

Abstract
Existing therapeutic approaches to treat cholangiocarcinoma (CCA) have limited effectiveness, prompting further study to develop novel therapies for CCA. Here we report a novel mechanistic role for the heparan sulfate editing enzyme sulfatase 2 (SULF2) in CCA pathogenesis. In silico analysis revealed elevated SULF2 expression in human CCA samples, occurring partly through gain of SULF2 copy number. We examined the effects of knockdown or overexpression of SULF2 on tumor growth, chemoresistance, and signaling pathway activity in human CCA cell lines in vitro. Upregulation of SULF2 in CCA leads to increased PDGFRβ-YAP signaling activity, promoting tumor growth and chemotherapy resistance. To explore the utility of targeting SULF2 in the tumor microenvironment for CCA treatment, we tested an anti-SULF2 mouse monoclonal antibody 5D5 in a mouse CCA xenograft model. Targeting SULF2 by monoclonal antibody 5D5 inhibited PDGFRβ-YAP signaling and tumor growth in the mouse xenograft model. Conclusion: These results suggest that SULF2 monoclonal antibody 5D5 or related agents may be potentially promising new therapeutic agents in CCA.
Keywords: Bile duct cancer, Tumor microenvironment, Preclinical experiment, Hippo pathway, Anticancer Target
Introduction
Cholangiocarcinoma (CCA) is an epithelial malignancy arising from the biliary duct. It is the most common biliary malignancy and the second most common primary hepatic malignancy after hepatocellular carcinoma (HCC) (1, 2). CCAs represent approximately 3–5% of all malignancies of the gastrointestinal system (3). In recent decades several studies report a rising incidence of CCAs globally (4, 5). The reason for this change is unclear. Risk factors associated with CCAs include cysts and stones in the bile ducts, cirrhosis, chronic hepatitis B and C virus infection, inflammatory bowel disease, primary sclerosing cholangitis, liver fluke infestation, diabetes, alcohol, and smoking (3, 6). However, these known risk factors account for only a minority of CCA cases. CCA is one of the most lethal cancers with a dismal 5-year survival rate of less than 10% (7). Surgical resection remains the mainstay of potentially curative treatment of CCA; however, the majority of patients present with advanced disease not amenable to surgical resection. Gemcitabine and platinum-based chemotherapy is the current standard first-line chemotherapy for unresectable or metastatic CCA. Unfortunately, the efficacy of chemotherapy is limited, with an overall survival rate of <1 year (8). Thus, there remains a critical need to develop novel therapies for treatment of CCA.
Heparan sulfate proteoglycans (HSPGs) are composed of a core protein and heparan sulfate (HS) side chains. They are present on the cell surface as well as in the extracellular matrix. HS chains bind to and modulate the availability and signaling of multiple growth factor and cytokine ligands and receptors, consequently, HSPGs regulate many key cell signaling pathways, including fibroblast growth factor (FGF), heparin-binding epidermal growth factor (HB-EGF), vascular endothelial growth factor (VEGF), Wnt/β-catenin, and transforming growth factor-β (TGF-β) pathways (9, 10). Sulfatase 1 (SULF1) and sulfatase 2 (SULF2) are heparan sulfatase editing sulfatases that remove 6-O sulfate moiety within HS chains and alter the affinity of the HS chains for growth factors and growth factor receptors, thus modulating HSPG function in cell signaling pathways (11). Our group has shown that the two human sulfatases SULF1 and SULF2 have differential effects on different receptor tyrosine kinase and Wnt/β-catenin signaling in hepatocellular carcinoma (HCC). In some HCCs, SULF1 suppresses tumor growth by inhibiting the co-receptor function of HSPGs in multiple receptor tyrosine kinase signaling pathways, particularly inhibiting the FGF2 signaling pathway (12–15). In contrast to this tumor suppressor effect of SULF1, in the majority of HCCs both SULF1 and SULF2 appear to enhance tumor growth and progression through activation of the Wnt and TGF-β signaling pathways (16, 17). SULF2 appears to have broad oncogenic effects in HCC, increasing HCC cell growth by upregulating glypican 3 (GPC3), which is the most highly expressed HSPG in HCC, promoting the release of FGF2 and Wnt from GPC3 to bind to their cognate receptors and enhancing both receptor tyrosine kinase and Wnt/β-catenin signaling (18, 19). Our previous studies have shown that SULF2 knockout or suppression of SULF2 levels or activity suppresses HCC tumorigenesis (18–20), while transgenic overexpression is associated with increased tumorigenesis (21). The oncogenic effects of SULF2 are also mediated in part through the tumor microenvironment (22).
The role of the SULF1 and SULF2 sulfatases in CCA is as yet unexplored, as is the potential for translation to therapy against CCA. So far no agent targeting sulfatases is available in the clinic. To address this gap, we performed this study to answer the questions: (1) What is the expression of SULF2 in CCA? (2) What is the effect of SULF2 on CCA growth? (3) Is there a mechanistic role for SULF2 in CCA? (4) Does anti-SULF2 antibody 5D5 suppress CCA tumor growth in vivo? (5) Does inhibition of SULF2 using the monoclonal antibody 5D5 abrogate signaling pathways mediating CCA progression? (6) Are other heparan sulfate related signaling molecules involved in regulation of CCA progression?
By in silico analysis of SULF2 expression in human CCA samples, we found elevated SULF2 expression in human CCA, occurring partly through the gain of SULF2 gene copy number. Next, we examined the effects of knockdown or overexpression of SULF2 in human CCA cell lines, and assessed their effects on tumor growth, chemoresistance, and signaling pathways in vitro. Finally, we tested the effects of SULF2 monoclonal antibody 5D5 on tumor signaling and growth in a mouse CCA xenograft model. Our findings indicate that SULF2 is upregulated in CCA, leading to increased activity of platelet-derived growth factor receptor beta (PDGFRβ)-Yes-associated protein (YAP) signaling and delineating a novel SULF2-PDGFRβ-YAP signaling axis that promotes CCA tumor growth and chemotherapy resistance. Targeting SULF2 by monoclonal antibody 5D5 inhibited PDGFRβ-YAP signaling and tumor growth in vivo in a mouse CCA xenograft model. Further studies exploring the clinical translation of this antibody in human CCA are indicated.
Experimental Procedures
Cell lines and patient derived xenografts
Established human intrahepatic cholangiocarcinoma cell lines HuCCT1 and CCLP1 were used for the experiments. The HuCCT1 cell line was cultured in Roswell Park Memorial Institute 1640 (RPMI1640; Gibco) supplemented with 5% fetal bovine serum and 0.1% primocin (Invitrogen); CCLP1 was cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 5% FBS and 0.1% primocin (Invitrogen). Both cell lines were grown at 37°C in a 5% CO2 incubator. SULF2 expression was examined in both HuCCT1 and CCLP1 cells before transfection with plasmids expressing either an shRNA targeting SULF2 or one encoding SULF2. The normal human cholangiocyte cell line (NHC) was a gift from Dr. Nicholas LaRusso and was cultured in H69 media. PAX165, LIV27, LIV31, LIV61 and LIV63 are patient derived xenografts (PDX) from CCA surgically resected at Mayo Clinic (Supplementary Table 4). Each of the cell lines was tested for mycoplasma using the Universal Mycoplasma Detection Kit from ATCC (#30–1012K, Manassas, VA).
Celigo imaging cytometer for cell proliferation and apoptosis assays
HuCCT1 scrRNA and HuCCT1 shSULF2 cells, or CCLP1 Vector and CCLP1 SULF2 cells were plated in 96-Well plates (#3603, Corning, NY) at the same density. Imaging and direct counting were performed on the Celigo imaging cytometer (Nexcelom Biosciences, Lawrence, MA) over multiple time points for the cell proliferation assay. For the apoptosis assay, cells were either untreated or treated with 1uM, 5uM, and 10uM of cisplatin for 24h or 48h (with the condition labeled in the Figure). Next, cells were incubated with Annexin V FITC (Nexcelom CS1–0114, 1:100), Propidium Iodide (PI) (Nexcelom CS1–0116, 1:500) and Hoechst 33342 (Nexcelom, CS1–0128, 4 μg/ml) in Annexin V binding buffer (Nexcelom CS1–0115) for 30 minutes in the dark at room temperature. Stained cells were then imaged and analyzed on the Celigo imaging cytometer. Annexin V is a member of the annexin family of intracellular proteins that binds to phosphatidylserine (PS) in a calcium dependent manner. PS is normally only found on the intracellular leaflet of the plasma membrane in healthy cells, but during early apoptosis, PS translocates to the external leaflet. Fluorochrome-labeled Annexin V can then be used to specifically target and identify the PS on the surface of apoptotic cells. Propidium Iodide (PI) solution is a membrane-exclusion dye that permeates cells with compromised cell membranes and binds to DNA. Early apoptotic and healthy cells with intact membranes will exclude PI, while dead cells (including late stage apoptotic and necrotic cells) with compromised membranes are stained. Hoechst 33342 stains all nuclei. Accordingly, the stained cells in three categories were quantified as early apoptotic (Annexin V stained only), late apoptotic (both Annexin V and PI stained) and dead (all PI stained).
Production of SULF2 monoclonal antibody
5D5 monoclonal antibody was produced by immunizing sulf2 null mice with recombinant human SULF2 and shown to specifically react with SULF2 but not SULF1 (23). An irrelevant mouse IgG (QED Bioscience Inc., San Diego, California) was used as the control.
Statistical analysis
RNA sequencing data with clinical information are available in Gene Expression Omnibus (accession number GSE107943), which was published in our previous paper (24). The Xena browser was used to compare transcript expression of the Cancer Genome Atlas (TCGA) tumor samples to corresponding Genotype-Tissue Expression (GTEx) normal samples (25). All statistical analyses were performed using GraphPad Prism 8 (San Diego, CA, USA). All data represent at least 3 independent experiments using cells from separate cultures and are expressed as the mean ± SEM. Two-tailed Student’s t-test or one-sample t-test was used for statistical analyses. When comparing percent of mice with a tumor volume of less than 500 mm3 in the two groups, differences between the curves were analyzed using the log-rank and Wilcoxon test. Second order polynomial test was used to compare the growth curves between groups. P<0.05 was statistically significant.
