

Abstract
Aims
Autophagy protects against the development of cardiac hypertrophy and failure. While aberrant Ca2+ handling promotes myocardial remodelling and contributes to contractile dysfunction, the role of autophagy in maintaining Ca2+ homeostasis remains elusive. Here, we examined whether Atg5 deficiency-mediated autophagy promotes early changes in subcellular Ca2+ handling in ventricular cardiomyocytes, and whether those alterations associate with compromised cardiac reserve capacity, which commonly precedes the onset of heart failure.
Methods and results
RT–qPCR and immunoblotting demonstrated reduced Atg5 gene and protein expression and decreased abundancy of autophagy markers in hypertrophied and failing human hearts. The function of ATG5 was examined using cardiomyocyte-specific Atg5-knockout mice (Atg5−/−). Before manifesting cardiac dysfunction, Atg5−/− mice showed compromised cardiac reserve in response to β-adrenergic stimulation. Consequently, effort intolerance and maximal oxygen consumption were reduced during treadmill-based exercise tolerance testing. Mechanistically, cellular imaging revealed that Atg5 deprivation did not alter spatial and functional organization of intracellular Ca2+ stores or affect Ca2+ cycling in response to slow pacing or upon acute isoprenaline administration. However, high-frequency stimulation exposed stunted amplitude of Ca2+ transients, augmented nucleoplasmic Ca2+ load, and increased CaMKII activity, especially in the nuclear region of hypertrophied Atg5−/− cardiomyocytes. These changes in Ca2+ cycling were recapitulated in hypertrophied human cardiomyocytes. Finally, ultrastructural analysis revealed accumulation of mitochondria with reduced volume and size distribution, meanwhile functional measurements showed impaired redox balance in Atg5−/− cardiomyocytes, implying energetic unsustainability due to overcompensation of single mitochondria, particularly under increased workload.
Conclusion
Loss of cardiac Atg5-dependent autophagy reduces mitochondrial abundance and causes subtle alterations in subcellular Ca2+ cycling upon increased workload in mice. Autophagy-related impairment of Ca2+ handling is progressively worsened by β-adrenergic signalling in ventricular cardiomyocytes, thereby leading to energetic exhaustion and compromised cardiac reserve.
Keywords: Cardiomyocytes, Calcium, Mitochondria, Autophagy, Beta-adrenergic signalling
Graphical Abstract

1. Introduction
Macroautophagy (hereafter referred to as autophagy) is a key protective process necessary for cardiac homeostasis.1,2 Under physiological conditions, constitutive autophagy maintains structure and function of the heart by eliminating damaged molecules and dysfunctional organelles from the cytoplasm, which might otherwise elicit harmful effects to cardiac cells, including long-lived cardiomyocytes.1,3,4 Autophagy-related proteins (ATG) are essential in this process, among which ATG5 is indispensable for the early phase of autophagosome formation. Mice with cardiomyocyte-restricted Atg5 deletion display increased ubiquitination, mitochondrial aggregates and disorganized sarcomere structure in cardiomyocytes.5 Despite early structural deteriorations within Atg5 null cardiomyocytes, cardiac function is remarkably normal until early adulthood. Hence, it has been argued that a mechanism, such as the Atg5/Atg7-independent alternative autophagy6 and/or chaperone-mediated autophagy7 may compensate for Atg5 deficiency to limit cardiac damage. However, such cardioprotective mechanism efficiently operates against adverse remodelling only temporarily, because cardiomyocyte-specific Atg5-deficient mice develop left ventricular hypertrophy that shortly thereafter progresses to heart failure, resulting ultimately in premature death.5
Mechanistically, long-term absence of basal autophagy is associated with reduced mitochondrial respiratory function and increased oxidative stress, which predisposes exhausted cardiomyocytes to apoptotic cell death and contractility decline. In addition, the absence of essential autophagy-related proteins may also cause perturbations of other inter-related cellular mechanisms,8 which in conjunction with damaged mitochondria initiate and drive adverse remodelling and contractile dysfunction. Notably, disturbed Ca2+ handling is one such early abnormality during adverse myocardial remodelling9 that causes heart failure.10,11 In this regard, autophagy-deprived hearts exhibit compromised energy and redox balance due to increased number of ill-functioning mitochondria. Such perturbations may cause shifts in ionic gradients and attenuate excitability by deregulating the energy-demanding process of intracellular Ca2+ homeostasis, especially under increased workload. Besides cytoplasmic and mitochondrial Ca2+ changes, altered nucleoplasmic Ca2+ signalling could be critical to functional remodelling of cardiomyocytes, which lack autophagy, as activation of nuclear Ca2+-mediated hypertrophic gene program affects protein expression, including components of the excitation–contraction coupling machinery.9 To this end, however, it remains unknown whether absence of cardiac autophagy alters subcellular Ca2+ cycling at early stages of remodelling that precede the development of heart failure.
In this study, we examined the ATG5 expression at the transcriptional and protein levels in non-failing, hypertrophic and failing human hearts. We then used cardiomyocyte-specific Atg5-deficient mice before they develop heart failure to identify the specific role of ATG5 in subcellular Ca2+ cycling under normal and stressed conditions. Finally, we functionally assessed the translational potential of our findings in hypertrophied human cardiomyocytes.
2. Methods
A detailed description of methods is available in Supplementary material online.
2.1 Ethics
Animal experiments were performed in accordance with European ethical regulation (Directive 2010/63/EU) and approved by the responsible Austrian government agency (BMBWF: 66.010/0038-WFN/3b/2017; BMBWF-66.010/0208-V/3b/2018). Procedures involving human samples were approved by the Ethical Committee of the Medical University of Graz (28-508 ex 15/16), and performed in accordance with principles outlined in the Declaration of Helsinki. Informed written consent was not feasible to obtain because the patients were not able to give the informed consent as a result of their underlying medical condition. The requirement for informed consent was waived by the ethical committee. Patients’ characteristics are described in Supplementary material online, Table S1. Donor hearts were explanted post-mortem together with other organs. Upon ice-cold cardioplegia, cardiac biopsies were harvested from the left ventricular free wall, quickly frozen in liquid nitrogen and stored at −80°C for future analysis.
2.2 Mice
Cardiomyocyte-specific Atg5 knock-out mice (Atg5−/− mice; N = 52) were generated from Atg5flox/flox mice crossed with heterozygous knock-in mice that express Cre+ recombinase driven by the cardiomyocyte-specific α-myosin light chain (MLC2a-Cre) gene promoter.12 We used Atg5−/− male mice between 12 and 15 weeks, because at this age they present a normal cardiac phenotype despite a complete block of autophagy.4,5 Age-matched Atg5+/+ male littermates served as controls (N = 45).
2.3 Haemodynamic measurements and cardiac stress testing
Haemodynamic assessment was performed according to the established protocol.4 Mice were sedated with isoflurane (5% for induction and 1–2% for maintenance), intubated, and mechanically ventilated (Harvard Mini-Vent, type 845, Harvard Apparatus). A pressure-conductance catheter (SPR-839; Millar Instruments) was inserted into the right carotid artery and advanced into the LV, where pressure signals were recorded (MPVS ultra, Millar Instruments), and analysed offline (LabChart 8 Pro, AD Instruments). Cardiac parameters were measured at baseline and after intraperitoneal isoprenaline administration (2 µg/kg body weight13) to evaluate cardiac reserve capacity. After assessment of haemodynamic parameters that were averaged from 10 consecutive beats with ventilation suspended at end-expiration, mice were euthanized by decapitation. Hearts were rapidly frozen in liquid nitrogen and stored at −80°C for molecular analysis.
2.4 Treadmill running coupled to indirect calorimetry
A subset of mice (N = 16) underwent peak effort testing on a motorized treadmill coupled to a calorimetric unit (TSE Systems, Germany). Mice were subjected to a ramp exercise protocol with an initial velocity of 3 m/min followed by a constant acceleration of 3 m/min2 at 0° inclination.14 The exercise ended at the maximal exhaustion, which was defined as the mouse’s inability to maintain running speed despite being in contact with the electrical grid for five consecutive seconds. Maximal oxygen consumption (VO2max) and run distance were determined at the point at which oxygen uptake reached a plateau during exhaustive workout/exercise.
