

Abstract
Calcific aortic valve disease (CAVD) is the end result of active cellular processes that lead to the progressive fibrosis and calcification of aortic valve leaflets. In western populations, CAVD is a significant cause of cardiovascular morbidity and mortality, and in the absence of effective drugs, it will likely represent an increasing disease burden as populations age. As there are currently no pharmacological therapies available for preventing, treating, or slowing the development of CAVD, understanding the mechanisms underlying the initiation and progression of the disease is important for identifying novel therapeutic targets. Recent evidence has emerged of an important causative role for reactive oxygen species (ROS)-mediated oxidative stress in the pathophysiology of CAVD, inducing the differentiation of valve interstitial cells into myofibroblasts and then osteoblasts. In this review, we focus on the roles and sources of ROS driving CAVD and consider their potential as novel therapeutic targets for this debilitating condition.
Keywords: Aortic valve, Calcification, Reactive oxygen species, Oxidative stress, NADPH oxidases
1. Introduction
Calcific aortic valve disease (CAVD) is a progressive condition in which a normal tricuspid aortic valve or a congenitally abnormal bicuspid aortic valve becomes thickened, fibrosed, and calcified. The nature of this remodelling results in a spectrum of disease ranging from mild aortic sclerosis to severe aortic stenosis (AS). CAVD is currently the third most prevalent cardiovascular disease after coronary artery disease and hypertension.1 The prevalence of CAVD increases with age. Aortic valve sclerosis affects a quarter of over 65 years old,1 and progression from sclerosis to stenosis occurs in ∼2% of individuals per year.2–4 Most patients with established AS then develop moderate-to-severe symptoms requiring treatment.1–3 It is expected that in the absence of any preventative therapy, CAVD will represent an increasing disease burden as populations in developed countries age5 and as the prevalence of the associated cardiometabolic risk factors for CAVD namely obesity, type 2 diabetes, dyslipidaemia, and hypertension increase.6,7 Consequently, understanding the mechanisms underlying the initiation and progression of CAVD is vital for developing novel therapeutic targets.8
Amongst the various reported mediators of fibrocalcific valvular changes, there is emerging evidence of a critical, causative role for reactive oxygen species (ROS).9–13 This review will focus on the specific sources of ROS or ROS-mediated stress signalling driving CAVD, and examine their potential as novel therapeutic targets for this debilitating disease.
2. ROS in normal physiology
ROS are a group of highly reactive chemical forms of molecular oxygen, divided into free radical species (with at least one free electron) and non-radical (two electron) species.14 The most important examples of these are superoxide and hydrogen peroxide, respectively. Superoxide anions derive from the reduction of molecular oxygen in a reaction mediated by a variety of different enzymes at the cell membrane, cytoplasm, and in organelles, such as mitochondria, peroxisome, and endoplasmic reticulum.14,15 Subsequent spontaneous dismutation of superoxide or dismutation by superoxide dismutase (SOD) enzymes produces hydrogen peroxide. In turn, catalase converts hydrogen peroxide to water, although when partially reduced, hydrogen peroxide is converted to hydroxide ion and hydroxyl radical.
Highlights.
Calcific aortic valve disease (CAVD) is the third most common cardiovascular disease.
There are currently no pharmacological therapies available for preventing, treating, or slowing the development of CAVD.
Reactive oxygen species (ROS) have vital roles in the initiation and propagation of the disease.
Targeting specific adverse sources of ROS including uncoupled nitric oxide synthases and reduced nicotinamide adenine dinucleotide phosphate oxidases 2 proteins represents a novel therapeutic avenue.
At physiological concentrations, ROS regulate cell growth, differentiation, senescence, migration, apoptosis, and autophagy.16–18 They also modulate a variety of metabolic processes including glycolysis, oxidative phosphorylation, and fatty acid synthesis.15,19 These responses are predominantly mediated via the oxidation of cysteine thiolate groups by hydrogen peroxide and of iron–sulphur clusters by superoxide on a wide-range of target proteins.20,21 These modifications regulate protein localization and function, as well as intermolecular and protein–protein interactions.14 Cysteine thiolate oxidation, in particular, leads to disulfide formation, cysteine persulfidation, and glutathionylation, which serve as important modulators of protein activity.22
Several mechanisms maintain ROS at physiological concentrations, including subcellular compartmentalization, SOD and catalase, peroxiredoxins, and the thioredoxin and glutathione systems which reverse the aforementioned cysteine residue modifications.23,24 The NRF2–KEAP1 system is also an important oxidant sensor in which oxidation of residues on KEAP1 leads to the up-regulation of NRF2 nuclear translocation where it acts as a transcription factor for a series of antioxidant proteins.25
3. ROS in cardiovascular diseases
Numerous cardiovascular disorders and diseases including endothelial dysfunction, hypertension, vascular calcification, atherosclerosis, cardiac remodelling, stroke, and diabetes are associated with an oxidative stress state in which ROS-producing enzyme activity and expression levels are up-regulated, whilst the expression and activity of antioxidant mechanisms are down-regulated.26–28 Within the cardiovascular system, three major sources of ROS are uncoupled nitric oxide synthases (NOS), reduced nicotinamide adenine dinucleotide phosphate oxidases (NOXs), and mitochondria.29–31 The roles of these sources of ROS in cardiovascular diseases, extensively reviewed elsewhere,15,26–28,30,32,33 are briefly described here.
3.1 Uncoupled NOS
NOS function as dimers which catalyse the transformation of L-arginine and molecular oxygen to nitric oxide (NO) and L-citrulline, requiring NADPH-derived electrons. NOS uncoupling occurs when there is a depletion of tetrahydrobiopterin (BH4), an obligatory co-factor that actions as an auxiliary electron donor in the above reaction.34,35 Uncoupling results in the production of superoxide as the enzyme switches from its classical NO synthase function to that of an NADPH-dependent oxidase.36 In turn, superoxide combines with NO to produce peroxynitrite, thereby reducing the bioavailability of NO.11,34,37,38 In the case of the endothelial NOS isoform (eNOS), in particular, this leads to impaired endothelial-derived NO-mediated relaxation of vascular smooth muscle with an associated increase in systemic vascular resistance and hypertension.39,40 Reduced NO bioavailability also results in detrimental vascular remodelling, impaired platelet aggregation, and leucocyte adhesion.37 Endothelial dysfunction secondary to BH4 deficiency is an early marker of, and a critical step in the development of atherosclerosis, as well as in diabetic micro- and macrovascular disease.15,41–49 Superoxide release from uncoupled eNOS and nNOS (neuronal NOS) has also been implicated in pressure-overload left ventricular hypertrophy and diastolic dysfunction.42,50–53
3.2 Nox proteins
NOXs generate ROS as their primary function, catalysing electron transfer from NADPH to O2.54–56 There are seven mammalian NOX homologues (NOX1 to NOX5, Duox1, and Duox2) with NOX2 (also termed gp91phox) and NOX4 most widely expressed within the cardiovascular system.31,55 NOX2 is acutely activated by agonists such as angiotensin II, mechanical stimulation, and metabolic factors in a process that requires intracellular association between the transmembrane NOX2-p22phox complex and the cytosolic subunits p47phox, p67phox, p40phox, and Rac1 to generate ROS.54,57 By contrast, NOX4 is constitutively active and regulated mainly by its own expression level,58–61 whilst it may also be activated by mechanical stretch and agonists such as TGF-β.62,63 Moreover, while NOX2 generates superoxide, NOX4 predominantly generates H2O2.31,61,64,65
Overwhelming evidence indicates that the pathophysiological roles of the NOXs are isoform and cell-type specific. NOX2 mediates the development of adverse cardiac fibrosis, cardiomyocyte hypertrophy, contractile dysfunction, and cardiomyocyte death induced by angiotensin II, pressure overload, or myocardial infarction.30,31,59,66–75 In the vasculature, NOX2 may also have important roles driving the initiation and progression of atherosclerosis.15,32,76–81
In contrast to the largely deleterious roles of NOX2, NOX4 may mediate protective signalling in the heart and vasculature.60,82–87 NOX4 protects against pressure overload-induced cardiac remodelling and dysfunction through paracrine preservation of myocardial capillary density,60,88 NRF2-dependent modulation of redox state,89 and enhancement of the integrated stress response.90 Cardiomyocyte NOX4 also maintains optimal mitochondrial function and cardiac performance during physiological exercise.91 Endothelial NOX4 protects against chronic pressure-overload induced cardiac remodelling,84,86 and AngII-stimulated myocardial fibrosis.92 In the vasculature, NOX4 protects against endothelial dysfunction, leucocyte adhesion, inflammation, and atherosclerosis.82,87 However, up-regulated vascular smooth muscle cell (VSMC) NOX4 correlates with VSMC dysfunction and plaque instability, whilst VSMC NOX4 deletion attenuates western-diet–induced atherosclerosis.93,94
3.3 Mitochondria
Mitochondria generate ROS (mitoROS) as natural by-products of oxidative phosphorylation and basal metabolic activity.95,96 MitoROS are tightly regulated by a number of mechanisms including the glutaredoxin, glutathione, and thioredoxin systems which support thiol redox homoeostasis, as well as by the anion carriers uncoupling protein 2 and 3 (UCP2 and 3) which, when activated by ROS, induce a proton leak that negatively influences ATP synthesis to diminish mitoROS production.97 In addition, mitochondrial SOD mediates dismutation of superoxide into hydrogen peroxide and subsequent decomposition into O2 and H2O occurs via the glutathione redox system.98 Additional regulation of mitoROS is mediated by the cytoplasmic GTPase dynamin-related protein-1 (DRP1), a key protein that also regulates mitochondrial fission.99
Several cardiovascular diseases are associated with dysfunctional mitochondrial activity, morphology, and localization, resulting in increased mitoROS production and impaired ROS-scavenging mechanisms. In cardiac tissue, mitoROS contribute significantly to post-ischaemic inflammatory infiltration, resulting in adverse cardiac hypertrophy, fibrosis and necrosis, and impaired excitation–contraction coupling.100–104 In atherosclerosis, mitoROS scavenging, genetic inhibition of mitoROS, or mito-targeted catalase each attenuate lesion progression and reduce inflammatory signalling and immune cell infiltration.105–108
4. ROS in the pathophysiology of CAVD
It is now widely-recognized that CAVD is the manifestation of an active disease process involving multiple cellular mechanisms rather than the output of a passive, degenerative process.1,8,109–111 Increasing evidence points to critical roles of ROS in the initiation and propagation phases of CAVD (Table 1 and Figure 1). Understanding these roles requires a brief review of the cellular mechanisms driving CAVD, that are extensively reviewed elsewhere.1,111–116
Table 1.
Summary of animal and human studies investigating ROS in CAVD
| Experimental models/tissues | In vitro/in vivo/ex vivo | Observations | Sources of ROS | Inhibitors tested | ROS assay | Ref |
|---|---|---|---|---|---|---|
| Animal studies | ||||||
| LDLr−/−/ApoB100/100 mice fed normal chow | In vivo and in vitro | Hypercholesterolaemia induces CAVD in a subset of mice. Stenotic valves in hypercholesterolaemic mice demonstrate increased superoxide levels. | – | – | DHE | 9 |
| LDLr−/−/ApoB100/100 mice fed a western-type diet | In vivo and in vitro | Western-type diet induces hypercholesterolaemia and aortic valve lipid infiltration and deposition, and apoptosis leading to CAVD. | NOX2 | Pioglitazone | – | 223 |
| Chronic Ang II infusion mice fed a hypercholesterolaemic diet | In vivo and in vitro | Chronic infusion of Ang II and hypercholesterolaemia induce oxidative stress within AVs, resulting in leaflet thickening, and ECM remodelling. | – | MnBuOE | – | 205 |
| Cultured porcine aortic valve interstitial cells | In vitro | TGF-β1 induces ROS production and calcium nodule formation via Smad and MAPK pathways. | – | – | DCF | 203 |
| Cultured porcine aortic valve interstitial cells | In vitro | TGF-β1 induces superoxide production which partly mediates in vitro calcification. Co-application with NO donors scavenges superoxide, reducing calcification. | – |
DETA-NONOate SNP peg-SOD |
DHE | 247 |
| Cultured porcine aortic valve interstitial cells | In vitro | Application of osteogenic medium induces valvular fibrosis and calcification mediated by ROS, collagen deposition, and up-regulation of fibronectin, OPN and Runx2 β-catenin accumulation. | NOX2 | Celastrol | DHE | 13 |
| Cultured porcine aortic VECs and porcine aortic valve tissue | Ex vivo and in vivo | TNFα increases VEC intracellular oxidative stress, as well as superoxide and H2O2 synthesis. TNFα or H2O2 decrease nitric oxide synthesis by reducing eNOS expression. This results in myofibroblastic activation, calcification and changes in ECM composition and structure. |
Uncoupled NOS and NOX2 Mitochondria |
L-NAME BH4 Apocynin peg-SOD |
DCF DHE Fluoro H2O2 MitoSOX RedTM |
209 |
| Rabbits fed high cholesterol diet + vitamin D | In vivo and in vitro | Superoxide and H2O2 levels are increased around calcifying foci and potentiate the progression of AV calcification. AV calcification improves by reducing H2O2 levels with lipoic acid. | NOX2, NOX4 |
Lipoic acid Tempol |
DHE DCF |
10 |
| Rabbits fed high cholesterol diet + vitamin D | In vivo and in vitro | High cholesterol + vitamin D induces celastrol-sensitive increases in ROS which drives the development of CAVD and maladaptive cardiac remodelling. | NOX2 | Celastrol | DHE | 13 |
| Cultured bovine aortic VICs | In vitro | Application of LPS induces oxidative stress, ALP overexpression, VIC calcification and ECM remodelling. | XOS |
Allopurinol L-arginine |
– | 236 |
| Human studies | ||||||
| AV tissue from patients undergoing valve replacement surgery for symptomatic AS | In vitro | Superoxide and H2O2 levels are increased near calcified regions. SOD activity, expression of all 3 SOD isoforms, and catalase are significantly decreased in peri-calcific regions. | Uncoupled NOS | L-NAME |
DHE DCF Lucigenin- chemiluminescence |
11 |
| AV tissues from patients with stenosis or sclerosis collected at surgery or autopsy | In vitro | Increases in ROS production are noted around calcifying foci in human sclerotic or stenotic AV. | NOX2 |
peg-SOD peg-Catalase |
DHE | 10 |
| AV tissue and isolated cultured human VICs from patients with CAVD | In vitro |
CAVD tissue demonstrates nitrotyrosine accumulation and increased peroxynitrite levels. SOD and catalase expression and activity are down-regulated. H2O2 induces impaired DNA-damage responses, profibrotic and pro-osteogenic signalling leading to osteogenic differentiation and calcification in isolated VICs |
– | Adenoviral delivery of SOD/Catalase | – | 12 |
| AVs and isolated VICs from patients undergoing heart transplantation, valve replacement, or from deceased donor hearts. | In vitro |
In sclerotic and stenotic valves, nitrotyrosine is diffusely distributed with areas of higher intensity. Dityrosine is only observed in stenotic tissue. TGF-β induces α-SMA up-regulation and aortic valve remodelling. |
– | MnBuOE | – | 205 |
| Human VICs obtained from a normal healthy donor and two donor patients with calcified aortic valves | In vitro | In isolated human VICs reduction in antioxidant enzymes results in H2O2-induced increases in RUNX2 and OPN mRNA expression. | – | CNPs | DCF | 204 |
| Calcified human aortic valves obtained from adults undergoing valve replacement surgery | Ex vivo | Superoxide accumulates in calcified regions of the valves and in the fibrosa endothelium. Fibrosa-specific loss of SOD1 expression confers the fibrosa as the preferential site of superoxide accumulation. | – | – | DHE | 209 |
| Isolated aortic valve endothelial cells isolated from bicuspid valves explanted from patients with AS undergoing valve replacement | In vitro | Reduced expression of the antioxidants GPX3 and SRXN1 are observed in ECs isolated from BAVs leading to increased oxidative stress susceptibility. | – | – | – | 206 |
| Human aortic VICs isolated from patients undergoing AVR | In vitro | VICs incubated with Lp(a) develop increased ROS formation and undergo significant calcium deposition. | Mitochondria | – |
DHE mitoSOXTM Red |
225 |
| Primary and cultured human VICs isolated from stenotic AVs from patients undergoing valve replacement | In vitro | DRP1 overexpression, indicative of mitochondrial dysfunction, is observed, mediating OGM-induced VIC calcification. | Mitochondria | – | – | 228 |
| Myocardial biopsies from patients with AS undergoing elective valve replacement | Ex vivo | Myocardial biopsies demonstrate increased markers of the mitochondrial UPR, potentially indicating the presence of oxidative stress | Mitochondria | – | – | 226 |
| Isolated human VICs obtained from valves explanted from patients undergoing aortic valve replacement | In vitro | Application of Inorganic phosphate induces calcification of isolated human VICs. | SSAO | LJP1586 | – | 234 |
Figure 1.