Results
SULF2 Is Up-Regulated in Human CCA
First, we performed in silico analysis of SULF2 mRNA expression in different human cancer types from the Cancer Genome Atlas (TCGA) project database, compared to the equivalent normal human tissues from the Genotype-Tissue Expression (GTEx) project portal. The results show that many cancers have higher SULF2 mRNA expression than their corresponding normal tissues, and that CCA is one of the highest SULF2 expressing cancers, with an absolute median expression of 53.49 (FPKM) and a 3.09-fold increase over the expression in benign liver tissue (Fig. 1A). In the TCGA cholangiocarcinoma project, RNA-sequencing (RNA-seq) analysis showed significant upregulation of SULF2 mRNA in 36 CCA samples in comparison with 9 adjacent normal tissue samples (Fig. 1B; P=0.0003). In a South Korea CCA RNA-seq dataset, we also observed higher SULF2 mRNA expression in 29 CCA samples in comparison with 28 adjacent normal tissues (Fig. 1C; P=0.0003). Finally, in a Mayo Clinic CCA RNA-seq dataset, the SULF2 mRNA expression in 48 CCA samples was upregulated compared with 37 adjacent normal tissues (Fig. 1D; P<0.0001). All three RNA-seq datasets showed upregulation of SULF2 in human CCA compared with adjacent normal tissue. Further, we examined the relationship of SULF2 copy number alterations with SULF2 mRNA expression. In the TCGA CCA samples, 13 out of 35 CCAs (37.1%) had gain of SULF2 copy number and also showed a significantly higher SULF2 mRNA expression than tumors with diploid SULF2 copy number (Fig. 1E; P=0.02). This indicates that gain of SULF2 copy number may be one mechanism for upregulated SULF2 expression in CCA. Finally, to confirm that SULF2 protein expression is consistent with the observed SULF2 mRNA expression, we performed Western immunoblotting of lysates from a normal human cholangiocyte cell line (NHC) as well as five different patient derived xenografts (PDX) established in immunodeficient mice using freshly obtained tissue from CCA surgical resections, designated PAX165, LIV27, LIV31, LIV61 and LIV63 (26, 27). All five PDX expressed high levels of SULF2 protein compared to NHC (Fig. 1F).
Figure 1: SULF2 Expression is Increased in Many Human Cancers, Including Cholangiocarcinomas, at mRNA and Protein Levels.
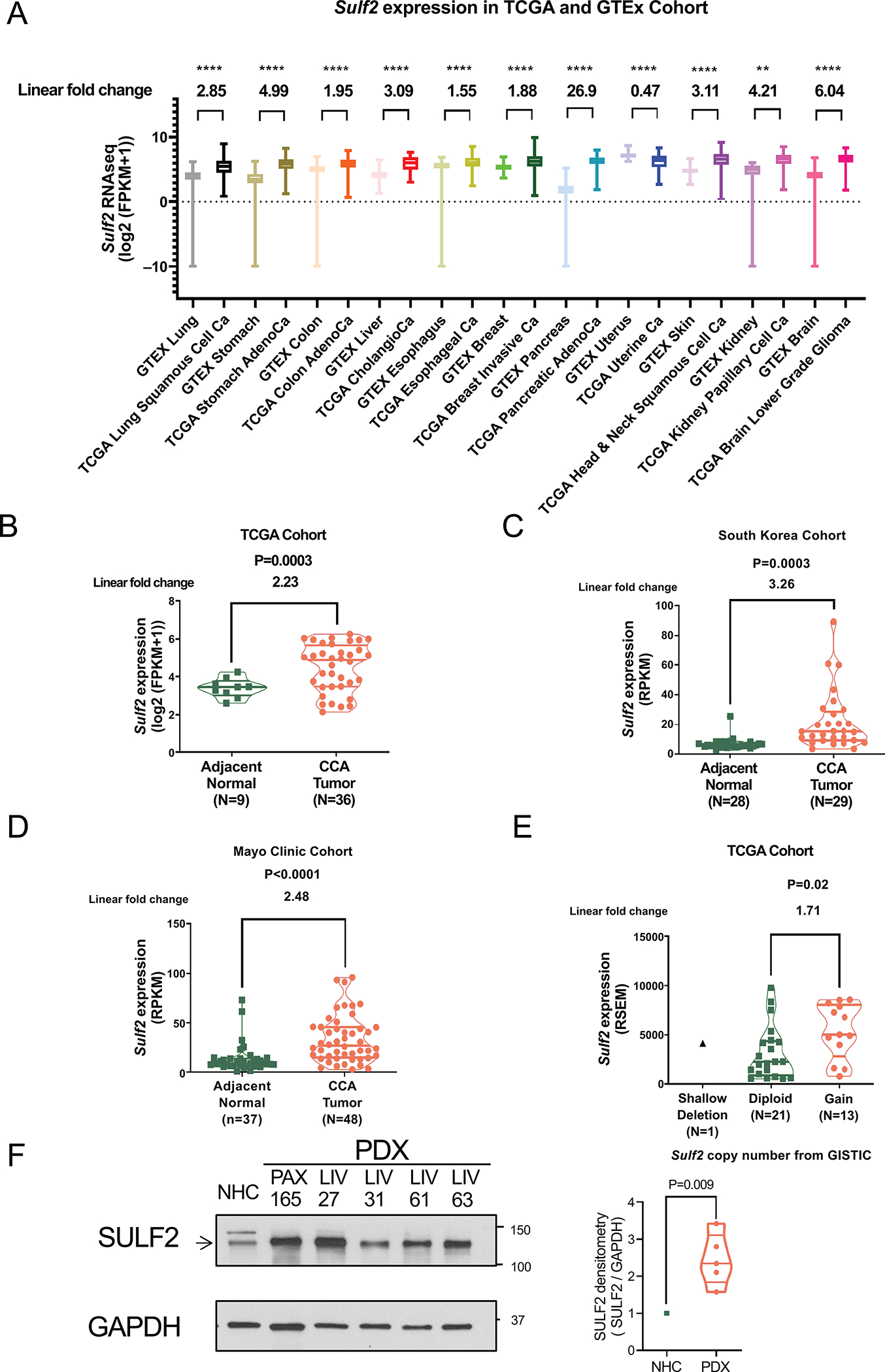
(A) In silico analysis of SULF2 expression in human normal tissue from the Genotype-Tissue Expression (GTEx) project and The Cancer Genome Atlas (TCGA) program cancer samples. SULF2 gene expression is in units of log2(FPKM+1). (B) In the TCGA dataset, SULF2 gene expression in CCA is compared to expression in adjacent normal tissue. Gene expression is in units of log2(FPKM+1). (C) In a South Korean RNA sequence dataset, SULF2 gene expression in CCA is compared to adjacent normal tissue. SULF2 gene expression is in units of RPKM. (D) In a Mayo Clinic RNA sequence dataset, SULF2 gene expression in CCA is compared to adjacent normal tissue. SULF2 gene expression is in units of RPKM. (E) Of the TCGA CCA samples, 13 out of 35 CCAs (37.1%) show gain of SULF2 copy number, which is associated with a significantly higher SULF2 mRNA expression level than is seen in diploid CCAs (F) Western immunoblotting shows that five CCA PDXs express high levels of SULF2 protein compared to normal human cholangiocytes (NHC). **: p<0.01, ****: p<0.0001.
Knockdown of SULF2 Decreases CCA Cell Proliferation and Resistance to Cisplatin Induced Apoptosis
Since most CCAs over-express SULF2, we examined the effects of downregulation of SULF2 on cell growth, apoptosis and cell death in a CCA cell line in vitro. After confirming that the HuCCT1 CCA cell line expresses high levels of SULF2 mRNA, we stably knocked down SULF2 in HuCCT1 cells using a plasmid construct expressing short hairpin RNA (shRNA) targeting the SULF2 mRNA. SULF2 mRNA expression in the stable clones was quantitated by qRT-PCR. SULF2 mRNA was significantly decreased in the HuCCT1 cells transfected with SULF2-targeting shRNA (HuCCT1 shSULF2) compared with the HuCCT1 transfected with scrambled shRNA (HuCCT1 scrRNA) (86.7% decreased, Supplementary Fig. 1A). Cell proliferation was evaluated by direct cell counting in the Celigo imaging cytometer. Decreased SULF2 expression was associated with substantially decreased cell growth of HuCCT1 cells, with a 32.3% reduction in cell number at 3 days (Fig. 2A; P<0.0001). Western immunoblotting for SULF2 and Ki-67 as a marker of cell proliferation showed that suppression of SULF2 protein expression was accompanied by decreased cell proliferation (Fig. 2B). Apoptosis was also assessed using the Celigo imaging cytometer. Knockdown of SULF2 did not impact the percentage of early-stage apoptotic, late-stage apoptotic, or dead cells (Fig. 2C, Supplementary Fig. 2A). Immunoblots also showed no difference in the expression of Caspase 9, Pro-Caspase 8, cleaved Caspase 8, Pro-Caspase 3, and cleaved Caspase 3 between the HuCCT1 scrRNA and HuCCT1 shSULF2 cells (Fig. 2D, Supplementary Fig. 2B). However, when cells were treated with cisplatin at different concentrations (1uM, 5uM, and 10uM) for 24h, the percentage of dead cells in the HuCCT1 shSULF2 group increased significantly compared to the HuCCT1 scrRNA group at all concentrations of cisplatin (Fig. 2E, Supplementary Fig. 3, 4A; P=0.005, P<0.0001, P<0.0001 respectively). The percentage of late-stage apoptotic cells was also significantly higher in the HuCCT1 shSULF2 group than the HuCCT1 scrRNA group at the concentration of 10uM cisplatin after 24h (Fig. 2E; P=0.0009). Further, when cells were treated with 10uM cisplatin for 48h, the percentages of early apoptotic, late-stage apoptotic, and dead cells all significantly increased in the HuCCT1 shSULF2 cells in comparison to the HuCCT1 scrRNA cells (Fig. 2E, Supplementary Fig. 4B; P=0.005, P<0.0001, P<0.0001 respectively). Immunoblot also showed increased expression of cleaved Caspase 8 and cleaved Caspase 3, with corresponding decreased expression of Pro-Caspase 8 and Pro-Caspase 3 in the HuCCT1 shSULF2 group compared to the HuCCT1 scrRNA group (Fig. 2F). These data reveal that knockdown of SULF2 decreases resistance of CCA cells to cisplatin induced apoptosis.
