

SUMMARY
Cells repair DNA double-strand breaks (DSBs) through a complex set of pathways critical for maintaining genomic integrity. To systematically map these pathways, we developed a high-throughput screening approach called Repair-seq that measures the effects of thousands of genetic perturbations on mutations introduced at targeted DNA lesions. Using Repair-seq, we profiled DSB repair products induced by two programmable nucleases (Cas9 and Cas12a) in the presence or absence of oligonucleotides for homology-directed repair (HDR) after knockdown of 476 genes involved in DSB repair or associated processes. The resulting data enabled principled, data-driven inference of DSB end joining and HDR pathways. Systematic interrogation of this data uncovered unexpected relationships among DSB repair genes and demonstrated that repair outcomes with superficially similar sequence architectures can have markedly different genetic dependencies. This work provides a foundation for mapping the complex pathways of DSB repair and for optimizing genome editing across diverse modalities.
Keywords: DNA repair, double-strand breaks, functional genomics, genome editing, CRISPR-Cas9
Graphical Abstract
IN-BRIEF
Measuring the effects of genetic perturbations on the spectrum of mutations produced at targeted DNA breaks in high-throughput allows systematic mapping of DNA repair pathways.
INTRODUCTION
Failure to properly repair DNA double-strand breaks (DSBs) can result in genome instability and cell death (Ciccia and Elledge, 2010). To deal with DSBs, eukaryotic cells have evolved a set of specialized repair mechanisms typically conceptualized as a decision tree, with “branches” representing the various molecular events leading to a repaired break and “nodes” indicating points of transition or commitment between processes (Scully et al., 2019). Within this framework, DSB repair has two main branches. The first, non-homologous end joining (NHEJ), comprises a set of flexible mechanisms that act throughout the cell cycle to facilitate the processing, synapsis, and direct ligation of DSB ends. The second, homology-directed repair (HDR), uses homologous regions of DNA to facilitate repair in S/G2 after 5'- to-3' nucleolytic resection of DSB ends. Beyond these high-level branches, however, descriptions of DSB repair as decision trees can oversimplify a complex mechanistic landscape (Paull, 2021) and mask gaps and ambiguities that remain in our understanding. For example, although well established to exist, alternative mechanisms of DSB end-joining have been difficult to fully characterize (Ramsden et al., 2021), leading to uncertainty in the number of possible “routes” from DSB to repaired DNA.
Limitations in our understanding of DSB repair are due in part to technical challenges associated with profiling the determinants of repair outcomes. While many genetic screens have been performed to identify DSB repair factors, such efforts have largely relied on phenotypes that conflate the activity of many processes (e.g., growth assays) (Hurov et al., 2010; Noordermeer et al., 2018; O’Connell et al., 2010; Olivieri et al., 2020; Smogorzewska et al., 2010; Zimmermann et al., 2018) or used low-resolution reporters (Adamson et al., 2012; Howard et al., 2015; Richardson et al., 2018), limiting their ability to unambiguously define the roles of identified genes. Alternatively, deep sequencing of mutations produced at enzymatically induced DSBs has implicated specific repair factors in defined DSB processing events by revealing shifts in repair product frequencies after genetic or chemical perturbations (Bothmer et al., 2017; van Overbeek et al., 2016; Wyatt et al., 2016), but to date such experiments have been limited to small numbers of perturbations.
We reasoned that simultaneously measuring the genetic determinants of a broad range of DSB repair outcomes would enable systematic studies of DSB repair. With this goal in mind, we developed Repair-seq, a high-throughput method that combines locus-specific deep sequencing with CRISPR-interference (CRISPRi)-based genetic screens to measure the effects of thousands of genetic perturbations on the spectrum of mutations produced at targeted DNA lesions. Using Repair-seq, we studied the repair of DSBs induced by two programmable nucleases (Cas9 and Cas12a). Systematic exploration of these maps isolated processes responsible for common nuclease-induced mutations and revealed unexpected roles for canonical DNA damage response genes. Additionally, we applied Repair-seq to study DSB repair with different exogenous repair templates (oligonucleotide donors). This effort identified a range of genetically distinct processes by which template sequences are incorporated at breaks and demonstrated that Repair-seq is a flexible approach for uncovering how diverse genome editing technologies interact with endogenous repair processes. Finally, in two companion manuscripts (Chen et al. 2021, Koblan et al., 2021), we used Repair-seq to identify the genetic determinants of base and prime editing outcomes, enabling the development of more efficient and precise editing tools. Together, these data demonstrate the broad utility of Repair-seq and provide a foundation for further exploration of DNA repair pathways.
RESULTS
A functional genomics platform for interrogating DSB repair pathways
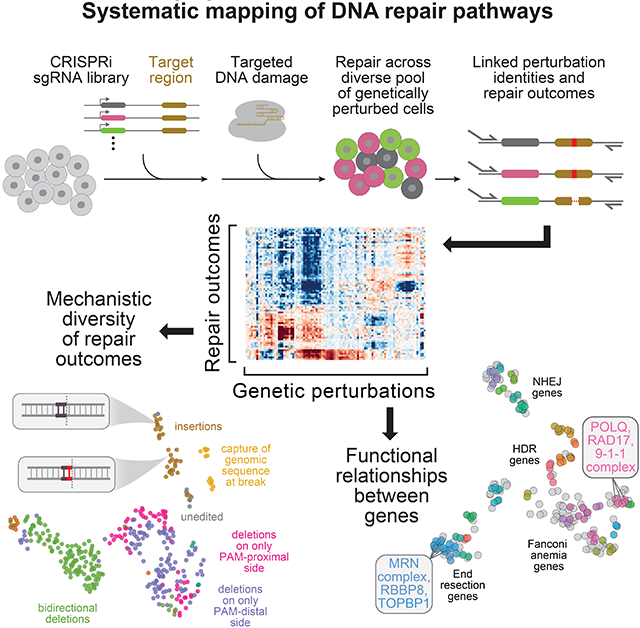
CRISPR-based screening approaches use libraries of perturbations directed by single-guide RNAs (sgRNAs) to enable massively parallel interrogation of gene function in scalable pooled experiments (Doench, 2018). To perform screens with targeted sequencing of DSB repair products, we engineered a lentiviral screening vector that links sgRNA expression to a nearby “target region” for DNA damage (Figures 1A and S1A). We reasoned that genomic integration of this vector would allow induction and repair of targeted damage across diverse pools of perturbed cells, followed by linked recovery of perturbation identities and repair outcomes from individual cells by paired-end sequencing (Figure 1B).
Figure 1. Repair-seq is a high-resolution screening platform for systematically interrogating DNA repair processes.

(A) Schematic of Repair-seq screening vector with linked CRISPRi sgRNA expression cassette and target region for induced DNA damage.
(B) Experimental workflow of a Repair-seq screen. Cells expressing a CRISPRi effector protein (dCas9-KRAB) are infected with CRISPRi sgRNAs linked to a targetable editing region. After allowing time for targeted gene repression, a programmable nuclease pre-complexed with a gRNA targeting the editing site is delivered to perturbed cells by electroporation. The genomic region containing the CRISPRi sgRNA and repair outcome is then isolated, ligated with a unique molecular identifier (UMI), and amplified. Paired-end sequencing of linked CRISPRi sgRNA identities and repair outcomes then measure perturbation-specific repair outcome distributions.
(C) Functional annotations for 476 genes targeted by 1,573 sgRNA CRISPRi library.
(D) Screen data viewed from perturbation-centric perspective. Diagrams (left) show the 30 most frequent indels observed. Green rectangles in the top row mark the protospacer and PAM of the Cas9 target site. Vertical dashed line marks the expected DSB location. Insertions are marked by purple vertical lines and deletions by grey horizontal lines. Line plots show frequencies of each outcome for indicated sgRNAs. Heatmaps show log2 fold changes in each outcome frequency for POLQ or 53BP1 sgRNAs relative to all non-targeting sgRNAs.
(E) Screen data viewed from outcome-centric perspective, focusing on the indicated microhomology-flanked 4 nt deletion. Scatter plot shows number of UMIs recovered for each sgRNA (y-axis) against the percentage of UMIs reporting this deletion (x-axis).
To test induction and repair of DSBs, we transduced our vector into the human myelogenous leukemia cell line K562 and induced DSBs at several sites within a target region derived from the human HBB locus via electroporation of three differently targeted Streptococcus pyogenes Cas9 (SpCas9) RNPs (Figure S1A and Table S1). Distributions of DSB repair outcome frequencies were highly reproducible at each site (Figure S1B) but differed considerably between sites, including two sites located only one base pair (bp) apart (Figures S1C). Because RNPs also matched sequences within the endogenous HBB locus, we next compared outcome distributions produced in the integrated vector to those in the endogenous context. Despite the potential influence of local chromatin (Schep et al., 2021), distributions from the two contexts were closely concordant with each other (Figures S1D and S1E). We concluded that repair outcomes generated within our integrated screening construct would produce reproducible and informative readouts of DSB repair.
We also considered how to accurately quantify the effects of genetic perturbations on a broad range of outcomes. Because many genes with known roles in DNA repair and replication are essential, we chose to perturb gene expression with CRISPRi (Figure 1B). CRISPRi uses a catalytically-inactive SpCas9 fusion protein (dCas9-KRAB) to inhibit transcription at targeted promoters (Gilbert et al., 2013) and has been shown to be well-suited for investigating genes whose inhibition causes adverse growth phenotypes (Gilbert et al., 2014). Additionally, because PCR amplification may distort the relative abundances of different repair outcome sequences, we developed a sequencing strategy in which unique molecular identifiers (UMIs) are attached to repair outcomes prior to PCR (Figures 1B and S1F). UMIs enable grouping of reads derived from the same starting molecule to ensure that repair events from each cell are counted only once (Shiroguchi et al., 2012). This process also allows for correction of errors introduced by PCR or sequencing to distinguish genuine repair outcomes from technical artifacts (Schmitt et al., 2012). Finally, we developed a computational outcome classification pipeline based on our previously described knock-knock approach (Canaj et al., 2019) to characterize both simple indels and more complex sequence arrangements. We note, however, that previously identified repair processes may remove flanking sequences necessary for sequencing library construction and therefore not be detected, including long deletions (Kosicki et al., 2018) (Figures S2A-S2E, Table S2, and STAR Methods), loss of chromosomal segments (Zuccaro et al., 2020), chromothripsis (Leibowitz et al., 2021), or translocations (Bothmer et al., 2020).
Repair-seq enables sequence-independent classification of DSB repair outcomes and systematic identification of functionally related genes
To perform an initial Repair-seq screen, we designed a CRISPRi library targeting 476 genes with previously-identified roles in DNA repair or associated processes (1,513 gene-targeting sgRNAs and 60 non-targeting controls; Figure 1C, Tables S3 and S4). We cloned this library into our screening vector, transduced the library into K562 cells expressing dCas9-KRAB (K562 CRISPRi cells), allowed 6 days for gene depletion, and electroporated the cells with one HBB-targeting Cas9 RNP. We then allowed 3 days for induction and repair of DSBs at the Cas9 target site, extracted genomic DNA, and sequenced the distribution of repair outcomes for each genetic perturbation.
A reduction in the frequency of an outcome upon knockdown of a gene is evidence that activity of the corresponding gene product promotes formation of the outcome, while an increase indicates that the gene’s product suppresses the outcome. To quantify such phenotypes from our screen, we compared the frequency of each repair outcome in the presence of a gene-targeting CRISPRi sgRNA to the average frequency of that outcome across all 60 non-targeting control sgRNAs (Figures 1D). Performing the screen at high coverage (median 6,312 UMIs recovered per sgRNA; Table S5) enabled statistically significant comparisons across many repair outcomes, including those produced at low frequency (Figure 1E).
To explore data from this initial Repair-seq screen, we considered two complementary perspectives. First, we examined the effects of many different gene knockdowns on specific repair outcomes, producing “parts lists” of genes involved in creating or preventing each outcome (Figure 1E). For example, a common deletion flanked by two nucleotides (nts) of microhomology (MH) was reduced by sgRNAs targeting POLQ, a gene known to promote deletions with such sequence features (Ramsden et al., 2021), and was increased by sgRNAs targeting 53BP1, a negative regulator of such events. Next, we examined the effects of specific CRISPRi sgRNAs across many repair outcomes, producing detailed views of the roles that each gene plays in different repair pathways (Figure 1D). From this perspective, the effect of POLQ knockdown was unexpectedly complex, with knockdown depleting a specific subset of MH-flanked deletions but increasing or having no effect on others. In another example, knockdown of genes that encode an NHEJ heterodimer, XRCC5 (encoding Ku80) and XRCC6 (encoding Ku70) (Fell and Schild-Poulter, 2015), produced strikingly similar redistributions of outcomes, including specific increases in two short insertions (Figure S2F and S2G). This example suggested that systematic comparison of sgRNA-induced outcome redistributions could identify other functionally related genes. Conversely, the observation that outcomes with related sequence characteristics sometimes shared genetic regulators suggested that comparing dependencies of repair outcomes could identify those produced by similar mechanisms.
To test these ideas, we formed an outcome-by-sgRNA matrix of the log2 fold changes in frequencies caused by the 100 most active sgRNAs on all outcomes above a baseline frequency of 0.2% and hierarchically clustered this matrix along both dimensions (Figure 2). Clustering of sgRNAs revealed a rich set of distinct gene signatures across our library, with sgRNAs targeting the same gene or genes with known functional relationships frequently clustering closely together. Clustering of repair outcomes, on the other hand, revealed that while some outcomes with similar sequence architectures shared consistent genetic dependencies, other sets of similar sequences exhibited substantial variability in dependencies (e.g., bidirectional deletions).
Figure 2. Repair-seq enables data-driven inference of the genetic organization of DSB repair pathways.

Central heatmap shows log2 fold changes in repair outcome frequencies for the 100 most active CRISPRi sgRNAs relative to the average of all non-targeting sgRNAs, hierarchically clustered along both dimensions. Rows correspond to outcomes depicted by diagrams on the left, and columns correspond to individual CRISPRi sgRNAs. Triangular heatmaps depict correlations between pairs of sgRNAs (above) and between pairs of outcomes (right). sgRNA cluster assignments are labeled below.
Repair-seq screens at multiple target sites produce a high-resolution map of DSB repair
Because local sequence context is a major determinant of DSB repair outcomes (van Overbeek et al., 2016), evaluating repair within any particular sequence may provide an incomplete or idiosyncratic view of repair processes. We therefore next applied Repair-seq to a broader set of Cas9-induced DSBs by screening three additional Cas9 target sites located across a 102 nt window of our screening construct (Figures 3A, Tables S1 and S5). To enable more efficient parallelization of screens, we built and validated a condensed CRISPRi library with 336 sgRNAs targeting 118 genes and 30 non-targeting control sgRNAs, prioritizing sgRNAs empirically determined to produce significant redistribution of outcome frequencies in initial analyses (Figures S3A-S3C, Tables S4 and S6).
Figure 3. A systematic map of the genetic dependencies of repair outcomes generated at Cas9-induced DSBs.

(A) Cas9 target sites. Data from screens at target site 4 are shown in panels B-E; data from other target sites are shown in Figure S3.
(B) Reproducibility of the effects of CRISPRi sgRNAs on individual outcome frequencies. Black line (middle) shows baseline percentages averaged across two replicates. Brown line (right) shows correlations between replicates in log2 fold changes to outcome frequencies across the 100 most active sgRNAs (right). Insets compare changes in frequency of a common insertion (top) or less common deletion (bottom) for all CRISPRi sgRNAs between two replicates.
(C) Correlations between pairs of distinct outcomes in log2 fold changes in frequency across active sgRNAs. Green points mark pairs of distinct bidirectional deletions; light grey points mark all other outcome pairs.
(D) Log2 fold changes in frequency of outcomes produced by indicated sgRNAs targeting MRE11 and XRCC5 in two replicate screens. All outcomes above baseline frequency of 0.5% are plotted.
(E) Correlations between pairs of distinct CRISPRi sgRNAs in log2 fold changes in frequency across outcomes. Blue points mark pairs of sgRNAs targeting the same gene; grey points mark pairs of sgRNAs targeting distinct genes.
(F) Composite matrix of changes in outcome frequencies in screens performed at all four Cas9 target sites. See also Figure S4.
(G) UMAP embedding summarizing relationships between outcomes at Cas9 target sites based on their genetic dependencies. Points represent outcomes from individual screen replicates; colors represent outcome sequence architecture categories.
(H+I) UMAP embedding of Cas9 outcomes colored by Cas9 target site of origin (H) or baseline frequency of outcome (I).
Replicate screens performed at four Cas9 target sites using either the original 1,573 sgRNA library or the 336 sgRNA sub-library allowed evaluation of the technical quality of screen data. Across replicates, the effects of different CRISPRi sgRNAs on individual repair outcomes were highly reproducible, even for lower frequency outcomes (Figures 3B and S3D-S3F). Furthermore, correlations between these “outcome signatures” for pairs of distinct outcomes reproducibly spanned a range from high (indicating many shared genetic dependencies) to intermediate (indicating partially-overlapping dependencies) to negative (indicating production by opposing pathways) (Figures 3C and S3G-S3I). These correlations therefore represent a high-resolution measurement of relatedness between outcomes. Similarly, the effects of individual sgRNAs on different repair outcomes were reproducible between replicates (Figure 3D), and pairs of sgRNAs targeting the same gene reproducibly produced highly correlated “sgRNA signatures” (Figures 3E and S3J-S3L).
We next combined the changes in outcome frequencies measured in each of our screens into a single composite matrix, with outcomes from each replicate screen at the same target site included separately (Figures 3F and S4). Hierarchical clustering of these data frequently grouped identical outcomes from different replicates closely together, indicating that the precise dependencies of each outcome are highly specific, while also revealing rich structure in patterns of outcomes with shared genetic dependencies and genes with similar effects. To better visualize groupings of outcomes, we performed dimensionality reduction on outcome signatures using uniform manifold approximation and projection (UMAP), creating an embedding based on the genetic dependencies of each outcome (Figure 3G). Examination of this embedding revealed three high-level features. First, although outcomes with similar sequence features were often grouped together, many were separated into distinct groups, including three clusters of short insertions (labeled I, II, and III in Figure 3G). Second, outcomes from different target sites were interspersed throughout the embedding (Figure 3H), supporting the idea that DSBs in different sequence contexts engage broadly similar repair mechanisms. An interesting exception to this observation was a cluster of genetically-related insertions and more complex insertion + deletion outcomes, which were observed predominantly at target site 4, suggesting that the mechanism underlying these outcomes is more sensitive to sequence context (Figures 3G and 3H). Finally, high-frequency outcomes clustered into specific areas of the embedding (Figure 3I), indicating that while a subset of repair processes are preferentially engaged at Cas9-induced breaks, the genetic dependencies of many individually rare outcomes collectively reveal a broader diversity of mechanisms.
