

Structural characterization of shikimate pathway multi-enzyme Aro1 from opportunistic fungal pathogen Candida albicans provides the first molecular image of all five domains. Only four of these domains are active and individually essential for C. albicans viability.
Abstract
In the human fungal pathogen Candida albicans, ARO1 encodes an essential multi-enzyme that catalyses consecutive steps in the shikimate pathway for biosynthesis of chorismate, a precursor to folate and the aromatic amino acids. We obtained the first molecular image of C. albicans Aro1 that reveals the architecture of all five enzymatic domains and their arrangement in the context of the full-length protein. Aro1 forms a flexible dimer allowing relative autonomy of enzymatic function of the individual domains. Our activity and in cellulo data suggest that only four of Aro1’s enzymatic domains are functional and essential for viability of C. albicans, whereas the 3-dehydroquinate dehydratase (DHQase) domain is inactive because of active site substitutions. We further demonstrate that in C. albicans, the type II DHQase Dqd1 can compensate for the inactive DHQase domain of Aro1, suggesting an unrecognized essential role for this enzyme in shikimate biosynthesis. In contrast, in Candida glabrata and Candida parapsilosis, which do not encode a Dqd1 homolog, Aro1 DHQase domains are enzymatically active, highlighting diversity across Candida species.
Introduction
Fungal, archaeal, bacterial, plant, and apicomplexan species have the capacity for de novo synthesis of aromatic amino acids from carbohydrate precursors. The linear portion of this biosynthetic cascade is known as the shikimate pathway. It combines seven consecutive enzymatic reactions to convert phosphoenolpyruvate and erythrose-4-phosphate into chorismate (Fig 1A). Chorismate is the branch point for synthesis of aromatic acids (L-tryptophan, L-phenylalanine, and L-tyrosine) and their derivatives, including vitamin K, ubiquinone, and p-aminobenzoate.
Figure 1. C. albicans shikimate pathway.
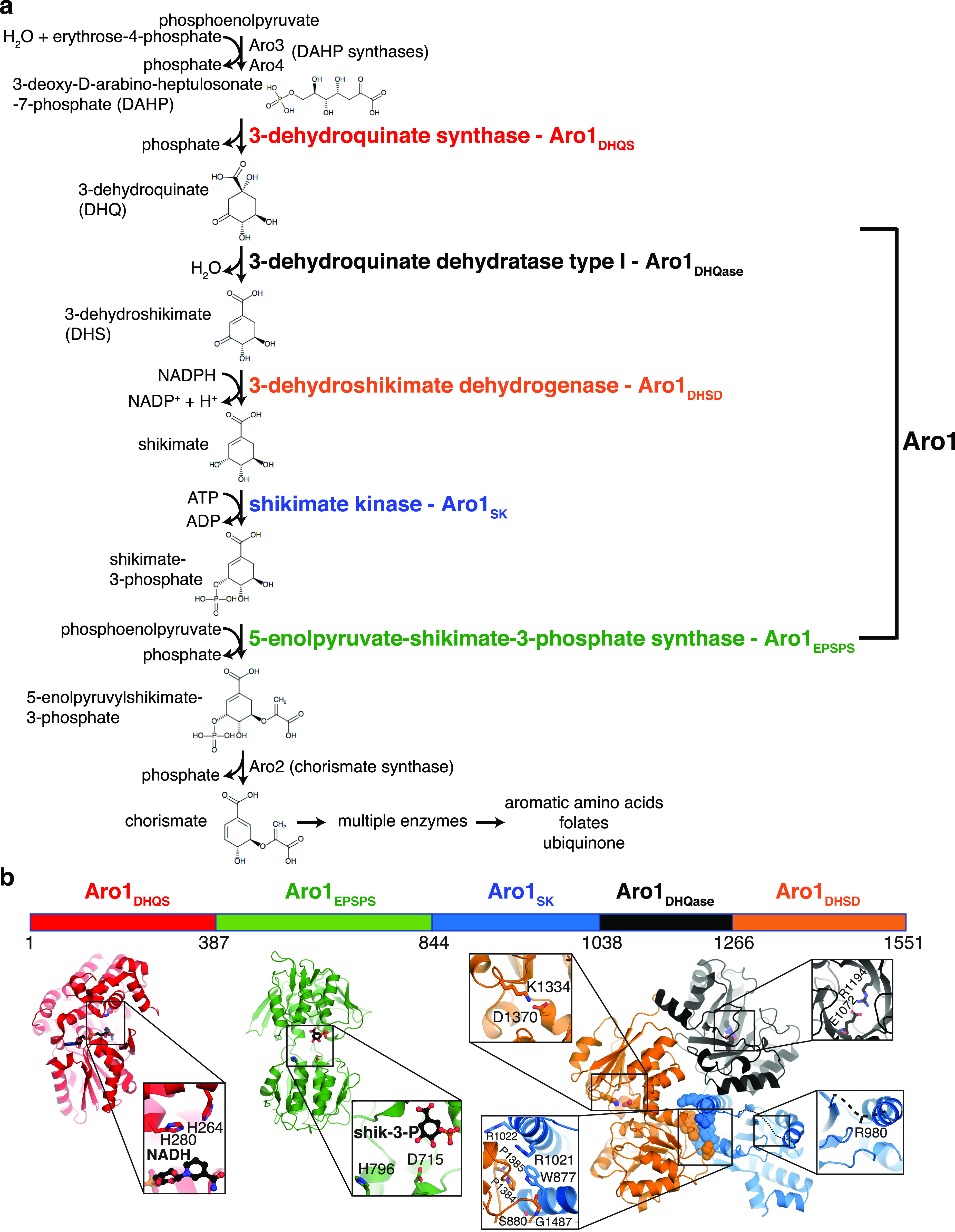
(A) Shikimate pathway, with enzyme names indicated to the right of each reaction. Aro1 individual domains are highlighted in colours. (B) Crystal structures of Aro1 fragments solved in this work, including Aro1DHQS, Aro1EPSPS, Aro1SK+DHQase+DHSD. Zooms show active sites, key catalytic residues, or domain–domain interactions in the Aro1SK+DHQase+DHSD structure.
Shikimate biosynthesis has been the focus of intense studies because of its essential role in microbial and plant cell metabolism and it serves as a segue pathway for the synthesis of biotechnologically important aromatic compounds (Gosset, 2009; Liu et al, 2020). This pathway is absent in metazoa, including humans, which instead require exogenous aromatic amino acids as part of their diet. This makes the shikimate pathway an attractive target for development of herbicides and antimicrobial agents (Coracini & de Azevedo, 2014; Duke, 2018; Nunes et al, 2020). Glyphosate, which inhibits the plant 5-enolpyruvylshikimate-3-phosphate synthase (EPSPS) that catalyses the sixth step in the shikimate pathway (Fig 1A), has been developed into one of the most successful agricultural herbicides widely used for weed management (Duke, 2018). Along the same lines, active programs have been mounted to identify and optimise inhibitors of the bacterial shikimate pathway enzymes (Coggins et al, 2003; Cheng et al, 2012; Blanco et al, 2013; Gordon et al, 2015; Sahu et al, 2020). However, antimicrobials based on shikimate pathway inhibition are yet to be introduced to the clinic.
Recognising their potential as antimicrobial targets, the shikimate biosynthesis enzymes have been characterised in multiple microbial species, revealing several conserved features (Mir et al, 2015). For example, 3-deoxy-D-arabinoheptulosonate-7-phosphate (DAHP) synthase, which catalyses the first step of the pathway, is represented by multiple isoenzymes that are subject to feedback inhibition by individual aromatic amino acid products. The Gram-negative bacterium Escherichia coli encodes three DAHP synthases that are feedback inhibited by L-phenylalanine, L-tyrosine, and L-tryptophan, respectively (Staub & Denes, 1969). The model yeast Saccharomyces cerevisiae and pathogenic fungus Candida albicans produce two DAHP synthases, which are inhibited by L-phenylalanine and L-tyrosine, respectively (Teshiba et al, 1986; Pereira & Livi, 1993; Sousa et al, 2002; Konig et al, 2004). Despite similarity in primary sequence, there is significant diversity in the organisation of enzymatic domains among shikimate pathway enabled species. In Fungi and Apicomplexa, the second to the sixth steps of the shikimate pathway are supported by five enzymatic domains combined in a single protein known as Aro1 or AroM (Graham et al, 1993; Campbell et al, 2004). In plants and bacteria these activities are typically performed by individual single-domain enzymes: 3-dehydroquinate synthase (DHQS), 3-dehydroquinate dehydratase type I (DHQase), 3-dehydroshikimate dehydrogenase (DHSD), shikimate kinase (SK), and 5-enolpyruvate-shikimate-3-phosphate synthase (EPSPS). DHQS (Liu et al, 2008; Neetu et al, 2020) and type I DHQase (Nichols et al, 2004b; Light et al, 2011).
The biological consequence(s) of encoding fungal and apicomplexan shikimate biosynthesis enzymes as a multi-domain protein remain poorly understood. AroM from the thermophilic soil-dwelling fungus Chaetomium thermophilum is the only fully structurally characterised representative of a penta-functional shikimate biosynthesis protein (Verasztó et al, 2020). This multi-enzyme forms a stable dimer with two of the domains (DHQS and DHQase/DHQD) mediating inter-subunit interactions, reminiscent of the interactions described for the corresponding single-domain shikimate pathway enzymes in bacteria. Surprisingly, C. thermophilum AroM showed no evidence for increased metabolic throughput in vitro when compared with isolated enzymatic domains (Verasztó et al, 2020). However, significant sequence variation exists between AroM/Aro1 homologs—specifically, 52% identity between C. thermophilum AroM and C. albicans Aro1—and limited enzymatic characterization necessitates further structural and functional study of multi-domain complexes involved in chorismate biosynthesis, particularly in clinically important species such as C. albicans (Verasztó et al, 2020).
C. albicans is a member of healthy human microbiota, but can also cause life-threatening systemic fungal infections, which occur predominantly in immunocompromised people (Low & Rotstein, 2011; Bongomin et al, 2017). Mortality from these infections remains alarmingly high at ∼40% despite current state-of-the-art treatments (Pfaller & Diekema, 2010). The limited selection of antifungal therapies is further threatened by the evolution of drug resistance in C. albicans (Lee et al, 2021), and emergence of other drug-resistant Candida species, such as Candida auris (Lockhart et al, 2017). To address these challenges, it is essential to identify targets to exploit in novel antifungal therapies.
Hundreds of genes essential for viability, morphogenesis, and host cell escape have been identified in C. albicans using genomic resources, including the C. albicans gene replacement and conditional expression (GRACE) library that covers close to half of the protein coding genes (Roemer et al, 2003; O’Meara et al, 2015; O’Meara et al, 2018; Fu et al, 2021). “Fungal-specific” genes with no identifiable mammalian homolog that encode proteins essential for growth and/or pathogenesis are of particular interest as targets for antifungal drug development (Roemer et al, 2003; Becker et al, 2010; O’Meara et al, 2015; Segal et al, 2018). These studies identified the genes ARO1, ARO2, and ARO7 encoding for non-redundant enzymes in the shikimate pathway as essential in cellulo and in a murine model of disseminated candidiasis (Roemer et al, 2003; Becker et al, 2010; Fu et al, 2021). A recent study demonstrated that transcriptional repression of ARO1 in C. albicans results in a complex phenotype including changes to cell wall integrity and biofilm formation, and attenuated virulence of C. albicans in the Galleria mellonella infection model (Yeh et al, 2020). Furthermore, ARO1 was classified as essential based on mapping transposon insertion sites in a stable haploid isolate of C. albicans (Segal et al, 2018), and confirmed in this study (Fig S1). These observations identify the Aro1 multi-enzyme as essential for C. albicans viability and pathogenesis; however, the role of individual enzymatic domains in this protein remained unknown until now.
Figure S1. GRACE library mutants for ARO1 and DQD1 do not grow in the presence of DOX.

(A) Dilutions of C. albicans CaSS1 and derived conditional expression strains for DQD1 and ARO1 were prepared as described in Fig 4. SC medium lacking arginine and supplemented with 150 μl NAT was inoculated with single colonies of C. albicans and incubated overnight at 30°C. Subcultures were prepared at 1:1,000 in SC medium supplemented with 50 ng/ml DOX and incubated overnight at 30°C. Cultures were normalized to 10 OD600 units/ml then 10-fold serial diluted in water. Diluted cultures were spotted onto SC Agar and SC Agar supplemented with 5 μg/ml DOX, then photographed after 48 h incubation at 30°C. (B) Growth curves were performed as described in Fig S12. (C) Cultures were prepared and imaged as described in Fig S10.
Characterization of the five enzymatic activities of Aro1 and their essentiality for fungal growth is critical to define target sites for small molecule inhibitors with potential therapeutic relevance. Here, we present the molecular structure of all five Aro1 domains across crystal structures of three Aro1 fragments and a cryo-EM–derived model of the full-length protein. These structures reveal a homodimer mediated by the DHQase domain and highlight extensive interdomain flexibility. Surprisingly, we identified that the C. albicans Aro1 (type I) DHQase domain harbors sequence substitutions in its catalytic site that render it inactive. We suggest that this step in the pathway may be supported instead by the type II DHQase Dqd1 enzyme in C. albicans, which showed robust activity against shikimate pathway intermediates in our in vitro assays. Finally, leveraging a conditional expression system, we show that all four functional domains of Aro1 are individually essential for growth of C. albicans. Therefore, we propose that each of these four enzyme domains of C. albicans Aro1 may be targeted by small molecule inhibitors for development of novel antifungal therapies.
Results
Crystal structures reveal active site architectures of all five domains of C. albicans Aro1
To provide molecular details into the essentiality of C. albicans Aro1, we first pursued its structural characterization. For this we designed expression constructs for full-length C. albicans Aro1 and fragments corresponding to individual enzymatic domains N-terminally fused to polyhistidine-TEV protease cleavage site (His6TEV) tag in E. coli. The expression construct for full-length C. albicans Aro1 was unstable in E. coli, likely due to toxicity that was abrogated through a H268K/H280K double mutation. We were unable to express the wild-type full-length Aro1 protein but we were able to express and purify three Aro1 fragments that together encompass all five enzymatic domains. These fragments included the N-terminal 387 residues corresponding to the DHQS domain (Aro1DHQS); residues 387–844, corresponding to the EPSPS domain (Aro1EPSPS); and the C-terminal residues 845–1,551, comprising the SK, DHQase, and DHSD domains (Aro1SK-DHQase-DHSD) (Fig 1). These three fragments were submitted to crystallisation and crystal structures of Aro1DHQS, Aro1EPSPS, and Aro1SK-DHQase-DHSD were determined to 1.85, 1.85, and 2.3 Å, respectively, by molecular replacement (x-ray crystallographic statistics in Table S1).
