

Abstract
Chromatin is a system of proteins and DNA that regulates chromosome organization and gene expression in eukaryotes. Essential features that support these processes include biochemical marks on histones and DNA, ‘writer’ enzymes that generate or remove these marks and proteins that translate the marks into transcriptional regulation: reader-effectors. Here, we review recent studies that reveal how reader-effectors drive chromatin-mediated processes. Advances in proteomics and epigenomics have accelerated the discovery of chromatin marks and their correlation with gene states, outpacing our understanding of the corresponding reader-effectors. Therefore, we summarize the current state of knowledge and open questions about how reader-effectors impact cellular function and human disease and discuss how synthetic biology can deepen our knowledge of reader-effector activity.
The discovery of reader-effectors advanced our understanding of epigenetics
A cornerstone concept of genetic inheritance is the relationship between the sequence of nucleotides in DNA and cell phenotypes. Scientists have also sought to understand how the physical behavior of DNA, rather than its sequence, impacts the expression of genetically encoded information. The system of proteins and DNA that organize eukaryotic genomes inside the nucleus is called chromatin (Greek chroma, ‘colored’; see Glossary), a term that was coined in 1882 by biologist Walther Flemming who used cytology to investigate the dynamic behavior of dye-stained nuclear material. Since then, advances in analytical techniques have helped reveal the specific components of chromatin and their impact on key cellular processes, including transcriptional regulation and genome organization.
The bulk of chromatin includes histones, a family of 10–15 kD proteins (H1, H2A, H2B, H3, and H4) that assemble into an octamer (two H2A:H2B dimers, one H32:H42 tetramer) encased by 1.7 turns of double-stranded DNA within a unit called a nucleosome (Figure 1A). Unwrapped linker DNA outside of the nucleosome, plus a bound histone H1, makes up the chromatosome. By 1955, biochemist Marie M. Daly had determined that a major component of DNA-binding histones is lysine [1] which is a major carrier of biochemical information in chromatin, in particular those found in the ‘tail’ of the histones. The disordered N-terminal regions of histones, called histone tails (Figure 1A), are substrates for an array of post-translational modifications (PTMs) that are mediated by chromatin-modifying enzymes, that is, writers and erasers (Figure 1B). Among the most common and biologically understood histone, PTMs are modifications of lysine (acetylation, Kac; methylation, Kme; sumoylation, Ksumo; crotonylation, Kcr; butyrylation, Kbu), arginine (methylation, Rme), serine (phosphorylation, Sp), tyrosine (phosphorylation, Yp), and threonine (phosphorylation, Tp) (Figure 1C). CpG dinucleotides are target sites for DNA methylation (5-methylcytosine, 5meC) and chemical intermediates that are generated during the removal of methylation (5-hydroxymethylcytosine, 5hmeC; 5-formylcytosine, 5fC; 5-carboxylcytosine, 5caC; 5-hydroxymethyluracil, 5hmU). Several excellent reviews have cataloged the growing number of characterized histone and DNA modifications, mechanisms of their addition and removal, and their roles in chromatin-mediated genome organization and gene regulation [2–5].
Figure 1. The flow of molecular information in chromatin from chromatin modifying enzymes (CMEs), to chromatin marks (signals), to reader-effectors and regulatory outputs.
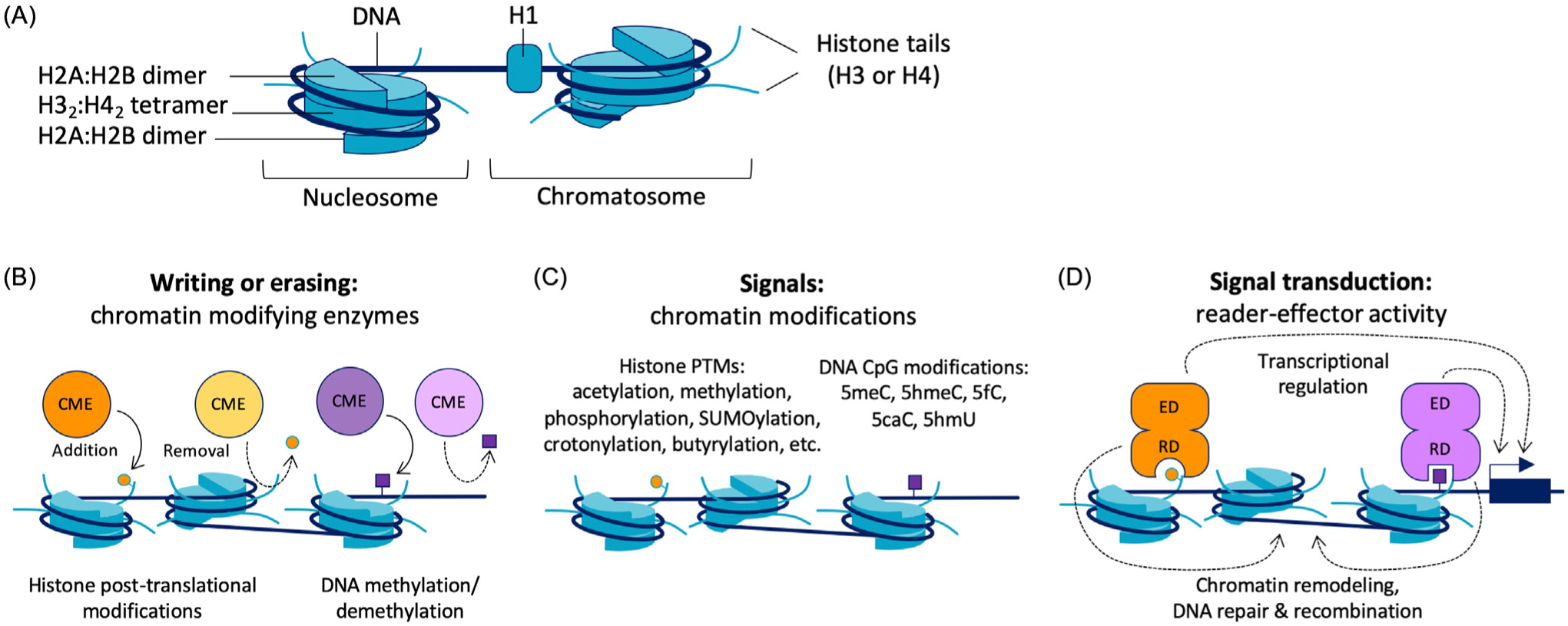
(A) Diagram of nucleosome and chromatosome structure. (B) CMEs add (write) or remove (erase) histone post-translational modifications (PTMs) and DNA methylation. (C) Chromatin modifications include histone PTMs and methylated DNA CpG dinucleotides. (D) Reader-effectors bind to chromatin modifications via a reader domain (RD) and drive processes such as transcriptional regulation, chromatin remodeling, and DNA repair and recombination through an effector domain (ED).
Eventually, antibody technology enabled more protein-specific staining than earlier work with dyes, and scientists began to identify the less abundant, non-histone nuclear proteins that bound to AT-rich DNA [6]. In 1986, Sarah Elgin reported the discovery of heterochromatin protein 1 (HP1) [7], which contains no ‘AT-hook’ DNA-binding motifs and has a repressive effect on gene transcription. In 1999, biochemical and 3-D structure studies led to the first characterization of a histone PTM reader domain, the C-terminal region of p300/CBP-associated factor (PCAF) that binds acetylated lysines in histone H4 (H4Kac) [8]. In 2001 the structure of the N-terminal reader domain of HP1 that interacts with histone H3 trimethylated at lysine 9 (H3K9me3) was reported [9,10]. The characterization of these chromatin proteins and dozens of other transcriptional regulators that interact with modified histones and methylated DNA established a class of proteins called reader-effectors (REs) [11,12]. An RE is a protein that contains two distinct regions, a reader domain that interacts with a chromatin modification, and an effector domain that is involved in downstream processes such as gene regulation, chromatin remodeling, or DNA repair (Figure 1D), either via intrinsic enzymatic activity or through interactions with other proteins. Therefore, REs drive epigenetic regulation by transducing chromatin modification signals into biological outputs (Figure 1D). The roles of different REs in cell development and the mechanisms of their activity are still being investigated today. In this review, we survey recent studies of REs, consider their importance in cell developmental diseases, and examine their function through the lens of protein engineering and synthetic biology.
Reader activity: processing chromatin marks as signals
Initiation of RE activity requires physical proximity with the genomic target site; therefore, chromatin modifications function as ‘signals’ that control the engagement or disengagement of the REs near genes or noncoding regulatory DNA elements. It should also be noted that reader domain-containing proteins often simply stabilize complexes at chromatin, and can bind PTMs on transcriptional regulators (e.g., BRD7 binds RelA acetylated at K310 [13]), otherwise serving a role outside of targeting histone PTMs and DNA methylation [12]. In the following section, we highlight the most recent evidence that recognition of specific chromatin signals via reader domains is an important step for RE protein function.