Distinct mechanisms generate insertions at Cas9-induced DSBs
A key strength of Repair-seq is the ability to classify repair outcomes by their genetic dependencies rather than assumptions based on their sequences. This strength is highlighted by the separation of Cas9-induced short insertions into distinct groups (Figure 3G). Consistent with a general requirement for DNA synthesis in generation of insertions, these groups exhibited strong dependence on different polymerases, with DNA polymerase theta (Pol θ, encoded by POLQ) promoting insertions in one group (III) (Figure 4A) and DNA polymerase lambda (Pol λ, encoded by POLL) promoting insertions within the other two (I and II) (Figures 4B-4D). Notably, while a previous study found that a Pol λ homolog was responsible for Cas9-induced insertions in yeast (Lemos et al., 2018), the existence of Pol λ-independent insertions in our data indicates that the genetic landscape of insertion mechanisms in human cells is more complex.
Figure 4. Insertions at Cas9-induced DSBs have distinct sets of dependencies on core NHEJ factors.

(A-C) Effects of gene knockdowns on the frequency of representative insertions from indicated outcome groups. Points depict the mean log2 fold change in frequency of the insertion relative to non-targeting sgRNAs for the two sgRNAs with the most extreme phenotypes targeting each gene. See also Figure 3G.
(D+E) Log2 fold changes in outcome frequencies produced by indicated CRISPRi sgRNAs overlaid on UMAP embedding of Cas9 outcomes.
(F) Effects of indicated CRISPRi sgRNAs on the most frequent insertions at four Cas9 target sites. Letters to the left of outcome diagrams mark insertions highlighted in panels (A-C). Black bars mark insertions that duplicate PAM-distal sequence adjacent to the canonical DSB location or insertions consistent with multiple iterated duplications.
(G) Effects of indicated CRISPRi sgRNAs on insertions at endogenous loci in HeLa cells.
(H) Model for generation of Cas9-induced insertions from 5′ overhangs.
Bifurcation of Pol λ-dependent insertions into two groups suggested that Cas9-induced DSBs can be repaired by distinct Pol λ-mediated mechanisms. Comparing the effects of all gene knockdowns on insertions from each group revealed striking differences for three mechanistically related NHEJ genes, XRCC5, XRCC6, and PRKDC, which encode the core NHEJ heterodimer Ku70/Ku80 and its binding partner DNA-dependent protein kinase catalytic subunit, DNA-PKcs, respectively. Remarkably, repression of these genes dramatically increased the frequency of insertions from group I while decreasing the frequency of insertions from group II (Figures 4B, 4C, and 4E). Additionally, across all four target sites, the insertions suppressed by these genes shared common sequence characteristics, consisting of three related sequence types: 1 nt insertions that duplicated the nucleotide on the side of the canonical Cas9 cut site opposite the protospacer adjacent motif (PAM-distal); 2 nt insertions that duplicated this same nucleotide twice (“iterated duplications”); and 2 nt insertions that duplicated two consecutive PAM-distal nucleotides adjacent to the cut site (Figure 4F). Validating this observation, CRISPRi-mediated repression of XRCC5 and POLL in HeLa cells produced similar results at five endogenous Cas9 target sites (Figures 4G and S5A).
Notably, the sequence features of insertions suppressed by Ku70/Ku80 and DNA-PKcs are consistent with products of staggered Cas9 cleavage. Although canonically blunt, Cas9-induced DSBs are variably generated with short 5' overhangs (Jones et al., 2021; Shou et al., 2018), and fill-in synthesis can create insertions of the overhang sequence (Lemos et al., 2018). Our results suggest that these fill-in events are mediated by Pol λ in human cells and that the resulting insertions, but not those generated by other Pol λ-directed processes, are suppressed by Ku70/Ku80 and DNA-PKcs. Intriguingly, biochemical assays have demonstrated that Ku70/80 and DNA-PKcs can protect DNA ends from processing by DNA polymerases, including Pol λ (Stinson et al., 2020). Synthesizing this observation with screen results suggests that when Cas9 produces a staggered cut, Ku70/80 and DNA-PKcs efficiently suppress fill-in of overhangs by POLL to favor direct rejoining of the compatible ends (Figure 4H). The counter-intuitive fact that knockdown of canonical mediators of NHEJ increases the frequency of these specific NHEJ-associated outcomes highlights the complexity of the interplay between DNA end configurations and repair factors.
Capture of human genomic sequence at DSBs is stimulated by loss of DNA2 and MCM10
In addition to short insertions, we observed repair outcomes in which longer stretches of sequence from the human genome were “captured” at breaks (Onozawa et al., 2014; Yu et al., 2018) (Figure S5B). The length distribution of these insertions in unperturbed cells consisted of partially-overlapping populations centered at ~75 and ~150 nts (Figure S5C). These populations exhibited dramatically different genetic dependencies, with a broad set of genes causing substantial changes in the shorter population but weak or no changes in the longer population (Figures S5D and S5E). This effect was most striking for two genes: DNA2, which encodes a nuclease with high affinity for single-stranded 5' flaps and multiple proposed roles in replication and DNA repair (Zheng et al., 2020), and MCM10, which encodes a replisome component with roles in replication initiation and elongation (Lõoke et al., 2017). Knockdown of these genes produced 4.5- to 13-fold increases in the short population but substantially weaker changes in the long population. Mirroring this effect, overexpression of DNA2 specifically reduced the frequency of the short population of captured human sequence fragments (Figure S5F).
Intriguingly, DNA2 deficiency has been shown to increase genomic capture events at induced breaks in yeast, potentially by increasing levels of overreplicated single stranded DNA produced during Okazaki fragment maturation or the processing of reversed replication forks (Yu et al., 2018). Our data indicate that this phenotype is recapitulated in human cells and further nominates a key role for a specific component of the replisome, MCM10, in suppressing these events. Notably, we also observed rare capture of sequence from the Bos taurus genome (Figure S5B), presumably stemming from DNA in fetal bovine serum used in culture media entering cells during electroporation (Ono et al., 2019). Capture of bovine sequences was not suppressed by knockdown of DNA2 or MCM10 (Figure S5G), suggesting that the longer population of captured human sequence fragments may also originate as extra-cellular DNA. Together, these results highlight how direct sequencing of outcomes allows Repair-seq to reveal the genetic determinants of a diverse range of repair events.
Repair-seq allows systematic identification of DSB repair complexes and pathways
We next explored the idea that Repair-seq could be used to identify DSB repair complexes and pathways. Supporting this idea, repair outcome redistributions produced by pairs of CRISPRi sgRNAs targeting different members of the Ku70/80 heterodimer (XRCC5 and XRCC6), the MRN complex (MRE11, NBN, and RAD50), and replication factor C complex (RFC2, RFC3, RFC4, and RFC5, across all four Cas9 target sites frequently correlated more highly with each other than with any other targeted genes (Figures 5A-5C). To further explore such relationships, we performed dimensionality reduction on the columns of our composite sgRNA-by-outcome matrix, creating an embedding based on the outcome redistribution profiles of each sgRNA (Figure 5D). This embedding revealed many groups of sgRNA-targeted genes with similar roles in DSB repair, including those involved in initiating DSB end resection (e.g., MRE11, NBN, RAD50, RBBP8), replication (e.g., RFC1, RFC2, RFC3, RFC4, RFC5, MCM2, MCM6, TIMELESS, TONSL, MMS22L, POLE, POLE2), the replication checkpoint (e.g., RAD17, HUS1, RAD9A), HDR (e.g., BRCA2, PALB2), and the Fanconi anemia pathway (e.g., FANCI, FANCE, FANCD2, FANCB, FANCM, FANCF, FANCA, FAAP24, UBE2T, BRCA1, and BARD1). This embedding also separated canonical NHEJ genes into multiple distinct clusters (e.g., 53BP1, RNF8, PAXIP1, and DCLRE1C; LIG4 and NHEJ1; XRCC5, XRCC6, PRKDC and APTX), highlighting the power of high-resolution phenotypes to capture specific gene functions. Finally, unexpected groupings of genes without known functional relationships nominated noncanonical roles for several DSB repair genes. Specifically, screen data identified phenotypic similarities between POLQ and a set of replication checkpoint genes (RAD17, HUS1, and RAD9A), as well as between the canonical replication and checkpoint gene TOPBP1 and genes that initiate DNA end resection (MRE11, NBN, RAD50, RBBP8).
Figure 5. Systematic inference of functional relationships between DNA repair genes.

(A-C) Correlations between outcome redistribution signatures for pairs of active sgRNAs targeting distinct genes where one or both genes is a member of a known complex: XRCC5 and XRCC6 (A), MRE11, RAD50, and NBN (B), or RFC2, RFC3, RFC4, and RFC5 (C).
(D) UMAP embedding of outcome redistribution signatures for individual sgRNAs (columns of the indicated composite matrix of Cas9 outcome frequency changes). sgRNAs are colored by cluster assignments prior to dimensionality reduction; grey points were not assigned to a specific cluster. sgRNAs are labeled with GeneCards gene symbols (www.genecards.org), except that a common alias 53BP1 is used for gene symbol TP53BP1. We note other common aliases: XLF (NHEJ1), ARTEMIS (DCLRE1C), PTIP (PAXIP1), KU80 (XRCC5), KU70 (XRCC6), DNAPK (PRKDC), H2AX (H2AXF), and NBS1 (NBN).
(E+F) Changes in the fraction of cells for each sgRNA assigned to S phase (E) or G2/M phase (F) relative to all non-targeting sgRNAs based on expression profiles measured by Perturb-seq.
Because DSB repair is regulated by the cell cycle (Hustedt and Durocher, 2017), effects of gene knockdowns on cell cycle progression may contribute indirectly to Repair-seq phenotypes. Most notably, perturbations that reduce the fraction of cells in S/G2 would be expected to decrease the frequency of resection-dependent repair outcomes. We therefore used direct capture Perturb-seq, a method for performing single-cell RNA-sequencing of CRISPR-based screens (Replogle et al., 2020), to measure the fraction of cells observed in S and G2/M phases for each CRISPRi sgRNA in our 366 sgRNA sub-library. Overlaying these phenotypes onto the Repair-seq embedding of outcome redistribution profiles revealed that sgRNAs with strong cell cycle effects were distributed both within and outside of distinct clusters (Figures 5E and 5F), suggesting that Repair-seq phenotypes capture signals of gene function beyond simple cell cycle effects. Clustering of TOPBP1 with end resection genes provided an interesting example of this logic. Intriguingly, TOPBP1 has been shown to physically interact with MRN (Duursma et al., 2013). TOPBP1 is also required for DNA replication initiation (Wardlaw et al., 2014), and consistent with this role, sgRNAs targeting TOPBP1 reduced the fraction of cells in S phase (Figure S6A). While these cell cycle effects likely contribute to the phenocopying of MRN genes by TOPBP1, other sgRNAs in the library, such as those targeting MCM2, produced substantially stronger reductions in S phase occupancy but weaker correlations with the outcome redistribution signatures of MRN genes. Cell cycle effects may therefore not be sufficient to explain the specificity of the observed TOPBP1-MRN relationship.
We next asked whether the functional relationships identified in our Cas9 screens are specific to DSBs produced by Cas9 or represent more general features of DSB repair. To survey an additional DSB configuration, we performed a Repair-seq screen using Acidaminococcus sp. Cas12a (H800A mutant; Table S1) with our 1,573 CRISPRi sgRNA library (Figure S1A). Cas12a-induced DSBs differ from Cas9-induced DSBs in two key ways: Cas12a produces staggered nicks that leave ~5 nt 5' overhangs and are located further from the PAM sequence (Zetsche et al., 2015). Consistent with these differences, screen data revealed several Cas12a-specific features. First, clustering of Cas12a outcomes based on their genetic signatures identified distinct groups of deletions spanning each of the two staggered nicks, with complex patterns of dependencies on different NHEJ factors between the groups (Figure S6B). Second, we observed a group of sgRNAs that specifically increased a set of outcomes that were either unmutated or contained mutations that did not disrupt the PAM or protospacer (Figure S6B). Because these outcomes are susceptible to re-targeting but will only be observed when they do not acquire additional mutations, increases in these outcomes may be driven by sgRNA-induced reductions in the overall rate of cutting. Finally, baseline frequencies of capture of human genomic sequences at Cas12a-induced DSBs were 3.6-fold lower than in Cas9 screens, suggesting that such events favor Cas9 end configurations.
Despite these differences, clustering of sgRNA signatures produced with Cas12a recapitulated many of the gene-gene relationships observed with Cas9. Specifically, we observed similar groupings of NHEJ genes, including one of end synapsis genes (LIG4 and NHEJ1), one with XRCC5 and XRCC6 next to PRKDC, and one featuring 53BP1, RNF8, PAXIP1, and DCLRE1C (Figure S6B). Furthermore, sgRNAs targeting POLQ again clustered closely with those targeting replication checkpoint genes (e.g., RAD17, HUS1), and TOPBP1 phenocopied DSB end resection genes (e.g., MRE11, NBN, RAD50, RBBP8).
Identification of genetically distinct pathways of microhomology-mediated end-joining
A major goal in building Repair-seq was to systematically define DSB repair pathways. We were therefore intrigued by the observation that POLQ, a key mediator of alternative end-joining, was phenocopied by a set of genes without characterized roles in POLQ-mediated repair (RAD17, RAD9A, RAD1, and HUS1, encoding the 9-1-1 complex and associated clamp loader) (Figures 6A, 6B, S4, and S6B). To quantify the similarity between these phenotypes, we calculated correlations between outcome redistribution profiles for all pairs of active sgRNAs in both Cas9 and Cas12a screens (Figure 6C). Correlations between POLQ sgRNAs and sgRNAs targeting RAD17, RAD9A, HUS1, and RAD1 were among the strongest observed for any pair of genes in our library. Validating this relationship, POLQ and RAD17 phenotypes were also similar at one endogenous Cas9 target site in K562 cells (Figure 6D) and at two lentivirally transduced target sites (Cas9 and Cas12a) in both K562 and HeLa cells, albeit with data from a third cell line (RPE1) suggesting possible cell type specificity (Figure S6C). Together, these results indicate that RAD17 and the 9-1-1 complex can promote and suppress similar subsets of DSB repair outcomes as Pol θ (the gene product of POLQ).
Figure 6. Repair-seq delineates microhomology-mediated end-joining pathways.

(A+B) Log2 fold changes in outcome frequencies produced by indicated CRISPRi sgRNAs overlaid on the UMAP embedding of Cas9 outcomes. See also Figure 3G.
(C) Comparison of correlations between outcome redistribution signatures for pairs of sgRNAs targeting distinct genes between composite Cas9 screens and Cas12a screen.
(D) Effects of indicated CRISPRi sgRNAs or of chemical MRE11 inhibitor on repair outcome frequencies at Cas9 target site 1 in the endogenous HBB locus. Heatmaps display log2 fold changes in outcome frequency for the 20 outcomes with highest baseline frequency, sorted by average log2 fold change across POLQ sgRNA replicates.
(E) Examples of Cas9-induced mutations consistent with known Pol θ-mediated architectures (insets). Top black lines represent the sequence of a repair outcome; bottom grey line represents the sequence flanking the targeted DSB in the integrated screening vector; lines between these depict local alignments; and red (top) and blue (bottom) rectangles mark the protospacer and PAM of Cas9 target sites.
(F+G) Deletions in UMAP embedding of Cas9 outcomes colored by the length of microhomology flanking the deletion junction (F) or the length of sequence removed by the deletion (G).
(H) Log2 fold changes in outcome frequencies produced by an MRE11-targeting sgRNA overlaid on the UMAP embedding of Cas9 outcomes. The region marked with a dotted line includes the vast majority of MRE11-promoted outcomes and is similarly indicated in panels (A, B, E, F, and G).
(I) Effects of indicated CRISPRi sgRNAs on outcome frequencies in cells ectopically expressing MRE11 + GFP, or nuclease-inactive MRE11 (H129N) + GFP, or control (GFP). Heatmaps (right) display log2 fold changes in the frequencies of the 20 outcomes with highest baseline frequency, sorted by average log2 fold change across MRE11-targeting sgRNAs in control cells.
(J) Model of genetically distinct sub-pathways of resection-initiated end joining.
We next examined the sequence characteristics of POLQ-dependent outcomes. To facilitate repair, Pol θ engages short stretches of complementary base pairing present in DSB repair intermediates (Ramsden et al., 2021; Sfeir and Symington, 2015). These microhomologies prime Pol θ for DNA synthesis and leave recognizable sequence characteristics at repaired breaks. Previous work has defined two categories of Pol θ-mediated mutations: (1) complex insertion outcomes in which DSBs are “filled-in” with sequences bearing microhomologies at the boundaries, sometimes also accompanied by deletions, and (2) deletions of typically <50 nts flanked by >2-3 nts of microhomology. Within our screen data, we found examples of each of these mutation types with clear POLQ dependence (Figures 6A and 6E); however, consistent with a recent observation (Carvajal-Garcia et al., 2020), we also identified many low-frequency deletions with lengths and microhomology usage consistent with expected Pol θ-mediated sequence architectures that were independent of, or even suppressed by, POLQ (Figures 3I, 6A, 6F and 6G).
Given this observation, we next examined how inhibition of DNA end resection, a defined step in Pol θ-mediated repair (Truong et al., 2013; Wyatt et al., 2016), affected outcome frequencies at our Cas9 and Cas12a target sites. Within screen data, genes responsible for initiating resection (i.e., MRE11, RAD50, NBN, and RBBP8) promoted a superset of POLQ-dependent outcomes, including both complex insertions and a broad set of bidirectional deletions flanked by microhomologies (Figures 6A, 6E-6H, S4, and S6B). Consistent with a general requirement for end resection across these outcomes, ectopic expression of MRE11 but not a nuclease inactive mutant (MRE11-H129N) rescued the frequencies of both POLQ-dependent and -independent bidirectional deletions after MRE11 knockdown (Figures 6I and S4). Bidirectional deletions across the spectrum of POLQ phenotypes were also reduced by treatment with an MRE11 exonuclease inhibitor (Figure 6D). These data demonstrate that not all resection-dependent end-joining requires Pol θ in human cells.
Altogether, these observations suggest that there exist two genetically distinct mechanisms of “microhomology-mediated end-joining” downstream of MRE11-mediated end processing (Figure 6J), each with different dependence on POLQ. Furthermore, the POLQ-dependent mechanism is promoted by RAD17 and the 9-1-1 complex, either through checkpoint signaling or alternative functions. Notably, while lack of dependence on NHEJ factors is a canonical characteristic of such alternative end-joining mechanisms (Ramsden et al., 2021; Sfeir and Symington, 2015), the effects of NHEJ factors on these outcomes in our screens were not always straightforward (Figure S6D). The phenotypes of one NHEJ regulator (53BP1), however, clearly opposed those of POLQ (Figures S4, S6B, S6E).
Repair-seq is a versatile platform for mapping the cellular determinants of genome editing systems
A broad range of genome editing approaches rely on endogenous DNA repair processes to install programmed sequence changes (Anzalone et al., 2020). To explore the flexibility of Repair-seq to study diverse editing modalities, we next performed screens in the presence of a panel of different homologous repair templates (donors), including four single-stranded oligodeoxyribonucleotides (ssODNs) and one linear double-stranded ODN with homology to our target region as well as a non-homologous single-stranded control (Figure 7A, Tables S2 and S5).
