

Abstract
Rapid and accurate assessment of protein structural models is essential for protein structure prediction and design. Great progress has been made in this regard, especially by recent application of “knowledge-based” potentials. Various machine learning based protein structural model quality assessment methods are also quite successful. However, performance of traditional “physics-based” models has not been as effective. Based on our analysis of the fundamental computational limitation behind unsatisfactory performance of “physics-based” models, we propose a generalized solvation free energy (GSFE) framework, which is intrinsically flexible for multi-scale treatments and is amenable for machine learning implementation. Finally, we implemented a simple example of backbone-based residue level GSFE with neural network, which was found to have competitive performance when compared with highly complex latest “knowledge-based” atomic potentials in distinguishing native structures from decoys.
A residue based implementation of GSFE where each residue is solvated by its specific local solvent (i.e. neighboring residues as shown in the figure, explicit or implicit water and/or ion may also be included).
1. Introduction
With time-consuming, expensive and sometimes extremely challenging high resolution experimental analysis, reliable and accurate computational protein structure prediction is highly desired due to the tremendous amount of protein sequences generated by present sequencing technologies. The long-lasting and steadily increasing interest in this regard is evidenced by the impressive development of the CASP (Critical Assessment of techniques for protein Structure Prediction, http://www.predictioncenter.org) community. The fundamental underlying assumption of protein folding is that native structures have the lowest free energy among all possible structural arrangements of residues/atoms in space.1 Unfortunately, rigorous and atomistically detailed calculation of free energy for typical realistic protein molecules is intractable and development of approximate free energy estimators, widely addressed as potentials or scoring functions, is essential. Two basic steps of protein structure prediction are proposal and assessment of structural models; the latter step is performed by comparing scores given by various methodologies, which can be classified into two major categories, namely “knowledge-based” (KB) and “physics-based” (PB).2 It is widely realized that KB scoring is much more effective than PB scoring in recognizing native structures from decoys.3
A number of KB potentials are based on atom pair distances, possibly with various forms of orientation consideration.4–8 Reference state definition was acknowledged to be a major source of differences among them.4,9 Additionally, definition of reference state was found to impact selection of cutoff distances.10 OPUS-CSF utilized some designed features extracted from backbone segments of various lengths (5 to 11 residues) to describe extent of nativeness11 and it was further developed to include side chains.12 Utilization of rotameric states besides distances13 and Voronoi contact surface14 were also found to be quite successful. Entropy was also explicitly considered in some models and found to be helpful.15,16 These carefully handcrafted potentials were relatively easy to explain physically.
Many machine learning protein structural model quality assessment protocols have been developed with great success. Both single quality assessment models3,17–22 and meta-models.23–25 have been utilized. A multiple object optimization approach was also investigated.26,27 Physical explanation is less straightforward for these machine learning based protocols.
In this paper, we analyzed the computational limitation of present theoretical formulations underlying PB scoring, and proposed an intrinsically multi-scale generalized solvation free energy (GSFE) framework to facilitate application of powerful machine learning optimization algorithms. Furthermore, we provide a simple neural network implementation of the GSFE framework at residue level for assessment of backbone protein structural models. Despite simplicity, this implementation was found to be competitive when compared with state-of-the-art KB methods that are significantly more complex. Further development of GSFE for quality assessment of protein structural models and protein design will be carried out in the near future. We emphasize that traditional solvation free energy formulations remain powerful tools in tackling many problems. Our formulation of GSFE is one possible alternative way to facilitate utilization of machine learning optimization capacity.