2.5 Real-time quantitative PCR (RT–qPCR)
Total purification of mouse and human RNA and measurements of RNA yield and quality were performed as described previously.15 cDNA was synthesized using the QuantiTect Reverse Transcription kit (for mouse; Qiagen, Hilden, Germany) and the HighCapacity cDNA RT Kit (for human; Thermo Fisher Scientific, Wilmington, USA), respectively. RT–qPCR was performed with cDNA at a concentration of 10 ng per 10 µL reaction volume. TaqMan Gene Expression Assays were used for all qPCR experiments following the protocol outlined in Supplementary material online, Tables S2 and S3. Primer efficiency correction was performed with GenEx 5.4.4. Software (MultiD, Sweden). RT–qPCR data analysis was done using the 2^-(ddct)-method.
2.6 Cardiomyocyte isolation
Mice were heparinized (500 IU/kg body weight), and anaesthetized with 5% isoflurane prior to cervical dislocation. Hearts were excised and the aorta was rapidly cannulated. Mouse and human ventricular cardiomyocytes were isolated by a standard liberase-based dissociation procedure using Langendorff perfusion protocol as described previously.9,16
2.7 Subcellular Ca2+ imaging and cardiomyocyte shortening
Intracellular Ca2+ stores were visualized in cardiomyocytes loaded with 10 µM Mag-fluo-4/AM (Life Technologies, Grand Island, NY, USA) using a confocal imaging system (Zeiss LSM 510 Meta).9 Ca2+ transients were recorded in cardiomyocytes loaded with 5 µM Indo-1/AM or 8 µM Fluo-4/AM (both Thermo Fisher Scientific, Vienna, Austria).2,9,10 Indo-1-loaded and field‐stimulated cardiomyocytes were subjected to a stress protocol to simulate an increased physiological workload. Cells were first paced at 0.5 Hz (baseline) followed by administration of 30 nM isoprenaline and a subsequent increase of the stimulation frequency to 5 Hz for 3 min. Stimulation rate was then set back to 0.5 Hz and isoprenaline was washed out. Ca2+ signals were acquired using a customized IonOptix system (Ionoptix, Dublin, Ireland).17 In a subset of dye-free cardiomyocytes that were subjected to the stress protocol, contraction was assessed using a SarcLen Sarcomere Length Acquisition Module (MyoCam‐S, IonOptix, Dublin, Ireland) at the acquisition rate of 240 Hz. At least 10 consecutive beats were averaged and analysed using IonWizard Software (IonOptix, Dublin, Ireland). Simultaneous imaging of nucleoplasmic and cytoplasmic Ca2+ transients was done in Fluo-4/AM-loaded cardiomyocytes using confocal microscopy as described.9 A subset of cardiomyocytes was acutely exposed to 10 nM isoprenaline to measure the response to β-adrenergic stimulation. Frequency-dependent changes in Ca2+ cycling were assessed by gradually increasing stimulation rate from 1 to 4 Hz (mouse) or from 0.25 to 1 Hz (human). Cytoplasmic and nucleoplasmic fluorescence signals were transformed into calibrated [Ca2+].18 Ca2+ spark were recorded in line-scan mode at a sampling rate of 1.54 or 1.92 ms per line within 30 s after the cessation of stimulation at 4 Hz, and analysed with ImageJ plugin SparkMaster (NIH, Bethesda, USA).19 Ca2+ spark frequency was normalized to cell length and recording time, meanwhile RyR2-mediated Ca2+ leak was calculated as described previously.20
2.8 Mitochondrial redox state measurements
The mitochondrial redox state of NAD(P)H/NAD(P)+ and FADH2/FAD was quantified as the autofluorescence of NAD(P)H and FAD, which was recorded in the same cell using excitation/emission wavelengths of 340/450 nm for NAD(P)H and 485/525 nm for FAD. Calibration was performed at the end of every experiment with the mitochondrial uncoupler FCCP (5 µM) and the complex IV inhibitor Na-cyanide (4 mM). Alternatively, the mitochondrial membrane potential (ΔΨm) was determined with 100 nM TMRM using excitation/emission wavelengths of 540/605 nm. For the semi‐quantitative assessment of ΔΨm, TMRM fluorescence was determined before and after the administration of 5 μM FCCP and 1.25 μM oligomycin, which is known to completely dissipate ΔΨm.21
2.9 Respiration measurements in isolated cardiac mitochondria
Freshly isolated cardiac mitochondria were used for high-resolution respirometry measurements that were performed as described previously.21 Oxygen consumption was assayed at 37°C with an Oxygraph-2k high-resolution respirometer and DatLab software was used for data acquisition and analysis (Oroboros Instruments, Innsbruck, Austria). Two mitochondrial respiration protocols were used to quantify oxygen consumption rate upon supplementation of pyruvate (for carbohydrate metabolism) or fatty acids (for β-oxidation) as a fuel. In the ‘carbohydrate’ protocol, measurements of complex I and II activity were performed with 5 mM Na-pyruvate and 5 mM Na-malate. The metabolites were added as reduced substrates after initially recording residual oxygen consumption resulting in leak respiration, followed by increasing concentrations of ADP (0.03, 0.1, 0.3, 1 mM). Complex I was inhibited by 0.5 µM rotenone to prevent reverse electron flux. Finally, oxygen consumption coupled to ADP phosphorylation was inhibited by adding 1.25 µM oligomycin, followed by titration with 10 µM DNP to determine uncoupled respiration state 3 u. In the ‘fatty acid’ protocol, respiration was measured using 1 mM carnitine , 3 mM malate , 10 µM palmitoyl-CoA , 10 µM oleoyl-l-carnitine. Similar to the ‘carbohydrate’ protocol, increasing amounts of ADP, 1.25 µM oligomycin or 10 µM DNP were added. Mitochondrial membrane potential was simultaneously probed using 1 µM TMRM and Smart Fluo-Sensor Green, as described before.21
2.10 Immunocytochemistry
Immunocytochemistry was performed using antibodies against SERCA2a (A010-20, Badrilla), RyR2 (MA3-925, Thermo Fisher Scientific) and phospho-Thr286-CaMKII (ab32678, Abcam).9 Western blotting and detailed immunostaining analyses were used to validate the specificity of antibodies. Cells pretreated with KN-93 served as a negative control for anti-phospho-Thr286-CaMKII antibody; meanwhile bona fide target staining was distinguished from the background by staining with a suitable secondary antibody alone.
2.11 Western blotting
Hearts were perfused with normal Tyrode solution containing or not 10 nM isoprenaline for 5 min, and were then flash frozen for further immunoblotting with primary antibodies recognizing ATG5, SERCA2a, phospholamban (PLB), Troponin I, phospho-PLB-Ser16, phospho-Troponin I-Ser23/24 and GAPDH (for details, see Supplementary material online).
2.12 Acute inhibition of autophagic flux
Autophagic flux in Atg5+/+ animals was blocked using leupeptin-based inhibition of LC3-II (microtubule-associated protein 1 light chain 3-II) turnover, as described previously.22 Atg5+/+ mice received intraperitoneal injections of leupeptin (40 mg/kg body weight, Sigma–Aldrich, Vienna, Austria) or vehicle (sterile 0.9% NaCl solution), and were sacrificed 50 min after the injection. Isolated cardiomyocytes were subjected to subcellular Ca2+ imaging and immunoblot analysis of LC3 and p62/SQSTM1.
2.13 Design-based stereology
Hearts were retrogradely perfused with 4% neutral-buffered formaldehyde and processed for the quantification of LV cardiomyocyte composition using electron microscopy and design-based stereology as described elsewhere.4
2.14 Statistical analysis
Statistical testing was performed using GraphPad Prism 8 (GraphPad Software, CA). Data are presented as bar graphs with error bars showing mean and SEM, respectively, with single data points superimposed. Indicated sample size was determined based on the means and variance of the studied parameters in our previous work on disease mouse models and failing human hearts.9,10 Data residuals distribution was determined using Shapiro–Wilk’s test, while homogeneity of variance was verified by Bartlett’s test. Comparisons between two groups were analysed using Student’s t-test or Mann–Whitney test, as appropriate. In case of multiple comparisons, ANOVA with Dunnett’s or Bonferroni’s post hoc test was applied. In case of Ca2+ transient measurements at different stimulation frequencies or time-lapse/serial measurements, two-way repeated measures ANOVA was used. Significant factorial designs were followed by multiple comparisons using Sidak’s or Bonferroni’s post hoc test. Association of Atg5 gene and ATG5 protein expression levels to interventricular septum was tested using Pearson correlation. Differences were considered significant, if two-tailed P-value was less than 0.05.