Table S1 X-ray crystallographic statistics. (17.3KB, docx)
Aro1DHQS was crystallised in the presence of its cofactor NADH. Accordingly, in addition to the two copies of this domain found in the asymmetric unit we identified unambiguous additional electron density corresponding to one NADH molecule bound to each copy of this fragment (Figs 1B and S2). The Aro1DHQS structure was most similar to the corresponding domains in previously characterised AroM proteins from Aspergillus nidulans (Carpenter et al, 1998; Nichols et al, 2003, 2004a) and from C. thermophilum (PDB 6HQV [Verasztó et al, 2020]) (Fig S2). Aro1DHQS also shared significant structural similarity with bacterial DHQS enzymes from Acinetobacter baumannii (PDB 5EKS), Vibrio cholerae (PDB 3OKF), Mycobacterium tuberculosis (PDB 3QBE), and Staphylococcus aureus (PDB 1XAL; [Nichols et al, 2004c]). Comparison of these structures with the Aro1DHQS-NADH complex suggests that the latter follows the “open” binding mode characteristic of DAHP-free structures of DHQS (Fig S2). Notably, the residues that interact with the 3-dehydroquinate analog carbaphosphonate, NADH, and Zn2+ as observed in the A. nidulans AroMDHQS structure are completely conserved in the Aro1DHQS active site (Fig 2A). Specifically, Aro1DHQS residues H264, H268, and H280 coordinate the catalytic Zn2+, allowing for identification of potential catalytic residues of Aro1. As mentioned above, the two Aro1DHQS chains in the asymmetric unit formed a dimer with over 1,100 Å2 buried surface area (Fig S2). The two corresponding domains in the fungal AroM multi-enzymes showed very similar dimeric arrangements (Fig S2), along with the single-domain DHQS enzymes listed above.
Figure S2. Structural analysis of domains of Aro1.

(A) Structure of dimeric Aro1DHQS, coloured by chain. NADH show as ball-and-stick. (B) Superposition of Aro1DHQS with DHQS domain from A. niger (PDB 1NRX) in the open conformation. (C) Structure of Aro1EPSPS. Shikimate-3-phosphate (S3P) shown in ball-and-stick. (D) Superposition of Aro1EPSPS and C. thermophilum AroM (PDB 6QHV). Shikimate (S) and S3P bound to C. thermophilum are shown in ball-and-stick. (E) Structure of Aro1SK from Aro1SK+DHQase+DHSD structure. Region with missing electron density indicated in dashed line (986-967). (F) Superposition of Aro1SK and C. thermophilum AroM (PDB 6QHV). Shikimate (S) and phosphate ions bound to AroM shown in ball-and-stick. (G) Structure of Aro1DHQase from Aro1SK+DHQase+DHSD structure. (H) Superposition of Aro1DHQase, C. thermophilum AroM (PDB 6QHV), and S. pombe AroM (PDB 5SVW). 3-dehydroshikimate (3-DS) bound to C. thermophilum AroM shown in ball-and-stick. (I) Structure of Aro1DHSD from Aro1SK+DHQase+DHSD structure. (J) Superposition of Aro1DHSD, C. thermophilum AroM (PDB 6QHV), and S. pombe AroM (PDB 5SVW). Shikimate (S) bound to C. thermophilum AroM shown in ball-and-stick.
Figure 2. Molecular architecture of active sites in C. albicans Aro1 domains.
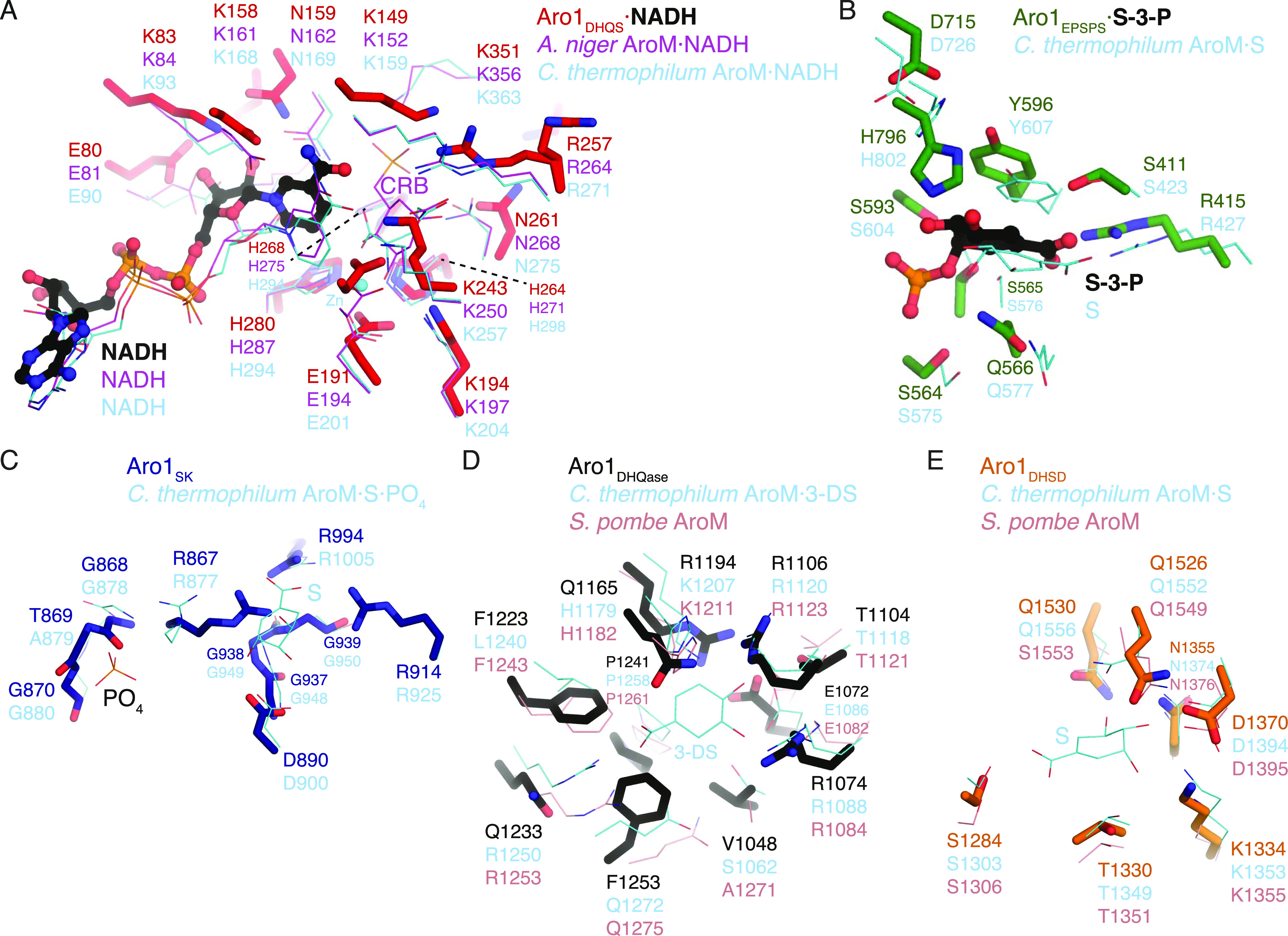
In all panels, Aro1 residues are shown as sticks and the comparative structure’s residues are shown as thin lines. (A) Aro1DHQS superposed with A. niger AroM DHQS domain (PDB 1NVB). NAD molecules bound to the respective structures are shown in sticks. Carbaphosphonate bound to AroM DHQS shown in sticks. Zn2+ ion bound to Aro1DHQS shown as a sphere. (B) Aro1EPSPS superposed with C. thermophilum AroM EPSPS domain (PDB 6HQV). Shikimate-3-phosphate and shikimate bound to Aro1DHQS and AroM, respectively, shown as sticks. (C) Aro1SK from Aro1SK+DHQase+DHSD structure superposed with C. thermophilum AroM SK domain (PDB 6HQV). Shikimate bound to AroM shown in sticks. (D) Aro1DHQase from Aro1SK+DHQase+DHSD structure superposed with C. thermophilum AroM DHQase domain (PDB 6HQV) and S. pombe AroM DHQase domain (PDB 5SWV). 3-dehydroshikimate bound to C. thermophilum AroM shown in sticks. (E) Aro1DHSD from Aro1SK+DHQase+DHSD structure superposed with C. thermophilum AroM DHSD (PDB 6HQV) and S. pombe AroM DHSD domain (PDB 5SWV).
For the Aro1EPSPS fragment structure, we identified two copies of this domain in the asymmetric unit and they adopt a near identical conformation. The regions corresponding to residues 784–794 and 819–823 were disordered and could not be modeled. The structure of Aro1EPSPS (Fig 1B) is composed of two distinct subdomains (I and II) connected by a hinge region. This architecture is consistent with many previously characterised EPSPS enzymes, including those from C. thermophilum (PDB 6HQV); (Verasztó et al, 2020) (Fig S2), V. cholerae (PDB 3NVS), E. coli (1G6T, [Schonbrunn et al, 2001]), and A. baumannii (PDB 5BS5). Because the Aro1EPSPS fragment was crystallised in the presence of shikimate-3-phosphate, we identified additional electron density corresponding to a molecule of this substrate in the cleft between the two subdomains (Figs 1B and S2). The lobes of EPSPS enzymes are known to transition from open to closed states upon substrate binding and our analysis confirmed that Aro1EPSPS-shikimate-3-phosphate complex structure adopted the closed conformation. The interactions with shikimate-3-phosphate in the Aro1EPSPS structure were primarily localised to subdomain II. These molecular interactions identify key catalytic residues of Aro1EPSPS such as D715 and H796. The equivalent residues have been implicated in catalysis of single-domain EPSPS enzymes (Sutton et al, 2016; Lee et al, 2017; Verasztó et al, 2020) as well as in the corresponding domain of C. thermophilum AroM (Fig 2B).
The structure of Aro1SK-DHQase-DHSD contained four copies of this fragment in the asymmetric unit. Aro1SK domain spanned residues 857–1,032 and was connected by a short linker (residues 1,033–1,038) to Aro1DHQase (residues 1,039–1,267), which is directly followed by Aro1DHSD (residues 1,268–1,551). Notably, each Aro1DHQase mediated homodimerization within the asymmetric unit and via crystal symmetry (Fig S3).
Figure S3. Crystal packing of Aro1SK+DHQase+DHSD structure showing dimerization via Aro1DHQase.

left, asymmetric unit; right, crystal lattice. Green arrows highlight dimerization via Aro1DHQase.
Aro1SK (Fig 1B) was highly structurally similar to other SK enzymes (Fig S2) (Gu et al, 2002; Hartmann et al, 2006; Sutton et al, 2015). The regions corresponding to residues 844–855 and 970–984 were not modeled because of poor electron density. The latter region corresponds to the “lid loop” or “P-loop” that is involved in positioning of ATP and shikimate in the active site (Gu et al, 2002; Hartmann et al, 2006; Sutton et al, 2015). Based on comparisons with other SK structures, residues 967–984 are expected to coordinate the phosphate groups of ATP and shikimate itself. Also, in accordance with its apoenzyme state, residues 1,011–1,021 were localised away from the active site cleft; these residues are expected to engage with the adenine moiety of ATP. Whereas the disordered regions did not allow for full structural analysis of the Aro1SK active site, all the ATP phosphate and shikimate-binding residues appear conserved with other characterised SK, including this domain in C. thermophilum AroM (Fig 2C). Based on this, we hypothesised that D890 in Aro1SK would interact directly with shikimate as observed in bacterial SKs (Gu et al, 2002; Hartmann et al, 2006; Sutton et al, 2015), whereas the R980 residue would be involved in positioning of ATP phosphates (Gu et al, 2002; Hartmann et al, 2006; Sutton et al, 2015). We predicted that both these residues would be critical for Aro1SK activity.
Aro1DHQase domain within the Aro1SK-DHQase-DHSD fragment adopts the TIM barrel fold typical of type I DHQase enzymes and also shows significant similarity with the corresponding domains of the two other structurally characterised fungal AroM proteins (Figs 1B and S2). However, the active site composition of Aro1DHQase shows important differences in comparison with previously characterised type I DHQase enzymes (Fig 2D). Notably, we failed to identify residues that could serve as the catalytic dyad typical of type I DHQase enzymes (Leech et al, 1998; Lee et al, 2002; Light et al, 2013). This catalytic dyad features a histidine that acts as a general acid/base and is conserved in most characterised members of this enzyme family including this domain in AroM proteins (i.e., H1179 and H1182 in C. thermophilum and Schizosaccharomyces pombe AroM’s, respectively, Fig 2D) (Verasztó et al, 2020) (PDB 5SWV). The corresponding position in Aro1DHQase is occupied by a glutamine (Q1165) (Fig 2D). The other residue of the dyad in type I DHQase enzymes is usually represented by a lysine that functions as a nucleophile and forms a covalent bond with 3-dehydroquinate (Light et al, 2014). With no suitable lysine residue in the Aro1DHQase active site, only the R1194 residue could be implicated for this role. As mentioned, the Aro1SK-DHQase-DHSD fragment structure shows that Aro1DHQase is actively involved in dimerization of this fragment (Fig S3).
Finally, Aro1DHSD within the Aro1SK-DHQase-DHSD fragment shows the two subdomain architecture typical of AroE-type shikimate dehydrogenase enzymes (Figs 1B and S2) (Bagautdinov & Kunishima, 2007; Gan et al, 2007; Hoppner et al, 2013). The N-terminal subdomain adopts an ɑ/β fold with a central 6-stranded β-sheet that usually harbors most of the catalytic apparatus, whereas the C-terminal subdomain adopts the Rossman fold that, in other homologs, provides residues involved in interactions with NAD(P)H. Comparative analysis with equivalent domains of the two fungal AroM enzymes suggested Aro1DHSD residues involved in the shikimate binding (Fig 2E). In particular, Aro1DHSD K1134 and D1370 residues are appropriately positioned to serve as the catalytic dyad identified in other DHSD enzymes, with the lysine acting as a general base and the aspartate stabilizing this residue as well as the 3-dehydroshikimate C4 hydroxyl (Peek & Christendat, 2015).
The crystal structure of Aro1SK-DHQase-DHSD provided the first indication of interdomain interactions in full-length Aro1. According to this structure, each of the three Aro1 domains forms extensive interactions with the other two neighboring domains, specifically involving residues W177, S880, R1021, R1022, P1384, P1385 and G1487 (Fig 1B). The Aro1SK and Aro1DHQase interface covered 1,096 Å2, whereas the Aro1SK-Aro1DHSD and Aro1DHQase-Aro1DHSD interfaces buried 557 and 542 Å2, respectively. The Aro1SK domain, in particular, made the most intimate contacts with the other two domains including interdigitation of its C-terminal α-helix seven via residues R1021 and R1022 into the space between the three domains (Fig 1B). A comparison of this fragment of Aro1 with the structures of full-length C. thermophilum AroM and the S. pombe AroM DHQase-DHSD fragment revealed significant differences in the relative arrangement of equivalent domains (Fig S4). Specifically, the location of Aro1SK relative to Aro1DHQase appears to be rotated ∼30° compared with the arrangement of the corresponding domains in the two AroM protein structures. Along the same lines, the C-terminal lobe of the Aro1DHSD domain also appears rotated by ∼30°. This differential positioning, however, could be due to the apo status of the Aro1SK domain, with its α-helix 7 lacking interactions with a phosphate group donor molecule allowing it to protrude into the core of the tri-domain structure.
Figure S4. Comparison of C. albicans Aro1SK+DHQase+DHSD and AroM SK+DHQase+DHSD regions.

S and 3-DS bound to AroM shown in ball-and-stick. Rotational differences between SK and DHSD domains differ by ∼30°.