2. Methodology
2.1. Computational limitation of present solvation free energy formulation
It has been widely accepted that hydrophobic interactions are among the most important driving forces for protein folding.28,29 Therefore, solvation free energy has been an important topic with a long history of theoretical development.30 Protein solvation free energy was itemized as an independently calculated contribution to the total free energy as indicated by the following equation:30
| −kBT logP(S) = Eintra(S) + Δμ(S) + constant | 1 |
with S being the configuration of protein atoms and Δμ being the solvation free energy when the protein is in configuration S, P(S) being the probability that the protein is in the S configuration. The constant term was determined by selection of a reference state. This framework is conceptually helpful for understanding and also amenable for theoretical derivation of large solute molecules like proteins. Most importantly, it is very convenient for us to explicitly treat many interesting and useful interfacial properties. These advantages explain its popularity. However, direct and reliable separation of free energy of a protein molecular system into internal and solvation free energy experimentally is not an easy task for biomolecular systems. Therefore, in development of computational models according to this classical formulation, experimental validation is not readily available. Meanwhile, due to the complexity of configurational sampling and electrostatic calculations, theoretical approximations are unavoidable and experimental validation therefore is essential for development of reliable computational methodologies. Present strategies for solving this dilemma have been to validate computational methods of solvation free energy with molecular dynamics (MD) simulations30 (and references therein), which have their own approximations. An additional caveat of this formulation is that the intramolecular packing term Eintra(S) and the solvation free energy term Δμ are calculated independently, thus making the variance of the free energy calculation the sum of these two parts. A formulation with unified calculation of both terms would be preferable, where firstly direct comparison with experimental data becomes more straightforward, and secondly variance increase due to independent calculations could be reduced.
Machine learning, especially deep learning has been demonstrated to facilitate understanding of complex molecular systems.31 The caveat of brute force utilization of deep learning is that a deep neural network works as a black box of function composers in many cases, and does not necessarily improve our fundamental physical understanding. Therefore, it is highly desirable to develop a theoretical framework that is amenable to machine (deep) learning on the one hand, and to achieve a good balance of explainability and prediction capacity on the other hand. With present solvation free energy frameworks, it is difficult to effectively utilize both the optimization capacity of machine learning algorithms and the available/expected high resolution structure data.
We therefore propose in this paper an alternative theoretical framework, a generalized solvation free energy framework. The aim is to provide convenience for taking advantage of optimization capacity of machine learning algorithms and available structural data on the one hand, and to support physical explanation on the other hand.
2.2. The generalized solvation free energy framework
The definition of solute and solvent in molecular systems, while it follows some intuition, is fundamentally arbitrary. The idea of the generalized solvation free energy (GSFE) framework is to define each basic physical comprising unit of a given complex system as solute and all its surrounding units as its specific solvent. The GSFE framework has the following basic properties:
(i) It is intrinsically multi-scale. Using protein molecular systems as examples, basic physical comprising units of which can be atoms, atomic groups, residues, clusters of residues, structural domains, individual protein chains or even small protein complexes in mega protein complexes.
(ii) Each basic comprising unit is both a solute and a comprising unit of solvent for its neighboring units simultaneously.
(iii) Solvent is specific for each solute unit, and is usually heterogeneous.
This is in contrast to traditional definition of solution where each basic unit/molecule is either solute or solvent, but usually not both. Solvent in the traditional solution concept is in most cases homogeneous. An illustration of the GSFE idea is presented in Fig. 1, where a residue in a protein molecule is solvated by its neighboring residues. This new way of defining solute and solvent seems to engender great difficulty in theoretical formulations. In contrast to homogeneous and consequently easy to describe solvent in traditional definition of solvation, here in GSFE each solute unit has its own specific solvent, rendering direct representation of almost infinitely complex combinations of solvent impossible in multiple component complex molecular systems. Pondering further, this seemingly difficulty provides great convenience for machine learning of the free energy as described below.
Fig. 1. Schematic illustration of the GSFE framework. Here a residue in a protein molecule is considered to be solvated by its neighboring residues.

