

Abstract
Background
Preeclampsia, a leading cause of maternal and fetal mortality and morbidity, is characterized by an increase in S‐nitrosylated proteins and reactive oxygen species, suggesting a pathophysiologic role for dysregulation in nitrosylation and nitrosative stress.
Methods and Results
Here, we show that mice lacking S‐nitrosoglutathione reductase (GSNOR−⁄− ), a denitrosylase regulating protein S‐nitrosylation, exhibit a preeclampsia phenotype, including hypertension, proteinuria, renal pathology, cardiac concentric hypertrophy, decreased placental vascularization, and fetal growth retardation. Reactive oxygen species, NO, and peroxynitrite levels are elevated. Importantly, mass spectrometry reveals elevated placental S‐nitrosylated amino acid residues in GSNOR−⁄− mice. Ascorbate reverses the phenotype except for fetal weight, reduces the difference in the S‐nitrosoproteome, and identifies a unique set of S‐nitrosylated proteins in GSNOR−⁄− mice. Importantly, human preeclamptic placentas exhibit decreased GSNOR activity and increased nitrosative stress.
Conclusions
Therefore, deficiency of GSNOR creates dysregulation of placental S‐nitrosylation and preeclampsia in mice, which can be rescued by ascorbate. Coupled with similar findings in human placentas, these findings offer valuable insights and therapeutic implications for preeclampsia.
Keywords: mouse model, NO, preeclampsia, pregnancy, S‐nitrosylation
Subject Categories: Animal Models of Human Disease, Basic Science Research, Endothelium/Vascular Type/Nitric Oxide, Oxidant Stress, Physiology
Nonstandard Abbreviations and Acronyms
- eNOS
endothelial nitric oxide synthase
- GSNOR
S‐nitrosogluathione reductase
- HPDP
N‐[6‐(biotinamido)hexyl]‐3′‐(2′‐pyridyldithio) propionamide
- ROS
reactive oxidative stress
- SOD
superoxide dismutase
Clinical Perspective
What Is New?
Preeclampsia is a major unmet need in maternal fetal medicine engendering substantial mortality and morbidity in both mothers and infants.
While nitroso‐redox imbalance is an implicated pathogenetic process, the full extent to which it plays a role in preeclampsia has remained controversial; in this study, we demonstrated that mice lacking the gene for S‐nitrosoglutathione reductase, a denitrosylase that regulates protein S‐nitrosylation, recapitulate the majority of preeclampsia maternal and fetal phenotypes.
Antioxidant treatment rescued preeclampsia phenotypes by reducing dysregulation of nitrosylation and nitrosative stress; importantly, placentas from women with preeclampsia exhibited S‐nitrosoglutathione reductase deficiency, suggesting that this enzyme plays a potentially key role in normal human pregnancy.
What Are the Clinical Implications?
Our findings support the idea that S‐nitrosoglutathione reductase is a central regulator of physiologic nitrosylation and is crucial to governing normal pregnancy.
These findings have important implications for understanding the pathogenesis of preeclampsia, developing novel preeclampsia therapies, and identifying clinically useful biomarkers for this unaddressed need in maternal‐fetal medicine.
Moreover, the S‐nitrosoglutathione reductase homozygous knockout mouse represents a valuable animal model for ongoing study of the numerous processes operative in preeclampsia.
Preeclampsia is a life‐threatening disorder of pregnancy, characterized by new‐onset hypertension, proteinuria, abnormal maternal cardiovascular and renal adaptations, poor placental vascularization, and fetal growth restriction. Preeclampsia affects up to 10% of pregnancies and is a leading cause of maternal and fetal/neonatal mortality and morbidity worldwide, 1 , 2 and maternal mortality rates have been steadily rising over the past 30 years in large part attributable to cardiovascular complications. 3 , 4 The pathogenesis of preeclampsia is incompletely understood, but emerging data support a role for impaired protein S‐nitrosylation, nitration, and increased reactive oxidative stress (ROS), contributing to alterations in NO bioavailability and nitroso‐redox imbalance. 5 , 6 , 7 , 8 , 9 , 10 A paradoxical finding in preeclampsia is the elevation in circulating S‐nitrosylated albumin in human pregnancy 6 , 7 because elevated S‐nitrosylated albumin would traditionally be considered a vasorelaxant and an antioxidant. 11 Furthermore, cellular S‐nitrosylation level is elevated despite factors that are assumed to abrogate the formation of nitroso‐thiols, including oxidative stress and defects in NO bioavailability. 7 , 12 These findings raise 2 alternative pathogenic possibilities: Either S‐nitrosylation increases to compensate for the increased ROS levels or elevated S‐nitrosylation directly reflects abnormal regulation of S‐nitrosylation in preeclampsia (nitrosative stress). To differentiate between these 2 possible roles, we studied the impact of the loss of an important denitrosylase, S‐nitrosogluathione reductase (GSNOR−⁄− ), in pregnant mice.
Protein S‐nitrosylation participates in numerous pregnancy‐related processes including placental trophoblast cell migration, apoptosis, angiogenesis, immunomodulation, and oxygen delivery. 13 , 14 Protein S‐nitrosylation is enhanced in a transnitrosylation reaction using S‐nitrosogluathione, which thus acts as a second messenger to transduce NO bioactivity. 15 This process is tightly regulated by GSNOR, which selectively metabolizes S‐nitrosogluathione, thereby depleting the levels of S‐nitrosylated proteins in equilibrium with S‐nitrosogluathione. Ascorbate, with its dual roles as an antioxidant and a reductant, is required for the release of biologically active NO from these nitrosylated thiols. Although GSNOR−⁄− mice have increased numbers of S‐nitrosylated proteins, 16 , 17 which can in some circumstances lead to favorable outcomes (eg, recovery from myocardial infarction), 18 deficiency of this enzyme can also disrupt physiological nitrosylation‐denitrosylation dynamic cycles, leading to unfavorable outcomes (eg, increase in oxidative stress). 17 , 19 , 20 , 21 Thus, to assess the role of S‐nitrosylation and GSNOR in pregnancy outcome, we examined multiple organ systems, including the heart, kidney, and placenta, and the offspring during pregnancy in GSNOR−⁄− mice. Initially, we anticipated that GSNOR−⁄− mice would exhibit favorable maternal and fetal adaptations to pregnancy and were surprised to discover that pregnant GSNOR−⁄− mice recapitulate many of the features of preeclampsia. As a result, we subsequently tested the predictions that placental nitrosylation would exhibit widespread derangement in the knockout compared with control mice and that ascorbate would restore the intact animal and nitroso‐proteome phenotypes toward normal.
Methods
All data are available in the main text or the supplemental materials.
Study Approval
All animal care was carried out in accordance with approval by the Institutional Animal Care and Use Committee. For the human study, tissue collection was approved by the University of Miami Institutional Review Board, and the patient and family privacy were assured under the conditions of the Health Insurance Portability and Accountability Act. Written informed consent was received from participants before inclusion in the study.
Breeding
C57Bl/6J (wild type [B6]) controls (stock No. 000664) were purchased from Jackson Laboratories (Bar Harbor, ME). GSNOR−⁄− mice were raised in house. GSNOR−⁄− mouse line was created from ES clones after 10 consecutive backcrosses with C57Bl/6J. 16 Females were bred at 3 to 4 months of age and were studied in their first pregnancies. The presence of a sperm plug was defined as day 0.5 of gestation. Experimental time points included before breeding (nonpregnant), and day 17.5 (late gestation, 2 days before normal‐term delivery). Fetal, placental, and maternal organ weights were also recorded at time of euthanasia. For breeding, B6 females were bred with B6 males and GSNOR−⁄− females were bred with GSNOR−⁄− males.
Blood Pressure Determination
Mice were anesthetized with isoflurane. A 1.4‐F micromanometer‐tipped catheter (SPR‐839; Millar Instruments, Houston, TX) was inserted into the right carotid artery and advanced retrograde into the aorta. All analyses were performed using LabChart Pro 7 software (Millar Instruments).
Urinary Protein Measurements
Urine samples were collected from nonpregnant and pregnant mice (17.5 days of gestation). Urinary protein levels were measured using dipstick. A score of 0 to 3 was given on the basis of the color change on the dipstick following urine analysis. Twenty‐four‐hour urine was collected using metabolic cages (Tecniplast). Urine samples were analyzed for macroglobulin levels using Coomassie blue staining (Fisher).
Echocardiography
Echocardiographic assessments were performed in anesthetized mice (1% isoflurane in oxygen) using a Vevo‐770 micro‐ultrasound (VisualSonics, Toronto, Ontario, Canada) equipped with a 30‐MHz transducer. Cardiac dimensions including left ventricular end‐diastolic diameter, left ventricular end‐systolic diameter, and anterior and posterior wall thickness at systole and diastole were recorded from M‐mode images; cardiac output and stroke volume were calculated from bidimensional long‐axis parasternal views from 3 consecutive cardiac cycles. Doppler waveforms in the umbilical vein and artery were obtained near the placental end of the umbilical cord. Area under the peak velocity–time curve and R‐R interval were measured from 3 consecutive cardiac cycles, and the results were averaged. Umbilical venous and arterial diameters were measured from B‐mode images. Mean velocity over the cardiac cycle was calculated by dividing the area under the peak velocity–time curve by the R‐R interval. A parabolic blood velocity distribution was assumed so that umbilical venous and arterial blood flows were determined by the formula: F=½ п MV (D/2)2 (where MV=mean peak velocity (cm/s); D=diameter [cm]; F=blood flow [mL/min]).