Analysis of full-length C. albicans Aro1 by cryogenic electron microscopy suggests significant interdomain mobility
Although we were unable to express recombinantly wild-type Aro1 in E. coli, we succeeded in purifying His6TEV-tagged Aro1 from a C. albicans engineered strain described in the next section (see also the Materials and Methods section). The size-exclusion chromatography indicates that Aro1 exists predominantly as a stable dimer under experimental conditions (Fig S5). Analysis of Aro1DHQS and the Aro1SK-DHQase-DHSD fragment X-ray structures presented above (Figs 1B and 2) suggested that Aro1 dimer can be stabilised via its DHQS and DHQase domains and interactions between the SK, DHQase, and DHSD domains. Although we were unable to obtain crystals of C. albicans Aro1, we used cryogenic electron microscopy single particle reconstruction (cryoEM SPR) to investigate the Aro1 structure (Figs 3 and S6 and Table S2). The analysis of this sample under cryogenic conditions revealed a combination of full-length and fragmented particles (Fig 3A), likely due to breakage by the water–air interface during blotting. Nevertheless, we were able to identify a subgroup of particles corresponding to dimers of full-length Aro1. Within these particles, there was further structural heterogeneity, due to the Aro1SK-DHQase-DHSD fragment tilting in various conformations relative to the Aro1DHQS domain. In all classifiable Aro1 particles, including the ones corresponding to Aro1 fragments, we also observed considerable blurring of one of the two Aro1EPSPS domains, indicating extensive mobility of this domain. After 3D classification, we identified 87,484 particles belonging to a stable conformation representing a dimer of full-length Aro1 at resolution of 4.19 Å. We performed local refinement on the dimer of the Aro1SK-DHQase-DHSD region, as well as the dimer of Aro1DHQS and Aro1EPSPS domains that resulted in molecular maps of resolutions of 3.16 Å for Aro1SK-DHQase-DHSD and 3.43 Å for Aro1DHQS-EPSPS (Figs 3B and S6).
Figure S5. Purification and size-exclusion profiles of Aro1.

(A) Size-exclusion chromatography profile of Aro1 purified from C. albicans. Fractions collected are demarcated in red. (B) SDS–PAGE of Aro1 SEC fractions.
Figure 3. Cryo-EM structure of the unliganded full-length Aro1 from C. albicans.
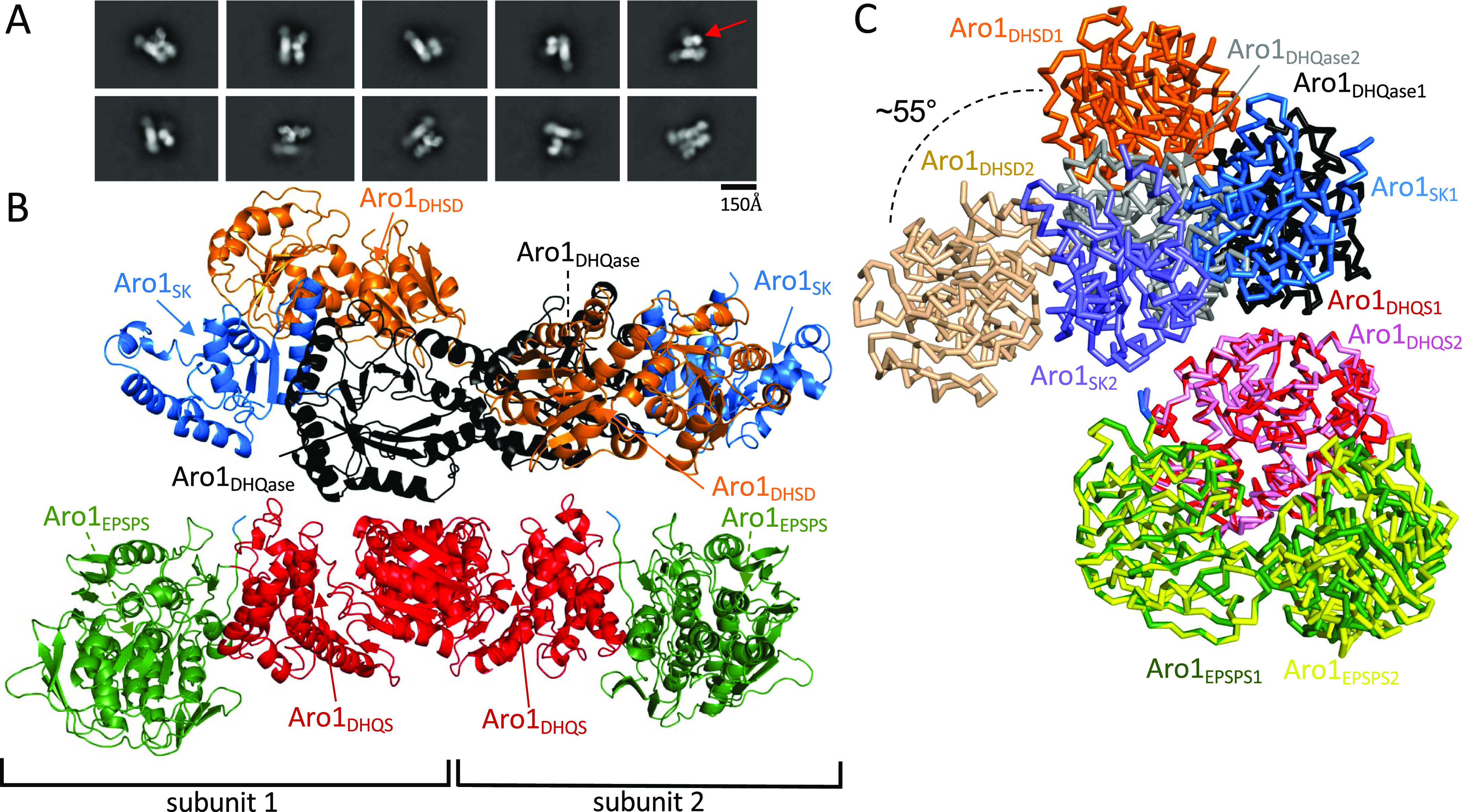
(A) Selected 2D class averages of the full-length Aro1 dimer. The red arrow points to the Aro1SK-DHQase-DHSD region, which is highly heterogeneous and found tilted at various angles relative to the Aro1DHQS-EPSPS region. (B) The structure of the full-length Aro1 from C. albicans. The final resolution for the Aro1SK-DHQase-DHSD domain dimer is 3.16 Å and the Aro1DHQS-EPSPS domain dimer is 3.43 Å. Coloured arrows point towards the ligand binding sites of each domain, which are unoccupied in our structure. The Aro1SK-DHQase-DHSD region of subunit 1 is tilted away from the Aro1DHQS-EPSPS region and is referred to as the “loose” monomer, whereas the Aro1SK-DHQase-DHSD region of subunit 2 is nearly parallel to the Aro1DHQS-EPSPS region and is referred to as the “tight” monomer. (C) Comparison of “tight” and “loose” monomers from the full-length Aro1 dimer superposed on the Aro1DHQS-EPSPS region. Although the backbones of each domain overlays with low RMSD, the relative orientations of the Aro1SK-DHQase-DHSD and Aro1DHQS-EPSPS regions change dramatically.
Figure S6. Cryo-EM image processing flow chart.

(A) Example high quality micrograph of Aro1 and selected 2D class averages showing broken particles consisting of Aro1DHQS and Aro1EPSPS as well as full-length Aro1. (B) FSC curves for homogeneous refinement of the full-length Aro1 dimer (red), local refinement of Aro1SK-DHQase-DHSD dimer (orange), local refinement of Aro1DHQS-EPSPS dimer (green), local refinement of Aro1DHQS-EPSPS subunit 1 after EPSPS domain 1 focused classification (blue), and local refinement of Aro1DHQS-EPSPS subunit 2 after EPSPS domain 1 focused classification (purple). (C) Image processing flow chart for structure determination of full-length Aro1.
Table S2 Cryo-EM data collection, refinement, and validation statistics. (14.8KB, docx)
The 4.19 Å map that corresponds to the homogeneous refinement based on 3D reconstruction of full-length Aro1 dimer (EMDB: 26357, PDB: 7U5S). In this map, we performed only a rigid body fit of two high-resolution models for Aro1DHQS-EPSPS and Aro1SK-DHQase-DHSD domains. These two models (Aro1DHQS-EPSPS and Aro1SK-DHQase-DHSD) were obtained by docking and refining against cryo-EM maps X-ray crystallographic models described earlier. We performed atomic refinement of X-ray crystallographic models with PHENIX and corrected structures manually with Coot. The molecular maps at lower contour level indicated where the linker between Aro1DHQS-EPSPS and Aro1SK-DHQase-DHSD is located (Fig S6). Analysis of the possible distances for 11 aa unmodeled linker excluded the possibility of domain swapping.
As implied by the crystal structure of isolated Aro1DHQS, dimerization of the full-length cryoEM SPR structure of Aro1 is mediated by extensive contacts between the Aro1DHQS domain, resulting in an arrangement similar to the one observed in C. thermophilum AroM homodimer. Notably, we did not add any ligands to C. albicans Aro1 sample used for our cryo-EM analysis, nor did we observe any evidence of endogenous ligands in the five active sites, in contrast to the crystal structure of C. thermophilum Aro1 that contained numerous ligands. The interface between the Aro1DHQS domains in the Aro1 dimer is the same as seen in the Aro1DHQS crystal structure. There are additional contacts between the Aro1DHQase domains in the Aro1 dimer that may also contribute to dimerization. Interestingly, within the Aro1 dimer we observed asymmetry between the monomers, with the Aro1SK-DHQase-DHSD region tilting ∼55° relative to the Aro1DHQS and Aro1EPSPS domains (Fig 3C). This large rearrangement of the Aro1SK-DHQase-DHSD region results in ∼35 Å shift in the Aro1SK domain positioning it directly above subdomain II of the Aro1EPSPS domain. In this conformation the Aro1SK-DHQase-DHSD region in one monomer is nearly parallel to the Aro1DHQS and Aro1EPSPS domains, whereas the other monomer has the Aro1SK-DHQase-DHSD region tilted ∼40° relative to the Aro1DHQS and Aro1EPSPS domains.
The analysis of the full-length Aro1 structure highlighted the high level of structural similarity between the specific domains in the Aro1 dimer and their individual crystal structures (Fig S7). The Aro1DHQS domain, Aro1EPSPS subdomain II, and Aro1SK-DHQase-DHSD backbones show nearly identical conformation within the core structure of each region with minor differences seen in some loop regions. This implies that the large movement of the Aro1SK-DHQase-DHSD region does not drive structural changes that modulate substrate binding or catalysis, but instead reorients and repositions the domains. When visualized in the molecular structure, the positioning of the domains of Aro1 matches the order of reactions in the pathway in a counter-clockwise fashion, with the domain that catalyses the first reaction (Aro1DHQS) positioned at the “bottom” of the dimer, with the next two domains (Aro1DHQase and Aro1DHSD) positioned sequentially above Aro1DHQS, followed by Aro1SK at the “top left” and terminating with Aro1EPSPS at the “bottom left” (Fig 3C). However, each individual active site is more than 40 Å from each other and each face outwards from the core of the dimer (Fig 3C). We also do not observe any obvious clefts or tunnels between active sites.
Figure S7. Superposition of individual domains and regions of the full-length monomers and crystal structures.

(A) Superposition of the Aro1DHQS dimer from the crystal structure (pink) and from the full-length dimer (red). (B) Superposition of Aro1EPSPS from the crystal structure (yellow) and from a monomer of the full-length dimer (green). (C) Superposition of the Aro1SK-DHQase-DHSD region from the crystal structure (cyan, light orange, and grey) and the Aro1SK-DHQase-DHSD region (blue, orange, and black) from a monomer of the full-length dimer.
Impairment of specific enzymatic activities of Aro1 has a dramatic effect on C. albicans growth and viability
To test the essentiality of Aro1’s individual enzymatic activities, we generated conditional expression strains in C. albicans in which mutations were introduced in a constitutively expressed allele of ARO1, whereas the other wild-type allele was transcriptionally repressible. First, we replaced the promoter of one allele of ARO1 with a tetracycline-repressible promoter (tetO) such that addition of the tetracycline analog doxycycline (DOX) represses transcription of this allele of ARO1. In this strain, in the absence of DOX, ARO1 was overexpressed at the transcript (Fig S8) and protein level (Fig S9), relative to the parental strain C. albicans SN95. To repress transcription of the ARO1 allele controlled by the tetO promoter, cultures were incubated overnight in a low concentration of DOX, then subcultured into media with (or without) a high concentration of DOX. Based on transcriptional analysis, the DOX treatment reduced ARO1 transcript abundance to ∼50% of wild-type levels (Fig S8), but did not alter growth on solid or liquid synthetic complete medium (Fig 4A and B). As anticipated, introduction of the tetO promoter at both alleles of ARO1 resulted in further overexpression in the absence of DOX, whereas DOX reduced ARO1 transcript abundance to ∼1% of that observed for the wild-type strain (Fig S8) and caused a severe growth defect on solid and liquid medium (Fig 4A and B). After 24 h of transcriptional repression, the cells were examined by fluorescence microscopy. As anticipated, DOX treatment resulted in abnormal cell morphology and separation defects only for the C. albicans tetO-ARO/tetO-ARO1 strain (Fig 4C). Furthermore, we observed a significant increase in cell death upon repression of ARO1, based on staining with the membrane-impermeable dye propidium iodide (Fig 4C).
Figure S8. Validation of tetO-driven overexpression and DOX-mediated repression of ARO1 and DQD1 transcripts by RT-qPCR.

(A) Cultures were grown in SC medium overnight at 30°C, subcultured into SC medium at 1:1,000 supplemented with 0.05 μg/ml DOX overnight, then subcultured 1:100 into SC medium supplemented with 5 μg/ml DOX for 4 h. RNA was extracted, DNAse-treated, and used to synthesize cDNA. Transcript abundance was quantified by real-time PCR. Data are mean relative ARO1 transcript abundance normalized to ACT1 ± SEM for three independent cultures, each assayed in technical triplicate. Left: conditional expression strains constructed for this study in C. albicans SN95. Right: conditional expression strain for ARO1 in C. albicans CaSS1, from the GRACE library. (B) Data are mean relative DQD1 transcript abundance normalized to ACT1 ± SEM for three independent cultures assayed in technical triplicate. Conditional expression strain for DQD1 in C. albicans CaSS1, from the GRACE library.
Figure S9. Aro1 and mutant derivatives are expressed in C. albicans.

(A) The tetO promoter drives DOX-repressible overexpression of Aro1. Panel depicts SDS–PAGE of whole-cell lysates prepared from C. albicans, stained with Coomassie Blue R-250. Cultures were grown in SC medium at 30°C overnight, then subcultured into SC medium and SC medium supplemented with 5 μg/ml DOX for 4 h at 30°C. (B) The tetO promoter drives DOX-dependent overexpression of N-terminally His6TEV tagged Aro1. The Western immunoblot (upper) was probed with anti-His5 monoclonal antibody. Equivalent loading was confirmed by Coomassie Blue R-250 stained SDS–PAGE of identical samples (lower). (A) Lysates were prepared as in (A). (C) His6TEV-tagged truncation/domain deletion mutants of Aro1 expressed under the ACT1 promoter are produced at equal or more abundant levels than the full-length protein in C. albicans. Cultures were grown in SC medium overnight at 30°C, subcultured into SC medium supplemented with 0.050 μg/ml DOX overnight, then subcultured into SC medium supplemented with 5 μg/ml DOX for 4 h. (D) Amino acid substitutions in PACT1-His6TEVAro1 do not alter abundance of Aro1 in C. albicans. (C) Lysates were prepared as in (C).
Figure 4. Effect of Aro1 active site mutations on C. albicans viability.