Figure 2: Knockdown of SULF2 Decreases CCA Cell Proliferation and Resistance to Cisplatin Induced Apoptosis.
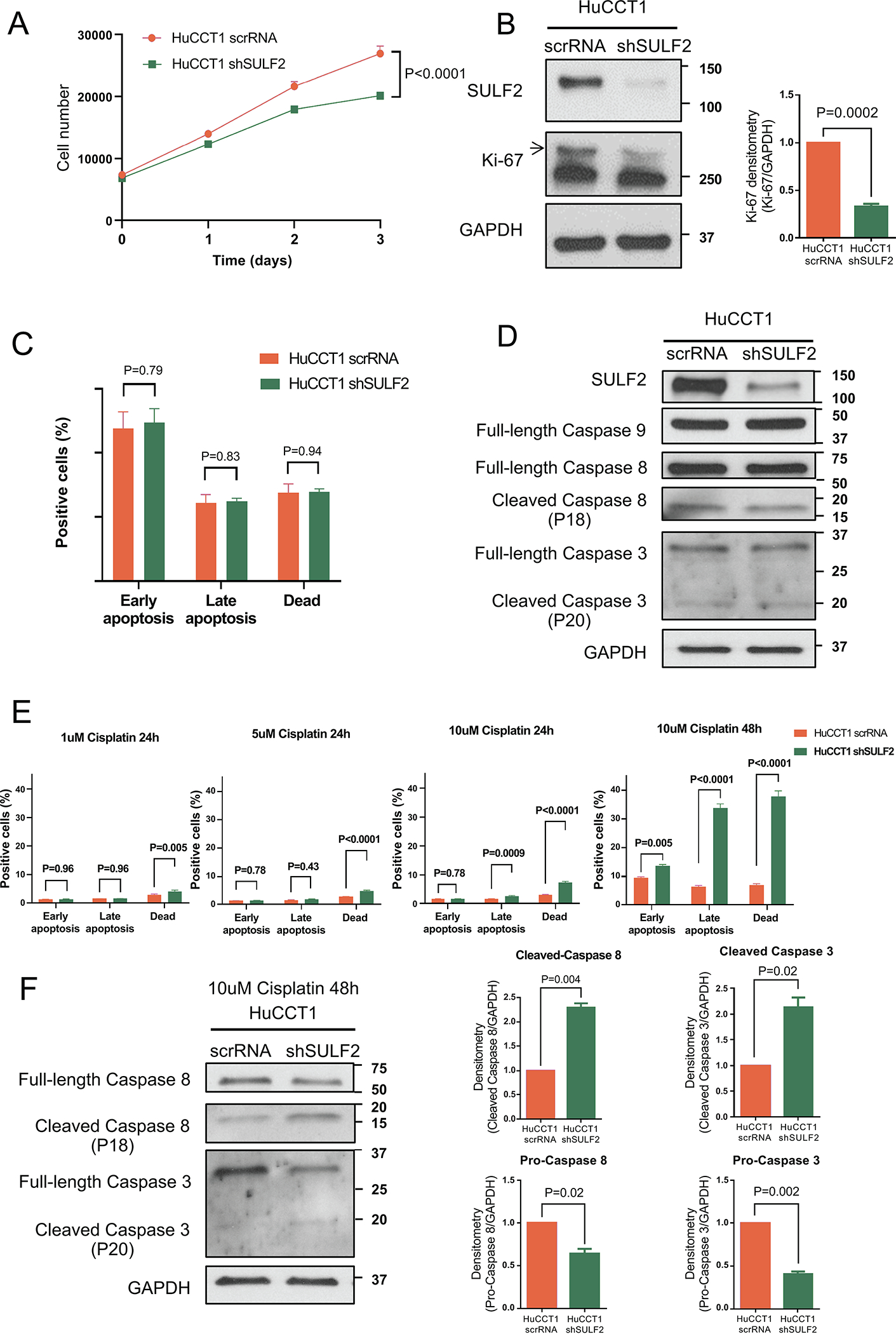
(A) HuCCT1 scrRNA or HuCCT1 shSULF2 were cultured counted by the Celigo imaging cytometer daily for 3 days. Decreased SULF2 expression was associated with substantially decreased HuCCT1 cell growth (n=10). (B) Western immunoblotting for SULF2 and Ki-67 showed that suppression of SULF2 protein expression was accompanied by decreased cell proliferation marker Ki-67. (C) Statistical analysis of HuCCT1 scrRNA or HuCCT1 shSULF2 apoptosis assay. NS, not significant (n=10). (D) Western immunoblotting showed no difference in the expression of Caspases 3, 8 and 9 between the HuCCT1 scrRNA group and HuCCT1 shSULF2 knockdown group. (E) Statistical analysis of HuCCT1 scrRNA or HuCCT1 shSULF2 apoptosis assays after treatment with cisplatin at concentrations of 1uM, 5uM, and 10uM for 24h or 48h (n=10 each). (F) HuCCT1 scrRNA or HuCCT1 shSULF2 cells were treated with 10uM cisplatin for 48h. Western immunoblotting showed increased cleaved Caspase 8 and cleaved Caspase 3, and decreased pro-caspase 8 and pro-caspase 3 in HuCCT1 shSULF2 cells compared to HuCCT1 scrRNA cells.
Forced Expression of SULF2 Promotes CCA Cell Proliferation and Resistance to Cisplatin Induced Apoptosis
To determine whether increased expression of SULF2 in SULF2-negative CCA cells produces opposing effects to those of suppression of SULF2 expression, we transfected a plasmid construct encoding SULF2 or control plasmid vector into the SULF2 negative CCLP1 cholangiocarcinoma cell line. Western immunoblotting and qRT-PCR confirmed that CCLP1 SULF2 cells express SULF2 at a high level compared with CCLP1 Vector cells (Fig. 3B; Supplementary Fig. 1B). To investigate the effect of forced expression of SULF2 on CCA cell growth, we quantitated cell proliferation by direct cell counting on the Celigo imaging cytometer. CCLP1 SULF2 cells grew significantly faster than CCLP1 Vector cells (29.7%, P<0.0001; Fig. 3A). Western immunoblotting for SULF2 and Ki-67 as a marker of cell proliferation showed that forced expression of SULF2 was accompanied by increased cell proliferation (Fig. 3B). Apoptosis assays showed that in the absence of cisplatin, forced expression of SULF2 slightly decreases CCLP1 apoptosis (Fig. 3C, E); however, on treatment of CCLP1 cells with 10uM cisplatin for 48h, the percentage of early apoptotic, late-stage apoptotic, and dead cells, which were all increased substantially by cisplatin treatment, all decreased significantly in the CCLP1 SULF2 group in comparison to the CCLP1 Vector group (Fig. 3D, E; P=0.006, P=0.005, P=0.003 respectively). These data revealed that forced expression of SULF2 promotes CCA cell proliferation and enhances resistance to cisplatin induced apoptosis.
Figure 3: Expression of SULF2 Promotes CCA Cell Proliferation and Resistance to Cisplatin Induced Apoptosis.
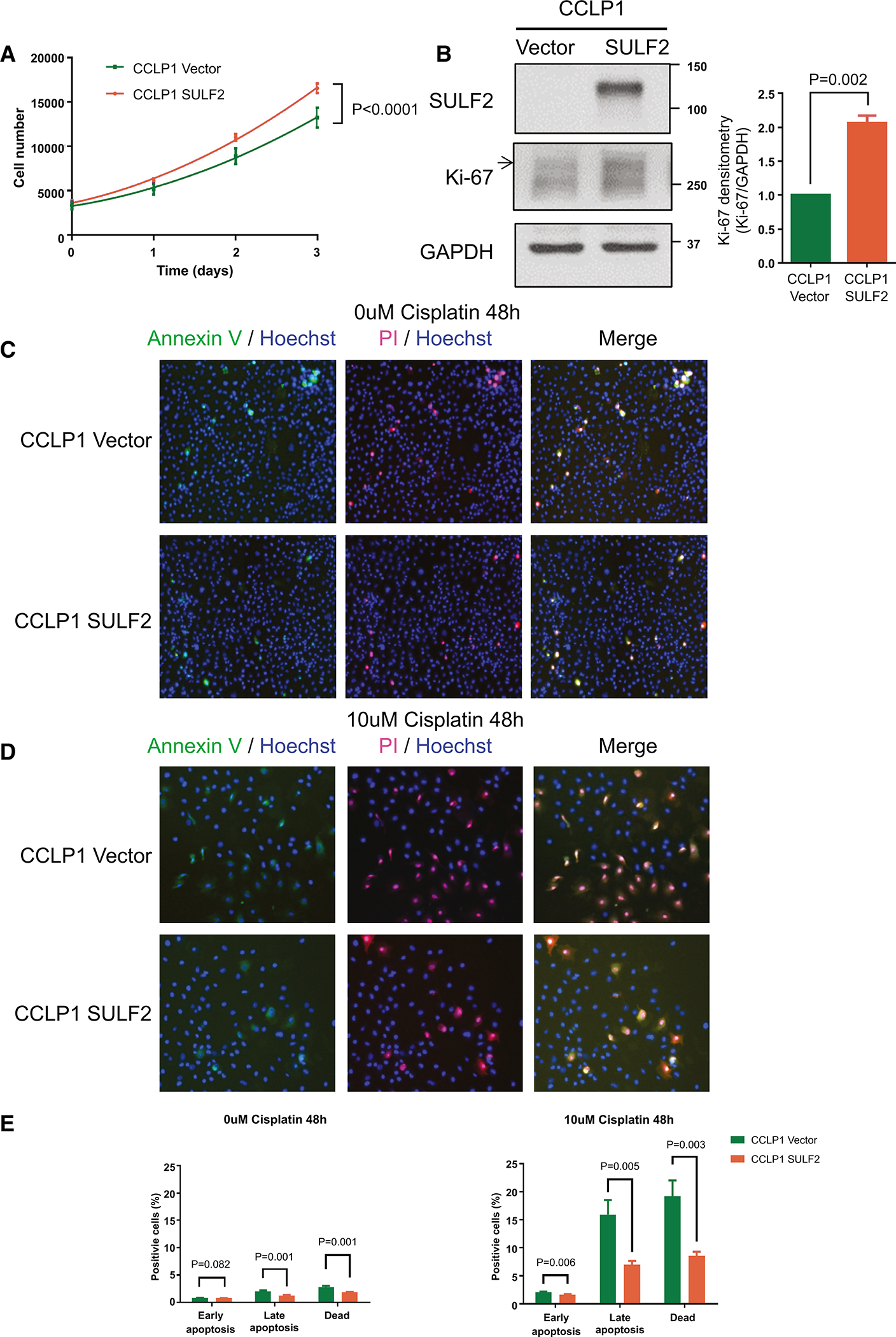
(A) CCLP1 Vector or CCLP1 SULF2 were cultured for 3 days and counted daily in the Celigo imaging cytometer (n=10). (B) Western immunoblotting for SULF2 and Ki-67 as a marker of cell proliferation showed that forced expression of SULF2 was accompanied by increased cell proliferation. (C) Representative images of apoptosis assays using the Celigo imaging cytometer. CCLP1 Vector or CCLP1 SULF2 without cisplatin treatment are stained by Fluorochrome-labeled Annexin V (green), Propidium Iodide (PI, red), and Hoechst 33342 (blue). The cells were cultured for 48h in 0uM cisplatin (D) Representative images of apoptosis assays using the Celigo imaging cytometer. CCLP1 Vector or CCLP1 SULF2 cells were treated with 10uM cisplatin for 48h. (E) Statistical analysis of CCLP1 Vector or CCLP1 SULF2 apoptosis assays of untreated cells or cells treated with 10uM cisplatin for 48h (n=10).