Figure 7. Repair-seq is a versatile platform for mapping the genetic determinants of diverse genome editing modalities.

(A) Repair-seq can be adapted to interrogate many DNA repair processes.
(B) Comparison of baseline outcome frequencies in screens performed with no donor, a ssODN homologous to the target site with or without PAGE-purification, or a ssODN with no homology to the target site. Outcomes (depicted, left) are sorted by average baseline frequency in no-donor screen. Data in line plot (middle) show mean baseline outcome frequencies (+/− s.d.) across replicates. Bottom two rows report the combined frequency of all outcomes with sequence architectures shown in (D) and (E); three rows above these show scarless HDR outcomes containing different subsets of donor-programmed SNVs as in (C).
(C+D+E) Examples of distinct categories of repair outcomes that incorporate sequence from ssODNs: scarless HDR outcome (C), “half-HDR” outcome (D), and outcome in which a fragment of the donor has been captured at the break without use of intended homology on either side (E). Middle black lines represent the sequence of a repair outcome; top orange lines represent the donor sequence; bottom grey lines represent the screen vector sequence; and lines between these represent alignments between an outcome and the relevant reference sequence. Xs mark any mismatches. Gray shaded boxes indicate regions of perfect homology between the donor and target region, which flank a homeologous region containing programmed SNVs.
(F) Effects of gene knockdown on the frequency of scarless HDR in replicate screens performed using a single-stranded donor with the 1,573 CRISPRi sgRNA library. Each dot depicts the mean log2 fold change in combined frequency of all scarless HDR outcomes for the two sgRNAs targeting each gene with the most extreme phenotypes.
(G) Comparison of the effects of gene knockdown on scarless HDR in a screen performed using a single-strand donor (x-axis) and a screen performed using a linear double-stranded donor with the same sequence (y-axis).
(H) Effects of gene knockdown on the frequency of half-HDR outcomes in two replicate screens performed using a single-strand donor with the 366 CRISPRi sgRNA library.
(I) Comparison of the effects of gene knockdown on the frequency of scarless HDR outcomes (x-axis) and half-HDR outcomes (y-axis) in a screen performed using a single-strand donor.
(J) Effects of gene knockdown on the frequency of capture of donor fragment outcomes in two replicate screens performed using a single-stranded donor with the 366 CRISPRi sgRNA library.
(K) The effects of repressing select genes on capture of donor fragments (left) and capture of genomic fragments >75 nts (right) in four screens performed using the same single-stranded donor.
Single-strand template repair (SSTR) is an exogenous form of HDR in which sequences from ssODNs are incorporated into nuclease-induced DSBs (Jasin and Haber, 2016). Previous work has identified regulators of SSTR in yeast and human cells (Bothmer et al., 2017; Canny et al., 2018; Gallagher et al., 2020; Maruyama et al., 2015; Richardson et al., 2018), but the processes responsible for SSTR remain incompletely defined. To investigate SSTR, we compared baseline outcome frequencies from Repair-seq screens performed with and without a 178 bp ssODN containing 8 programmed single-nucleotide variants (SNVs) near the DSB surrounded by regions of perfect homology to the corresponding genomic sequence (Figure 7B). Inclusion of this template reshaped repair outcome distributions in two informative ways. First, the frequencies of a set of microhomology-flanked bidirectional deletions dramatically decreased in the presence of homologous ssODN, while the presence of non-homologous donor produced more modest decreases, suggesting that SSTR competes with specific donor-independent outcomes. Second, we observed three major categories of outcomes in which donor sequence was incorporated at the break: HDR outcomes in which donor-encoded SNVs were scarlessly installed (Figure 7C); “half-HDR” outcomes in which the perfectly homologous region on the 3' end of the ssODN was correctly engaged but the transition back to genomic sequence on the other side of the break did not occur as intended (Rouet et al., 1994) (Figure 7D); and capture of fragments of donor sequence at the break without using intended homology on either side (Figure 7E). Baseline frequencies of each outcome type were similar between screens performed with and without purified donors, arguing that contamination by truncated donor molecules does not contribute substantially to these outcomes (Figure 7B).
We examined genetic regulators of each category of donor-dependent outcome, starting with scarless HDR events. We first calculated the effect of each gene knockdown on the combined frequency of all outcomes containing scarless installation of any subset of donor-encoded SNVs in single-stranded donor screens. Consistent with previous results (Richardson et al., 2018), this frequency was increased by knockdown of NHEJ genes (e.g., 53BP1, PRKDC, and LIG4) and decreased by knockdown of many HDR genes (e.g., BRCA1, MRN genes, RBBP8, Fanconi anemia (FA) genes, and gene encoding components of the POLD and RFC complexes) (Figure 7F). We then compared these phenotypes to corresponding phenotypes from a screen performed with a linear double-strand donor of the same sequence. While HDR from these two donor configurations shared many genetic regulators, knockdown of several genes, including DNA2 and multiple genes involved in RAD51-mediated strand invasion (e.g., BRCA2 and RAD51) reduced HDR from single-stranded donors but not from the double-stranded donor (Figure 7G and S7A).
We next examined outcome categories in which single-stranded donor sequence was incorporated at the break without perfect use of encoded homologies, producing two interesting observations. First, knockdown of several groups of genes that substantially reduced ssODN-templated scarless HDR did not reduce the frequency of half-HDR outcomes in which the 3' region of perfect homology but not the 5' region were correctly engaged, including RAD51-related genes, FA genes, and DNA2 (Figures 7H, 7I, S7A, and S7B). This observation suggests that activity of these genes may specifically promote second-end capture of a donor-extended DSB through pairing of donor-templated 5' homology to the other DSB end. Second, capture of partial ssODN fragments without the use of homology was promoted by LIG4, strongly suppressed by DNA2, and modestly suppressed by MCM10, suggesting that these events arise from similar but not identical mechanisms as capture of shorter human genomic sequence fragments (Figures 7J, 7K, and S7C; see discussion).
Finally, we reasoned that examining the genetic dependencies of individual SSTR outcomes in which different subsets of SNVs were scarlessly installed could reveal additional mechanistic diversity. In a set of screens performed with ssODNs carrying a variety of programmed SNV patterns, we found that genes encoding components of the MutSα/MutLα DNA mismatch repair (MMR) complexes (MSH2, MSH6, MLH1, and PMS2) promoted some scarless SSTR outcomes while suppressing others, with suppressed outcomes typically missing SNVs from the 3' ends of ssODNs (Figures S7D-S7F). These observations support a model of scarless SSTR in which MMR activity installs SNVs encoded on the 3' ends of ssODNs by “fixing” mismatches established upon donor annealing (Gallagher et al., 2020; Harmsen et al., 2018; Kan et al., 2017) (Figure S7G). Extending this model, we found that scarless SSTR outcomes within the MMR-suppressed set also generally exhibited stronger MRN dependence (Figures S7D-S7F), suggesting that incorporation of donor-encoded SNVs by MMR relieves a requirement for end resection and, more broadly, demonstrating that scarless SSTR can occur via distinct mechanistic paths.
Collectively, these observations highlight the complex range of pathways by which oligonucleotide donors are incorporated at DSBs and demonstrate the utility of Repair-seq for delineating the genetic determinants of both intended and unintended genome editing outcomes.
DISCUSSION
Repair-seq pairs CRISPR-based genetic screens with locus-specific deep sequencing to profile the spectrum of mutations produced at targeted DNA lesions across many genetic perturbations, enabling systematic interrogation of DNA repair pathways. Here, we applied Repair-seq to survey DSBs produced by 2 different programmable enzymes (Cas9 and Cas12a) within 5 different sequences contexts in the presence and absence of homologous repair templates. Data from these experiments produced a high-resolution atlas of the genetic dependencies of DSB repair outcomes. Systematic exploration of this atlas revealed that repair outcomes with superficially similar sequence architectures can show marked differences in genetic dependencies. For example, we observed that a subset of short Pol λ-mediated insertions at Cas9-induced DSBs were suppressed by canonical NHEJ factors (Ku70/80 and DNA-PKcs). Additionally, we found that deletions flanked by microhomologies can be produced by distinct mechanisms. Comparisons of signatures of each gene across outcomes then revealed associations between a canonical mediator of these deletions (POLQ) and several other genes (e.g., RAD17). Altogether, these observations deepen our understanding of how cells repair enzymatically induced DSBs and demonstrate the power of classifying of DNA mutations without a priori registration of sequence features.
Beyond simple insertions and deletions, Repair-seq enables the study of more complex repair outcomes. As examples, we identified genes that modulate the capture of fragments of genomic sequence or exogenous ssDNA at DSBs, including DNA2 and MCM10. Consistent with our results, DNA2 has been proposed to suppress such outcomes in yeast by preventing generation of ssDNA and/or by degrading intracellular ssDNA (Yu et al., 2018). Intriguingly, across our data sets, MCM10 suppressed capture of genomic sequence with comparable or greater magnitude as DNA2, but DNA2 had a much stronger impact on capture of exogenous ssDNA than MCM10 (Figures S5E and 7K). Together, these phenotypes suggest a model in which loss of MCM10 increases generation of genomic fragments, while DNA2 also shapes repair outcomes downstream of this step, either through degradation of ssDNA or via more direct activity of the enzyme at DSBs. These findings demonstrate how the ability to compare the effects of genetic perturbations across different repair outcomes can suggest and constrain models of the mechanisms behind DNA repair events.
Finally, the ability of Repair-seq to simultaneously measure the effects of genetic perturbations on a broad range of mutations makes it well-suited to study how DNA damage produced by genome editing tools is repaired in both intended and unintended ways. Our studies of HDR with oligonucleotide repair templates serve as proof of principle for this concept. Furthermore, in companion studies (Chen et al, 2021 and Koblan et al., 2021), we applied Repair-seq to identify the cellular determinants of base and prime editing and used the insights gained from those screens to develop editing tools with improved efficiency and precision. Although the complexity of DNA repair remains a formidable challenge for precise genome editing, these studies collectively demonstrate that Repair-seq is a powerful tool for unravelling this complexity.
LIMITATIONS OF THE STUDY
Due to its reliance on short-range amplicon sequencing, Repair-seq produces rich but not comprehensive profiles of DNA repair outcomes. In particular, repair processes that regenerate the original target sequence or that remove specific flanking sequences required for library preparation cannot be directly detected. The development of alternative sequencing strategies may enable screening of additional repair mechanisms. Alternatively, unobservable repair outcomes can be indirectly studied by comparing CRISPRi sgRNA abundances to matched control screens in which no on-target DNA damage was induced (Table S7).
Technical aspects of screen design may also impact repair outcomes. Random genomic integration of the screen vector masks the influence of chromatin context on repair, and the context of the screen vector itself may influence some repair processes. Additionally, nucleofection of RNPs into cells may introduce elevated levels of extracellular nucleic acids. We note that while Repair-seq is a powerful tool for nominating roles of genes in specific repair processes, understanding the exact mechanisms underlying each phenotype will require targeted validation and follow-up.
Applying Repair-seq in different cell types, at additional target sites, to DSBs produced by additional nucleases, and with alternative methods of delivering damage-induced agents will be helpful to understand the roles of these variables in DSB repair. Moving forward, genome-wide Repair-seq screens will be of interest to identify additional modulators of repair, but scaling the number of perturbations screened while maintaining sufficient coverage of cells per perturbation to resolve low frequency outcomes may be challenging. Determining ideal coverage levels will require balancing experimental practicality with the complexity of the outcome landscape produced by a given editing modality. Finally, we note for clarity that while this manuscript was in preparation, an unrelated method also called Repair-seq was published (Reid et al., 2021).
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Britt Adamson (badamson@princeton.edu).
Materials Availability.
Plasmids and CRISPRi sgRNA libraries generated in this study have been deposited to Addgene.
Data and Code Availability.
Raw sequencing data has been deposited at the NCBI Sequence Read Archive database under Bioproject PRJNA746980. Code is available at https://github.com/jeffhussmann/repair-seq (DOI: https://doi.org/10.5281/zenodo.5534778). Processed data and notebooks are available at DOI: https://doi.org/10.5281/zenodo.5555287. Data can be interactively explored at https://seq.repair. Any additional information required to reanalyze the data reported in this paper is available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines used in this study were 293T, K562, K562 CRISPRi, K562 CRISPRa, HeLa CRISPRi, RPE1 CRISPRi cell lines. K562 CRISPRi and CRISPRa cell lines were constructed as previously described (Gilbert et al., 2014). The K562 CRISPRi cell line expressed dCas9-BFP-KRAB (pHR-SFFV-dCas9-BFP-KRAB; Addgene, 46911), and the K562 CRISPRa cell line expressed both dCas9-SunTag (pHRdSV40-dCas9-10xGCN4_v4-P2A-BFP; Addgene, 60903) and scFV-sfGFP-VP64 (pHRdSV40-scFv-GCN4-sfGFP-VP64-GB1-NLS; Addgene, 60904). HeLa CRISPRi and RPE1 CRISPRi cells also expressed dCas9-BFP-KRAB and were constructed as previously described (Gilbert et al., 2013; Jost et al., 2017). During this study, K562, K562 CRISPRi, K562 CRISPRa, and HeLa CRISPRi cell lines were authenticated by analysis of short tandem repeats as exact matches to the corresponding line, CCL-243 (K-562, female) or CCL-2 (HeLa, female), from ATCC. One 293T cell line, used for packaging lentivirus, was replaced mid-study due to mutational events that reduced those cells to only a near perfect match to CRL-3216 (293-T). Cells tested negative for micoplasma. K562 cells were grown in RPMI 1640 with L-glutamine and 25 mM HEPES (Corning or Gibco) supplemented with 100 U/mL penicillin, 100 ug/mL streptomycin, and 0.292 mg/mL L-glutamine. 293T and HeLa cell lines were grown in DMEM with 4.5 g/L glucose and sodium pyruvate with or without L-glutamine (Corning or Gibco) supplemented with 100 U/mL penicillin and 100 ug/mL streptomycin. RPE1 cell lines were grown in DMEM/F12 with L-glutamine and 15 mM HEPES (Gibco) supplemented with 100 U/mL penicillin and 100 ug/mL streptomycin. Cells were grown at 37°C. Mirin (Millipore Sigma, M9948-5MG) was dissolved in DMSO.
METHOD DETAILS
Plasmid construction.
Our CRISPRi sgRNA libraries were delivered to cells using a custom expression vector (pAX198). We constructed this vector from pU6-sgRNA EF1Alpha-puro-T2A-BFP (Gilbert et al., 2014) (Addgene, 60955) as follows: We added two restriction sites (NotI and HindIII) and spacer sequence to pU6-sgRNA EF1Alpha-puro-T2A-BFP to construct an intermediate vector (pMJ468). We then cloned a “target region” for enzyme-induced DNA damage between the sgRNA and selection marker expression cassettes (the latter expresses both BFP and puromycin resistance) by Gibson assembly at the NsiI and XhoI sites. This target region comprised sequence from the human HBB gene—specifically the second and third exons of ENST00000647020.1 (no intron) and part of the 3' UTR—and was flanked by a NotI site. As part of this cloning, a second NotI site (adjacent to the target site) was removed and the termination signal for the U6-sgRNA expression cassette was reduced to a stretch of 6 thymines. Finally, to enable sgRNA cloning, a BstXI site was removed from the target site by site-directed mutagenesis. In arrayed experiments, individual sgRNAs were delivered using pU6-sgRNA EF1Alpha-puro-T2A-BF, including XRCC5_+_216974121.23-P1P2, POLL_−_103347937.23-P1P2, POLQ_+_121264772.23-P1P2, MRE11A_−_94227004.23-P1P2, RAD17_+_68665692.23-P1P2 (Table S3). pU6-sgRNA EF1Alpha-puro-T2A-BFP was used as a negative control in experiments depicted in Figures 4G, S5A, and 6D.
For the experiment depicted in Figure 6I, we built expression vectors using pInducer20 (Addgene, 44012) as follows: pInducer20 was first digested with AgeI and religated to remove unwanted functional cassettes. EF1A promoter was then inserted between the BamHI and XhoI sites. The resulting plasmid contains bicistronic expression cassettes with distinct promoters (EF1A and UBC) for each cassette. To generate MRE11 expression vectors (pJL056 and pJL057), superfolder GFP was inserted downstream of the UBC promoter between the KpnI and NotI sites, and MRE11 cDNA sequence, amplified from pICE-HA-MRE11-WT (Addgene, 82033) using primer pairs oJL162 and oJL163 (Table S2), was inserted downstream of the EF1A promoter at the XhoI site. The nuclease-inactivating mutation (H129N) was then introduced by PCR with primer pairs oJL079 and oJL080 (Table S2). To generate a GFP expression vector control (pJL059), superfolder GFP was inserted downstream of the UBC promoter between the KpnI and NotI sites. MREA11-targeting sgRNAs used in this experiment (1, 3, 8) were MRE11A_−_94227004.23-P1P2, MRE11A_−_94226994.23-P1P2, MRE11A_+_94226703.23-P1P2 (Tables S3 and S6). These sgRNAs and one non-targeting sgRNA (non-targeting_01332) were cloned into pAX198.
In Figure S6C we used a condensed Repair-seq library. In this library, individual sgRNAs were cloned into pAX198, including MRE11A_−_94227004.23-P1P2, MRE11A_−_94226994.23-P1P2, MRE11A_+_94226703.23-P1P2, POLQ_+_121264772.23-P1P2, RAD17_+_68665692.23-P1P2, non-targeting_00026, non-targeting_00566, and non-targeting_01332. The resulting plasmids were pooled in equal molar ratio for virus packaging.
For the experiment depicted in Figure S5F, individual sgRNAs were cloned into pAX198, including DNA2_−_70231468.23-P1P2 for CRISPRi and DNA2_+_70231809.23-P1P2 for CRISPRa (Gilbert et al., 2014). A non-targeting sgRNA (non-targeting_00026) was used as a negative control in both CRISPRi and CRISPRa assays.
Oligonucleotides.
Oligonucleotides (unless otherwise indicated) and TaqMan probes were obtained from Integrated DNA Technologies. Single-strand oligonucleotides for SSTR experiments (oBA701, oJAH158, oJAH159, oJAH160, and oJAH165; Table S2) were ordered with phosphorothioate bonds connecting the 3' and 5' terminal nucleotides in order to limit nuclease degradation (represented by asterisks in Table S2). One single-stranded oligonucleotide (oBA701) was PAGE-purified by IDT or as follows: 25 μL of 100 μM oBA701 ultramer (10 nmol total) were combined with 25 uL 2x Novex TBE-Urea sample buffer (LC6876) and run on a 6% TBE-Urea gel (UreaGel System, National Diagnostics, EC-833). The gel was stained by washing 10X SYBR Gold (10000x concentrate, catalog number S11494, diluted in 1X TBE) over the surface of the gel and visualized using a Safe Imager 2.0 Blue Light Transilluminator (ThermoFisher Scientific). A high intensity band corresponding to the full length ultramer was cut out of the gel. Gel slices were pulverized using Gel Breaker tubes (Ist Engineering/Fisher NC0462125), and DNA was extracted from pulverized gel by soaking in 750 uL of extraction buffer (300 mM NaCl, 10 mM Tris 8, 1 mM EDTA) overnight. Extracted DNA was isopropanol precipitated, and the resulting pellet was cleaned up by ethanol precipitation. Double-strand oligo donor (oBA701-PCR) was prepared by PCR using oBA701 as a template with oBA736 and oBA737 (Table S2). We note that these amplification primers did not have phosphorothioate bonds.