Pathogenesis of initiation and propagation of CAVD and potential roles of ROS. CAVD is initiated by valve endothelial cells (VEC) damage, triggered by diverse disease stimuli such as systemic inflammation, hyperlipidaemia, mechanical and shear stress, and other risk factors including ageing, hypertension, obesity, and diabetes. These permit the infiltration and oxidation of lipoproteins, and extravasation of inflammatory cells and subsequently innate and adaptive immune responses. In the propagation phase of the disease fibrosis and calcification occur, triggered by the release of cytokines that induce the transition of VICs into myofibroblasts and osteoblasts via the up-regulation of a number of fibrotic and osteogenic genes, respectively. Differentiation of VICs into myofibroblasts leads to leaflet thickening and fibrosis, due to extracellular matrix remodelling containing increased collagen and decreased elastin, forming a scaffold upon which amorphous deposition occurs. VIC transformation into osteoblasts is promoted further by a number of mechanisms including the down-regulation of and/or mutations in NOTCH1, which impair the ability of NOTCH1 to suppress osteogenic gene expression, up-regulated WNT/β-catenin signalling, and increased receptor activator of nuclear factor kappa B (RANK)/RANK ligand (RANKL) interactions. These differentiated VICs secrete calcifying microvesicles that promote calcium phosphate nucleation within valve leaflets. Apoptotic bodies released from VICs driven to programmed cell death by inflammatory cytokines and up-regulated ectonucleotidase expression act as a nidus for calcium and phosphorous crystal deposition. Adverse accumulation of ROS from a variety of sources mediates the oxidation of infiltrating lipids and inflammatory response in the initiation phase of CAVD. ROS are also critically implicated in the propagation phase of the disease, mediating the differentiation of VICs by up-regulating the expression of a series of osteogenic and fibrotic genes via a number of intracellular signalling cascades outlined in the main text. ATP, adenosine triphosphate; AMP, adenosine monophosphate; BAV, bicuspid aortic valve; BMP2, bone morphogenic protein2; ENPP1, ectonucleotide pyrophosphatase/phosphodiesterase family member 1; IL, interleukin; LDL, low-density lipoprotein; Lp(a), lipoprotein a; ROS, reactive oxygen species; NOTCH1, notch homolog 1, translocation-associated; PPi, inorganic pyrophosphate; RUNX2, runt-related transcription factor 2; SOX9, SRY-box 9; TGF-β, transforming growth factor beta; TNFα, tumour necrosis factor alpha.
4.1 Initiation of CAVD
The progressive processes of leaflet fibrosis and calcification are initiated by damage to the endothelial cells lining the aortic valve.117,118 This damage, triggered by diverse risk factors including ageing, obesity and hypertension, systemic inflammation, and mechanical and shear stress, permits the infiltration, deposition, retention, and subsequent oxidation of lipoproteins, such as low-density lipoprotein (LDL) and lipoprotein(a) (Lp(a)).119–123 These events result in a chronic inflammatory response mediated by innate and adaptive immune responses,118,124–127 sustained and promoted by local neovascularization induced by vascular endothelial growth factor secreted from infiltrating mast cells.128–132 These vessels display augmented expression of vascular and intercellular adhesion molecules and endothelial selectin that further promote the inflammatory response.133,134
4.2 Propagation of CAVD
In the propagation phase of the disease fibrosis and calcification become the driving forces.112,116 Cytokines secreted from macrophages and T cells, such as TNFα, IL-1β, IL-6, IL-8α, insulin-like growth factor-1, and TGF-β,1,112,135–139 induce the transition of the predominant aortic valve cell type, namely valve interstitial cells (VICs), into myofibroblasts and osteoblasts.1,140–146 VICs are a heterogeneous population of fibroblast-like cells which are important physiological regulators of valve structure.114 Cytokines induce VIC differentiation by up-regulating the expression of several osteogenesis pathway genes including runt-related transcription factor 2 (Runx2), low-density lipoprotein receptor-related protein 5 (Lrp5), distal-less homeobox 5 (Dlx5), SRY-box 9 (SOX9), and muscle homeobox protein MSX2, as well as bone morphogenic protein 2 (BMP2)—a potent osteogenic differentiation factor, and the osteoblast marker proteins osteopontin (OPN), osteocalcin, and osteonectin.137,144,145,147–157 Direct activation of Toll-like receptors 2 and 4 by oxidized LDL also induces VIC BMP2 expression.124,158,159
Osteogenic VIC transformation is then promoted further by the down-regulation of and/or mutations in NOTCH1, which impair the ability of NOTCH1 to suppress Runx2 and SOX9 expression.143,144,159,160 Furthermore, up-regulated WNT/β-catenin signalling, reductions in anti-osteogenic microRNAs, such as miRNA-30b, and increased receptor activator of nuclear factor kappa B (RANK)/RANK ligand interactions all serve to promote the differentiation of VICs into osteoblast-like cells by up-regulating the expression of osteoblast-related genes.112,143,144,146,152,156,159,161–168
Differentiated VICs then secrete microvesicles containing ectonucleotidases, promoting calcium phosphate nucleation within valve leaflets.1,169,170 VIC overexpression of ectonucleotidases, such as Ectonucleotide pyrophosphatase 1 (ENPP1), alkaline phosphatase (ALP) and 5′-nucleotidase also drive valvular mineralization by generating inorganic phosphate and adenosine,171–173 and in the case of ALP, by hydrolyzing inorganic pyrophosphate and de-phosphorylating OPN, natural inhibitors of calcium phosphate deposition.113,148
Apoptotic bodies released from VICs driven to programmed cell death by inflammatory cytokines, such as TGF-β1 and ENPP1-mediatiated depletion of the key-cell survival signal ATP, also act as a nidus for calcium and phosphorous crystal deposition.138,154,171,174 In a subset of patients, heterotopic ossification mediated by the recruitment of bone-marrow-derived circulating osteogenic progenitor cells and endothelial progenitor cells occurs resulting in the formation of lamellar bone within the aortic valve leaflets.175–178
Alongside this mineralization process, leaflet thickening and fibrosis occur. This is mediated by the differentiation of VICs into myofibroblasts that induce extracellular matrix (ECM) remodelling by secreting excess collagen and increasing their expression of matrix metalloproteinases.179 These changes form a vital scaffold upon which hydroxyapatite nucleation and progressive amorphous deposition occur.109,113,156,176,180–184 Fibrotic changes are driven, in particular, by RANK/RANKL stimulation and by angiotensin II which is produced by chymase released from mast cells and by ACE delivered via LDL infiltration.162,185–189 The resulting extracellular matrix has relatively decreased elastin content and excessive disorganized collagen content which significantly alters leaflet biomechanics, leading to a higher mechanical load that in itself directly encourages further myofibroblastic differentiation in a positive feedback loop.113,114,190
4.3 Systemic inflammation and oxidative stress in CAVD
Analysis of plasma from patients with known AS reveals evidence of a systemic, pro-inflammatory state within which oxidative stress occurs. Accordingly, reduced levels of C4 indicating active complement pathway activation are observed alongside increased circulating pyroglutamic acid, an intermediate only generated when levels of the antioxidant glutathione are depleted.191
In a recent cohort study using thiobarbituric acid to assess for systemic lipid peroxidation and 2,4-dinitrophenylhydrazine to assess for the oxidative modification of plasma proteins, elevated oxidative stress was observed to correlate with the severity of AS, as reflected by mean aortic valve area and mean and maximum aortic gradients.192 Levels of oxidative stress also correlate with impaired systemic fibrinolysis, again suggesting that the oxidative stress in CAVD patients might not simply reflect a localized phenomenon.192 Evidence from animal models of CAVD also supports a putative role for systemic inflammation in CAVD. In both wild-type mice and in the atherosclerotic ApoE*3Leiden mouse model, intraperitoneal LPS induces AV thickening, but not calcification, suggesting a role for systemic inflammation in the early stages of CAVD.193–195
There are conflicting data on the association between serum levels of C-reactive protein (CRP), a marker of systemic inflammation, and CAVD. Several relatively small cohort studies (n ≤ 141) have identified a correlation between CRP levels and the presence and, in some studies, the severity of CAVD.196–199 However, a significantly larger cohort study (n = 5621) performed over a 5-year period, found that C-reactive protein is in fact not associated with baseline incidence of AS, progression to aortic sclerosis, or progression to AS.200
4.4 Localized valvular ROS signalling in CAVD
The majority of the evidence demonstrating roles for oxidative stress in CAVD derives from studies using aortic valve tissue and isolated VICs that together indicate a localized inflammatory response within which ROS signalling occurs. Indeed, a study that used computed tomography–positron emission tomography on patients with varying degrees of CAVD, observed localized valvular uptake of the tracers 18F-sodium fluoride and 18F-fluorodeoxyglucose that assessed for valvular calcification and inflammation, respectively.201 The degree of local valvular tracer uptake was observed to be correlated with the severity of disease.201
Pre-clinical and human studies reportedly demonstrate increased local ROS levels in CAVD. Many of these use fluorescent dyes to indicate ROS accumulation which possess varying degrees of specificity. Thus, whilst dihydroethidium (DHE) is a relatively specific indicator of superoxide, 2′7′-dichlorofluorescein diacetate (DCFH-DA) reacts with hydrogen peroxide and itself induces superoxide production, dismutation of which leads to self-amplification of DCF fluorescence.202 Caution is, therefore, required when interpreting results using this assay. The specificity of the commonly used chemiluminescence-based techniques, such as lucigenin-enhanced chemiluminescence for superoxide, have also been questioned, although when low concentrations are used, concerns regarding redox cycling in which lucigenin can react with oxygen to produce superoxide, are mitigated.202 The variety of assays used by the studies reviewed in this article are summarized in Table 1.
Around 30% of hypercholesterolaemic LDLr−/−ApoB100/100 mice fed a normal diet develop AS, with elevated aortic valve superoxide present both prior to CAVD developing, and more abundant in mice that subsequently develop AS.9 In isolated cultured porcine VICs, TGF-β1 induces ROS production subsequently leading to calcium nodule formation in a signalling cascade involving P38 MAPK and MEK1/2/ERK1/2 pathways.203 These findings are replicated by exogenous ROS application, which promotes VIC calcification by up-regulating fibrotic and osteogenic gene expression, indicating that oxidative stress may precede the differentiation of VICs to an osteoblastic phenotype. In rabbits fed a high cholesterol diet, both superoxide and H2O2 levels are increased in and around calcifying aortic valve, with AV calcification reversed by reducing H2O2 levels with lipoic acid.10
Importantly, ROS have also been implicated in human CAVD. In explanted valves from patients with established AS, superoxide and H2O2 levels are markedly increased near the calcified regions of the valve.11 This accumulation is in part due to a marked reduction in the peri-calcific activity and expression of all three SOD isoforms as well as catalase.11,12 Along similar lines, reduction in antioxidant enzymes results in hydrogen peroxide-induced increases in Runx2 and OPN mRNA expression in isolated human VICs.204
Increased ROS levels are also seen in valves from patients with aortic sclerosis. Here, peroxynitrite and nitrogen dioxide-generated nitrotyrosine levels are increased alongside superoxide and hydrogen peroxide.12 Together, these induce DNA damage, trigger dysfunctional DNA-repair mechanisms, and promote early VIC phenotypic alteration via up-regulation of AKT signalling leading to Runx2 and MSX2 overexpression as well as in vitro calcification. These changes are reversed with adenoviral delivery of superoxide dismutase and catalase.12 In explanted AV valves from patients with CAVD, nitrotyrosine is distributed throughout the sclerotic leaflets, with localized areas of high intensity also noted.205 In contrast, dityrosine is only observed in stenotic valves indicating more advanced oxidation occurring as disease progresses.205
Finally, in bicuspid aortic valves (BAV) from patients with AS undergoing surgical replacement, application of hydrogen peroxide triggers DNA damage and apoptosis in isolated valvular endothelial cells (ECs).206 Interestingly, increased levels of DNA damage and sustained apoptosis signalling are observed in bicuspid compared with tricuspid valves, secondary to molecular differences in oxidative stress susceptibility with reduced expression of the antioxidants glutathione peroxidase 3 and sulfiredoxin noted in BAV ECs.206
4.5 NOS-derived ROS in CAVD
Since uncoupled NOS primarily generate superoxide, it is feasible that the pro-inflammatory milieu in the initiation of CAVD may drive NOS uncoupling and thereby contribute to increases in superoxide levels observed in CAVD.10,11,29,207 Indeed, proteomic analyses of calcified aortic valves reveal decreased expression of HSP90, part of a complex with endothelial NOS, the dissociation of which can cause uncoupling of eNOS, leading to the production of ROS and endothelial dysfunction.208
In support of this hypothesis, the arginine analogue L-NAME, a NOS inhibitor, reduces superoxide production by over 50% in calcified human aortic valves.11 Data from animal models likewise support a role for uncoupled NOS-derived ROS in CAVD. In cultured porcine aortic valve ECs and in porcine aortic valve leaflets, exogenous TNFα and H2O2 induce eNOS uncoupling leading to increases in superoxide and H2O2 levels.209 Application of TNFα and H2O2 also reduces eNOS expression resulting in a reduction in NO synthesis as measured using the Griess assay. This in turn drives disorganization of the extracellular matrix and valvular calcification.209 These changes are partially reversed in the presence of the NOS inhibitor L-NAME, NOX inhibitor apocynin, or PEG-SOD and are fully reversed by the eNOS co-factor BH4, confirming NOS uncoupling as a key mediator in this model of CAVD.
Reduced NO bioavailability within diseased valve leaflets further increases the burden of ROS by decreasing NO-mediated quenching of superoxide. Indeed, alongside eNOS uncoupling, several other mechanisms are responsible for reducing NO synthesis in CAVD including mechanical stress-induced down-regulation of eNOS expression,210,211 and valvular endothelial-mesenchymal transition which occurs as a result of TGF-β1 stimulation,212 the constituent elements of the ECM,213 shear and mechanical stress,214 and protein S-glutathionylation—the consequence of an imbalance between reduced and oxidized glutathione.215 Hypercholesteraemia also decreases the expression and activity of eNOS,216,217 and therefore taken together these mechanisms reduce the availability of NO, resulting in increased superoxide levels driving valvular myofibroblast proliferation and extracellular matrix production.216–220 Moreover, enhanced levels of superoxide from other sources such as NOX2 (described in detail below) result in increased peroxynitrite formation as it reacts with residual NO. In a positive feedback loop, this induces further eNOS uncoupling via peroxynitrite-mediated oxidation of BH4.10,207,221,222 Peroxynitrite is also implicated in evoking DNA damage and dysregulated DNA repair responses within cultured human aortic VICs leading to the up-regulated expression of Runx2 and MSX2 and subsequent in vitro calcification12 (Figure 2).
Figure 2.
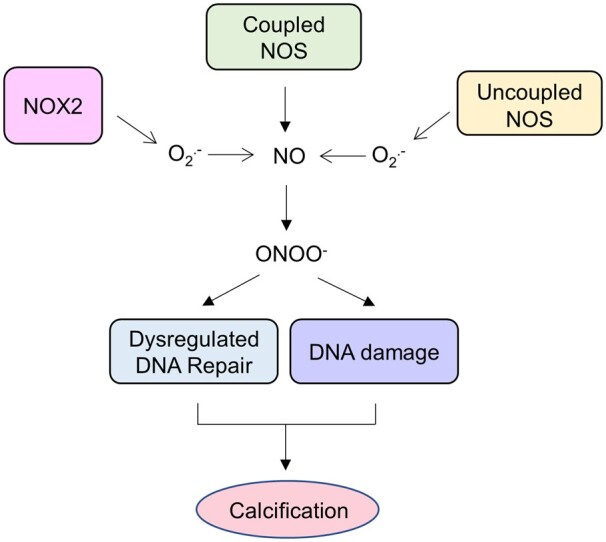
Nitric oxide synthase in the pathophysiology of calcific aortic valve disease. Uncoupling of endothelial nitric oxide synthase (eNOS) reduces nitric oxide bioavailability and increases superoxide (O2) generation. In combination with superoxide generated from other sources, such as nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2), there is increased peroxynitrite (ONOO−) formation as it reacts with residual NO. In a positive feedback loop, this induces further NOS uncoupling via peroxynitrite-mediated oxidation of the obligatory nitric oxide synthase co-factor tetrahydrobiopterin. Peroxynitrite is also implicated in evoking DNA damage and dysregulated DNA repair responses leading to the up-regulated expression of osteogenic genes and valvular calcification.