Knockdown of SULF2 downregulates PDGFRβ-YAP signaling
Next, we examined the potential mechanism of regulation of CCA growth and apoptosis by SULF2. The Hippo pathway effector Yes-associated protein (YAP) is transcriptionally active in many tumor types, including a majority of human CCA. YAP activity has been linked to proliferation and chemotherapy resistance of CCA. phosphorylation of the serine127 residue of YAP by a kinase module consisting of MST1/2 and LATS1/2 sequesters YAP in the cytosol and limits transcriptional activity (28). Previous work by Dr. R. Smoot and colleagues suggests that PDGFR signaling regulates YAP nuclear localization and cognate gene expression via Src family kinase (SFK)-mediated YAP tyrosine357 phosphorylation (29). Tyrosine357 phosphorylation of YAP also appears to promote its association with the transcription factor TBX5, promoting CCA cell viability by inducing expression of the anti-apoptotic regulator Mcl-1 (29). To determine whether the effects of SULF2 are mediated through PDGFR-YAP signaling, we examined the effect of knockdown or forced expression of SULF2 on PDGFRβ and YAP phosphorylation in CCA cells.
We found that knockdown of SULF2 in the HuCCT1 cell line decreased the level of phospho-PDGFRβ, phospho-YAPY357, and Cyclin D1 as assessed by Western immunoblotting, but did not change the levels of phospho-YAPS127 or phospho-ERK (Fig. 4A, B, Supplementary Fig. 5A). By contrast, forced expression of SULF2 in the CCLP1 cell line increased the levels of phospho-PDGFRβ, phospho-YAPY357, and Cyclin D1, but did not change the levels of phospho-YAPS127 or phospho-ERK (Fig. 4A, B, Supplementary Fig. 5A). Knockdown of SULF2 in HuCCT1 cells also significantly decreased the mRNA levels of four transcriptional targets of YAP signaling, CTGF, MCL1, CYR61 and PDGFB (p<0.05) (Figure 4C). By contrast, forced expression of SULF2 in the CCLP1 cell line significantly increased the mRNA levels of CTGF, MCL1, CYR61 and PDGFB (Figure 4C). Additionally, immunofluorescence showed significantly decreased phospho-YAPY357 in HuCCT1 shSULF2 cells compared to HuCCT1 scrRNA cells (Figure 4D, Supplementary Fig. 5B). These data suggest that SULF2 regulates CCA proliferation and chemotherapy resistance at least in part via regulation of PDGFRβ-YAP signaling.
Figure 4: The knockdown of SULF2 downregulates PDGFRβ-YAP signaling while upregulation of SULF2 enhances YAP signaling.
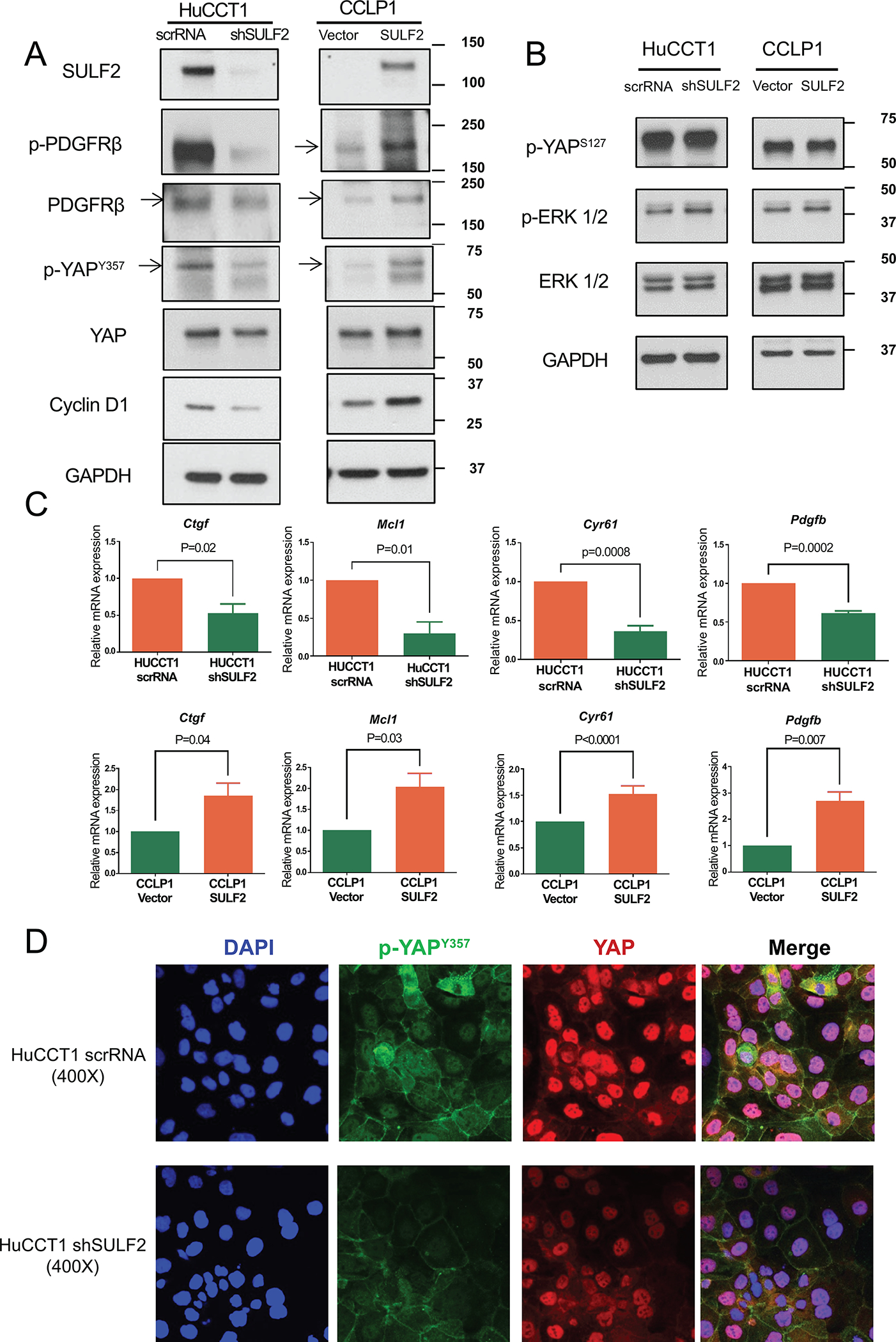
(A) Western immunoblotting showed that knockdown of SULF2 in HuCCT1 cells decreased the levels of phospho-PDGFRβ, phospho-YAPY357, and Cyclin D1. In contrast, forced expression of SULF2 in CCLP1 cells increased the levels of phospho-PDGFRβ, phospho-YAPY357, and Cyclin D1. (B) knockdown of SULF2 in HuCCT1 cells or forced expression of SULF2 in CCLP1 cells did not change the levels of phospho-YAPS127 or phospho-ERK. (C) knockdown of SULF2 in HuCCT1 cells significantly decreased the mRNA levels of CTGF, MCL1, CYR61, and PDGFB; by contrast, forced expression of SULF2 in the CCLP1 cell line significantly increased the mRNA levels of CTGF, MCL1, CYR61 and PDGFB. (D) Representative IF images of HuCCT1 scrRNA or HuCCT1 shSULF2 cells stained for phospho-YAPY357 (green), total YAP (red), and DAPI (blue). 400×.