3. Results
3.1 Reduced expression of ATG5 and autophagy markers in human hearts with compensated hypertrophy and heart failure
Given that acute pressure overload reduces ATG5 expression and overall pro-autophagic flux in mice,23 we assessed the clinical relevance of these findings by quantifying gene and protein expression of ATG5 in human left ventricular (LV) biopsies of non-failing, hypertrophied, and failing hearts from donors, who underwent echocardiographic examination prior to heart extraction (Supplementary material online, Figure S1). Importantly, both Atg5 gene and protein expression were significantly reduced in hypertrophied and failing human hearts as compared to non-failing controls (Figure 1A and C). Furthermore, mRNA and protein expression of ATG5 inversely correlated with the thickness of the interventricular septum, a measure of cardiac hypertrophy (Figure 1B and D). Notably, ATG5 down-regulation during early and late LV remodelling coincided with a decreased LC3-IIB expression and LC3B-II/LC3B-I ratio, and an increased expression of p62/SQSTM1 in hypertrophied and failing myocardium (Figure 1E and F). These results indicate that decreased Atg5 gene and protein expression along with the reduced amount of autophagy markers appear as an early event in human cardiac remodelling, likely contributing to its transition towards heart failure.
Figure 1.

Reduced gene and protein expression of Atg5 and impaired cardiac autophagy in hypertrophic and failing human hearts. (A) ATG5 expression in left ventricular tissue from non-failing, hypertrophied, moderately failing and end-stage failing human myocardium. Number of hearts per group is shown in each bar. (B) Regression analysis of ATG5 expression levels and thickness of interventricular septum (IVS) from non-failing, hypertrophied, and moderately failing human hearts (N = 23/12/6 hearts, respectively). (C) ATG5 protein expression in left ventricular tissue from non-failing, hypertrophied, and end-stage failing human ventricular tissue (N = 10/17/11 hearts, respectively). (D) Regression analysis of ATG5 protein expression levels and thickness of the interventricular septum (IVS) from non-failing and hypertrophied human hearts (N = 7/16 hearts, respectively). (E) Representative original immunoblots and, (F) expression of the autophagy-related protein markers in non-failing (NF), hypertrophied (Hyp), and end-stage failing hearts (F) (N = 6/3/5 hearts, respectively). (A–F) Data show mean ± SEM. Indicated P-values were calculated using ANOVA followed by Dunnett’s post hoc test. (B) and (D) Association of Atg5 gene and ATG5 protein expression with IVS thickness was computed using Pearson correlation.
3.2 Atg5−/− mice exhibit reduced contractility and exercise capacity in response to stress
To assess the effect of myocardial Atg5 deficiency on cardiac function in vivo, we performed baseline and acute β-adrenergic stress testing in Atg5−/− mice before the development of congestive heart failure. These mice had inhibited cardiomyocyte autophagy (Supplementary material online, Figure S2), which was associated with a mild increase in hypertrophy, as determined by elevated heart-to-body weight ratio and myocardial NppB gene expression (Figure 2A–C). At baseline, Atg5-deficient mice showed no cardiac abnormalities (Supplementary material online, Table S4), as also denoted by similar heart rates (Figure 2D) and systolic function, measured invasively by maximal LV pressure development rate (dPdtmax) (Figure 2E). Isoprenaline administration expectedly evoked increased heart rate and contractility in both groups. However, the increase in dPdtmax upon isoprenaline was lower in Atg5−/− mice than in control animals (Figure 2E), despite similar maximum heart rate development (Figure 2D), indicating reduced cardiac reserve capacity. To examine the outcome of such attenuated inotropic response in the physiological context of increased cardiac demand, we exposed Atg5-deficient mice to exercise tolerance testing. Atg5+/+ and Atg5−/− mice displayed a similar level of exhaustion as determined by comparable blood lactate concentrations (15 ± 6 and 17.3 ± 5.7 mM, respectively; P = 0.45). However, Atg5−/− mice reached exhaustion faster than control mice and, thus, had shorter maximum run distance (Figure 2F). Most importantly, Atg5−/− mice had significantly reduced total workload (Figure 2G), which coincided with lower maximal aerobic capacity (Figure 2H), collectively indicating effort intolerance.
Figure 2.

Atg5-deprived mice exhibit mild hypertrophy, reduced cardiac contractility and exercise capacity. (A) Photomicrographs of hearts from 12-week-old Atg5+/+ (left) and Atg5−/− mice (right). (B) Heart weight-to-body weight ratio (HW/BW) and, (C) NppB expression in Atg5+/+ vs. Atg5−/− hearts (N = 10 vs. 9 mice for HW/BW, and N = 5 mice per group for NppB expression). (D) Heart rate (bpm, beats per minute) and, (E) maximal positive pressure development (dP/dtmax) in the left ventricle at baseline and upon intraperitoneal administration of isoprenaline (ISO, 2 µg/kg body weight) in Atg5+/+ and Atg5−/− mice. N = 4/5 mice, respectively. (F) Maximal run distance, (G) workload and, (H) maximal oxygen consumption (VO2max) in Atg5−/− mice and their control littermates during peak effort testing on a motorized treadmill coupled to indirect calorimetry. N = 8 mice per group. (B–H) Data show mean ± SEM. Indicated P-values were calculated using Mann–Whitney test comparing Atg5−/− or ISO treatment to the respective control.
3.3 Preserved Ca2+ homeostasis in Atg5−/− cardiomyocytes during baseline and β-adrenergic stimulation
Since intracellular Ca2+ homeostasis is the major determinant of excitation–contraction coupling and, thus, determines cardiac performance, we evaluated the functional impact of impaired ATG5-dependent autophagy on intracellular Ca2+ handling by quantifying cytosolic and nuclear Ca2+ transients under normal and stressed conditions. At baseline, Indo-1 wide-field and Fluo-4 confocal imaging revealed similar intracellular Ca2+ concentrations ([Ca2+]i) at diastole and systole, as also kinetic parameters in both groups (Figure 3A–F). As alterations in Ca2+ cycling occur rapidly during sympathetic activation, β-adrenergic stimulation might unmask subtle changes that are indiscernible at baseline. To that end, we investigated whether acute isoprenaline exposure alters intracellular Ca2+ handling in Atg5-deprived cardiomyocytes. While isoprenaline increased the amplitude and reduced the decay time of global Ca2+ transients similarly in both groups (Figure 3A–F), Atg5−/− cardiac myocytes displayed reduced sarcomere length during diastole and systole, which coincided with normal fractional shortening and contractile kinetics (Figure 3C), suggesting an early onset of diastolic dysfunction in Atg5−/− mice. We showed previously that nucleoplasmic and cytoplasmic Ca2+ homeostasis can be regulated independently, and that nucleoplasmic Ca2+ signalling is an early contributor to the development of cardiac hypertrophy.9 However, at slow pacing rates Atg5−/cardiomyocytes displayed preserved nucleoplasmic and cytoplasmic Ca2+ handling at baseline and upon isoprenaline administration (Figure 3D–F), indicating that subcellular Ca2+ handling was preserved during normal and stressed conditions despite the loss of Atg5-dependent autophagy.
Figure 3.