CRISPRi library design.
For our 1,573 sgRNA CRISPRi library (AX227), we curated a set of 476 genes enriched for those involved in DNA repair and associated processes (e.g., DNA replication, repair, recombination) (Figure 1C and Table S4). We then selected 1,513 sgRNAs targeting these genes from a previously published CRISPRi library, hCRISPRi-v2.1 (Horlbeck et al., 2016) (Table S3). Selected sgRNAs were those ranked as the top 3 per transcript per gene. A minority of sgRNAs were annotated as targeting multiple gene promoters. We also included 60 non-targeting control sgRNAs. These were selected from hCRISPRi-v2 randomly and filtered using data from a genome-scale growth screen in K562 cells (Horlbeck et al., 2016).
For our 366 sgRNA CRISPRi library (AC001), we selected 336 sgRNAs targeting 118 genes and 30 non-targeting controls (Table S6). To select the 118 genes targeted by this library (Figure S3A and Table S4), we considered data from Repair-seq screens performed with our 1,573 sgRNA CRISPRi library. Of the 118 genes, 114 are a subset of those targeted by our large library (AX227), while 4 were included based on interest from the literature (EXD2, HELB, RBBP6, ZBTB38). Each gene in this library was targeted with 2-6 sgRNAs (26 with 2, 86 with 3, 5 with 4, and 1 with 6). Among sgRNAs that target genes also included in AX227, we excluded 19 from AC001. A minority of sgRNAs included in AC001 have been annotated as targeting multiple gene promoters.
CRISPRi library cloning.
Oligonucleotides containing sgRNA targeting sequences were synthesized by Twist Bioscience (Q-15620=AX227; Q-28859=AC001). These sequences were cloned into pAX198 with standard protocols (details available at https://weissmanlab.ucsf.edu/CRISPR/Pooled_CRISPR_Library_Cloning.pdf). Briefly, library sequences were amplified by PCR, purified (column-based), and digested with BlpI and BstXI (ThermoFisher Scientific). Digested fragments were then isolated by gel purification, precipitated using isopropanol (AC001) or ethanol (AX227), and ligated into a similarly digested vector (BlpI and BstXI). Ligations were performed at an insert to backbone ratio of 1:1 for 16 hours at 16 °C and subsequently electroporated into MegaX DH10B T1R Electrocomp™ cells (ThermoFisher Scientific). Cells were grown in liquid culture (~1L) supplemented with carbenicillin (AC001) or on agar plates supplemented with carbenicillin and then scraped into liquid for plasmid purification (AX227). Final plasmid libraries were isolated using column-based purification.
Perturb-seq.
The relatively compact size of our 366 sgRNA CRISPRi library (AC001) allowed for characterization of transcriptional phenotypes produced by all sgRNAs in the library using direct capture Perturb-seq (Figures S3B and S3C), a method for multiplexed single-cell RNA-sequencing of CRISPR-based screens (Replogle et al., 2020). To perform this experiment, we transduced the 366 sgRNA library (AC001) into K562 CRISPRi cells (9e6 cells) with centrifugation (2 hours at 1000 x g). Cells were grown for ~6 days and selected with puromycin (3 μg/mL, starting on day 2). During selection, cells were split and media was replaced as needed. Selected cells were prepared for single-cell RNA-sequencing (scRNA-seq) as follows: Cells were centrifuged at 100 x g for 3 minutes to remove media, washed in PBS supplemented with 0.04% sterile-filtered Bovine Serum Albumin (Sigma Aldrich), centrifuged again at 100 x g for 3 minutes to remove wash, diluted with wash buffer, and then kept on ice before loading into droplets for scRNA-seq using the Chromium Single Cell 5' Library & Gel Bead Kit (10x Genomics, PN-1000006) and the Chromium Controller (10x Genomics). Altogether, we performed 8 reactions of scRNA-seq (aiming for ~14,000 cells per reaction). Cells were 88% viable and 94% BFP+ (sgRNA marker) before loading into droplets for scRNA-seq.
For scRNA-seq, we followed instructions in the Chromium Single Cell V(D)J Reagent Kits with Feature Barcode technology for Cell Surface Protein user guide (CG000186) with modifications to enable recording of sgRNAs as previously described (Replogle et al., 2020). Briefly, we added 5 pmol of a sgRNA-specific primer (oJR160, Table S2) to each reaction Master Mix prior to droplet formation. During cDNA amplification, we performed 11 cycles of PCR (according to manufacturer’s instructions) and after cDNA amplification separated the sample as follows: We performed a 0.6X left-sided cleanup reaction with SPRIselect (Beckman Coulter) and collected both the beads (which carry material for preparing the gene expression library) and the supernatant (containing sgRNA-derived cDNA amplicons). Using the supernatant, we then completed a 0.6X-1.35X double-sided cleanup. Eluate from the 0.6X cleanup was used to complete the gene expression library (according to manufacturer’s instructions), and eluate from the 0.6X-1.35X reaction was used to produce a library of sgRNA sequences containing scRNA-seq indexes.
Perturb-seq data from this experiment showed that sgRNAs in the 366 sgRNA CRISPRi library (AC001) produced a median of 86% depletion of target gene expression (Figure S3B), with 113 of 118 genes targeted by at least one sgRNA achieving >70% knockdown (Figure S3C). Additionally, using the Perturb-seq data, we quantified the effect of each sgRNA on relative occupancy of each cell cycle phase (Figures 5E and 5F).
Ribonucleoprotein (RNP) complexes.
Streptococcus pyogenes Cas9 and Acidaminococcus sp. BV3L6 (A.s.) Cas12a with an RNase inactivating mutation (H800A) were complexed with guide RNAs obtained from Integrated DNA Technologies (Cas9, Alt-R® CRISPR-Cas9 crRNA and Alt-R® CRISPR-Cas9 tracrRNA; Cas12a, Alt-R® CRISPR-Cpf1 crRNA). Enzymes were obtained from Integrated DNA Technologies (Alt-R® spCas9 Nuclease V3, 1081059) or from Editas Medicine (made in-house or ordered from supervised vendor, Aldevron; sequences included in Table S1). RNPs obtained from Editas Medicine were complexed at a ratio of 2:1 (RNA:protein) as follows: Cas9 complexes were prepared to 50 μM by diluting Cas9 to 100 μM in complete 1X HG300 buffer (50 mM N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES), 300 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 20% glycerol (% v/v), pH 7.5) and mixing 1:1 with guide RNAs diluted to 200 μM in 1X H150 buffer (10 mM N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES), 150 mM NaCl, pH 7.5). Components were allowed to complex for at least 45 minutes at room temperature and a differential scanning fluorimetry assay (Maeder et al., 2019) was performed to confirm complexation occurred. Cas12 complexes were prepared to 66 μM by resuspending guide RNAs in H150:HG300 buffer to 200 μM and mixing with Cas12a (400 μM) and 1X H150 at a ratio of 4:1:1 (RNA:protein:buffer) by volume. Components were allowed to complex for at least 45 minutes at room temperature and a differential scanning fluorimetry assay was performed to confirm complexation occurred. Other RNPs were complexed as follows: crRNA and tracrRNA oligos were mixed in equimolar concentrations (1:1) to a final duplex concentration of 100 μM (16 μl total), incubated at 95 °C for 5 minutes, and allowed to cool to room temperature (15-25 °C) for ~45 minutes. Duplexed guide RNAs were mixed with Cas9 enzyme and PBS (Thermo Fisher Scientific) to a final 12.2 μM RNP concentration and incubated at room temperature for ~1 hr.
Electroporation.
For experiments depicted in Figures 4G, 6D, 6I, S1B-S1E, S2C, S2E, S5A, S5F, and S6C, indicated cells were electroporated with RNPs as follows: Cells (1.25-6e5 or 2e6-3e6 per sample) were spun out of media by centrifugation (1000-1500 rpm for 5 minutes) and washed once with PBS (Thermo Fisher Scientific) or directly resuspended in electroporation buffer. Using an SE Cell Line 4D X Kit S (Lonza Bioscience), cells were electroporated on a 4D-Nucleofector (Lonza Bioscience) according to manufacturer’s instructions (CN-114 reagent code for HeLa cells, FF120 reagent code for K562 cells, and EA-104 reagent code for RPE1 cells) using 50 pmol of Cas9 RNP. Details of electroporation during screens are described below (see Repair-seq Screens—cell culture).
Virus preparation.
Lentivirus was produced in 293T cells by co-transfection of transfer plasmids (single or library) and packaging plasmids for expression of HIV-1 gag/pol and rev (+/−tat) and VSV-G envelope protein using either TransIT®-LT1 Transfection Reagent (Mirus) or Polyethylenimine (Polysciences, Inc.) with or without ViralBoost Reagent (Alstem, Inc.). For screening, virus-containing supernatant was either used directly, or filtered using either syringe filters (0.45 μm) or a vacuum filtration system (0.20 μm). Viral titers were determined by test transductions prior to screening (except for screen 1). For individual experiments, virus-containing supernatants were frozen prior to use.
Repair-seq—cell culture.
K562 CRISPRi cells were transduced with an sgRNA library (AX227 or AC001) in large-scale infections (140e6 or 640e6 cells for AX227; 300e6 or 406e6 cells for AC001) (Table S5). Infections were supplemented with 8 μg/mL polybrene and conducted in many wells of multiple 6 well plates with centrifugation (~2-3 hours at 1000 x g). After centrifugation, cells were pooled and resuspended in fresh, complete RPMI to ~0.5e6 cells per mL. Screens 1-15 were spun out of virus-containing supernatant prior to resuspension. Cells were then grown with or without agitation for 6 days and selected with 1-3 μg / mL puromycin (added 2-3 days post transduction). For each screen, cells were less than 30% BFP+ (sgRNA library marker) 2 days post transduction (Table S5). To remove dead cells, cultures were periodically spun out of media +/− PBS wash.
Screens were performed in the following groups: 1, 2-6, 7, 8-15, 16-21 (Table S5). Screens that were performed concurrently were derived from a single transduction culture and split prior to RNP electroporation. Cells were electroporated with RNPs, with or without oligonucleotides, as follows: First, cells were spun out of media by centrifugation and washed with PBS. Next, using an SE Cell Line 4D X Kit L (Lonza Bioscience), cells were electroporated on a 4D-Nucleofector (Lonza Bioscience) with the FF120 reagent code, according to manufacturer’s instructions, with the following exceptions: (1) Post wash, cell pellets were mixed with Buffer SE (100 μL per reaction) and RNP (13 μL of 66 μM Cas12a or 5 μL of 50 μM Cas9 per reaction) with or without oligonucleotides (2.5 μL of 100 μM ssODN or 5.3 μL of 4.7 μM dsODN per reaction). (2) These mixtures were then loaded into cuvettes, typically using as many cuvettes as necessary to process the entire volume of cell mixture (~100 μl per cuvette; numbers of reactions and cuvettes per screen are indicated in Table S5). We note that practical constraints on nucleofection limit how many sgRNAs can be screened at any given level of per-sgRNA coverage. Prior to performing Repair-seq experiments, we recommend that nucleofection procedures be piloted to ensure good cell recovery.
All RNPs used in screening were prepared and provided by Editas Medicine (see Ribonucleoprotein (RNP) complexes). Following nucleofection, cells were allowed to recover for ~10 minutes at 37 °C (except for screen 9). Cells were then resuspended with media and transferred for growth. For screens 1 and 8-15 cells were spun out of media and washed with PBS two days post electroporation to help remove dead cells. After 3 days, cells were collected and cell pellets were processed to generate sequencing libraries (numbers of cells collected per screen, viability of cells at collection, and cell population doublings between nucleofection and collection are included in Table S5). Throughout screening, cells were counted and viabilities were determined using an Accuri™ C6 flow cytometer (BD Biosciences) or Countess II Automated Cell Counter (Thermofisher Scientific). Cells were typically maintained at densities between ~0.5e6 and ~le6 cells per mL (splitting as necessary).
Repair-seq—sequencing library preparation.
Sequencing libraries were prepared from the cells collected at the end of the screens as follows: Genomic DNA was extracted using the NucleoSpin Blood XL, Maxi kit for DNA from blood (Macherey-Nagel) with a condition of 50e6-100e6 cells per column. Genomic DNA was then digested with NotI-HF (NEB) and run on large 0.8% agarose gels (Owl™ A1 Large Gel System, Thermo Fisher Scientific) prepared with custom 3D printed combs capable of producing wells large enough for loading 1.5 mL volume per well (for more information on these gels, see https://weissmanlab.ucsf.edu/CRISPR/IlluminaSequencingSamplePrep_old.pdf) with individual samples typically spread across multiple wells. Fragments of genomic DNA within the size range of those containing both edited sequences and sgRNA expression cassettes (target fragments) were then excised with wide boundaries, purified using NucleoSpin® Gel and PCR Clean-up kit (Macherey-Nagel), digested with HindIII-HF (NEB) to remove a NotI overhang from one end of the target fragments, and isolated using SPRIselect Reagent (Beckman Coulter) in a 0.8X reaction. We note that fragment recovery throughout our library preparation protocol is lossy and thus limits how many sgRNAs can be screened at any given level of per-sgRNA coverage (see Sequencing section for estimate of UMIs recovered per cell). When piloting Repair-seq experiments, we recommend that efficiency of recovery (at least through the gel electrophoresis step) be empirically determined before scaling to a full screen.
Custom adapters containing 12 nt unique molecular identifiers (UMIs) were then ligated onto target fragments using the remaining 4 nt NotI overhang. These adapters (oBA676 and oBA677; Table S2) were obtained as individual DNA oligonucleotides (HPLC purified) from Integrated DNA Technologies and then annealed. For the annealing reaction, an equimolar amount of each oligo was mixed and were run on a C1000 touch thermal cycler with the following program: 1 cycle of 5 minutes at 95 °C; 70 cycles of 1 minute each with gradual cooling from 95 °C to 25 °C; 1°C hold. For the ligation reactions, we used enzyme and buffer from the KAPA HyperPrep Kit (PCR-free) (Roche). Ligation reactions were assembled as follows: 30 μL ligation buffer, 10 μL ligase, adapter at 200:1 adaptor:insert ratio, 1 μg of HindIII digested product, and PCR-grade water to 110 μL total volume. These reactions were incubated at 4 °C overnight on a thermocycler with lid temperature set to 30 °C. Following ligation, DNA was purified using SPRIselect Reagent (Beckman Coulter) in two steps: First, we performed a 0.65X reaction as calculated; however, as the ligation buffer contains PEG, the reaction ratio was effectively higher. Then, we performed a 0.8X reaction.
To enrich our target fragments for sequencing, we next amplified those sequences by PCR, performing enough PCR reactions to use nearly the entirety of each sample obtained from the ligation and subsequent clean-up reactions (number of PCR reactions per screen indicated in Table S5). We assembled PCR reactions with 30-50 ng of template into 50 μL total volume. These reactions contained amplification primers at 0.6 μM final concentration (each), 3% dimethyl sulfoxide, and 1X KAPA HiFi HotStart ReadyMix PCR Kit (Roche) and were run on a thermocycler with the following program: 1 cycle of 3 minutes at 95 °C; 16 or 20 cycles of 15 seconds at 98 °C, followed by 15 seconds at 70 °C; 1 cycle of 1 minute at 72 °C; 4 °C hold. Amplification primers used were against the Illumina P7 sequence (included in the adapter) and an indexed version of the Screenindexing primer (Table S2). Amplified DNA was purified for sequencing using SPRIselect Reagent (Beckman Coulter) in a 0.8X reaction, and index samples were mixed. Throughout the sample preparation, samples were checked for quality and yield using either a NanoDrop Spectrophotometers (Thermo Fisher Scientific), Agilent 2100 Bioanalyzer system, or by running on a Novex™ TBE Gel.
Repair-seq—control arms.
Alongside screens 16-21, we performed control arms wherein cells were electroporated without RNP. Cell culture and library preparation were performed as described above. Counts of UMIs recovered for each CRISPRi sgRNA in these controls are provided in Table S7.. Comparing sgRNA counts between these controls and screens performed with RNP identifies genes with multiple sgRNAs whose relative abundance is increased or decreased in the presence of induced on-target DSBs. Further interpretation of these signals however is limited by the fact that such phenotypes represent an unknown mixture of contributions from many possible mechanisms. Changes in sgRNA abundance may reflect indirect effects of gene deficiency on the cytotoxicity of DSBs, on the kinetics of DSB repair or cell doublings after damage, or on the formation of repair outcomes that cannot be sequenced by short-read amplicon sequencing (e.g., long deletions or more extreme on-target rearrangements).
Arrayed DSB repair experiments.
For arrayed experiments depicted in Figures 4G, 6D, S1B-S1E, S5A, and S5F, cells were electroporated with indicated RNPs (as described above). Three days post electroporation, cells were collected and cell pellets were processed to generate sequencing libraries as follows. Genomic DNA was extracted using the NucleoSpin® Blood, Mini kit for DNA from blood (Macherey-Nagel). The targeted loci were amplified with primers (oJAH262 and oJAH263 for HEK3_3a target site, oJAH264 and oJAH265 for RNF2 target site, oJAH258 and oJAH259 for Cas9 target sites 1, 3, and 4 at the endogenous locus, oBA991 and oBA992 for Cas9 target sites 1, 3, and 4 within a genomically integrated screening construct; Table S2) using NEBNext Ultra II Q5 Master Mix (New England Biolabs). Each reaction (100 μL total volume) contained 1 μg of genomic DNA and 1 μM of each primer and was run on a thermocycler with the following program: 1 cycle of 30 seconds at 98 °C; 22 cycles of 10 seconds at 98 °C, followed by 75 seconds at 65 °C; 1 cycle of 5 minute at 65 °C; 4 °C hold. The resulting products were purified using SPRIselect Reagent (Beckman Coulter) in 1.2X to 1.8X reactions and used as templates for a second PCR to add indexes to the amplified fragments. The indexing primers used were versions of Indexing_REV and Indexing_FOR in Table S2. Each of these reactions (50 μL total volume) contained 6 ng or 10 ng of template DNA and 0.6 μM of each primer with 1x KAPA HiFi HotStart ReadyMix PCR Kit (Roche) and was run on a thermocycler with the following program: 1 cycle of 3 minutes at 95 °C or 98 °C; 8 cycles of 20 seconds at 98 °C, followed by 15 seconds at 65 °C and 15 seconds at 72 °C; 1 cycle of 1 minute at 72 °C; 4 °C hold. Amplified DNA was purified for sequencing using SPRIselect Reagent (Beckman Coulter) in 1.2X to 1.8X reactions, and index samples were mixed prior to sequencing.