Monovalent REs engage with a single chromatin modification (Figure 2A) to regulate the transcription of nearby target genes or modify local chromatin features. For instance, methyl-CpG-binding protein 2 (MeCP2) is a transcriptional repressor that contains a methyl-binding domain (MBD) that binds to a single methyl-CpG (meCpG; Figure 2A, top left), and is not influenced by the flanking sequences. A large body of evidence has established that the MBD, which also appears in other REs such as MBD1, MBD2, and MBD4, is necessary and sufficient to bind meCpG [14,15]. While the classical view of methylation-mediated RE interactions is that only proteins with an MBD can interact with methylated DNA, mounting evidence is starting to reveal that other types of domains can interact with methylated DNA, albeit in a DNA sequence-specific manner [16]. Furthermore, genome-wide protein binding studies suggest that intermediate methylation states, for example, 5hmC and 5fC, may be bound by transcriptional regulators MeCP2 and forkhead box proteins, respectively.
Figure 2. https://elsevier.proofcentral.com/en-us/landing-page.html?token=2f29704b3d8907fef00c5766c86361Reader-effectors (REs) engage with chromatin marks via monovalent or multivalent interactions.
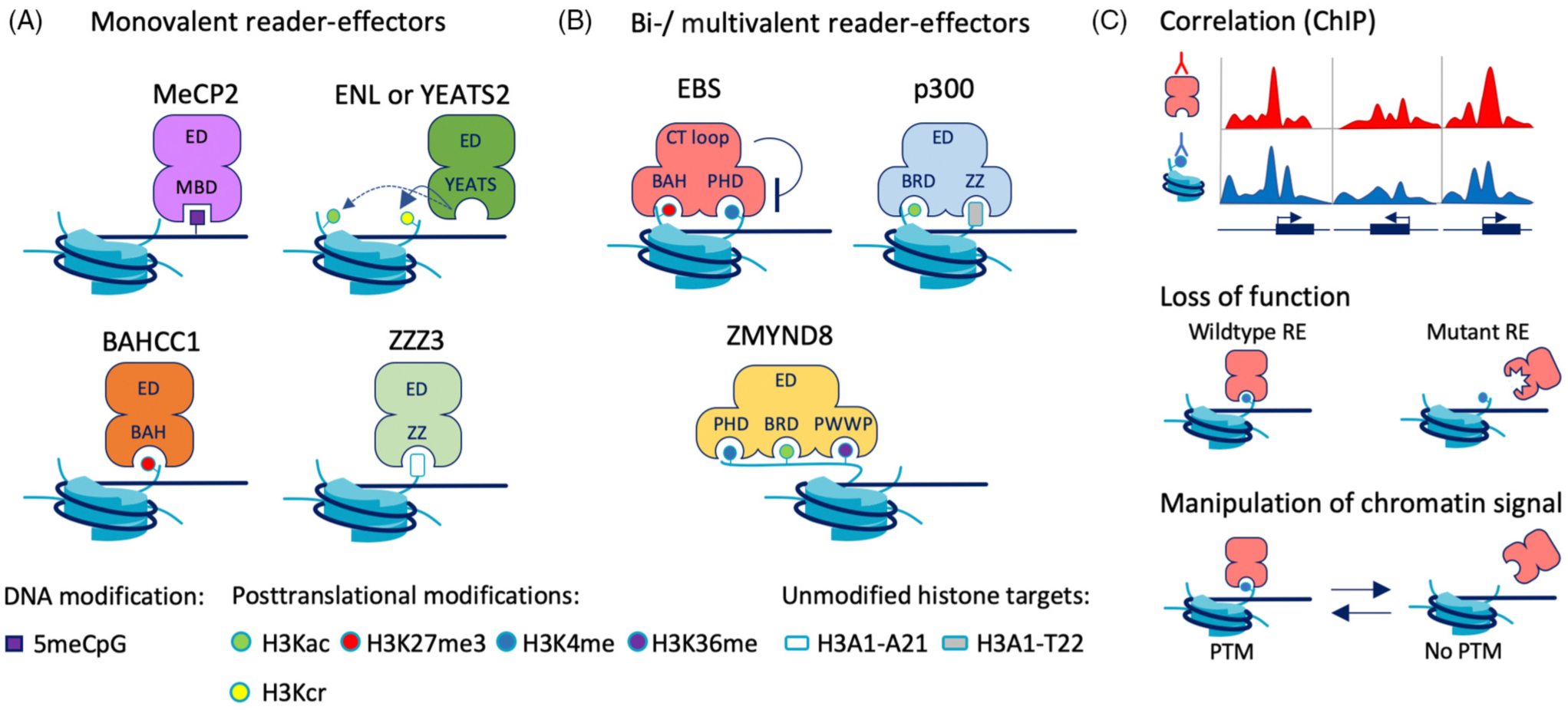
(A) Monovalent REs, such as MeCP2 (methyl-CpG-binding protein 2), ENL (eleven–nineteen leukemia), YEATS2 (YEATS domain-containing protein 2), BAHCC1 (BAH and coiled-coil domain-containing protein 1), and ZZZ3 (ZZ-type zinc finger-containing protein 3) contain a single reader domain that interacts with one chromatin signal. Recent data suggest that YEATS preferentially binds K27cr [22,23]. (B) Bi- and multivalent REs EBS (EARLY BOLTING IN SHORT DAYS), p300, and ZMYND8 (protein kinase C-binding protein 1) interact with two or three chromatin signals through multiple reader domains within the protein. EBS contains a C-terminal loop that may inhibit PHD-H3K4me3 interactions to favor BAH-H3K27me3 binding [27]. (C) Experimental approaches to determine how chromatin marks regulate RE activity in cells are illustrated: correlative mapping of REs and their target signals via ChIP, loss of function via mutation of the reader domain, and erasure or addition of chromatin marks. Abbreviations: BAH, bromo-adjacent homology; BRD, bromodomain, ED, effector domain; PHD, plant homeodomain; YEATS, Yaf9 ENL AF9 Taf14 Sas5; ZZ, ZZ-type zinc finger.
Other examples of monovalent transcriptional regulators include eleven–nineteen leukemia (ENL), a super elongation complex (SEC) unit, and YEATS2, a member of the Ada2A-containing (ATAC) histone acetyltransferase complex. Each contains a YEATS domain, an eight-stranded protein fold that binds to H3K9ac and to a lesser extent H3K27ac and H3K18ac [17] (Figure 2A, top right). Before the discovery of YEATS, bromodomains (BRDs), double plant homeodomain fingers (DPFs), and double pleckstrin homology (PH) domains were the only protein folds known to specifically recognize histone acetyllysines [18]. ENL ChIP signals overlap with acetylated histones. Mutations introduced into the YEATS domain abolish ENL and YEATS2 binding and transcriptional regulation at H3K27ac-enriched target genes [19–21]. Emerging structural data suggest that the YEATS domain may differ from BRD in that YEATS preferentially binds H3Kcr versus Kac [22,23].
It was recently discovered that BAHCC1, a transcription-silencing RE, binds H3K27me3 through its bromo-adjacent homology (BAH) domain as evidenced by crystallography and colocalization of BAHCC1 with H3K27me3 (ChIP) at target genes in Jurkat leukemia cells [24] (Figure 2A, bottom left). The introduction of a point mutation in the BAH domain abrogated its genomic distribution and resulted in similar gene misregulation as a BAHCC1 knockdown with shRNA. Previously, H3K27me3 was only known to recruit polycomb repressive complexes to chromatin. Therefore, the BAH domain sheds new light on how H3K27me3 affects gene regulation. The most recently discovered histone reader mechanism, binding of unmodified histone H3A1–-A21, was reported for the ZZ-type zinc finger (ZZ) domain [25] of the RE ZZZ3, a member of the ATAC complex (Figure 2A, bottom right). ZZ-domain binding is not entirely specific as it can interact with lysine-methylated and K4-acetylated H3A1-A21 [26]. Rescue of a ZZZ3 mutant with wildtype ZZZ3, and not a ZZ-domain mutant, restored gene expression levels of target genes. These studies collectively demonstrate that recognizing chromatin signals via reader domains is a critical step for the function of RE proteins.
Multiple chromatin modifications within a single histone, nucleosome, or genomic region are recognized by bivalent or multivalent RE proteins that contain more than one histone-binding domain (Figure 2B). In arabidopsis, modulators of flowering time EARLY BOLTING IN SHORT DAYS (EBS) and SHORT LIFE (SHL) each contain adjacent reader domains, BAH that binds H3K27me3, and PHD that binds H3K4me3 [27,28]. Mutations in either histone binding pocket of EBS result in the same phenotype (early flowering) as a loss-of-function mutation, ebs [27]. A unique feature of EBS is a C-terminal loop that blocks the PHD domain in crystal structure, suggesting an autoinhibitory mechanism that might switch the target preference of EBS from H3K4me3 to H3K27me3 (Figure 2B, top left). Recently, the chromatin-modifying RE p300, that has been known for a while to contain a BRD that binds acetylated histone lysines, was found to also contain a functional ZZ domain that binds histone resides 1–22 (H3A1-T22; Figure 2B, top right) [29]. The BRD and ZZ domains show cooperative binding in pull-down assays, and ZZ is necessary for p300 to bind and modify chromatin at genes and enhancers in human lung carcinoma cells. A tripartite histone reader called ZMYND8, a subunit of DNA repair complexes, contains a PHD-BRD-PWWP domain that binds histone H3K4me-K14ac-K36me (Figure 2B, bottom). Mutational analyses confirmed that each histone binding domain is necessary for optimal recruitment of ZMYND8 to sites of DNA damage in human U2-OS cells [30].
