N., Nain V., J. Biomol. Struct. Dyn. 2021, 0, 1. 10.1080/07391102.2021.1886174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rolta R., Yadav R., Salaria D., Trivedi S., Imran M., Sourirajan A., Baumler D. J., Dev K., J. Biomol. Struct. Dyn. 2021, 39, 7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ogunyemi O. M., Gyebi G. A., Elfiky A. A., Afolabi S. O., Ogunro O. B., Adegunloye A. P., Ibrahim I. M., Antiviral Chem. Chemother. 2020, 28, 204020662098407. [DOI] [PMC free article] [PubMed] [Google Scholar]