Cardiomyocyte Isolation
The isolation of cardiomyocytes was performed as previously described. 22 Briefly, hearts were perfused with Ca2+‐free bicarbonate buffer containing 120 mmol/L NaCl, 5.4 mmol/L KCl, 1.2 mmol/L MgSO4, 1.2 mmol/L NaH2PO4, 5.6 mmol/L glucose, 20 mmol/L NaHCO3, 20 mmol/L 2,3 butanedione monoxime (Sigma‐Aldrich, St. Louis, MO), and 5 mmol/L taurine (Sigma‐Aldrich), gassed with 95%O2/5% CO2, followed by enzymatic digestion with collagenase type II (1 mg/mL) (Worthington, Lakewood, NJ) and protease type XIV (0.1 mg/mL; Sigma‐Aldrich). Cardiomyocytes were obtained from digested hearts followed by mechanical disruption, filtration, centrifugation, and resuspension in a Tyrode solution containing 0.125 mmol/L CaCl2 Tyrode buffer containing 144 mmol/L NaCl, 1 mmol/L MgCl2, 10 mmol/L HEPES, 1.2 mmol/L NaH2PO4, 5.6 mmol/L glucose, and 5 mmol/L KCl, adjusted to pH 7.4 with NaOH.
ROS by DCF, NO by DAF, Peroxynitrite by DHR 123
ROS, intracellular NO, and peroxynitrite were measured by epifluorescence using 2′,7′‐dichlorodihydrofluoresceine (10 μmol/L; Molecular Probes), 4,5‐diaminofluorescein (10 mmol/L; Cayman Chemical Co., Ann Arbor, MI) and dihydrorhodamine 123 (DHR 123, 25 mmol/L; Sigma‐Aldrich), respectively. These dyes have been well validated. 23 , 24 Briefly, fresh isolated mouse cardiomyocytes were placed in the chamber of an IonOptix spectrofluorometer and the background fluorescence (F 0 ) was acquired with an excitation wavelength of 488 nm and emission fluorescence collected at 510±15 nm. Cardiomyocytes were incubated for 40 minutes at room temperature (23 °C) with 2′,7′‐dichlorodihydrofluoresceine or 4,5‐diaminofluorescein or 20 minutes with DHR 123 and washed by superfusing fresh Tyrode (1.8 mmol/L CaCl2) solution for 10 minutes. Fluorescence (F) was acquired at 37 °C every 1 minute for 10 minutes. Myocytes were stimulated at 1 Hz during the 10‐minute experiment. ROS, NO or peroxynitrite levels were expressed as:
Superoxide Dismutase Measurement
Sample preparation: frozen placentas were ground up in a Dounce homogenizer on liquid nitrogen. Then, pulverized tissue was homogenized in ice‐cold PBS (10 µL per mg of tissue). The suspension was strained through a 250 µm‐pore mesh. Then, samples were assessed for protein content by BCA (Pierce, Thermo Scientific) and diluted 1:10 in PBS for superoxide measurement. Superoxide was assessed by lucigenin‐enhanced chemiluminescence.
For superoxide dismutase (SOD) assay, superoxide was assessed with the Superoxide Anion Assay Kit CS1000 (Sigma‐Aldrich). Briefly, a superoxide‐generator system (xanthine–xanthine oxidase [XO]) was used as a source of superoxide. Xanthine–xanthine oxidase (25 mU/mL final activity) plus 5 µL of [SOD‐containing] samples and 94.5 µL of PBS were added to the multiwell plate (in duplicate). Then, the luminol plus the enhancer salutation and xanthine were added. For positive control, a mix of 1 µL of 4 U/µL SOD (Sigma‐Aldrich) with 99 µL of PBS was used. As negative control for SOD, the superoxide generation system alone (without the sample) was used.
Luminescence was acquired for 10 minutes every 30 seconds and the integrated luminescence units were used for calculations.
Ascorbate Treatment
Ascorbate (sodium l‐ascorbate, 3.3 g/L, Sigma, St. Louis, MO) 25 was given in the drinking water starting from day 0.5 (time of plug detection). The water was changed every 2 days. In isolated cardiomyocytes, ascorbate (0.1 mmol/L, 0.5 mmol/L, or 1 mmol/L) was incubated for 30 minutes before the start of the epifluorescence experiments.
Tissue Preparation and Histology and Immunohistochemistry
At the end of the study, maternal organs (heart, kidney) and placenta were harvested, weighed, and processed for further analysis. Tissues were either flash‐frozen in liquid nitrogen for total RNA isolation and protein analysis, while some tissues were fixed with 10% formalin for histology. Slides were stained with hematoxylin and eosin and Masson’s trichrome staining for heart and kidney and periodic acid–Schiff staining for kidney. Glomerular size was quantified using Image J (National Institutes of Health).
Scanning Electron Microscopy of the Kidney
Tissue was fixed in 2% glutaraldehyde in 0.05 M phosphate buffer and 100mM sucrose, post‐fixed overnight in 1% osmium tetroxide in 0.1 M phosphate buffer, dehydrated through a series of cold graded ethanols, and embedded in a mixture of EM‐bed/Araldite (Electron Microscopy Sciences). 1 μm thick sections were stained with Richardson’s stain for observation under a light microscope. 100 nmol/L sections were cut on a Leica Ultracut‐R ultramicrotome and stained with uranyl acetate and lead citrate. The grids were viewed at 80 kV in a Philips cardiomyocyte‐10 transmission electron microscope and images captured by a Gatan ES1000W digital camera. N=2 samples were examined per each group at 17.5 d of gestation.
Isolectin Immunofluorescence for Paraffin‐Embedded Tissues and Analysis of Placental Capillary Density
Paraffin‐embedded placental sections were deparaffinized and rehydrated by immersion in xylene followed by a graded series of ethanol. Antigen retrieval was performed by a heat‐induced method with citrate buffer (Dako, Carpinteria, CA). The slides were then blocked for 1 hour in 10% normal donkey serum to reduce background. Sections were then incubated with DyLight 594‐GSL I‐isolectin B4 (Vector Laboratories, Burlingame, CA) primary antibody for 1 hour. at 37 °C. After washing with PBS, nuclei were counterstained with DAPI (Invitrogen, Carlsbad, CA). A stereological grid consisting of crosses was superimposed on images of placental sections stained with isolectin. The relative capillary density in the tissue was calculated on each section by dividing the number of crosses falling on capillary structure by the total number of points falling on the sampling area using Image J. For each placenta, 4 sections were analyzed.
Human Placental Tissue Collection
Placentas were collected immediately after delivery from normotensive (N=8) and preeclamptic (N=6) pregnancies at Jackson Memorial Hospital and Jackson North (Miami, FL). Tissue collection was approved by the University of Miami Institutional Review Board, and patient and family privacy were assured under conditions of the Health Insurance Portability and Accountability Act. Written informed consent was received from participants before inclusion in the study. Placental tissue was isolated by sterile dissection. The tissue was washed with cold PBS to remove blood from the intervillous spaces and then snap frozen in liquid nitrogen for storage at −80 °C. The demographic data are summarized in Table 2. Preeclampsia was defined as maternal systolic blood pressure at least 140 mm Hg or higher or diastolic blood pressure at least 90 mm Hg or higher and 1 additional factor such as proteinuria, impaired liver function, or thrombocytopenia was also observed.
Table 2.
Demographic Data of the Pregnant Study Human Subjects Used in the Study
| Variables | Normotensive | Preeclampsia | P value |
|---|---|---|---|
| N=8 | N=6 | ||
| Maternal age, y | 33±2 | 32±3 | … |
| Race or ethnicity | |||
| Non‐Hispanic Black | 8 | 5 | … |
| Hispanic | 0 | 1 | … |
| Gestational age, wk | 38±1 | 36±2 | … |
| Mode of delivery | |||
| Vaginal | 5 | 3 | … |
| Cesarean section | 3 | 3 | … |
| Blood pressure, mm Hg | |||
| Systolic | 116±6 | 166±5 | <0.001 |
| Diastolic | 67±3 | 89±4 | <0.01 |
Statistical significance between the 2 groups was determined by Student’s t test; Mean±SEM.
GSNOR Activity in the Mouse Heart and Mouse and Human Placenta
Heart and placental homogenate (100 µg/mL) were incubated with Tris‐HCl (2 mmol/L, pH 8.0), EDTA (0.5 mmol/L) and NADH (200 µmol/L). The reaction was started by adding S‐nitrosogluathione (400 µmol/L) and activity was measured as S‐nitrosogluathione–dependent NADH consumption at absorbance of 340 nm for 5 minutes in the mouse tissue and area under the curve for 8 minutes in the human placenta.
Protein Immunoanalysis
Samples were electrophoresed using a NuPAGE 10% Bis‐Tris gel (Invitrogen) and transferred to polyvinylidene membranes (Bio‐Rad Laboratories). Immunoblot detection was performed for vascular endothelial growth factor (VEGF) (ab46154, 1:1000; Abcam, Cambridge, MA), VEGFR2 (55B11, 1:1000; Cell Signaling, Danvers, MA), eNOS (1:1000; BD Bioscience, BD Bioscience, San Jose, CA) in mouse tissue, GSNOR in (1:1000, 11051‐1‐AP, Proteintech, Rosemont, IL) in human tissues, B‐actin (4957S, 1:1000, Cell Signaling, Danvers, MA), GAPDH (G8795, 1:1000, Sigma, St Louis, MO) and subsequent reaction with a goat anti‐rabbit horseradish peroxidase–conjugated antibody (1:1000; Cell Signaling). Then, membranes were developed by enhanced chemiluminescence (Super Signal West Pico, Thermo Scientific, Hampton, NH) and analyzed by the QuantityOne software (Bio‐Rad, Hercules, CA).
VEGF Nitrosylation
To assess S‐nitrosylation, biotin‐switch assay was used following methods described. Hearts were homogenized in HEN buffer (250 mmol/L Hepes (pH 7.7), 1 mmol/L EDTA, and 0.1 mmol/L neocuproine). Free cysteine (Cys) residues were blocked with S‐methyl methanethiosulfonate and labeled with N‐[6‐(biotinamido)hexyl]‐3′‐(2′‐pyridyldithio) propionamide (HPDP‐biotin) with or without sodium ascorbate. Biotinylated VEGF was individually immunoprecipitated with protein G‐Sepharose beads, electrophoretically resolved, and immunoblotted with anti‐biotin antibody. Blotted membranes were re‐probed with related antibody for detection of protein load.