Much of what we understand about the recognition of chromatin marks by REs in cells is based on data from chromatin profiling (correlation) and reader-domain mutations (loss-of-function) (Figure 2C). To directly demonstrate that chromatin modifications function as control signals, RE activity needs to be observed as chromatin marks are written and erased at genomic loci. Chemical inhibition and genetic knockdown of chromatin modifying enzymes have been used to deplete cells of specific chromatin marks, and loss of binding of the corresponding REs (e.g., BAHCC1 [24] and ZMYND8 [30]) was observed via ChIP. Chromatin marks can also be masked to perturb RE engagement, as demonstrated by an isolated BRD domain that competed away ZMYND8 binding of H3K14ac [30]. Chromatin marks can also be manipulated with fusion proteins built from chromatin-modifying proteins, such as the Gal4-EED/EZH2 system reported by Hansen et al. in 2008 [31]. In more recent work, the ‘epigenome editing’ approach has been substantially expanded to include other chromatin writer enzymes [3,32–36] but has yet to be used extensively in experiments to study the dynamic engagement and activity of REs. We discuss epigenome editing further in the section ‘Reader-effectors as substrates for protein engineering and synthetic biology.’
Effector activity: transcriptional regulation and chromatin remodeling
Once an RE engages with a chromatin mark (signal) at a specific genomic region, the effector domain within the protein can act within the local chromatin environment to modulate transcriptional regulation, chromatin organization and remodeling, and DNA repair. Compared with reader domains, effector domains are often poorly defined structurally and are less evolutionarily conserved. In this review, effector domains are generally defined as the region of the RE that lies outside of the reader domain. These regions are typically annotated as ‘disordered’, lacking any predicted secondary structure motifs. Effector domains often contain post-translationally modified sites and charged patches, which indicates a scaffolding function that may explain why REs are often found in multiprotein complexes. Here, we highlight recent investigations that have helped to generate a clearer understanding of effector mechanisms.
REs can modulate transcriptional initiation by affecting the recruitment and behavior of RNA polymerase II (RNA Pol II). The preinitiation complex at transcription start sites (TSSs) includes a set of transcription factors that directly position RNA Pol II, whereas RE proteins play a modulatory role in the transcription initiation and elongation steps [37]. ENL protein contains a YEATS reader domain and 338 amino acid disordered, highly charged C-terminal effector domain (Uniprot Q03111), and is a member of the SEC (Figure 3A). Depletion of ENL via the dTAG system [20,38] in an acute myeloid leukemia cell line (MV4–11) led to increased pausing of RNA Pol II at TSSs of active genes, as evidenced by ChIP-seq of Pol II. ChIP data also show that members of SEC such as AFF4 and CDK9 are lost from gene promoters after ENL is degraded, suggesting that ENL functions as a recruiter to promote effective transcriptional elongation at tumor-promoting genes such as MYC, MYB, MEIS1, and others [20].
Figure 3. Reader-effectors modulate transcriptional regulation, chromatin remodeling, and DNA repair through effector domains (EDs) and protein binding partners.
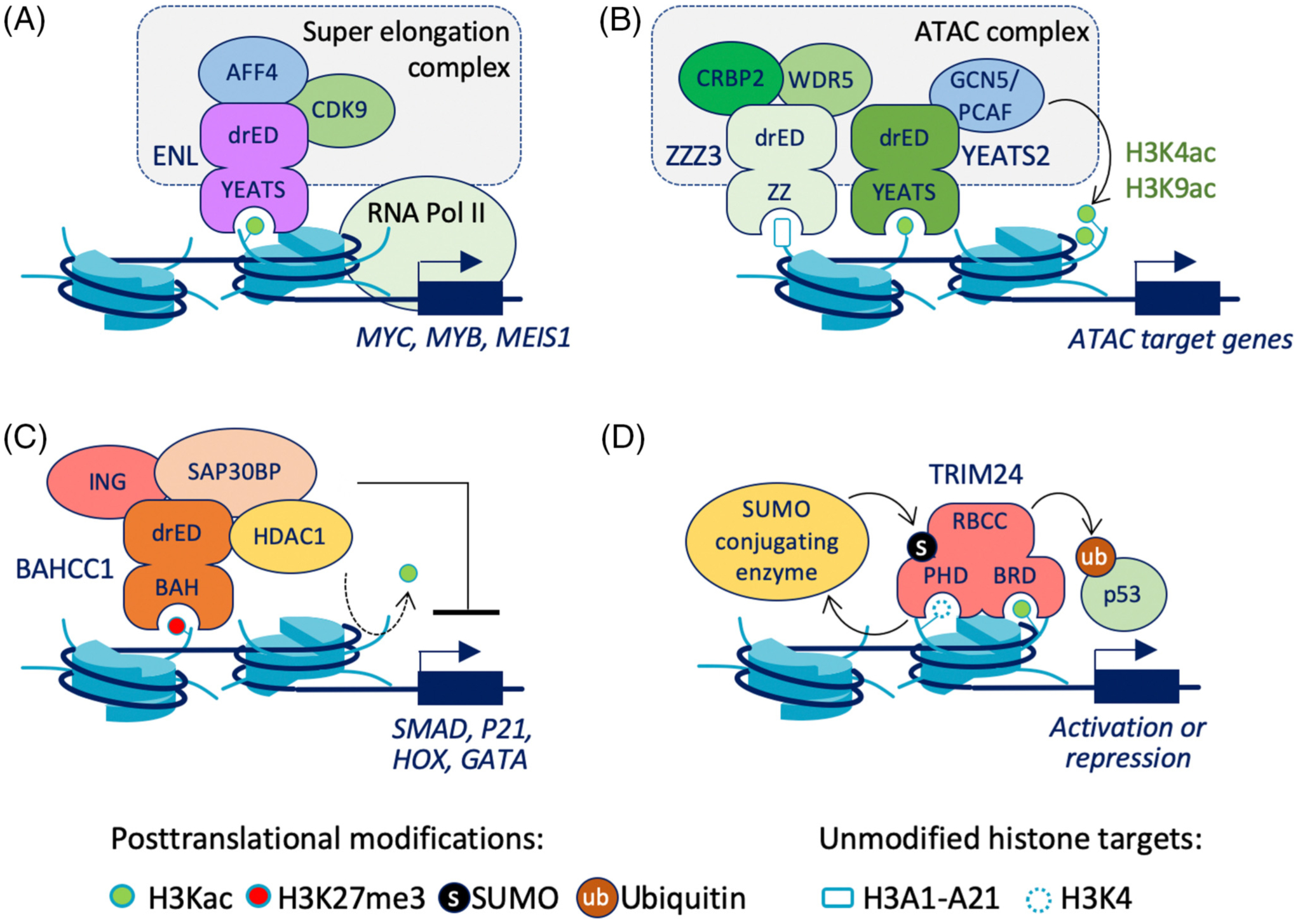
ENL (eleven–nineteen leukemia), a part of the super elongation complex (SEC), binds to H3Kac and then initiates transcriptional initiation and elongation of ENL target genes. (B) YEATS2 (YEATS domain-containing protein 2) and ZZZ3 (ZZ-type zinc finger-containing protein 3) bind to acetylated lysines and lead to the formation of the ATAC complex to transcribe ATAC target genes. (C) The binding of BAHCC1 (BAH and coiled-coil domain-containing protein 1) to H3K4me3 recruits HDAC1 (Histone deacetylase 1), ING (Inhibitor of growth) proteins, and SAP30BP (SAP30-binding protein) leading to repression of target genes. (D) TRIM24 recruitment to H3K4-H3K23ac via PHD-BRD stimulates SUMOylation of TRIM24 via SUMO-conjugating enzymes. TRIM24 ubiquitinates the transcription factor p53. Abbreviations: AFF4, AF4/FMR2 family member 4; ATAC, Ada2a-containing; CRBP2, cellular retinol-binding protein II; CDK9, cyclin-dependent kinase 9; drED, disordered region effector domain; GCN5, general control of amino acid synthesis protein 5-like 2; PCAF, histone acetyltransferase PCAF; RNA Pol II, RNA polymerase II; WDR5, WD repeat-containing protein 5.
REs can also modify and organize chromatin to generate transcriptionally permissive or repressive states. For instance, transcriptionally active chromatin is supported by YEATS2 and ZZZ3 RE proteins, which both interact with the ATAC histone acetyltransferase complex that generates H3K4ac and H3K9ac via GCN5 (KAT2A) or PCAF (KAT2B) (Figure 3B) [21,26]. In a recent study, ZZZ3 knockdown led to a loss of YEATS2 binding to ATAC target genes and loss of H3K9Ac and H3K4Ac, indicating that ZZZ3 and YEATS2 have complementary roles in chromatin modification as subunits of the ATAC complex.