Again we use a typical protein molecular system as an example with protein residues, water molecules and various ions as the basic unit. For a given residue, its specific solvent includes all other surrounding residues, possibly water molecules and ions for residues close to surface. Fortunately, it is quite well understood that in liquids and non-crystalline condensed soft matter, predominant molecular interactions are usually quite short-ranged. We therefore may safely restrict our attention to a limited spatial range by limiting the specific solvent of a given solute to a certain cutoff distance. The task of properly describing the solvent of a given solute unit remains daunting after this cutoff simplification. Specifically, two layers of neighboring residues for a fully buried residue solute may amount up to 20 or even more, resulting in a maximum of 2020 possible amino acid identity combinations for a given positional configuration with immense additional spatial variations. For solute residues that are close to the surface, inclusion of complex possible combinations of water molecules and various other molecular entities are essential when explicit water/ion representation is necessary. Putting aside the complexity of the specific solvent environment for a given solute unit for the time being, a great property emerges immediately. The probability of a given unit existing in a given solvent is solely determined by that solvent. Thus, for a given protein structure model (e.g. a model in PDB), the probability of a given residue having native sequence identity is solely determined by the specific local solvent specified by the given structure model, and this is true for all residues. When such a conditional probability (p(xi|solventxi)) is properly accounted for, no additional explicit correlation between probabilities of different residues taking respective amino acid identity and positions is necessary. Consequently, all correlations among various comprising solvent units and between the solute unit and collections of solvent units are covered by this conditional probability term, and therefore need not to be accounted for explicitly! The free energy of a given n-residue protein sequence X = {x1, x2, …, xn} adopting a given structure (that defines solvent for each solute residue) may be formulated as the following:
| F = −ln P(structure|X) | 2 |
By the Bayes formula:
 |
3 |
| ∝ P(X|structure)P(structure) | 4 |
When considering a maximum likelihood treatment, we focus our attention to the likelihood, which may be expanded as the following:
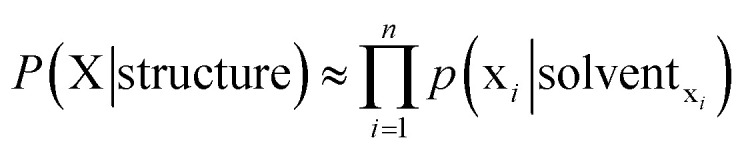 |
5 |
 |
6 |
Besides the maximum likelihood approximation introduced in eqn (5) and (6), two additional approximations introduced are the following: (1) Neglect of interactions between the ith residue and all molecular systems comprising units not accounted for by solventxi. (2) The over counting of implicit correlations by p(xi|solventxi). Specifically, let B, C and D be neighbors of solute A, then correlations among (B, C, D) are implicitly accounted for in p(A|B, C, D). Conversely, A is a neighboring solvent unit for B, C and D; let us focus on D and assume that D has four neighbors (A, B, C, X), then correlation between B and C is counted again in p(D|A, B, C, X). The extent of implicit over counting is determined by the number of neighbors (i.e. cutoff distance). Even with the above-stated approximations, this simple model performs competitively (see Results section). We aim to improve GSFE in this respect in our future work.
Given all the above mentioned approximations, p(xi|solventxi), which we have no idea in terms of explicit functional forms, needs to solved for each residue. Fortunately, a neural network may approximate any functions without knowing explicit symbolic mathematical formulae as long as sufficient data are available. p(xi|solventxi) can be learned from the training dataset directly. Identities and positions of neighboring residues constitute input for the neural network approximating this complex conditional probability. All possible complex correlations between solute xi and its environment solventxi may be implicitly captured by a neural network that is extremely good at solving non-linear mapping. Pairwise interaction approximation has been widely utilized in both KB and PB potentials. In the GSFE framework, both pairwise and higher ordered correlations within the selected solvent were included and the relative importance of specific ordered interactions (pair, triple and higher ordered) were determined by the optimization process of the chosen neural network.
2.3. A simple neural network implementation
2.3.1. Datasets
We selected the Cullpdb dataset32 generated on 2018.11.26 as our training dataset. The percentage identity cutoff is 25%, the resolution cutoff is 2.0 angstroms, and the R-factor cutoff is 0.25. For the 9311 chains in the Cullpdb list, we downloaded the corresponding structures from PDB. We culled the Cullpdb dataset and CASP13 dataset using the CD-HIT server,33 sequences that had more than 25% identity to any sequences in the two datasets were removed. 8129 structures of the Cullpdb dataset and 16 structures of the CASP13 dataset were kept after these processes. The biopython library was used to process and get features from these structures (for details see definition of solvent environment). We further divided the Cullpdb dataset into a training set (7316 structures), a validation set (406 structures) and a test set (407 structures) randomly. 16 proteins of CASP13 and their decoy structures are another test set.