RNA Preparation and Quantitative Real‐Time Polymerase Chain Reaction
Total RNA was extracted from tissues using the TRIzol method, and then reverse transcribed to complementary DNA using High‐Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol. The quantitative reverse transcriptase polymerase chain reaction for indicated genes was performed in TaqMan Universal PCR Master Mix (Applied Biosystems). Quantitation of mRNAs was performed using Applied Biosystems TaqMan Gene Expression Assays according to the manufacturer’s protocol. Samples were analyzed using the BIORAD sequence detection system. All polymerase chain reactions were performed in triplicate, and the specificity of the reaction was determined by melting curve analysis at the dissociation stage. The relative quantitative method was used for the quantitative analysis. The calibrator was the averaged ΔCt from the untreated cells. The endogenous control was GAPDH.
Mass Spectrometry Sample Preparation
All blocking and labeling steps were performed protected from light. Frozen placentas were individual minced in 0.9 mL of cold homogenization buffer (PEN: PBS pH 8.0, 1mM ETDA, 0.1 mmol/L Neocupine; supplemented with 20 mmol/L n‐ethylmaleimide). The tissue was then disrupted in 1ml mixer mill (Mixer Mill MM 400, retsch.com) for 5 minutes and then subjected to probe sonication. Samples were adjusted to 2.5% SDS and clarified by centrifugation for 5 minutes at 2000g. The resulting supernatant was incubated for 10 minutes at 50 °C to completing the blocking step. The unreacted n‐ethylmaleimide was removed using a Zeba spin column (Thermo Fisher Scientific, Waltham, MA) equilibrated with PEN buffer supplemented with 0.5% SDS. Each sample was divided and labeled with either 1 mmol/L biotin‐HPDP (Thermo Fisher Scientific) or 0.3 mmol/L iodoTMT6 (Thermo Fisher Scientific) in the presence of 5 mmol/L sodium ascorbate. As a labeling control, five pooled samples consisting of one replicate from each of the biological samples were prepared and reacted with each label in the absence of ascorbate. Samples were incubated for 1 hour. (HPDP) or 2 hour. (iodoTMT6) at 37 °C. Excess label was removed from the HPDP‐treated samples by adding 2 volumes of cold acetone and incubating for 20 minutes at −20 °C. Precipitated protein was pelleted by centrifugation and washed with 2 additional volumes of cold acetone. The pellets were resuspended 200 µL of PBS containing 1% (w/v) SDS aided by sonication. The excess iodoTMT6 label was removed by adding 5 volumes of cold acetone and precipitating as above. Samples were resuspended in 600 µL of PBS containing 1% (w/v) SDS aided by sonication. IodoTMT6 samples were then further reduced and alkylated using DTT and iodacetamide. Residual reagents were removed by Zeba spin column equilibrated with PBS. The protein concentration of each labeled sample was determined by BCA assay.
For HPDP‐labeled samples, 500 µg was digested overnight using 0.02 µg trypsin/µg of protein (Promega). iodoTMT6 labeled samples were combined according to the label’s isotope. Five‐plexes were prepared containing 350 µg of each of the different biological samples and the pooled control. An additional set of 6‐plexes was prepared containing 250 µg of each biological replicates and a pooled control. The mixtures were digested overnight with 0.02 µg trypsin/µg of protein. Digestions were halted with 0.25 mmol/L PMSF. The resulting peptides were captured using either streptavidin (HPDP) or TMT affinity resin (iodoTMT6). Peptides were enriched, washed, and eluted according to the manufacture’s protocol or as described here (PMID: 21036925). In the case of HPDP, eluted peptides were further alkylated with iodoacetamide.
Mass Spectrometry Analysis
The resulting peptides were desalted using Oasis HLB μ‐elution plates (Waters, Milford, MA). Samples were eluted with 300 µL of 50% ACN, 0.1% FA dried in speedvac, then resuspended in 0.1% FA for liquid chromatography–tandem mass spectrometry analysis. Liquid chromatography–tandem mass spectrometry analysis was performed using an Ultimate 3000 nano LC (Thermo Scientific) connected to an Orbitrap LUMOS mass spectrometer (Thermo Scientific) equipped with an EasySpray ion source. Peptides were loaded onto a PepMap RSLC C18 column (2 µm, 100 Å, 75 µm i.d. ×250 mm, Thermo Scientific) using a flow rate of 300 nL/min for 15 minutes at 1% B (mobile phase A was 0.1% formic acid in water and mobile phase B was 0.1% formic acid in acetonitrile) after which point they were separated with a linear gradient of 2% to 20%B for 90 minutes, 20% to 32%B for 20 minutes, 32% to 95%B for 2 minutes, holding at 95%B for 8 minutes and reequilibrating at 1%B for 5 minutes. Each sample was followed by a blank injection to both clean the column and reequilibrated at 1%B. MS1 scans were acquired at a resolution of 240 000 Hz from mass range 400 to 1600 m/z. For MS1 scans the automatic gain control target was set to 4×105 ions with a max fill time of 50 ms. MS2 spectra were acquired using the TopSpeed method with a total cycle time of 3 seconds and an automatic gain control target of 1×104 and a max fill time of 100 ms, and an isolation width of 1.6 Da in the quadrapole. Precursor ions were fragmented using higher‐energy C‐trap dissociation with normalized collision energy of 30% and analyzed using rapid scan rates in the ion trap. Monoisotopic precursor selection was enabled and only MS1 signals exceeding 5000 counts triggered the MS2 scans, with +1 and unassigned charge states not being selected for MS2 analysis. Dynamic exclusion was enabled with a repeat count of 1 and exclusion duration of 15 seconds.
Mass Spectrometry Data Analysis
Raw data were searched using a uniprot reviewed mouse database (09/18) with the X!Tandem (PMID: 14976030) algorithm version 2013.06.15.1 and Comet (PMID:23148064) algorithm version 2014.02 rev.2 search engines with the following parameters: full trypsin cleavage allowing for up to 2 missed cleavages, variable modifications +16 Da on methionine (oxidation), +57 Da, +125 Da and, in the case of IodoTMT6 labeled samples, +329 on cysteine (carbamidomethylation, n‐ethylmaleimide, TMT). Mass tolerance of MS1 error of 10 ppm, MS2 error of 1 Da were used. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (PMID: 30395289) partner repository with the data set identifier PXD012706 (reviewer account details: Username: reviewer52954@ebi.ac.uk; Password: MkOzBjKi).
Differences in S‐nitrosylation modification for HPDP‐labeled samples were determined by label‐free quantitation using MS1 extracted ion chromatograms in Skyline (version 4.1) using a dot product cutoff 0.8 (PMID: 22454539). Signal for each modified cysteine was summed within a replicate and normalized against the average of the pooled controls for that site. For iodoTMT6 labeled samples, only peptides with an iprofit score >0.95 were considered. Reporter ion intensities were determined using the Libra module of the trans‐proteomic pipeline (PMID: 20101611). Replicates were normalized against the pooled control present in the 5‐ or 6‐plex. For all analysis, replicates need to be at least 1.5‐fold greater than control and present in at least 40% of the replicates per group. Bioinformatic analysis was performed using Ingenuity Pathway Analysis (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity‐pathway‐analysis) and using gene ontology annotations in the Uniprot database (uniprot.org).
String v11 software was used to create pathway analysis of the data shown in Figure S3. 26
Statistical Analysis
The results are expressed as mean±SEM. Differences between groups were examined for statistical significance using Student’s t test or 1‐way or 2‐way ANOVA, with Newman‐Keuls for multiple comparisons for post hoc analysis where appropriate. Results with P<0.05 were considered significant. All analysis were performed using SPSS and Prism Statistical software.
Results
Pregnant GSNOR−⁄− Mice Exhibit Hallmark Features of Preeclampsia, Hypertension, and Proteinuria
Development of hypertension and proteinuria are hallmarks of preeclampsia, and GSNOR−⁄− mice are hypotensive compared with wild‐type mice before pregnancy, 16 consistent with GSNOR regulation of endothelium‐dependent vasodilation (Figure 1A). At day 17.5 of pregnancy, GSNOR−⁄− mice develop hypertension (Figure 1A, Figure S1) and proteinuria (Figure 1B), characterized by elevated urine macroglobulin levels, associated with renal pathology including enlarged glomeruli, swelling of the endothelial cells and loss of fenestration and corrugation of the glomerular basement membrane (Figures 1C through 1F), changes that recapitulate human preeclampsia. 27 , 28
Figure 1. Pregnant GSNOR−⁄− mice exhibit hallmark features of preeclampsia including hypertension, proteinuria, and concentric hypertrophy in the heart.