Recently, H3K27me3-binding protein BAH Domain and Coiled-Coil Containing 1 (BAHCC1) was found to support transcriptionally repressive chromatin through the recruitment of histone deacetylases and polycomb proteins (Figure 3C) [24]. Genetic knockdown of BAHCC1 and UNC1999 treatment in Jurkat cells followed by combined RNA-seq and ChIP-seq analyses showed upregulation of tumor-suppressive genes like CDKN1A (p21), as well as crucial developmental transcription factors HOX, GATA, and SMAD. Mass spectrometric analyses of pulldowns of BAHCC1 in HeLa and co-immunoprecipitations in Jurkat cells revealed that BAHCC1 binds to SAP30BP, HDAC1, and Inhibitor of Growth (ING) proteins. Most of BACCH1 (~95%) is an uncharacterized region that contains disordered and coiled-coil motifs (Uniprot Q9P281), and may encompass an effector domain that acts as a scaffold for its binding partners. BAH-containing transcriptional repressors have also been found in plant cells (Arabidopsis thaliana). EBS and SHL mediate protein–protein interactions that support a condensed chromatin structure [27,28]. It is unclear whether a distinct effector domain exists in EBS apart from the histone PTM-binding domains. The histone-binding domains BAH and PHD span ~70% of EBS. The remaining C-terminal loop is essential for histone PTM binding, but is not required for interactions with MSI4 (MULTICOPY SUPPRESSOR OF IRA 4) [27], a member of the PRC2-like complex in plants. Recent work has also shed new light on a long-studied repressive effector, the Kruppel-associated box (KRAB). A clever high-throughput magnetic separation-based system called HT-Recruit was used to screen hundreds of KRAB variants, and revealed that evolutionarily old KRAB domains have transcriptional activation, rather than repression, activity [98].
A different type of RE output that has recently come to light is covalent RE modification induced by the engagement of reader domains with histone PTMs. A recent study of TRIM24 showed that engagement of the PHD-BRD with H3K4me0-H3K23ac leads to SUMOylation of TRIM24 at lysines 723 and 741 (Figure 3D). Inactivation of the BRD by mutation or with a small-molecule inhibitor (IACS-9571) abolished TRIM24 SUMOylation, which is proposed to be mediated by SUMO-conjugating enzymes within the local chromatin environment. The biological importance of TRIM24 SUMOylation is supported by results from an experiment where a double mutant K723R/K741R abolished SUMOylation and led to misregulation of several target genes in MCF7 breast cancer cells [39].
How reader-effectors impact disease states
Aberrant expression of REs and mutations in their chromatin-binding and effector domains have been recognized as key contributors to disease pathology. A large number of studies underscore the potential of REs as targets for new therapies. Here, we highlight recent studies of RE dysfunction in cancer, neurological disease, and immune disease. The multitude of known examples does not allow us to provide a comprehensive survey. Several more examples of misregulated REs are provided in Table 1, and readers may find even more examples for the diseases listed above, plus cardiovascular disease, in other reviews [15,40–42].
Table 1.
Reader-effector proteinsa, their functional domains, biological activity, and misregulation in human diseaseb
| RE (Uniprot) | Chromatin signal(s) | Reader domain(s) | Effector domain(s) | Interacting effector(s) | Misregulation in human disease |
|---|---|---|---|---|---|
| Positive regulation of transcription and/ or elongation | |||||
| BRD4 (O60885) | H3/H4Kac | BRD |
|
|
Cancer: fusion [81,82] |
| BRD4S, short isoform | H3/H4Kac | BRD |
|
|
Cancer: overexpressed [83] |
| PcTF | H3K27me3 | CD | VP64 | MED15, MED17, TFII, TBP [84] | Not applicable |
| TRIM24 (O15164) |
|
|
RBCC E3 ubiquitin ligase domain | Coactivators (e.g., NCOA2, CARM1), corepressors (e.g., CBX1/3/5) | Cancer: overexpressed [85] |
| AF9 (P42568) | H3K9cr, H3K9ac, H3K9bu | YEATS | Disordered C-terminal region | SEC (EAF1, EAF2, CDK9, AFF), P-TEFb complex, ELL | Cancer: fusion [86] |
| ENL (Q03111) | H3K9ac, H3K9cr | YEATS | Disordered C-terminal region | SEC (AFF4, CDK9), ALKBH4, EBAFb complex | Cancer: fusion [86], mutated [87], overexpressed [19,20] |
| Negative regulation of transcription | |||||
| BAHCC (Q9P281) | H3K27me3 H3K4me3 |
BAH PHD |
Disordered N-terminal region | SAP30BP, HDAC1, ING | Cancer: overexpressed [24] |
| EBSc (F4JL28) |
|
|
Undetermined | MSI4, HDACs | Not applicable |
| SHLc (Q9FEN9) |
|
|
Disordered C-terminal region | MSI4, HDACs | Not applicable |
| CBX1/HP1a (P83916) | H3K9me3 | CD | Disordered region, chromoshadow domain | HMTs (SUV39H1, SETDB1, KMT5B, KMT5C) | Cancer: overexpressed [88] |
| CBX2 (Q14781) | H3K27me3 | CD | Polycomb box | Polycomb repressive complex 1: RING1A/B, BMI1, PHC | Cancer: overexpressed [43,44] |
| HP1ad (P05205) | H3K9me3 | CD | Chromoshadow domain | ||
| MeCP2 (P51608) | 5meCpG | MBD | NID | Nuclear receptor corepressor 2 | Neurological: mutated [47–50], underexpressed [51] |
| MBD1 (Q9UIS9) | 5meCpG | MBD | TRD | Cancer: overexpressed [89] | |
| MBD2 (Q9UBB5) | 5meCpG | MBD | TRD | Nucleosome remodeling and deacetylase (Mi-2/NuRD) complex, Sin3A | Cancer: overexpressed [90–92] Immune: overexpressed [56,58], underexpressed [57] |
| MBD4 (O95243) | 5meCpG | MBD | TRD | Sin3A, HDAC1, DNMT1, DNMT3B | Cancer: overexpressed [92] Immune: overexpressed [56,58], underexpressed [57] Neurological: mutated [93,94], overexpressed [58,95], underexpressed [57] |
| Chromatin remodeling | |||||
| BPTF (Q12830) |
|
|
Coiled-coil and disordered regions | Nucleosome remodeling factor complex | Cancer: overexpression [45] Neurological: mutated [96] |
| p300 (Q09472) |
|
|
Acetyltransferase targeting histones and non-histone proteins | Cancer: fusion [97] | |
| AIRE (O43918) | H3K4 | PHD | Caspase recruitment domain, SAND domain | Immune: mutated [54,55] | |
| RAG2 (P55895) | H3K4me3 | PHD | Disordered central region | RAG complex (RAG1, HMGB1/2) | Immune: mutated [52] |
| YEATS2 (Q9ULM3) | H3K27ac, H3K27cr | YEATS | Disordered N- and C-terminal regions | GCN5, PCAF | Cancer: overexpressed [21] |
| ZZZ3 (Q8IYH5) | H3A1-A21 | ZZ | Disordered N-terminal region | CRBP2; ATAC (KAT14, KAT2A, TADA2L, TADA3L, MBIP, WDR5, YEATS2, CCDC101, DR1) | Not applicable |
The table includes RE proteins mentioned in this review and select additional REs that are associated with human diseases. REs are grouped by their biological outputs and by reader domain type. The corresponding chromatin signals, reader domains, effector domains, and other effector proteins that interact with the RE are described in the literature cited by the Uniprot database entry for each RE, and by work cited in this review.
Abbreviations: ALKBH4, AlkB homolog 4, lysine demethylase; ATAC, ADA2A-containing complex; ATAD5, ATPase family AAA domain containing 5; BMI1, polycomb complex protein BMI-1; CARM1, coactivator associated arginine methyltransferase 1; CHD4, chromodomain-helicase-DNA-binding protein 4; CRBP2, cellular retinol binding protein 2; CTM, C-terminal motif; DNMT1, DNA methyltransferase 1; DNMT3B, DNA methyltransferase 3 beta; ELL, elongation factor for RNA polymerase II; ET, extraterminal; GLTSCR1, glioma tumor suppressor candidate region gene 1; HDAC, histone deacetylase; HMGB1/2, high mobility group box 1 protein 1/2; HMT, histone methyltransferase; JMJD6, Jumonji domain containing 6; KMT5B, lysine methyltransferase 5B; KMT5C, lysine methyltransferase 5C; MED15, mediator complex subunit 15; MED17, mediator complex subunit 17; MSI4, WD-40 repeat-containing protein MSI4; NCOA2, nuclear receptor coactivator 2; NID, NCoR/SMRT interaction domain; n/a, not available; NSD3, nuclear receptor binding SET domain protein 3; P-TEFb, positive transcription elongation factor b; RAG, recombination activating gene; RBCC, ring, B-Box and coiled-coil; RING1A/B, ring finger protein 1A/B; SAND, human Sp100, Aire1, NucP41/P75, and Drosophila DEARF1; SEC, super elongation complex; SETDB1, SET domain bifurcated histone lysine methyltransferase 1; SIN3A, SIN3 transcription regulator family member A; SUV39H1, SUV39H1 histone lysine methyltransferase); TBP, TATA-binding protein; TP53, tumor protein 53; TRD, transcription repression domain.
Arabidopsis thaliana (plant).
Drosophila melanogaster (fly).
Clinical and experimental evidence show that misregulated and mutated RE proteins support cancer progression. Dysregulation and mutations of the polycomb chromatin protein EZH2 (a writer enzyme) has been a strong center of focus for cancer epigenetic research. Recently, scientists have reported elevated levels of the polycomb protein CBX2, a reader of H3K27me3, and its binding partners BMI1 and RING1B in aggressive basal-like and triple-negative breast cancer tissues [43,44], bringing more focus to REs as central factors in neoplasia. Another RE protein BAHCC1 was recently discovered to also interact with H3K27me3, and BAHCC1 is overexpressed in acute myeloid leukemias, and acute lymphoblastic leukemia of T cell or B cell lineages (T-ALL or B-ALL) [24]. BRD PHD finger transcription factor (BPTF), an RE that contains tandem PHD and BRD reader motifs, is overexpressed in high-grade gliomas (HGG) in children and adults. An shRNA screen identified BPTF as a vulnerability for HGG growth [45], pointing toward BPTF as a priority target for therapy.