4.6 NOX-derived ROS in CAVD
Recent evidence has emerged that NOX-derived ROS are critically involved in the development of CAVD. This is particularly so in animal models of CAVD, although conflicting evidence emerges with regards to the specific isoforms involved. In hypercholesteraemic mice fed a Western-type diet in order to induce CAVD, increases in NOX2 and p22phox mRNA levels are observed within harvested valve leaflets, whilst no changes are observed for NOX4, eNOS, catalase, or SOD.223
Our recent studies with cultured primary porcine aortic VICs reveal that application of osteogenic medium (OGM) results in marked fibrosis and calcification associated with increased expression of NOX2, collagen, fibronectin, pro-osteogenic β-catenin accumulation, OPN, and Runx2.13 Additionally, in vivo evidence for NOX2-mediated CAVD is also shown in a rabbit model of CAVD fed cholesterol-enriched chow and vitamin D (HC + VitD). HC + VitD-treated rabbits develop thickened and fibrosed valve leaflets with increased calcium deposits.10,13 Phenotypically, this translates into significant decreases in AV area accompanied by enhanced transvalvular peak and mean jet velocity, ventricular dilatation, and contractile impairment as well as exaggerated cardiac hypertrophy.13 NOX2, p22phox, and its regulator protein disulfide isomerase are strongly expressed around calcification foci in HC+VitD rabbit aortic valves, coincident with the sites of highest hydrogen peroxide levels and the osteoblast differentiation marker OPN.10 Analyses of NOX1 and NOX4 mRNA expression also demonstrate increases in NOX4 expression in HC+VitD rabbits. Taken together therefore, these data support roles for NOX, especially NOX2-derived ROS in animal models of CAVD.
Nevertheless, in human CAVD, roles for NOX-derived ROS are less clear. In one study using stenotic valve tissue acquired from patients undergoing aortic valve replacement surgery or healthy tissue acquired post-mortem, NOX1 mRNA expression was below detectable limits, whilst expression of NOX2 and NOX4 mRNA did not differ significantly between normal valves and non-calcified regions of stenotic valves.11 Moreover, NOX2 and NOX4 mRNA levels in calcified regions of stenotic valves were significantly decreased compared with normal valve tissue. In addition, using a lucigenin-enhanced chemiluminescence assay to assess superoxide production, application of exogenous NADPH also produced similar increases in superoxide in tissue homogenates derived from calcified and non-calcified valves.11 Importantly, recent data has revealed that NADPH–dependent lucigenin chemiluminescence is an imperfect method to examine NOX activity.224
In contrast to these findings, a subsequent study revealed markedly increased NOX2 and p22phox subunit expression around calcifying foci in human sclerotic and, to a greater degree, in human stenotic AV leaflets collected <6 hours post-mortem and surgery, respectively.10 Indeed, these findings have been confirmed in our recent study that clearly demonstrates NOX2 up-regulation in human CAVD.13 In normal valve tissue, immunohistology reveals relatively low expression of NOX2 whereas diseased valves exhibit intense NOX2 accumulation around the calcified regions. Western blotting demonstrates increased levels of NOX2 protein compared with non-calcified valves, accompanied by higher expression of Runx2.13
4.7 Mitochondrial ROS in CAVD
Recent evidence indicates a potential role for mitoROS in the development of CAVD. In isolated cultured human VICs, application of Lp(a) generates superoxide production from mitochondria as indicated by an increase in mitoSOX™ Red fluorescence, with Lp(a) application subsequently inducing in vitro VIC calcification.225 Notably MitoSOX™ Red fluorescence transiently increases after 1 h incubation with Lp(a), and then normalizes at 4 h. Thus, the significance of this observation is unclear given the temporary nature of the finding, although mitoROS might be important in the initiation of VIC calcification. Moreover, whether inhibiting mitoROS abrogates Lp(a)-induced VIC calcium deposition was not examined.
In isolated porcine aortic VICs, application of TNFα induces acute, peg-SOD-sensitive increases in mitochondrial ROS indicated by an increase in MitoSOX Red fluorescence, with the peak effect observed at 30 min.209 Moreover, in ex vivo porcine aortic valve leaflets, 21-day treatment with TNFα likewise induces peg-SOD-sensitive increases in mitoROS production in VECs located in the ventricular endothelium.209 Importantly, this study did not produce any direct experimental evidence implicating mitoROS as a propagator of CAVD and thus the observed increases in mitoROS may simply represent associations.
Myocardial biopsies from patients with AS demonstrate increased markers of the mitochondrial unfolded protein response, indicating a dysfunctional mitochondrial protein-folding environment. This environment is likely the result of cellular stress conditions, including up-regulated ROS and oxidative stress that results from and contributes to mitochondrial dysfunction in CAVD.226 Myocardial biopsies from patients with AS also demonstrate reduced expression of fatty acid translocase—a key enzyme involved in fatty acid oxidation, and the increased expression of glucose transporters 1 and 4. Decreases are also observed in the expression of the fatty acid binding proteins FABPpm and H-FABP, the β-oxidation protein medium chain acyl-coenzyme A dehydrogenase, the Krebs cycle protein α-ketoglutarate dehydrogenase, and the oxidative phosphorylation protein ATP synthase.227 Complex I of the electron transport chain is also down-regulated. Together these changes suggest a metabolic shift from fatty acid to glucose utilization, and, more generally, suggest further evidence of general mitochondrial dysfunction in patients with CAVD.227 However, the extent to which this metabolic shift results from and/or contributes to mitoROS in initiating CAVD remains unclear with no direct data linking the two. Moreover, it is important to point out that these data derive from myocardial tissue alone and thus whether similar abnormalities occur in the aortic valve tissue of these patients likewise requires further investigation.
In primary human VICs obtained from patients with established AS undergoing valve replacement surgery, DRP1 immunoreactivity is detected in calcified valve tissue. Similarly, in cultured human VICs treated with OGM containing inorganic phosphate and L-ascorbic acid for three weeks, DRP1 mRNA is significantly increased.228 DRP1 siRNA reduces human osteogenic medium-induced VIC calcification, indicating an important role for this mitochondrial regulator protein in regulating VIC calcification in vitro.228 These findings together intimate that mitochondrial dysfunction is an important feature of CAVD.
4.8 Other sources of ROS in CAVD
Several studies have identified semicarbazide-sensitive amine oxidase (SSAO), also known as vascular adhesion protein-1 as an additional deleterious source of ROS in CAVD. SSAO generates H2O2 from endogenous amines such as histamine and dopamine, with their activity and expression up-regulated in atherosclerosis, obesity, and diabetes.229–231
SSAO expression and activity are also increased in human CAVD where serum and valvular expression levels correlate with disease severity.232,233 Here, enzyme activity is up to seven times higher than it is in healthy parts of the same valves as assessed by H2O2 production following the application of the SSAO substrate benzylamine.234 In addition, a significant correlation of SSAO expression with oxidative stress is observed, where the enzyme is co-localized with calcified regions of aortic valve tissue. SSAO mRNA levels are positively correlated with mRNA levels of the NOX subunit p22phox and the nuclear enzyme poly(ADP-ribose) polymerase which is activated following DNA damage and oxidative stress.235 In addition, inhibition of SSAO activity with LJP1586 attenuates calcification induced by high concentrations of phosphate in primary cultures of VICs isolated from aortic valves of CAVD patients.234
In bovine aortic VICs, application of LPS induces increased ALP expression and VIC calcification.236 This is associated with increased expression of xanthine oxidase (XOS), which, under oxidative stress, produces superoxide.237 Co-application of allopurinol which inhibits XOS, reverses LPS-induced ALP overexpression.236
5. Targeting ROS in CAVD
There is a major unmet clinical need for novel pharmacological treatments capable of preventing or slowing the progression of CAVD. Several avenues using existing therapies have failed to show benefit in large randomized controlled trials (RCTs) including treatment with statins, antihypertensives, and drugs targeting phosphate and calcium metabolism.1,111,115,238–243 This failure partially results from the fact that by the time patients present with CAVD, the multifactorial, and self-perpetuating cellular mechanisms driving the disease have already been set in motion.113,117,244–246 As described above, specific ROS sources and ROS-mediated signalling may represent novel therapeutic targets given their roles in the initiation and propagation of the cellular mechanisms driving CAVD. Proposed strategies for inhibiting ROS-induced oxidative stress either systemically or locally in CAVD are therefore outlined below and summarized in Table 1 and Figure 3.
Figure 3.
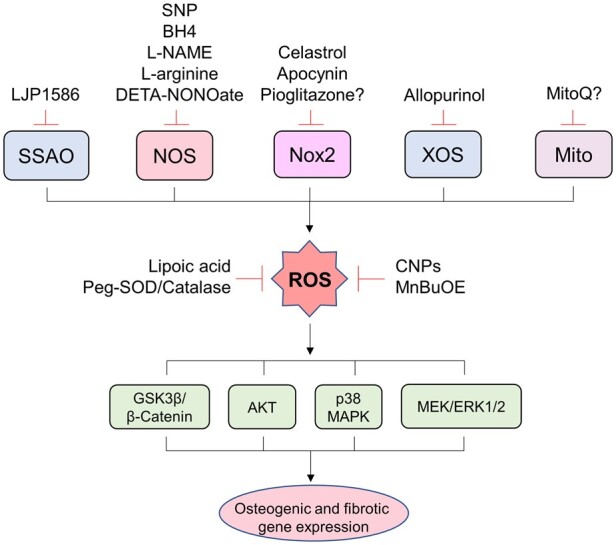
The adverse sources of ROS and potential therapeutic implications in CAVD. Pre-clinical and human studies implicate several adverse sources of ROS in the pathophysiology of CAVD. Intracellular ROS accumulation induces the activation of a variety of signalling cascades that increase the expression of osteogenic and fibrotic genes, phenotypically transforming aortic valvular interstitial cells and leading to valvular calcification and fibrosis. Several potential therapeutic strategies that seek to inhibit ROS-mediated oxidative stress have been suggested. AKT, protein kinase B; BH4, tetrahydrobiopterin; CNPs, cerium oxide nanoparticles; DETA-NONOate, 2,2'-(2-Hydroxy-2 nitrosohydrazinylidene)bis-ethanamine; ERK, extracellular signal-regulated kinases; GS3β, glycogen synthase kinase 3 beta; L-NAME, L-NG-Nitroarginine methyl ester; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase; Mito, mitochondria; MnBuOE, Mn(III) meso-tetrakis (N-n-butoxyethylpyridinium-2-yl) porphyrin; NOS, nitric oxide synthase; NOX2, reduced nicotinamide adenine dinucleotide phosphate oxidase 2; Peg-Catalase, polyethylene glycol-catalase; Peg-SOD, polyethylene glycol-superoxide dismutase; ROS, reactive oxygen species; SSAO, semicarbazide-sensitive amine oxidase; SNP, sodium nitroprusside; XOS, xanthine oxidase.
5.1 Impairing lipid oxidation
As described, lipid oxidation is a key trigger of inflammation in the initiation of CAVD.248 Oxidized LDLs are increased in valves removed from patients with AS, with an association between the level of oxidized LDL and the extent and pace of aortic valve fibrosis and calcification.189,245,249–251 As carriers of oxidized phospholipids (OxPL), Lp(a) also have an important role in the initiation of inflammation seen in CAVD. Approximately one-third of patients with AS have elevated plasma Lp(a), with large genetic and cohort studies revealing that the higher the level, the faster CAVD progresses, with marked increases in the risk of requiring valve replacement or death from the disease.140,141,225,245,252,253
In vitro studies have demonstrated that LDL and Lp(a) oxidation promote the expression of osteogenic differentiation genes but also ROS-mediated VIC calcification. Isolated human aortic VICs cells incubated with either LDL or Lp(a) demonstrate calcification, with those treated with Lp(a) displaying a higher burden of calcium deposition and ROS formation.225 Moreover, progression of CAVD is significantly impaired in transgenic Ldlr−/− mice that express a single-chain variable fragment of E06, a natural antibody which binds to the phosphocholine headgroup of OxPL, and blocks the uptake of oxidized low-density lipoprotein by macrophages.254
These data, therefore, suggest that diminishing the extent of lipid/phospholipid oxidation by lowering plasma LDL and/or Lp(a) levels might represent a therapeutic avenue for treating CAVD. Indeed, in Ldlr−/−Apob100/100 mice, inactivation of the mttp gene significantly impairs hypercholesterolaemia-induced oxidative stress, thereby reducing aortic valve lipid deposition, osteogenic signalling, and valvular calcification.255
Nevertheless, despite promising associations between statin use and lower prevalence of CAVD in observational studies,256–262 RCT data demonstrate no benefit for statin use in CAVD.240,241,244–246,263–266 Randomization to statin use is not only of no benefit in halting the progression of established CAVD but also does not reduce the incidence of CAVD in patients with no established diagnosis at the start of the clinical trials.266 In addition, as yet there are no RCT data on specific Lp(a)-lowering therapy, although an antisense oligonucleotide that specifically lowers Lp(a) levels is currently under investigation.267
The synthetic compound probucol, purported to inhibit LDL oxidation, has also been proposed as a novel treatment for CAVD.268,269 Indeed, probucol has been clinically proven to arrest the progression of atherosclerosis in the vasculature and in coronary artery restenosis following angioplasty.12,270–273 However, it is now clear that probucol likely mediates most of its effects via the induction of HO-1, rather than through direct inhibition of lipid oxidation.274
5.2 Antioxidants
Broad ROS scavenging with natural and synthetic ‘antioxidant’ compounds has been attempted by several groups for attenuating atherosclerosis and vascular calcification. These studies, reviewed extensively elsewhere,275 have investigated a variety of compounds, such as diosgenin, vitamins A–E, quercetin, 10-DHGD, and curcumin, each of which have been shown in vitro to scavenge ROS and/or up-regulate endogenous antioxidant mechanisms, subsequently attenuating inflammation and vascular calcification.275
Moreover, several large observational studies have suggested an inverse relationship between dietary ‘antioxidant’ intake such as α-tocopherol, β-carotene, and vitamins C and E, with cardiovascular morbidity and mortality.273,276,277 However, meta-analyses of randomized control trial data on the effects of vitamins B, C, E, and S or β-carotene have found identical rates of cardiovascular morbidity and mortality in the placebo and antioxidant groups.273,278,279
A number of reasons for these disappointing findings have been proposed including the inadequate length of follow-up periods and the dosing of antioxidants tested. In addition, given that ROS also mediate important physiological processes,273 generalized scavenging of ROS is unlikely to prove beneficial if it interrupts cellular homeostasis and cardiovascular physiology such as endothelial-mediated control of vascular tone by superoxide and hydrogen peroxide as well as platelet aggregation, angiogenesis, and immune cell activity.15 It is, therefore, plausible to hypothesize that given the failure of these compounds in atherosclerosis and vascular calcification, that they are unlikely to prove beneficial for those with CAVD.
5.3 Enhancing SOD and catalase activity
Reductions in antioxidant enzyme levels are observed in calcified aortic valve leaflets.11,12 Adenoviral delivery of SOD or catalase significantly reduces ROS-induced human VIC DNA damage, osteoblastic differentiation, and valvular calcification.12 Similarly, application of the cell-permeable polyethylene glycol-SOD (peg-SOD) or peg-catalase significantly decrease superoxide and H2O2 levels respectively in human stenotic AVs and in rabbits on a high cholesterol and vitamin D diet.10 Peg-SOD also inhibits TGF-β1-induced calcium nodule formation by 70% in porcine aortic VICs.247 Intriguingly, in porcine aortic VICs, peg-SOD actually promotes OGM-induced AVIC calcification,13 suggesting that this approach may only work in certain models of CAVD, or at a certain range of dosage.
Cerium oxide nanoparticles (CNPs) act as ROS scavengers by switching between Ce3+ and Ce4+ oxidation states238,280 and by acting as catalase and SOD-mimetics.204,280–282 In calcified human VICs, rod- and sphere-shaped CNPs scavenge H2O2 in vitro and decrease VIC differentiation as assessed by osteoblast marker expression.204 Recently, the manganese porphyrin‐based compound and SOD mimic MnTnBuOE‐2‐PyP5+ (MnBuOE) has been shown to inhibit aortic valve remodelling in human VICs and mouse models of CAVD.205 MnBuOE prevents TGF‐β-induced α‐SMA up-regulation in human VICs, and in hypercholesterolaemic mice with a chronic infusion of Ang II, co-treatment with MnBuOE reduces AV thickening and ECM remodelling by reducing collagen deposition and improving fibre alignment.205
It is important to point out that clinical trials adopting this approach, i.e. increasing SOD and catalase expression and/or activity must carefully consider the pleiotropic roles for ROS in regulating a wide-range normal physiological processes.14,15,18 It is reasonable to assume that any intervention which overcorrects for oxidative stress will unlikely prove beneficial, particularly so when considering the balance between local adverse ROS signalling and systemic increases in ROS driving CAVD.