Targeting SULF2 by monoclonal antibody 5D5 suppresses growth of CCA tumor xenografts in nude mice
Based on these in vitro findings, we next explored the effect of an anti-SULF2 mouse monoclonal antibody designated 5D5 in the treatment of CCA cell line xenografts in nude mice. 1×106 HuCCT1 cells were injected subcutaneously into the flanks of nude mice. When the tumor volume reached 100 to 200 mm3, the mice were randomized into 2 groups of 10 mice each. The active treatment group received an intraperitoneal injection of SULF2 monoclonal antibody 5D5 at 40mg/kg three times a week while the control group received mouse IgG antibody at 40mg/kg three times a week for 35 days. The tumor size and the weight of the mice were measured three times a week during the treatment period. Treatment with the SULF2 monoclonal antibody 5D5 significantly reduced the CCA xenograft tumor size in mice compared with mouse IgG antibody (P<0.0001, Fig. 5B). The body weights of the two groups showed no differences (Supplementary Fig. 6B). At the end of the study, tumor xenograft weights were significantly decreased by SULF2 monoclonal antibody 5D5 in comparison with mouse IgG antibody (P=0.005, Fig. 5D). H&E slides of the xenografts were analyzed by the pathologist Dr. Torbenson. The percentage of necrosis was scored on each slide, rounded to the nearest 10%. 1% was used when there was necrosis, but very focal. The percentage of tumor necrosis was significantly increased by SULF2 monoclonal antibody 5D5 in comparison with mouse IgG antibody (P=0.04, Fig. 5E). Thus, targeting SULF2 by monoclonal antibody 5D5 suppressed CCA cell line xenograft growth in nude mice.
Figure 5: Targeting SULF2 using anti-SULF2 monoclonal antibody 5D5 suppresses tumor growth in nude mice.
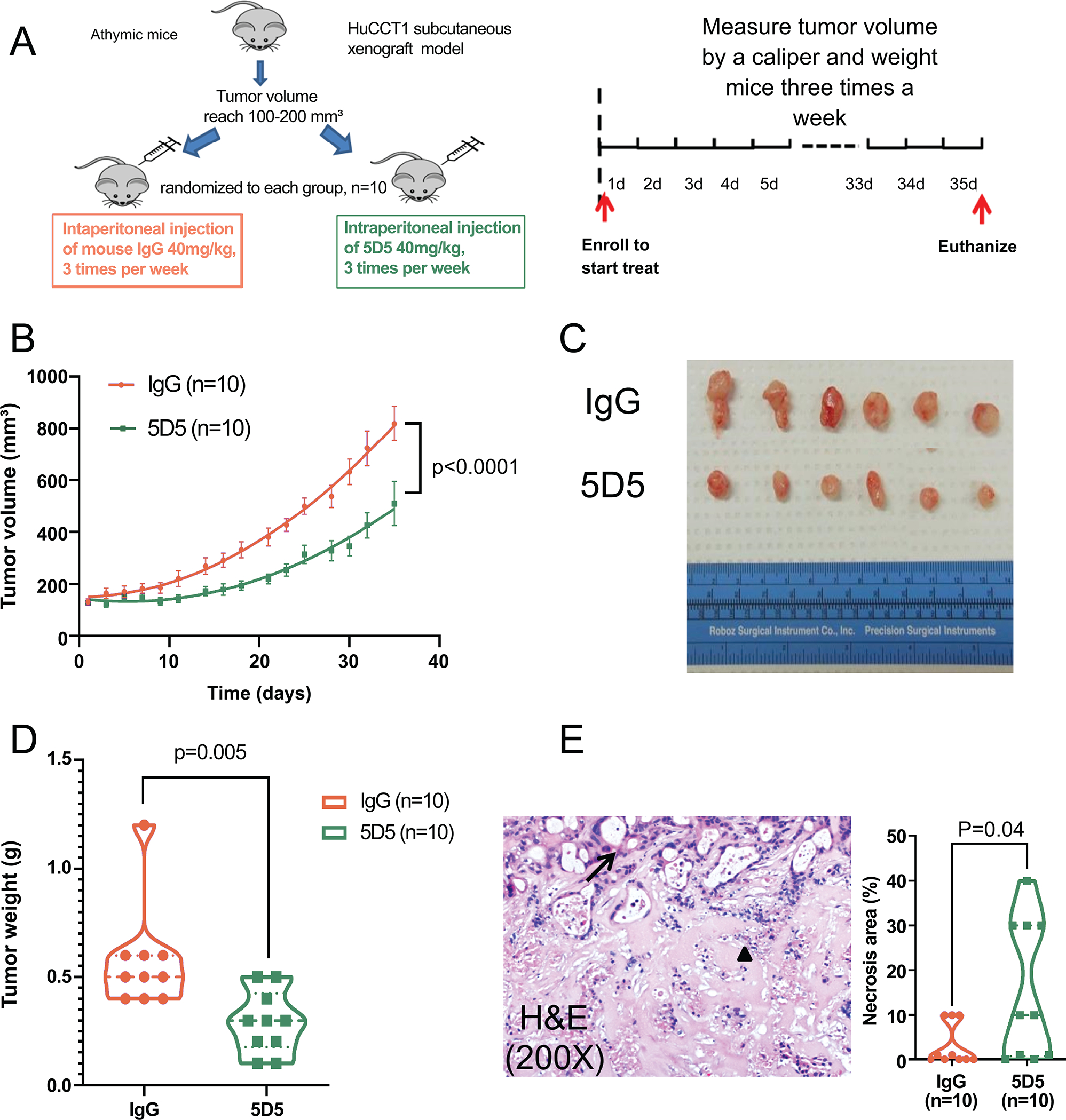
(A) Illustration of mouse xenograft experiment. (B) Treatment with the 5D5 antibody significantly reduced tumor size compared to mouse IgG antibody (n=10). (C) Representative image of tumor xenografts at the end of the study. (D) Tumor xenograft weight was significantly decreased by 5D5 compared to mouse IgG (n=10). (E) Representative H&E staining of tumor xenografts showed viable (black arrow) and necrotic (black triangle) glands. (200×). The percentage of necrosis was scored on each slide. SULF2 monoclonal antibody 5D5 significantly increased the percentage of tumor necrosis in comparison with mouse IgG antibody.
SULF2 monoclonal antibody 5D5 inhibits PDGFRβ-YAP signaling in vivo
Finally, we determined whether the inhibition of CCA tumor growth by the anti-SULF2 specific monoclonal antibody 5D5 was associated with effects on cell proliferation, apoptosis, and PDGFRβ-YAP signaling in vivo. To confirm that SULF2 monoclonal antibody treatment suppressed CCA tumor proliferation in vivo, we performed immunohistochemistry using antibodies against Ki67. The Ki67 positive nuclear percentage in the SULF2 monoclonal antibody 5D5 treated group was significantly decreased by 48.3% compared to the mouse IgG antibody control group (P<0.0001, Fig. 6A, B). By contrast, Cleaved caspase-3 immunohistochemistry and TUNEL assays showed no differences in caspase 3 activation or apoptosis between the SULF2 monoclonal antibody 5D5 treated group and mouse IgG antibody control group (Fig. 6A, B). Consistent with our in vitro experiment, Western blotting of tumor tissue protein extracts showed that 5D5 treatment significantly reduced the levels of phospho-PDGFRβ, phospho-YAPY357, total YAP and Cyclin D1 (Fig. 7A). Additionally, immunofluorescence revealed that phospho-YAPY357 was substantially lower in tumors from 5D5 treated mice than in tumors from mouse IgG treated mice (Fig. 7B). These data suggest that the anti-SULF2 monoclonal antibody 5D5 mediates suppression of CCA xenograft growth through inhibition of PDGFRβ-YAP signaling in vivo.
Figure 6: SULF2 monoclonal antibody 5D5 inhibits tumor proliferation in vivo.
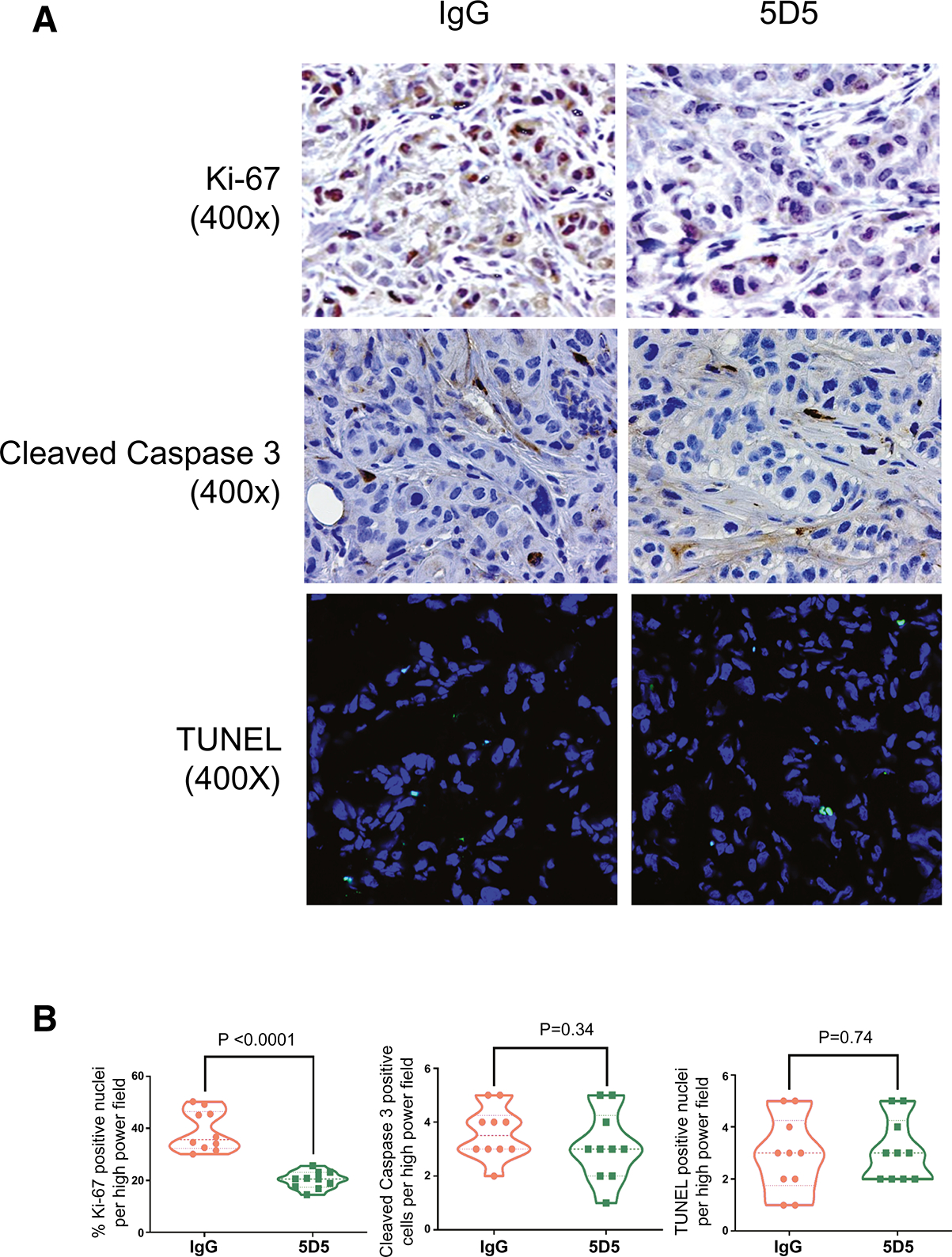
(A) Representative immunohistochemical staining of HuCCT1 tumor xenografts treated with control IgG and anti-SULF2 5D5 antibody for Ki67 (upper panel ), Cleaved Caspase 3 (middle panel), and apoptosis by TUNEL labeling (lower panel); 400×. (B) Statistical analyses of quantification for Ki67 staining, Cleaved Caspase 3 staining, and TUNEL labeling (n=8–10).