RE dysfunction has also been implicated in the etiology of neurological diseases. MeCP2 is necessary for human brain development and cognition [46]. Point mutations clustered in the MBD domain of MeCP2 lessen its affinity for methylated DNA, causing severe loss of function leading to Rett syndrome, a severe neurodevelopmental disorder [47]. MeCP2 downregulation, SNPs, and 3′ UTR variants have been linked to autism and autism spectrum disorders [48–51]. MBD4, another methyl CpG-binding protein, is upregulated in the hippocampus and prefrontal cortex of patients with schizophrenia.
Mutation and misregulation underlie RE dysfunction in immunodeficiency and autoimmune diseases. The recombination activating gene 2 (RAG2) RE interacts with the DNA endonuclease RAG1 to initiate V(D)J recombination, which allows B and T cells to generate receptor diversity and an adaptive immune response. Mutations in the RAG2 PHD domain lead to severe combined immunodeficiency (SCID), a condition where patients lack functional B or T cells, or Omenn syndrome (OS), a form of SCID with deficiency of B cells and elevated T cells [52,53]. Point mutations in the two PHD domains of the autoimmune regulator (AIRE) are linked to the rare disease autoimmune polyglandular syndrome type I (APS-1) [54,55]. MBD2 and MBD4 are overexpressed in CD4+ T cells in systemic lupus erythematosus (SLE) patients, while underexpression and loss of DNA methylation is seen in patients with primary immune thrombocytopenia (ITP) [56–58].
Reader-effectors as substrates for protein engineering and synthetic biology
Epigenetic engineering is becoming increasingly recognized as a powerful approach for cell engineering. Three central features of the chromatin machinery, chromatin modifying enzymes, chromatin marks, and REs, have been used to build molecular tools for manipulating the flow of information in chromatin or building ectopic chromatin de novo in a variety of organisms. Chromatin modifying enzymes have been used for epigenome editing, a method that uses DNA-binding module fused to chromatin modifying enzyme to alter local chromatin features at genomic sites such as pericentromeric chromatin [32], therapeutic genes [33], and noncoding enhancer elements [34], and other loci [3,35,36]. Chromatin marks can also be manipulated by using engineered tRNAs to incorporate modified residues into histones during translation [59] (reviewed in [2]) or by inserting premethylated DNA into specific chromosomal sites [60]. In the following sections, we describe emerging epigenetic engineering technologies inspired by the structure and function of RE proteins.
Engineered probes to track chromatin marks in cells
Histone binding domains from REs are being developed as alternatives to antibodies for in vitro assays. This is particularly useful for studying chromatin marks that cannot be targeted by specific antibodies. Wildtype proteins, including alpha-thalassemia/mental retardation, X-linked (ATRX) ATRX-DNMT3-DNMT3L (ADD) domain, CBX7 chromodomain (CD), and the TAF3 PHD, have been used against H3K9me3, H3K27me3, and H3K4me3, respectively, in peptide arrays, western blot, and ChIP assays [61,62]. The wildtype MPP8 CD and DNMT3A PWWP were fused to generate ‘histone modification interacting domains’ (HiMID), a single pull-down probe for H3K9me3 and H3K36me2/3 (Figure 4A) that obviates the need for sequential two-step ChIP to study bivalent chromatin regions [63]. Reader domains have also been altered at the amino acid level to generate new affinity agents. For instance, the ‘eReader’ system (Figure 4B) uses mutant CBX1 CD with ~five-fold enhanced affinity for H3K9me3 (Kd = 0.13 μM vs. Kd = 0.6 μM for wildtype CBX1). When eReader-MBP (maltose-binding protein) fusion proteins were used instead of primary antibodies to probe a blot of mononucleosomes from HeLa cells and then detected with an antibody against the MBD, the eReader probe showed stronger signal than a western blot with a commercial anti-H3K9me3 primary antibody (Active Motif, 39161) [64]. This result could be due in part to the differences in secondary detection methods for eReader (anti-MBP) versus anti-H3K9me3 (anti-rabbit IgG). Also, binding of commercial antibodies can be affected by additional modifications of the histone tail, for example, reduced binding of anti-H3K9me3 (Active Motif 39161) with H3R8me1-K9me3 [65]. To fully understand how reader domains differ from anti-bodies, further studies with sets of more complex histone ligands should be performed.
Figure 4. Emerging epigenetic engineering technologies leverage the structure and function of reader-effector proteins.

(A) The HiMID (histone modification interacting domains) probe combines a CD from MPP8 and PWWP domain from DNMT3A to identify regions of transcriptional elongation with both H3K9me3 and H3K36me3 via ChIP. (B) The eReader chromatin probe contains mutated chromodomains (CDs) from the reader-effector protein CBX1, enabling enhanced detection of H3K9me3 on a far western blot. (C) Modified reader domains that contain a photoactive amino acid in the binding pocket enable UV light-induced covalent linkage with histone PTMs so that complexes can be extracted and analyzed via ChIP, western blot, or liquid chromatography–mass spectrometry (LC-MS). A photoactive BRD derived from BRD4 is shown here as an example. (D) ChromID couples a CD and a biotin ligase to tag neighboring proteins near histone marks of interest. Abbreviations: GST, glutathione S-transferase; MBP, maltose-binding protein.
Genetically expressed reader-fluorescent protein fusions are useful for tracking gains and losses of histone marks at subnuclear scale in living cells [66–68], as we have discussed in a previous review [69]. Biochemical tagging is the latest read-out mechanism that has been incorporated into chromatin probes to enhance the sensitivity and specificity of detection. The acetyl lysine-binding domain of BRD4 and H3K27me3-binding CD of CBX1 were each engineered to include a photo-sensitive amino acid that upon exposure to UV light rapidly forms a covalent bond between the reader domain and the histone PTM. This stabilizes the reader–signal interaction so that complexes can be extracted and analyzed by ChIP, western blot, or liquid chromatography–mass spectrometry (Figure 4C) [70–72]. Probes built from an engineered reader domain fused to a biotin ligase were used to tag proteins within a 35-nm radius of H3K27me3, H3K9me3, H3K4me3, and mCpG in mouse stem cells (Figure 4D) [73]. These new probes enable the capture of weak and transient interactions between specific readers and their binding partners at high temporal precision.
Epigenome actuation: genetic control via synthetic reader-effectors
Epigenome actuation is a new method in epigenetic engineering that uses synthetic RE proteins to control gene transcription. In contrast to epigenome editing, which alters histone modifications and DNA methylation at specific sites, epigenome actuation operates downstream of the editing step by sensing the marks (signals) and then regulating gene expression (output). In the same way that chromatin probes like Bimolecular Anchor Detector (BiAD) [67], chromatin-sensing multivalent probes (cMAP) [66] and HiMID [63] have been designed to target combinations of chromatin marks, synthetic REs could be designed to target and act upon a subset of genes or enhancers that share a specific chromatin modification profile.
In the epigenome actuation framework, a specific type of chromatin modification acts as a binary input signal, that is, present = 1, or absent = 0, and REs act as logic gates that mediate how certain inputs are transduced to output, that is, gene regulation states (Figure 5A). This approach is similar to the general Boolean logic framework that has been used in other synthetic biology applications [74]. REs can be classified by the type of operation they perform, such as NOT, AND, OR, and so on. For instance, a transcriptionally repressive RE such as CBX2/4/6/7/8 from polycomb repressive complex 1, mediates a NOT operation where the presence of a signal (H3K27me3) is translated into a transcriptional ‘off’ state (Figure 5B). Using H3K27me3 as input, we were the first to demonstrate that a synthetic RE, polycomb transcription factor (PcTF) built from a CBX8 CD reader and a VP64 transcription-activating effector, could transduce H3K27me3 signal into a transcriptional ‘on’ state [75]. Later, Park et al. used a synthetic RE, DPN-VP64, to activate a gene near artificially generated DNA methylation (6meA) in mammalian cells [76]. Both synthetic REs are examples of a buffer gate, an if-then logic operator that simply passes an input to an output. Buffer gates can be designed to amplify the effect of an input, as we have demonstrated with a bivalent PcTF that contained two tandem CD readers, showed enhanced avidity for H3K27me3 in vitro, and showed about twofold-enhanced activation of a target gene in cells [77].
Figure 5. Epigenome actuation implemented with synthetic reader-effector (RE) proteins.