In this work we use four decoy datasets to test our model, CASP5-CASP8,34 CASP10-13,35 I-TASSER36 and 3DRobot37 decoy sets. All the datasets are used to test ANDIS by Yu,35 the Rosetta dataset was also used by Yu35 but the link is no longer available.
2.3.2. Neural network architecture and input tensor
We define the environment of a target residue by identity and position of its neighboring residues. Different input features are tested and the results are shown in the Results section. Here we use the input features that produce the best performance as an example. Pairwise distances of all residues in a protein are calculated. For a target residue we sort the distances and choose 19 nearest residues as its neighbors. The relative orientation for a pair of residues is defined by six angles. For residue A and residue B, we construct Cα(A) − Cα(B), Cα(A) − N(A), Cα(A) − Cβ(A) and Cα(A) − C(A) vectors. Three angles formed by three pair of vectors, (Cα(A) − Cα(B), Cα(A) − N(A)), (Cα(A) − Cα(B), Cα(A) − Cβ(A)) and (Cα(A) − Cα(B), Cα(A) − C(A)) are calculated. For residue B and residue A we also construct three angles with corresponding vectors, so for a pair of residues we have six angles to represent their orientation. For GLY (that has no Cβ atom) we guess its HA2 position by the positions of its Cα, C and N. For a neighbor residue we use its residue type (one-hot encoding), distance to target residue and orientation as its features. We further divide the neighbor residues into two parts, sequence adjacent (abs(id(T) − id(N)) ≤ 6) and sequence non-adjacent (abs(id(T) − id(N)) > 6) ones. id(T) is the primary sequence position of the target residue and id(N) is the primary sequence position of a neighbor residue, abs function returns the absolute value. Adjacent residues are sorted with the order id(T) − id(N) (−6, −5, −4, −3, −2, −1, 1, 2, 3, 4, 5, 6). Not all adjacent residues are necessarily neighbor residues as specified by the distance criteria above. If a residue in this list is not a neighbor residue the features of this residue is padded with zero. Non-adjacent residues are sorted with their distance to target residue. We use a list (1, 2, 3, 4, 5, 6, 7, 8, 9) to store the features of these residues, for each position we store the features of a residue. If there are not enough neighbor residues, we will pad zeros as features in the corresponding place. Cα depth and half sphere exposure (HSE) of the target residue are also used as input features. The Cα depth is the distance of a residue’s Cα atom to the solvent accessible surface. HSE is a 2-dimensional measure of solvent exposure. Basically, it counts the number of Cα atoms around a residue in the direction of its side chain, and in the opposite direction (within a radius of 13 Å). HSE comes in two flavors: HSEα and HSEβ. The former only uses Cα atom position, while the latter uses both Cα and Cβ positions. So for a given target residue its input includes adjacent residue features, non-adjacent residue features, its Cα depth and HSE values. All features are calculated with the biopython library. For each neighbor residue we use a 22-dimensional one-hot vector to encode its residue type (20 natural amino acids, 1 for other non-natural amino acids and an additional type for non-neighboring residues for indexing convenience), a 6-dimensional vector to store angles and a 1-dimensional vector to store distance. For a target residue we have 21 neighbor residues (12 adjacent and 9 non-adjacent), and HSEα (2 dimension), HSEβ (2 dimension), and Cα depth (1 dimension) together as a 5-dimensional feature. All features are put together to form a 614-dimensional (21 × 29 + 5) feature vector as our input (Fig. 2). We adjust distances, angles and HSE values so that they fall approximately in the range (0, 1).
Fig. 2. Schematic representation of the vector organization for neural network input features.