Impaired subcellular Ca2+ cycling in response to increased workload in Atg5−/− cardiomyocytes. (A) Schematic representation of the stress protocol that simulates increased physiological workload by increasing stimulation frequency from 0.5 to 5 Hz during exposure to 10 nM isoprenaline (ISO). (B) Quantification of global calcium transients using Indo-1/AM. From left to right: Systolic and diastolic [Ca2+], amplitude, decay time (DT) and time-to-peak (ttp) in isolated cardiac myocytes subjected to a physiological stress protocol (A). Data show mean ± SEM. n = 28/18 cells from 3/2 Atg5+/+/Atg5−/− mice, respectively. (C) Quantification of myocyte contractility in unstained cells using the stress protocol in (A). From left to right: Sarcomere length, fractional shortening (FS), relaxation time (RT) and time-to-peak (ttp). Data show mean ± SEM (n = 35/32 cells from 3/3 Atg5+/+/Atg5−/− mice, respectively). (D) Original line-scan Fluo-4/AM fluorescence recordings (top) of intracellular Ca2+ transients at baseline and upon acute administration of 10 nM isoprenaline (ISO) in a representative control (left) and Atg5-deficient (right) cardiomyocyte, and corresponding calibrated cytoplasmic (black) and nucleoplasmic (red) Ca2+ transients (bottom). Scale bar, 20 µm. Mean values of cytoplasmic (E) and nucleoplasmic (F) Ca2+ transient parameters displaying diastolic (dia) [Ca2+], Ca2+ transient amplitude, time-to-peak (ttp), and time from peak [Ca2+] to 50% decline (DT50). Data show mean ± SEM (n = 24/20–22 cells from 5/4 Atg5+/+/Atg5−/− mice, respectively). (G) Original line-scan confocal images of cytoplasmic and nucleoplasmic Ca2+ transients at 1 and 4 Hz stimulation in a ventricular myocyte isolated from Atg5+/+ (left) and Atg5−/− (right) mice. Note the area of increased fluorescence intensity displaying increased nuclear Ca2+ (nuc). Scale bar, 20 µm. (H) Averaged original recordings of electrically stimulated Ca2+ transients in the nucleus (red) vs. cytoplasm (black) of ventricular myocytes isolated from Atg5+/+ (left) and Atg5−/− (right) mice in response to a gradual increase of stimulation frequency from 1 to 4 Hz. (I) Frequency-dependent changes in peak systolic amplitude in electrically stimulated Ca2+ transient in the cytoplasm (left) vs. nucleus (right) of ventricular myocytes isolated from control and Atg5-deficient mice. (J) Cytoplasmic (left) and nucleoplasmic (right) Ca2+ load was calculated as a time-integral area under Ca2+ transient–time curve within 1 s in Atg5+/+ and Atg5−/− cardiomyocytes. (H–J) Data show mean ± SEM (n = 19/25 cells from 3/3 Atg5+/+/Atg5−/− mice, respectively). (B and C) and (I and J) P-values were calculated using Bonferroni’s or Sidak’s post hoc test (vs. Atg+/+ control), following significant two-way repeated-measures ANOVA. (E and F) P-values were calculated using Mann–Whitney test comparing Atg5−/− or ISO to the respective control. Average data and group comparisons related to Figure 3 are summarized in Supplementary material online, Table S5.
3.4 Impaired subcellular Ca2+ cycling and signalling in Atg5−/− mice at high-pacing frequencies
Unlike in vitro, β-adrenergic stimulation in vivo exerts multiple effects on the heart, including positive inotropy and chronotropy. Therefore, we assessed the impact of Atg5 deficiency at high heart rates by exposing cardiomyocytes to increasing pacing frequencies (1–4 Hz), but in the absence of isoprenaline. Notably, the frequency-dependent increase in peak systolic Ca2+ amplitude that was observed in Atg5+/+ cells was blunted in Atg5−/− cardiomyocytes in both the cytoplasm and nucleus (Figure 3G–I). However, although the time-averaged cytoplasmic Ca2+ transient was identical between Atg5−/− and Atg5+/+, time-averaged nucleoplasmic Ca2+ load was elevated in Atg5-deficient cells (Figure 3J). Such increase in nucleoplasmic [Ca2+] is known to activate Ca2+/calmodulin-dependent protein kinase II (CaMKII) at high-pacing frequencies and stimulate cardiomyocyte growth,24 as indicated by increased cell and nucleus width-to-length ratio (Supplementary material online, Figure S3). Therefore, we opted to measure CaMKII phosphorylation in Atg5−/− cardiomyocytes subjected to 1 and 4 Hz stimulation. Although the extent of CaMKII phosphorylation was comparable in the cytoplasm and nucleoplasm between Atg5−/− and Atg5+/+ cells, CaMKII activation was more pronounced on the nuclear envelope of Atg5−/− cardiomyocytes already at low pacing rates as compared to control cells (Figure 4D). At high frequencies, CaMKII phosphorylation was elevated also in the cytoplasm and nucleoplasm of Atg5−/− cells (Figure 4A–C). To examine whether acute or chronic autophagy inhibition impairs subcellular Ca2+ handling, we then acutely blocked autophagic flux in Atg5+/+ mice, as determined by LC3 and p62 immunoblotting after blocking lysosome function with intraperitoneal injection of leupeptin (Supplementary material online, Figure S4). Pharmacological autophagy inhibition failed to elicit direct changes in time-averaged Ca2+ transients in the cytoplasm and nucleoplasm. Therefore, loss of Atg5-dependent autophagy seems to induce adverse cardiac remodelling that involves increased CaMKII activity due to the imbalances in nuclear [Ca2+].
Figure 4.
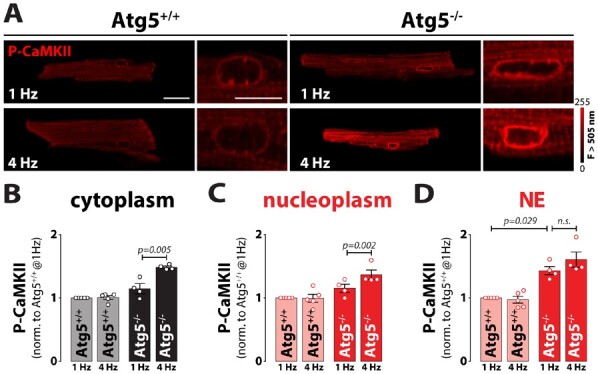
Intact intracellular Ca2+ stores and impaired Ca2+ signalling in response to increased workload in Atg5-deprived hearts. (A) Representative fluorescent images of cardiomyocytes isolated from Atg5+/+ and Atg5−/− mice immunostained against phospho-CaMKII at 1 or 4 Hz stimulation frequency. Scale bar indicates 20 µm for whole cell images or 10 µm for magnified images of the nucleus. (B) Mean cytosolic, (C) nucleoplasm and, (D) nuclear envelope (NE) fluorescence values normalized to Atg5+/+ control cardiomyocytes paced at 1 Hz. Data show mean ± SEM (n = 153/159 cells from 5/4 Atg5+/+/Atg5−/− mice, respectively). Indicated P-values were calculated using Mann–Whitney test comparing Atg5−/− or 4 Hz to the respective control.
3.5 Atg5 deficiency does not alter intracellular Ca2+ stores but reduces the phosphorylation of troponin I
Because sarcoplasmic reticulum (SR) is the major determinant of the Ca2+ transient in cardiomyocytes, we examined whether Atg5 deficiency alters the properties of the SR and, consequently, has an impact on the SR Ca2+ load and/or spontaneous SR Ca2+ release. To this end, we performed imaging of Ca2+ in intracellular stores, which revealed no organizational alterations or fragmentation of the SR or NE, and no obvious reduction in the ability of Mag-Fluo-4 to diffuse through the SR and NE lumen (Supplementary material online, Figure S5A), suggesting that both intracellular Ca2+ stores were efficiently connected in Atg5−/− cardiomyocytes. This finding was corroborated with a similar SR Ca2+ load in Atg5−/− and Atg5+/+ cardiomyocytes, as assessed by the peak amplitude of the caffeine-induced Ca2+ transient (Supplementary material online, Figure S5B). Loss of ATG5 also failed to increase RyR2-mediated SR Ca2+ leak measured as a product of Ca2+ spark frequency and Ca2+ spark mass (Supplementary material online, Figure S5C). These effects coincided with unaltered expression of SR Ca2+ ATPase 2a (SERCA2a) and phosphorylation of its main regulator phospholamban (PLB) between both groups, meanwhile phosphorylation of troponin I (TnI) at serines 23/24 was significantly reduced in Atg5−/− mice as compared to controls (Figure 5A–D). To assess the effect of acute β-adrenergic stimulation on PLB and TnI phosphorylation, we then perfused a subset of mouse hearts ex vivo with 10 nM isoprenaline. As isoprenaline only moderately augments the beat rate of ex vivo perfused hearts,25 the effects of frequency-mediated β-adrenergic signalling in vivo are essentially blunted. Hence, this approach enabled us to study primarily the impact of protein kinase A (PKA)-dependent phosphorylation on PLB and TnI. Notably, isoprenaline-perfused hearts of Atg5+/+ and Atg5−/− mice displayed increased PLB and Tnl phosphorylation (Figure 5B, C, and E). However, despite fully preserved PLB phosphorylation, Atg5-deficient hearts showed a markedly attenuated increase in PKA-dependent phosphorylation of TnI at serines 23/24 (Figure 5E).