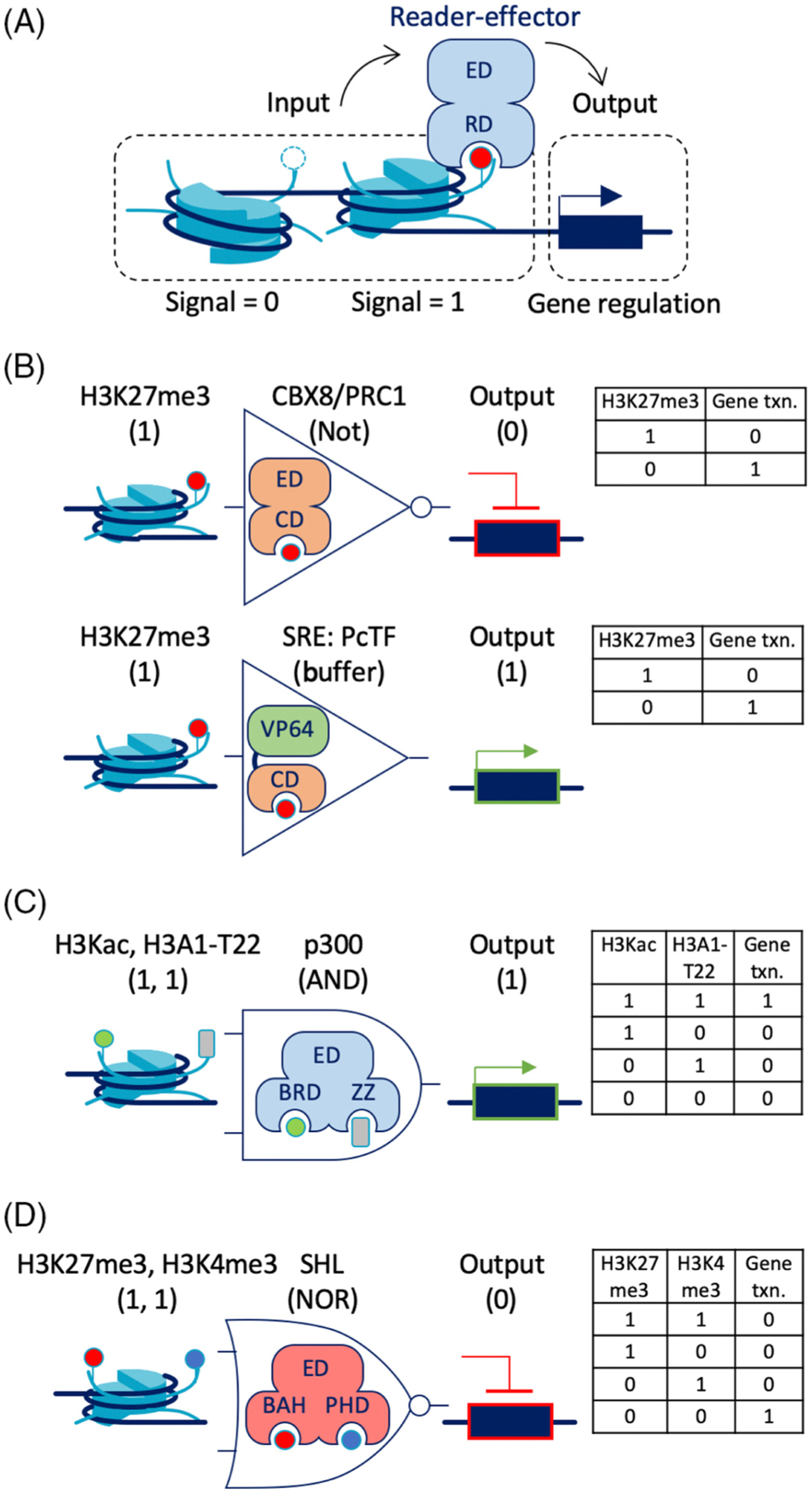
(A) The simplified diagram shows the flow of epigenetic information from a chromatin signal (0 = absence, 1 = presence), to an RE, to output (regulation of gene transcription). Sensor actuators can read these signals and translate them into functional output. (B) CBX8 (a member of the PRC1 complex) acts as a one-input operator (NOT) that transduces H3K27me3 into a negative output (gene repression). The synthetic RE PcTF also acts as buffer gate for H3K27me3, but inverts the signal into activation. (C) Co-recognition of H3Kac and H3A1-T22 by the natural bivalent RE p300 suggests that synthetic REs could be designed as two-input AND operators. (D) The RE SHL binds equally well to H4 that carries K27me3 or K4me3 or both, and therefore represents an OR operator. Abbreviations: BRD, bromodomain; CBX8, chromobox protein homolog 8; CD, chromodomain; gene txn., gene transcription; PcTF, polycomb transcription factor; PHD, plant homeodomain; PRC1, polycomb repressive complex 1; TRIM24, tripartite motif-containing protein 24.
So far, synthetic REs have only been used as one-input gates. Certain natural REs show the potential to build two-input gates. For instance, a two-input AND operator would bind two distinct chromatin signals to activate a target gene. Binding must require the presence of both, that is, the presence of one or the other signal would result in no or very little effector activity. The RE p300 may meet these criteria, as suggested by the cooperative binding observed for its BRD and ZZ reader domains [29] (Figure 5C). It may be possible to build a NAND (‘not and’) gate by replacing the p300 effector domain with a module like Krüppel associated box (KRAB), a popular choice for synthetic transcriptional repressors. Bivalent REs such as SHL that bind equally well in the presence of H3K27me3, H3K4me3, or both [28] may represent a NOR operation where the output is gene repression (Figure 5D). Switching the output to gene activation by replacing the effector domain with a transcriptional activator would result in an OR gate. Synthetic RE design based on ZMYND8 [30] might enable three-input signal integration using a single protein, whereas other synthetic biological systems rely upon layered gene transcription cascades to achieve multisignal processing. Studies of ZMYND8 also suggest that specific recognition of a linear arrangement of chromatin marks (i.e., H3K4me-H3K14ac-H3K36me) could be achieved with a rigid, linear arrangement of chromatin-binding modules.
Beyond exploring the potential to rationally design chromatin signal integration, epigenome actuation with synthetic REs is useful for basic research. Synthetic REs can be used as simplified models to deconvolute the mechanism of chromatin signal transduction, that is, chromatin binding followed by gene regulation, by determining which RE domains are necessary and/or sufficient for a certain biological effect. The overall size of large REs, and their primary and secondary interactions within multiprotein chromatin complexes make it challenging to perform experimental manipulations to distinguish the roles of each protein and subdomain in the regulation of specific genes and phenotypes. Recently, a pared-down MecP2 protein that contained only the MBD and the NCoR/SMRT interaction domain (NID) was used to rescue normal neurological function in an MeCP2-deficient mouse model [78]. This breakthrough shed light on why mutations in Rett syndrome occur most frequently in MBD and NID, although other regions in MeCP2 are highly conserved. Synthetic REs could also be used to determine the mechanistic role of reader domain valency and physical arrangement of multiple reader domains within REs. For instance, do multiple reader domains have an additive or synergistic effect on RE-mediated gene regulation?
Further development of epigenome actuation could fill a critical gap in epigenetic cellular engineering. Current epigenome editing approaches operate at the chromatin signal step (Figure 1B), and rely upon endogenous regulators such as REs, DNA-binding regulators, and chromatin remodelers to act downstream. However, genes in cells that lack these regulators because of cell-specific silencing or a disease-related mutation [e.g., SWItch/Sucrose NonFermentable (SWI/SNF)] remodeling complex proteins are mutated in 25% of cancers [79]) might not respond to epigenome editing. Synthetic REs could be used to complement epigenome editing by compensating for deficient regulation machinery to drive gene regulation at the target site. An inspiring example of using synthetic REs (synthetic read-out synR modules) in conjunction with epigenome editing (artificial, targeted methylation of adenine in DNA) is presented in work by Park et al. [76]. Furthermore, epigenome actuation alone can counteract transcriptional misregulation of aberrant REs in disease. In breast cancer cells that overexpress the EZH2 chromatin modifying enzyme and CBX proteins, overexpression of the synthetic RE PcTF restored the expression of epigenetically silenced genes [80].
Concluding remarks
REs play a central role in chromatin structure and function as drivers of epigenetic regulation by transducing chromatin modification signals into transcriptional regulation, chromatin reorganization, and feedback through self-modification. As more misregulated and mutated REs are becoming identified in cancer, neurological diseases, and immune diseases, the importance of REs in cell development and disease is becoming more pronounced. Many mechanistic features, such as signal recognition and effector function, remain untapped for drug design (see Outstanding questions). Our mechanistic understanding could be further expanded by using bioengineering approaches like epigenome editing to determine how chromatin mark dynamics affect RE engagement and regulatory activity at target sites.
Outstanding questions.
Small molecule inhibitors have been used to block the reader function of hyperactive BRD-containing reader-effectors in cancer. How can the growing mechanistic understanding of other reader domains, as well as effector domains, be further leveraged for drug design and development?
Could epigenome editing, the addition or erasure of chromatin signals from a specific site, be used to validate reader-effectors’ recognition of chromatin signals in living cells? Would the results support or change conclusions from studies that have used chromatin profiling (correlative) and reader-effector mutations (loss of function)?
Reader domains are typically tested in isolation to determine their affinity and specificity for histone modifications and DNA methylation, and these data have been used to infer their activity within whole reader-effector proteins that sometimes contain multiple tandem reader domains. Would in vitro studies with fused or scaffolded reader domains produce new insights into higher-dimensional ligand specificity (e.g., recognition of a linear array of histone modifications)?
Can the full complement of Boolean operations be carried out by any known or unknown natural reader-effector proteins? If not, can synthetic reader-effectors be designed to carry out these operations?