5.4 Increasing NO bioavailability
Several studies have attempted to reduce the overall burden of ROS within the aortic valve by increasing the bioavailability of NO. Increasing NO bioavailability not only reduces the burden of ROS but also regulates Notch1 signalling and its nuclear localization to reduce the expression of osteogenic markers in VICs.283,284
In isolated porcine VICs, application of TGF-β1 induces superoxide production which partly mediates valve calcification.247 However, co-application with the NO donors DETA-NONOate and sodium nitroprusside (SNP) scavenges superoxide, reducing in vitro calcification.247 DETA-NONOate also inhibits OGM-induced VIC differentiation and matrix calcification in isolated porcine VICs.285 Moreover, pre-treatment with L-arginine, the precursor for NO synthesis, significantly attenuates the osteogenic differentiation of bovine aortic VICs exposed to the endotoxin LPS.236 In the presence of L-arginine, LPS-induced ALP expression and subsequent matrix calcification are reduced, alongside reductions in LPS-induced TNF-alpha, IL-6, and IL-1β expression. Pre-treatment with L-arginine also reduces xanthine oxidase expression, and markers of ECM remodelling including ADAMTSL4, basigin, and COL3A1.236 Finally, in calcified ex vivo human aortic valves exposed to TNFα, co-treatment with BH4 mitigates eNOS uncoupling, increasing NO bioavailability which reduces superoxide levels, thereby attenuates the expression of osteogenic genes.209
The above findings support, at least in principle, the proposal that increasing NO bioavailability might represent a novel approach for treating and/or slowing the progression of CAVD. Nevertheless, there is currently no clinical trial evidence on the effectiveness of this approach. In addition, part of the pleiotropic nature of statins are their ability to increase NO bioavailability via the stabilization of eNOS mRNA,216,286–289 and in a rabbit model of CAVD, statins reduce AV calcification via this mechanism.216 Given the aforementioned failure of statin therapy in clinical trials, different approaches to increasing NO bioavailability require further investigation. These might include organic nitrate therapy, administration of L-arginine or BH4, or using the KATP channel opener nicorandil, which also possesses a nitrate moiety. Renin-angiotensin system inhibitors such as ACE inhibitors might also slow the progression of CAVD by increasing NO bioavailability via up-regulating eNOS expression, reducing bradykinin breakdown, and by supressing NOX-generated superoxide.290–294
5.5 Inhibiting NOX2
Specific inhibition of only the deleterious sources of ROS in CAVD represents a more nuanced approach than the broad ROS scavenging approaches described above. To this end, inhibitors of NOX2 signalling have been used in animal models of CAVD, but with varying results. In porcine aortic valves, apocynin, which blocks phosphorylation of the obligatory NOX2 cytosolic component p47phox 58,295,296 but may also have non-specific antioxidant activity, only partly reduces TNFα-induced increases in superoxide and hydrogen peroxide.209 This is in contrast to isolated mice aortic myofibroblasts where apocynin markedly impairs TNF- and IL-1β-induced NOX2 ROS generation, a finding that is recapitulated with antisense oligonucleotides targeted against NOX2.137
In hypercholesterolaemic LDLr−/−/apoB100/100 mice fed a western diet, pioglitazone reduces aortic valve osteogenic signalling, aortic valve calcification, and improves cusp mobility, possibly due to a reduction of inflammation and oxidative stress that is associated with down-regulated NOX2 expression.223 The specific contribution of NOX2 to this improvement is not however clear, given that multiple other inflammatory mediators including TNF and IL-6 are also down-regulated by pioglitazone therapy.223
Interestingly, in isolated porcine VICs, treatment with celastrol, a pentacyclic triterpene naturally extracted from the roots of Tripterygium wilflordii, and a potent NOX inhibitor with higher potency against NOX2,297 decreases NOX2 and Runx2 protein levels, VIC ROS levels, and reduces calcium deposition.13 Pro-osteogenic accumulation of β-catenin is also reversed with celastrol, impairing NOX2-mediated inactivation of GSK3β, which in turn enables β-catenin degradation. These findings are replicated when isolated aortic VICs are transfected with adenoviral vectors expressing a short hairpin sequence targeted against NOX2,13 an important observation given that NOX-independent effects of celastrol are also described in the cultured cell line PLB-985.298 Of note, celastrol may have other global beneficial effects, such as anti-obesity and anti-inflammation properties,299,300 which are comorbidities of CAVD likewise characterized by an increase in ROS production mediated, at least in part, by NOX2 activation.17
Moreover, in a rabbit model of CAVD fed with HC + VitD, dietary celastrol markedly alleviates the degree of AS and improves cardiac dilatation, contractility, and function.13 Celastrol treatment also improves rabbit AV fibro-calcification, as indicated by decreases in collagen deposition, the expression of fibronectin, OPN, and the number of calcium deposits.13
Recently, celastrol has also been shown to suppress Runx2 and OPN expression and reverse calcification in cultured porcine aortic VICs exposed to a medium containing high concentrations of calcium and phosphate.301 Here, celastrol reverses calcium-induced up-regulation of BMP2‐BMPRII-Smad1/5 and Wnt/β‐catenin signalling pathways, preventing their respective nuclear translocation and modulation of osteogenic gene expression. These findings are replicated in vivo by intraperitoneal injection of celastrol in mice fed adenine to induce CKD and intraperitoneal vitamin D to induce valvular calcification.301 However, NOX2 involvement in this model of CAVD remains to be investigated.301
Together, these data suggest that NOX2 represents a novel therapeutic target in the treatment of CAVD with further work required to delineate its role in human CAVD. Given the role played by NOX2 in neutrophil function,302 future work targeting NOX2 in CAVD will need to consider whether this compromises innate immune responses, although previous work demonstrates that this only occurs with substantial NOX2 inhibition,303 thus safe therapeutic targeting of cardiovascular NOX2 could be feasible.
6. Conclusions and perspectives
CAVD is the end result of multiple active cellular and molecular mechanisms converging on the phenotypic change of aortic VICs. Oxidative stress is an important driver of these processes and targeted inhibition of the deleterious sources of ROS, particularly NOX2 and uncoupled eNOS, represent important avenues in the search for novel therapies. This targeted approach theoretically avoids interfering with the wide variety of normal cellular processes dependent on ROS signalling and may therefore yield better results for preventing or slowing the progression of CAVD than approaches non-specifically inhibiting redox signalling trialled thus far. Notably, in cancer medicine, a number of drugs targeting excessive ROS signalling that broadly up-regulate antioxidant mechanisms have thus far proved unsuccessful with unexpected toxic side effects often observed in normal tissues.304
Presently there is a lack of in vivo and clinical data on human CAVD investigating roles and sources of adverse ROS signalling driving the disease. Specifically, there is a need for adequately powered cohort observational studies that might establish if there is a relationship between the extent of valvular oxidative stress, the pace of disease progression, and its relation to clinical severity. Novel imaging modalities that assess the in vivo burden of valvular oxidative stress using redox-sensitive probes may therefore represent important tools.305,306 In addition, larger scale in vitro studies examining roles and sources of ROS in human CAVD will also be necessary given the relatively small number of patients recruited to each of the studies described in this review. These in vitro studies may wish to make use of novel, and more-specific fluorescent ROS probes307 and ROS-targeting nanotechnologies,308,309 when exploring roles of ROS signalling in human CAVD.
Despite evidence of a role for mitoROS in a range of cardiovascular diseases,15,27,105 there remains no direct evidence of a pathophysiological role for these in CAVD. Future work might seek to identify whether mitoROS indeed play a role and whether targeting these ROS with recently developed site-specific mitochondrial ROS inhibitors, such as MitoQ effects the development and/or progression of the disease whilst maintaining the range of normal metabolic functions reliant on normal levels of mitoROS.310 Importantly, excessive ROS production by non-mitochondrial sources (such as uncoupled NOS enzymes or NOXs) can trigger mitochondrial dysfunction and further ROS production—sometimes termed ROS-induced ROS release.311,312 Therefore, further work is needed to explore the cross-talk between ROS at distinct subcellular compartment in aortic valve cells and its contribution to CAVD.
Finally, it would appear likely that any effective therapy preventing or slowing the progression of CAVD needs to be initiated early in the evolution of the disease, rather than at the stage where a patient presents with severe AS. Identifying and targeting pivotal molecular pathways especially ROS-mediated canonical redox signalling at an early stage will prove challenging, and may require the use of state-of-the-art technologies, such as network medicine analysis and multi-omics mapping that generate transcriptional and protein expression signatures for the different stages of CAVD.313 Indeed, the feasibility of this approach has recently been demonstrated for CAVD,314 potentially an important step in characterizing the large number of cellular and molecular changes occurring as the disease develops and progresses.
Conflict of interest: none declared.
Funding
This study was supported by the British Heart Foundation (PG/17/39/33027 to M.Z.; CH/1999001/11735 to A.M.S.), the National Natural Science Foundation of China (81470506), and Key Research Project of the Heart Center of Xinxiang Medical University (2017360).
Contributor Information
Harry Z E Greenberg, Department of Cardiology, Cardiovascular Division, King's College London British Heart Foundation Centre of Research Excellence, James Black Centre, 125 Coldharbour Lane, London SE5 9NU, UK.
Guoan Zhao, Department of Cardiology, The First Affiliated Hospital of Xinxiang Medical University, Heart Center of Xinxiang Medical University, Henan, China.
Ajay M Shah, Department of Cardiology, Cardiovascular Division, King's College London British Heart Foundation Centre of Research Excellence, James Black Centre, 125 Coldharbour Lane, London SE5 9NU, UK.
Min Zhang, Department of Cardiology, Cardiovascular Division, King's College London British Heart Foundation Centre of Research Excellence, James Black Centre, 125 Coldharbour Lane, London SE5 9NU, UK.
References
- 1. Lindman BR, Clavel MA, Mathieu P, Iung B, Lancellotti P, Otto CM, Pibarot P. Calcific aortic stenosis. Nat Rev Dis Prim 2016;2:16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coffey S, Cox B, Williams MJA. The prevalence, incidence, progression, and risks of aortic valve sclerosis: a systematic review and meta-analysis. J Am Coll Cardiol 2014;63:2852–2861. [DOI] [PubMed] [Google Scholar]
- 3. Eveborn GW, Schirmer H, Heggelund G, Lunde P, Rasmussen K. The evolving epidemiology of valvular aortic stenosis. the Tromsø Study. Heart 2013;99:396–400. [DOI] [PubMed] [Google Scholar]
- 4. Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet 2006;368:1005–1011. [DOI] [PubMed] [Google Scholar]
- 5. Danielsen R, Aspelund T, Harris TB, Gudnason V. The prevalence of aortic stenosis in the elderly in Iceland and predictions for the coming decades: the AGES-Reykjavík study. Int J Cardiol 2014;176:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Owens DS, Katz R, Takasu J, Kronmal R, Budoff MJ, O'Brien KD. Incidence and progression of aortic valve calcium in the multi-ethnic study of atherosclerosis (MESA). Am J Cardiol 2010;105:701–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Katz R, Wong ND, Kronmal R, Takasu J, Shavelle DM, Probstfield JL, Bertoni AG, Budoff MJ, O’Brien KD. Features of the metabolic syndrome and diabetes mellitus as predictors of aortic valve calcification in the multi-ethnic study of atherosclerosis. Circulation 2006;113:2113–2119. [DOI] [PubMed] [Google Scholar]
- 8. Hutcheson JD, Aikawa E, Merryman WD. Potential drug targets for calcific aortic valve disease. Nat Rev Cardiol 2014;11:218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation 2006;114:2065–2069. [DOI] [PubMed] [Google Scholar]
- 10. Liberman M, Bassi E, Martinatti MK, Lario FC, Wosniak J, Pomerantzeff PMA, Laurindo FRM. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol 2008;28:463–470. [DOI] [PubMed] [Google Scholar]
- 11. Miller JD, Chu Y, Brooks RM, Richenbacher WE, Peña-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol 2008;52:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Branchetti E, Sainger R, Poggio P, Grau JB, Patterson-Fortin J, Bavaria JE, Chorny M, Lai E, Gorman RC, Levy RJ, Ferrari G. Antioxidant enzymes reduce DNA damage and early activation of valvular interstitial cells in aortic valve sclerosis. Arterioscler Thromb Vasc Biol 2013;33:e66–e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu H, Wang L, Pan Y, Wang X, Ding Y, Zhou C, Shah AM, Zhao G, Zhang M. Celastrol alleviates aortic valve calcification via inhibition of NADPH oxidase 2 in valvular interstitial cells. JACC Basic Transl Sci 2020;5:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 2020;21:363–383. [DOI] [PubMed] [Google Scholar]
- 15. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res 2018;122:877–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 2014;15:411–421. [DOI] [PubMed] [Google Scholar]
- 17. Zhang L, Wang X, Cueto R, Effi C, Zhang Y, Tan H, Qin X, Ji Y, Yang X, Wang H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol 2019;26:101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012;48:158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paik JY, Jung KH, Lee JH, Park JW, Lee KH. Reactive oxygen species-driven HIF1α triggers accelerated glycolysis in endothelial cells exposed to low oxygen tension. Nucl Med Biol 2017;45:8–14. [DOI] [PubMed] [Google Scholar]
- 20. Winterbourn CC. Biological production, detection, and fate of hydrogen peroxide. Antioxidants Redox Signal 2018;29:541–551. [DOI] [PubMed] [Google Scholar]
- 21. Castro L, Tórtora V, Mansilla S, Radi R. Aconitases: non-redox iron-sulfur proteins sensitive to reactive species. Acc Chem Res 2019;52:2609–2619. [DOI] [PubMed] [Google Scholar]
- 22. Zeida A, Trujillo M, Ferrer-Sueta G, Denicola A, Estrin DA, Radi R. Catalysis of peroxide reduction by fast reacting protein thiols. Chem Rev 2019;119:10829–10855. [DOI] [PubMed] [Google Scholar]
- 23. Rhee SG, Kil IS. Multiple functions and regulation of mammalian peroxiredoxins. Annu Rev Biochem 2017;86:749–775. [DOI] [PubMed] [Google Scholar]
- 24. Hanschmann EM, Godoy JR, Berndt C, Hudemann C, Lillig CH. Thioredoxins, glutaredoxins, and peroxiredoxins-molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxidants Redox Signal 2013;19:1539–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 2018;98:1169–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sugamura K, Keaney JF. Reactive oxygen species in cardiovascular disease. Free Radic Biol Med 2011;51:978–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol 2018;100:1–19. [DOI] [PubMed] [Google Scholar]