Figure 5.

Expression and phosphorylation of Ca2+-regulatory proteins in Atg5-deprived hearts. (A and B) Representative original immunoblots and, (C–E) densitometric analysis of expression and phosphorylation of Ca2+-regulatory proteins in lysates from Atg5+/+ and Atg5−/− mouse hearts in the absence or presence of ISO. From left to right: total SERCA2a, phospholamban (PLB) and Troponin I (TnI), PLB phosphorylated on serine at site 16 (PLB-Ser16), TnI phosphorylated on serine at sites 23 and 24 (TnI-Ser23/24), and relative increase in PLB and TnI phosphorylation upon isoprenaline (ISO) administration are shown. Data show mean ± SEM of N = 6–12 samples per group, indicated P-values were calculated using Mann–Whitney test comparing Atg5−/− and Atg5+/+.
3.6 Reduced abundance of mitochondria leads to their functional overcompensation in Atg5−/− mice
Both reduced TnI phosphorylation and blunted increase of Ca2+ transient amplitude at high-pacing frequencies contribute to the reduction of systolic function in Atg5−/− mice under conditions of increased cardiac demand. This may result from mitochondrial abnormalities, such as reduced respiratory activity,5 and/or impaired cardiomyocyte composition. Therefore, we systematically assessed the structural characteristics and quantified ultrastructural composition of Atg5-deficient cardiomyocytes. Unlike in Atg5+/+ cells (Figure 6A and B), we observed accumulation of concentric layers of membrane sheaths as well as cisternae of membranes resembling SR (Figure 6C and D) in the sarcoplasm of autophagy-incompetent cardiomyocytes. In addition, myofibrillar disarray and atypical aggregates of mitochondria were clearly distinguishable (Figure 6E and F). Atg5−/− cardiomyocytes had slightly, but significantly increased volume of myofibrils (Figure 6G) and decreased total and also individual mitochondrial volume (i.e. volume-weighted mean volume of mitochondria) (Figure 6H and J), resulting in reduced mitochondrial abundance and myofibril-to-mitochondria relative volume ratio (Figure 6I).
Figure 6.

Atg5-deficiency causes aberrant cardiomyocyte composition, decreased mitochondrial abundance and functional overcompensation of individual mitochondria. (A–F) Representative transmission electron micrographs of longitudinal sections of cardiomyocytes depict ultrastructural changes due to the absence of basal autophagy. Note onion-like membrane aggregation (red arrows in C and D), membrane cisternae (red arrows in E), sarcomere disarray, misalignment of myofibrils (red arrows in F), and aggregation of mitochondria (F). Scale bar, 2 µm; Cap, capillary; mf, myofibril; mi, mitochondria; cm, cardiomyocytes. Relative volume of (G) myofibrils Vv(mf/cm) and, (H) mitochondria Vv(mito/cm) per cardiomyocyte from Atg5+/+ and Atg5−/− mouse hearts. (I) Ratio between the relative volume of mitochondria and myofibrils, Vv(mito/cm)/Vv(mf/cm). (J) Volume-weighted mean volume of mitochondria, meanVv(mi), was used to quantify mitochondrial volume and size distribution in Atg5+/+ vs. Atg5−/− mouse hearts. (G–J) Data show mean ± SEM of N = 11–13 mice per group, indicated P-values were calculated using Mann–Whitney test comparing Atg5−/− to the Atg5+/+ control. Isolated Atg5−/− and Atg5+/+ cardiomyocytes were exposed to the stress protocol as in Figure 3A. (K) Mitochondrial membrane potential (ΔΨm; determined by TMRM fluorescence normalized to resting fluorescence, F/F0), (L) NAD(P)H/NAD(P)+ and FADH2/FAD (autofluorescence determined in the same cells) and, (M) ratio of reduced NAD(P)H to oxidized FAD (data from L) were determined. (K–M) Data show mean ± SEM, n = 28/18 cells from 3/2 Atg5+/+/Atg5−/− mice, respectively, for TMRM measurements, and n = 33/32 cells from 3/3 Atg5+/+/Atg5−/− mice, respectively, for NAD(P)H/NAD(P)+ and FADH2/FAD measurements. P-values were calculated using Bonferroni’s post hoc test, following significant two-way repeated-measures ANOVA.
Atypical accumulation of mitochondria with decreased volume prompted us to test the functional properties of cardiac mitochondria from Atg5−/− mice. Surprisingly, we found no difference in oxygen consumption rate and mitochondrial membrane potential (ΔΨm) upon pyruvate or fatty acids supplementation between both groups (Supplementary material online, Figure S6). As a considerable fraction of dysfunctional mitochondria can be lost during mitochondria isolation by gradient centrifugation, we assessed mitochondrial redox state in intact Atg5−/− cardiomyocytes using a physiological stress protocol (Figure 3A). Despite preserved mitochondrial membrane potential (Figure 6K), the overall redox state, measured as the NAD(P)H/FAD fluorescence intensity ratio, was increased in Atg5−/− cardiomyocytes (Figure 6L and M). Furthermore, absolute fluorescence intensities of NAD(P)H and FAD were comparable between both groups (NAD(P)H: 1404 ± 105 a.u. vs. 1432 ± 63 a.u., P = 0.82; FAD: 270 ± 21 a.u. vs. 270 ± 15, P = 0.99; respectively). However, considering that Atg5-deficient cardiomyocytes were hypertrophied and had reduced mitochondrial volume, these results suggest that individual mitochondria are likely in an overcompensated state, which can lead to mitochondrial exhaustion and energy deficit, especially under increased workload.
3.7 Human cardiomyocytes from hypertrophied hearts phenocopy impaired subcellular Ca2+ cycling in Atg5−/− cardiomyocytes
Finally, we tested whether reduced ATG5 expression in hypertrophied human myocardium (Figure 1A and C) is related to alterations in Ca2+ handling, which resemble those in Atg5−/− mice. Indeed, human cardiomyocytes displayed blunted frequency-dependent increase of Ca2+ amplitude and increased nucleoplasmic Ca2+ load at higher pacing frequencies (Figure 7A–D). As β-adrenergic stimulation accelerates Ca2+ transient decay and improves frequency-dependent changes in Ca2+ cycling,26 we hypothesized that isoprenaline diminishes or even prevents the disturbances of Ca2+ cycling in hypertrophied cardiomyocytes exposed to increasing pacing frequencies alone. Isoprenaline expectedly provoked a robust increase of both nucleoplasmic and cytoplasmic Ca2+ transients in both non-failing and hypertrophied cardiomyocytes. Interestingly, the increase in peak amplitude was preserved in hypertrophied compared to control cells at high-stimulation frequencies only in the presence of isoprenaline (Figure 7E–G), indicating an overproportioned reliance on β-adrenergic stimulation. Overall, frequency-dependent changes of subcellular Ca2+ transients in hypertrophied human cardiomyocytes were remarkably similar to those observed in Atg5−/− cells.
Figure 7.