For experiment depicted in Figures S1B-S1E, K562 cells transduced with pAX198 were electroporated with RNPs against target sites 1, 3, and 4 (Table S1) (as described above). For experiments depicted in Figures 4G, S5A, and 6D, K562 CRISPRi cells or HeLa CRISPRi cells were transduced with indicated sgRNAs, selected with puromycin (1-3 μg/mL) prior to electroporation, and electroporated on day 6 or 7 post transduction. For experiments depicted in Figure 6D, right after electroporation, each sample was divided into two portions, CRISPRi data are from cells with indicated sgRNAs treated with DMSO (normalized to non-targeting sgRNA sample treated with DMSO) and MRE11 chemical inhibitor data are from cells carrying non-targeting sgRNA treated with 50 μM Mirin (normalized to non-targeting sgRNA sample treated with DMSO). For experiments depicted in Figure S5F, K562 CRISPRi cells or K562 CRISPRa cells were transduced with indicated sgRNAs, selected with puromycin (3 μg/mL) and pooled for electroporation on day 5 post transduction. CRISPRi cells with DNA2-targeting sgRNA and non-targeting sgRNA were pooled in a 2:1 ratio to account for the depletion effect caused by DNA2 knockdown. CRISPRa cells with DNA2-targeting sgRNA and non-targeting sgRNA were pooled in a 1:1 ratio.
MRE11 rescue experiment.
For the experiment depicted in Figure 6I, constructs expressing GFP, MRE11, and a nuclease-inactive mutant (MRE11-H129N) were transduced into K562 CRISPRi cells alongside one of three MRE11-targeting sgRNAs or one negative control (see Plasmid construction). Cells infected with the same expression construct (different sgRNAs) were pooled together and, after 3 days, cells expressing GFP (expression construct marker) and BFP (sgRNA marker) were isolated by fluorescence-activated cell sorting on a FACSAria Fusion (BD Biosciences). On day 6, cells were electroporated with RNP (Cas9 against target site 1) as described above. After 3 days, cells were collected and cell pellets were processed to generate sequencing libraries as described above for arrayed experiments, except that 1.8 μg genomic DNA was used for the first PCR amplification. Notably, all of the MRE11-targeting sgRNAs used in this experiment were designed to target endogenous MRE11 and are not expected to deplete exogenous MRE11 expressed from the EF1A promoter.
Condensed Repair-seq library Experiment.
For experiment depicted in Figure S6C, K562 CRISPRi cells, HeLa CRISPRi, or RPE1 CRISPRi cells were transduced with small pools of sgRNAs (see Plasmid construction) and selected with puromycin (3 μg/mL for K562, 1 μg/mL for HeLa, and 10 μg/mL for RPE1, starting on day 2). Cells were no more than 10% BFP+ (sgRNA library marker) without selection three days post transduction. On day 7 post transduction, cells were electroporated with indicated RNPs as described above. Three days post electroporation, cells were collected and cell pellets were processed to generate sequencing libraries as described above for the MRE11 rescue experiment. As this was initially a control arm for a treated experiment, cells were grown in DMSO-containing medium.
Sequencing.
Sequencing libraries from Repair-seq screens were sequenced on an Illumina NovaSeq 6000 System with 4 total reads per cluster (paired end reads plus two additional indexing reads) with the following read lengths: I1 = 12 nts (UMI sequence); I2 = 8 nts (sample index); R1 = 45 nts (CRISPRi sgRNA); R2 = 258 nts (editing outcome) (Figure S1F). Reads were demultiplexed based on sample index and CRISPRi sgRNA, allowing up to one mismatch between observed sequences and expected sample indices or spacer sequences. Reads reporting a CRISRPi spacer sequence of GACCAGGATGGGCACCACCC represented delivery of the sgRNA present in pAX198 before sgRNA library cloning; these reads were disregarded. Across screens, we obtained ~0.03 UMIs per cell collected at the end of the screen. Number of sequencing reads, total UMIs, and median UMIs per sgRNA obtained from each screen are indicated in Table S5. Sequencing for Perturb-seq experiment was performed on an Illumina NovaSeq 6000 system with paired end reads (I1 = 8nts; R1 = 28 nts; R2 = 98 nts). Sequencing of arrayed experiments, MRE11 rescue, and experiment depicted in Figure S6C were performed on an Illumina MiSeq System with single end reads (I1 = 8 nts; I2 = 8 nts; R1 = 300 nts) or paired end reads (I1 = 8 nts; I2 = 8 nts; R1 = 44 nts; R2 = 240 nts or 256nts). Sequencing of experiment depicted in Figure S5F was performed on an Illumina MiSeq System with paired end reads (I1 = 8 nts; I2 = 8 nts; R1 = 44 nts; R2 = 350nts).
Measurement of large deletions.
Some DSB repair products can remain “unseen” when analyzed by short-read sequencing. We observed that, after RNP electroporation, a fraction of electroporated cells lost expression of a BFP marker encoded on our screening construct (~2000 nt away from the target region) (Figure S2A). Further investigation of this phenomenon by droplet digital PCR (ddPCR) analysis showed that BFP− populations isolated by fluorescence-activated cell sorting (FACS) were enriched for cells lacking sequence within a 4069 nt window around the targeted DSB, as compared to BFP+ populations isolated from the same electroporation reaction (Figures S2B and S2C). This observation is consistent with reports of large deletions induced by endonuclease cutting (Kosicki et al., 2018); however, we also observed that some of these repair products represented repair junctions wherein the flanking lentiviral long terminal repeats (LTRs) collapsed, indicative of repair through single-strand annealing (SSA) (Figures S2D and S2E). Given the relative positions of sequences important for Repair-seq sequencing library preparation (Figure S2B), these events would not be detected.
For the experiments depicted in Figures S2B-S2E, we constructed 3 cell lines. First, we transduced K562 CRISPRi cells with pAX198 and selected them with puromycin. Cells expressing BFP were then isolated by FACS (FACSAria Fusion, BD Biosciences) to establish (1) a BFP+ cell pool and (2) two clonal BFP+ lines. To ensure homogeneous expression of BFP, both clonal lines were checked by flow cytometry (LSR II Flow Cytometer, BD Biosciences), and we verified single-copy lentivirus integration for each by ddPCR (QX200 AutoDG Droplet Digital PCR system, Bio-Rad). For clonal line 2, we determined the lentiviral integration site using a protocol adapted from Lenti-X Integration Site Analysis Kit (Takara Bio) and oligonucleotides oJY0104-oJY0109. Briefly, genomic DNA (gDNA) was extracted from clonal lines with single lentiviral vector integration and 2.5 μg of gDNA was digested with 80 U HpaI (New England Biolabs) for 18 hr at 37°C. Digestion product (120 ng) was then ligated with 48 pmol annealed genome walker adaptor (oJY0104 and oJY0105) and 100 U T4 ligase (New England Biolabs) in an 8 μL reaction at 16°C overnight. After T4 ligase was heat inactivated (65°C for 10 min) and diluted with 32 μL IDTE pH 7.5 (Integrated DNA Technologies), 1 μL of the product was PCR amplified with OJY0106 and OJY0108. Product from this reaction was then diluted and 1 μL of the diluted product was used for a second round of PCR using oJY0107 and oJY0109. The nested PCR reaction was checked by 1% SYBR safe agarose gel and the major band was excised and extracted for Sanger sequencing with oJY097 and oJY0109.
ddPCR experiments were then performed as follows: First, our established BFP+ cell pool and/or BFP+ clonal line(s) were electroporated with Cas9 RNP against target site 1 (Table S1) or Cas9 programmed with a negative control gRNA (gRNA-neg in Table S1) as described above. Four days post nucleofection, BFP+ and BFP− cell populations were separated by FACS (FACSAria Fusion, BD Biosciences). Genomic DNA was then extracted from each population using the NucleoSpin Blood, Mini kit for DNA from blood (Macherey-Nagel) according to manufacturer’s instructions. ddPCR was performed using this DNA, a forward primer, a reverse primer, and a TaqMan probe for each amplicon indicated in Figures S2B and S2D, as well as an ACTB control amplicon (oBA074, oBA075, oJY012, oJY0117, oJY033, oJY0034, oJY0037-39, oJY0089, oJY0090, and oJY040; Table S2). Amplicons used to quantify large deletions surrounding the edit site were 298 nt upstream, 126 nt downstream, and 3453 nt downstream of the DSB (designed using Primer3Plus; Figure S2B). For our SSA assay, we designed two amplicons specific to clonal line 2, using the sequence of the lentiviral integration site: (1) the “LTR amplicon” (designed using Primer3Plus), which was amplified by primers that bound immediately upstream and downstream of the 5′ LTR (oJY0110, oJY0115; Table S2), and (2) the “SSA amplicon”, which was amplified by oJY0110 and a primer that binds immediately downstream of the 3′ LTR (oJY0113; Table S2). A single TaqMan probe (oJY0099; Table S2) was used for both amplicons. For each amplicon, raw concentration measurements were normalized to the ACTB control amplicon from the same sample. Comparison of the normalized concentration of the “SSA amplicon” to that of the “LTR amplicon” in cells electroporated with Cas9 and gRNA-neg (Table S1) allowed quantification of the former. ddPCR reactions were performed using ddPCR Supermix for Probes (no dUTP) (Bio-Rad) following manufacturer’s instructions with a QX200 AutoDG Droplet Digital PCR system (Bio-Rad).
Flow cytometry.
Flow cytometry data was collected with an LSR II Flow Cytometer (BD Biosicences) and FACSDiva™ Software (BD Biosicences). Data in Figure S2A was processed with FCS Express 7 Research software (Version 7.04.0014).
QUANTIFICATION AND STATISTICAL ANALYSIS
UMI consensus calling.
Within each sample index and CRISPRi sgRNA, reads are grouped by UMI sequence. Distinct reads with the same sample index, CRISPRi sgRNA, and UMI may represent amplification products from the same initial UMI-ligated molecule (possibly modified by point mutations or short indels from PCR or sequencing errors) but may also represent chimeric products generated by recombination during PCR or by index switching during cluster generation on Illumina platforms that use exclusion amplification for cluster generation (Sinha et al., 2017). The goals of processing each group of reads with the same UMI are to cluster together reads likely to represent the same repair event and use the redundant information present across such reads to correct sequencing or PCR errors by identifying a consensus sequence, and to filter out reads likely to represent recombination/index switching artifacts.
To accomplish these goals, a greedy clustering algorithm consisting of the following iterative process is applied to each UMI read group. First, the most common sequence in the group is identified. Then, all reads for which no base calls with quality score greater than 20 disagreed with the identified common sequence are removed from the read group to form a cluster. A consensus sequence of the cluster is called by identifying the most frequent base call across all reads at each position. If more than one base call is equally frequent, the consensus base is set to ‘N’ and consensus quality score is set to 2. Positions at which a single base call is present in more than 50% of reads for which at least one such base call had a quality score greater than or equal to 30 are assigned a consensus quality score of 31. All other positions are assigned a consensus quality score of 10. This consensus formation process is applied to both the repair outcome sequence and the CRISPRi sgRNA sequence. This process is iteratively repeated on remaining reads in the UMI read group until all reads have been assigned to clusters. Any cluster containing fewer than 4 reads is discarded. Of the remaining clusters, the cluster containing the highest number of reads is retained and all others are discarded. Finally, clusters for which the consensus CRISPRi sgRNA sequence did not perfectly match the designed protospacer may represent sgRNAs that experienced an error during synthesis, cloning, or lentiviral packaging. Since such errors may result in weaker knockdown of the targeted gene, any such clusters are discarded. All remaining clusters are advanced to outcome categorization.
Categorization of repair outcome sequences.
Consensus sequences of editing outcomes are classified using a modified version of knock-knock. Conceptually, the process of classifying a given sequence outcome consists of first generating a comprehensive set of local alignments between the outcome sequence and all relevant references sequences: the lentiviral screen vector, any donor sequences supplied in the screen, the human (hg19) genome, and the cow (bosTau7) genome. Alignments to the screen vector and donor sequences are generated using BLASTN version 2.10.1+ and using a custom Smith-Waterman implementation. Alignments to genomes are generated using STAR version 2.6.0a. All local alignments for a given read are then jointly processed to identify the arrangement of segments of the screen vector and any other relevant sequence sources that make up the outcome,identify any mismatches, insertion, deletions, or other rearrangements of these sequences, and to assign a classification to the type of editing outcome represented by the read.
A summary of the portions of the decision tree relevant to results presented in the manuscript is presented below. The categorization process assigns a “category”, “subcategory”, and “details” to each outcome. The process first attempts to identify a single alignment to the screen vector, potentially containing a single deletion or short insertion but not containing any mismatches, that begins by pairing the beginning of the read with the expected sequencing primer and that extends to the end of the sequencing read. If such an alignment exists with no insertion or deletion, the outcome is classified as category “wild type”. If the alignment contains an insertion, the outcome is classified as category “insertion” and subcategory “insertion”, with the inserted sequence and the position in the target after which the insertion begins recorded as details. If the alignment contains a deletion, the outcome is classified as category “deletion” and subcategory “near cut” if the deleted region overlaps a window of 5 nts around the expected cut site and subcategory “far from cut” if it does not, with the length of the deletion and the position in the target at which it begins recorded as details. The exact insertion or deletion assigned by the alignment process may represent an arbitrary representative from a class of equivalent insertions or deletions that would have produced indistinguishable resulting sequences. To account for this, the screen vector sequence is preprocessed to identify classes of degenerate insertions (that is, distinct combinations of inserted sequence plus insertion location that result in identical post-insertion sequences) and degenerate deletions (distinct starting locations of a given deletion length that result in identical post-deletion sequences). A lookup table from each member of a degenerate class to the representation of the full class is populated, and the observed insertion or deletion is looked up in this table to possibly replace its details with the details of the degenerate class of which it is a member.
If a single read-spanning alignment contains mismatches relative to the screen vector, these mismatches are compared to the expected SNVs programmed by a donor sequence. If all mismatches correspond to donor SNVs, the alignment contains no insertions, and the alignment contains no deletions longer than 1 nt long, the outcome is assigned category “HDR”. If there are no deletions, the outcome is assigned subcategory “clean”. Empirically, in reads that contain donor SNVs, we frequently observe 1 nt deletions throughout the region corresponding to the donor sequence. Deletion of 1 nt is a common error mode of oligonucleotide synthesis, so we conservatively attribute such deletions as likely existing in the starting oligonucleotide donors rather than being introduced by an error-prone repair process. If there are 1 or more 1 nt long deletions in the alignment, the outcome is therefore assigned subcategory “synthesis errors”. These outcomes correspond to the scarless HDR category in Figures 7 and S7.
If no single alignment to the target covering the whole read is identified, the longest alignments to the target that reach each edge of the read are identified. Note that while the start of the read should always be covered by such an alignment because of the amplification strategy, the end of the read may not be covered if a sufficiently long sequence fragment has been captured at the DSB. The gap between these target edge alignments on the read is determined, and the remaining local alignments are searched to attempt to explain the gap. If no gap-covering alignments exist, a more permissive application of Smith-Waterman is applied to specifically search for additional local alignments between the gap region and the screen vector or the sequence of any donor used.
If the gap can be covered by an alignment to donor sequence, the outcome is assigned category “donor misintegration”. Donor misintegrations are sub-categorized based on how the donor sequence lines up with the target edge alignments on each side of the cut site. The two sides of the cut site relative to the read (left and right) are separately classified. For each side, if the homology arm in the donor alignment and in the target edge alignment are aligned to the same portion of the read, that side is classified as “intended”. If this is not true and if the donor alignment on a side extends all the way to an edge of the donor, the side is classified as “blunt”. If neither of these conditions is true, the side is classified as “unintended”. An additional wrinkle exists on the right side of the read, where the sequencing read may not be long enough to reach the end of the donor alignment and therefore may not be able to resolve how the outcome transitioned from donor sequence back to target sequence on the right side. If this is the case, the right side is classified as “ambiguous”. The full subcategory assigned to the outcome is the pair of classifications assigned to its two break sides. Half-HDR outcomes analyzed in Figures 7 and S7 consist of outcomes classified as “intended” on the side corresponding to the 3′ end of the donor and “unintended” on the 5′ end of the donor. Capture of donor fragment outcomes consist of outcomes classified as “unintended” on both sides.
If the gap can be covered by a single alignment to the human genome or to the cow genome, the outcome is assigned category “genomic insertion”, with the subcategory indicating which organism the gap covering alignment was from.
If none of the above conditions are satisfied but the outcome architecture contains two target edge alignments with an unexplained gap in between, the outcome is classified as “insertion” if there is no missing reference sequence on the target between the target edge alignments, and as “insertion with deletion” if there is.
If none of these conditions are satisfied, the outcome is assigned to category “uncategorized”.
Finally, because many of the individual repair outcome sequences seen in a screen will occur many times each, performing the categorization process described above on each outcome sequence would result in substantial computational redundancy. To improve performance, data from a screen is first preprocessed to identify any sequences that occur more than once across all UMIs for all CRISPRi sgRNAs. The categorization process described above is applied to each common sequence to populate a lookup table of common sequence to outcome. Then, all sequences for each UMIs for each CRISPRi sgRNA are categorized by first checking this lookup table for each sequence and only carrying out the categorization process on sequences not present in the table.
Following categorization of all reads from a screen, the number of occurrences of each combination of outcome category, subcategory, and details is counted for each CRISPRi sgRNA. Outcome counts for the entire screen are then collected into a matrix with rows for each outcome (category, subcategory, and details) combination and columns for each CRISPRi perturbation.
Analysis of Repair-seq reproducibility.
For the purposes of all analyses that involve comparisons between screen replicates, replicates consisted of independent nucleofections of RNPs that were subsequently independently cultured and processed for sequencing. As noted above, when replicates were performed concurrently, they were derived from the same CRISPRi sgRNA library lentivirus transduction culture, split prior to nucleofection. Note that some such comparisons are between a screen performed with the 1,573 CRISPRi sgRNA library and a screen performed with the 366 sgRNA sub-library but with otherwise matched conditions. In these cases, analyses were restricted to sgRNAs present in both libraries.
Ranking sgRNAs by outcome redistribution.
To rank CRISPRi sgRNAs in a screen by overall outcome redistribution activity, a chi-squared-like statistic is calculated to quantify the statistical strength of the extent to which editing outcomes are redistributed by each sgRNA relative to all non-targeting sgRNAs. Given a targeting sgRNA t and a set of n relevant outcomes, let nt, i be the number of UMIs for outcome i in sgRNA t and nnt, i by the number of UMIs for outcome i collectively across all non-targeting sgRNAs. Let nt, total = nt,i and nnt, total = nnt,i Then the expected number of counts for outcome i in sgRNA t if sgRNA t has no effect is et, i = nnt, i / nnt, total * nt, total. The chi-squared statistic for sgRNA t is then (nt,i – et,i)2 / et,i
Clustering.