Figure 7: SULF2 monoclonal antibody 5D5 inhibits PDGFRβ-YAP signaling in vivo.
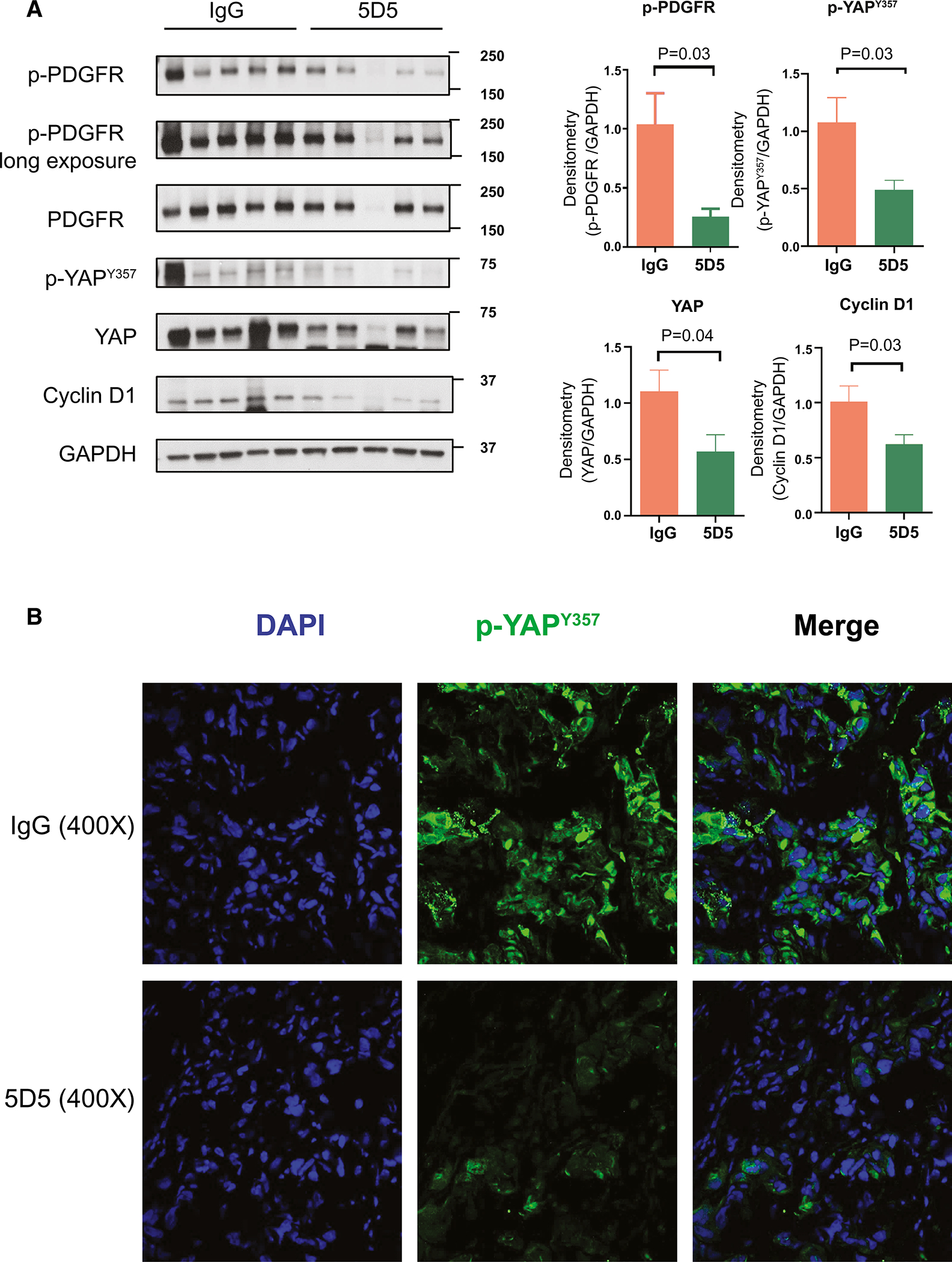
(A) Western blot of xenograft tissue protein extracts showed that treatment with anti-SULF2 5D5 antibody significantly reduced the levels of phospho-PDGFRβ, phospho-YAPY357, total YAP and Cyclin D1 compared to IgG treated xenografts (n=5,5). (B) Representative IF images of HuCCT1 tumor xenografts treated with control IgG and anti-SULF2 5D5 antibody stained for phospho-YAPY357 (green), and DAPI (Blue). 400×.
Effects of SULF2 in extrahepatic cholangiocarcinoma
Since most of the data we acquired was from patients with intrahepatic CCA, we explored the potential effects of SULF2 in extrahepatic CCA. We abstracted the extrahepatic CCA data from the Mayo Clinic RNA sequence dataset and compared SULF2 gene expression in extrahepatic CCA to adjacent normal tissue (Supplementary Figure 7A). In a proportion of extrahepatic CCA, SULF2 mRNA expression was upregulated compared to paired adjacent normal tissue. The P value was not significant, likely due to the small sample size (N=9). For SULF2 protein expression, western immunoblotting also showed that the extrahepatic CCA cell line WITT expresses a higher level of SULF2 protein compared to normal human cholangiocytes (NHC). In contrast, another extrahepatic CCA cell line, EGI-1, has lower SULF2 levels than NHC (Supplementary Figure 7B). Similar to our observation in the intrahepatic CCA cell line CCLP1, forced expression of SULF2 in EGI-1 cells increased the levels of phospho-YAPY357 and Cyclin D1 (Supplementary Figure 7C).
Effects of other heparan sulfate related signaling molecules in cholangiocarcinoma
To determine whether there is a general effect of modulation of heparan sulfate related signaling molecules in progression of cholangiocarcinoma, we examined the levels of heparanase, which cleaves the heparan sulfate side chains of heparan sulfate proteoglycans, VEGFR1, encoded by the FLT1 gene, and EGFR. Western blotting of PDX protein extracts showed that compared with NHC, CCA PDXs have significantly decreased heparanase levels, but no significant difference in the levels of VEGFR1 and EGFR (Supplementary Figure 8A). Western blotting of protein extracts from HuCCT1 xenografts showed that compared to IgG treatment, 5D5 treatment significantly reduced the levels of heparanase, VEGFR1 and EGFR (Supplementary Figure 8B). These results suggest that other signaling pathways modulated by the sulfatases are also potential targets for anti-SULF2 antibodies and other therapeutics in CCA.
Taken together, our results suggest that SULF2 increases the activity of the PDGFRβ-YAP pathway to promote tumor growth and chemotherapy resistance. Targeting SULF2 by monoclonal antibody 5D5 inhibits the PDGFRβ-YAP pathway and CCA growth in a mouse model (Fig. 8).
Figure 8: Working model for the effect of SULF2 monoclonal antibody 5D5 on PDGFRβ-YAP signaling in CCA.
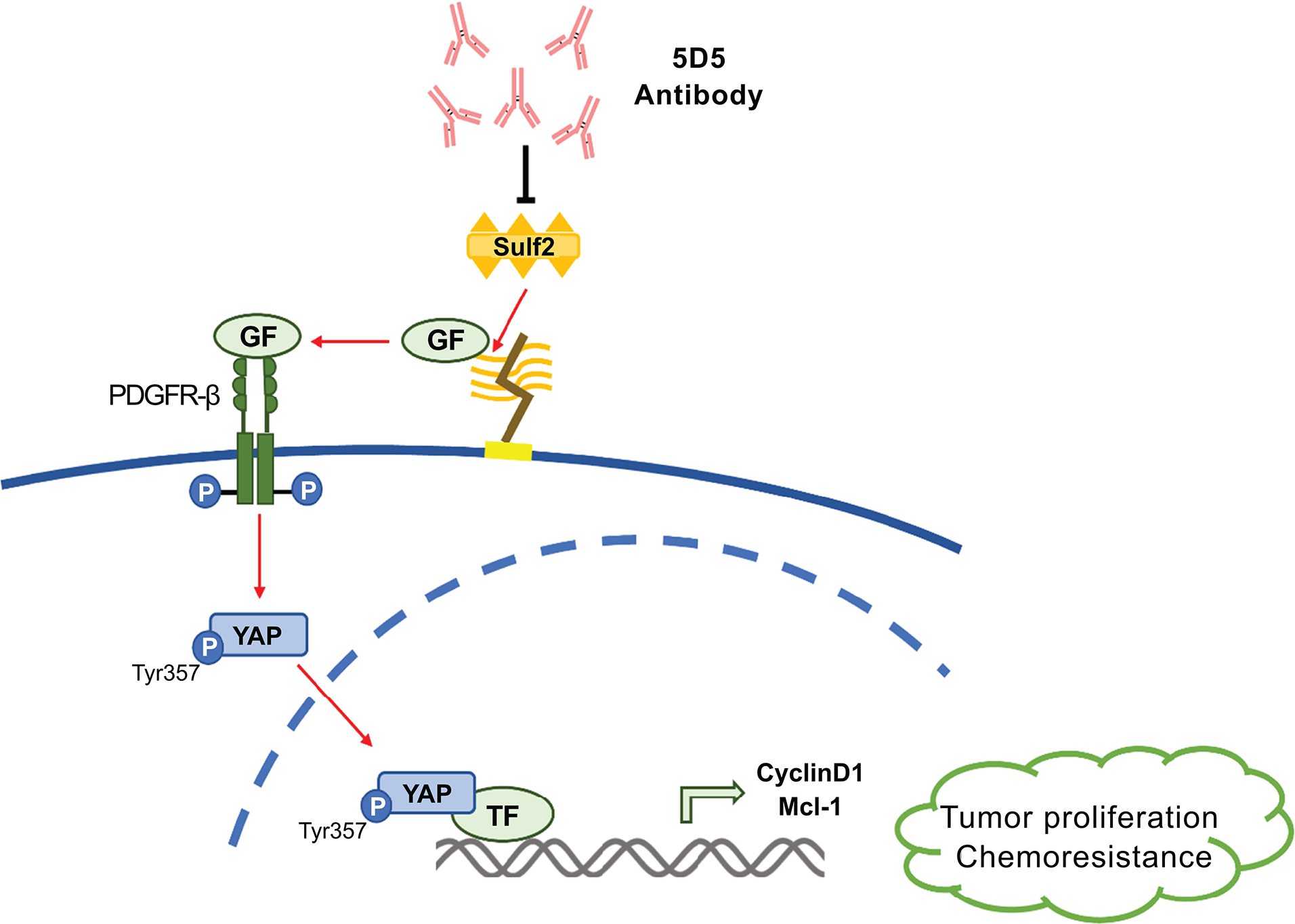
SULF2 increases the activity of the PDGFRβ-YAP pathway to promote tumor growth and chemotherapy resistance. Targeting SULF2 by monoclonal antibody 5D5 showed inhibition of the PDGFRβ-YAP pathway and CCA growth in a mouse model.