Impaired subcellular Ca2+ cycling in response to increased workload in hypertrophied human cardiomyocytes. (A) Original line-scan confocal images of cytoplasmic and nucleoplasmic Ca2+ transients recorded at 0.25 and 1 Hz stimulation in a ventricular myocyte isolated from non-failing (left) and hypertrophied (right) human hearts. Scale bar, 10 µm. (B) Averaged original recordings of electrically stimulated Ca2+ transients in the nucleus (red) vs. cytoplasm (black) of ventricular myocytes isolated from non-failing (left) and hypertrophied (right) human hearts in response to gradual increase of stimulation frequency from 0.25 to 1 Hz. (C) Frequency-dependent changes in peak systolic amplitude in electrically stimulated Ca2+ transient in the cytoplasm (left) vs. nucleus (right) of ventricular myocytes isolated from control and hypertrophied human hearts. (D) Cytoplasmic (left) and nucleoplasmic (right) Ca2+ load calculated as a time-integral area under Ca2+ transient-time curve over 1 s in control and hypertrophied cardiomyocytes. (C and D) Data show mean ± SEM of n = 7 cells from N = 3 hearts per group, P-values were calculated using Sidak’s post hoc test (vs. non-failing at 0.25 Hz), following significant two-way repeated-measures ANOVA. (E) Original line-scan Fluo-4/AM fluorescence recordings (top) of intracellular Ca2+ transients at baseline and upon acute application of 10 nM isoprenaline (ISO) in a representative control (left) and hypertrophied (right) cardiomyocyte, and corresponding calibrated cytoplasmic (black) and nucleoplasmic (red) Ca2+ transients (bottom). Mean values of cytoplasmic (F) and nucleoplasmic (G) Ca2+ transient parameters displaying diastolic (dia) [Ca2+], Ca2+ transient amplitude, time-to-peak (ttp), and time from peak [Ca2+] to 50% decline (DT50). Data show mean ± SEM of n = 5–6 cells from N = 3 hearts per group. Scale bar, 10 µm. Average data and group comparisons related to Figure 7 are summarized in Supplementary material online, Table S6.
4. Discussion
Beta-adrenergic receptor signalling is the fundamental physiological mechanism underlying acute modulation of Ca2+ handling and contractility in cardiomyocytes.27 However, sympathetic hyperactivity is a hallmark of heart failure development,28 and previous work demonstrated that autophagy protects mice against the development of heart failure induced by chronic β-adrenergic stress.2 Our results demonstrate that the non-selective β-adrenergic agonist, isoprenaline, induced only a moderately positive inotropic response in autophagy-deficient mouse hearts during increased cardiac demand. Reduced positive inotropy in Atg5−/− mice coincided with compromised cardiopulmonary functional reserve capacity during exercise. Our finding that reduced cardiac reserve and exercise intolerance in Atg5−/− mice preceded the onset of heart failure might be clinically relevant as exercise capacity is known to be a major predictor of all-cause mortality.29 In addition, our study demonstrates that a failure to reinstate or increase basal autophagy in the heart, and possibly also in other tissues (e.g. skeletal muscle), could be associated with the low ability of some individuals to sustain or enhance cardiac reserve in response to exercise.30 Of note, physical activity is a non-pharmacological intervention that consistently re-establishes autophagic flux and mitochondrial quality control in experimental heart failure.31 This underscores the importance of basal autophagy in cardiac response to acute exercise, and extends previous reports on skeletal muscle function, in which Atg7 gene deletion was shown to compromise muscle mass, force generation and mitochondrial function.32 Indeed, isoprenaline-mediated β-adrenergic stress unmasked cardiac decompensation associated with exercise intolerance, a hallmark of heart failure, arising from cardiomyocyte abnormalities, including alterations in myofilament properties (e.g. TnI) and mitochondrial inability to maintain redox balance under acute stress. In line with this notion, aged hearts display a decline in autophagic activity, altered redox state, attenuated response to sympathetic stimulation, and reduced contractility.4 In the absence of β-adrenergic stimulation, however, high-frequency pacing stunted the amplitude of Ca2+ transients, while it augmented nucleoplasmic Ca2+ load and CaMKII activity, especially in the nuclear region, suggesting that Atg5 null cardiomyocytes rely considerably on β-adrenergic signalling during acute stress.
Cardiomyocyte contraction and relaxation depend on the conversion of rise and fall of cytosolic [Ca2+] into force production.27 Increases in contraction and relaxation under conditions of elevated workload are primarily determined by a combination of increased SERCA2a activity that is regulated via phosphorylation of PLB, and TnI phosphorylation, which links Ca2+-troponin C binding with the activation of crossbridge reactions with the actin filament. Increased phosphorylation of PLB augments the rate and the maximal Ca2+ uptake by increasing the affinity of SERCA2a for Ca2+ and its turnover, whereas TnI Ser-23/24 phosphorylation contributes to Ca2+ regulation of the myofilament, resulting in decreased Ca2+-sensitive force production and accelerated myofilament relaxation.33,34 Notably, we found that Atg5−/− mice exposed to isoprenaline showed attenuated positive inotropic responsiveness to β‐adrenergic stimulation in vivo, which was associated with reduced increase TnI phosphorylation, but intact PLB phosphorylation. Although previous studies35–37 that proposed a predominance of the phosphorylation of TnI over that of PLB to mediate positive inotropy in response to isoprenaline may partly explain the reduced cardiac reserve in vivo, reasons for these differences are not entirely clear. At least part of the difference may reflect high pre‐existing sympathetic activity during in situ assessment (e.g. by catheterization), whereas the use of isolated cardiomyocytes minimizes these influences. In support of this idea, Layland et al.38 demonstrated that reduced phosphorylation of TnI is linked to contractile dysfunction in conditions similar to those occurring in vivo but much less so in the unloaded cardiomyocyte.
ATG5-mediated autophagy failed to directly regulate the subcellular Ca2+ handling. Along these lines, the properties of the major intracellular Ca2+ store, SR, including its spatial organization, Ca2+ content and RyR2-mediated SR Ca2+ leak, were unchanged in Atg5−/− cardiomyocytes. In this regard, it is important to mention that mammalian cells lacking Atg5, including Atg5−/− cardiomyocytes, can still form a fraction of autophagosomes/autolysosomes and perform autophagy-mediated protein degradation via the Atg5/Atg7-independent non-canonical pathway.6 Interestingly, this alternative autophagy process was also proposed to play a predominant role in mediating mitochondrial degradation in the heart during energy deprivation (e.g. ischaemia).39 Therefore, it is possible that the non-conventional form of autophagy acts as a compensatory mechanism that maintains, at least temporarily, the Ca2+-regulatory machinery in young Atg5−/− mice. However, Atg5−/− cardiomyocytes displayed accumulated mitochondria aggregates due to impaired mitochondrial clearance,2,40 which intriguingly coincided with reduced relative mitochondrial mass. This seemingly counterintuitive finding results from decreased individual mitochondrial volume in Atg5−/− cardiomyocytes and, albeit not measured in our study, can be attributed to balanced mitochondrial degradation vs. biogenesis. In support of this notion, Sun et al.41 recently reported that insufficient autophagy causes reduction in mitochondria content in the heart of a mouse model of lipopolysaccharide-induced sepsis, meanwhile enhanced autophagy in Beclin-1 transgenic hearts is associated with unexpectedly high mitochondrial abundance.
Apart from rapid Ca2+ regulation by β-adrenergic signalling, integrative subcellular changes in Ca2+ levels are critical for cardiac remodelling by modifying Ca2+-dependent pathways mediating transcriptional regulation in cardiomyocytes. The selective increase in nucleoplasmic Ca2+ load at high-pacing frequencies in Atg5-deprived mice activated Ca2+-dependent signalling involving CaMKII that is known to induce cardiac hypertrophy.18 Given that persistent overstimulation desensitizes the β-adrenergic signalling pathway, the progression of hypertrophy to heart failure in older Atg5−/− mice is expected to depend, at least in part, on sustained CaMKII activity. Activated nuclear CaMKII can trigger up-regulation of well-established marker genes linked to pathological hypertrophy, such as NppB, RCAN-1, IL-6 receptor, and transforming growth factor (TGF)-β1.24 Therefore, it is possible that CaMKII regulates Ca2+-mediated gene expression that results from early anomalies in Ca2+ handling in Atg5-deficient mice. Importantly, future studies are warranted to unveil the mechanistic link between autophagy and CaMKII-dependent gene regulation.