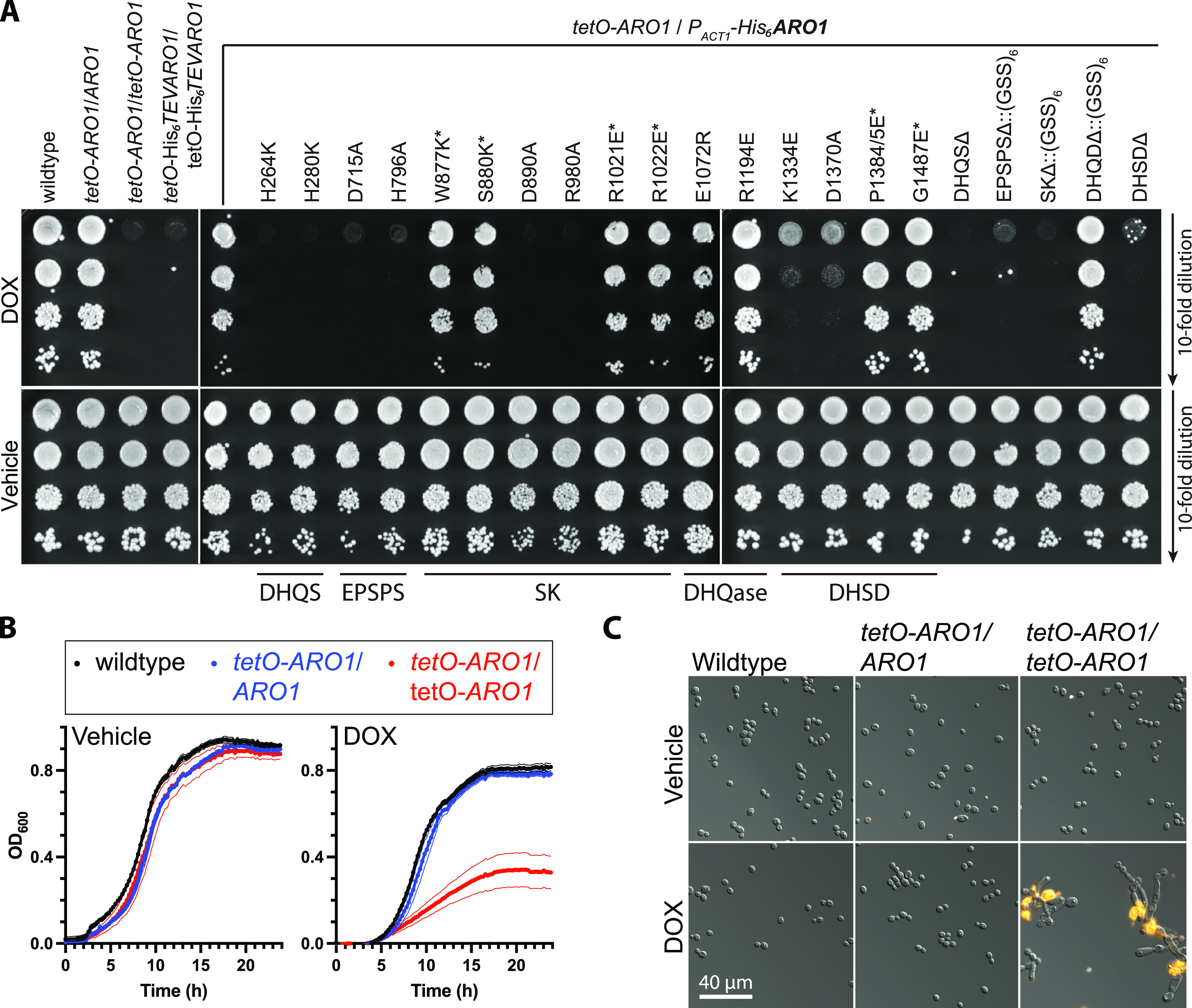
(A) ARO1 activities are required for growth of C. albicans. SC medium lacking arginine and supplemented with 150 μl NAT was inoculated with single colonies of C. albicans and incubated overnight at 30°C. Subcultures were prepared at 1:1,000 in SC medium supplemented with 50 ng/ml DOX and incubated overnight at 30°C. Cultures were normalized to 10 OD600 units/ml then 10-fold serial diluted in water. Diluted cultures were spotted onto SC Agar and SC Agar supplemented with 5 μg/ml DOX, then photographed after 48 h incubation at 30°C. Asterisk indicates substitutions to amino acids identified to mediate interdomain contacts. (B) ARO1 is required for growth of C. albicans. SC medium lacking arginine and supplemented with 150 μl NAT was inoculated with single colonies of C. albicans and incubated overnight at 30°C. 80 μl SC cultures were prepared at 0.001 OD600 unit/ml, in the presence and absence of 5 μg/ml DOX, then incubated at 30°C for 24 h. Data are mean optical densities for quadruplicate cultures ± SD for a representative experiment. (B, C) ARO1 is required for viability of C. albicans. Representative fluorescence micrographs of cultures from (B), stained with 1 μg/ml propidium iodide to label inviable cells.
To determine the essentiality of individual amino acids in the active sites of the five domains of Aro1 revealed by our structural analysis, we altered the remaining wild-type allele of ARO1 in C. albicans tetO-ARO1/ARO1. To monitor expression of the mutant Aro1 derivatives, we introduced a TEV protease cleavable hexahistidine tag (His6TEV) to the N-terminus of Aro1. To first confirm that this tag did not interfere with Aro1 activity, we introduced a tetO promoter and His6TEV tag at both ARO1 alleles in C. albicans. Similarly to C. albicans tetO-ARO1/tetO-ARO1, C. albicans tetO-His6TEVARO1/tetO-His6TEVARO1 had a growth defect on solid (Fig 4A) and in liquid medium (Fig S12) when doxycycline was added. Overexpression and DOX-dependent depletion of His6TEVAro1 was confirmed by Western blotting (Fig S9). This indicated that both alleles of ARO1 were tagged and functional in vivo. We then generated a DNA construct in which His6TEVAro1 was under the control of the constitutive ACT1 promoter (PACT1). This construct was introduced into the wild-type allele of C. albicans tetO-ARO1/ARO1, creating C. albicans tetO-ARO1/PACT1-His6TEVARO1. As expected, this strain did not present a growth defect on solid (Fig 4) or liquid medium (Fig S12), because of expression of functional His6TEV-tagged Aro1 from the ACT1 promoter. Next, we generated isogenic strains encoding site-specific amino acid substitutions, truncations, and internal domain deletions to PACT1-His6TEVARO1. For deletion of the DHQS domain (Aro1DHQSΔ) the His6TEV tag was fused to the N-terminus of Aro1388-1551; for deletion of the DHSD domain (Aro1DHSDΔ), a stop codon was introduced at Ile879; and the three internal domains were deleted and replaced with a (GlySerSer)6 linker (Aro1EPSPSΔ::(GSS)6, residues 387–857; Aro1SKΔ::(GSS)6, residues 849–1,040; Aro1DHQaseΔ::(GSS)6, residues 1,036–1,284). All truncation and deletion containing Aro1 derivatives appeared at expected molecular weight and at equal abundance when compared with the wild-type His6TEVAro1 based on Western blotting of C. albicans whole-cell lysates (Fig S9); Aro1DHQSΔ appeared at greater abundance. Introduction of amino acid substitutions did not alter protein abundance in Western blots (Fig S9).
Figure S12. Catalytically inactive derivatives of ARO1 have domain-specific effects on growth of C. albicans in liquid culture.

Cultures were grown in SC medium overnight at 30°C, subcultured into SC medium and SC medium supplemented with 0.05 μg/ml DOX overnight. Cultures were normalized to an OD600 of 1, then diluted at 1:1,000 into 96-well plates containing 80 μl of SC medium (left) or SC medium supplemented with 5 μg/ml DOX (right). Growth at 30°C was monitored by optical density at 15-min intervals for 24 h. Data are mean OD600 ± SD for quadruplicate cultures in a representative experiment of three performed.
DOX-dependent depletion of tetO-ARO1 in strains carrying His6TEVAro1 with predicted inactivating substitutions in the DHQS domain of Aro1 as informed by structural analysis (Aro1H264K, Aro1H280K, and Aro1DHQSΔ) conferred severe DOX-dependent growth defects on solid (Fig 4A) and liquid medium (Fig S12). Almost all cells had abnormal morphology and were dead after 24 h growth in liquid medium with DOX, based on fluorescence micrographs (Fig S10). The same growth phenotype was observed for EPSPS domain mutants (Aro1D715A, Aro1H796A, and Aro1EPSPSΔ::(GSS)6) (Figs 4A and S12), except no cell death was evident in DOX-treated cultures after 24 h (Fig S10). SK mutants (Aro1D890A, Aro1R980A, and Aro1SKΔ::(GSS)6) had a severe growth defect on solid medium (Fig 4A), and reduced growth in liquid medium (Fig S12); cells were rounded with a minor separation defect, and a minority of dead cells were identified. DHSD mutants (Aro1K1334E, Aro1D1370A, and Aro1DHSDΔ) had reduced growth on solid (Fig 4A) and liquid medium (Figs 4A and S12); cells were enlarged, and a minority of dead cells were identified (Fig S10). No effects on growth or viability were identified for DHQase mutants (Aro1E1072R, Aro1R1194E, and Aro1DHQaseΔ::(GSS)6).
Figure S10. Catalytically inactive derivatives of ARO1 have domain-specific effects on viability of C. albicans in liquid culture.

C. albicans CaLC6598 and derivative strains encoding amino acid substitutions, deletions, and truncations of His6TEV-Aro1 were examined by fluorescence microscopy to assess morphology and viability. Cultures were grown in SC medium overnight at 30°C, subcultured into SC medium and SC medium supplemented with 0.05 μg/ml DOX overnight. Cultures were normalized to an OD600 of 1, then diluted at 1:1,000 into 96-well plates containing 80 μl of SC medium (top) or SC medium supplemented with 5 μg/ml DOX (bottom). After 24-h incubation at 30°C, propidium iodide was added to 1 μg/ml (final). Cells were spotted on glass slides after 10 min incubation at room temperature and then examined by fluorescence microscopy. Panels depict representative fields of view for wells from three independent experiments. Scale bar is 40 μm.
We also investigated the effect of Aro1 interdomain interactions captured by our structural analysis. However, the amino acid substitutions at residues that mediate interdomain interactions (Aro1W877K, Aro1S880K, Aro1R1021E, Aro1R1022E, Aro1P1384EP1385E, and Aro1G1487E) showed no effect on growth or viability of corresponding C. albicans strains (Fig 4A).
Enzymatic characterization of Aro1 active site mutants
To define how site-specific substitutions in each of the five active sites of Aro1 affect their enzymatic activities, we tested the five enzymatic domains of this multi-domain protein in vitro using previously established protocols (see the Materials and Methods section for details). In contrast to wild-type Aro1, the version of this protein carrying H268K/H280K double mutation in the N-terminal DHQS domain was successfully expressed and purified from E. coli as an N-terminal His6TEV-tag fusion. Aro1H268K/H280K was catalytically inactive in a DHQS activity assay as compared with wild-type His6TEV-tagged Aro1 purified from C. albicans (Table 1). This result was consistent with previous studies that showed mutation to His residues involved in divalent metal ion coordination resulted in a complete loss of enzymatic activity in an AroM protein (Park et al, 2004). This suggested that the severe growth defect we observed for the C. albicans strain expressing this Aro1 mutant is due to a complete loss of one of the individual enzymatic activities of this protein (Fig 4A).
Table 1.
Relative enzymatic activity of Aro1 active site mutants.
| Enzymatic activity relative to WT Aro1 | Active Site Mutant | ||||||
|---|---|---|---|---|---|---|---|
| DHQS | EPSPS | SK | SDH | ||||
| H268K/H280K | D715A | H796A | D890A | R980A | K1334E | D1370A | |
| N.d. | 1.6% | 20.6% | 1.9% | 2.9% | N.d. | N.d. | |
n.d. none detected.
Next, we used the expression construct for Aro1H268K/H280K as a base for introduction of mutations in other Aro1 domain active sites. Catalytic activity in other Aro1 domains was unaffected by this mutation (Fig S11). As above, the corresponding Aro1 mutants were purified from E. coli and their enzymatic activities were compared to wild-type His6TEV-tagged Aro1 purified from C. albicans (Table 1). We found that active site mutations in SK and DHSD domains resulted in near-complete and complete losses of corresponding activities for Aro1, respectively. Similarly, a near-complete loss of activity was observed for the D715A mutant localised to the EPSPS domain. Introduction of the H796A mutation in this domain had a less severe effect, with Aro1 retaining ∼20% of EPSPS activity relative to the wild-type protein. Overall, our enzymatic analysis demonstrated that selected active site mutations severely abrogated the DHQS, EPSPS, SK, and DHSD activities of C. albicans Aro1 multi-domain enzyme. Taken together with the growth defect phenotypes that we observed for C. albicans strains expressing the corresponding Aro1 active site mutants, these results suggested that the enzymatic activity of each of these four Aro1 domains is critical for C. albicans growth and viability.
Figure S11. Comparison of enzymatic activity between ARO1 purified from C. albicans and recombinant ARO1 purified from E. coli.

Enzymatic activity, expressed as percent relative to wild type, is shown. The data depicted for the EPSPS, SK, and DHSD domains is from recombinant full-length enzyme purified from E. coli. The wild-type data are from C. albicans Aro1 and normalized to 100%.
The DHQase domain of Aro1 is catalytically inactive
Our structural analysis revealed significant deviation in active site composition of the Aro1 DHQase domain and the C. albicans strain expressing an Aro1 derivative lacking this domain (Aro1DHQaseΔ::(GSS)6) showed no detectable growth defect in contrast to other ARO1 mutants (Figs 4A and S12). These data led us to hypothesize that the DHQase domain of Aro1 in C. albicans is catalytically inactive. To investigate this, we purified wild-type Aro1 from C. albicans tetO-His6TEVARO1/tetO-His6TEVARO1 and tested it for DHQase activity in vitro (Fig 5A). The Aro1 enzyme showed no detectable activity in this assay, whereas the E. coli type I DHQase, AroD, which we used as a control had 1.3 × 103 U/mg DHQase activity under the same conditions. Because Aro1 is a multi-domain enzyme, we considered the possibility that the 3-dehydroshikimate product measured in our assay may have been rapidly converted to shikimate via the activity of the DHSD domain. In such a scenario, our assay may have failed to capture product formation. To account for this, we expressed and purified the isolated DHQase domain of Aro1 (residues 1,041–1,249). Consistent with full-length Aro1, we observed no DHQase activity for this fragment (Fig 5A).
Figure 5. Diversity of type I DHQase active site composition in Candida species.

(A) C. albicans Aro1 is catalytically inactive in a DHQase activity assay. Candida glabrata and C. parapsilosis Aro1 were active, along with C. glabrata Dqd1. E. coli AroD was included as a positive control. (B) Multiple sequence alignment of the region of DHQase containing residues that comprise the catalytic dyad. Positions where residues involved in catalysis are expected are denoted with a (*), and the position where the catalytic His is expected is highlighted in blue. If a DQD1 ortholog was found in the genome, it is listed on the right and given as percent shared amino acid sequence identity.
A catalytic His is conserved in type I DHQases and its substitution to Ala in E. coli AroD severely abrogated DHQase activity (Leech et al, 1995). Because C. albicans Aro1 features a Gln at this position, we sought to determine if this substitution (His to Gln) would eliminate enzymatic activity in a functionally active DHQase. Indeed, the E. coli AroDH143Q completely lost DHQase activity in our assay (Fig 5) in line with our hypothesis that DHQase domain in C. albicans Aro1 carries active site substitutions incompatible with catalytic activity typical for this class of enzymes.