The scope of the epigenetic engineering field is expanding from a focus on chromatin modifying enzymes and their marks to REs. Reader domains are gaining popularity as substrates for engineering research tools to study chromatin structure and function. Epigenome actuation is a new area where synthetic REs are designed by linking chromatin signal inputs to custom regulatory outputs. Insights from research and advances in engineering of RE proteins will produce complementary knowledge to understand and control cell behavior.
Highlights.
Mounting evidence associates reader-effectors to disease pathology and progression, and has brought more focus to chromatin reader-effectors as targets for therapies.
Ongoing investigation of reader-effectors provides new insights into chromatin signaling mechanisms, such as recognition of histone H3(1–22) by the p300 ZZ domain.
Reader-effectors have recently been discovered to modify other reader-effector proteins or generate covalent self-modifications.
Wildtype and engineered reader domains have been repurposed as affinity agents for chromatin research, making them suitable alternatives to commercial antibodies.
Synthetic covalent modification is the latest output that has been used to extend the utility of reader-effector-derived probes, and enables enhanced sensitivity and specificity for mapping the chromatin landscape.
Epigenome actuation, the use of synthetic reader-effectors to transduce chromatin signals into gene regulation outputs, complements epigenome editing and addresses unmet needs in epigenetic engineering.
Acknowledgments
K.A.F. is supported by the President’s Fellowship from Georgia Tech and the Centennial Scholar Fellowship from the Laney Graduate School at Emory. She is also supported by NIH NCI (R21CA232244) and start-up funds from the joint Georgia Tech and Emory School of Medicine, Wallace H. Coulter Department of Biomedical Engineering.
Glossary
- Bivalent chromatin region
a region of chromatin that contains two or more chromatin signals, where each signal is typically associated with opposing gene activity states, such as transcriptional activation (H3K36me2/3) and transcriptional silencing (H3K9me3).
- Chromatin
a system of nucleic acids (DNA and RNA) and proteins that physically organize the genome and regulate gene transcription within the nuclei of eukaryotes. Chromatin features are also present in archaebacteria, which lack nuclei.
- ChIP
a method to investigate the genomic regions where a protein of interest binds to DNA through precipitating the protein using an antibody and amplifying/sequencing the DNA.
- Chromatin modifying enzymes
a group of enzymes that covalently modify chromatin, typically referred to as writers and erasers. Writers catalyze the attachment of small molecules (e.g., acetyl, methyl, or phosphate groups) to histone residues or cytosines in DNA. Erasers catalyze the removal of these modifications.
- Chromatosome
a unit of chromatin structure that includes a nucleosome and unwrapped linker DNA bound by histone H1.
- Methylated DNA
a chromatin signal where cytosines at cytosine-guanine dinucleotides (5′-CpG-3′) are methylated to form 5-methylcytosine. This modification, 5meCpG, is often associated with transcriptional silencing.
- Effector domain
a domain within a reader-effector protein that mediates a biological action such as transcriptional activation, transcriptional repression, recruitment of other proteins/cofactors, or modification of chromatin.
- Epigenome editing
a bioengineering technique that uses synthetic chromatin modifying enzymes to generate chromatin signals. A fusion protein that contains a DNA-binding module and a chromatin modifying enzyme is used to alter local chromatin features at specific genomic sites.
- Epigenome actuation
a bioengineering technique that uses synthetic reader-effectors to transduce chromatin signals into biological outputs. A fusion protein that contains a reader domain and an effector domain is used to regulate genes that are located near specific chromatin signals.
- Epigenetic engineering
a specialized area within cellular engineering or bioengineering that focuses on manipulating features of the chromatin machinery such as chromatin modifying enzymes, chromatin marks, and reader-effectors.
- Histones
a family of basic proteins that associate with DNA to form nucleosomes and affect chromatin organization and gene expression.
- Histone post-translational modification (PTM)
a chromatin signal that consists of covalent modifications of histone proteins, including but not limited to methylation, acetylation, crotonylation, ubiquitination, and phosphorylation.
- Nucleosome
the simplest repeating unit of chromatin. A nucleosome consists of two copies each of histones H2A, H2B, H3, and H4, 147 bp of DNA wrapped in 1.7 turns around an [H2A: H2B]2,H32:H42 core octamer.
- Reader domain
a domain within a reader-effector protein that physically binds to methylated DNA or one or more specific histone PTMs.
Footnotes
Declaration of interests No interests are declared.
References
- 1.Daly MM and Mirsky AE (1955) Histones with high lysine content. J. Gen. Physiol 38, 405–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tekel SJ and Haynes KA (2017) Molecular structures guide the engineering of chromatin. Nucleic Acids Res. 45, 7555–7570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holtzman L and Gersbach CA (2018) Editing the epigenome: reshaping the genomic landscape. Annu. Rev. Genomics Hum. Genet 19, 43–71 [DOI] [PubMed] [Google Scholar]
- 4.Huang H et al. (2015) Quantitative proteomic analysis of histone modifications. Chem. Rev 115, 2376–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu X and Zhang Y (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet 18, 517–534 [DOI] [PubMed] [Google Scholar]
- 6.Hsieh T and Brutlag DL (1979) A protein that preferentially binds Drosophila satellite DNA. Proc. Natl. Acad. Sci. U. S. A 76, 726–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.James TC and Elgin SC (1986) Identification of a nonhistone chromosomal protein associated with heterochromatin in Drosophila melanogaster and its gene. Mol. Cell. Biol 6, 3862–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhalluin C et al. (1999) Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496 [DOI] [PubMed] [Google Scholar]
- 9.Fischle W et al. (2003) Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 17, 1870–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bannister AJ et al. (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120–124 [DOI] [PubMed] [Google Scholar]
- 11.Yun M et al. (2011) Readers of histone modifications. Cell Res. 21, 564–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Musselman CA et al. (2012) Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol 19, 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang B et al. (2009) Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell. Biol 29, 1375–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bienvenu T and Chelly J (2006) Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet 7, 415–426 [DOI] [PubMed] [Google Scholar]
- 15.Du Q et al. (2015) Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics 7, 1051–1073 [DOI] [PubMed] [Google Scholar]
- 16.Zhu H et al. (2016) Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet 17, 551–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y et al. (2014) AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 159, 558–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrews FH et al. (2016) The essential role of acetyllysine binding by the YEATS domain in transcriptional regulation. Transcription 7, 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan L et al. (2017) ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erb MA et al. (2017) Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mi W et al. (2017) YEATS2 links histone acetylation to tumorigenesis of non-small cell lung cancer. Nat. Commun 8, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao D et al. (2016) YEATS2 is a selective histone crotonylation reader. Cell Res. 26, 629–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y et al. (2016) Molecular coupling of histone crotonylation and active transcription by AF9 YEATS Domain. Mol. Cell 62, 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan H et al. (2020) BAHCC1 binds H3K27me3 via a conserved BAH module to mediate gene silencing and oncogenesis. Nat. Genet 52, 1384–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y et al. (2019) The ZZ domain as a new epigenetic reader and a degradation signal sensor. Crit. Rev. Biochem. Mol. Biol 54, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mi W et al. (2018) The ZZ-type zinc finger of ZZZ3 modulates the ATAC complex-mediated histone acetylation and gene activation. Nat. Commun 9, 3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Z et al. (2018) EBS is a bivalent histone reader that regulates floral phase transition in Arabidopsis. Nat. Genet 50, 1247–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian S et al. (2018) Dual recognition of H3K4me3 and H3K27me3 by a plant histone reader SHL. Nat. Commun 9, 2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y et al. (2018) The ZZ domain of p300 mediates specificity of the adjacent HAT domain for histone H3. Nat. Struct. Mol. Biol 25, 841–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savitsky P et al. (2016) Multivalent histone and DNA engagement by a PHD/BRD/PWWP Triple Reader Cassette Recruits ZMYND8 to K14ac-Rich Chromatin. Cell Rep. 17, 2724–2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hansen KH et al. (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol 10, 1291–1300 [DOI] [PubMed] [Google Scholar]
- 32.Yamazaki T et al. (2017) Targeted DNA methylation in pericentromeres with genome editing-based artificial DNA methyltransferase. PLoS One 12, e0177764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mlambo T et al. (2018) Designer epigenome modifiers enable robust and sustained gene silencing in clinically relevant human cells. Nucleic Acids Res. 46, 4456–4468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li K et al. (2020) Interrogation of enhancer function by enhancer-targeting CRISPR epigenetic editing. Nat. Commun 11, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakamura M et al. (2021) CRISPR technologies for precise epigenome editing. Nat. Cell Biol 23, 11–22 [DOI] [PubMed] [Google Scholar]
- 36.Cano-Rodriguez D and Rots MG (2016) Epigenetic editing: on the verge of reprogramming gene expression at will. Curr. Genet. Med. Rep 4, 170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vannini A and Cramer P (2012) Conservation between the RNA polymerase I, II, and III transcription initiation machineries. Mol. Cell 45, 439–446 [DOI] [PubMed] [Google Scholar]