A simple four layer perceptron is constructed here with PyTorch. For a target residue we present its input feature vector to the neural network and predict its residue type. There are three hidden layers and 512 neurons per layer. The activation function used for hidden layers is ReLU. We use 21 neurons in the output layer, 20 common residues are classified as 20 classes and all other residues are classified as the 21st class. Softmax activation function is used for the output layer and cross entropy is selected as the loss function. The probability of each residue type is predicted from the softmax layer. We train the network with our training dataset. A stochastic gradient descent optimizer is utilized with a learning rate of 0.1. For each batch we train all target residues in a protein. We train the network for 30 epochs, after each epoch we test the network with the validation set. Best performing parameters in the validation set are saved as our final parameters. Finally we test the model with the test dataset and the test accuracy for native sequence identity prediction is 35.2%. Loss value and accuracy of each epoch are shown in Fig. 3.
Fig. 3. Loss value and accuracy of training dataset and validation dataset.

3. Results
To approximate local conditional probability p(xi|solventxi), we investigated four progressively more complex sets of input features. The most complex (also the one with the best performance) has been presented in detail above. Three simpler sets of input features are listed below: (1) Identity of neighbor residues and distances from them to the target residue are chosen as input features, neighbor residues are sorted according to their respective distance to the target residue. (2) HSE values and Cα depth of target residue are added to the features. (3) Relative orientations of neighboring residues are added. Finally, on the basis of the third model, we divide neighboring residues into two groups, sequence adjacent and sequence non-adjacent, and organize as illustrated in Fig. 2. The architecture of the neural network stays fixed for all different input features, and investigation in this regard will be performed in future studies. We train our model using the four different sets of input features, and test them on decoy sets. These datasets have been used by Yu to compare ANDIS with other 8 methods.35 To evaluate a structural model with our neural network, we first compute the conditional probability of each residue being in its native sequence identity, and subsequently taking summation of logarithm probabilities to obtain the maximum likelihood score, with higher likelihood scores corresponding to lower approximate free energy. The obtained scores are used to distinguish native structures from decoy models. As indicated by the results in Table 1, our best performing model is competitive when compared with other KB potentials, most of which takes atomic positions. We also find that progressively better performance is obtained when more information is added to input features.
Performance comparison in native structure recognition. Models with input feature sets 1 through 4 are trained with different input features as mentioned in the text. The number of proteins whose native structure is given the lowest energy score (our method uses largest score) by the potential is listed outside the parentheses. The average Z-scores of native structures are listed in parentheses. Z-score is defined as (〈Edecoy〉 − Enative)/δ (our method (Enative − 〈Edecoy〉)/δ), where Enative is the energy score of the native structure, 〈Edecoy〉 and δ are respectively the average and the standard deviation of energy scores for all decoys in the set.
| Decoy sets | CASP5-8 | CASP10-13 | I-TASSER | 3DRobot |
|---|---|---|---|---|
| No. of targets | 143 (2759) | 175 (13 474) | 56 (24 707) | 200 (60 200) |
| Model1 | 99 (1.35) | 80 (1.13) | 12 (1.54) | 66 (1.85) |
| Model2 | 114 (1.52) | 96 (1.20) | 28 (2.32) | 120 (2.09) |
| Model3 | 140 (1.83) | 132 (1.75) | 43 (3.95) | 200 (3.33) |
| Model4 | 140 (1.76) | 135 (1.83) | 48 (4.21) | 200 (3.43) |
We further compare the results of our best performing model with other state-of-the-art potentials as reported by Yu in their ANDIS potential paper;35 the results are presented in Table 2. We further present details of a test on 16 proteins of the CASP13 decoy set and the results are presented in Table 3. Their sequence identity with our training set is less than 25%. We recognize 8/16 target structures from decoy structures, and the average Z-score is 1.27. So our model can work with proteins significantly different from our training set in terms of sequence identity.