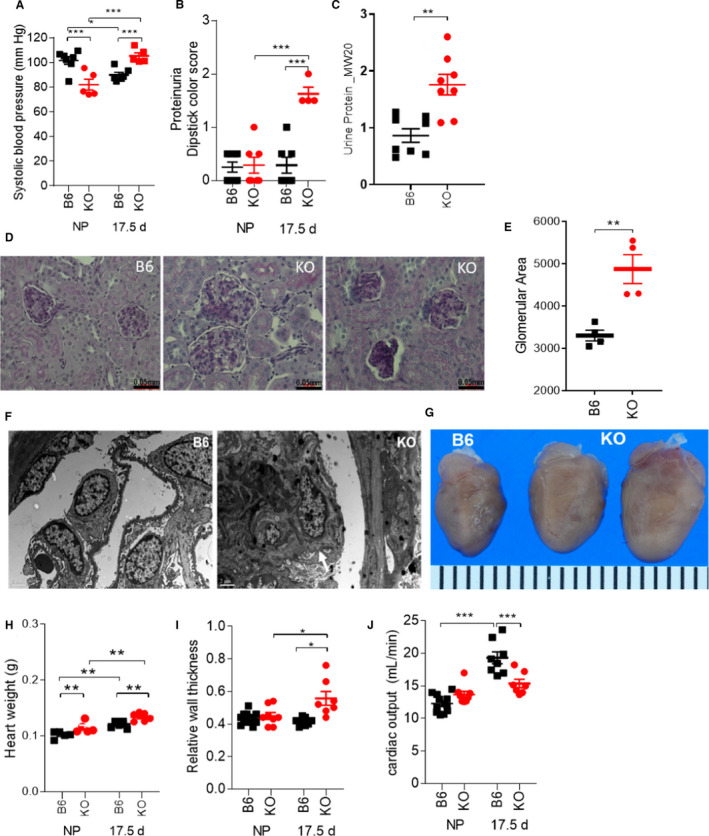
Nonpregnant and pregnant (17.5 d) (N=4–12 mothers per group) C57Bl/6J (B6) and GSNOR−⁄− (KO) mice were examined. At late gestation, knockout (KO) mothers exhibited (A) hypertension, (B) proteinuria, and elevated (C) urine macroglobulin levels. D, E, Kidney sections at late gestation stained with periodic acid–Schiff showed enlarged glomeruli and focal and sclerosis with collapsed glomerular capillaries. F, Electron microscopy on renal tissues showed that GSNOR–/–kidneys exhibited glomerular endotheliosis comprised of endothelial cell swelling, along with loss in fenestration and corrugation of the glomerular basement membrane (N=2 was examined per group). G, H, Heart weight and (I) relative wall thickness were significantly bigger in KO mice as compared with controls, indicating the presence of concentric hypertrophy. J, The normal increase in cardiac output was absent in KO mice at late gestation. For systolic blood pressure, proteinuria, heart weight, relative wall thickness, and cardiac output, a 2‐way ANOVA with Newman‐Keuls for post hoc analysis was performed. For the other variables, Student’s t test was performed. Results are shown as mean±SEM. ***P<0.001, **P<0.01, *P<0.05. B6 indicates C57Bl/6J mice; KO, GSNOR−⁄− mice; and NP, non‐pregnant.
GSNOR−⁄− Hearts Exhibit Concentric Hypertrophy During Pregnancy
We next studied cardiac responses to pregnancy in the GSNOR−⁄− mouse. The heart responds to sustained hypertension by an increase in wall thickness leading to concentric hypertrophy, and concentric hypertrophy independently predicts adverse outcome in preeclamptic pregnancies. 29 At late gestation, left ventricular end‐diastolic dimension was lower, whereas the anterior wall at diastole (Table 1) was thicker, contributing to higher relative wall thicknesses in pregnant GSNOR−⁄− mice as compared with controls (Figures 1G through 1I, Table 1). Using isolated cardiomyocytes we observed that width was greater in GSNOR−⁄− mice, whereas length was not different between the 2 strains at late gestation (Table 1). These factors likely contributed to the enlarged GSNOR−⁄− hearts at late gestation, even when normalized to tibia length (Figure 1G and 1H, Table 1). Furthermore, the normal physiologic increases in maternal cardiac output and stroke volume were completely abrogated at late gestation in GSNOR−⁄− mice (Figure 1J, Table 1), consistent with the phenotype of lower cardiac output and stroke volume in preeclamptic patients with concentric heart geometry. 29 Heart rate remained unchanged between groups (Table 1). These data suggest that S‐nitrosylation homeostasis plays an important role in limiting pathological hypertrophy. 30
Table 1.
KO Mice Exhibited Concentric Hypertrophy and Abnormal Maternal Cardiovascular Adaptation to Pregnancy, Which Was Rescued With Ascorbate Treatment
| Nonpregnant | Pregnant | Ascorbate‐treated pregnant | ||||
|---|---|---|---|---|---|---|
| B6 | Knockout | B6 | Knockout | B6 | Knockout | |
| Maternal body weight, g | 20.3±0.36* | 20.3±0.70* | 35.4±0.56 | 31.1±1.55 † | 36.24±0.78 | 34.2±0.55* |
| Heart rate, bpm | 454±9 | 497±18 | 494±14 | 498±12 | 484±7 | 517±14 |
| LV end‐diastolic dimension, diastole, mm | 3.40±0.04* | 3.50±0.05 | 3.78±0.04 | 3.52±0.12 † | 3.67±0.07 | 4.00±0.09* , † |
| LV anterior wall thickness, diastole, mm | 0.80±0.02 | 0.82±0.04* | 0.91±0.03 | 1.07±0.07 † | 0.84±0.03 | 0.88±0.04* |
| Relative wall thickness | 0.43±0.01 | 0.45±0.02* | 0.41±0.01 | 0.56±0.04 † | 0.44±0.01 | 0.45±0.03* |
| Myocyte length, µmol/L | 138±2* | 164±4* , † | 158±2 | 154±2 | 158±7 | 150±2 |
| Myocyte width, µmol/L | 25.7±0.28 | 25.1±0.46* | 25.2±0.53 | 29.4±0.67 † | 25.3±0.34 | 22.4±0.29* , † |
| Stroke volume, µL | 27.1±0.50* | 27.8±1.16 | 39.2±1.96 | 30.9±1.27 † | 34.5±1.54* | 40.1±1.60* , † |
| Heart weight normalized to tibal length, mg | 5.88±0.13* | 7.09±0.31* , † | 7.07±0.10 | 7.98±0.34 † | 7.01±0.63 | 9.98±3.38 † |
Results are shown as mean±SEM. Two‐way ANOVA with Newman‐Keuls for post hoc analysis. LV indicates left ventricular.
P<0.05 vs pregnancy (same strain).
Vs strain difference.
GSNOR−⁄− Mice Exhibited Placental Insufficiency During Pregnancy
Placental insufficiency (fetal weight to placental weight ratio) is believed to contribute directly to the development of preeclampsia. The placentas of preeclamptic pregnancies often exhibit fetoplacental hypovascularity and decreased fetoplacental perfusion, 31 both of which decrease transfer of oxygen and nutrients to the placenta, thereby limiting fetal growth. Fetal litter size was significantly lower in GSNOR−⁄− mice at 17.5 d of gestation (Figure 2A). Fetal body weights were significantly lower in GSNOR−⁄− mice at 17.5 d of gestation (Figure 2B). To evaluate the role of GSNOR on placental vascularization and fetoplacental perfusion, we examined the placentas at late gestation. GSNOR activity was present in the control placentas, and as anticipated was absent in the knockout mice (Figure 2C). Placental weight (Figure 2D) and umbilical arterial blood flow (Figure S2) were not significantly different between the 2 strains. However, placental efficiency and umbilical venous blood flow were significantly lower in GSNOR−⁄− placentas as compared with controls at late gestation (Figures 2E, Figure S2). In addition, placental vascularization was decreased in GSNOR−⁄− placentas (Figures 2F and 2G). These findings suggest that GSNOR plays an essential role in placental development and function during pregnancy.
Figure 2. GSNOR−⁄− mice exhibited placental insufficiency during pregnancy and showed alteration of VEGF pathway.
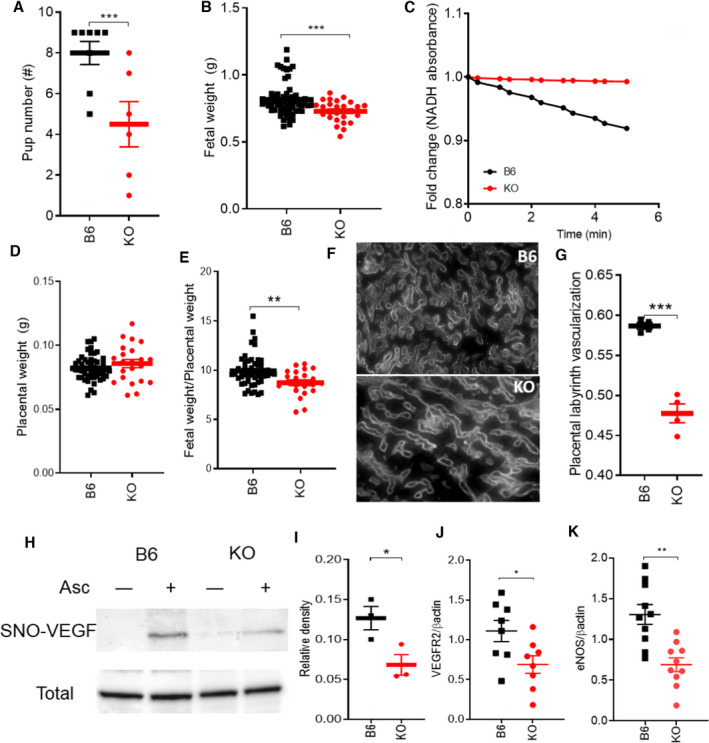
Nonpregnant and pregnant (17.5 d) (N=4–12 mothers per group) C57Bl/6J (B6) and GSNOR−⁄− (knockout [KO]) mice were examined. A, Fetal number and (B) weight were significantly lower in KO mice. C, NADH‐dependent GSNOR enzymatic activity was determined in placental tissue at 17.5 days of gestation. GSNOR activity is enriched in B6 placentas, whereas it is completely absent in the KO placentas. D, E, Placental weight was not significantly different between the 2 strains, whereas placental efficiency was significantly lower in KO mice as compared with control. F, G, Placental vascularization determined using isolectin immunostaining, was significantly lower in KO as compared with B6 at late gestation. SNO‐VEGF was measured using Biotin‐switch assay. H, Representative blots shown for S‐nitrosylated and total VEGF in the placenta. Omission of ascorbate was used as the negative control. I, Nitrosylation of VEGF was significantly lower in KO placentas as compared with B6 at late gestation. J, K, VEGFR2 and eNOS protein levels were determined in the placentas at 17.d of gestation. Both VEGFR2 and eNOS protein levels were significantly lower in in GSNOR−⁄− placentas as compared with B6 placentas at 17.5 days of gestation. For statistical significance, 2‐way ANOVA with Newman‐Keuls for post hoc analysis or Student’s t test were performed. Results are shown as mean±SEM. ***P<0.001, **P<0.01, *P<0.05. B6 indicates C57Bl/6J mice; GSNOR, S‐nitrosoglutathione reductase; KO, GSNOR−⁄− mice; and NP, nonpregnant.