In hierarchical clustering of data from a single screen or composite data from multiple screens, sgRNAs were ranked by total outcome redistribution activity across all outcomes above indicated baseline frequency thresholds as described above, and the indicated number of most active sgRNAs were retained. Clustering of outcomes was performed using the scipy.cluster.hierarchy.linkage function from scipy 1.5.4 using the ‘correlation’ metric and single linkage with the optimal_ordering argument set to True. Clustering of sgRNAs was performed using HDBSCAN version 0.8.26 using pre-computed cosine distances.
Dimensionality reduction.
Dimensionality reduction of composite Cas9 repair maps was performed using umap-learn version 0.4.6 (McInnes et al., 2018).
Perturb-seq analysis.
Data from the Perturb-seq experiment was analyzed using custom Python scripts built on SCANPY (Wolf et al., 2018). Assignments of CRISPRi sgRNAs to cells was performed using the mixture model approach described in (Replogle et al., 2020). Assignment of cells to cell cycle stages was performed as described in the SCANPY documentation at https://nbviewer.jupyter.org/github/theislab/scanpy_usage/blob/master/180209_cell_cycle/cell_cycle.ipynb. Briefly, each cell is scored for expression of a set of S-phase marker genes and a set of G2/M-phase marker genes and assigned to S or G2/M if either of these sets are highly expressed, or to G1 if neither are. Marker gene sets for cell cycle stages were taken from https://github.com/hbc/tinyatlas/blob/master/cell_cycle/Homo_sapiens.csv. which is derived from gene lists originally describe in (Tirosh et al., 2016).
ADDITIONAL RESOURCES
Repair-seq data can be interactively explored at https://seq.repair.
Supplementary Material
Figure S1. Design and validation of the Repair-seq screening vector and sequencing strategy, Related to Figure 1.
(A) Schematic of Repair-seq screening vector. LTR indicates long terminal repeat. Elements along top line are not represented at scale.
(B) Plots depict the frequencies of individual DSB repair outcome sequences generated at indicated target sites (top, Cas9 target site 1; middle, Cas9 target site 3; bottom, Cas9 target site 4) compared between replicates. Outcome distributions were collected within the context of the genomically integrated Repair-seq construct from K562 cells after electroporation with corresponding RNPs. In each panel, each dot represents a single sequence outcome, with dots colored according to sequence architecture category. Locations of relevant target sites are indicated in panel A.
(C) Plots depict the frequencies of individual DSB repair outcome sequences compared at 2 different Cas9 target sites that are offset from each other by 1 nt. Outcome distributions were collected within the context of the genomically integrated Repair-seq construct from K562 cells after electroporation with corresponding RNPs.
(D) Plots depict the frequencies of individual DSB repair outcomes at indicated target sites (top, Cas9 target site 1; middle, Cas9 target site 3; bottom, Cas9 target site 4) compared between replicates. Outcome distributions were collected from the endogenous HBB locus in K562 cells after electroporation with corresponding RNPs. In each panel, each dot represents a single sequence outcome, with dots colored according to sequence architecture category. Locations of relevant target sites are indicated in panel A.
(E) Plots depict the frequencies of individual DSB repair outcomes at indicated target sites (top, Cas9 target site 1; middle, Cas9 target site 3; bottom, Cas9 target site 4) compared between those generated within the genomically integrated Repair-seq construct (x-axis, mean of 3 replicates) and the endogenous HBB locus (y-axis, mean of 3 replicates). Outcome distributions were collected from K562 cells after electroporation with corresponding RNPs.
(F) Schematic of Repair-seq sequencing strategy. Sequencing reads are indicated in red. Read 1 (R1) records the CRISPRi sgRNA sequence, Read 2 (R2) records the DSB repair outcome, one short index read records a unique molecular identifier (UMI) sequence, and another short index read records sample indices (i5) that allow multiplexing of libraries from multiple screens. Elements not represented at scale.
Figure S2. Characterization of observed and unobserved DSB repair products, Related to Figure 1 and STAR Methods.
(A) Plots show the distributions of BFP fluorescence in K562 CRISPRi cells after transduction with Repair-seq screening constructs and electroporation with Cas9 RNP against target site 1 (measured by flow cytometry). Data were collected from uncut control cells and cells used for screen number 14 (Table S5) after 3 (top) and 10 days (bottom) of recovery from electroporation.
(B) Schematic depicts the locations of sequences required for Repair-seq library preparation (PCR primer annealing site, NotI site) as well as amplicons used for droplet digital PCR (ddPCR) detection of large deletions generated within the Repair-seq screening construct (amplicons 1, 2, and 3). Indicated distances are relative to the “Cas9 target 1” shown in Figure S1A. Elements are not represented at scale.
(C) Data represent the percentages of K562 CRISPRi cells with intact amplicons (as detected by ddPCR using amplicons depicted in Figure S2B) in BFP+ and BFP− populations isolated by fluorescence activated cell sorting 4 days after electroporation of a Cas9 RNP targeting HBB site 1 (left, amplicon 1; middle, amplicon 2; right, amplicon 3). Cells were transduced with the Repair-seq screening construct prior to electroporation and were analyzed as a pool or isolated clones. Depicted results were calculated by comparing the normalized amplicon concentration from indicated sample to that of cells electroporated with a non-targeting RNP (Table S1).
(D) Schematic depicts the relative locations of the “LTR amplicon” and the “SSA amplicon”. To amplify the former, primers were designed that bind immediately upstream and downstream of the 5′ LTR in a single clonal cell line (2). To amplify the latter, primers were designed that bind immediately upstream of the 5′ LTR and downstream of the 3′ LTR. The “SSA amplicon” allows detection of an inferred single-strand annealing (SSA) repair event. Indicated distances are relative to the “Cas9 target 1” shown in Figure S1A. Elements are not represented at scale.
(E) Data represent the percentages of K562 CRISPRi cells (clonal cell line 2) with the “SSA amplicon” (as detected by ddPCR using amplicons depicted in Figure S2D) in BFP+ and BFP− populations isolated by fluorescence activated cell sorting 4 days after electroporation of indicated RNPs. Measurements were normalized to an ACTB control amplicon from each sample and then compared to the normalized concentration of the “LTR amplicon” in cells electroporated with a non-targeting RNP (Table S1).
(F) Data from initial Repair-seq screen highlighting the phenotypes of XRCC5 (encoding Ku80) and XRCC6 (encoding Ku70), including diagrams of the 30 most frequent indels observed in cells receiving non-targeting CRISPRi sgRNAs (left), the frequency of each outcome in the presence of individual non-targeting sgRNAs (middle, 60 grey lines) or sgRNAs targeting XRCC5 (middle, 3 green lines), and heatmaps of log2 fold changes in the frequency of each outcome caused by XRCC5- and XRCC6-targeting sgRNAs relative to the average frequency across all non-targeting sgRNAs.
(G) Plot shows the effect of every sgRNA in initial Repair-seq screen on the frequency of the indicated 2 nt insertion. Each dot plots the number of UMIs recovered for one sgRNA (y-axis) against the percentage of UMIs reporting the indicated insertion. Green and orange dots mark sgRNAs targeting XRCC5 and XRCC6; dark grey dots mark individual non-targeting sgRNAs; light grey dots mark all other sgRNAs. Black vertical line indicates the average percentage across all non-targeting sgRNAs. Dark grey density plot above the scatter plot shows a kernel density estimate (KDE) of the distribution of frequencies in non-targeting sgRNAs, and light grey density plots above and to the right show KDEs of the distribution of frequencies (top) and UMI counts (right) for all sgRNAs.
Figure S3. Validation of CRISPRi sub-library and of the reproducibility of outcome redistribution profiles, Related to Figure 3.
(A) Functional annotation classes for genes targeted by CRISPRi sub-library (366 sgRNAs).
(B) Histogram depicts the distribution of fractional knockdowns produced by sgRNAs in CRISPRi sub-library as measured by Perturb-seq. Results were calculated as follows: For each sgRNA, we assembled a pseudo-bulk expression profile by summing UMIs counts from all cells that received a given sgRNA. The expression of the targeted gene from each profile was then compared to expression of the same gene in a profile for cells that received non-targeting sgRNAs.
(C) Plot shows the highest fractional knockdown achieved for each gene across all sgRNAs targeting that gene in our CRISPRi sub-library. Genes are ordered by highest fractional knockdown. The most extreme outliers for which effective knockdown was not achieved by any sgRNA were ZBTB38 and RAD52.
(D+E+F) Diagrams (left) represent the most frequent repair outcomes observed at Cas9 target site 1 (D), Cas9 target site 2 (E), and Cas9 target site 3 (F). Plots (middle) show the baseline frequencies of each outcome across all non-targeting sgRNAs, averaged across two independent screens. Plots (right) depict correlations between replicates in log2 fold changes in the frequency of each outcome caused by the 100 sgRNAs that produced the most substantial overall redistribution of outcomes.
(G+H+I) Plots show correlations between outcome signatures for pairs of distinct outcomes in replicate screens performed at Cas9 target site 1 (G), Cas9 target site 2 (H), and Cas9 target site 3 (I). Correlations were calculated between the log2 fold changes in frequency caused by the 100 sgRNAs that produced the strongest overall redistribution of outcomes. Green points mark pairs of distinct bidirectional deletions; light grey points mark all other outcome pairs.
(J+K+L) Plots show correlations between CRISPRi sgRNA signatures for pairs of distinct sgRNAs in replicate screens performed at Cas9 target site 1 (J), Cas9 target site 2 (K), and Cas9 target site 3 (L). Correlations were calculated between the log2 fold changes in frequency for all outcomes present above baseline frequency of 0.5%. Blue points mark pairs of sgRNAs targeting the same gene; grey points mark pairs of sgRNAs targeting distinct genes.
Figure S4. Hierarchical clustering of sgRNA signatures and profiles of genetic dependencies for individual outcomes across screens performed at four Cas9 target sites, Related to Figures 3 and 5.
Heatmap represents a composite matrix of log2 fold changes in outcome frequencies produced by the most active CRISPRi sgRNAs in screens performed at four Cas9 target sites (Cas9 target sites 1, 2, 3, and 4). Screens represented include two replicates at Cas9 target sites 1, 3, and 4 and one replicate at Cas9 target site 2, all performed with the 366 CRISPRi sgRNA sub-library. Rows represent outcomes from individual screen replicates, and columns represent individual CRISPRi sgRNAs, with data hierarchically clustered along both dimensions. All individual outcomes present above baseline frequency of 0.2% in each screen were included, as well as two composite outcome categories per screen representing outcomes in which genomic sequence fragments ≤75 nts or >75 nts were captured at breaks. All CRISPRi sgRNAs that were among the 100 most active (i.e., that caused the strongest overall redistribution of outcome frequencies) in any screen were included. Color shading of outcome diagrams indicates target site of origin (green, Cas9 target site 1; orange, Cas9 target site 2; blue, Cas9 target site 3; red, Cas9 target site 4). Outcome sequence architecture categories are labelled to the left of diagrams. sgRNAs are labeled by cluster assignments produced by HDBSCAN clustering.
Figure S5. Validation of insertion phenotypes at endogenous loci and characterization of genomic capture outcomes, Related to Figures 3 and 4.
(A) The effects of XRCC5- and POLL-targeting sgRNAs on insertion frequencies at the endogenous HBB loci in HeLa CRISPRi cells. Endogenous target sites correspond to Cas9 target sites 1, 2, and 4 as depicted in Figure S1A, except that Cas9 target site 2 differs by one nucleotide at the endogenous locus with a PAM sequence of GGG instead of AGG. Diagrams (left) depict all insertions observed at baseline frequencies of at least 0.1%. Line plots (middle) show baseline frequencies of each insertion. Heatmaps (right) show log2 fold changes in insertion frequencies produced by one CRISPRi sgRNA targeting each of XRCC5 or POLL in three biological replicates.
(B) Diagrams depict example repair outcomes in which segments of human genomic sequence (top) or Bos taurus genomic sequence (bottom) were observed at DSBs. In these diagrams, the bottom blue line represents the target region of the integrated Repair-seq construct, with the DSB site, Cas9 target site 3 protospacer and PAM, and sequencing primer locations indicated. The black line represents the sequencing read of the repair outcome; blue lines between the target site and the sequencing read represent local alignments between the two; the grey line above the read represents a local alignment of the read to the relevant reference genome.
(C) Length distributions of captured human genomic sequence in a screen performed at Cas9 target site 3. Among target sites screened in this paper, this one was located closest to the sequencing primer and therefore maximized the range of captured sequence lengths that could be resolved before reaching the end of the sequencing read. Lines in the top panel plot the percentage of repair outcomes for which genomic sequence of the indicated length was captured (averaged over 21 nt windows) for all non-targeting sgRNAs (black), all DNA2-targeting sgRNAs (red), all MCM10-targeting sgRNAs (green), or all LIG4-targeting sgRNAs (purple). Lower detection limit indicates the length below which insertion sequence cannot be unambiguously assigned a source. Upper detection limit indicates the length above which the sequencing read is not long enough to resolve the end of an insertion directly at the DSB. Total percentages of outcomes with captured sequence above the upper detection limit are plotted to the far right, with a different y-axis scale. Lines in bottom panel plot log2 fold changes in length-specific insertion frequency for sgRNAs targeting DNA2, MCM10, and LIG4 relative to non-targeting sgRNAs (averaged over 21 nt windows).
(D) Comparison of the effects of gene knockdowns on frequencies of capture of human genomic sequence of different length ranges in screens performed at Cas9 target site 3 with the 366 CRISPRi sgRNA library. To maximize the ability to detect differential genetic modulators of potentially overlapping populations, separated length ranges of ≤75 nts and ≥125 nts were considered. Gene-level effects were calculated as the mean log2 fold change in frequency from non-targeting for the two sgRNAs targeting each gene with most extreme log2 fold changes.
(E) Reproducibility of the effects of knockdown of DNA2, MCM10, and LIG4 on the frequency of capture of human genomic sequence of indicated length ranges across nine screens performed at the indicated Cas9 target sites with either the 1,573 sgRNA or 366 sgRNA CRISPRi library. Data represent the average of log2 fold changes in frequency from non-targeting for the two sgRNAs targeting each gene with the most extreme phenotypes.
(F) Length distributions of captured human genomic sequence in focused experiments performed at Cas9 target site 1 with CRISPR-activation (CRISPRa) and CRISPRi in K562 cells. CRISPRa is a system for upregulating transcription of targeted genes. In these experiments, longer sequencing reads were used to allow resolution of longer insertions than in screens. Lines in the top panel plot the average percentage of repair outcomes for which genomic sequence of the indicated lengths was captured (averaged over 21 nt windows) across three replicates (shading represents +/− standard deviation) for single non-targeting or DNA2-targeting sgRNAs. Total percentages of outcomes with captured sequence above the upper detection limit are plotted to the far right, with a different y-axis scale. Each line in the bottom panel plots the mean of the log2 fold changes in length-specific insertion frequency for the DNA2-targeting sgRNA relative to the non-targeting control (averaged over 21 nt windows; shading represents +/− standard deviation).
(G) The effects of gene knockdowns on the frequency of capture of Bos taurus genomic sequence in replicate screens performed at Cas9 target site 3 with the 366 CRISPRi sgRNA sub-library. Data represent the average of log2 fold changes in frequency from non-targeting for the two sgRNAs targeting each gene with the most extreme phenotypes.
Figure S6. Repair-seq analysis of Cas12a-induced DSBs and evaluation of select phenotypes across cell lines, Related to Figures 5 and 6.
(A) Plot shows the fraction of all cells containing each sgRNA in our CRISPRi sub-library (366 sgRNAs) assigned to S phase based on transcriptional profiles in Perturb-seq data (x-axis) compared to the correlation of outcome redistribution profiles for each sgRNA across all Cas9 target sites with the average profile for sgRNAs targeting MRE11, RAD50, or NBN in Repair-seq data (y-axis). Select gene are highlighted with colored dots.
(B) Repair-seq analysis of Cas12a-induced DSBs. Central heatmap depicts log2 fold changes in repair outcome frequencies produced by the 100 most active CRISPRi sgRNAs relative to the average of all non-targeting CRISPRi sgRNAs. Rows represent individual outcomes (depicted in diagrams on the left), and columns represent individual CRISPRi sgRNAs (columns). Data is hierarchically clustered along both dimensions. Analyzed outcomes include all those present above a baseline frequency of 0.2%, as well as one composite group representing the combined frequency of all outcomes in which stretches of genomic sequence >75 nts were captured at the break. Triangular heatmaps depict correlations between pairs of sgRNAs (above) and between pairs of outcomes (right). sgRNA cluster assignments generated by HDBSCAN are labeled below.
(C) The effects of repressing select genes (RAD17, POLQ, MRE11) on the frequencies of repair outcomes in focused experiments performed at Cas9 target site 1 and with Cas12a in K562, HeLa, and RPE1 cells. Each point represents the mean of log2 fold changes in frequency of a specific outcome relative to non-targeting sgRNAs across three replicates (error bars indicate +/− standard deviation).
(D+E) Log2 fold changes in outcome frequencies produced by the indicated CRISPRi sgRNAs overlaid on UMAP embedding of Cas9 outcomes. See also Figure 3G.
Figure S7. Repair-seq analysis of DSB repair with oligonucleotide repair templates, Related to Figure 7.
(A) Comparison of the effects of select gene knockdowns on the frequency of scarless HDR outcomes across ten screens performed with different donor configurations. Diagrams indicate whether a donor was single-stranded or double-stranded (single or double lines); the strand orientations of single-stranded donors; the pattern of SNVs encoded, with orange indicating unevenly spaced as in Figure 7B and green indicating evenly spaced as in panel (D) (Table S2); the Cas9 target site; and the CRISPRi library used. Data displayed are the average log2 fold changes in frequency from non-targeting of the two most extreme sgRNAs targeting each gene. Note that all single-strand donors contain terminal phosphorothioate linkages, but the double-strand donor does not.
(B) Comparison of the effects of select gene knockdowns on the frequency of half-HDR outcomes across nine different screens performed with single-stranded donors. Screen conditions are indicated as in (A). Data calculated as in (A).
(C) Comparison of the effects of select gene knockdowns on the frequency of repair outcomes with captured donor fragments (as in Figure 7E) across ten different screens performed with single-stranded donors. Screen conditions are indicated as in (A), with the addition of a black donor diagram indicating a non-homologous donor. Data calculated as in (A).
(D) Repair-seq analysis of Cas9-induced DSB repair in the presence of a single-stranded donor with 6 programmed SNVs evenly spaced around Cas9 target site 1. Heatmap (central) depicts log2 fold changes in repair outcome frequencies produced by the 100 most active CRISPRi sgRNAs relative to the average of all non-targeting CRISPRi sgRNAs. Rows represent individual outcomes (depicted in diagrams on the left), and columns represent individual CRISPRi sgRNAs (columns). Data is hierarchically clustered along both dimensions. Analyzed outcomes include all those present above a baseline frequency of 0.5%, as well as composite groups representing the combined frequency of all outcomes in which stretches of genomic sequence >75 nts were captured at the break, half-HDR outcomes (as in Figure 7D), and capture of fragments of donor sequence (Figure 7E). Cluster assignments produced by HDBSCAN are labelled above.