Performance comparison in native structure recognition. The number of proteins whose native structure is given the lowest energy score (our method uses largest score) by the potential is listed outside the parentheses. The average Z-scores of native structures are listed in parentheses. Z-score is defined as (〈Edecoy〉 − Enative)/δ (our method (Enative − 〈Edecoy〉)/δ), where Enative is the energy score of the native structure, 〈Edecoy〉 and δ are respectively the average and the standard deviation of energy scores for all decoys in the set.
| Decoy sets | CASP5-8 | CASP10-13 | I-TASSER | 3DRobot |
|---|---|---|---|---|
| No. of targets | 143 (2759) | 175 (13 474) | 56 (24 707) | 200 (60 200) |
| Dfire | 64 (0.61) | 56 (0.72) | 43 (2.80) | 1 (0.83) |
| RW | 65 (1.01) | 36 (0.86) | 53 (4.42) | 0 (−0.30) |
| GOAP | 106 (1.67) | 89 (1.62) | 45 (4.98) | 94 (1.85) |
| DOOP | 135 (1.96) | 121 (1.99) | 52 (6.18) | 197 (3.53) |
| ITDA | 71 (1.15) | 117 (1.67) | 52 (4.98) | 196(3.83) |
| VoroMQA | 132 (2.00) | 111 (1.77) | 48 (5.11) | 114 (1.89) |
| SBROD | 88 (1.62) | 119 (2.32) | 33 (3.25) | 49 (1.76) |
| AngularQA | 59 (1.26) | 24 (1.11) | 29 (1.82) | 9 (0.99) |
| ANDIS | 138 (2.16) | 129 (2.32) | 47 (6.45) | 200 (4.99) |
| GSFE | 140 (1.76) | 135 (1.83) | 48 (4.21) | 200 (3.43) |
CASP13 result; native structure ranks in decoy structures.
| Structure id | Rank (Z-score) | Structure id | Rank (Z-score) |
|---|---|---|---|
| T0950-D1 | 1/39 (2.65) | T0966-D1 | 1/87 (1.18) |
| T0953s1-D1 | 4/90 (1.92) | T0968s1-D1 | 1/94 (1.42) |
| T0954-D1 | 1/87 (1.33) | T0968s2-D1 | 2/95 (1.07) |
| T0955-D1 | 18/92 (0.68) | T1003-D1 | 8/89 (1.13) |
| T0957s1-D1 | 2/92 (1.01) | T1005-D1 | 1/83 (1.23) |
| T0957s2-D1 | 1/91 (1.60) | T1008-D1 | 25/91 (0.84) |
| T0958-D1 | 4/90 (1.22) | T1009-D1 | 1/85 (1.04) |
| T0960-D1 | 22/84 (0.71) | T1011-D1 | 1/82 (1.33) |
4. Discussion
In this simple neural network implementation of the GSFE framework, we utilized residue level spatial resolution. Despite the simplicity, the best performing trained model is competitive in selecting native structures from decoys when compared with sophisticated atomic potentials. The weakness of the trained model as reflected by the relatively small values of Z-scores is likely due to the fact that only native structures are utilized for the training, and this issue will be tackled in our future work.
It is important to note that the GSFE formulation has its own limitation. Like all data driven representations, the availability of sufficient data is critical in determining the accuracy of the model. In this specific implementation, the work horse of GSFE is the amino acid identity conditional probability p(xi|solventxi). When a local structure very different from all available structural data is presented to the neural network, its prediction is apparently not as reliable as in the case of familiar input features. It is trained on available structures and therefore is likely to be not as effective under physical conditions that are far from these data. The optimistic perspective is that more and higher quality data will be generated and the model may continuously advance based on further data input. Traditional formulation is more powerful in many interesting schemes. For example, if we were interested in investigating the response of a protein under periodically varying electric fields; or if we were interested in interfacial properties.
The free energy resolution and spatial range of molecular interactions of GSFE is apparently limited by the specific basic unit selected and available data. If we chose to carry out atomistic GSFE training with a large cutoff distance, the complexity of such local solvent would require excessively large datasets and training time. However, due to the intrinsic multi-scale definition, it is hopeful that we may train hierarchical GSFE models to account for both relatively long-range interactions and local chemical details. This is also a future direction of our GSFE development.