Next, we performed a comparative analysis of Aro1 sequences across seven clinically important Candida species including C. albicans, C. auris, Candida glabrata, Candida lusitaniae, Candida parapsilosis, Candida tropicalis, and Candida. viswanathii (Fig 5B). All seven Aro1 sequences showed conservation in all residues established to be essential for activity of the four C. albicans Aro1 enzymatic domains described above; however, five of seven Aro1 sequences analysed showed substitution to the DHQase domain’s catalytic His residue, whereas three of the seven also contained substitutions at the position corresponding to catalytically essential Lys residue in this domain. Only the C. glabrata and C. parapsilosis Aro1 sequences retained the conserved catalytic dyad (Fig 5B). Based on this analysis we predicted that these two Aro1 enzymes feature catalytically active DHQase domains. To test this, we expressed and purified C. glabrata and C. parapsilosis Aro1 proteins as N-terminal His6TEV fusions in E. coli. Both multi-enzymes showed robust DHQase activities of 23.7 U/mg and 22.3 U/mg, respectively. Thus, our analysis revealed an important divergence in activity of Aro1 among Candida species, with the multi-domain enzyme in C. albicans showing no DHQase activity in contrast to its homologues found in C. glabrata and C. parapsilosis.
The discovery that C. albicans Aro1 does not have DHQase activity prompted us to search the genome for genes encoding other enzymes that might perform this reaction. We did not identify any genes encoding type I DHQase homologs, so we searched for type II DHQases, which perform the same reaction but share no sequence or structural similarity and use a different catalytic mechanism (Roszak et al, 2002). We identified DQD1, which encodes a type II DHQase homolog that adopts the canonical fold and dodecamerization expected for type II DHQases (Trapani et al, 2010). Notably, DQD1 was identified as an essential gene in C. albicans by the GRACE strategy (Fu et al, 2021) and by mapping transposon insertions in a stable haploid isolate of C. albicans (Segal et al, 2018). Purified Dqd1 showed 1.5 × 103 U/mg DHQase activity in vitro, which is comparable with that observed for E. coli AroD and higher than DHQase activity we observed for C. glabrata and C. parapsilosis Aro1 enzymes (Fig 5A). This suggests that the activity of Dqd1 in C. albicans may be sufficient to support the shikimate pathway and proposes a new role for this enzyme. We then searched each of the clinically relevant Candida genomes mentioned above for the presence of a gene encoding a type II DHQase. In all instances where mutations have occurred in the catalytic dyad of the Aro1DHQase domain, there was a DQD1 ortholog present in the genome; however, no type II DHQase was found in the genome of either C. glabrata or C. parapsilosis (Fig 5B). This analysis demonstrates a correlation between the presence of a type II DHQase and the loss of DHQase activity in the Aro1 multi-domain enzyme.
Discussion
AroM/Aro1 multi-enzymes that catalyse five consecutive reactions of the shikimate pathway are a distinctive feature of chorismate biosynthesis in apicomplexan and fungal species, such as Candida. Aro1 is essential for C. albicans viability and pathogenesis, opening the possibility to target its enzymatic activities in new antifungal therapies. This study uncovers the molecular features of C. albicans Aro1 that paves the way for design of such inhibitors.
To gain molecular insight into this protein’s activities, we visualized C. albicans Aro1’s domains and the full-length protein using a combination of protein crystallography and single-molecule cryoEM SPR approaches. These analyses revealed that C. albicans Aro1 forms a dimer, the oligomeric state shared by AroM from C. thermophilum; the only other fully structurally characterised representative of this multienzyme family (Verasztó et al, 2020). Our structural analysis suggests the Aro1 dimer is primarily maintained through interactions between N-terminal DHQS domains and supported by additional interactions between DHQase domains of the protomers. However, an Aro1 construct containing a deletion of the DHQase domain was able to support wild-type growth of C. albicans, suggesting that this domain is not required for folding of the rest of Aro1 protein into an active conformation(s).
Wheeras the Aro1 dimer is reminiscent of that observed in C. thermophilum AroM, Aro1 monomers adopt distinct conformations in the dimer which were not observed in the structure of AroM. We refer to these distinct conformations of Aro1 chains in the dimer as the “tight” and “loose” monomers, respectively (Fig 3). Notably, the “tight” monomer does not permit direct interaction between the Aro1SK and Aro1EPSPS domains, despite cross-linking mass spectrometry data for C. thermophilum AroM suggesting that the AroMSK domain and AroMEPSPS subdomain II can co-localise in solution (Verasztó et al, 2020). This discrepancy may be due to significant interdomain movement observed by cryoEM SPR in truncated Aro1 fragments and also reported for the AroM protein. Accordingly, observed asymmetry in Aro1 protomers may represent stable intermediates within the conformational cycle of Aro1 that is also implied for the AroM protein.
The impact of assembly of multiple enzymatic domains into a single Aro1/AroM protein for their catalytic activity remains elusive. In vitro characterization of C. thermophilum AroM concluded that concatenation of domains catalysing consecutive enzymatic reactions into a single protein does not increase the flux through the pathway (Verasztó et al, 2020). As in C. thermophilum AroM, our cryo-EM SPR structure of C. albicans Aro1 did not reveal that the individual active sites are spatially co-localized, nor is there evidence of any structural features connecting them. These observations suggest that there is no exchange of substrates/products between active sites and concatenation of the domains in C. albicans Aro1 should not increase the flux through the pathway; however, this remains to be experimentally deduced. Given our observation that the DHQase domain in this multi-enzyme is inactive, there are clearly differences between fungal Aro1/AroM that remain to be explored. The relative orientation between the Aro1SK and Aro1EPSPS domains changes dramatically between “tight” and “loose” conformations of Aro1 protomers (Fig 3), which may facilitate diffusion of substrates between these domains and support flux between consecutive steps supported by these Aro1 domains. The “blurring” of the subdomain I of Aro1EPSPS in our cryoEM SPR analysis suggests local movement of this fragment. The exchange from “loose” to “tight” conformation may restrict the movement of this portion of Aro1EPSPS and promote the catalytically active positioning between the two Aro1EPSPS subdomains.
DHQS, EPSPS, SK, and DHSD domains of Aro1 (Fig 1) shared strong structural similarity with single-domain bacterial and fungal shikimate pathway enzymes, including in the molecular architecture of their active sites. This allowed us to identify key residues important for individual catalytic activities of Aro1. We constructed a collection of Aro1 mutants with impaired activity in one of the domains, which was verified by in vitro enzymatic assays. C. albicans strains expressing these Aro1 mutants demonstrated severe growth defects, particularly in those carrying active site substitutions in the DHQS and EPSPS domains. Notably, we observed a severe growth defect in the H796A mutant in the EPSPS domain despite this mutant retaining ∼20% of corresponding activity in vitro, suggesting that this level of EPSPS activity is insufficient to support viability of C. albicans (Table 1). Strong growth defects and cell death were also observed in case of C. albicans strains carrying active site mutations in the SK and DHSD domains. The difference in severity of observed effects between the growth on solid and liquid media may be due to significant clumping of the cells due to cell lysis that would interfere with optical density reading used for the latter assay. These data suggest that C. albicans lacks enzymes that could provide compensatory activities to support growth in the absence of the DHQS, EPSPS, SK, and DHSD activities of Aro1, at least under the cultivation conditions tested in this study. Combined, this data clearly demonstrates that the activity of each of these four Aro1 domains is essential for the viability of C. albicans. Whereas the effect of essentiality of these four individual domains of Aro1 remains to be tested in infection models, our data suggest an important novel direction for antifungal therapy can be directed to targeting of one of these four enzymatic activities of Aro1.
Unlike the other Aro1 domains, the DHQase domain in C. albicans Aro1 lacked the catalytic residues typical of homologous type I DHQase enzymes. We demonstrated that this domain was not active in vitro, both as part of Aro1 or when expressed as a single-domain fragment. We identified similar active site aberrations in several other Candida species, including C. auris, C. lusitaniae, C. tropicalis, and C. viswanathii. C. glabrata and C. parapsilosis Aro1 retained the typical DHQase active site composition and purified Aro1 enzymes from these organisms had robust DHQase activity in vitro. Because the deletion of the DHQase domain of ARO1 did not have any effect on C. albicans viability, this raised the question of which enzyme was responsible for supporting this reaction in the shikimate pathway in these species. Our analysis identified that this function in C. albicans is fulfilled by the type II DHQase encoded by DQD1. We demonstrated that this essential gene product shows robust activity for conversion of 3-dehydroquinate to 3-dehydroshikimate, and this activity is even higher than that of corresponding domains in C. glabrata and C. parapsilosis Aro1 enzymes under the same assay conditions. Interestingly, these two Candida species do not encode for DQD1 orthologs, whereas we identified DQD1 orthologs in all Candida species encoding DHQase-inactive variants of Aro1. Type II DHQases are typically involved in quinate catabolism in fungi; however, the type II DHQase from the Gram-negative bacterium Acidomonas methanolica participates in both quinate catabolism and shikimate biosynthesis (Euverink et al, 1992). We hypothesize that the presence of a more efficient type II DHQase in C. albicans may have alleviated selective pressure for maintaining DHQase activity within Aro1. Similarly, the lack or loss of a type II DHQase ortholog in C. glabrata and C. parapsilosis necessitated maintenance of the catalytic dyad and consequently, enzymatic activity, in the DHQase domain of Aro1 in these species.
Because type I and type II DHQases are structurally and mechanistically distinct, the observation that C. albicans, and other clinically important Candida species such as C. auris, rely on a type II DHQase rather than the type I DHQase domain present in Aro1 informs on potential therapeutic strategies targeting this step of the shikimate pathway. Further analysis is warranted into other possible deviations in the shikimate pathway of other species.
Materials and Methods
Recombinant protein purification
E. coli BL21(DE3) Gold was used for overexpression of C. albicans Aro1 coding for residues 1–387 residues corresponding to the DHQS domain (Aro1DHQS); residues 387–844, corresponding to the EPSPS domain (Aro1EPSPS); and the C-terminal residues 845–1,551, comprising the SK, DHQase, and DHSD domains (Aro1SK-DHQase-DHSD). 1 liter LB media containing 100 μg/ml ampicillin was inoculated with 3 ml of overnight culture and incubated at 37°C with shaking. Cultures were induced with IPTG at 17°C once the OD600 reached 0.6–0.8. Cell pellets were collected by centrifugation at 7,000g. Ni-NTA affinity chromatography was used for protein purification. Cells were resuspended in a binding buffer (100 mM Hepes, pH 7.5, 300 mM NaCl, 5 mM imidazole, and 5% glycerol [vol/vol]) and lysed with a sonicator. Then insoluble cell debris was removed by centrifugation at 30,000g. The soluble cell lysate fraction was loaded on a 4 ml Ni-NTA column (QIAGEN) pre-equilibrated with binding buffer, washed with 150 ml washing buffer (100 mM Hepes, pH 7.5, 300 mM NaCl, 30 mM imidazole, and 5% glycerol [vol/vol]), and N-terminal His6-tagged protein was eluted with elution buffer (100 mM Hepes, pH 7.5, 300 mM NaCl, 250 mM imidazole, and 5% glycerol [vol/vol]). The His6-tagged proteins were then subjected to overnight TEV cleavage using 50 μg of TEV per mg of His6-tagged protein and dialyzed overnight against the binding buffer. The His-tag and TEV were removed by rerunning the protein over the Ni-NTA column. The tag-free protein was then dialyzed against crystallisation buffer (50 mM Hepes, pH 7.5, 300 mM NaCl) and the purity of the protein was analysed by SDS-polyacrylamide gel electrophoresis.
Crystallization and X-ray structure determination
All crystals were grown at room temperature using the vapor diffusion sitting drop method. For the Aro1DHQS-NADH crystal, 25 mg/ml protein was mixed with 2 mM NADH and 0.5 mM ZnCl2, and then with reservoir solution 0.2 M sodium thiocyanate and 20% (wt/vol) PEG3350; for the Aro1EPSPS-shikimate-3-phosphate crystal, 25 mg/ml protein was mixed with 1 mM shikimate-3-phosphate, and then with reservoir solution 0.4 M sodium chloride, 0.1 M Tris, pH 8, and 26% (wt/vol) PEG3350; for the Aro1SK-DHQase-DHSD crystal, 25 mg/ml protein was mixed with 2 mM shikimate-3-phosphate, 1 mM ATP, 0.1 M sodium formate, and 12% (2/v) PEG3350. Cryoprotection solutions for the Aro1DHQS, Aro1EPSPS and the Aro1SK-DHQase-DHSD crystals were: 8% glycerol, 8% ethylene glycol, 8% sucrose; 22% ethylene glycol, Hepes, pH 7.8; and 20% glycerol plus 4% tacsimate.
Diffraction data at 100 K were collected at beamline 19-ID of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory. Diffraction data were processed using HKL3000 (Minor et al, 2006). Structures were solved by Molecular Replacement using Phenix.phaser and the following models: DHQS domain from AroM from A. nidulans (PDB 1SG6), (Nichols et al, 2004a); EPSPS from V. cholerae (PDB 3NVS); DHQase+DHSD domains from S. pombe AroM (PDB 5SWV). Both model building and refinement were performed using Phenix.refine (Afonine et al, 2012) and Coot (Emsley et al, 2010). Atomic coordinates have been deposited in the Protein Data Bank with accession codes 6C5C, 7TBU and 7TBV. Structural homologs were identified in the PDB using the Dali-lite server (Holm, 2020).
Dehydroquinate synthase (DHQS) assay
DHQS reactions were performed in 250-μl volumes containing 50 mM Tris–HCl, pH 7.5, 20 nM NAD+, 0.2–0.8 mM DAHP, and 10 nM of Aro1 or Aro1 mutant protein. Enzyme activity was allowed to proceed for 5 min at room temperature (22°C), after which the reaction was terminated via the addition of 100 μl of malachite green assay solution (Echelon Biosciences). After incubation for 30 min post termination, absorbance was read at 620 nm using a Victor 1,420 plate reader (Perkin-Elmer), and the levels of inorganic phosphate were determined by comparison to a phosphate standard curve generated according to the manufacturer’s protocol.
EPSP synthase assay
EPSPS activity was assayed at 22°C in a reaction buffer containing 50 mM Hepes, pH 7.5, 100 mM KCl, 2 mM DTT, 1 mM shikimate-3-phosphate, and 1 mM phospoenol pyruvate. Aro1 and Aro1 mutant protein was added to a final concentration of 10 nM, and the reaction was allowed to proceed for 5 min, then terminated with the addition of 200 μl of malachite green assay solution (Echelon Biosciences). The remainder of the procedure was performed as described above for the DHQS assay. Enzyme activity was expressed in terms of production of Pi over time, and the activity of Aro1 mutants was expressed relative to wild-type Aro1.
Shikimate kinase assay
SK activity was measured using the ADP-Glo Kinase Assay kit (Promega). The assay was performed in a 25-μl volume containing 100 mM Tris–HCl, pH 7.5, 10 μM ATP, 20 mM MgCl2, and 100 μM shikimic acid. Catalysis was initiated by the addition of Aro1 or Aro1 mutant protein to a final concentration of 10 nM and allowed to proceed for 30 min at 22°C, then terminated via the addition of 25 μl of ADP-Glo Reagent. After 40 min of incubation, 50 μl of the kinase detection reagent was added, samples were incubated for 30 min, and luminescence was quantified using a Victor 1,420 plate reader (Perkin-Elmer). The amount of ATP converted to ADP by Aro1 was determined by comparing luminescence readings to a standard curve generated according to the manufacturer’s instructions.