Discussion
CCA is one of the most lethal cancers with limited and relatively ineffective treatment options. Novel molecular targeted therapies are in critical need for CCA treatment. The results of this study provide novel insights into the role of SULF2 in the pathogenesis of CCA. The principal findings of this study are as follows: (i) SULF2 is upregulated in human CCA compared with adjacent normal tissue, in some tumors through the gain of SULF2 copy number; (ii) SULF2 modulates CCA cell proliferation and resistance to cisplatin induced apoptosis in vitro; (iii) SULF2 activates the PDGFRβ-YAP pathway signaling, which may explain the dependency on SULF2 for CCA proliferation and resistance to cisplatin induced apoptosis; (iv) Targeting SULF2 by monoclonal antibody 5D5 inhibits the activity of the PDGFRβ-YAP pathway and suppresses CCA xenograft growth in vivo.
HSPGs in the extracellular matrix or on the cell surface can sequester growth factor ligands and cytokines in a sulfation-dependent manner and release the factors when desulfated by heparan sulfate-editing endosulfatases. SULF1 and SULF2 are human heparan sulfate-editing endosulfatases shown to be involved in carcinogenesis in several tissues, including the liver (12, 30, 31). However, a functional role for these sulfatases in CCA has not been elucidated. In the present study, analysis of three different human CCA and adjacent normal tissue RNA sequence datasets showed that SULF2 was upregulated in CCAs compared to adjacent normal tissue. Further analysis showed 37.1% of CCAs in the TCGA dataset show gain of SULF2 copy number and a significantly higher SULF2 mRNA expression level than CCAs that are diploid at the SULF2 locus. This indicates that gain of SULF2 copy number may be one mechanism for upregulated SULF2 expression in CCA. Modulation of SULF2 expression by shRNA mediated knock-down of SULF2 in SULF2-expressing HuCCT1 CCA cells and forced expression by transfection of SULF2 encoding constructs into SULF2-negative CCLP1 CCA cells showed that SULF2 promotes cell proliferation and chemoresistance in CCA cells. This is the first study to demonstrate a tumorigenic effect of SULF2 in CCA.
To determine the specific mechanistic role of SULF2 in CCA, we investigated the effects of SULF2 on PDGFRβ-YAP signaling in CCA. Although previous studies from our group and others have implicated SULF2 in the regulation of several signaling pathways, the key downstream pathway mediating SULF2 signaling is still unknown and is likely to be different in different cellular contexts (18, 32, 33). YAP, the effector of the Hippo pathway, is a transcriptional co-activator that has increased activity in many cancers including CCA. YAP activity has been linked to tumor growth and chemotherapy resistance. Interestingly, the Hippo pathway does not have a dedicated cell-surface receptor and consequently is regulated via cross-talk with other signaling pathways (34, 35). Previous studies have demonstrated that a PDGFR-SFK cascade regulates YAP activation via tyrosine phosphorylation in CCA (29). Since PDGFR signaling is regulated by heparan sulfate, we developed the hypothesis that SULF2 regulates CCA tumorigenesis through effects on PDGFRβ-YAP signaling. In experiments exploring this hypothesis, we found that knockdown of SULF2 in the SULF2-overexpressing HuCCT1 CCA cell line downregulated the levels of phospho-PDGFRβ, phospho-YAPY357, and transcriptional targets of YAP signaling. Consistent with this result, forced expression of SULF2 up-regulated the levels of phospho-PDGFR-β, phospho-YAPY357, and CyclinD1. Thus, we demonstrated for the first time that the effects of SULF2 appear to be exerted in part through regulation of PDGFRβ/YAP signaling in CCA. In terms of the machinery by which SULF2 regulates phosphorylation of PDGFRβ, SULF2 may alter PDGFRβ activation via its potential effects on PDGF bioavailability, given that PDGFs interact with HSPGs and this interaction is influenced by 6-O sulfation (36–38). However, other mechanisms may also exist. Indeed, VEGF-A, a factor known to bind HSPGs in a sulfatase-dependent manner, can directly activate PDGFRβ (39). Furthermore, we observed a slight but consistent change in the total levels of PDGFRβ. This change could be due to the composition of the ECM, given that PDGFR levels are affected by alterations in the composition or amount of HSPGs (40). Thus, further studies will explore the machinery by which SULF2 regulates PDGFRβ activity in CCA.
Similar to our observation in the intrahepatic CCA, in a proportion of extrahepatic CCA, SULF2 mRNA and protein expression is upregulated. Forced expression of SULF2 in the extrahepatic CCA cell line EGI-1 increased the levels of phospho-YAPY357 and Cyclin D1. Thus it appears that SULF2 is also upregulated and oncogenic in a proportion of extrahepatic CCA.
Accumulating evidence has shown that the complex interplay between cancer cells and their microenvironment can have profound implications on tumorigenesis and progression (33, 41). Sulfatases modify HSPGs in the tumor microenvironment and influence exogenous ligand availability (42, 43). Interestingly, the HS mimetic PI-88 (muparfostat), originally designed as an inhibitor of heparanase in the late 1990s, strongly inhibits SULF2 function (44), and has antiproliferative and antiangiogenic properties in preclinical animal models. Unfortunately, PI-88 was not successfully translated into clinical use (22, 45). To explore the utility of targeting SULF2 for treatment of CCA, we employed the SULF2 monoclonal antibody 5D5 to treat subcutaneous CCA xenografts in nude mice. In this preclinical animal model, we showed for the first time that the 5D5 monoclonal antibody suppresses tumor growth, and consistent with our previous experiments, suppresses PDGFRβ-YAP activity in vivo. These findings confirm SULF2 monoclonal antibody therapy as a potentially promising new therapeutic agent in CCA. Western blotting of HuCCT1 xenograft protein extracts showed that compared to IgG treatment, 5D5 treatment also significantly reduced the levels of the heparan sulfate related proteins VEGFR1, EGFR, and heparanase. Our future studies will pursue the mechanistic understanding of the effects of the sulfatases and anti-SULF agents on these additional signaling pathways.
In summary, we have demonstrated SULF2 as a potential therapeutic target by which CCA increases the activity of the PDGFRβ-YAP pathway to promote tumor growth and chemotherapy resistance. Targeting SULF2 by monoclonal antibody 5D5 showed inhibition of the PDGFRβ-YAP pathway and CCA growth in a mouse model. In the future, targeting SULF2 in combination with conventional cancer therapies may improve the clinical outcome of CCA patients.
Supplementary Material
Acknowledgements:
The authors thank Jianbo Huang, Qianqian Guo, Jingchun Yang, Eugene Krueger, Maria E. Guicciardi, Christy E. Trussoni, Amy S. Mauer, Nathan W. Werneburg, Anuradha krishnan and the Mayo Clinic Animal Facilties for their assistance with the experiments.
Financial Support:
NIH grants CA128633 to LRR and CA165076 to LRR, the Mayo Clinic Center for Cell Signaling in Gastroenterology (NIDDK P30 DK084567), the Mayo Clinic Cancer Center (P30 CA015083), the Mayo Clinic Hepatobiliary SPORE (P50 CA210964), The Cholangiocarcinoma Foundation and National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No 2018R1C1B3004435).
Abbreviations
- CCA
cholangiocarcinoma
- HCC
hepatocellular carcinoma
- HSPG
heparan sulfate proteoglycan
- HS
heparan sulfate
- FGF
fibroblast growth factor
- HB-EGF
heparin-binding epidermal growth factor
- VEGF
vascular endothelial growth factor
- TGF-β
transforming growth factor-β
- SULF1
sulfatase 1
- SULF2
sulfatase 2
- GPC3
glypican 3
- PDGFRβ
platelet-derived growth factor receptor beta
- YAP
Yes-associated protein
- PBS
phosphate-buffered saline
- qRT-PCR
quantitative real time PCR
- TCGA
The Cancer Genome Atlas
- shRNA
small hairpin RNA
- PDGF
platelet-derived growth factor
- SFK
Src family kinase
- TBST
Tris-buffered saline and Tween 20
- WB
western blotting
Footnotes
Conflicts of Interest: LRR has received grant funding from BTG International, Exact Sciences, Gilead Sciences, Redhill, TARGET PharmaSolutions and FUJIFILM Medical Systems.