Finally, the clinical relevance of our study is presented by the observation that cardiac expression of ATG5 at both transcriptional and protein levels was markedly reduced in hypertrophied and end-stage failing human hearts, while gene and protein expression of ATG5 inversely correlated with the extent of hypertrophy in donor hearts. Importantly, frequency-dependent and isoprenaline-induced changes of subcellular Ca2+ transients in hypertrophied human cardiomyocytes closely resembled those observed in Atg5−/− ventricular myocytes. Hence, progressive loss of ATG5 protein and concomitant autophagy impairment parallels the development of hypertrophy and heart failure in humans. Along this line, early impairment of autophagy may cause energy deprivation in stressed myocardium, thereby initiating similar compensatory mechanisms followed by maladaptive events potentially resembling those observed in Atg5−/− mice. However, although we present a large body of phenomenological evidence that points to a role for ATG5-induced autophagy in Ca2+ handling in cardiomyocytes, we acknowledge that a direct mechanistic link between impaired autophagy and Ca2+ cycling abnormalities requires further investigation.
In summary, this study demonstrates reduced mitochondrial abundance and capability to maintain redox balance in Atg5-deprived cardiomyocytes under stress. Functional alterations of the cytoplasmic and nucleoplasmic Ca2+ handling were associated with increased CaMKII activity and concomitant Ca2+-mediated transcriptional regulation, and resulted in compromised cardiac functional reserve capacity, which precedes the development of heart failure in both mice and humans.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
S.S. conceptualized the study; S.S. and S.L.-H. designed the study; S.L.-H., S.K., N.D., M.A., J.V., V.H., M.S., M.K., A.N., J.S., K.-M.K., G.F. and J.E. performed the experiments and analysed data; S.L.-H., M.A., C. Mühlfeld, C. Maack, and S.S. interpreted the data; A.Z., D.v.L., P.P.R. and D.S. characterized the patients or provided human cardiac tissue and data; S.L.-H. and S.S. wrote the manuscript. All authors revisited the work critically for important intellectual content and approved the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supplementary Material
Acknowledgements
The authors thank Noboru Mizushima (University of Tokyo, Japan) and Kenneth Chien (Harvard University, USA) for their generosity in providing Atg5flox/flox mice and MLC2a-Cre+ mice, respectively. We are grateful for the excellent support by the animal facility staff of the Institute of Biomedical Research (IBF, Medical University of Graz) for animal wellbeing.
Conflict of interest: none declared.
Contributor Information
Senka Ljubojević-Holzer, Department of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria; BioTechMed Graz, Mozartgasse 12/II, 8010 Graz, Austria.
Simon Kraler, Department of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria.
Nataša Djalinac, Department of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria.
Mahmoud Abdellatif, Department of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria.
Julia Voglhuber, Department of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria; BioTechMed Graz, Mozartgasse 12/II, 8010 Graz, Austria.
Julia Schipke, Institute of Functional and Applied Anatomy, Hannover Medical School, Carl-Neuberg-Str. 1, 30625 Hannover, Germany.
Marlene Schmidt, Department of Translational Research, Comprehensive Heart Failure Center (CHFC), University Clinic Würzburg, Am Schwarzenberg 15, Haus A15, 97078 Würzburg, Germany.
Katharina-Maria Kling, Institute of Functional and Applied Anatomy, Hannover Medical School, Carl-Neuberg-Str. 1, 30625 Hannover, Germany.
Greta Therese Franke, Institute of Functional and Applied Anatomy, Hannover Medical School, Carl-Neuberg-Str. 1, 30625 Hannover, Germany.
Viktoria Herbst, Department of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria.
Andreas Zirlik, Department of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria.
Dirk von Lewinski, Department of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria.
Daniel Scherr, Department of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria.
Peter P Rainer, Department of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria.
Michael Kohlhaas, Department of Translational Research, Comprehensive Heart Failure Center (CHFC), University Clinic Würzburg, Am Schwarzenberg 15, Haus A15, 97078 Würzburg, Germany.
Alexander Nickel, Department of Translational Research, Comprehensive Heart Failure Center (CHFC), University Clinic Würzburg, Am Schwarzenberg 15, Haus A15, 97078 Würzburg, Germany.
Christian Mühlfeld, Institute of Functional and Applied Anatomy, Hannover Medical School, Carl-Neuberg-Str. 1, 30625 Hannover, Germany.
Christoph Maack, Department of Translational Research, Comprehensive Heart Failure Center (CHFC), University Clinic Würzburg, Am Schwarzenberg 15, Haus A15, 97078 Würzburg, Germany.
Simon Sedej, Department of Cardiology, Medical University of Graz, Auenbruggerplatz 15, 8036 Graz, Austria; BioTechMed Graz, Mozartgasse 12/II, 8010 Graz, Austria; Institute of Physiology, Faculty of Medicine, University of Maribor, 2000 Maribor, Slovenia.
Funding
This work was supported by the Austrian Science Fund (FWF) (grants P27637-B28 and I3301-MINOTAUR to S.S. and V-530 to S.L.-H.), and BioTechMed-Graz [Young Researcher Groups (YRG) to S.L.-H.]. S.K. was a recipient of the Medical University of Graz scholarship for talented students. M.A. is supported by research grants from the European Society of Cardiology and Austrian Society of Cardiology (Präsidentenstipendium-ÖKG).
Data availability
The raw data are available from the corresponding author upon reasonable request.
Translational perspective
Autophagy is a cytoprotective process ensuring homeostasis of the heart. Here, we report that maladaptive hypertrophy and heart failure associate with reduced expression of autophagy-related protein 5 (ATG5) in humans. In mice, ATG5-dependent autophagy disrupts subcellular Ca2+ homeostasis and Ca2+-mediated transcriptional regulation, coinciding with impaired mitochondrial redox balance, especially during stress and increased cardiac demand. These effects translate into reduced cardiac functional reserve capacity and the development of premature heart failure. Hence, reinstating autophagy at early stages of cardiac remodelling may protect from heart failure through preserving mitochondrial abundance and nuclear Ca2+ homeostasis.
References
- 1. Abdellatif M, Sedej S, Carmona-Gutierrez D, Madeo F, Kroemer G. Autophagy in cardiovascular aging. Circ Res 2018;123:803–824. [DOI] [PubMed] [Google Scholar]
- 2. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007;13:619–624. [DOI] [PubMed] [Google Scholar]
- 3. Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and autophagy in the heart. Circ Res 2016;118:1563–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A, Tong M, Ruckenstuhl C, Dammbrueck C, Gross AS, Herbst V, Magnes C, Trausinger G, Narath S, Meinitzer A, Hu Z, Kirsch A, Eller K, Carmona-Gutierrez D, Büttner S, Pietrocola F, Knittelfelder O, Schrepfer E, Rockenfeller P, Simonini C, Rahn A, Horsch M, Moreth K, Beckers J, Fuchs H, Gailus-Durner V, Neff F, Janik D, Rathkolb B, Rozman J, de Angelis MH, Moustafa T, Haemmerle G, Mayr M, Willeit P, von Frieling-Salewsky M, Pieske B, Scorrano L, Pieber T, Pechlaner R, Willeit J, Sigrist SJ, Linke WA, Mühlfeld C, Sadoshima J, Dengjel J, Kiechl S, Kroemer G, Sedej S, Madeo F. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 2016;22:1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, Nishida K, Shimizu T, Hori M, Komuro I, Takuji Shirasawa TS, Mizushima N, Otsu K. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010;6:600–606. [DOI] [PubMed] [Google Scholar]
- 6. Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009;461:654–658. [DOI] [PubMed] [Google Scholar]