Shikimate dehydrogenase assay
The assay for shikimate dehydrogenase activity was performed as described by Kubota et al (2013) and measured the conversion of NADPH to NADP+ by reading absorbance at 340 nm (ε = 6.2 × 104 M−1 cm−1). All reactions were performed using a final volume of 1 ml containing 100 mM Tris–HCl, pH 7.5, 200 μM NADPH, and 500 μM 3-dehydroshikimate (MilliporeSigma). The reaction was initiated by adding the Aro1 enzyme to a final concentration of 5 nM, then monitored for 2 min at 37°C.
DHQase assay
DHQase activity was tested as previously described by Chaudhuri et al (1986). Briefly, formation of 3-dehydroshikimate was measured by reading absorbance at 234 nm (ε = 1.2 × 104 M−1 cm−1). The assay was performed in 1-ml quartz cuvettes at 37°C in a buffer containing 100 mM Tris–HCl, pH 7.5, and 0.5 mM 3-dehydroquinate. The assay was initiated by the addition of protein and monitored for 2 min. Protein concentration ranges tested were C. albicans Aro1 (1–50 nM), E. coli AroD (1–25 nM), C. albicans Dqd1 (0.25–10 nM), C. glabrata, and C. parapsilosis Aro1 (1–10 nM).
Purification of Aro1 from C. albicans
C. albicans CaLC6830 was grown overnight at 30°C, 200 rpm to late log phase, then harvested via centrifugation (15 min, 5,000g, 4°C). The cell pellet was flash frozen in liquid N2, and then ground under liquid N2 using a mortar and pestle until the consistency reached a fine powder. The powder was resuspended in Binding Buffer (300 mM NaCl, 50 mM Hepes, pH 7.5, 5 mM imidazole, and 5% glycerol). His6-tagged Aro1 was then purified using Ni-NTA chromatography. After purification, the sample was incubated overnight at 4°C with TEV protease to remove the 6His tag and passaged over a second Ni-NTA column. The flowthrough was concentrated and fractionated using a Superdex 200 column (150 mM NaCl and 10 mM Hepes, pH 7.5). The eluate was concentrated and flash frozen in liquid N2 for cryo-EM analysis, or diluted with glycerol (final concentration 50%) and stored at −20°C for use in enzymatic assays.
Cryo-EM grid preparation, data collection, and image processing
Quantifoil R1.2/1.3 300 mesh Au grids were glow-discharged with air at 30 mA for 80 s in a PELCO easiGlow Glow Discharge Cleaning System. Freshly glow-discharged grids were put into an FEI Vitrobot Mark IV at 4°C and 100% humidity. Aro1 protein was thawed and 3 μl of sample at 2.2 or 0.55 mg/ml was applied to the grids. Grids were blotted for 5 s before vitrification by plunging into liquid ethane cooled by liquid N2. Vitrified samples were stored in liquid N2 until imaging. Movies were acquired on a 300 keV Titan Krios G2 microscope (Thermo Fisher Scientific) using a K3 Summit direct electron detector (Gatan) in super-resolution mode with a nominal magnification of 1,050,00× and pixel size of 0.4165 Å/pixel. SerialEM was used for automated data collection in beam-image shift mode with nine images collected per stage movement and a defocus range from −1.5 to −3 μm. The slit width of the GIF Quantum Energy Filter was set to 25 eV. Movies were dose-fractionated into 100 or 120 frames with a total dose of 83.5 or 72.4 electrons/Å2, respectively.
All movies were imported into CryoSPARC for motion correction using patch motion correction and binned to a pixel size of 0.833 Å/pixel followed by patch CTF estimation. Post-processing of all maps was performed using DeepEMhancer. Processing was performed on an initial dataset consisting of 932 manually curated micrographs. Initial particle picking was performed using Topaz autopicking with the pretrained model, resulting in 334,664 particles. Particles were extracted with a box size of 600 pixels and Fourier cropped to a pixel size of 2.50 Å. Multiple rounds of 2D classification were performed to identify 158,808 high quality particles. Ab-initio model building was performed in C1 symmetry using four classes to sort out broken and full-length particles, leading to one high quality class representing the dimer of the Aro1DHQS and Aro1EPSPS domain. A second dataset consisting of 1,623 high quality movies was collected and template-based autopicking was performed using templates generated from the Aro1DHQS and Aro1EPSPS dimer map. Particles were extracted in 600 pixel boxes and Fourier cropped to 400 pixels with a pixel size of 1.2495 Å/pixel. Multiple rounds of 2D classification were performed and the high quality particles were combined with the particles from the previous dataset, resulting in 740,854 total particles. The particles were exported to star format using Csparc2star.py for further classification in Relion 3.1. This group of particles was subjected to 3D classification with eight classes using the Aro1DHQ and Aro1EPSPS dimer map as the initial model. After classification, one class consisting of 50,390 particles with features consistent with the full-length Aro1 dimer were identified. Another round of 3D classification with 10 classes was performed on the full 740,854 particle dataset using the Aro1 full-length dimer as the initial model. After classification, one class of 63,494 particles was imported into cryoSPARC for homogeneous refinement, resulting in a map of the full-length Aro1 protein at 4.52 Å resolution.
The particle set resulting in the 4.52 Å map was relatively inhomogeneous after 2D classification, so the dataset was reclassified using heterogeneous refinement in cryoSPARC. Heterogeneous refinement was run on the 740,854 particle dataset using the Aro1DHQS and Aro1EPSPS dimer map, the full-length Aro1 map, and two noise maps generated from ab-initio reconstruction until the noise classes contained less than 5% of the total particles. A total of 87,484 particles corresponded to the full-length dimer of Aro1 and after homogeneous refinement the final map resolution was 4.16 Å. Tight masks were generated around the Aro1SK-DHQase-DHSD dimer and Aro1DHQS and Aro1EPSPS dimer for local refinement in cryoSPARC. Local refinement of the Aro1SK-DHQase-DHSD dimer and Aro1DHQS and Aro1EPSPS dimer resulted in 3.16 and 3.43 Å maps, respectively. The particles from the final homogeneous refinement were exported to Relion 3.1 and subjected to focused classification without image alignment with masks around Aro1EPSPS domain 1 from subunit 1 and subunit 2. After classification particles were exported to cryoSPARC and local refinement was performed with a mask around Aro1DHQS-EPSPS of subunit 1 and subunit 2, leading to maps at final resolutions of 4.23 and 4.63 Å, respectively.
Cryo-EM model building full-length Aro1 dimer
The crystal structures for the Aro1DHQS (PDB ID: 6C5C) and Aro1EPSPS domains (PDB ID: 7TBU) were rigid body fit into the maps Aro1DHQS-EPSPS from subunit 1 and subunit 2. The monomer models were then rigid body fit into each protomer of the symmetric map of the Aro1DHQ and Aro1EPSPS dimer from the broken particles using Chimera. The dimer map was then fit into the focused map of the Aro1DHQS and Aro1EPSPS domains from the full-length particles using Chimera. The crystal structure of Aro1SK-DHQ-DHSD was a rigid body fit into the focused map of the Aro1SK-DHQ-DHSD dimer from the full-length particles using Chimera. Model building was performed on each focused map using Coot with real space refinement performed with Phenix.
Alignment of ARO1 orthologs
Aro1 sequences from C. auris (acc. No. XP_018172187.1), C. glabrata (acc. No. XP_449840), C. lusitaniae (acc. No. OVF04495.1), C. parapsilosis (acc. No. CCE44200), C. tropicalis (acc. No. XP_002545280.1), C. viswanathii (acc. No. RCK59526.1) were obtained from GenBank and aligned using Muscle (Edgar, 2004).
Cloning of recombinant AROD, DQD1, C. glabrata ARO1, and C. parapsilosis ARO1
AROD (acc. no. NP_416208) was PCR-amplified from E. coli gDNA using Phusion polymerase (NEB). C. albicans DQD1 (acc. no. XP_714872), C. glabrata ARO1 (acc. no. XP_449840), and C. parapsilosis ARO1 (acc. no. CCE44200) were codon-optimized for E. coli expression and synthesized (Codex DNA). All genes were cloned into the pMCSG53 expression vector via Gibson assembly (Codex DNA).
Expression and purification of AroD, Dqd1, C. glabrata Aro1, and C. parapsilosis Aro1
Plasmids were transformed into the E. coli strain BL21(DE3)-Gold. Cells were grown at 37°C and 200 rpm to an OD600 of 0.8, cooled to 20°C then induced with 1 mM IPTG and incubated overnight. Cells were collected via centrifugation at 5,000g, resuspended in binding buffer (300 mM NaCl, 50 mM Hepes, pH 7.5, 5 mM imidazole, 5% glycerol) then sonicated. Proteins were purified using Ni-NTA chromatography, and treated with TEV protease after elution for removal of the His6 tag.
C. albicans strain construction
C. albicans strains used in this work are summarized in Table S3. Liquid cultures were grown in YPD (1% [wt/vol] yeast extract, 2% [wt/vol] bactopeptone, 2% [wt/vol] glucose), or synthetic defined (SD) medium (1.74 g/l yeast nitrogen base without amino acids without ammonium sulfate [BioShop], 2% [wt/vol] glucose, and 0.1% [wt/vol] sodium L-glutamate [Millipore Sigma]), or synthetic complete (SC) medium (SD medium supplemented with 1.57 g/l SC-Arg-His-Leu-Ura powder [Sunrise Science Products], 85.6 g/l L-arginine, 85.6 g/l L-histidine, 173.4 g/l L-leucine, and 85.6 g/l uridine [Millipore Sigma]). For solid medium, Agar was added to 2% (wt/vol). For selections with SAT1, nourseothricin (NAT; Jena Bioscience GmbH) was supplemented to 150 μg/ml. DNA constructs were PCR-amplified using phusion polymerase (NEB) using custom oligonucleotide primers (Thermo Fisher Scientific) described in Table S4. PCR products were cleaned using QIAquick PCR Purification Kit (QIAGEN) or purified using QIAquick Gel Extraction Kit (QIAGEN). Genomic DNA was purified using the PureLink Genomic DNA Mini Kit (Invitrogen). Overnight YPD cultures of C. albicans cultures were diluted 1:1 in 50% (wt/vol) glycerol and archived at −80°C. Recombinant plasmids used in cloning are described in Table S5 and were maintained in E. coli Top10.
Table S3 Strains used in this work. (14.7KB, docx)
Table S4 Plasmids used in this study. (13.5KB, docx)
Table S5 Oligonucleotide primers used in this study. (19.5KB, docx)
CaLC5759 and CaLC5761
An integrating repair cassette encoding frt-PSAP2-FLP-SAT-frt-tetO with 72 bp 5ʹ homology to 250 bp upstream of the ARO1 ORF, and 3ʹ homology to the first 72 bp of the ARO1 ORF was PCR-amplified from pLC605 using oligonucleotide primers oLC7059/oLC7060. A cassette for transient expression of an sgRNA targeting PARO1 was constructed by fusion of two PCR products amplified from plasmid pLC963 (Veri et al, 2018). Product 1 was amplified using oLC5979/oLC7048, and Product 2 using oLC7047/oLC5981. These PCR products were gel-purified and then fused by PCR using primers oLC5979/oLC5981. Integration of the frt-PSAP2-FLP-SAT-frt-tetO construct eliminates 250 bp of PARO1 including the sgRNA target sequence. A cassette for transient expression of Cas9 was amplified from plasmid pLC963 using oLC6924/oLC6925. The three PCR constructs were transformed into C. albicans SN95 (Noble & Johnson, 2005) and transformants were selected by 2 d growth at 30°C on YPD NAT agar. Downstream integration of the tetO-ARO1 construct was confirmed by colony PCR using oLC274/oLC7074. Presence of the wild-type junction between PARO1 and ARO1 was tested by colony PCR with oLC7071/oLC7074. CaLC5759 was heterozygous for integration of the tetO-ARO1 construct and CaLC5761 was homozygous.
CaLC6830
An integrating repair cassette encoding frt-PSAP2-FLP-SAT-frt-tetO-His6TEV with 72 bp 5ʹ homology to 250 bp upstream of the ARO1 ORF, and 3ʹ homology to the first 72 bp of the ARO1 ORF was PCR-amplified from pLC605 using oligonucleotide primers oLC7059/oLC8510 (Veri et al, 2018). Cas9 and sgRNA transient expression cassettes were prepared as described for CaLC5759. The three PCR products were transformed into CaLC239, and transformants were selected by 2 d growth at 30°C on YPD NAT agar. Absence of the wild-type junction between PARO1 and ARO1 was confirmed by colony PCR with oLC7071/oLC7074. Downstream integration of the tetO-ARO1 construct was confirmed by colony PCR using oLC274/oLC7074. Protein expression was confirmed by Western immunoblotting of whole-cell lysates.
CaLC7800, CaLC7801, and CaLC7803
An integrating repair cassette encoding frt-PSAP2-FLP-SAT-frt-tetO with 72 bp 5ʹ homology to 250 bp upstream of the DQD1 ORF and 3ʹ homology to the first 72 bp of the DQD1 ORF was amplified from pLC605 using oLC9711/oLC9712. The PCR product was transformed into C. albicans SN95 and transformants were selected by 2 d growth at 30°C on YPD NAT agar. Downstream integration of the tetO-DQD1 construct was confirmed by colony PCR using oLC274/oLC9714. Presence of the wild-type junction between PDQD1 and DQD1 was identified by colony PCR using oLC9713/oLC9714. This heterozygous strain was designated CaLC7800. A single colony of CaLC7800 was used to inoculate 5 ml of SD medium lacking glucose and supplemented with 0.4% (wt/vol) bovine serum albumin and 2% maltose, to induce expression of FLP recombinase and drive excision of SAT1. After 48 h incubation at 30°C, cells were diluted in water and ∼100 cells plated on YPD Agar. After 2 d incubation at 30°C, NAT sensitive colonies were identified by replica plating onto YPD NAT agar. SAT1 excision was confirmed by colony PCR using oLC275/oLC9714. The resultant strain was designated CaLC7801. CaLC was then transformed with the same frt-PSAP2-FLP-SAT-frt-tetO cassette to introduce the tetO promoter at the second DQD1 allele. Absence of the wild-type junction between PDQD1 and DQD1 was confirmed by colony PCR using oLC9713/oLC9714.
CaLC6598
An integrating repair cassette encoding PACT1-His6TEVARO1-ARG4 with 70 bp 5ʹ homology to 250 bp upstream of the ARO1 ORF and 3ʹ homology to the first 70 bp after the ARO1 ORF was constructed from three PCR products. First, PACT1 with 5ʹ homology to PARO1 was PCR-amplified from pLC620 using oLC7384/oLC7383 (Shapiro et al, 2012). Second, His6TEVAro1 was amplified from pLC1335 using oLC7385/oLC7482. Third, a PCR product containing the ARG4 selectable marker with 3ʹ homology to the ARO1 locus was amplified from pLC576 using oLC7387/oLC738 (Lavoie et al, 2008). These three PCR products were gel-purified, then 0.35 pmol of each was combined and fused using NEBuilder HiFi DNA Assembly Kit according to the manufacturer’s instructions (New England Biolabs). The fusion product was amplified using primers oLC7384/oLC7388, then gel-purified. Transient expression cassettes for Cas9 and the PARO1-targeting sgRNA were prepared as described for CaLC5759. The three PCR products were transformed into CaLC5759, and transformants were selected by 2 d growth at 30°C on SC agar lacking arginine and supplemented with 150 μg/ml NAT. Absence of the wild-type junction between PARO1 and ARO1 was confirmed by colony PCR with oLC7071/oLC7074, upstream integration of the PACT1-His6TEVARO1-ARG4 cassette with oLC7073/oLC1118, downstream integration of the PACT1-His6TEVARO1-ARG4 cassette with oLC4879/oLC7072, presence of the junction between ARO1 and ARG4 with oLC7075/oLC5893, and presence of the tetO-ARO1 construct with oLC274/oLC7074. The His6TEVARO1 allele was PCR-amplified from genomic DNA (PureLink Genomic DNA Mini Kit; Invitrogen) using oLC5385/oLC5893, and verified by Sanger sequenced using oLC7601, oLC7602, oLC7603, oLC7604, oLC7605, oLC7606, oLC7607, oLC7608, oLC7074, and oLC7076.