- 28. Münzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC. Impact of oxidative stress on the heart and vasculature: part 2 of a 3-part series. J Am Coll Cardiol 2017;70:212–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vásquez-Vivar J, Kalyanaraman B, Martásek P, Hogg N, Masters BSS, Karoui H, Tordo P, Pritchard KA. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A 1998;95:9220–9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang M, Perino A, Ghigo A, Hirsch E, Shah AM. NADPH oxidases in heart failure: poachers or gamekeepers? Antioxidants Redox Signal 2013;18:1024–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 2012;110:1364–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Konior A, Schramm A, Czesnikiewicz-Guzik M, Guzik TJ. NADPH oxidases in vascular pathology. Antioxidants Redox Signal 2014;20:2794–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brandes RP, Weissmann N, Schröder K. NADPH oxidases in cardiovascular disease. Free Radic Biol Med 2010;49:687–706. [DOI] [PubMed] [Google Scholar]
- 34. Gebhart V, Reiß K, Kollau A, Mayer B, Gorren ACF. Site and mechanism of uncoupling of nitric-oxide synthase: uncoupling by monomerization and other misconceptions. Nitric Oxide Biol Chem 2019;89:14–21. [DOI] [PubMed] [Google Scholar]
- 35. Alp NJ, Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol 2004;24:413–420. [DOI] [PubMed] [Google Scholar]
- 36. Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MAH, Chen YR, Druhan LJ, Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010;468:1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kietadisorn R, Juni RP, Moens AL. Tackling endothelial dysfunction by modulating NOS uncoupling: new insights into its pathogenesis and therapeutic possibilities. Am J Physiol Endocrinol Metab 2012;302:E481–E495. [DOI] [PubMed] [Google Scholar]
- 38. Pou S, Keaton L, Surichamorn W, Rosen GM. Mechanism of superoxide generation by neuronal nitric-oxide synthase. J Biol Chem 1999;274:9573–9580. [DOI] [PubMed] [Google Scholar]
- 39. Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest 2005;115:1221–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J 2012;5:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vanhoutte PM, Shimokawa H, Tang EHC, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol 2009;196:193–222. [DOI] [PubMed] [Google Scholar]
- 42. Daiber A, Steven S, Weber A, Shuvaev VV, Muzykantov VR, Laher I, Li H, Lamas S, Münzel T. Targeting vascular (endothelial) dysfunction. Br J Pharmacol 2017;174:1591–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hattori Y, Hattori S, Wang X, Satoh H, Nakanishi N, Kasai K. Oral administration of tetrahydrobiopterin slows the progression of atherosclerosis in apolipoprotein E-knockout mice. Arterioscler Thromb Vasc Biol 2007;27:865–870. [DOI] [PubMed] [Google Scholar]
- 44. Schmidt TS, McNeill E, Douglas G, Crabtree MJ, Hale AB, Khoo J, O’Neill CA, Cheng A, Channon KM, Alp NJ. Tetrahydrobiopterin supplementation reduces atherosclerosis and vascular inflammation in apolipoprotein E-knockout mice. Clin Sci 2010;119:131–142. [DOI] [PubMed] [Google Scholar]
- 45. Wilcox JN, Subramanian RR, Sundell CL, Ross Tracey W, Pollock JS, Harrison DG, Marsden PA. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler Thromb Vasc Biol 1997;17:2479–2488. [DOI] [PubMed] [Google Scholar]
- 46. Tiefenbacher CP, Bleeke T, Vahl C, Amann K, Vogt A, KüBler W. Endothelial dysfunction of coronary resistance arteries is improved by tetrahydrobiopterin in atherosclerosis. Circulation 2000;102:2172–2179. [DOI] [PubMed] [Google Scholar]
- 47. Zhou ZW, Xie XL, Zhou SF, Li CG. Mechanism of reversal of high glucose-induced endothelial nitric oxide synthase uncoupling by tanshinone IIA in human endothelial cell line EA.hy926. Eur J Pharmacol 2012;697:97–105. [DOI] [PubMed] [Google Scholar]
- 48. Weidig P, McMaster D, Bayraktutan U. High glucose mediates pro-oxidant and antioxidant enzyme activities in coronary endothelial cells. Diabetes Obes Metab 2004;6:432–441. [DOI] [PubMed] [Google Scholar]
- 49. Cosentino F, Hishikawa K, Katusic ZS, Lüscher TF. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation 1997;96:25–28. [DOI] [PubMed] [Google Scholar]
- 50. Moens AL, Kietadisorn R, Lin JY, Kass D. Targeting endothelial and myocardial dysfunction with tetrahydrobiopterin. J Mol Cell Cardiol 2011;51:559–563. [DOI] [PubMed] [Google Scholar]
- 51. Münzel T, Gori T, Keaney JF, Maack C, Daiber A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur Heart J 2015;36:555–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roe ND, Ren J. Nitric oxide synthase uncoupling: a therapeutic target in cardiovascular diseases. Vascul Pharmacol 2012;57:168–172. [DOI] [PubMed] [Google Scholar]
- 53. Silberman GA, Fan THM, Liu H, Jiao Z, Xiao HD, Lovelock JD, Boulden BM, Widder J, Fredd S, Bernstein KE, Wolska BM, Dikalov S, Harrison DG, Dudley SC. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 2010;121:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 2004;4:181–189. [DOI] [PubMed] [Google Scholar]
- 55. Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med 2009;47:1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol 2003;35:851–859. [DOI] [PubMed] [Google Scholar]
- 57. Ambasta RK, Kumar P, Griendling KK, Schmidt HHHW, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem 2004;279:45935–45941. [DOI] [PubMed] [Google Scholar]
- 58. Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Fórró L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 2007;406:105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nabeebaccus A, Zhang M, Shah AM. NADPH oxidases and cardiac remodelling. Heart Fail Rev 2011;16:5–12. [DOI] [PubMed] [Google Scholar]
- 60. Zhang M, Brewer AC, Schröder K, Santos CXC, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A 2010;107:18121–18126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Martyn KD, Frederick LM, Loehneysen K, Von Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 2006;18:69–82. [DOI] [PubMed] [Google Scholar]
- 62. Montenegro MF, Valdivia A, Smolensky A, Verma K, Taylor WR, San Martín A. Nox4-dependent activation of cofilin mediates VSMC reorientation in response to cyclic stretching. Free Radic Biol Med 2015;85:288–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Martin-Garrido A, Brown DI, Lyle AN, Dikalova A, Seidel-Rogol B, Lassègue B, Martín AS, Griendling KK. NADPH oxidase 4 mediates TGF-β-induced smooth muscle α-actin via p38MAPK and serum response factor. Free Radic Biol Med 2011;50:354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, Shah AM. NADPH oxidases in cardiovascular health and disease. Antioxidants Redox Signal 2006;8:691–728. [DOI] [PubMed] [Google Scholar]
- 65. Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HHHW, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med 2008;45:1340–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Qin F, Simeone M, Patel R. Inhibition of NADPH oxidase reduces myocardial oxidative stress and apoptosis and improves cardiac function in heart failure after myocardial infarction. Free Radic Biol Med 2007;43:271–281. [DOI] [PubMed] [Google Scholar]
- 67. Bauersachs J, Galuppo P, Fraccarollo D, Christ M, Ertl G. Improvement of left ventricular remodeling and function by hydroxymethylglutaryl coenzyme A reductase inhibition with cerivastatin in rats with heart failure after myocardial infarction. Circulation 2001;104:982–985. [DOI] [PubMed] [Google Scholar]
- 68. Hayashidani S, Tsutsui H, Shiomi T, Suematsu N, Kinugawa S, Ide T, Wen J, Takeshita A. Fluvastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 2002;105:868–873. [DOI] [PubMed] [Google Scholar]
- 69. Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 2003;41:2164–2171. [DOI] [PubMed] [Google Scholar]
- 70. Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ, Vanhoutte D, Heymans S, Grieve DJ, Cave AC, Brewer AC, Zhang M, Shah AM. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial- mesenchymal transition. J Am Coll Cardiol 2014;63:2734–2741. [DOI] [PubMed] [Google Scholar]
- 71. Sirker A, Murdoch CE, Protti A, Sawyer GJ, Santos CXC, Martin D, Zhang X, Brewer AC, Zhang M, Shah AM. Cell-specific effects of Nox2 on the acute and chronic response to myocardial infarction. J Mol Cell Cardiol 2016;98:11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Doerries C, Grote K, Hilfiker-Kleiner D, Luchtefeld M, Schaefer A, Holland SM, Sorrentino S, Manes C, Schieffer B, Drexler H, Landmesser U. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ Res 2007;100:894–903. [DOI] [PubMed] [Google Scholar]
- 73. Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 2002;105:293–296. [DOI] [PubMed] [Google Scholar]
- 74. Looi YH, Grieve DJ, Siva A, Walker SJ, Anilkumar N, Cave AC, Marber M, Monaghan MJ, Shah AM. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension 2008;51:319–325. [DOI] [PubMed] [Google Scholar]
- 75. He BJ, Joiner MLA, Singh MV, Luczak ED, Swaminathan PD, Koval OM, Kutschke W, Allamargot C, Yang J, Guan X, Zimmerman K, Grumbach IM, Weiss RM, Spitz DR, Sigmund CD, Blankesteijn WM, Heymans S, Mohler PJ, Anderson ME. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med 2011;17:1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Agharazii M, St-Louis R, Gautier-Bastien A, Ung RV, Mokas S, Larivière R, Richard DE. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcification. Am J Hypertens 2015;28:746–755. [DOI] [PubMed] [Google Scholar]
- 77. Kirk EA, Dinauer MC, Rosen H, Chait A, Heinecke JW, LeBoeuf RC. Impaired superoxide production due to a deficiency in phagocyte NADPH oxidase fails to inhibit atherosclerosis in mice. Arterioscler Thromb Vasc Biol 2000;20:1529–1535. [DOI] [PubMed] [Google Scholar]
- 78. Barry-Lane PA, Patterson C, M Van Der M, Hu Z, Holland SM, Yeh ETH, Runge MS. p47phox is required for atherosclerotic lesion progression in ApoE-/- mice. J Clin Invest 2001;108:1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Judkins CP, Diep H, Broughton BRS, Mast AE, Hooker EU, Miller AA, Selemidis S, Dusting GJ, Sobey CG, Drummond GR. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE -/- mice. Am J Physiol Heart Circ Physiol 2010;298:H24–H32. [DOI] [PubMed] [Google Scholar]
- 80. Douglas G, Bendall JK, Crabtree MJ, Tatham AL, Carter EE, Hale AB, Channon KM. Endothelial-specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE -/- mice. Cardiovasc Res 2012;94:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Quesada IM, Lucero A, Amaya C, Meijles DN, Cifuentes ME, Pagano PJ, Castro C. Selective inactivation of NADPH oxidase 2 causes regression of vascularization and the size and stability of atherosclerotic plaques. Atherosclerosis 2015;242:469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schürmann C, Rezende F, Kruse C, Yasar Y, Löwe O, Fork C, B Van De S, Bremer R, Weissmann N, Shah AM, Jo H, Brandes RP, Schröder K. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur Heart J 2015;36:3447–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Schröder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 2012;110:1217–1225. [DOI] [PubMed] [Google Scholar]
- 84. Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, Shibata R, Sato K, Walsh K, Keaney JF. NADPH Oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011;124:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Brewer AC, Murray TVA, Arno M, Zhang M, Anilkumar NP, Mann GE, Shah AM. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic Biol Med 2011;51:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhang M, Mongue-Din H, Martin D, Catibog N, Smyrnias I, Zhang X, Yu B, Wang M, Brandes RP, Schröder K, Shah AM. Both cardiomyocyte and endothelial cell Nox4 mediate protection against hemodynamic overload-induced remodelling. Cardiovasc Res 2018;114:401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Langbein H, Brunssen C, Hofmann A, Cimalla P, Brux M, Bornstein SR, Deussen A, Koch E, Morawietz H. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J 2016;37:1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007;446:444–448. [DOI] [PubMed] [Google Scholar]
- 89. Smyrnias I, Zhang X, Zhang M, Murray TVA, Brandes RP, Schröder K, Brewer AC, Shah AM. Nicotinamide adenine dinucleotide phosphate oxidase-4-dependent upregulation of nuclear factor erythroid-derived 2-like 2 protects the heart during chronic pressure overload. Hypertension 2015;65:547–553. [DOI] [PubMed] [Google Scholar]
- 90. Santos CX, Hafstad AD, Beretta M, Zhang M, Molenaar C, Kopec J, Fotinou D, Murray TV, Cobb AM, Martin D, Zeh Silva M, Anilkumar N, Schröder K, Shanahan CM, Brewer AC, Brandes RP, Blanc E, Parsons M, Belousov V, Cammack R, Hider RC, Steiner RA, Shah AM. Targeted redox inhibition of protein phosphatase 1 by Nox4 regulates eIF 2α‐mediated stress signaling. EMBO J 2016;35:319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hancock M, Hafstad AD, Nabeebaccus AA, Catibog N, Logan A, Smyrnias I, Hansen SS, Lanner J, Schröder K, Murphy MP, Shah AM, Zhang M. Myocardial NADPH oxidase-4 regulates the physiological response to acute exercise. Elife 2018;7:e41044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wang M, Murdoch CE, Brewer AC, Ivetic A, Evans P, Shah AM, Zhang M. Endothelial NADPH oxidase 4 protects against angiotensin II-induced cardiac fibrosis and inflammation. ESC Heart Fail 2021;8:1427–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tong X, Khandelwal AR, Wu X, Xu Z, Yu W, Chen C, Zhao W, Yang J, Qin Z, Weisbrod RM, Seta F, Ago T, Lee KSS, Hammock BD, Sadoshima J, Cohen RA, Zeng C. Pro-atherogenic role of smooth muscle Nox4-based NADPH oxidase. J Mol Cell Cardiol 2016;92:30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Xu S, Chamseddine AH, Carrell S, Miller FJ. Nox4 NADPH oxidase contributes to smooth muscle cell phenotypes associated with unstable atherosclerotic plaques. Redox Biol 2014;2:642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res 2010;106:1253–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pryde KR, Hirst J. Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J Biol Chem 2011;286:18056–18065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Mailloux RJ, Harper ME. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med 2011;51:1106–1115. [DOI] [PubMed] [Google Scholar]
- 98. Ribas V, García-Ruiz C, Fernández-Checa JC. Glutathione and mitochondria. Front Pharmacol 2014;5:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J 2011;278:941–954. [DOI] [PubMed] [Google Scholar]
- 100. Maack C, Dabew ER, Hohl M, Schäfers HJ, Böhm M. Endogenous activation of mitochondrial KATP channels protects human failing myocardium from hydroxyl radical-induced stunning. Circ Res 2009;105:811–817. [DOI] [PubMed] [Google Scholar]
- 101. Prabu SK, Anandatheerthavarada HK, Raza H, Srinivasan S, Spear JF, Avadhani NG. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J Biol Chem 2006;281:2061–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest 2005;115:3527–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zweier JL, Talukder MAH. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res 2006;70:181–190. [DOI] [PubMed] [Google Scholar]
- 104. Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res 2014;114:524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wang Y, Wang W, Wang N, Tall AR, Tabas I. Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arterioscler Thromb Vasc Biol 2017;37:e99–e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 2012;15:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-κB-mediated inflammation in macrophages. Circ Res 2014;114:421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mercer JR, Yu E, Figg N, Cheng KK, Prime TA, Griffin JL, Masoodi M, Vidal-Puig A, Murphy MP, Bennett MR. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM +/-/ApoE -/- mice. Free Radic Biol Med 2012;52:841–849. [DOI] [PubMed] [Google Scholar]
- 109. Gomez-Stallons MV, Tretter JT, Hassel K, Gonzalez-Ramos O, Amofa D, Ollberding NJ, Mazur W, Choo JK, Smith JM, Kereiakes DJ, Yutzey KE. Calcification and extracellular matrix dysregulation in human postmortem and surgical aortic valves. Heart 2019;105:1616–1621. [DOI] [PubMed] [Google Scholar]
- 110. Yutzey KE, Demer LL, Body SC, Huggins GS, Towler DA, Giachelli CM, Hofmann-Bowman MA, Mortlock DP, Rogers MB, Sadeghi MM, Aikawa E. Calcific aortic valve disease: a consensus summary from the alliance of investigators on calcific aortic valve disease. Arterioscler Thromb Vasc Biol 2014;34:2387–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Cho KI, Sakuma I, Sohn IS, Jo SH, Koh KK. Inflammatory and metabolic mechanisms underlying the calcific aortic valve disease. Atherosclerosis 2018;277:60–65. [DOI] [PubMed] [Google Scholar]