At first sight, the possible local chemical environment for any residue/atom (and other possible definitions of the basic unit) is an immense space and the available datasets (structures in PDB) seems hopelessly small. However, evolution in billions of years by huge number of organisms have sampled and found at least a significant fraction, if not all, of the important local chemical environment spaces that were partially represented in the presently available datasets. Nevertheless, the approximations introduced in our formulation have room to be improved. The P(structure) term in eqn (4) was left out in the maximum likelihood treatment. Physically, the P(structure) may be expressed as the following:
| P(structure) = P(structure|fold)P(fold) | 7 |
With P(fold) being the probability of the concerned protein fold in the manifold of all possible protein folds. Apparently, accurate quantization of this equation needs understanding of the whole protein fold space on the one hand, and structural variation within a specific fold on the other hand. To address this issue properly so that we may move from present maximum likelihood estimation to a full Bayesian treatment is a long term goal of GSFE development.
5. Conclusions
We proposed the GSFE framework to achieve a balance of physical interpretability and powerful non-linear optimization capacity of neural networks. This framework is intrinsically multi-scale and amenable to machine learning optimization. One distinctive feature of GSFE is that implicitly considered local correlations were not limited to pairwise interactions, which is a widely utilized practice in many KB and PB potentials. A simple neural network implementation of GSFE at residue level for protein structural model assessment was found to be competitive with sophisticated state-of-the-art atomic KB potentials in distinguishing native structures from decoys. In future work, we plan to carry out investigations on more sophisticated neural network architectures and training strategies to achieve stronger capability in ranking decoys besides distinguishing native structures from decoys, and to develop hierarchical implementations of GSFE.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work has been supported by the National Key Research and Development Program of China (2017YFB0702500), by the postdoctoral start up fund from Jilin University (801171020439), by National Natural Science Foundation of China (31270758), and by the Fundamental Research Funds for the Central Universities (451170301615).
Notes and references
- Anfinsen C. B. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- Skolnick J. Curr. Opin. Struct. Biol. 2006;16:166–171. doi: 10.1016/j.sbi.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Pei J. Zheng Z. Merz K. M. J. Chem. Inf. Model. 2019;59:1919–1929. doi: 10.1021/acs.jcim.8b00734. [DOI] [PubMed] [Google Scholar]
- Zhang J. Zhang Y. PLoS One. 2010;5:1–13. [Google Scholar]
- Chae M. H. Krull F. Knapp E. W. Proteins: Struct., Funct., Bioinf. 2015;83:881–890. doi: 10.1002/prot.24782. [DOI] [PubMed] [Google Scholar]
- Hoque M. T. Yang Y. Mishra A. Zhou Y. J. Comput. Chem. 2016;37:1119–1124. doi: 10.1002/jcc.24298. [DOI] [PubMed] [Google Scholar]
- Xu G. Ma T. Zang T. Sun W. Wang Q. Ma J. J. Mol. Biol. 2017;429:3113–3120. doi: 10.1016/j.jmb.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z. Yao Y. Deng H. BMC Bioinf. 2019;20:299. doi: 10.1186/s12859-019-2898-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Zeng J. Gong H. Proteins: Struct., Funct., Bioinf. 2014;82:2383–2393. doi: 10.1002/prot.24600. [DOI] [PubMed] [Google Scholar]