CaLC6599
CaLC6599 was constructed as per CaLC6598, except His6TEVAro1388-1551 was amplified from pLC1349 using oLC7385/oLC7482.
C. albicans strains encoding amino acid substitutions to, truncations to, or domain deletions within His6TEVARO1
Isogenic strains to CaLC6598 were constructed using the following methodology. Construction of CaLC7016 is provided as a representative example. An integrating repair cassette was constructed encoding PACT1-His6TEVARO1H264K-ARG4 with 70 bp of 5ʹ homology 250 bp upstream of the ARO1 ORF and 70 bp 3ʹ homology immediately after the ARO1 ORF. This construct was generated by amplification of two PCR products from purified CaLC6598 genomic DNA using oligonucleotide primer pairs oLC7384/oLC7109 (H264K-SDM-Rv) and oLC7108(H264K-SDM-Fw)/oLC7388. These PCR products were gel-purified and fused by PCR using primers oLC8966/oLC8967. For other mutant derivatives oLC7109 and oLC7108 were replaced with primers corresponding to the desired amino acid substitution or domain deletion described in Table S3. Transient expression cassettes for Cas9 and the PARO1-targeting sgRNA were prepared as described for CaLC5759. The three PCR products were transformed into CaLC5759 and transformants were selected by 2 d growth at 30°C on SC agar lacking arginine and supplemented with 150 μg/ml NAT. Absence of the wild-type junction between PARO1 and ARO1 was confirmed by colony PCR with oLC7071/oLC7074, upstream integration of the PACT1-His6TEVARO1H264K-ARG4 cassette with oLC7073/oLC1118, downstream integration of the PACT1-His6TEVARO1H264K-ARG4 cassette with oLC4879/oLC7072, and presence of the tetO-ARO1 construct with oLC274/oLC7074. Genomic DNA was purified, the His6TEVARO1H264K allele was PCR-amplified using oLC5385/oLC5893 and verified by Sanger sequencing using oLC7601, oLC7602, oLC7603, oLC7604, oLC7605, oLC7606, oLC7607, oLC7608, oLC7074, and oLC7076.
pLC1335
His6TEV-tagged ARO1 was PCR-amplified from C. albicans SC5314 (Jones et al, 2004) genomic DNA using oLC7479/oLC7332. Both this PCR product and pBAD24 (Guzman et al, 1995) were digested with Sal1 and Sma1 (NEB), PCR purified, and then ligated using T4 DNA ligase (NEB). Plasmids RE mapped using EcoR1 and EcoRV (NEB) and then verified by Sanger sequencing using oLC7075, oLC7076, oLC7077, and oLC7385. QuikChange (Stratagene) site-directed mutagenesis with oLC8080/oLC8081 was used to correct a frameshift mutation in ARO1 and silently removed a ClaI restriction site. This plasmid was verified by Sanger sequencing using oLC7601, oLC7602, oLC7603, oLC7604, oLC7605, oLC7606, oLC7607, oLC7608, oLC7074, and oLC7076.
pLC1349
His6TEV-tagged C. albicans ARO1DHQSΔ (amino acid residues 388–1,551) was PCR-amplified from pLC1335 using oLC7397/oLC7332. Both this PCR product and pBAD24 were digested with Sal1 and Sma1 (NEB), PCR purified, ligated using T4 DNA ligase (NEB), and transformed into E. coli Top10. Plasmid was verified by Sanger sequencing using oLC7602.
qRT-PCR
10 ml liquid cultures were grown in SC medium overnight at 30°C, subcultured into 10 ml SC medium or SC medium supplemented with 0.05 μg/ml DOX overnight, then subcultured into 10 ml SC medium or SC medium supplemented with 5 μg/ml DOX for 4 h. Cells were collected by centrifugation at 3,000g for 10 min at 4°C, washed once with 1 ml of ice-cold water, then flash frozen in liquid N2. RNA was extracted using RNeasy Mini Kit (QIAGEN). For cell lysis, the frozen pellet was resuspended in 650 μl ice-cold Buffer RLT supplemented with 10 μl/ml 2-mercaptoethanol, then disrupted with 0.5 mm acid-washed glass beads (Sigma-Aldrich) for 4 × 1 min using a Mini-BeadBeater-24 (BioSpec Products Inc.). Contaminating DNA was removed from 4 μg of the purified RNA using the DNA-free DNA Removal Kit (Invitrogen), according to the manufacturer’s instructions. 1 μg of the DNAse-treated RNA was used to synthesize cDNA using the iScript cDNA Synthesis Kit (Bio-Rad). Transcript abundance was quantified from cDNA by real-time PCR using a CFX384 Touch Real-Time PCR Detection System (Bio-Rad), Fast SYBR Green Master Mix (Applied Biosystems), and oligonucleotide primers described in Table S4. Data were analysed using CFX Manager (Bio-Rad; v3.1). Amplicons were ∼150 bp, had single peaks in melt curves, and abundances were within the linear range of six point 10-fold dilution standard curves.
Growth assays
10 ml liquid cultures were grown in SC medium lacking arginine and supplemented with 150 μg/ml NAT with 200 rpm shaking overnight at 30°C then subcultured at 1:1,000 into 10 ml SC medium or SC medium supplemented with 0.05 μg/ml DOX overnight. Cultures were diluted to an OD600 of 1, then diluted at 1:80 into 96-well plates containing 80 μl/well SC medium or SC medium supplemented with 5 μg/ml DOX. Plates were sealed with an adhesive plate seal (AB-0580; Thermo Fisher Scientific), and placed in a custom-built growth curve robot (S&P Robotics Inc.). Within, cultures were incubated at 30°C and OD600 was measured at 15 min intervals for 24 h.
Microscopy
After growth curves described above, propidium iodide (Sigma-Aldrich) was added to cultures to 1 μg/ml (final). 5 μl of cell suspension was placed on a glass slide under a cover slip. Fluorescence micrographs were acquired on a Zeiss Axio Imager.M1 at 400 × magnification (40 × oil objective). Propidium iodide fluorescence was observed using an X-Cite series 120Q light source (Excelitas Technologies Corp.) and ET HQ DSRed (TRITC/Cy3) filter (Chroma Technology Corp.).
Protein extraction and Western blotting
At mid-log phase, 10 ml cultures of C. albicans were collected by centrifugation at 3,000g for 10 min at 4°C. Cell pellets were washed with 1 ml ice-cold water then flash frozen in liquid N2. Whole-cell lysates were prepared by mild alkaline lysis, trichloroacetic acid precipitation, and solubilized in SDS/urea loading buffer as previously described, other than omission of bromophenol blue dye. Protein concentration was roughly normalized by absorbance at 280 nm. Lysates were separated in NuPAGE 3–8% acrylamide Tris-acetate gels using NuPAGE Tris-acetate SDS running buffer (Thermo Fisher Scientific). Proteins were transferred to PVDF membranes (Bio-Rad Immun-Blot, 0.2 μm). Membranes were blocked with 3% (wt/vol) bovine serum albumin in Tris-buffered saline with 0.5% (vol/vol) Tween-20 (TBS-T). Membranes were probed with mouse–anti-His5 monoclonal primary antibody (QIAGEN; 1:3,000) and horseradish peroxidase-conjugated goat-anti mouse secondary antibody (Bio-Rad; 1:5,000). Chemiluminescence images were acquired on a Chemidoc Touch imaging system (Bio-Rad) using Clarity western ECL substrate (Bio-Rad).
Supplementary Material
Acknowledgements
SD Liston was supported by a Postdoctoral Fellowship from the Natural Sciences and Engineering Research Council of Canada (516840-2018). Crystal and cryo-EM structures solved in this work were funded in whole or in part with US federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract No. HHSN272201700060C (Center for Structural Genomics of Infectious Diseases (CSGID); http://csgid.org). LE Cowen is funded by a Canadian Institutes of Health Research (CIHR) Foundation grant (FDN-154288) and is a Canada Research Chair (Tier 1) in Microbial Genomics & Infectious Disease and co-director of the CIFAR Fungal Kingdom: Threats & Opportunities program. We thank Merck and Genome Canada for making the GRACE C. albicans mutant collection available and Andre Nantel and the National Research Council of Canada for their distribution. We thank all members of Savchenko and Cowen laboratories for their help and assistance.
Author Contributions
PJ Stogios: conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, and writing—original draft, review, and editing.
SD Liston: data curation, validation, methodology, and writing—original draft.
C Semper: conceptualization, data curation, formal analysis, validation, methodology, and writing—original draft.
B Quade: data curation, validation, methodology, and writing—original draft.
K Michalska: data curation and validation.
E Evdokimova: data curation.
S Ram: data curation.
Z Otwinowski: validation and methodology.
D Borek: data curation, methodology, and writing—original draft.
LE Cowen: conceptualization, supervision, funding acquisition, methodology, and writing—original draft.
A Savchenko: conceptualization, data curation, formal analysis, supervision, funding acquisition, validation, investigation, methodology, project administration, and writing—original draft, review, and editing.
Conflict of Interest Statement
LE Cowen is a co-founder and shareholder in Bright Angel Therapeutics, a platform company for development of novel antifungal therapeutics. LE Cowen is a consultant for Boragen, a small-molecule development company focused on leveraging the unique chemical properties of boron chemistry for crop protection and animal health. SD Liston was supported by a Mitacs postdoctoral fellowship funded in partnership with Amplyx Pharmaceuticals Inc., now a subsidiary of Pfizer. D Borek and Z Otwinowski are co-founders of Ligo Analytics, a company that develops a software for cryogenic electron microscopy. Z Otwinowski is a co-founder of HKL Research, a company that develops software for X-ray diffraction data analysis.
References
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68: 352–367. 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagautdinov B, Kunishima N (2007) Crystal structures of shikimate dehydrogenase AroE from thermus thermophilus HB8 and its cofactor and substrate complexes: Insights into the enzymatic mechanism. J Mol Biol 373: 424–438. 10.1016/j.jmb.2007.08.017 [DOI] [PubMed] [Google Scholar]
- Becker JM, Kauffman SJ, Hauser M, Huang L, Lin M, Sillaots S, Jiang B, Xu D, Roemer T (2010) Pathway analysis of Candida albicans survival and virulence determinants in a murine infection model. Proc Natl Acad Sci U S A 107: 22044–22049. 10.1073/pnas.1009845107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco B, Prado V, Lence E, Otero JM, Garcia-Doval C, van Raaij MJ, Llamas-Saiz AL, Lamb H, Hawkins AR, González-Bello C (2013) Mycobacterium tuberculosis shikimate kinase inhibitors: Design and simulation studies of the catalytic turnover. J Am Chem Soc 135: 12366–12376. 10.1021/ja405853p [DOI] [PubMed] [Google Scholar]
- Bongomin F, Gago S, Oladele RO, Denning DW (2017) Global and multi-national prevalence of fungal diseases-estimate precision. J Fungi (Basel) 3: 57. 10.3390/jof3040057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SA, Richards TA, Mui EJ, Samuel BU, Coggins JR, McLeod R, Roberts CW (2004) A complete shikimate pathway in toxoplasma gondii: An ancient eukaryotic innovation. Int J Parasitol 34: 5–13. 10.1016/j.ijpara.2003.10.006 [DOI] [PubMed] [Google Scholar]
- Carpenter EP, Hawkins AR, Frost JW, Brown KA (1998) Structure of dehydroquinate synthase reveals an active site capable of multistep catalysis. Nature 394: 299–302. 10.1038/28431 [DOI] [PubMed] [Google Scholar]
- Chaudhuri S, Lambert JM, McColl LA, Coggins JR (1986) Purification and characterization of 3-dehydroquinase from Escherichia coli. Biochem J 239: 699–704. 10.1042/bj2390699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng WC, Chen YF, Wang HJ, Hsu KC, Lin SC, Chen TJ, Yang JM, Wang WC (2012) Structures of Helicobacter pylori shikimate kinase reveal a selective inhibitor-induced-fit mechanism. PLoS One 7: e33481. 10.1371/journal.pone.0033481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins JR, Abell C, Evans LB, Frederickson M, Robinson DA, Roszak AW, Lapthorn AP (2003) Experiences with the shikimate-pathway enzymes as targets for rational drug design. Biochem Soc Trans 31: 548–552. 10.1042/bst0310548 [DOI] [PubMed] [Google Scholar]
- Coracini JD, de Azevedo WF Jr. (2014) Shikimate kinase, a protein target for drug design. Curr Med Chem 21: 592–604. 10.2174/09298673113206660299 [DOI] [PubMed] [Google Scholar]
- Duke SO (2018) The history and current status of glyphosate. Pest Manag Sci 74: 1027–1034. 10.1002/ps.4652 [DOI] [PubMed] [Google Scholar]
- Edgar RC (2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euverink GJ, Hessels GI, Vrijbloed JW, Coggins JR, Dijkhuizen L (1992) Purification and characterization of a dual function 3-dehydroquinate dehydratase from Amycolatopsis methanolica. J Gen Microbiol 138: 2449–2457. 10.1099/00221287-138-11-2449 [DOI] [PubMed] [Google Scholar]
- Fu C, Zhang X, Veri AO, Iyer KR, Lash E, Xue A, Yan H, Revie NM, Wong C, Lin ZY, et al. (2021) Leveraging machine learning essentiality predictions and chemogenomic interactions to identify antifungal targets. Nat Commun 12: 6497. 10.1038/s41467-021-26850-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan J, Wu Y, Prabakaran P, Gu Y, Li Y, Andrykovitch M, Liu H, Gong Y, Yan H, Ji X (2007) Structural and biochemical analyses of shikimate dehydrogenase AroE from Aquifex aeolicus: Implications for the catalytic mechanism. Biochemistry 46: 9513–9522. 10.1021/bi602601e [DOI] [PubMed] [Google Scholar]
- Gordon S, Simithy J, Goodwin DC, Calderón AI (2015) Selective Mycobacterium tuberculosis shikimate kinase inhibitors as potential antibacterials. Perspect Medicin Chem 7: 9–20. 10.4137/PMC.S13212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosset G (2009) Production of aromatic compounds in bacteria. Curr Opin Biotechnol 20: 651–658. 10.1016/j.copbio.2009.09.012 [DOI] [PubMed] [Google Scholar]
- Graham LD, Gillies FM, Coggins JR (1993) Over-expression of the yeast multifunctional arom protein. Biochim Biophys Acta 1216: 417–424. 10.1016/0167-4781(93)90009-3 [DOI] [PubMed] [Google Scholar]