- 112. Pawade TA, Newby DE, Dweck MR. Calcification in aortic stenosis: the skeleton key. J Am Coll Cardiol 2015;66:561–577. [DOI] [PubMed] [Google Scholar]
- 113. Towler DA. Molecular and cellular aspects of calcific aortic valve disease. Circ Res 2013;113:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bowler MA, Merryman WD. In vitro models of aortic valve calcification: solidifying a system. Cardiovasc Pathol 2015;24:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Alushi B, Curini L, Christopher MR, Grubitzch H, Landmesser U, Amedei A, Lauten A. Calcific aortic valve disease-natural history and future therapeutic strategies. Front Pharmacol 2020;11:685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Goody PR, Hosen MR, Christmann D, Niepmann ST, Zietzer A, Adam M, Bönner F, Zimmer S, Nickenig G, Jansen F. Aortic valve stenosis: from basic mechanisms to novel therapeutic targets. Arterioscler Thromb Vasc Biol 2020;40:885–900. [DOI] [PubMed] [Google Scholar]
- 117. Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O’Brien KD, Schoen FJ, Towler DA, Yoganathan AP, Otto CM. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute aortic stenosis working group. Circulation 2011;124:1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis: histological and immunohistochemical studies. Circulation 1994;90:844–853. [DOI] [PubMed] [Google Scholar]
- 119. O’Brien KD, Reichenbach DD, Marcovina SM, Kuusisto J, Alpers CE, Otto CM. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler Thromb Vasc Biol 1996;16:523–532. [DOI] [PubMed] [Google Scholar]
- 120. Hung MY, Witztum JL, Tsimikas S. New therapeutic targets for calcific aortic valve stenosis: the lipoprotein(a)-lipoprotein-associated phospholipase A 2-oxidized phospholipid axis. J Am Coll Cardiol 2014;63:478–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Thanassoulis G, Massaro JM, Cury R, Manders E, Benjamin EJ, Vasan RS, Cupple LA, Hoffmann U, O'Donnell CJ, Kathiresan S. Associations of long-term and early adult atherosclerosis risk factors with aortic and mitral valve calcium. J Am Coll Cardiol 2010;55:2491–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Stritzke J, Linsel-Nitschke P, Markus MRP, Mayer B, Lieb W, Luchner A, Döring A, Koenig W, Keil U, Hense HW, Schunkert H; for the MONICA/KORA Investigators. Association between degenerative aortic valve disease and long-term exposure to cardiovascular risk factors: results of the longitudinal population-based KORA/MONICA survey. Eur Heart J 2009;30:2044–2053. [DOI] [PubMed] [Google Scholar]
- 123. Smith JG, Luk K, Schulz CA, Engert JC, Do R, Hindy G, Rukh G, Dufresne L, Almgren P, Owens DS, Harris TB, Peloso GM, Kerr KF, Wong Q, Smith AV, Budoff MJ, Rotter JI, Cupples LA, Rich S, Kathiresan S, Orho-Melander M, Gudnason V, O’Donnell CJ, Post WS, Thanassoulis G. Association of low-density lipoprotein cholesterol - related genetic variants with aortic valve calcium and incident aortic stenosis. JAMA 2014;312:1764–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Venardos N, Nadlonek NA, Zhan Q, Weyant MJ, Reece TB, Meng X, Fullerton DA. Aortic valve calcification is mediated by a differential response of aortic valve interstitial cells to inflammation. J Surg Res 2014;190:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Winchester R, Wiesendanger M, O’Brien W, Zhang H-Z, Maurer MS, Gillam LD, Schwartz A, Marboe C, Stewart AS. Circulating activated and effector memory T cells are associated with calcification and clonal expansions in bicuspid and tricuspid valves of calcific aortic stenosis. J Immunol 2011;187:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wallby L, Janerot-Sjöberg B, Steffensen T, Broqvist M. T lymphocyte infiltration in non-rheumatic aortic stenosis: a comparative descriptive study between tricuspid and bicuspid aortic valves. Heart 2002;88:348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Song R, Zeng Q, Ao L, Yu JA, Cleveland JC, Zhao KS, Fullerton DA, Meng X. Biglycan induces the expression of osteogenic factors in human aortic valve interstitial cells via toll-like receptor-2. Arterioscler Thromb Vasc Biol 2012;32:2711–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Coté N, Mahmut A, Bosse Y, Couture C, Pagé S, Trahan S, Boulanger MC, Fournier D, Pibarot P, Mathieu P. Inflammation is associated with the remodeling of calcific aortic valve disease. Inflammation 2013;36:573–581. [DOI] [PubMed] [Google Scholar]
- 129. Yoshioka M, Yuasa S, Matsumura K, Kimura K, Shiomi T, Kimura N, Shukunami C, Okada Y, Mukai M, Shin H, Yozu R, Sata M, Ogawa S, Hiraki Y, Fukuda K. Chondromodulin-I maintains cardiac valvular function by preventing angiogenesis. Nat Med 2006;12:1151–1159. [DOI] [PubMed] [Google Scholar]
- 130. Charest A, Pépin A, Shetty R, Côté C, Voisine P, Dagenais F, Pibarot P, Mathieu P. Distribution of SPARC during neovascularisation of degenerative aortic stenosis. Heart 2006;92:1844–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Bossé Y, Miqdad A, Fournier D, Pépin A, Pibarot P, Mathieu P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ Cardiovasc Genet 2009;2:489–498. [DOI] [PubMed] [Google Scholar]
- 132. SyväRanta S, Helske S, Laine M, Lappalainen J, Kupari M, MäYräNpäÄ MI, Lindstedt KA, Kovanen PT, Vascular endothelial growth factor-secreting mast cells and myofibroblasts: a novel self-perpetuating angiogenic pathway in aortic valve stenosis. Arterioscler Thromb Vasc Biol 2010;30:1220–1227. [DOI] [PubMed] [Google Scholar]
- 133. Mazzone A, Epistolato MC, Caterina R, De Storti S, Vittorini S, Sbrana S, Gianetti J, Bevilacqua S, Glauber M, Biagini A, Tanganelli P. Neoangiogenesis, T-lymphocyte infiltration, and heat shock protein-60 are biological hallmarks of an immunomediated inflammatory process in end-stage calcified aortic valve stenosis. J Am Coll Cardiol 2004;43:1670–1676. [DOI] [PubMed] [Google Scholar]
- 134. Ghaisas NK, Foley JB, O’Briain DS, Crean P, Kelleher D, Walsh M. Adhesion molecules in nonrheumatic aortic valve disease: endothelial expression, serum levels and effects of valve replacement. J Am Coll Cardiol 2000;36:2257–2262. [DOI] [PubMed] [Google Scholar]
- 135. New SEP, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res 2011;108:1381–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Isoda K, Matsuki T, Kondo H, Iwakura Y, Ohsuzu F. Deficiency of interleukin-1 receptor antagonist induces aortic valve disease in BALB/c Mice. Arterioscler Thromb Vasc Biol 2010;30:708–715. [DOI] [PubMed] [Google Scholar]
- 137. Lai CF, Shao JS, Behrmann A, Krchma K, Cheng SL, Towler DA. TNFR1-activated reactive oxidative species signals up-regulate osteogenic Msx2 programs in aortic myofibroblasts. Endocrinology 2012;153:3897–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Galeone A, Brunetti G, Oranger A, Greco G, Benedetto A, Di Mori G, Colucci S, Zallone A, Paparella D, Grano M. Aortic valvular interstitial cells apoptosis and calcification are mediated by TNF-related apoptosis-inducing ligand. Int J Cardiol 2013;169:296–304. [DOI] [PubMed] [Google Scholar]
- 139. Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation 2007;116:2841–2850. [DOI] [PubMed] [Google Scholar]
- 140. Boffa MB, Koschinsky ML. Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease. Nat Rev Cardiol 2019;16:305–318. [DOI] [PubMed] [Google Scholar]
- 141. Tsimikas S. Potential causality and emerging medical therapies for lipoprotein(a) and its associated oxidized phospholipids in calcific aortic valve stenosis. Circ Res 2019;124:405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Rajamannan NM, Bonow RO, Rahimtoola SH. Calcific aortic stenosis: an update. Nat Clin Pract Cardiovasc Med 2007;4:254–262. [DOI] [PubMed] [Google Scholar]
- 143. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005;437:270–274. [DOI] [PubMed] [Google Scholar]
- 144. Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol 2009;47:828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Nagy E, Andersson DC, Caidahl K, Eriksson MJ, Eriksson P, Franco-Cereceda A, Hansson GK, Bäck M. Upregulation of the 5-lipoxygenase pathway in human aortic valves correlates with severity of stenosis and leads to leukotriene-induced effects on valvular myofibroblasts. Circulation 2011;123:1316–1325. [DOI] [PubMed] [Google Scholar]
- 146. Albanese I, Yu B, Al-Kindi H, Barratt B, Ott L, Al-Refai M, Varennes B, De Shum-Tim D, Cerruti M, Gourgas O, Rhéaume E, Tardif JC, Schwertani A. Role of noncanonical Wnt signaling pathway in human aortic valve calcification. Arterioscler Thromb Vasc Biol 2017;37:543–552. [DOI] [PubMed] [Google Scholar]
- 147. O’Brien KD, Kuusisto J, Reichenbach DD, Ferguson M, Giachelli C, Alpers CE, Otto CM. Osteopontin is expressed in human aortic valvular lesions. Circulation 1995;92:2163–2168. [DOI] [PubMed] [Google Scholar]
- 148. Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation 2003;107:2181–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kaden JJ, Kiliç R, Sarikoç A, Hagl S, Lang S, Hoffmann U, Brueckmann M, Borggrefe M. Tumor necrosis factor alpha promotes an osteoblast-like phenotype in human aortic valve myofibroblasts: a potential regulatory mechanism of valvular calcification. Int J Mol Med 2005;16:869–872. [PubMed] [Google Scholar]
- 150. Yu Z, Seya K, Daitoku K, Motomura S, Fukuda I, Furukawa KI. Tumor necrosis factor-α accelerates the calcification of human aortic valve interstitial cells obtained from patients with calcific aortic valve stenosis via the BMP2-Dlx5 pathway. J Pharmacol Exp Ther 2011;337:16–23. [DOI] [PubMed] [Google Scholar]
- 151. Yang X, Fullerton DA, Su X, Ao L, Cleveland JC, Meng X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol 2009;53:491–500. [DOI] [PubMed] [Google Scholar]
- 152. Yang X, Meng X, Su X, Mauchley DC, Ao L, Cleveland JC, Fullerton DA. Bone morphogenic protein 2 induces Runx2 and osteopontin expression in human aortic valve interstitial cells: role of Smad1 and extracellular signal-regulated kinase 1/2. J Thorac Cardiovasc Surg 2009;138:1008–1015. [DOI] [PubMed] [Google Scholar]
- 153. Guauque-Olarte S, Messika-Zeitoun D, Droit A, Lamontagne M, Tremblay-Marchand J, Lavoie-Charland E, Gaudreault N, Arsenault BJ, Dubé M-P, Tardif J-C, Body SC, Seidman JG, Boileau C, Mathieu P, Pibarot P, Bossé Y. Calcium signaling pathway genes RUNX2 and CACNA1C are associated with calcific aortic valve disease. Circ Cardiovasc Genet 2015;8:812–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Husseini D, El Boulanger MC, Mahmut A, Bouchareb R, Laflamme MH, Fournier D, Pibarot P, Bossé Y, Mathieu P. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: implication for calcific aortic valve disease. J Mol Cell Cardiol 2014;72:146–156. [DOI] [PubMed] [Google Scholar]
- 155. Grau JB, Poggio P, Sainger R, Vernick WJ, Seefried WF, Branchetti E, Field BC, Bavaria JE, Acker MA, Ferrari G. Analysis of osteopontin levels for the identification of asymptomatic patients with calcific aortic valve disease. Ann Thorac Surg 2012;93:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, Spelsberg TC, McCarthy PM, Rahimtoola SH, Rajamannan NM. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol 2006;47:1707–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Li X, Lim J, Lu J, Pedego TM, Demer L, Tintut Y. Protective role of Smad6 in inflammation-induced valvular cell calcification. J Cell Biochem 2015;116:2354–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Derbali H, Bossé Y, Côté N, Pibarot P, Audet A, Pépin A, Arsenault B, Couture C, Després JP, Mathieu P. Increased biglycan in aortic valve stenosis leads to the overexpression of phospholipid transfer protein via toll-like receptor 2. Am J Pathol 2010;176:2638–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Zeng Q, Song R, Ao L, Weyant MJ, Lee J, Xu D, Fullerton DA, Meng X. Notch1 promotes the pro-osteogenic response of human aortic valve interstitial cells via modulation of erk1/2 and nuclear factor-κb activation. Arterioscler Thromb Vasc Biol 2013;33:1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Acharya A, Hans CP, Koenig SN, Nichols HA, Galindo CL, Garner HR, Merrill WH, Hinton RB, Garg V. Inhibitory role of Notch1 in calcific aortic valve disease. PLoS One 2011;6:e27743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lerman DA, Prasad S, Alotti N. Calcific aortic valve disease: molecular mechanisms and therapeutic approaches. Eur Cardiol Rev 2015;10:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kaden JJ, Bickelhaupt S, Grobholz R, Haase KK, Sarikoç A, Kiliç R, Brueckmann M, Lang S, Zahn I, Vahl C, Hagl S, Dempfle CE, Borggrefe M. Receptor activator of nuclear factor κB ligand and osteoprotegerin regulate aortic valve calcification. J Mol Cell Cardiol 2004;36:57–66. [DOI] [PubMed] [Google Scholar]
- 163. Weiss RM, Lund DD, Chu Y, Brooks RM, Zimmerman KA, Accaoui R, El Davis MK, Hajj GP, Zimmerman MB, Heistad DD. Osteoprotegerin inhibits aortic valve calcification and preserves valve function in hypercholesterolemic mice. PLoS One 2013;8:e65201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Rajamannan NM. The role of Lrp5/6 in cardiac valve disease: experimental hypercholesterolemia in the ApoE -/-/Lrp5 -/- mice. J Cell Biochem 2011;112:2987–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Albanese I, Khan K, Barratt B, Al-Kindi H, Schwertani A. Atherosclerotic calcification: Wnt is the hint. J Am Heart Assoc 2018;7:e007356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Zhang M, Liu X, Zhang X, Song Z, Han L, He Y, Xu Z. MicroRNA-30b is a multifunctional regulator of aortic valve interstitial cells. J Thorac Cardiovasc Surg 2014;147:1073–1080. [DOI] [PubMed] [Google Scholar]
- 167. Chen JH, Chen WLK, Sider KL, Yip CYY, Simmons CA. Β-catenin mediates mechanically regulated, transforming growth factor-Β1-induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler Thromb Vasc Biol 2011;31:590–597. [DOI] [PubMed] [Google Scholar]
- 168. Sterbakova G, Vyskocil V, Linhartova K. Bisphosphonates in calcific aortic stenosis: association with slower progression in mild disease - a pilot retrospective study. Cardiology 2010;117:184–189. [DOI] [PubMed] [Google Scholar]
- 169. Bouchareb R, Boulanger MC, Fournier D, Pibarot P, Messaddeq Y, Mathieu P. Mechanical strain induces the production of spheroid mineralized microparticles in the aortic valve through a RhoA/ROCK-dependent mechanism. J Mol Cell Cardiol 2014;67:49–59. [DOI] [PubMed] [Google Scholar]
- 170. Bertazzo S, Gentleman E, Cloyd KL, Chester AH, Yacoub MH, Stevens MM. Nano-analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nat Mater 2013;12:576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Côté N, Husseini D, El Pépin A, Guauque-Olarte S, Ducharme V, Bouchard-Cannon P, Audet A, Fournier D, Gaudreault N, Derbali H, McKee MD, Simard C, Després JP, Pibarot P, Bossé Y, Mathieu P. ATP acts as a survival signal and prevents the mineralization of aortic valve. J Mol Cell Cardiol 2012;52:1191–1202. [DOI] [PubMed] [Google Scholar]
- 172. Mahmut A, Boulanger MC, Bouchareb R, Hadji F, Mathieu P. Adenosine derived from ecto-nucleotidases in calcific aortic valve disease promotes mineralization through A2a adenosine receptor. Cardiovasc Res 2015;106:109–120. [DOI] [PubMed] [Google Scholar]
- 173. Mathieu P, Voisine P, Pépin A, Shetty R, Savard N, Dagenais F. Calcification of human valve interstitial cells is dependent on alkaline phosphatase activity. J Heart Valve Dis 2005;14:353–357. [PubMed] [Google Scholar]
- 174. Jian B, Narula N, Li QY, Mohler ER, Levy RJ. Progression of aortic valve stenosis: TGF-β1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg 2003;75:457–465. [DOI] [PubMed] [Google Scholar]
- 175. Mohler ER, Kaplan FS, Pignolo RJ. Boning-up on aortic valve calcification. J Am Coll Cardiol 2012;60:1954–1955. [DOI] [PubMed] [Google Scholar]
- 176. Mohler ER, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation 2001;103:1522–1528. [DOI] [PubMed] [Google Scholar]