- Yao Y. Gui R. Liu Q. Yi M. Deng H. BMC Bioinf. 2017;18:1–12. doi: 10.1186/s12859-017-1983-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G. Ma T. Zang T. Wang Q. Ma J. Protein Sci. 2018;27:286–292. doi: 10.1002/pro.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G. Ma T. Wang Q. Ma J. Protein Sci. 2019;28:1157–1162. doi: 10.1002/pro.3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. Saitou K. BMC Bioinf. 2014;15:1–16. doi: 10.1186/1471-2105-15-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olechnovič K. Venclovas Č. Proteins: Struct., Funct., Bioinf. 2017;85:1131–1145. doi: 10.1002/prot.25278. [DOI] [PubMed] [Google Scholar]
- Sankar K. Jia K. Jernigan R. L. Proc. Natl. Acad. Sci. U. S. A. 2017;114:2928–2933. doi: 10.1073/pnas.1613331114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Zhang D. Huang S. Y. J. Chem. Inf. Model. 2018;58:724–732. doi: 10.1021/acs.jcim.7b00601. [DOI] [PubMed] [Google Scholar]
- Cao R. Cheng J. Sci. Rep. 2016;6:1–8. doi: 10.1038/s41598-016-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R. Bhattacharya D. Hou J. Cheng J. BMC Bioinf. 2016;17:1–9. doi: 10.1186/s12859-015-0844-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R. Adhikari B. Bhattacharya D. Sun M. Hou J. Cheng J. Bioinformatics. 2017;33:586–588. doi: 10.1093/bioinformatics/btw694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavalan B. Lee J. Bioinformatics. 2017;33:2496–2503. doi: 10.1093/bioinformatics/btx222. [DOI] [PubMed] [Google Scholar]
- Gao J. Yang Y. Zhou Y. BMC Bioinf. 2018;19:1–8. doi: 10.1186/s12859-017-2006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derevyanko G. Grudinin S. Bengio Y. Lamoureux G. Bioinformatics. 2018;34:4046–4053. doi: 10.1093/bioinformatics/bty494. [DOI] [PubMed] [Google Scholar]
- Cao R. Bhattacharya D. Adhikari B. Li J. Cheng J. Bioinformatics. 2015;31:i116–i123. doi: 10.1093/bioinformatics/btv235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing X. Dong Q. BMC Bioinf. 2017;18:1–8. doi: 10.1186/s12859-017-1691-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulnaes D. Gohlke H. J. Chem. Theory Comput. 2018;14:6117–6126. doi: 10.1021/acs.jctc.8b00690. [DOI] [PubMed] [Google Scholar]
- Song S. Ji J. Chen X. Gao S. Tang Z. Todo Y. Applied Soft Computing. 2018;72:539–551. doi: 10.1016/j.asoc.2018.07.042. [DOI] [Google Scholar]
- Song S. Gao S. Chen X. Jia D. Qian X. Todo Y. Knowl. Based Syst. 2018;146:58–72. doi: 10.1016/j.knosys.2018.01.028. [DOI] [Google Scholar]
- Dill K. A. MacCallum J. L. Science. 2012;338:1042–1046. doi: 10.1126/science.1219021. [DOI] [PubMed] [Google Scholar]
- Bellissent-Funel M. C. Hassanali A. Havenith M. Henchman R. Pohl P. Sterpone F. Van Der Spoel D. Xu Y. Garcia A. E. Chem. Rev. 2016;116:7673–7697. doi: 10.1021/acs.chemrev.5b00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matubayasi N. Curr. Opin. Struct. Biol. 2017;43:45–54. doi: 10.1016/j.sbi.2016.10.005. [DOI] [PubMed] [Google Scholar]
- Pérez A. Martínez-Rosell G. De Fabritiis G. Curr. Opin. Struct. Biol. 2018;49:139–144. doi: 10.1016/j.sbi.2018.02.004. [DOI] [PubMed] [Google Scholar]
- Wang G. Dunbrack R. PISCES: a protein sequence culling server. Bioinformatics. 2003;19:1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- Ying H. Beifang N. Ying G. Limin F. Weizhong L. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rykunov D. Fiser A. BMC Bioinf. 2010;11:128. doi: 10.1186/1471-2105-11-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z. Yao Y. Deng H. Yi M. BMC Bioinf. 2019;20:299. doi: 10.1186/s12859-019-2898-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Zhang Y. PLoS One. 2010;5:e15386. doi: 10.1371/journal.pone.0015386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H. Jia Y. Zhang Y. Bioinformatics. 2016;32:378–387. doi: 10.1093/bioinformatics/btv601. [DOI] [PMC free article] [PubMed] [Google Scholar]