VEGF Pathway Was Blunted in GSNOR−⁄− Placentas at Late Gestation
We next examined the VEGF pathway as a potential mechanism for the impaired placental vascularization as VEGFR2 levels are decreased in human preeclamptic pregnancies. 32 Whereas VEGF protein abundance in the placentas was not different between the 2 strains at late gestation (Figure 2H), nitrosylation of VEGF protein was significantly lower in GSNOR−⁄− placentas (Figures 2H and 2I). VEGF binds to its receptor VEGFR2 and signals through the endothelial nitric oxide synthase (eNOS) pathway. 33 Similar to human preeclampsia, 10 , 32 we found that total VEGFR2 (P<0.05) and eNOS (P<0.01) protein quantities were lower in the GSNOR−⁄− placentas as compared with controls (Figures 2J and 2K). Thus, alterations in VEGF signaling may represent a contributory mechanism for impaired placental development in GSNOR−⁄− mice.
GSNOR−⁄− Mice Exhibit Nitroso‐Redox Imbalance and Nitrosative Stress During Pregnancy
Aberrant ROS and NO signaling has been implicated in the pathogenesis of preeclampsia. 34 , 35 To address this issue, we measured the cellular ROS levels in GSNOR−⁄− mice. Before pregnancy, cellular ROS generation was higher in cardiomyocytes isolated from GSNOR−⁄− mice as compared with control mice (Figure 3A), yet ROS levels were not significantly different between the 2 groups (Figure 3B), suggesting increased involvement of antioxidant scavengers. During pregnancy, ROS generation and levels were significantly higher in GSNOR−⁄− cardiomyocyte as compared with controls (Figures 3A and 3B) confirming the presence of oxidative stress. Increased oxidative stress can lead to alterations in NO/S‐nitrosylation signaling; therefore, we next measured NO levels. As expected, there was a significant increase in NO levels in isolated cardiomyocytes of control mice during pregnancy (Figure 3C). This increase likely plays a critical role in vasodilation leading to the normal adaptations to pregnancy. NO levels were also significantly higher in cardiomyocytes isolated from pregnant as compared with nonpregnant GSNOR−⁄− mice. With the presence of elevated ROS and NO/S‐nitrosylation levels in GSNOR−⁄− mice, we predicted an increase production of the potent prooxidant peroxynitrite. Before pregnancy, peroxynitrite levels were significantly higher in GSNOR−⁄− cardiomyocytes as compared with controls (Figure 3D). This preexisting elevation in peroxynitrite levels suggests that the GSNOR−⁄− mice were less able to respond to the stress of pregnancy. At late gestation, peroxynitrite levels were, indeed, significantly higher in isolated cardiomyocytes of GSNOR−⁄− mice (Figure 3D), suggesting the presence of nitrosative stress. Furthermore, superoxide dismutase (SOD) levels were significantly lower in GSNOR−⁄− placentas as compared with controls, suggesting decreased antioxidant capacities (Figure 3E). NO signaling via S‐nitrosylated proteins may exert a protective role against an oxidative environment by competing with other posttranslational modifications and shielding critical cysteine residues from the damaging effects of irreversible oxidation as shown in ischemic preconditioning. 36 , 37 These findings suggests that S‐nitrosylation–based mechanisms that protect physiologic signaling may be impaired in pregnant GSNOR−⁄− mice, contributing to the preeclampsia phenotype. Alternatively, increased levels of NO and ROS can form peroxynitrite, which can irreversibly lead to protein nitration. 8 Protein nitration plays a relevant role in posttranslational modification of protein and increased nitration of proteins has been implicated in human preeclampsia pregnancies. 8 Thus, protein nitration may be another mechanism involved in the development of preeclampsia‐like conditions in GSNOR−⁄− animals.
Figure 3. GSNOR−⁄− mice exhibit nitroso‐redox imbalance and nitrosative stress that is rescued with ascorbate treatment.
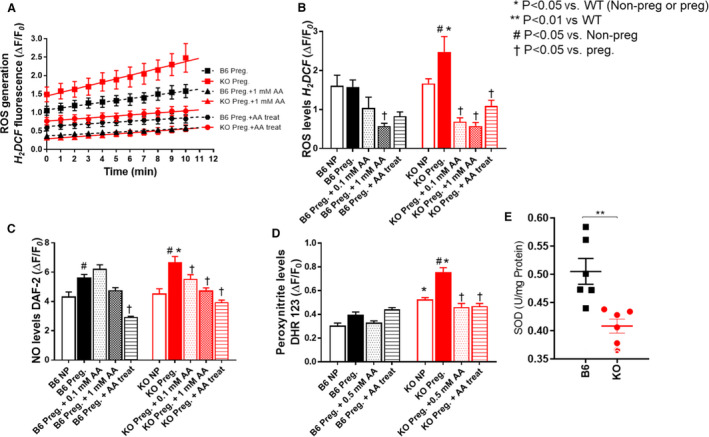
In nonpregnant (NP) and pregnant (17.5 d) C57Bl/6J (B6) and GSNOR−⁄− (knockout [KO]) mice, reactive oxygen species, NO and peroxynitrite levels were determined in isolated cardiomyocyte using epifluorescence by 2’,7’dichlorodihydro‐fluoresceine (H2DCF‒DA, 10mM), 4,5‐diaminofluorescein (DAF‐2DA, 10 mmol/L) and dihydrorhodamine 123 (DHR 123, 25 mmol/L), respectively. Ascorbate (AA) studies included both acute (0.1 mmol/L, 0.5 mmol/L, 1 mmol/L) and chronic (AA provided in drinking water from day 0.5) treatments. A and B, ROS generation and levels, and (C) NO and (D) peroxynitrite levels were significantly higher in cardiomyocyte isolated from pregnant GSNOR−⁄− mice. This increase was prevented with acute and chronic AA treatment. E, Superoxide dismutase (SOD) levels were measured in the placenta using lucigenin‐enhanced chemiluminescence. SOD levels were significantly lower in the KO placentas as compared with controls. Results are shown as mean±SEM. N=3–5 animals per experiment. One‐way ANOVA with Newman Keuls post hoc test.B6 indicates C57Bl/6J mice; KO, GSNOR−⁄− mice; NP, nonpregnant; and SNO, S‐nitrosylated.
Antioxidant Treatment Rescued the Preeclampsia Phenotype in GSNOR−⁄− Mice During Pregnancy
To test whether antioxidant treatment can rescue the preeclampsia phenotype, we treated animals or isolated cardiomyocytes with ascorbate. In addition to being an antioxidant, ascorbate also functions as a reductant, promoting the release of biologically active NO from nitrosylated thiols, and plasma ascorbate levels are commonly diminished in preeclamptic patients. 38 Importantly, ascorbate treatment rescued the onset of hypertension, proteinuria, and urinary macroglobulin levels (Figures 4A through 4C). The enlarged anterior wall thickness, relative wall thickness and cardiomyocyte width, all indicative of concentric hypertrophy, returned to normal levels in ascorbate treated pregnant GSNOR−⁄− mice (Table 1). Ascorbate also significantly improved cardiac output and stroke volume in GSNOR−⁄− mothers at late gestation, whereas it lowered both parameters in pregnant B6 controls (Figure 4D, Table 1). Ascorbate treatment significantly improved placental vascularization along with placental VEGFR2 and eNOS protein levels (Figures 4G, 4J and 4K). In addition, ascorbate improved umbilical venous blood flow and litter size but did not improve placental efficiency, which may account for the failure of fetal weights to improve at late gestation in the GSNOR−⁄− mice (Figure 4E through 4I, Figure S2). Acute and chronic treatment of ascorbate similarly reduced ROS, NO, and peroxynitrite levels in isolated cardiomyocytes from pregnant GSNOR−⁄− mice (Figure 3A through 3D). Thus, ascorbate may work as a potent scavenger of free radicals as seen in experimental models of hypertension 39 , 40 and in rat models of nephrotoxicity 41 to balance the nitroso‐redox system and in turn rescue the preeclampsia‐like phenotype in pregnant GSNOR−⁄− mice.
Figure 4. Preeclampsia phenotype in the mother and litter size is rescued with ascorbate.