- 38.Nabet B et al. (2018) The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Appikonda S et al. (2018) Cross-talk between chromatin acetylation and SUMOylation of tripartite motif-containing protein 24 (TRIM24) impacts cell adhesion. J. Biol. Chem 293, 7476–7485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeon YH et al. (2022) Heterochromatin protein 1: a multiplayer in cancer progression. Cancers 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin S and Du L (2020) The therapeutic potential of BRD4 in cardiovascular disease. Hypertens. Res 43, 1006–1014 [DOI] [PubMed] [Google Scholar]
- 42.Conteduca G et al. (2018) Beyond APECED: an update on the role of the autoimmune regulator gene (AIRE) in physiology and disease. Autoimmun. Rev 17, 325–330 [DOI] [PubMed] [Google Scholar]
- 43.Chan HL et al. (2018) Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat. Commun 9, 3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng S et al. (2019) Overexpression of CBX2 in breast cancer promotes tumor progression through the PI3K/AKT signaling pathway. Am. J. Transl. Res 11, 1668–1682 [PMC free article] [PubMed] [Google Scholar]
- 45.Green AL et al. (2020) BPTF regulates growth of adult and pediatric high-grade glioma through the MYC pathway. Oncogene 39, 2305–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonzales ML and LaSalle JM (2010) The role of MeCP2 in brain development and neurodevelopmental disorders. Curr. Psychiatry Rep 12, 127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amir RE et al. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet 23, 185–188 [DOI] [PubMed] [Google Scholar]
- 48.Shibayama A et al. (2004) MECP2 structural and 3′-UTR variants in schizophrenia, autism and other psychiatric diseases: a possible association with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet 128B, 50–53 [DOI] [PubMed] [Google Scholar]
- 49.Loat CS et al. (2008) Methyl-CpG-binding protein 2 polymorphisms and vulnerability to autism. Genes Brain Behav. 7, 754–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carney RM et al. (2003) Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr. Neurol 28, 205–211 [DOI] [PubMed] [Google Scholar]
- 51.Nagarajan RP et al. (2006) Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 1, e1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwarz K et al. (1996) RAG mutations in human B cell-negative SCID. Science 274, 97–99 [DOI] [PubMed] [Google Scholar]
- 53.Matthews AGW et al. (2007) RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature 450, 1106–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Björses P et al. (2000) Mutations in the AIRE gene: effects on sub-cellular location and transactivation function of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy protein. Am. J. Hum. Genet 66, 378–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bottomley MJ et al. (2005) NMR structure of the first PHD finger of autoimmune regulator protein (AIRE1). Insights into autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) disease. J. Biol. Chem 280, 11505–11512 [DOI] [PubMed] [Google Scholar]
- 56.Qin H-H et al. (2013) Associations between aberrant DNA methylation and transcript levels of DNMT1 and MBD2 in CD4+ T cells from patients with systemic lupus erythematosus. Australas. J. Dermatol 54, 90–95 [DOI] [PubMed] [Google Scholar]
- 57.Chen Z-P et al. (2011) Decreased expression of MBD2 and MBD4 gene and genomic-wide hypomethylation in patients with primary immune thrombocytopenia. Hum. Immunol 72, 486–491 [DOI] [PubMed] [Google Scholar]
- 58.Balada E et al. (2007) Transcript overexpression of the MBD2 and MBD4 genes in CD4+ T cells from systemic lupus erythematosus patients. J. Leukoc. Biol 81, 1609–1616 [DOI] [PubMed] [Google Scholar]
- 59.Elsässer SJ et al. (2016) Genetic code expansion in stable cell lines enables encoded chromatin modification. Nat. Methods 13, 158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cali CP et al. (2019) Targeted DNA methylation of neurodegenerative disease genes via homology directed repair. Nucleic Acids Res. 47, 11609–11622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kungulovski G et al. (2016) Application of recombinant TAF3 PHD domain instead of anti-H3K4me3 antibody. Epigenetics Chromatin 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kungulovski G et al. (2014) Application of histone modification-specific interaction domains as an alternative to antibodies. Genome Res. 24, 1842–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mauser R et al. (2017) Application of dual reading domains as novel reagents in chromatin biology reveals a new H3K9me3 and H3K36me2/3 bivalent chromatin state. Epigenetics Chromatin 10, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Albanese KI et al. (2020) Engineered reader proteins for enhanced detection of methylated lysine on histones. ACS Chem. Biol 15, 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rothbart SB et al. (2015) An interactive database for the assessment of histone antibody specificity. Mol. Cell 59, 502–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Delachat AM-F et al. (2018) Engineered multivalent sensors to detect coexisting histone modifications in living stem cells. Cell Chem. Biol 25, 51–56.e6 [DOI] [PubMed] [Google Scholar]
- 67.Lungu C et al. (2017) Modular fluorescence complementation sensors for live cell detection of epigenetic signals at endogenous genomic sites. Nat. Commun 8, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanchez OF et al. (2017) Engineering recombinant protein sensors for quantifying histone acetylation. ACS Sens. 2, 426–435 [DOI] [PubMed] [Google Scholar]
- 69.Haynes KA (2019) Chromatin research and biological engineering: an evolving relationship poised for new biomedical impacts. Curr. Opin. Syst. Biol 14, 73–81 [Google Scholar]
- 70.Arora S et al. (2020) Engineering a methyllysine reader with photoactive amino acid in mammalian cells. Chem. Commun 56, 12210–12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wagner S et al. (2020) Engineering bromodomains with a photoactive amino acid by engaging ‘Privileged’ tRNA synthetases. Chem. Commun 56, 3641–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sudhamalla B et al. (2017) Site-specific azide-acetyllysine photochemistry on epigenetic readers for interactome profiling. Chem. Sci 8, 4250–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Villaseñor R et al. (2020) ChromID identifies the protein interactome at chromatin marks. Nat. Biotechnol 38, 728–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Groseclose TM et al. (2020) Engineered systems of inducible anti-repressors for the next generation of biological programming. Nat. Commun 11, 4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haynes KA and Silver PA (2011) Synthetic reversal of epigenetic silencing. J. Biol. Chem 286, 27176–27182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park M et al. (2019) Engineering epigenetic regulation using synthetic read-write modules. Cell 176, 227–238.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tekel SJ et al. (2018) Tandem histone-binding domains enhance the activity of a synthetic chromatin effector. ACS Synth. Biol 7, 842–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tillotson R et al. (2017) Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature 550, 398–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Orlando KA et al. (2019) Remodeling the cancer epigenome: mutations in the SWI/SNF complex offer new therapeutic opportunities. Expert. Rev. Anticancer. Ther 19, 375–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Olney KC et al. (2018) The synthetic histone-binding regulator protein PcTF activates interferon genes in breast cancer cells. BMC Syst. Biol 12, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Filippakopoulos P et al. (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grayson AR et al. (2014) MYC, a downstream target of BRD-NUT, is necessary and sufficient for the blockade of differentiation in NUT midline carcinoma. Oncogene 33, 1736–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu S-Y et al. (2020) Opposing functions of BRD4 isoforms in breast cancer. Mol. Cell 78, 1114–1132.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nyer DB et al. (2017) Regulation of cancer epigenomes with a histone-binding synthetic transcription factor. NPJ Genom. Med 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Groner AC et al. (2016) TRIM24 is an oncogenic transcriptional activator in prostate cancer. Cancer Cell 29, 846–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Daser A and Rabbitts TH (2004) Extending the repertoire of the mixed-lineage leukemia gene MLL in leukemogenesis. Genes Dev. 18, 965–974 [DOI] [PubMed] [Google Scholar]
- 87.Perlman EJ et al. (2015) MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nat. Commun 6, 10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De Koning L et al. (2009) Heterochromatin protein 1alpha: a hallmark of cell proliferation relevant to clinical oncology. EMBO Mol. Med 1, 178–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xu J et al. (2013) Up-regulation of MBD1 promotes pancreatic cancer cell epithelial-mesenchymal transition and invasion by epigenetic down-regulation of E-cadherin. Curr. Mol. Med 13, 387–400 [PubMed] [Google Scholar]
- 90.Sapkota Y et al. (2012) A two-stage association study identifies methyl-CpG-binding domain protein 2 gene polymorphisms as candidates for breast cancer susceptibility. Eur. J. Hum. Genet 20, 682–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu D et al. (2011) Overexpression of MBD2 in glioblastoma maintains epigenetic silencing and inhibits the antiangiogenic function of the tumor suppressor gene BAI1. Cancer Res. 71, 5859–5870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schlegel J et al. (2002) Expression of the genes of methyl-binding domain proteins in human gliomas. Oncol. Rep 9, 393–395 [PubMed] [Google Scholar]
- 93.Cukier HN et al. (2010) Novel variants identified in methyl-CpG-binding domain genes in autistic individuals. Neurogenetics 11, 291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li H et al. (2005) Mutation analysis of methyl-CpG binding protein family genes in autistic patients. Brain and Development 27, 321–325 [DOI] [PubMed] [Google Scholar]
- 95.Benes FM et al. (2009) Site-specific regulation of cell cycle and DNA repair in post-mitotic GABA cells in schizophrenic versus bipolars. Proc. Natl. Acad. Sci. U. S. A 106, 11731–11736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stankiewicz P et al. (2017) Haploinsufficiency of the chromatin remodeler BPTF causes syndromic developmental and speech delay, postnatal microcephaly, and dysmorphic features. Am.J. Hum. Genet 101, 503–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kitabayashi I et al. (2001) Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation. Leukemia 15, 89–94 [DOI] [PubMed] [Google Scholar]
- 98.Tycko J et al. (2020) High-throughput discovery and characterization of human transcriptional effectors. Cell 183, 2020–2035. e16 [DOI] [PMC free article] [PubMed] [Google Scholar]