- 7. Kaushik S, Massey AC, Mizushima N, Cuervo AM. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 2008;19:2179–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell 2019;176:11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ljubojevic S, Radulovic S, Leitinger G, Sedej S, Sacherer M, Holzer M, Winkler C, Pritz E, Mittler T, Schmidt A, Sereinigg M, Wakula P, Zissimopoulos S, Bisping E, Post H, Marsche G, Bossuyt J, Bers DM, Kockskamper J, Pieske B. Early remodeling of perinuclear Ca2+ stores and nucleoplasmic Ca2+ signaling during the development of hypertrophy and heart failure. Circulation 2014;130:244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sedej S, Schmidt A, Denegri M, Walther S, Matovina M, Arnstein G, Gutschi EM, Windhager I, Ljubojevic S, Negri S, Heinzel FR, Bisping E, Vos MA, Napolitano C, Priori SG, Kockskamper J, Pieske B. Subclinical abnormalities in sarcoplasmic reticulum Ca(2+) release promote eccentric myocardial remodeling and pump failure death in response to pressure overload. J Am Coll Cardiol 2014;63:1569–1579. [DOI] [PubMed] [Google Scholar]
- 11. Lehnart SE, Maier LS, Hasenfuss G. Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail Rev 2009;14:213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wettschureck N, Rutten H, Zywietz A, Gehring D, Wilkie TM, Chen J, Chien KR, Offermanns S. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat Med 2001;7:1236–1240. [DOI] [PubMed] [Google Scholar]
- 13. Hoit BD, Khoury SF, Kranias EG, Ball N, Walsh RA. In vivo echocardiographic detection of enhanced left ventricular function in gene-targeted mice with phospholamban deficiency. Circ Res 1995;77:632–637. [DOI] [PubMed] [Google Scholar]
- 14. Ayachi M, Niel R, Momken I, Billat VL, Mille-Hamard L. Validation of a ramp running protocol for determination of the true VO2max in mice. Front Physiol 2016;7:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Djalinac N, Ljubojevic-Holzer S, Matzer I, Kolesnik E, Jandl K, Lohberger B, Rainer P, Heinemann A, Sedej S, von Lewinski D, Bisping E. The role of stretch, tachycardia and sodium-calcium exchanger in induction of early cardiac remodelling. J Cell Mol Med 2020;24:8732–8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sedej S, Heinzel FR, Walther S, Dybkova N, Wakula P, Groborz J, Gronau P, Maier LS, Vos MA, Lai FA, Napolitano C, Priori SG, Kockskamper J, Pieske B. Na+-dependent SR Ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human CPVT mutation. Cardiovasc Res 2010;87:50–59. [DOI] [PubMed] [Google Scholar]
- 17. Kohlhaas M, Nickel AG, Bergem S, Casadei B, Laufs U, Maack C. Endogenous nitric oxide formation in cardiac myocytes does not control respiration during β-adrenergic stimulation. J Physiol 2017;595:3781–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ljubojevic S, Walther S, Asgarzoei M, Sedej S, Pieske B, Kockskamper J. In situ calibration of nucleoplasmic versus cytoplasmic Ca(2)+ concentration in adult cardiomyocytes. Biophys J 2011;100:2356–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol 2007;293:C1073–C1081. [DOI] [PubMed] [Google Scholar]
- 20. Kolstad TR, van den Brink J, MacQuaide N, Lunde PK, Frisk M, Aronsen JM, Norden ES, Cataliotti A, Sjaastad I, Sejersted OM, Edwards AG, Lines GT, Louch WE. Ryanodine receptor dispersion disrupts Ca(2+) release in failing cardiac myocytes. Elife 2018;7:e39427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nickel AG, von Hardenberg A, Hohl M, Löffler JR, Kohlhaas M, Becker J, Reil JC, Kazakov A, Bonnekoh J, Stadelmaier M, Puhl SL, Wagner M, Bogeski I, Cortassa S, Kappl R, Pasieka B, Lafontaine M, Lancaster CR, Blacker TS, Hall AR, Duchen MR, Kästner L, Lipp P, Zeller T, Müller C, Knopp A, Laufs U, Böhm M, Hoth M, Maack C. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab 2015;22:472–484. [DOI] [PubMed] [Google Scholar]
- 22. Haspel J, Shaik RS, Ifedigbo E, Nakahira K, Dolinay T, Englert JA, Choi AM. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy 2011;7:629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xie X, Bi HL, Lai S, Zhang YL, Li N, Cao HJ, Han L, Wang HX, Li HH. The immunoproteasome catalytic beta5i subunit regulates cardiac hypertrophy by targeting the autophagy protein ATG5 for degradation. Sci Adv 2019;5:eaau0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ljubojevic-Holzer S, Herren AW, Djalinac N, Voglhuber J, Morotti S, Holzer M, Wood BM, Abdellatif M, Matzer I, Sacherer M, Radulovic S, Wallner M, Ivanov M, Wagner S, Sossalla S, von Lewinski D, Pieske B, Brown JH, Sedej S, Bossuyt J, Bers DM. CaMKIIδC drives early adaptive Ca(2+) change and late eccentric cardiac hypertrophy. Circ Res 2020;127:1159–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karczewski P, Bartel S, Krause EG. Differential sensitivity to isoprenaline of troponin I and phospholamban phosphorylation in isolated rat hearts. Biochem J 1990;266:115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Briston SJ, Caldwell JL, Horn MA, Clarke JD, Richards MA, Greensmith DJ, Graham HK, Hall MC, Eisner DA, Dibb KM, Trafford AW. Impaired beta-adrenergic responsiveness accentuates dysfunctional excitation-contraction coupling in an ovine model of tachypacing-induced heart failure. J Physiol 2011;589:1367–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bers DM. Cardiac excitation-contraction coupling. Nature 2002;415:198–205. [DOI] [PubMed] [Google Scholar]
- 28. Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol 2009;54:1747–1762. [DOI] [PubMed] [Google Scholar]
- 29. Kodama S, Saito K, Tanaka S, Maki M, Yachi Y, Asumi M, Sugawara A, Totsuka K, Shimano H, Ohashi Y, Yamada N, Sone H. Cardiorespiratory fitness as a quantitative predictor of all-cause mortality and cardiovascular events in healthy men and women: a meta-analysis. JAMA 2009;301:2024–2035. [DOI] [PubMed] [Google Scholar]
- 30. Bouchard C, Rankinen T. Individual differences in response to regular physical activity. Med Sci Sports Exerc 2001;33:S446–S451; discussion S452–S443. [DOI] [PubMed] [Google Scholar]
- 31. Campos JC, Queliconi BB, Bozi LHM, Bechara LRG, Dourado PMM, Andres AM, Jannig PR, Gomes KMS, Zambelli VO, Rocha-Resende C, Guatimosim S, Brum PC, Mochly-Rosen D, Gottlieb RA, Kowaltowski AJ, Ferreira JCB. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy 2017;13:1304–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab 2009;10:507–515. [DOI] [PubMed] [Google Scholar]
- 33. Zhang R, Zhao J, Potter JD. Phosphorylation of both serine residues in cardiac troponin I is required to decrease the Ca2+ affinity of cardiac troponin C. J Biol Chem 1995;270:30773–30780. [DOI] [PubMed] [Google Scholar]
- 34. Ramirez-Correa GA, Cortassa S, Stanley B, Gao WD, Murphy AM. Calcium sensitivity, force frequency relationship and cardiac troponin I: critical role of PKA and PKC phosphorylation sites. J Mol Cell Cardiol 2010;48:943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. England PJ. Correlation between contraction and phosphorylation of the inhibitory subunit of troponin in perfused rat heart. FEBS Lett 1975;50:57–60. [DOI] [PubMed] [Google Scholar]
- 36. Resink TJ, Gevers W. Dephosphorylation of myofibrillar proteins in actomyosin preparations and in isolated perfused rat hearts after beta-agonist withdrawal. J Mol Cell Cardiol 1982;14:329–337. [DOI] [PubMed] [Google Scholar]
- 37. Rapundalo ST, Solaro RJ, Kranias EG. Inotropic responses to isoproterenol and phosphodiesterase inhibitors in intact guinea pig hearts: comparison of cyclic AMP levels and phosphorylation of sarcoplasmic reticulum and myofibrillar proteins. Circ Res 1989;64:104–111. [DOI] [PubMed] [Google Scholar]
- 38. Layland J, Grieve DJ, Cave AC, Sparks E, Solaro RJ, Shah AM. Essential role of troponin I in the positive inotropic response to isoprenaline in mouse hearts contracting auxotonically. J Physiol 2004;556:835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saito T, Nah J, Oka SI, Mukai R, Monden Y, Maejima Y, Ikeda Y, Sciarretta S, Liu T, Li H, Baljinnyam E, Fraidenraich D, Fritzky L, Zhai P, Ichinose S, Isobe M, Hsu CP, Kundu M, Sadoshima J. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest 2019;129:802–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005;169:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun Y, Yao X, Zhang QJ, Zhu M, Liu ZP, Ci B, Xie Y, Carlson D, Rothermel BA, Sun Y, Levine B, Hill JA, Wolf SE, Minei JP, Zang QS. Beclin-1-dependent autophagy protects the heart during sepsis. Circulation 2018;138:2247–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data are available from the corresponding author upon reasonable request.