(E+F) Effects of knockdown of genes encoding components of the MRN complex and mismatch repair genes on the frequency of individual scarless HDR outcomes observed in screens performed at Cas9 target site 1 in the presence of two different single-stranded donors with 6 programmed SNVs evenly spaced around the DSB site with opposite strand orientations. Panels show data from separate screens. Diagrams (top left of each panel) depict the integrated target site along with the strand orientation and programmed edit patterns of the donor. Diagrams (bottom left of each panel) depict all scarless HDR outcomes observed at a baseline frequency above 0.5%. Black line plots show baseline frequencies of each outcome. Brown and green line plots and heatmaps (right of each panel) show log2 fold changes in outcome frequency produced by indicated CRISPRi sgRNAs.
(G) Model for roles of MRN complex and mismatch repair (MMR) in mediating scarless HDR templated by a single-strand donor. Single-strand donors with homology to the sequence surrounding a DSB can anneal to only one side of the break. Consistent with previous models, Repair-seq phenotypes suggest that the resulting heteroduplexes can be processed by MMR to install encoded SNVs on the annealed side, causing concomitant suppression of scarless SSTR outcomes lacking these SNV. Phenotypes of genes encoding the MRN complex (MRE11, NBN, RAD50) suggest an extension to this model wherein suppressed SSTR outcomes have a stronger requirement for resection.
Table S1. gRNAs and sequences of proteins used for DSB-induction, Related to all Figures.
Table S2. Oligonucleotides and TaqMan probes used in this study, Related to all Figures.
In this table, * denotes phosphorothioated bond, N’s denote index nucleotides, /3IABkFQ/ denotes Iowa Black® FQ quencher, /56-FAM/ denotes a single isomer derivative of fluorescein, /ZEN/ denotes a quencher, /5HEX/ denotes 5′ hexachlorofluorescein, /5Phos/ denotes 5′ phosphorylation, and /3ddC/ denotes 3′ dideoxycytidine (ddC).
Table S3. CRISPRi library with 1,573 sgRNAs targeting 476 genes, Related to all Figures except S1 S3, and S4.
Table S4. Functional annotation of genes targeted by 1,573 sgRNA and 366 sgRNA library, Related to Figures 1C and S3B and Table S3 and S6.
Table S5. Details of Repair-seq screens, Related to all Figures.
Table S6. CRISPRi library with 366 sgRNAs targeting 118 genes, Related to all Figures except 1, 2, S1, and S2.
Table S7. UMI counts recovered for each CRISPRi sgRNA in each Repair-seq screen performed, Related to STAR Methods and Table S5.
In this table, Screen_Name labels are composed of cell line, editing protein, DSB target site, exogenous repair template, CRISPRi sgRNA library, replicate of conditions.
HIGHLIGHTS.
Repair-seq maps the genetic dependencies of DNA repair outcomes
High-resolution signatures of gene function identify unexpected gene relationships
DSB-induced mutations with similar sequences can result from distinct mechanisms
Repair-seq can be adapted to study a broad range of genome editing tools
ACKNOWLEDGMENTS
We thank M. Jost, J. Replogle, M. Leonetti, H. Canaj, B. Conklin, M. Levine, S.J. Elledge, J. Haber, and members of the Adamson and Weissman labs for helpful discussions. We thank M. Demozzi, K.W. Gareau, H.S. Abdulkerim, C. Wilson, and M. Donepudi (Editas Medicine). We thank E. Chow (UCSF Center for Advanced Technology), M. Tan (Chan Zuckerberg Biohub), W. Wang (Genomics Core Facility of Princeton University), and C. DeCoste and K. Rittenbach (Princeton University Flow Cytometry Resource Facility; NCI-CCSG P30CA072720-5921). Research was supported by the National Institutes of Health (NIH) under award numbers 1RM1HG009490 (J.S.W.), 1R35GM138167-01 (B.S.A.), 5P30CA072720-22 (B.S.A.), and T32HG003284 (Princeton QCB training grant). J.S.W. is supported by HHMI. B.S.A. is supported by the Searle Scholars Program. A.C. is supported by the National Science Foundation GRFP (DGE-2039656). J.A.H. was the Rebecca Ridley Kry Fellow of the Damon Runyon Cancer Research Foundation (DRG-2262-16). J.Y. is supported by the China Scholarship Council (CSC) based on the April 2015 Memorandum of Understanding between the CSC and Princeton University.
Footnotes
DECLARATION OF INTERESTS
Editas Medicine was involved in this work and provided reagents. B.A. was a member of a ThinkLab Advisory Board for, and holds equity in, Celsius Therapeutics. J.A.H. is a consultant for Tessera Therapeutics. JSW declares outside interest in 5 AM Venture, Amgen, Chroma Medicine, KSQ Therapeutics, Maze Therapeutics, Tenaya Therapeutics, Tessera Therapeutics, and Third Rock Ventures. A.B. and C.C.R. are former employees and shareholders of Editas Medicine and were employed by Editas at the time this work was conducted. J.A.H. and B.A. have filed patent applications on related work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adamson B, Smogorzewska A, Sigoillot FD, King RW, and Elledge SJ (2012). A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol 14, 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen F, Crepaldi L, Alsinet C, Strong AJ, Kleshchevnikov V, De Angeli P, Páleníková P, Khodak A, Kiselev V, Kosicki M, et al. (2019). Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol 37, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone AV, Koblan LW, and Liu DR (2020). Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol [DOI] [PubMed] [Google Scholar]
- Bothmer A, Phadke T, Barrera LA, Margulies CM, Lee CS, Buquicchio F, Moss S, Abdulkerim HS, Selleck W, Jayaram H, et al. (2017). Characterization of the interplay between DNA repair and CRISPR/Cas9-induced DNA lesions at an endogenous locus. Nat. Commun 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Bothmer A, Gareau KW, Abdulkerim HS, Abdulkerim HS, Buquicchio F, Buquicchio F, Buquicchio F, Cohen L, Cohen L, et al. (2020). Detection and Modulation of DNA Translocations during Multi-Gene Genome Editing in T Cells. Cris. J 3, 177–187. [DOI] [PubMed] [Google Scholar]
- Canaj H, Hussmann JA, Li H, Beckman KA, Goodrich L, Cho Nathan, H., Li YJ, Santos DA, McGeever A, Stewart EM, et al. (2019). Deep profiling reveals substantial heterogeneity of integration outcomes in CRISPR knock-in experiments. BioRxiv. [Google Scholar]
- Canny MD, Moatti N, Wan LCK, Fradet-Turcotte A, Krasner D, Mateos-Gomez PA, Zimmermann M, Orthwein A, Juang Y-C, Zhang W, et al. (2018). Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol 36, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Garcia J, Cho JE, Carvajal-Garcia P, Feng W, Wood RD, Sekelsky J, Gupta GP, Roberts SA, and Ramsden DA (2020). Mechanistic basis for microhomology identification and genome scarring by polymerase theta. Proc. Natl. Acad. Sci. U. S. A 117, 8476–8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti AM, Henser-Brownhill T, Monserrat J, Poetsch AR, Luscombe NM, and Scaffidi P (2019). Target-Specific Precision of CRISPR-Mediated Genome Editing. Mol. Cell 73, 699–713.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, McKenna A, Schreiber J, Haeussler M, Yin Y, Agarwal V, Noble WS, and Shendure J (2019). Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 47, 7989–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG (2018). Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet 19, 67–80. [DOI] [PubMed] [Google Scholar]
- Duursma AM, Driscoll R, Elias JE, and Cimprich KA (2013). A Role for the MRN Complex in ATR Activation via TOPBP1 Recruitment. Mol. Cell 50, 116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell VL, and Schild-Poulter C (2015). The Ku heterodimer: function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res 763, 15–29. [DOI] [PubMed] [Google Scholar]
- Gallagher DN, Pham N, Tsai AM, Janto AN, Choi J, Ira G, and Haber JE (2020). A Rad51-independent pathway promotes single-strand template repair in gene editing. PLOS Genet. 16, e1008689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L. a, Horlbeck M. a, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen T, Klaasen S, van de Vrugt H, and Te Riele H (2018). DNA mismatch repair and oligonucleotide end-protection promote base-pair substitution distal from a CRISPR/Cas9-induced DNA break. Nucleic Acids Res. 46, 2945–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, et al. (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard SM, Yanez DA, and Stark JM (2015). DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 11, e1004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurov KE, Cotta-Ramusino C, and Elledge SJ (2010). A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev. 24, 1939–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N, and Durocher D (2017). The control of DNA repair by the cell cycle. Nat. Cell Biol 19, 1–9. [DOI] [PubMed] [Google Scholar]
- Jasin M, and Haber JE (2016). The democratization of gene editing: Insights from site-specific cleavage and double-strand break repair. DNA Repair (Amst). 44, 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SK, Hawkins JA, Johnson NV, Jung C, Hu K, Rybarski JR, Chen JS, Doudna JA, Press WH, and Finkelstein IJ (2021). Massively parallel kinetic profiling of natural and engineered CRISPR nucleases. Nat. Biotechnol 39, 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost M, Chen Y, Gilbert LA, Horlbeck MA, Krenning L, Menchon G, Rai A, Cho MY, Stern JJ, Prota AE, et al. (2017). Combined CRISPRi/a-Based Chemical Genetic Screens Reveal that Rigosertib Is a Microtubule-Destabilizing Agent. Mol. Cell 68, 210–223.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Y, Ruis B, Takasugi T, and Hendrickson EA (2017). Mechanisms of precise genome editing using oligonucleotide donors. Genome Res. 27, 1099–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblan LW, Arbab M, Shen MW, Hussmann JA, Anzalone AV, Doman JL, Newby GA, Yang D, Mok B, Replogle JM, et al. (2021). Efficient C•G-to-G•C base editors developed using CRISPRi screens, target-library analysis, and machine learning. Nat. Biotechnol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M, Tomberg K, and Bradley A (2018). Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol 36, 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenay RT, Aghazadeh A, Hiatt J, Tse D, Roth TL, Apathy R, Shifrut E, Hultquist JF, Krogan N, Wu Z, et al. (2019). Large dataset enables prediction of repair after CRISPR–Cas9 editing in primary T cells. Nat. Biotechnol 37, 1034–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz ML, Papathanasiou S, Doerfler PA, Blaine LJ, Sun L, Yao Y, Zhang C-Z, Weiss MJ, and Pellman D (2021). Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet 53, 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos BR, Kaplan AC, Bae JE, Ferrazzoli AE, Kuo J, Anand RP, Waterman DP, and Haber JE (2018). CRISPR/Cas9 cleavages in budding yeast reveal templated insertions and strand-specific insertion/deletion profiles. Proc. Natl. Acad. Sci 115, E2040–E2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lõoke M, Maloney MF, and Bell SP (2017). Mcm10 regulates DNA replication elongation by stimulating the CMG replicative helicase. Genes Dev. 31, 291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, Bumcrot D, Chao H, Ciulla DM, DaSilva JA, et al. (2019). Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med 25, 229–233. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, and Ploegh HL (2015). Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol 33, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Álvarez-Quilón A, Moatti N, Zimmermann M, et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell BC, Adamson B, Lydeard JR, Sowa ME, Ciccia A, Bredemeyer AL, Schlabach M, Gygi SP, Elledge SJ, and Harper JW (2010). A Genome-wide Camptothecin Sensitivity Screen Identifies a Mammalian MMS22L-NFKBIL2 Complex Required for Genomic Stability. Mol. Cell 40, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivieri M, Cho T, Álvarez-Quilón A, Li K, Schellenberg MJ, Zimmermann M, Hustedt N, Rossi SE, Adam S, Melo H, et al. (2020). A Genetic Map of the Response to DNA Damage in Human Cells. Cell 182, 481–496.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono R, Yasuhiko Y, Aisaki K, Kitajima S, Kanno J, and Hirabayashi Y (2019). Exosome-mediated horizontal gene transfer occurs in double-strand break repair during genome editing. Commun. Biol 2, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onozawa M, Zhang Z, Kim YJ, Goldberg L, Varga T, Bergsagel PL, Kuehl WM, and Aplan PD (2014). Repair of DNA double-strand breaks by templated nucleotide sequence insertions derived from distant regions of the genome. Proc. Natl. Acad. Sci. U. S. A 111, 7729–7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Overbeek M, Capurso D, Carter MM, Thompson MS, Frias E, Russ C, Reece-Hoyes JS, Nye C, Gradia S, Vidal B, et al. (2016). DNA Repair Profiling Reveals Nonrandom Outcomes at Cas9-Mediated Breaks. Mol. Cell 63, 633–646. [DOI] [PubMed] [Google Scholar]
- Paull TT (2021). Reconsidering pathway choice: a sequential model of mammalian DNA double-strand break pathway decisions. Curr. Opin. Genet. Dev 71, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, and Lim WA (2013). Repurposing CRISPR as an RNA-γuided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden DA, Carvajal-Garcia J, and Gupta GP (2021). Mechanism, cellular functions and cancer roles of polymerase-theta-mediated DNA end joining. Nat. Rev. Mol. Cell Biol 0123456789. [DOI] [PubMed] [Google Scholar]
- Replogle JM, Norman TM, Xu A, Hussmann JA, Chen J, Cogan JZ, Meer EJ, Terry JM, Riordan DP, Srinivas N, et al. (2020). Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol 38, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CD, Kazane KR, Feng SJ, Zelin E, Bray NL, Schäfer AJ, Floor SN, and Corn JE (2018). CRISPR-Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat. Genet 50, 1132–1139. [DOI] [PubMed] [Google Scholar]
- Rouet P, Smih F, and Jasin M (1994). Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol 14, 8096–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep R, Brinkman EK, Leemans C, Vergara X, van der Weide RH, Morris B, van Schaik T, Manzo SG, Peric-Hupkes D, van den Berg J, et al. (2021). Impact of chromatin context on Cas9-induced DNA double-strand break repair pathway balance. Mol. Cell 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, and Loeb L. a (2012). Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A 109, 14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Panday A, Elango R, and Willis NA (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol 20, 698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, and Symington LS (2015). Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci 40, 701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MW, Arbab M, Hsu JY, Worstell D, Culbertson SJ, Krabbe O, Cassa CA, Liu DR, Gifford DK, and Sherwood RI (2018). Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiroguchi K, Jia TZ, Sims P. a, and Xie XS (2012). Digital RNA sequencing minimizes sequence-dependent bias and amplification noise with optimized single-molecule barcodes. Proc. Natl. Acad. Sci. U. S. A 109, 1347–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou J, Li J, Liu Y, and Wu Q (2018). Precise and Predictable CRISPR Chromosomal Rearrangements Reveal Principles of Cas9-Mediated Nucleotide Insertion. Mol. Cell 71, 498–509.e4. [DOI] [PubMed] [Google Scholar]
- Sinha R, Stanley G, Gulati GS, Ezran C, Travaglini KJ, Wei E, Chan CKF, Nabhan AN, Su T, Morganti RM, et al. (2017). Index switching causes “spreading-of-signal” among multiplexed samples in Illumina HiSeq 4000 DNA sequencing. BioRxiv. [Google Scholar]
- Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiácovo MP, et al. (2010). A Genetic Screen Identifies FAN1, a Fanconi Anemia-Associated Nuclease Necessary for DNA Interstrand Crosslink Repair. Mol. Cell 39, 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinson BM, Moreno AT, Walter JC, and Loparo JJ (2020). A Mechanism to Minimize Errors during Non-homologous End Joining. Mol. Cell 77, 1080–1091.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohkendl I, Saifuddin FA, Rybarski JR, Finkelstein IJ, and Russell R (2018). Kinetic Basis for DNA Target Specificity of CRISPR-Cas12a. Mol. Cell 71, 816–824.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong LN, Li Y, Shi LZ, Hwang PY-H, He J, Wang H, Razavian N, Berns MW, and Wu X (2013). Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. U. S. A 110, 7720–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw CP, Carr AM, and Oliver AW (2014). TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair (Amst). 22, 165–174. [DOI] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt DW, Feng W, Conlin MP, Yousefzadeh MJ, Roberts SA, Mieczkowski P, Wood RD, Gupta GP, and Ramsden DA (2016). Essential Roles for Polymerase θ-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 63, 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Pham N, Xia B, Papusha A, Wang G, Yan Z, Peng G, Chen K, and Ira G (2018). Dna2 nuclease deficiency results in large and complex DNA insertions at chromosomal breaks. Nature 564, 287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, Van Der Oost J, Regev A, et al. (2015). Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 163, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Meng Y, Campbell JL, and Shen B (2020). Multiple roles of DNA2 nuclease/helicase in DNA metabolism, genome stability and human diseases. Nucleic Acids Res. 48, 16–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Ž, Muir M, Fluteau A, Aregger M, McEwan A, et al. (2018). CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccaro MV, Xu J, Mitchell C, Marin D, Zimmerman R, Rana B, Weinstein E, King RT, Palmerola KL, Smith ME, et al. (2020). Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos. Cell 183, 1650–1664.e15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Design and validation of the Repair-seq screening vector and sequencing strategy, Related to Figure 1.
(A) Schematic of Repair-seq screening vector. LTR indicates long terminal repeat. Elements along top line are not represented at scale.
(B) Plots depict the frequencies of individual DSB repair outcome sequences generated at indicated target sites (top, Cas9 target site 1; middle, Cas9 target site 3; bottom, Cas9 target site 4) compared between replicates. Outcome distributions were collected within the context of the genomically integrated Repair-seq construct from K562 cells after electroporation with corresponding RNPs. In each panel, each dot represents a single sequence outcome, with dots colored according to sequence architecture category. Locations of relevant target sites are indicated in panel A.
(C) Plots depict the frequencies of individual DSB repair outcome sequences compared at 2 different Cas9 target sites that are offset from each other by 1 nt. Outcome distributions were collected within the context of the genomically integrated Repair-seq construct from K562 cells after electroporation with corresponding RNPs.
(D) Plots depict the frequencies of individual DSB repair outcomes at indicated target sites (top, Cas9 target site 1; middle, Cas9 target site 3; bottom, Cas9 target site 4) compared between replicates. Outcome distributions were collected from the endogenous HBB locus in K562 cells after electroporation with corresponding RNPs. In each panel, each dot represents a single sequence outcome, with dots colored according to sequence architecture category. Locations of relevant target sites are indicated in panel A.
(E) Plots depict the frequencies of individual DSB repair outcomes at indicated target sites (top, Cas9 target site 1; middle, Cas9 target site 3; bottom, Cas9 target site 4) compared between those generated within the genomically integrated Repair-seq construct (x-axis, mean of 3 replicates) and the endogenous HBB locus (y-axis, mean of 3 replicates). Outcome distributions were collected from K562 cells after electroporation with corresponding RNPs.