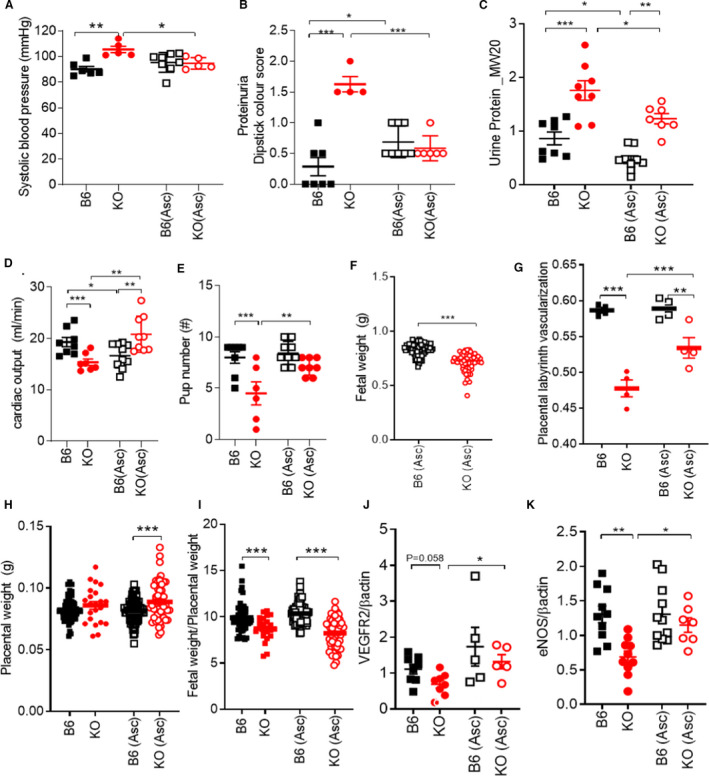
Late pregnant (17.5 d) C57Bl/6J (B6) and GSNOR−⁄− (knockout [KO]) mice were examined. N=4–10 mothers per group. A, Hypertension, (B) proteinuria, and (C) urine macroglobulin levels were rescued with ascorbate treatment. Ascorbate treatment increased (D) cardiac output in knockout (KO) mice at late gestation. E, Pup number was improved, whereas (F) fetal weight remained significantly lower in KO mice treated with ascorbate. G, Impaired placental vascularization was rescued with ascorbate treatment in KO placentas. With ascorbate treatment, (H) placental weight was significantly higher in the KO treated animals as compared with B6‐treated animals. Whereas (I) placental efficiency remained significantly lower in treated KO mice as compared with controls. J, K, VEGFR2 and endothelial nitric oxide synthase placental protein levels were significantly increased in KO (Asc) treated animals as compared with nontreated KO animals. Results are shown as mean±SEM. ***P<0.001, **P<0.01, *P<0.05. One‐way or 2‐way ANOVA with Newman Keuls post hoc test and Student’s t test were performed. Asc, ascorbate; B6, C57Bl/6J mice; KO, GSNOR−⁄− mice; and VEGF, vascular endothelial growth factor.
Mass Spectrometry Revealed Elevated Placental S‐Nitrosylated Amino Acid Residues in GSNOR−⁄− Mice
To directly test the prediction that S‐nitrosylation is dysregulated in GSNOR−⁄− placentas, we performed mass spectrometry using both thiol reactive biotin–HPDP and cysTMT labeling to maximize coverage. 42 Consistent with our prediction, this analysis revealed a marked increased number of S‐nitrosylated residues in GSNOR−⁄− placentas (459 corresponding to 351 proteins) compared with controls (264 S‐nitrosylated residues corresponding to 198 proteins) (Figure 5A through 5C, Table S1 through S3). Importantly, placentas from ascorbate‐treated mice exhibited an increased net number of S‐nitrosylated proteins in both control and GSNOR−⁄− placentas (consistent with the transnitrosylation properties of chronic ascorbate 43 ), but this increase was less in the GSNOR−⁄− mice (Figure 5D) decreasing the difference in number of S‐nitrosylated proteins between the 2 groups. Furthermore, we examined subcellular compartmentalization of the total proteins detected in the 4 groups. Similar to our previous study, 44 we found majority of the nitrosylated proteins were located in the cytoplasm and the nucleus (Figure 5E). To gain insights into the most important signaling pathways affected by excess S‐nitrosylation, we examined the subset of S‐nitrosylated proteins found exclusively in GSNOR versus B6, and which exhibited denitrosylation with ascorbate. Of all the detected peptides, there were 50 S‐nitrosylation residues unique to the GSNOR−⁄− placentas, but only 16 residues unique to the B6 placentas (Figures 5C and 5F, Table S1 through S3). All 50 S‐nitrosylation residues were reversed by ascorbate (Table S2). From these 50 proteins, 14 have been linked to important roles in processes essential in pregnancy, including angiogenesis, inflammation, cell migration, and apoptosis (Figure 5F), supporting the pathophysiological relevance of these proteins.
Figure 5. Duel‐labeling mass spectrometry revealed an increased number of SNOylated proteins in the placentas from GSNOR−⁄− animals.
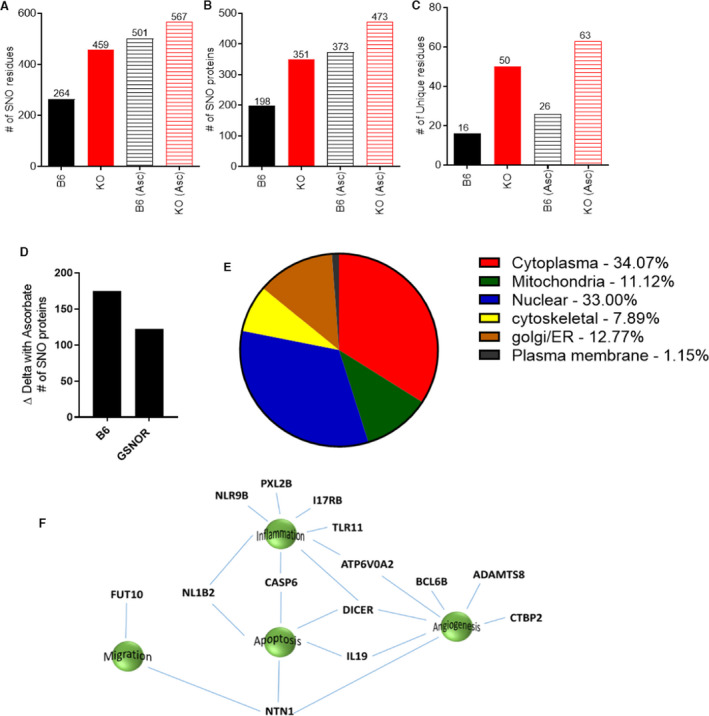
A and B, An increased number of S‐nitrosylated proteins were detected in the placentas of the knockout (KO) animals, and of these there were more unique S‐nitrosylated residues (C) in GSNOR−⁄− placentas as compared with controls. Ascorbate treatment increased S‐nitrosylated proteins in both groups, but this increase was less in the KO group as compared with control (D). E, The majority of the nitrosylated proteins were detected in the cytoplasm and in the nucleus in the 4 different groups analyzed. F, Schematic showing proteins unique in GSNOR−⁄− placentas but absent in the B6 placentas, and B6‐ and GSNOR−⁄− placentas treated with ascorbate. ADAMTS8 indicates A disintegrin and metalloproteinase with thrombospondin motifs 8; ATP6V0A2, V‐type proton ATPase 116 kDa subunit a isoform 2; BCL6B, B‐cell CLL/lymphoma 6 member B protein; CASP6, Caspase‐6 (CASP‐6); CASP6, Caspase‐6; CTBP2, C‐terminal‐binding protein 2; DICER, Endoribonuclease Dicer; FUT10, Alpha‐(1,3)‐fucosyltransferase 10; IL17RB, IL‐17 receptor B; IL‐19, interleukin‐19; NL1B2, NACHT, LRR and PYD domains‐containing protein 1b allele 2; NLR9B, NACHT, LRR and PYD domains‐containing protein 9B; NTN1, Netrin‐1; PXL2B, Prostamide/prostaglandin F; and TLR11, Toll‐like receptor 11.
GSNOR Activity Is Reduced in Human Preeclampsia Placentas
To examine the potential relevance of this pathway in human preeclampsia, we measured GSNOR mRNA, protein, and activity levels in the placenta of patients with pregnancies complicated with preeclampsia (Figure 6, Table 2). Whereas GSNOR mRNA and protein abundance were similar (data not shown), GSNOR activity was significantly lower in placentas of human pregnancies complicated with preeclampsia compared with placentas from normal pregnancies (Figure 6). Furthermore, affected placentas also had increased nitrosative stress as indicated by increased protein expression of nitrotyrosine and decreased antioxidant capacity as shown by decreased SOD levels (Figure 6). These findings support the physiologic importance of GSNOR in regulating human placental homeostasis and that the GSNOR−⁄− mouse represents a model of preeclampsia.
Figure 6. GSNOR activity is reduced and contributes to nitroso‐redox balance in the human preeclamptic placenta.
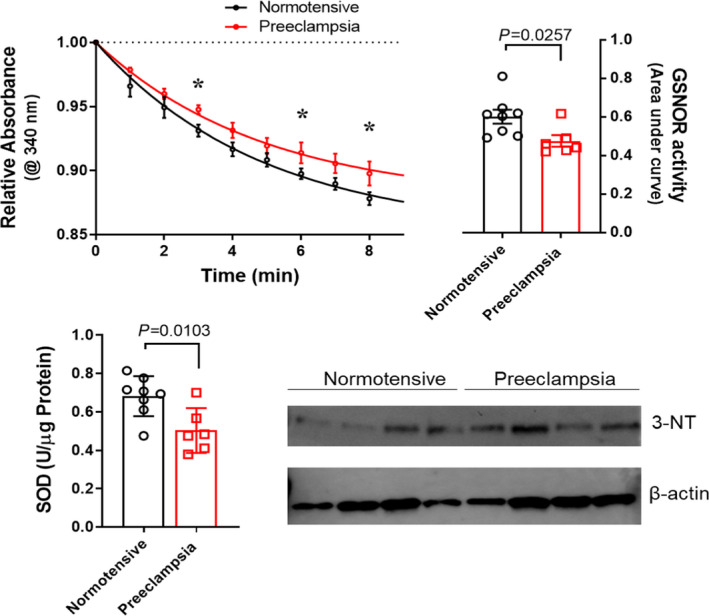
Human preeclamptic placentas exhibited decreased GSNOR activity, decreased antioxidant capacity (determined by superoxide dismutase levels), and increased nitrosative stress (determined by increased protein expression of nitrotyrosine). Significance for relative absorbance of GSNOR activity was determined by 2‐way ANOVA with Newman‐Keuls post hoc test. For other variables, Student’s t test was used. Results are shown as mean±SEM. *P<0.05, N=6–8 mothers. GSNOR indicates S‐nitrosoglutathione reductase; and SOD, superoxide dismutase.