References
- 1.Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2018;15:95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, Cardinale V, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol 2020;17:557–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clements O, Eliahoo J, Kim JU, Taylor-Robinson SD, Khan SA. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: A systematic review and meta-analysis. J Hepatol 2020;72:95–103. [DOI] [PubMed] [Google Scholar]
- 4.Saha SK, Zhu AX, Fuchs CS, Brooks GA. Forty-Year Trends in Cholangiocarcinoma Incidence in the U.S.: Intrahepatic Disease on the Rise. Oncologist 2016;21:594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang JD, Kim B, Sanderson SO, Sauver JS, Yawn BP, Larson JJ, Therneau TM, et al. Biliary tract cancers in Olmsted County, Minnesota, 1976–2008. Am J Gastroenterol 2012;107:1256–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaiteerakij R, Yang JD, Harmsen WS, Slettedahl SW, Mettler TA, Fredericksen ZS, Kim WR, et al. Risk factors for intrahepatic cholangiocarcinoma: association between metformin use and reduced cancer risk. Hepatology 2013;57:648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertuccio P, Malvezzi M, Carioli G, Hashim D, Boffetta P, El-Serag HB, La Vecchia C, et al. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J Hepatol 2019;71:104–114. [DOI] [PubMed] [Google Scholar]
- 8.Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–1281. [DOI] [PubMed] [Google Scholar]
- 9.Pye DA, Vives RR, Hyde P, Gallagher JT. Regulation of FGF-1 mitogenic activity by heparan sulfate oligosaccharides is dependent on specific structural features: differential requirements for the modulation of FGF-1 and FGF-2. Glycobiology 2000;10:1183–1192. [DOI] [PubMed] [Google Scholar]
- 10.Lin X Functions of heparan sulfate proteoglycans in cell signaling during development. Development 2004;131:6009–6021. [DOI] [PubMed] [Google Scholar]
- 11.David G, Bai XM, Van der Schueren B, Cassiman JJ, Van den Berghe H. Developmental changes in heparan sulfate expression: in situ detection with mAbs. J Cell Biol 1992;119:961–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai JP, Chien JR, Moser DR, Staub JK, Aderca I, Montoya DP, Matthews TA, et al. hSulf1 Sulfatase promotes apoptosis of hepatocellular cancer cells by decreasing heparin-binding growth factor signaling. Gastroenterology 2004;126:231–248. [DOI] [PubMed] [Google Scholar]
- 13.Lai J, Chien J, Staub J, Avula R, Greene EL, Matthews TA, Smith DI, et al. Loss of HSulf-1 up-regulates heparin-binding growth factor signaling in cancer. J Biol Chem 2003;278:23107–23117. [DOI] [PubMed] [Google Scholar]
- 14.Lai JP, Sandhu DS, Shire AM, Roberts LR. The tumor suppressor function of human sulfatase 1 (SULF1) in carcinogenesis. J Gastrointest Cancer 2008;39:149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shire A, Lomberk G, Lai JP, Zou H, Tsuchiya N, Aderca I, Moser CD, et al. Restoration of epigenetically silenced SULF1 expression by 5-aza-2-deoxycytidine sensitizes hepatocellular carcinoma cells to chemotherapy-induced apoptosis. Med Epigenet 2015;3:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang JD, Sun Z, Hu C, Lai J, Dove R, Nakamura I, Lee JS, et al. Sulfatase 1 and sulfatase 2 in hepatocellular carcinoma: associated signaling pathways, tumor phenotypes, and survival. Genes Chromosomes Cancer 2011;50:122–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhanasekaran R, Nakamura I, Hu C, Chen G, Oseini AM, Seven ES, Miamen AG, et al. Activation of the transforming growth factor-beta/SMAD transcriptional pathway underlies a novel tumor-promoting role of sulfatase 1 in hepatocellular carcinoma. Hepatology 2015;61:1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai JP, Oseini AM, Moser CD, Yu C, Elsawa SF, Hu C, Nakamura I, et al. The oncogenic effect of sulfatase 2 in human hepatocellular carcinoma is mediated in part by glypican 3-dependent Wnt activation. Hepatology 2010;52:1680–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai JP, Sandhu DS, Yu C, Han T, Moser CD, Jackson KK, Guerrero RB, et al. Sulfatase 2 up-regulates glypican 3, promotes fibroblast growth factor signaling, and decreases survival in hepatocellular carcinoma. Hepatology 2008;47:1211–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng X, Gai X, Han S, Moser CD, Hu C, Shire AM, Floyd RA, et al. The human sulfatase 2 inhibitor 2,4-disulfonylphenyl-tert-butylnitrone (OKN-007) has an antitumor effect in hepatocellular carcinoma mediated via suppression of TGFB1/SMAD2 and Hedgehog/GLI1 signaling. Genes Chromosomes Cancer 2013;52:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carr RM, Romecin Duran PA, Tolosa EJ, Ma C, Oseini AM, Moser CD, Banini BA, et al. The extracellular sulfatase SULF2 promotes liver tumorigenesis by stimulating assembly of a promoter-looping GLI1-STAT3 transcriptional complex. J Biol Chem 2020;295:2698–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen G, Nakamura I, Dhanasekaran R, Iguchi E, Tolosa EJ, Romecin PA, Vera RE, et al. Transcriptional Induction of Periostin by a Sulfatase 2-TGFbeta1-SMAD Signaling Axis Mediates Tumor Angiogenesis in Hepatocellular Carcinoma. Cancer Res 2017;77:632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singer MS, Phillips JJ, Lemjabbar-Alaoui H, Wang YQ, Wu J, Goldman R, Rosen SD. SULF2, a heparan sulfate endosulfatase, is present in the blood of healthy individuals and increases in cirrhosis. Clin Chim Acta 2015;440:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn KS, O’Brien D, Kang YN, Mounajjed T, Kim YH, Kim TS, Kocher JA, et al. Prognostic subclass of intrahepatic cholangiocarcinoma by integrative molecular-clinical analysis and potential targeted approach. Hepatol Int 2019;13:490–500. [DOI] [PubMed] [Google Scholar]
- 25.Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, Banerjee A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 2020;38:675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding X, Chaiteerakij R, Moser CD, Shaleh H, Boakye J, Chen G, Ndzengue A, et al. Antitumor effect of the novel sphingosine kinase 2 inhibitor ABC294640 is enhanced by inhibition of autophagy and by sorafenib in human cholangiocarcinoma cells. Oncotarget 2016;7:20080–20092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leiting JL, Murphy SJ, Bergquist JR, Hernandez MC, Ivanics T, Abdelrahman AM, Yang L, et al. Biliary tract cancer patient-derived xenografts: Surgeon impact on individualized medicine. JHEP Rep 2020;2:100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 2014;94:1287–1312. [DOI] [PubMed] [Google Scholar]
- 29.Smoot RL, Werneburg NW, Sugihara T, Hernandez MC, Yang L, Mehner C, Graham RP, et al. Platelet-derived growth factor regulates YAP transcriptional activity via Src family kinase dependent tyrosine phosphorylation. J Cell Biochem 2018;119:824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai JP, Chien J, Strome SE, Staub J, Montoya DP, Greene EL, Smith DI, et al. HSulf-1 modulates HGF-mediated tumor cell invasion and signaling in head and neck squamous carcinoma. Oncogene 2004;23:1439–1447. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Kleeff J, Abiatari I, Kayed H, Giese NA, Felix K, Giese T, et al. Enhanced levels of Hsulf-1 interfere with heparin-binding growth factor signaling in pancreatic cancer. Mol Cancer 2005;4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med 2006;12:410–416. [DOI] [PubMed] [Google Scholar]
- 33.Uchimura K, Morimoto-Tomita M, Bistrup A, Li J, Lyon M, Gallagher J, Werb Z, et al. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochem 2006;7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan D Hippo signaling in organ size control. Genes Dev 2007;21:886–897. [DOI] [PubMed] [Google Scholar]
- 35.Fan R, Kim NG, Gumbiner BM. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci U S A 2013;110:2569–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith EM, Mitsi M, Nugent MA, Symes K. PDGF-A interactions with fibronectin reveal a critical role for heparan sulfate in directed cell migration during Xenopus gastrulation. Proc Natl Acad Sci U S A 2009;106:21683–21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abramsson A, Kurup S, Busse M, Yamada S, Lindblom P, Schallmeiner E, Stenzel D, et al. Defective N-sulfation of heparan sulfate proteoglycans limits PDGF-BB binding and pericyte recruitment in vascular development. Genes Dev 2007;21:316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rolny C, Spillmann D, Lindahl U, Claesson-Welsh L. Heparin amplifies platelet-derived growth factor (PDGF)- BB-induced PDGF alpha -receptor but not PDGF beta -receptor tyrosine phosphorylation in heparan sulfate-deficient cells. Effects on signal transduction and biological responses. J Biol Chem 2002;277:19315–19321. [DOI] [PubMed] [Google Scholar]
- 39.Ball SG, Shuttleworth CA, Kielty CM. Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J Cell Biol 2007;177:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malmstrom J, Westergren-Thorsson G. Heparan sulfate upregulates platelet-derived growth factor receptors on human lung fibroblasts. Glycobiology 1998;8:1149–1155. [DOI] [PubMed] [Google Scholar]
- 41.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19:1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dhoot GK, Gustafsson MK, Ai X, Sun W, Standiford DM, Emerson CP, Jr. Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science 2001;293:1663–1666. [DOI] [PubMed] [Google Scholar]
- 43.Xu D, Fuster MM, Lawrence R, Esko JD. Heparan sulfate regulates VEGF165- and VEGF121-mediated vascular hyperpermeability. J Biol Chem 2011;286:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hossain MM, Hosono-Fukao T, Tang R, Sugaya N, van Kuppevelt TH, Jenniskens GJ, Kimata K, et al. Direct detection of HSulf-1 and HSulf-2 activities on extracellular heparan sulfate and their inhibition by PI-88. Glycobiology 2010;20:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chhabra M, Ferro V. PI-88 and Related Heparan Sulfate Mimetics. Adv Exp Med Biol 2020;1221:473–491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.