- 177. Gössl M, Khosla S, Zhang X, Higano N, Jordan KL, Loeffler D, Enriquez-Sarano M, Lennon RJ, Lerman LO, Lerman A. Role of circulating osteogenic progenitor cells in calcific aortic stenosis. J Am Coll Cardiol 2012;60:1945–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Egan KP, Kim JH, Mohler ER, Pignolo RJ. Role for circulating osteogenic precursor cells in aortic valvular disease. Arterioscler Thromb Vasc Biol 2011;31:2965–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Kaden JJ, Dempfle CE, Grobholz R, Fischer CS, Vocke DC, Kiliç R, Sarikoç A, Piñol R, Hagl S, Lang S, Brueckmann M, Borggrefe M. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc Pathol 2005;14:80–87. [DOI] [PubMed] [Google Scholar]
- 180. Pohjolainen V, Taskinen P, Soini Y, Rysä J, Ilves M, Juvonen T, Ruskoaho H, Leskinen H, Satta J. Noncollagenous bone matrix proteins as a part of calcific aortic valve disease regulation. Hum Pathol 2008;39:1695–1701. [DOI] [PubMed] [Google Scholar]
- 181. Hinton RB, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, Yutzey KE. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res 2006;98:1431–1438. [DOI] [PubMed] [Google Scholar]
- 182. Rattazzi M, Bertacco E, Iop L, D’Andrea S, Puato M, Buso G, Causin V, Gerosa G, Faggin E, Pauletto P. Extracellular pyrophosphate is reduced in aortic interstitial valve cells acquiring a calcifying profile: implications for aortic valve calcification. Atherosclerosis 2014;237:568–576. [DOI] [PubMed] [Google Scholar]
- 183. Lei Y, Masjedi S, Ferdous Z. A study of extracellular matrix remodeling in aortic heart valves using a novel biaxial stretch bioreactor. J Mech Behav Biomed Mater 2017;75:351–358. [DOI] [PubMed] [Google Scholar]
- 184. Satta J, Melkko J, Pöllänen R, Tuukkanen J, Pääkkö P, Ohtonen P, Mennander A, Soini Y. Progression of human aortic valve stenosis is associated with tenascin-C expression. J Am Coll Cardiol 2002;39:96–101. [DOI] [PubMed] [Google Scholar]
- 185. Helske S, Lindstedt KA, Laine M, Mäyränpää M, Werkkala K, Lommi J, Turto H, Kupari M, Kovanen PT. Induction of local angiotensin II-producing systems in stenotic aortic valves. J Am Coll Cardiol 2004;44:1859–1866. [DOI] [PubMed] [Google Scholar]
- 186. Fujisaka T, Hoshiga M, Hotchi J, Takeda Y, Jin D, Takai S, Hanafusa T, Ishizaka N. Angiotensin II promotes aortic valve thickening independent of elevated blood pressure in apolipoprotein-E deficient mice. Atherosclerosis 2013;226:82–87. [DOI] [PubMed] [Google Scholar]
- 187. Schoen FJ. Evolving concepts of cardiac valve dynamics: the continuum of development, functional structure, pathobiology, and tissue engineering. Circulation 2008;118:1864–1880. [DOI] [PubMed] [Google Scholar]
- 188. O’Brien KD, Shavelle DM, Caulfield MT, McDonald TO, Olin-Lewis K, Otto CM, Probstfield JL. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation 2002;106:2224–2230. [DOI] [PubMed] [Google Scholar]
- 189. Côté N, Pibarot P, Pépin A, Fournier D, Audet A, Arsenault B, Couture C, Poirier P, Després JP, Mathieu P. Oxidized low-density lipoprotein, angiotensin II and increased waist cirumference are associated with valve inflammation in prehypertensive patients with aortic stenosis. Int J Cardiol 2010;145:444–449. [DOI] [PubMed] [Google Scholar]
- 190. Fernández Esmerats J, Heath J, Jo H. Shear-sensitive genes in aortic valve endothelium. Antioxidants Redox Signal 2016;25:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Mourino-Alvarez L, Baldan-Martin M, Gonzalez-Calero L, Martinez-Laborde C, Sastre-Oliva T, Moreno-Luna R, Lopez-Almodovar LF, Sanchez PL, Fernandez-Aviles F, Vivanco F, Padial LR, Akerstrom F, Alvarez-Llamas G, la Cuesta FD, Barderas MG. Patients with calcific aortic stenosis exhibit systemic molecular evidence of ischemia, enhanced coagulation, oxidative stress and impaired cholesterol transport. Int J Cardiol 2016;225:99–106. [DOI] [PubMed] [Google Scholar]
- 192. Siudut J, Natorska J, Wypasek E, Wiewiórka Ł, Ostrowska-Kaim E, Wiśniowska-Śmiałek S, Plens K, Legutko J, Undas A. Impaired fibrinolysis in patients with isolated aortic stenosis is associated with enhanced oxidative stress. J Clin Med 2020;9:2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Broekhoven A. V, Krijnen PAJ, Fuijkschot WW, Morrison MC, Zethof IPA, Wieringen WN, van Smulders YM, Niessen HWM, Vonk ABA. Short-term LPS induces aortic valve thickening in ApoE3Leiden mice. Eur J Clin Invest 2019;49:e13121. [DOI] [PubMed] [Google Scholar]
- 194. Zeng Q, Song R, Fullerton DA, Ao L, Zhai Y, Li S, Ballak DB, Cleveland JC, Reece TB, McKinsey TA, Xu D, Dinarello CA, Meng X. Interleukin-37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc Natl Acad Sci U S A 2017;114:1631–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation 2007;115:377–386. [DOI] [PubMed] [Google Scholar]
- 196. Gunduz H, Akdemir R, Binak E, Tamer A, Keser N, Uyan C. Can serum lipid and CRP levels predict the ‘severity’ of aortic valve stenosis? Acta Cardiol 2003;58:321–326. [DOI] [PubMed] [Google Scholar]
- 197. Galante A, Pietroiusti A, Vellini M, Piccolo P, Possati G, Bonis M, De Grillo RL, Fontana C, Favalli C. C-reactive protein is increased in patients with degenerative aortic valvular stenosis. J Am Coll Cardiol 2001;38:1078–1082. [DOI] [PubMed] [Google Scholar]
- 198. Skowasch D, Schrempf S, Preusse CJ, Likungu JA, Welz A, Lüderitz B, Bauriedel G. Tissue resident C reactive protein in degenerative aortic valves: correlation with serum C reactive protein concentrations and modification by statins. Heart 2005;92:495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Jeevanantham V, Singh N, Izuora K, D'souza JP, Hsi DH. Correlation of high sensitivity C-reactive protein and calcific aortic valve disease. Mayo Clin Proc 2007;82:171–174. [DOI] [PubMed] [Google Scholar]
- 200. Novaro GM, Katz R, Aviles RJ, Gottdiener JS, Cushman M, Psaty BM, Otto CM, Griffin BP. Clinical factors, but not C-reactive protein, predict progression of calcific aortic-valve disease. The Cardiovascular Health Study. J Am Coll Cardiol 2007;50:1992–1998. [DOI] [PubMed] [Google Scholar]
- 201. Dweck MR, Jones C, Joshi NV, Fletcher AM, Richardson H, White A, Marsden M, Pessotto R, Clark JC, Wallace WA, Salter DM, McKillop G, Beek EJR, Van Boon NA, Rudd JHF, Newby DE. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation 2012;125:76–86. [DOI] [PubMed] [Google Scholar]
- 202. Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension 2007;49:717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Das D, Holmes A, Murphy GA, Mishra K, Rosenkranz AC, Horowitz JD, Kennedy JA. TGF-beta1-Induced MAPK activation promotes collagen synthesis, nodule formation, redox stress and cellular senescence in porcine aortic valve interstitial cells. J Heart Valve Dis 2013;22:621–630. [PubMed] [Google Scholar]
- 204. Xue Y, Hilaire CS, Hortells L, Phillippi JA, Sant V, Sant S. Shape-specific nanoceria mitigate oxidative stress-induced calcification in primary human valvular interstitial cell culture. Cell Mol Bioeng 2017;10:483–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Anselmo W, Branchetti E, Grau JB, Li G, Ayoub S, Lai EK, Rioux N, Tovmasyan A, Fortier JH, Sacks MS, Batinic-Haberle I, Hazen SL, Levy RJ, Ferrari G. Porphyrin-based SOD mimic MnTnBuOE-2-PyP5+ inhibits mechanisms of aortic valve remodeling in human and murine models of aortic valve sclerosis. J Am Heart Assoc 2018;7:e007861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Poggio P, Songia P, Moschetta D, Valerio V, Myasoedova V, Perrucci GL, Pompilio G. MiRNA profiling revealed enhanced susceptibility to oxidative stress of endothelial cells from bicuspid aortic valve. J Mol Cell Cardiol 2019;131:146–154. [DOI] [PubMed] [Google Scholar]
- 207. Milstien S, Katusic Z. Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem Biophys Res Commun 1999;263:681–684. [DOI] [PubMed] [Google Scholar]
- 208. Weisell J, Ohukainen P, Näpänkangas J, Ohlmeier S, Bergmann U, Peltonen T, Taskinen P, Ruskoaho H, Rysä J. Heat shock protein 90 is downregulated in calcific aortic valve disease. BMC Cardiovasc Disord 2019;19:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Farrar EJ, Huntley GD, Butcher J. Endothelial-derived oxidative stress drives myofibroblastic activation and calcification of the aortic valve. PLoS One 2015;10:e0123257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Liu M, Luo M, Sun H, Ni B, Shao Y. Integrated bioinformatics analysis predicts the key genes involved in aortic valve calcification: from hemodynamic changes to extracellular remodeling. Tohoku J Exp Med 2017;243:263–273. [DOI] [PubMed] [Google Scholar]
- 211. Choi B, Lee S, Kim SM, Lee EJ, Lee SR, Kim DH, Jang JY, Kang SW, Lee KU, Chang EJ, Song JK. Dipeptidyl peptidase-4 induces aortic valve calcification by inhibiting insulin-like growth factor-1 signaling in valvular interstitial cells. Circulation 2017;135:1935–1950. [DOI] [PubMed] [Google Scholar]
- 212. Hjortnaes J, Shapero K, Goettsch C, Hutcheson JD, Keegan J, Kluin J, Mayer JE, Bischoff J, Aikawa E. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis 2015;242:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Dahal S, Huang P, Murray BT, Mahler GJ. Endothelial to mesenchymal transformation is induced by altered extracellular matrix in aortic valve endothelial cells. J Biomed Mater Res Part A 2017;105:2729–2741. [DOI] [PubMed] [Google Scholar]
- 214. Mahler GJ, Frendl CM, Cao Q, Butcher JT. Effects of shear stress pattern and magnitude on mesenchymal transformation and invasion of aortic valve endothelial cells. Biotechnol Bioeng 2014;111:2326–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Valerio V, Myasoedova VA, Moschetta D, Porro B, Perrucci GL, Cavalca V, Cavallotti L, Songia P, Poggio P. Impact of oxidative stress and protein S-glutathionylation in aortic valve sclerosis patients with overt atherosclerosis. J Clin Med 2019;8:552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Rajamannan NM, Subramaniam M, Stock SR, Stone NJ, Springett M, Ignatiev KI, McConnell JP, Singh RJ, Bonow RO, Spelsberg TC. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart 2005;91:806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Blair A, Shaul PW, Yuhanna IS, Conrad PA, Smart EJ. Oxidized low density lipoprotein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. J Biol Chem 1999;274:32512–32519. [DOI] [PubMed] [Google Scholar]
- 218. Accaoui RE, Gould ST, Hajj GP, Chu Y, Davis MK, Kraft DC, Lund DD, Brooks RM, Doshi H, Zimmerman KA, Kutschke W, Anseth KS, Heistad DD, Weiss RM. Aortic valve sclerosis in mice deficient in endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol 2014;306:H1302–H1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Rajamannan NM. Bicuspid aortic valve disease: the role of oxidative stress in Lrp5 bone formation. Cardiovasc Pathol 2011;20:168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Aicher D, Urbich C, Zeiher A, Dimmeler S, Schäfers HJ. Endothelial nitric oxide synthase in bicuspid aortic valve disease. Ann Thorac Surg 2007;83:1290–1294. [DOI] [PubMed] [Google Scholar]
- 221. Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 2003;111:1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem 2003;278:22546–22554. [DOI] [PubMed] [Google Scholar]
- 223. Chu Y, Lund DD, Weiss RM, Brooks RM, Doshi H, Hajj GP, Sigmund CD, Heistad DD. Pioglitazone attenuates valvular calcification induced by hypercholesterolemia. Arterioscler Thromb Vasc Biol 2013;33:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Rezende F, Prior KK, Löwe O, Wittig I, Strecker V, Moll F, Helfinger V, Schnütgen F, Kurrle N, Wempe F, Walter M, Zukunft S, Luck B, Fleming I, Weissmann N, Brandes RP, Schröder K. Cytochrome P450 enzymes but not NADPH oxidases are the source of the NADPH-dependent lucigenin chemiluminescence in membrane assays. Free Radic Biol Med 2017;102:57–66. [DOI] [PubMed] [Google Scholar]
- 225. Yu B, Khan K, Hamid Q, Mardini A, Siddique A, Aguilar-Gonzalez LP, Makhoul G, Alaws H, Genest J, Thanassoulis G, Cecere R, Schwertani A. Pathological significance of lipoprotein(a) in aortic valve stenosis. Atherosclerosis 2018;272:168–174. [DOI] [PubMed] [Google Scholar]
- 226. Smyrnias I, Gray SP, Okonko DO, Sawyer G, Zoccarato A, Catibog N, López B, González A, Ravassa S, Díez J, Shah AM. Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J Am Coll Cardiol 2019;73:1795–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Heather LC, Howell NJ, Emmanuel Y, Cole MA, Frenneaux MP, Pagano D, Clarke K. Changes in cardiac substrate transporters and metabolic proteins mirror the metabolic shift in patients with aortic stenosis. PLoS One 2011;6:e26326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Rogers MA, Maldonado N, Hutcheson JD, Goettsch C, Goto S, Yamada I, Faits T, Sesaki H, Aikawa M, Aikawa E. Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ Res 2017;121:220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Weiss HG, Klocker J, Labeck B, Nehoda H, Aigner F, Klingler A, Ebenbichler C, Föger B, Lechleitner M, Patsch JR, Schwelberger HG. Plasma amine oxidase: a postulated cardiovascular risk factor in nondiabetic obese patients. Metabolism 2003;52:688–692. [DOI] [PubMed] [Google Scholar]
- 230. Boomsma F, Meiracker AH, Van Den Winkel S, Aanstoot HJ, Batstra MR, Man In’t Veld AJ, Bruining GJ. Circulating semicarbazide-sensitive amine oxidase is raised both in type I (insulin-dependent), in type II (non-insulin-dependent) diabetes mellitus and even in childhood type I diabetes at first clinical diagnosis. Diabetologia 1999;42:233–237. [DOI] [PubMed] [Google Scholar]
- 231. Zhang M, Liu L, Zhi F, Niu P, Yang M, Zhu X, Diao Y, Li Y, Wang J, Zhao Y. Inactivation of semicarbazide-sensitive amine oxidase induces the phenotypic switch of smooth muscle cells and aggravates the development of atherosclerotic lesions. Atherosclerosis 2016;249:76–82. [DOI] [PubMed] [Google Scholar]
- 232. Anger T, Pohle FK, Kandler L, Barthel T, Ensminger SM, Fischlein T, Weyand M, Stumpf C, Daniel WG, Garlichs CD. VAP-1, eotaxin3 and MIG as potential atherosclerotic triggers of severe calcified and stenotic human aortic valves: effects of statins. Exp Mol Pathol 2007;83:435–442. [DOI] [PubMed] [Google Scholar]
- 233. Cakmak HA, Aslan S, Erturk M, Ornek V, Tosu AR, Kalkan AK, Ozturk D, Tasbulak O, Avci Y, Gul M. Assessment of the relationship between serum vascular adhesion protein (Vap)-1 and severity of calcific aortic valve stenosis. J Am Coll Cardiol 2016;67:2186. [PubMed] [Google Scholar]
- 234. Mercier N, Pawelzik SC, Pirault J, Carracedo M, Persson O, Wollensack B, Franco-Cereceda A, Bäck M. Semicarbazide-sensitive amine oxidase increases in calcific aortic valve stenosis and contributes to valvular interstitial cell calcification. Oxid Med Cell Longev 2020;2020:5197376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Nagy E, Caidahl K, Franco-Cereceda A, Bäck M. Increased transcript level of poly(ADP-ribose) polymerase (PARP-1) in human tricuspid compared with bicuspid aortic valves correlates with the stenosis severity. Biochem Biophys Res Commun 2012;420:671–675. [DOI] [PubMed] [Google Scholar]
- 236. Rattazzi M, Donato M, Bertacco E, Millioni R, Franchin C, Mortarino C, Faggin E, Nardin C, Scarpa R, Cinetto F, Agostini C, Ferri N, Pauletto P, Arrigoni G. L-Arginine prevents inflammatory and pro-calcific differentiation of interstitial aortic valve cells. Atherosclerosis 2020;298:27–35. [DOI] [PubMed] [Google Scholar]
- 237. Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase-derived reactive species: physiological and pathological effects. Oxid Med Cell Longev 2016;2016:3527579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Walkey C, Das S, Seal S, Erlichman J, Heckman K, Ghibelli L, Traversa E, McGinnis JF, Self WT. Catalytic properties and biomedical applications of cerium oxide nanoparticles. Environ Sci Nano 2015;2:33–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Rajeshkumar S, Naik P. Synthesis and biomedical applications of Cerium oxide nanoparticles – a review. Biotechnol Reports 2018;17:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Novo G, Fazio G, Visconti C, Carita P, Maira E, Fattouch K, Novo S. Atherosclerosis, degenerative aortic stenosis and statins. Curr Drug Targets 2010;12:115–121. [DOI] [PubMed] [Google Scholar]
- 241. Rosenhek R, Baumgartner H. Aortic sclerosis, aortic stenosis and lipid-lowering therapy. Expert Rev Cardiovasc Ther 2008;6:385–390. [DOI] [PubMed] [Google Scholar]