Discussion
We have identified that mice lacking S‐nitrosoglutathione reductase (GSNOR−⁄−), a denitrosylase regulating S‐nitrosylation, exhibit most of the clinical features of preeclampsia including hypertension, proteinuria, renal pathology, cardiac concentric hypertrophy, decreased placental vascularization and fetal growth restriction. The primary mechanisms involved in this preeclampsia phenotype appears to be nitrosative stress attributable to aberrant S‐nitrosylation leading to the presence of nitroso‐redox imbalance. In addition, we showed that antioxidant, ascorbate, rescued the nitrosative stress and preeclampsia phenotype in the mother (Figure 7). Our findings demonstrate that the absence of a single gene, GSNOR, alters large numbers of downstream signaling pathway. Ascorbate, which creates a net increase in the number of S‐nitrosylated proteins, decreases the differences between the GSNOR−⁄− and B6 mice. As such, these results suggest that the regulation of the S‐nitrosylation–integrated posttranslational modification system may account in large part for the phenotype of preeclampsia in the GSNOR−⁄− mice. Together, these results suggest that this system‐wide alteration in S‐nitrosylated proteins has a detrimental effect on the function of these pathways and, as such, could be a key mechanism involved in the pathological phenotype seen in multiple organ systems, including the heart, kidney, and placenta, and the offspring of the knockout animals with preeclampsia. Furthermore, our data revealed that GSNOR plays an essential role in normal human pregnancies, as human placentas from pregnancies complicated with preeclampsia showed a significant decrease in GSNOR activity with the presence of nitrosative stress and decrease in antioxidant capacities.
Figure 7. Dysregulation in nitrosylation contributes to nitroso‐redox imbalance and nitrosative stress contributing to clinical features of preeclampsia including hypertension, proteinuria, concentric hypertrophy in the heart, decrease placental vascularization, and fetal growth restriction.
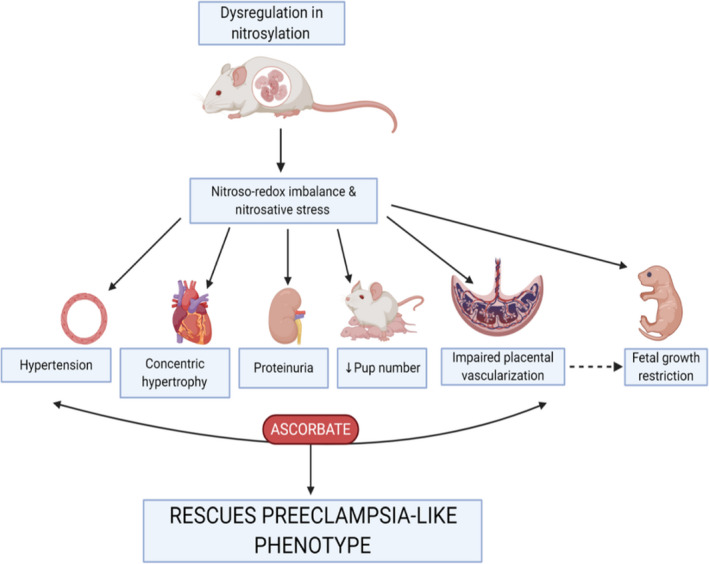
Antioxidant treatment rescued the preeclampsia‐like phenotype in the mother.
One of the primary clinical features of preeclampsia is an elevation in blood pressure and number of different mechanisms may account for the alterations in blood pressure in GSNOR−⁄− animals. Elevated plasma S‐nitrosylation can lead to adverse cardiovascular outcomes in patients with end‐stage renal disease, which correlates with elevated blood pressure. 45 Gandley et al 7 postulated that the buffering function of S‐nitrosylated albumin was impaired in patients with preeclampsia, where the thiol of albumin acts as a sink for NO, therefore lowering NO bioavailability and thus raising blood pressure. Alternatively, denitrosylation of S‐nitrosylated albumin may be regulated by glutathione. In preeclampsia, plasma glutathione levels are low, 46 most likely attributable to oxidative stress, and this decrease may effect NO‐dependent vasodilation of red blood cells, as glutathione may facilitate their export of S‐nitrosylations. 47 In addition, ROS which is increased in preeclampsia, potentiates protein S‐nitrosylation. 48 Therefore, an altered redox state, which influences the thiol/nitrosothiol balance, may convey NO bioactivity, regulating free NO, thereby affecting blood pressure in GSNOR−⁄− mice.
It is well established that VEGF mediates endothelium‐dependent vasodilation and angiogenesis, in part via the NO pathway. Furthermore, our laboratory previously showed that mesenchymal stem cells isolated from GSNOR–/– mice exhibited markedly diminished capacity for vasculogenesis, 49 suggesting that GSNOR plays a role in part working through the VEGF/NO pathway. VEGF binds to its receptor VEGFR2 and signals through the eNOS pathway, 33 and a decrease in VEGFR2 and eNOS levels are reported in human preeclamptic pregnancies. 10 , 32 During preeclampsia, impaired placental vascularization contributes to chronic ischemia and increases oxidative stress causing the placental release of factors such as soluble Flt1 (sFlt1). In turn, sFlt1 acts as a decoy receptor for VEGF, 50 and also antagonizes autocrine VEGF signaling, 51 rendering endothelial cells more sensitive to proinflammatory factors released by the placenta and decreasing the bioavailability of VEGF. 50 , 51 Accordingly, our current data suggest that the decrease in VEGF nitrosylation could further contribute to the downregulation of VEGF function, decreased eNOS protein levels, in turn affecting both vasodilation and angiogenesis during pregnancy. VEGF has not been previously reported to be nitrosylated, although it bears 4 potential cysteine sites; future experiments will be conducted to identify the exact nitrosylation posttranslational sites.
Several clinical trials have examined the effectiveness of antioxidants (ascorbate) in preventing preeclampsia. Small, randomized placebo‐controlled trials showed reduced preeclampsia with ascorbate treatment, whereas all large multicenter randomized trials have yielded disappointing results. These large trials showed that ascorbate did not decrease the risk of preeclampsia in either high‐risk (women with type 1 diabetes, nutritionally deficient, poor social economic status) or low‐risk populations. 52 , 53 , 54 In turn, increased incidence of low birthweight, gestational hypertension, fetal loss, stillbirth, or premature rupture of membranes have been reported in some trials, but these findings were not confirmed across all studies; therefore, their significance remains uncertain. 52 , 53 , 54 The dosage and/or timing of the antioxidant treatment (8–22 weeks) may have played a role in the unsuccessful outcomes in these large clinical trials. Alternatively, oxidative stress may be relevant to the pathogenesis in only a subgroup of women, with no appreciable benefit of antioxidant therapy for the overall population. Preeclampsia is a multisystemic/multifactorial disease, and based on the heterogeneity of the clinical presentation, there may be different “subtypes” of preeclampsia, 55 which may explain, in part, why many of the clinical trials using ascorbate have yet to show favorable outcomes. Therefore, identifying women showing dysregulation in nitrosylation and/or altered GSNOR activity levels may be the ideal target subpopulation for treatment with ascorbate, permitting a precision medicine approach for future clinical trials.
This study has several limitations. All the features of the murine phenotype may not translate into human pathophysiology given its heterogenous nature and involvement of multiple organ systems. We did not examine some of the clinical features of preeclampsia such as thrombocytopenia, abnormal liver enzymes or abnormal spiral artery remodeling in the placenta. Furthermore, blood pressure was measured in unconscious animals.
Development of effective therapies for preeclampsia is hampered by a failure to understand the causative mechanisms involved and the absence of robust animal models that exhibit essentially all the clinical features of preeclampsia. Therefore, the identification of the GSNOR−⁄− mice as such a model with the causative role of dysregulation in nitrosylation contributing to nitrosative stress and dysregulation of physiologic posttranslational modification in a large number of signaling pathways as one of the primary mechanisms contributing to this disorder, has important implications for developing novel therapies and identifying clinically useful biomarkers for this difficult to treat maternal‐fetal syndrome.
Sources of Funding
This study was funded by R01 HL09489 and R01 HL137355 to Dr Hare and by Canadian Institute of Health Research postdoctoral fellowship and American Heart Association Career Development Award (19CDA34660102) to SK. Dr Hare is also supported by National Institutes of Health grants R01 HL134558, R01 HL101110, and 5UM 1HL113460 and by the Starr and Soffer Family Foundations.
Disclosures
Dr Hare reported having a patent for cardiac cell‐based therapy. He holds equity in Vestion Inc. and maintains a professional relationship with Vestion Inc. as a consultant and member of the Board of Directors and Scientific Advisory Board. Dr Hare is the chief scientific officer, a compensated consultant and advisory board member for Longeveron, and equity holder in Longeveron. He is also the coinventor of intellectual property licensed to Longeveron. Longeveron LLC and Vestion Inc. did not participate in funding this work. Dr Hare’s relationships are disclosed to the University of Miami, and a management plan is in place. The remaining authors have no disclosures to report.
Supporting information
Tables S1–S3
Figures S1–S3
Acknowledgments
We acknowledge Vania Almeida and the UM Transmission Electron Microscopy Core, Dr Wen Ding for urine collection and Infant Kidney Project for collection of human placentas.
Dr Kulandevlu conceived, designed, and executed the experiments and wrote the manuscript. Drs Dulce, Bellio, Fritsch, Kanashiro‐Takeuchi, Paulino, and Arora performed experiments. Drs Murray, Soetkamp, and Van Eyk performed mass spectrometry experiment and analysis. Dr Balkan assisted in manuscript writing. Dr Hare conceived of and designed experiments, cowrote the manuscript, and provided funding.
Preprint posted on BioRxiv June 21, 2021. doi: https://doi.org/10.1101/2020.07.01.183012.
Supplemental Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.024008
For Sources of Funding and Disclosures, see page 17.