- Gu Y, Reshetnikova L, Li Y, Wu Y, Yan H, Singh S, Ji X (2002) Crystal structure of shikimate kinase from Mycobacterium tuberculosis reveals the dynamic role of the LID domain in catalysis. J Mol Biol 319: 779–789. 10.1016/S0022-2836(02)00339-X [DOI] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177: 4121–4130. 10.1128/jb.177.14.4121-4130.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann MD, Bourenkov GP, Oberschall A, Strizhov N, Bartunik HD (2006) Mechanism of phosphoryl transfer catalyzed by shikimate kinase from Mycobacterium tuberculosis. J Mol Biol 364: 411–423. 10.1016/j.jmb.2006.09.001 [DOI] [PubMed] [Google Scholar]
- Holm L (2020) Using Dali for protein structure comparison. Methods Mol Biol 2112: 29–42. 10.1007/978-1-0716-0270-6_3 [DOI] [PubMed] [Google Scholar]
- Höppner A, Schomburg D, Niefind K (2013) Enzyme-substrate complexes of the quinate/shikimate dehydrogenase from Corynebacterium glutamicum enable new insights in substrate and cofactor binding, specificity, and discrimination. Biol Chem 394: 1505–1516. 10.1515/hsz-2013-0170 [DOI] [PubMed] [Google Scholar]
- Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, Newport G, Thorstenson YR, Agabian N, Magee PT, et al. (2004) The diploid genome sequence of Candida albicans. Proc Natl Acad Sci U S A 101: 7329–7334. 10.1073/pnas.0401648101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- König V, Pfeil A, Braus GH, Schneider TR (2004) Substrate and metal complexes of 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase from Saccharomyces cerevisiae provide new insights into the catalytic mechanism. J Mol Biol 337: 675–690. 10.1016/j.jmb.2004.01.055 [DOI] [PubMed] [Google Scholar]
- Kubota T, Tanaka Y, Hiraga K, Inui M, Yukawa H (2013) Characterization of shikimate dehydrogenase homologues of Corynebacterium glutamicum. Appl Microbiol Biotechnol 97: 8139–8149. 10.1007/s00253-012-4659-y [DOI] [PubMed] [Google Scholar]
- Lavoie H, Sellam A, Askew C, Nantel A, Whiteway M (2008) A toolbox for epitope-tagging and genome-wide location analysis in Candida albicans. BMC Genomics 9: 578. 10.1186/1471-2164-9-578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Choi JM, Kim HJ (2017) Crystal structure of 5-enolpyruvylshikimate-3-phosphate synthase from a psychrophilic bacterium, Colwellia psychrerythraea 34H. Biochem Biophys Res Commun 492: 500–506. 10.1016/j.bbrc.2017.08.063 [DOI] [PubMed] [Google Scholar]
- Lee WH, Perles LA, Nagem RA, Shrive AK, Hawkins A, Sawyer L, Polikarpov I (2002) Comparison of different crystal forms of 3-dehydroquinase from Salmonella typhi and its implication for the enzyme activity. Acta Crystallogr D Biol Crystallogr 58: 798–804. 10.1107/s0907444902003918 [DOI] [PubMed] [Google Scholar]
- Lee Y, Puumala E, Robbins N, Cowen LE (2021) Antifungal drug resistance: Molecular mechanisms in Candida albicans and beyond. Chem Rev 121: 3390–3411. 10.1021/acs.chemrev.0c00199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech AP, Boetzel R, McDonald C, Shrive AK, Moore GR, Coggins JR, Sawyer L, Kleanthous C (1998) Re-evaluating the role of His-143 in the mechanism of type I dehydroquinase from Escherichia coli using two-dimensional 1H,13C NMR. J Biol Chem 273: 9602–9607. 10.1074/jbc.273.16.9602 [DOI] [PubMed] [Google Scholar]
- Leech AP, James R, Coggins JR, Kleanthous C (1995) Mutagenesis of active site residues in type I dehydroquinase from Escherichia coli. Stalled catalysis in a histidine to alanine mutant. J Biol Chem 270: 25827–25836. 10.1074/jbc.270.43.25827 [DOI] [PubMed] [Google Scholar]
- Light SH, Anderson WF, Lavie A (2013) Reassessing the type I dehydroquinate dehydratase catalytic triad: Kinetic and structural studies of Glu86 mutants. Protein Sci 22: 418–424. 10.1002/pro.2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light SH, Antanasijevic A, Krishna SN, Caffrey M, Anderson WF, Lavie A (2014) Crystal structures of type I dehydroquinate dehydratase in complex with quinate and shikimate suggest a novel mechanism of Schiff base formation. Biochemistry 53: 872–880. 10.1021/bi4015506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light SH, Minasov G, Shuvalova L, Duban ME, Caffrey M, Anderson WF, Lavie A (2011) Insights into the mechanism of type I dehydroquinate dehydratases from structures of reaction intermediates. J Biol Chem 286: 3531–3539. 10.1074/jbc.M110.192831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JS, Cheng WC, Wang HJ, Chen YC, Wang WC (2008) Structure-based inhibitor discovery of Helicobacter pylori dehydroquinate synthase. Biochem Biophys Res Commun 373: 1–7. 10.1016/j.bbrc.2008.05.070 [DOI] [PubMed] [Google Scholar]
- Liu Q, Liu Y, Chen Y, Nielsen J (2020) Current state of aromatics production using yeast: Achievements and challenges. Curr Opin Biotechnol 65: 65–74. 10.1016/j.copbio.2020.01.008 [DOI] [PubMed] [Google Scholar]
- Lockhart SR, Etienne KA, Vallabhaneni S, Farooqi J, Chowdhary A, Govender NP, Colombo AL, Calvo B, Cuomo CA, Desjardins CA, et al. (2017) Simultaneous emergence of multidrug-resistant Candida auris on 3 continents confirmed by whole-genome sequencing and epidemiological analyses. Clin Infect Dis 64: 134–140. 10.1093/cid/ciw691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low CY, Rotstein C (2011) Emerging fungal infections in immunocompromised patients. F1000 Med Rep 3: 14. 10.3410/M3-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M (2006) HKL-3000: The integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62: 859–866. 10.1107/S0907444906019949 [DOI] [PubMed] [Google Scholar]
- Mir R, Jallu S, Singh TP (2015) The shikimate pathway: Review of amino acid sequence, function and three-dimensional structures of the enzymes. Crit Rev Microbiol 41: 172–189. 10.3109/1040841X.2013.813901 [DOI] [PubMed] [Google Scholar]
- Neetu N, Katiki M, Dev A, Gaur S, Tomar S, Kumar P (2020) Structural and biochemical analyses reveal that chlorogenic acid inhibits the shikimate pathway. J Bacteriol 202: e00248-20. 10.1128/JB.00248-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CE, Hawkins AR, Stammers DK (2004. a) Structure of the [L8S2Q1M6]open[R8S2Q1M7] form of Aspergillus nidulans 3-dehydroquinate synthase at 1.7 A resolution from crystals grown following enzyme turnover. Acta Crystallogr D Biol Crystallogr 60: 971–973. 10.1107/S0907444904004743 [DOI] [PubMed] [Google Scholar]
- Nichols CE, Lockyer M, Hawkins AR, Stammers DK (2004. b) Crystal structures of Staphylococcus aureus type I dehydroquinase from enzyme turnover experiments. Proteins 56: 625–628. 10.1002/prot.20165 [DOI] [PubMed] [Google Scholar]
- Nichols CE, Ren J, Lamb HK, Hawkins AR, Stammers DK (2003) Ligand-induced conformational changes and a mechanism for domain closure in Aspergillus nidulans dehydroquinate synthase. J Mol Biol 327: 129–144. 10.1016/s0022-2836(03)00086-x [DOI] [PubMed] [Google Scholar]
- Nichols CE, Ren J, Leslie K, Dhaliwal B, Lockyer M, Charles I, Hawkins AR, Stammers DK (2004. c) Comparison of ligand-induced conformational changes and domain closure mechanisms, between prokaryotic and eukaryotic dehydroquinate synthases. J Mol Biol 343: 533–546. 10.1016/j.jmb.2004.08.039 [DOI] [PubMed] [Google Scholar]
- Noble SM, Johnson AD (2005) Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot Cell 4: 298–309. 10.1128/EC.4.2.298-309.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes JES, Duque MA, de Freitas TF, Galina L, Timmers LFSM, Bizarro CV, Machado P, Basso LA, Ducati RG (2020) Mycobacterium tuberculosis shikimate pathway enzymes as targets for the rational design of anti-tuberculosis drugs. Molecules 25: 1259. 10.3390/molecules25061259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Meara TR, Duah K, Guo CX, Maxson ME, Gaudet RG, Koselny K, Wellington M, Powers ME, MacAlpine J, O’Meara MJ, et al. (2018) High-throughput screening identifies genes required for Candida albicans induction of macrophage pyroptosis. mBio 9: e01581-18. 10.1128/mBio.01581-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Meara TR, Veri AO, Ketela T, Jiang B, Roemer T, Cowen LE (2015) Global analysis of fungal morphology exposes mechanisms of host cell escape. Nat Commun 6: 6741. 10.1038/ncomms7741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A, Lamb HK, Nichols C, Moore JD, Brown KA, Cooper A, Charles IG, Stammers DK, Hawkins AR (2004) Biophysical and kinetic analysis of wild-type and site-directed mutants of the isolated and native dehydroquinate synthase domain of the AROM protein. Protein Sci 13: 2108–2119. 10.1110/ps.04705404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek J, Christendat D (2015) The shikimate dehydrogenase family: Functional diversity within a conserved structural and mechanistic framework. Arch Biochem Biophys 566: 85–99. 10.1016/j.abb.2014.12.006 [DOI] [PubMed] [Google Scholar]
- Pereira SA, Livi GP (1993) Cloning and expression of the ARO3 gene encoding DAHP synthase from Candida albicans. Gene 132: 159–165. 10.1016/0378-1119(93)90191-5 [DOI] [PubMed] [Google Scholar]
- Pfaller MA, Diekema DJ (2010) Epidemiology of invasive mycoses in North America. Crit Rev Microbiol 36: 1–53. 10.3109/10408410903241444 [DOI] [PubMed] [Google Scholar]
- Roemer T, Jiang B, Davison J, Ketela T, Veillette K, Breton A, Tandia F, Linteau A, Sillaots S, Marta C, et al. (2003) Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol Microbiol 50: 167–181. 10.1046/j.1365-2958.2003.03697.x [DOI] [PubMed] [Google Scholar]
- Roszak AW, Robinson DA, Krell T, Hunter IS, Fredrickson M, Abell C, Coggins JR, Lapthorn AJ (2002) The structure and mechanism of the type II dehydroquinase from Streptomyces coelicolor. Structure 10: 493–503. 10.1016/s0969-2126(02)00747-5 [DOI] [PubMed] [Google Scholar]
- Sahu PK, Mohapatra PK, Rajani DP, Raval MK (2020) Structure-based discovery of narirutin as a shikimate kinase inhibitor with anti-tubercular potency. Curr Comput Aided Drug Des 16: 523–529. 10.2174/1573409915666191025112150 [DOI] [PubMed] [Google Scholar]
- Schönbrunn E, Eschenburg S, Shuttleworth WA, Schloss JV, Amrhein N, Evans JN, Kabsch W (2001) Interaction of the herbicide glyphosate with its target enzyme 5-enolpyruvylshikimate 3-phosphate synthase in atomic detail. Proc Natl Acad Sci U S A 98: 1376–1380. 10.1073/pnas.98.4.1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal ES, Gritsenko V, Levitan A, Yadav B, Dror N, Steenwyk JL, Silberberg Y, Mielich K, Rokas A, Gow NAR, et al. (2018) Gene essentiality analyzed by in vivo transposon mutagenesis and machine learning in a stable haploid isolate of Candida albicans. mBio 9: e02048-18. 10.1128/mBio.02048-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro RS, Sellam A, Tebbji F, Whiteway M, Nantel A, Cowen LE (2012) Pho85, Pcl1, and Hms1 signaling governs Candida albicans morphogenesis induced by high temperature or Hsp90 compromise. Curr Biol 22: 461–470. 10.1016/j.cub.2012.01.062 [DOI] [PubMed] [Google Scholar]
- Sousa S, McLaughlin MM, Pereira SA, VanHorn S, Knowlton R, Brown JR, Nicholas RO, Livi GP (2002) The ARO4 gene of Candida albicans encodes a tyrosine-sensitive DAHP synthase: Evolution, functional conservation and phenotype of Aro3p-, Aro4p-deficient mutants. Microbiology (Reading) 148: 1291–1303. 10.1099/00221287-148-5-1291 [DOI] [PubMed] [Google Scholar]
- Staub M, Dénes G (1969) Purification and properties of the 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase (phenylalanine sensitive) of Escherichia coli K12. II. Inhibition of activity of the enzyme with phenylalanine and functional group-specific reagents. Biochim Biophys Acta 178: 599–608. 10.1016/0005-2744(69)90228-9 [DOI] [PubMed] [Google Scholar]
- Sutton KA, Breen J, MacDonald U, Beanan JM, Olson R, Russo TA, Schultz LW, Umland TC (2015) Structure of shikimate kinase, an in vivo essential metabolic enzyme in the nosocomial pathogen Acinetobacter baumannii, in complex with shikimate. Acta Crystallogr D Biol Crystallogr 71: 1736–1744. 10.1107/S139900471501189X [DOI] [PubMed] [Google Scholar]
- Sutton KA, Breen J, Russo TA, Schultz LW, Umland TC (2016) Crystal structure of 5-enolpyruvylshikimate-3-phosphate (EPSP) synthase from the ESKAPE pathogen Acinetobacter baumannii. Acta Crystallogr F Struct Biol Commun 72: 179–187. 10.1107/S2053230X16001114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teshiba S, Furter R, Niederberger P, Braus G, Paravicini G, Hütter R (1986) Cloning of the ARO3 gene of Saccharomyces cerevisiae and its regulation. Mol Gen Genet 205: 353–357. 10.1007/BF00430450 [DOI] [PubMed] [Google Scholar]
- Trapani S, Schoehn G, Navaza J, Abergel C (2010) Macromolecular crystal data phased by negative-stained electron-microscopy reconstructions. Acta Crystallogr D Biol Crystallogr 66: 514–521. 10.1107/S0907444910002763 [DOI] [PubMed] [Google Scholar]
- Verasztó HA, Logotheti M, Albrecht R, Leitner A, Zhu H, Hartmann MD (2020) Architecture and functional dynamics of the pentafunctional AROM complex. Nat Chem Biol 16: 973–978. 10.1038/s41589-020-0587-9 [DOI] [PubMed] [Google Scholar]
- Veri AO, Miao Z, Shapiro RS, Tebbji F, O’Meara TR, Kim SH, Colazo J, Tan K, Vyas VK, Whiteway M, et al. (2018) Tuning Hsf1 levels drives distinct fungal morphogenetic programs with depletion impairing Hsp90 function and overexpression expanding the target space. PLoS Genet 14: e1007270. 10.1371/journal.pgen.1007270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh YC, Wang HY, Lan CY (2020) Candida albicans Aro1 affects cell wall integrity, biofilm formation and virulence. J Microbiol Immunol Infect 53: 115–124. 10.1016/j.jmii.2018.04.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 X-ray crystallographic statistics. (17.3KB, docx)
Table S2 Cryo-EM data collection, refinement, and validation statistics. (14.8KB, docx)
Table S3 Strains used in this work. (14.7KB, docx)
Table S4 Plasmids used in this study. (13.5KB, docx)
Table S5 Oligonucleotide primers used in this study. (19.5KB, docx)