(F) Schematic of Repair-seq sequencing strategy. Sequencing reads are indicated in red. Read 1 (R1) records the CRISPRi sgRNA sequence, Read 2 (R2) records the DSB repair outcome, one short index read records a unique molecular identifier (UMI) sequence, and another short index read records sample indices (i5) that allow multiplexing of libraries from multiple screens. Elements not represented at scale.
Figure S2. Characterization of observed and unobserved DSB repair products, Related to Figure 1 and STAR Methods.
(A) Plots show the distributions of BFP fluorescence in K562 CRISPRi cells after transduction with Repair-seq screening constructs and electroporation with Cas9 RNP against target site 1 (measured by flow cytometry). Data were collected from uncut control cells and cells used for screen number 14 (Table S5) after 3 (top) and 10 days (bottom) of recovery from electroporation.
(B) Schematic depicts the locations of sequences required for Repair-seq library preparation (PCR primer annealing site, NotI site) as well as amplicons used for droplet digital PCR (ddPCR) detection of large deletions generated within the Repair-seq screening construct (amplicons 1, 2, and 3). Indicated distances are relative to the “Cas9 target 1” shown in Figure S1A. Elements are not represented at scale.
(C) Data represent the percentages of K562 CRISPRi cells with intact amplicons (as detected by ddPCR using amplicons depicted in Figure S2B) in BFP+ and BFP− populations isolated by fluorescence activated cell sorting 4 days after electroporation of a Cas9 RNP targeting HBB site 1 (left, amplicon 1; middle, amplicon 2; right, amplicon 3). Cells were transduced with the Repair-seq screening construct prior to electroporation and were analyzed as a pool or isolated clones. Depicted results were calculated by comparing the normalized amplicon concentration from indicated sample to that of cells electroporated with a non-targeting RNP (Table S1).
(D) Schematic depicts the relative locations of the “LTR amplicon” and the “SSA amplicon”. To amplify the former, primers were designed that bind immediately upstream and downstream of the 5′ LTR in a single clonal cell line (2). To amplify the latter, primers were designed that bind immediately upstream of the 5′ LTR and downstream of the 3′ LTR. The “SSA amplicon” allows detection of an inferred single-strand annealing (SSA) repair event. Indicated distances are relative to the “Cas9 target 1” shown in Figure S1A. Elements are not represented at scale.
(E) Data represent the percentages of K562 CRISPRi cells (clonal cell line 2) with the “SSA amplicon” (as detected by ddPCR using amplicons depicted in Figure S2D) in BFP+ and BFP− populations isolated by fluorescence activated cell sorting 4 days after electroporation of indicated RNPs. Measurements were normalized to an ACTB control amplicon from each sample and then compared to the normalized concentration of the “LTR amplicon” in cells electroporated with a non-targeting RNP (Table S1).
(F) Data from initial Repair-seq screen highlighting the phenotypes of XRCC5 (encoding Ku80) and XRCC6 (encoding Ku70), including diagrams of the 30 most frequent indels observed in cells receiving non-targeting CRISPRi sgRNAs (left), the frequency of each outcome in the presence of individual non-targeting sgRNAs (middle, 60 grey lines) or sgRNAs targeting XRCC5 (middle, 3 green lines), and heatmaps of log2 fold changes in the frequency of each outcome caused by XRCC5- and XRCC6-targeting sgRNAs relative to the average frequency across all non-targeting sgRNAs.
(G) Plot shows the effect of every sgRNA in initial Repair-seq screen on the frequency of the indicated 2 nt insertion. Each dot plots the number of UMIs recovered for one sgRNA (y-axis) against the percentage of UMIs reporting the indicated insertion. Green and orange dots mark sgRNAs targeting XRCC5 and XRCC6; dark grey dots mark individual non-targeting sgRNAs; light grey dots mark all other sgRNAs. Black vertical line indicates the average percentage across all non-targeting sgRNAs. Dark grey density plot above the scatter plot shows a kernel density estimate (KDE) of the distribution of frequencies in non-targeting sgRNAs, and light grey density plots above and to the right show KDEs of the distribution of frequencies (top) and UMI counts (right) for all sgRNAs.
Figure S3. Validation of CRISPRi sub-library and of the reproducibility of outcome redistribution profiles, Related to Figure 3.
(A) Functional annotation classes for genes targeted by CRISPRi sub-library (366 sgRNAs).
(B) Histogram depicts the distribution of fractional knockdowns produced by sgRNAs in CRISPRi sub-library as measured by Perturb-seq. Results were calculated as follows: For each sgRNA, we assembled a pseudo-bulk expression profile by summing UMIs counts from all cells that received a given sgRNA. The expression of the targeted gene from each profile was then compared to expression of the same gene in a profile for cells that received non-targeting sgRNAs.
(C) Plot shows the highest fractional knockdown achieved for each gene across all sgRNAs targeting that gene in our CRISPRi sub-library. Genes are ordered by highest fractional knockdown. The most extreme outliers for which effective knockdown was not achieved by any sgRNA were ZBTB38 and RAD52.
(D+E+F) Diagrams (left) represent the most frequent repair outcomes observed at Cas9 target site 1 (D), Cas9 target site 2 (E), and Cas9 target site 3 (F). Plots (middle) show the baseline frequencies of each outcome across all non-targeting sgRNAs, averaged across two independent screens. Plots (right) depict correlations between replicates in log2 fold changes in the frequency of each outcome caused by the 100 sgRNAs that produced the most substantial overall redistribution of outcomes.
(G+H+I) Plots show correlations between outcome signatures for pairs of distinct outcomes in replicate screens performed at Cas9 target site 1 (G), Cas9 target site 2 (H), and Cas9 target site 3 (I). Correlations were calculated between the log2 fold changes in frequency caused by the 100 sgRNAs that produced the strongest overall redistribution of outcomes. Green points mark pairs of distinct bidirectional deletions; light grey points mark all other outcome pairs.
(J+K+L) Plots show correlations between CRISPRi sgRNA signatures for pairs of distinct sgRNAs in replicate screens performed at Cas9 target site 1 (J), Cas9 target site 2 (K), and Cas9 target site 3 (L). Correlations were calculated between the log2 fold changes in frequency for all outcomes present above baseline frequency of 0.5%. Blue points mark pairs of sgRNAs targeting the same gene; grey points mark pairs of sgRNAs targeting distinct genes.
Figure S4. Hierarchical clustering of sgRNA signatures and profiles of genetic dependencies for individual outcomes across screens performed at four Cas9 target sites, Related to Figures 3 and 5.
Heatmap represents a composite matrix of log2 fold changes in outcome frequencies produced by the most active CRISPRi sgRNAs in screens performed at four Cas9 target sites (Cas9 target sites 1, 2, 3, and 4). Screens represented include two replicates at Cas9 target sites 1, 3, and 4 and one replicate at Cas9 target site 2, all performed with the 366 CRISPRi sgRNA sub-library. Rows represent outcomes from individual screen replicates, and columns represent individual CRISPRi sgRNAs, with data hierarchically clustered along both dimensions. All individual outcomes present above baseline frequency of 0.2% in each screen were included, as well as two composite outcome categories per screen representing outcomes in which genomic sequence fragments ≤75 nts or >75 nts were captured at breaks. All CRISPRi sgRNAs that were among the 100 most active (i.e., that caused the strongest overall redistribution of outcome frequencies) in any screen were included. Color shading of outcome diagrams indicates target site of origin (green, Cas9 target site 1; orange, Cas9 target site 2; blue, Cas9 target site 3; red, Cas9 target site 4). Outcome sequence architecture categories are labelled to the left of diagrams. sgRNAs are labeled by cluster assignments produced by HDBSCAN clustering.
Figure S5. Validation of insertion phenotypes at endogenous loci and characterization of genomic capture outcomes, Related to Figures 3 and 4.
(A) The effects of XRCC5- and POLL-targeting sgRNAs on insertion frequencies at the endogenous HBB loci in HeLa CRISPRi cells. Endogenous target sites correspond to Cas9 target sites 1, 2, and 4 as depicted in Figure S1A, except that Cas9 target site 2 differs by one nucleotide at the endogenous locus with a PAM sequence of GGG instead of AGG. Diagrams (left) depict all insertions observed at baseline frequencies of at least 0.1%. Line plots (middle) show baseline frequencies of each insertion. Heatmaps (right) show log2 fold changes in insertion frequencies produced by one CRISPRi sgRNA targeting each of XRCC5 or POLL in three biological replicates.
(B) Diagrams depict example repair outcomes in which segments of human genomic sequence (top) or Bos taurus genomic sequence (bottom) were observed at DSBs. In these diagrams, the bottom blue line represents the target region of the integrated Repair-seq construct, with the DSB site, Cas9 target site 3 protospacer and PAM, and sequencing primer locations indicated. The black line represents the sequencing read of the repair outcome; blue lines between the target site and the sequencing read represent local alignments between the two; the grey line above the read represents a local alignment of the read to the relevant reference genome.
(C) Length distributions of captured human genomic sequence in a screen performed at Cas9 target site 3. Among target sites screened in this paper, this one was located closest to the sequencing primer and therefore maximized the range of captured sequence lengths that could be resolved before reaching the end of the sequencing read. Lines in the top panel plot the percentage of repair outcomes for which genomic sequence of the indicated length was captured (averaged over 21 nt windows) for all non-targeting sgRNAs (black), all DNA2-targeting sgRNAs (red), all MCM10-targeting sgRNAs (green), or all LIG4-targeting sgRNAs (purple). Lower detection limit indicates the length below which insertion sequence cannot be unambiguously assigned a source. Upper detection limit indicates the length above which the sequencing read is not long enough to resolve the end of an insertion directly at the DSB. Total percentages of outcomes with captured sequence above the upper detection limit are plotted to the far right, with a different y-axis scale. Lines in bottom panel plot log2 fold changes in length-specific insertion frequency for sgRNAs targeting DNA2, MCM10, and LIG4 relative to non-targeting sgRNAs (averaged over 21 nt windows).
(D) Comparison of the effects of gene knockdowns on frequencies of capture of human genomic sequence of different length ranges in screens performed at Cas9 target site 3 with the 366 CRISPRi sgRNA library. To maximize the ability to detect differential genetic modulators of potentially overlapping populations, separated length ranges of ≤75 nts and ≥125 nts were considered. Gene-level effects were calculated as the mean log2 fold change in frequency from non-targeting for the two sgRNAs targeting each gene with most extreme log2 fold changes.
(E) Reproducibility of the effects of knockdown of DNA2, MCM10, and LIG4 on the frequency of capture of human genomic sequence of indicated length ranges across nine screens performed at the indicated Cas9 target sites with either the 1,573 sgRNA or 366 sgRNA CRISPRi library. Data represent the average of log2 fold changes in frequency from non-targeting for the two sgRNAs targeting each gene with the most extreme phenotypes.
(F) Length distributions of captured human genomic sequence in focused experiments performed at Cas9 target site 1 with CRISPR-activation (CRISPRa) and CRISPRi in K562 cells. CRISPRa is a system for upregulating transcription of targeted genes. In these experiments, longer sequencing reads were used to allow resolution of longer insertions than in screens. Lines in the top panel plot the average percentage of repair outcomes for which genomic sequence of the indicated lengths was captured (averaged over 21 nt windows) across three replicates (shading represents +/− standard deviation) for single non-targeting or DNA2-targeting sgRNAs. Total percentages of outcomes with captured sequence above the upper detection limit are plotted to the far right, with a different y-axis scale. Each line in the bottom panel plots the mean of the log2 fold changes in length-specific insertion frequency for the DNA2-targeting sgRNA relative to the non-targeting control (averaged over 21 nt windows; shading represents +/− standard deviation).
(G) The effects of gene knockdowns on the frequency of capture of Bos taurus genomic sequence in replicate screens performed at Cas9 target site 3 with the 366 CRISPRi sgRNA sub-library. Data represent the average of log2 fold changes in frequency from non-targeting for the two sgRNAs targeting each gene with the most extreme phenotypes.
Figure S6. Repair-seq analysis of Cas12a-induced DSBs and evaluation of select phenotypes across cell lines, Related to Figures 5 and 6.
(A) Plot shows the fraction of all cells containing each sgRNA in our CRISPRi sub-library (366 sgRNAs) assigned to S phase based on transcriptional profiles in Perturb-seq data (x-axis) compared to the correlation of outcome redistribution profiles for each sgRNA across all Cas9 target sites with the average profile for sgRNAs targeting MRE11, RAD50, or NBN in Repair-seq data (y-axis). Select gene are highlighted with colored dots.
(B) Repair-seq analysis of Cas12a-induced DSBs. Central heatmap depicts log2 fold changes in repair outcome frequencies produced by the 100 most active CRISPRi sgRNAs relative to the average of all non-targeting CRISPRi sgRNAs. Rows represent individual outcomes (depicted in diagrams on the left), and columns represent individual CRISPRi sgRNAs (columns). Data is hierarchically clustered along both dimensions. Analyzed outcomes include all those present above a baseline frequency of 0.2%, as well as one composite group representing the combined frequency of all outcomes in which stretches of genomic sequence >75 nts were captured at the break. Triangular heatmaps depict correlations between pairs of sgRNAs (above) and between pairs of outcomes (right). sgRNA cluster assignments generated by HDBSCAN are labeled below.
(C) The effects of repressing select genes (RAD17, POLQ, MRE11) on the frequencies of repair outcomes in focused experiments performed at Cas9 target site 1 and with Cas12a in K562, HeLa, and RPE1 cells. Each point represents the mean of log2 fold changes in frequency of a specific outcome relative to non-targeting sgRNAs across three replicates (error bars indicate +/− standard deviation).
(D+E) Log2 fold changes in outcome frequencies produced by the indicated CRISPRi sgRNAs overlaid on UMAP embedding of Cas9 outcomes. See also Figure 3G.
Figure S7. Repair-seq analysis of DSB repair with oligonucleotide repair templates, Related to Figure 7.
(A) Comparison of the effects of select gene knockdowns on the frequency of scarless HDR outcomes across ten screens performed with different donor configurations. Diagrams indicate whether a donor was single-stranded or double-stranded (single or double lines); the strand orientations of single-stranded donors; the pattern of SNVs encoded, with orange indicating unevenly spaced as in Figure 7B and green indicating evenly spaced as in panel (D) (Table S2); the Cas9 target site; and the CRISPRi library used. Data displayed are the average log2 fold changes in frequency from non-targeting of the two most extreme sgRNAs targeting each gene. Note that all single-strand donors contain terminal phosphorothioate linkages, but the double-strand donor does not.
(B) Comparison of the effects of select gene knockdowns on the frequency of half-HDR outcomes across nine different screens performed with single-stranded donors. Screen conditions are indicated as in (A). Data calculated as in (A).
(C) Comparison of the effects of select gene knockdowns on the frequency of repair outcomes with captured donor fragments (as in Figure 7E) across ten different screens performed with single-stranded donors. Screen conditions are indicated as in (A), with the addition of a black donor diagram indicating a non-homologous donor. Data calculated as in (A).
(D) Repair-seq analysis of Cas9-induced DSB repair in the presence of a single-stranded donor with 6 programmed SNVs evenly spaced around Cas9 target site 1. Heatmap (central) depicts log2 fold changes in repair outcome frequencies produced by the 100 most active CRISPRi sgRNAs relative to the average of all non-targeting CRISPRi sgRNAs. Rows represent individual outcomes (depicted in diagrams on the left), and columns represent individual CRISPRi sgRNAs (columns). Data is hierarchically clustered along both dimensions. Analyzed outcomes include all those present above a baseline frequency of 0.5%, as well as composite groups representing the combined frequency of all outcomes in which stretches of genomic sequence >75 nts were captured at the break, half-HDR outcomes (as in Figure 7D), and capture of fragments of donor sequence (Figure 7E). Cluster assignments produced by HDBSCAN are labelled above.
(E+F) Effects of knockdown of genes encoding components of the MRN complex and mismatch repair genes on the frequency of individual scarless HDR outcomes observed in screens performed at Cas9 target site 1 in the presence of two different single-stranded donors with 6 programmed SNVs evenly spaced around the DSB site with opposite strand orientations. Panels show data from separate screens. Diagrams (top left of each panel) depict the integrated target site along with the strand orientation and programmed edit patterns of the donor. Diagrams (bottom left of each panel) depict all scarless HDR outcomes observed at a baseline frequency above 0.5%. Black line plots show baseline frequencies of each outcome. Brown and green line plots and heatmaps (right of each panel) show log2 fold changes in outcome frequency produced by indicated CRISPRi sgRNAs.
(G) Model for roles of MRN complex and mismatch repair (MMR) in mediating scarless HDR templated by a single-strand donor. Single-strand donors with homology to the sequence surrounding a DSB can anneal to only one side of the break. Consistent with previous models, Repair-seq phenotypes suggest that the resulting heteroduplexes can be processed by MMR to install encoded SNVs on the annealed side, causing concomitant suppression of scarless SSTR outcomes lacking these SNV. Phenotypes of genes encoding the MRN complex (MRE11, NBN, RAD50) suggest an extension to this model wherein suppressed SSTR outcomes have a stronger requirement for resection.
Table S1. gRNAs and sequences of proteins used for DSB-induction, Related to all Figures.
Table S2. Oligonucleotides and TaqMan probes used in this study, Related to all Figures.
In this table, * denotes phosphorothioated bond, N’s denote index nucleotides, /3IABkFQ/ denotes Iowa Black® FQ quencher, /56-FAM/ denotes a single isomer derivative of fluorescein, /ZEN/ denotes a quencher, /5HEX/ denotes 5′ hexachlorofluorescein, /5Phos/ denotes 5′ phosphorylation, and /3ddC/ denotes 3′ dideoxycytidine (ddC).
Table S3. CRISPRi library with 1,573 sgRNAs targeting 476 genes, Related to all Figures except S1 S3, and S4.
Table S4. Functional annotation of genes targeted by 1,573 sgRNA and 366 sgRNA library, Related to Figures 1C and S3B and Table S3 and S6.
Table S5. Details of Repair-seq screens, Related to all Figures.
Table S6. CRISPRi library with 366 sgRNAs targeting 118 genes, Related to all Figures except 1, 2, S1, and S2.
Table S7. UMI counts recovered for each CRISPRi sgRNA in each Repair-seq screen performed, Related to STAR Methods and Table S5.
In this table, Screen_Name labels are composed of cell line, editing protein, DSB target site, exogenous repair template, CRISPRi sgRNA library, replicate of conditions.
Data Availability Statement
Raw sequencing data has been deposited at the NCBI Sequence Read Archive database under Bioproject PRJNA746980. Code is available at https://github.com/jeffhussmann/repair-seq (DOI: https://doi.org/10.5281/zenodo.5534778). Processed data and notebooks are available at DOI: https://doi.org/10.5281/zenodo.5555287. Data can be interactively explored at https://seq.repair. Any additional information required to reanalyze the data reported in this paper is available upon request.