- 242. Marquis-Gravel G, Redfors B, Leon MB, Généreux P. Medical treatment of aortic stenosis. Circulation 2016;134:1766–1784. [DOI] [PubMed] [Google Scholar]
- 243. Lindman BR, Bonow RO, Otto CM. Current management of calcific aortic stenosis. Circ Res 2013;113:223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med 2005;352:2389–2397. [DOI] [PubMed] [Google Scholar]
- 245. Capoulade R, Chan KL, Yeang C, Mathieu P, Bossé Y, Dumesnil JG, Tam JW, Teo KK, Mahmut A, Yang X, Witztum JL, Arsenault BJ, Després JP, Pibarot P, Tsimikas S. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J Am Coll Cardiol 2015;66:1236–1246. [DOI] [PubMed] [Google Scholar]
- 246. Farmer JA. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis (the SEAS trial). Curr Atheroscler Rep 2009;11:82–83. [PubMed] [Google Scholar]
- 247. Kennedy JA, Hua X, Mishra K, Murphy GA, Rosenkranz AC, Horowitz JD. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur J Pharmacol 2009;602:28–35. [DOI] [PubMed] [Google Scholar]
- 248. Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: methods, models, and mechanisms. Circ Res 2011;108:1392–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Olsson M, Thyberg J, Nilsson J. Presence of oxidized low density lipoprotein in nonrheumatic stenotic aortic valves. Arterioscler Thromb Vasc Biol 1999;19:1218–1222. [DOI] [PubMed] [Google Scholar]
- 250. Mohty D, Pibarot P, Després J-P, CôTé C, Arsenault B, Cartier A, Cosnay P, Couture C, Mathieu P, Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Arterioscler Thromb Vasc Biol 2008;28:187–193. [DOI] [PubMed] [Google Scholar]
- 251. Stewart CR, Stuart LM, Wilkinson K, Gils JM, Van Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, Khoury JE, Golenbock DT, Moore KJ. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 2010;11:155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Zheng KH, Tsimikas S, Pawade T, Kroon J, Jenkins WSA, Doris MK, White AC, Timmers NKLM, Hjortnaes J, Rogers MA, Aikawa E, Arsenault BJ, Witztum JL, Newby DE, Koschinsky ML, Fayad ZA, Stroes ESG, Boekholdt SM, Dweck MR. Lipoprotein(a) and oxidized phospholipids promote valve calcification in patients with aortic stenosis. J Am Coll Cardiol 2019;73:2150–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Arsenault BJ, Boekholdt SM, Dubé MP, Rhéaume É, Wareham NJ, Khaw KT, Sandhu MS, Tardif JC. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis a prospective mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet 2014;7:304–310. [DOI] [PubMed] [Google Scholar]
- 254. Que X, Hung MY, Yeang C, Gonen A, Prohaska TA, Sun X, Diehl C, Määttä A, Gaddis DE, Bowden K, Pattison J, MacDonald JG, Ylä-Herttuala S, Mellon PL, Hedrick CC, Ley K, Miller YI, Glass CK, Peterson KL, Binder CJ, Tsimikas S, Witztum JL. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018;558:301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Miller JD, Weiss RM, Serrano KM, Brooks RM, Berry CJ, Zimmerman K, Young SG, Heistad DD. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation 2009;119:2693–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Bellamy MF, Pellikka PA, Klarich KW, Tajik AJ, Enriquez-Sarano M. Association of cholesterol levels, hydroxymethylglutaryl coenzyme-A reductase inhibitor treatment, and progression of aortic stenosis in the community. J Am Coll Cardiol 2002;40:1723–1730. [DOI] [PubMed] [Google Scholar]
- 257. Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, Kitzman DW, Otto CM. Clinical factors associated with calcific aortic valve disease. J Am Coll Cardiol 1997;29:630–634. [DOI] [PubMed] [Google Scholar]
- 258. Liebe V, Brueckmann M, Borggrefe M, Kaden JJ. Statin therapy of calcific aortic stenosis: hype or hope? Eur Heart J 2006;27:773–778. [DOI] [PubMed] [Google Scholar]
- 259. Novaro GM, Tiong IY, Pearce GL, Lauer MS, Sprecher DL, Griffin BP. Effect of hydroxymethylglutaryl coenzyme A reductase inhibitors on the progression of calcific aortic stenosis. Circulation 2001;104:2205–2209. [DOI] [PubMed] [Google Scholar]
- 260. Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, Schemper M, Binder T, Maurer G, Baumgartner H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation 2004;110:1291–1295. [DOI] [PubMed] [Google Scholar]
- 261. Aronow WS, Ahn C, Kronzon I, Goldman ME. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. Am J Cardiol 2001;88:693–695. [DOI] [PubMed] [Google Scholar]
- 262. Moura LM, Ramos SF, Zamorano JL, Barros IM, Azevedo LF, Rocha-Gonçalves F, Rajamannan NM. Rosuvastatin affecting aortic valve endothelium to slow the progression of aortic stenosis. J Am Coll Cardiol 2007;49:554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Teo KK, Corsi DJ, Tam JW, Dumesnil JG, Chan KL. Lipid lowering on progression of mild to moderate aortic stenosis: meta-analysis of the randomized placebo-controlled clinical trials on 2344 patients. Can J Cardiol 2011;27:800–808. [DOI] [PubMed] [Google Scholar]
- 264. Chan KL, Teo K, Dumesnil JG, Ni A, Tam J. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (Astronomer) trial. Circulation 2010;121:306–314. [DOI] [PubMed] [Google Scholar]
- 265. Linde DVD, Yap SC, Dijk APJ, Van Budts W, Pieper PG, Burgh PH, Van Der Mulder BJM, Witsenburg M, Cuypers JAAE, Lindemans J, Takkenberg JJM, Roos-Hesselink JW. Effects of rosuvastatin on progression of stenosis in adult patients with congenital aortic stenosis (PROCAS Trial). Am J Cardiol 2011;108:265–271. [DOI] [PubMed] [Google Scholar]
- 266. Arsenault BJ, Boekholdt SM, Mora S, Demicco DA, Bao W, Tardif JC, Amarenco P, Pedersen T, Barter P, Waters DD. Impact of high-dose atorvastatin therapy and clinical risk factors on incident aortic valve stenosis in patients with cardiovascular disease (from TNT, IDEAL, and SPARCL). Am J Cardiol 2014;113:1378–1382. [DOI] [PubMed] [Google Scholar]
- 267. Viney NJ, Capelleveen JC, van Geary RS, Xia S, Tami JA, Yu RZ, Marcovina SM, Hughes SG, Graham MJ, Crooke RM, Crooke ST, Witztum JL, Stroes ES, Tsimikas S. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016;388:2239–2253. [DOI] [PubMed] [Google Scholar]
- 268. Stacker R. Molecular mechanisms underlying the antiatherosclerotic and antidiabetic effects of probucol, succinobucol, and other probucol analogues. Curr Opin Lipidol 2009;20:227–235. [DOI] [PubMed] [Google Scholar]
- 269. Lönn ME, Dennis JM, Stocker R. Actions of antioxidants in the protection against atherosclerosis. Free Radic Biol Med 2012;53:863–884. [DOI] [PubMed] [Google Scholar]
- 270. Levonen AL, Vähäkangas E, Koponen JK, Ylä-Herttuala S. Antioxidant gene therapy for cardiovascular disease: current status and future perspectives. Circulation 2008;117:2142–2150. [DOI] [PubMed] [Google Scholar]
- 271. Tardif J-C, Côté G, Lespérance J, Bourassa M, Lambert J, Doucet S, Bilodeau L, Nattel S, Guise PD. Probucol and multivitamins in the prevention of restenosis after coronary angioplasty. N Engl J Med 1997;337:365–372. [DOI] [PubMed] [Google Scholar]
- 272. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. J Am Med Assoc 2007;297:842–857. [DOI] [PubMed] [Google Scholar]
- 273. Steinhubl SR. Why have antioxidants failed in clinical trials? Am J Cardiol 2008;101:14D–19D. [DOI] [PubMed] [Google Scholar]
- 274. Wu BJ, Kathir K, Witting PK, Beck K, Choy K, Li C, Croft KD, Mori TA, Tanous D, Adams MR, Lau AK, Stocker R. Antioxidants protect from atherosclerosis by a heme oxygenase-1 pathway that is independent of free radical scavenging. J Exp Med 2006;203:1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Chao CT, Yeh HY, Tsai YT, Chuang PH, Yuan TH, Huang JW, Chen HW. Natural and non-natural antioxidative compounds: potential candidates for treatment of vascular calcification. Cell Death Discov 2019;5:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Knekt P, Reunanen A, Jävinen R, Seppänen R, Heliövaara M, Aromaa A. Antioxidant vitamin intake and coronary mortality in a longitudinal population study. Am J Epidemiol 1994;139:1180–1189. [DOI] [PubMed] [Google Scholar]
- 277. Saremi A, Arora R. Vitamin E and cardiovascular disease. Am J Ther 2010;17:e56–e65. [DOI] [PubMed] [Google Scholar]
- 278. Bleys J, Miller ER, Pastor-Barriuso R, Appel LJ, Guallar E. Vitamin-mineral supplementation and the progression of atherosclerosis: a meta-analysis of randomized controlled trials. Am J Clin Nutr 2006;84:880–887. [DOI] [PubMed] [Google Scholar]
- 279. Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet 2003;361:2017–2023. [DOI] [PubMed] [Google Scholar]
- 280. Nelson BC, Johnson ME, Walker ML, Riley KR, Sims CM. Antioxidant cerium oxide nanoparticles in biology and medicine. Antioxidants 2016;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Rzigalinski BA, Carfagna CS, Ehrich M. Cerium oxide nanoparticles in neuroprotection and considerations for efficacy and safety. Wires Nanomed Nanobiotechnol 2017;9:e1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Xue Y, Balmuri SR, Patel A, Sant V, Synthesis SS. Physico-chemical characterization, and antioxidant effect of PEGylated cerium oxide nanoparticles. Drug Deliv Transl Res 2018;8:357–367. [DOI] [PubMed] [Google Scholar]
- 283. Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, LaHaye S, Tao G, Lucchesi PA, Lincoln J, Lilly B, Garg V. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol 2013;60:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Majumdar U, Manivannan S, Basu M, Ueyama Y, Blaser MC, Cameron E, McDermott MR, Lincoln J, Cole SE, Wood S, Aikawa E, Lilly B, Garg V. Nitric oxide prevents aortic valve calcification by S-nitrosylation of USP9X to activate NOTCH signaling. Sci Adv 2021;7:eabe3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Richards J, El-Hamamsy I, Chen S, Sarang Z, Sarathchandra P, Yacoub MH, Chester AH, Butcher JT. Side-specific endothelial-dependent regulation of aortic valve calcification: interplay of hemodynamics and nitric oxide signaling. Am J Pathol 2013;182:1922–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Laufs U, Fata VL, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 1998;97:1129–1135. [DOI] [PubMed] [Google Scholar]
- 287. Laufs U, Liao JK. Isoprenoid metabolism and the pleiotropic effects of statins. Curr Atheroscler Rep 2003;5:372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res 2017;120:229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Dichtl W, Alber HF, Feuchtner GM, Hintringer F, Reinthaler M, Bartel T, Süssenbacher A, Grander W, Ulmer H, Pachinger O, Müller S. Prognosis and risk factors in patients with asymptomatic aortic stenosis and their modulation by atorvastatin (20 mg). Am J Cardiol 2008;102:743–748. [DOI] [PubMed] [Google Scholar]
- 290. Ancion A, Tridetti J, Nguyen Trung M-L, Oury C, Lancellotti P. A review of the role of bradykinin and nitric oxide in the cardioprotective action of angiotensin-converting enzyme inhibitors: focus on perindopril. Cardiol Ther 2019;8:179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Goel SS, Kleiman NS, Zoghbi WA, Reardon MJ, Kapadia SR. Renin-angiotensin system blockade in aortic stenosis: implications before and after aortic valve replacement. J Am Heart Assoc 2020;9:e016911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Comini L, Bachetti T, Cargnoni A, Bastianon D, Gitti GL, Ceconi C, Ferrari R. Therapeutic modulation of the nitric oxide: all ace inhibitors are not equivalent. Pharmacol Res 2007;56:42–48. [DOI] [PubMed] [Google Scholar]
- 293. Wassmann S, Hilgers S, Laufs U, Böhm M, Nickenig G. Angiotensin II type 1 receptor antagonism improves hypercholesterolemia-associated endothelial dysfunction. Arterioscler Thromb Vasc Biol 2002;22:1208–1212. [DOI] [PubMed] [Google Scholar]
- 294. Bull S, Loudon M, Francis JM, Joseph J, Gerry S, Karamitsos TD, Prendergast BD, Banning AP, Neubauer S, Myerson SG. A prospective, double-blind, randomized controlled trial of the angiotensin-converting enzyme inhibitor Ramipril in Aortic Stenosis (RIAS trial). Eur Heart J Cardiovasc Imaging 2015;16:834–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol. Metab 2014;25:452–463. [DOI] [PubMed] [Google Scholar]
- 296. Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 1994;11:95–102. [DOI] [PubMed] [Google Scholar]
- 297. Jaquet V, Marcoux J, Forest E, Leidal KG, McCormick S, Westermaier Y, Perozzo R, Plastre O, Fioraso-Cartier L, Diebold B, Scapozza L, Nauseef WM, Fieschi F, Krause KH, Bedard K. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br J Pharmacol 2011;164:507–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Reis J, Massari M, Marchese S, Ceccon M, Aalbers FS, Corana F, Valente S, Mai A, Magnani F, Mattevi A. A closer look into NADPH oxidase inhibitors: validation and insight into their mechanism of action. Redox Biol 2020;32:101466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Liu J, Lee J, Hernandez MAS, Mazitschek R, Ozcan U. Treatment of obesity with celastrol. Cell 2015;161:999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Hu M, Luo Q, Alitongbieke G, Chong S, Xu C, Xie L, Chen X, Zhang D, Zhou Y, Wang Z, Ye X, Cai L, Zhang F, Chen H, Jiang F, Fang H, Yang S, Liu J, Diaz-Meco MT, Su Y, Zhou H, Moscat J, Lin X, Zhang X. K. Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol Cell 2017;66:141–153.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Su Z, Zong P, Chen J, Yang S, Shen Y, Lu Y, Yang C, Kong X, Sheng Y, Sun W. Celastrol attenuates arterial and valvular calcification via inhibiting BMP2/Smad1/5 signalling. J Cell Mol Med 2020;24:12476–12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Winterbourn CC, Kettle AJ, Hampton MB. Reactive oxygen species and neutrophil function. Annu Rev Biochem 2016;85:765–792. [DOI] [PubMed] [Google Scholar]
- 303. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, Uzel G, DeRavin SS, Priel DAL, Soule BP, Zarember KA, Malech HL, Holland SM, Gallin JI. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med 2010;363:2600–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Kirkpatrick DL, Powis G. Clinically evaluated cancer drugs inhibiting redox signaling. Antioxidants Redox Signal 2017;26:262–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Shah SA, Cui SX, Waters CD, Sano S, Wang Y, Doviak H, Leor J, Walsh K, French BA, Epstein FH. Nitroxide-enhanced MRI of cardiovascular oxidative stress. NMR Biomed 2020;33:e4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Lazarova D, Semkova S, Zlateva G, Tatsuya H, Aoki I, Bakalova R. Quantum sensors to track total redox-status and oxidative stress in cells and tissues using electron-paramagnetic resonance, magnetic resonance imaging, and optical imaging. Anal Chem 2021;93:2828–2837. [DOI] [PubMed] [Google Scholar]
- 307. Fernández-Puente E, Sánchez-Martín MA, Andrés J. D, Rodríguez-Izquierdo L, Méndez L, Palomero J. Expression and functional analysis of the hydrogen peroxide biosensors HyPer and HyPer2 in C2C12 myoblasts/myotubes and single skeletal muscle fibres. Sci Rep 2020;10:871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Yang B, Chen Y, Shi J. Reactive oxygen species (ROS)-based nanomedicine. Chem. Rev 2019;119:4881–4985. [DOI] [PubMed] [Google Scholar]
- 309. Adhikari A, Mondal S, Darbar S, Kumar Pal S. Role of nanomedicine in redox mediated healing at molecular level. Biomol Concepts 2019;10:160–174. [DOI] [PubMed] [Google Scholar]
- 310. Murphy MP, Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov 2018;17:865–886. [DOI] [PubMed] [Google Scholar]
- 311. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 2014;94:909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Koju N, Taleb A, Zhou J, Lv G, Yang J, Cao X, Lei H, Ding Q. Pharmacological strategies to lower crosstalk between nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondria. Biomed Pharmacother 2019;111:1478–1498. [DOI] [PubMed] [Google Scholar]
- 313. Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet 2011;12:56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Schlotter F, Halu A, Goto S, Blaser MC, Body SC, Lee LH, Higashi H, Delaughter DM, Hutcheson JD, Vyas P, Pham T, Rogers MA, Sharma A, Seidman CE, Loscalzo J, Seidman JG, Aikawa M, Singh SA, Aikawa E. Spatiotemporal multi-omics mapping generates a molecular atlas of the aortic valve and reveals networks driving disease. Circulation 2018;138:377–393. [DOI] [PMC free article] [PubMed] [Google Scholar]