References
- 1. Sibai B, Dekker G, Kupferminc M. Pre‐eclampsia. Lancet. 2005;365:785–799. doi: 10.1016/S0140-6736(05)17987-2 [DOI] [PubMed] [Google Scholar]
- 2. Ives CW, Sinkey R, Rajapreyar I, Tita ATN, Oparil S. Preeclampsia‐pathophysiology and clinical presentations: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2020;76:1690–1702. doi: 10.1016/j.jacc.2020.08.014 [DOI] [PubMed] [Google Scholar]
- 3. Creanga AA, Syverson C, Seed K, Callaghan WM. Pregnancy‐related mortality in the United States, 2011–2013. Obstet Gynecol. 2017;130:366–373. doi: 10.1097/AOG.0000000000002114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Molina RL, Pace LE. A renewed focus on maternal health in the United States. N Engl J Med. 2017;377:1705–1707. doi: 10.1056/NEJMp1709473 [DOI] [PubMed] [Google Scholar]
- 5. Al‐Gubory KH, Fowler PA, Garrel C. The roles of cellular reactive oxygen species, oxidative stress and antioxidants in pregnancy outcomes. Int J Biochem Cell Biol. 2010;42:1634–1650. doi: 10.1016/j.biocel.2010.06.001 [DOI] [PubMed] [Google Scholar]
- 6. Tyurin VA, Liu SX, Tyurina YY, Sussman NB, Hubel CA, Roberts JM, Taylor RN, Kagan VE. Elevated levels of S‐nitrosoalbumin in preeclampsia plasma. Circ Res. 2001;88:1210–1215. doi: 10.1161/hh1101.092179 [DOI] [PubMed] [Google Scholar]
- 7. Gandley RE, Tyurin VA, Huang W, Arroyo A, Daftary A, Harger G, Jiang J, Pitt B, Taylor RN, Hubel CA, et al. S‐nitrosoalbumin‐mediated relaxation is enhanced by ascorbate and copper: effects in pregnancy and preeclampsia plasma. Hypertension. 2005;45:21–27. doi: 10.1161/01.HYP.0000150158.42620.3e [DOI] [PubMed] [Google Scholar]
- 8. Webster RP, Roberts VH, Myatt L. Protein nitration in placenta – functional significance. Placenta. 2008;29:985–994. doi: 10.1016/j.placenta.2008.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Possomato‐Vieira JS, Khalil RA. Mechanisms of endothelial dysfunction in hypertensive pregnancy and preeclampsia. Adv Pharmacol. 2016;77:361–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Osol G, Ko NL, Mandala M. Altered endothelial nitric oxide signaling as a paradigm for maternal vascular maladaptation in preeclampsia. Curr Hypertens Rep. 2017;19:82. doi: 10.1007/s11906-017-0774-6 [DOI] [PubMed] [Google Scholar]
- 11. Stamler JS, Jaraki O, Osborne J, Simon DI, Keaney J, Vita J, Singel D, Valeri CR, Loscalzo J. Nitric oxide circulates in mammalian plasma primarily as an S‐nitroso adduct of serum albumin. Proc Natl Acad Sci USA. 1992;89:7674–7677. doi: 10.1073/pnas.89.16.7674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Foster MW, Pawloski JR, Singel DJ, Stamler JS. Role of circulating S‐nitrosothiols in control of blood pressure. Hypertension. 2005;45:15–17. doi: 10.1161/01.HYP.0000150160.41992.71 [DOI] [PubMed] [Google Scholar]
- 13. Harris LK, McCormick J, Cartwright JE, Whitley GS, Dash PR. S‐nitrosylation of proteins at the leading edge of migrating trophoblasts by inducible nitric oxide synthase promotes trophoblast invasion. Exp Cell Res. 2008;314:1765–1776. doi: 10.1016/j.yexcr.2008.02.010 [DOI] [PubMed] [Google Scholar]
- 14. Iyer AK, Rojanasakul Y, Azad N. Nitrosothiol signaling and protein nitrosation in cell death. Nitric Oxide. 2014;42:9–18. doi: 10.1016/j.niox.2014.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lima B, Forrester MT, Hess DT, Stamler JS. S‐nitrosylation in cardiovascular signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, et al. Essential roles of S‐nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/S0092-8674(04)00131-X [DOI] [PubMed] [Google Scholar]
- 17. Beigi F, Gonzalez DR, Minhas KM, Sun Q‐A, Foster MW, Khan SA, Treuer AV, Dulce RA, Harrison RW, Saraiva RM, et al. Dynamic denitrosylation via S‐nitrosoglutathione reductase regulates cardiovascular function. Proc Natl Acad Sci USA. 2012;109:4314–4319. doi: 10.1073/pnas.1113319109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hatzistergos KE, Paulino EC, Dulce RA, Takeuchi LM, Bellio MA, Kulandavelu S, Cao Y, Balkan W, Kanashiro‐Takeuchi RM, Hare JM. S‐nitrosoglutathione reductase deficiency enhances the proliferative expansion of adult heart progenitors and myocytes post myocardial infarction. J Am Heart Assoc. 2015;4:e001974. doi: 10.1161/JAHA.115.001974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O'Rourke B, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a [DOI] [PubMed] [Google Scholar]
- 20. Wei W, Li B, Hanes MA, Kakar S, Chen X, Liu L. S‐nitrosylation from GSNOR deficiency impairs DNA repair and promotes hepatocarcinogenesis. Sci Transl Med. 2010;2:19ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonzalez DR, Treuer AV, Castellanos J, Dulce RA, Hare JM. Impaired S‐nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J Biol Chem. 2010;285:28938–28945. doi: 10.1074/jbc.M110.154948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khan SA, Skaf MW, Harrison RW, Lee K, Minhas KM, Kumar A, Fradley M, Shoukas AA, Berkowitz DE, Hare JM. Nitric oxide regulation of myocardial contractility and calcium cycling: independent impact of neuronal and endothelial nitric oxide synthases. Circ Res. 2003;92:1322–1329. doi: 10.1161/01.RES.0000078171.52542.9E [DOI] [PubMed] [Google Scholar]
- 23. Crow JP. Dichlorodihydrofluorescein and dihydrorhodamine 123 are sensitive indicators of peroxynitrite in vitro: implications for intracellular measurement of reactive nitrogen and oxygen species. Nitric Oxide. 1997;1:145–157. doi: 10.1006/niox.1996.0113 [DOI] [PubMed] [Google Scholar]
- 24. Strijdom H, Muller C, Lochner A. Direct intracellular nitric oxide detection in isolated adult cardiomyocytes: flow cytometric analysis using the fluorescent probe, diaminofluorescein. J Mol Cell Cardiol. 2004;37:897–902. doi: 10.1016/j.yjmcc.2004.05.018 [DOI] [PubMed] [Google Scholar]
- 25. Kim H, Bae S, Kim Y, Cho CH, Kim SJ, Kim YJ, Lee SP, Kim HR, Hwang YI, Kang JS, et al. Vitamin C prevents stress‐induced damage on the heart caused by the death of cardiomyocytes, through down‐regulation of the excessive production of catecholamine, TNF‐alpha, and ROS production in Gulo(‐/‐)Vit C‐Insufficient mice. Free Radic Biol Med. 2013;65:573–583. [DOI] [PubMed] [Google Scholar]
- 26. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta‐Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stillman IE, Karumanchi SA. The glomerular injury of preeclampsia. J Am Soc Nephrol: JASN. 2007;18:2281–2284. doi: 10.1681/ASN.2007020255 [DOI] [PubMed] [Google Scholar]
- 28. Kronborg C, Vittinghus E, Allen J, Knudsen UB. Excretion patterns of large and small proteins in pre‐eclamptic pregnancies. Acta Obstet Gynecol Scand. 2011;90:897–902. doi: 10.1111/j.1600-0412.2011.01164.x [DOI] [PubMed] [Google Scholar]
- 29. Novelli GP, Valensise H, Vasapollo B, Larciprete G, Altomare F, Di Pierro G, Casalino B, Galante A, Arduini D. Left ventricular concentric geometry as a risk factor in gestational hypertension. Hypertension. 2003;41:469–475. doi: 10.1161/01.HYP.0000058001.67791.0A [DOI] [PubMed] [Google Scholar]
- 30. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351–356. doi: 10.1038/s41586-019-1100-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karsdorp VH, van Vugt JM, van Geijn HP, Kostense PJ, Arduini D, Montenegro N, Todros T. Clinical significance of absent or reversed end diastolic velocity waveforms in umbilical artery. Lancet. 1994;344:1664–1668. doi: 10.1016/S0140-6736(94)90457-X [DOI] [PubMed] [Google Scholar]
- 32. Groten T, Gebhard N, Kreienberg R, Schleussner E, Reister F, Huppertz B. Differential expression of VE‐cadherin and VEGFR2 in placental syncytiotrophoblast during preeclampsia–new perspectives to explain the pathophysiology. Placenta. 2010;31:339–343. doi: 10.1016/j.placenta.2010.01.014 [DOI] [PubMed] [Google Scholar]
- 33. Grummer MA, Sullivan JA, Magness RR, Bird IM. Vascular endothelial growth factor acts through novel, pregnancy‐enhanced receptor signalling pathways to stimulate endothelial nitric oxide synthase activity in uterine artery endothelial cells. Biochem J. 2009;417:501–511. doi: 10.1042/BJ20081013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sanchez‐Aranguren LC, Prada CE, Riano‐Medina CE, Lopez M. Endothelial dysfunction and preeclampsia: role of oxidative stress. Front Physiol. 2014;5:372. doi: 10.3389/fphys.2014.00372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Phipps EA, Thadhani R, Benzing T, Karumanchi SA. Pre‐eclampsia: pathogenesis, novel diagnostics and therapies. Nat Rev Nephrol. 2019;15:275–289. doi: 10.1038/s41581-019-0119-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, Steenbergen C. Simultaneous measurement of protein oxidation and S‐nitrosylation during preconditioning and ischemia/reperfusion injury with resin‐assisted capture. Circ Res. 2011;108:418–426. doi: 10.1161/CIRCRESAHA.110.232173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly‐S‐nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234 [DOI] [PubMed] [Google Scholar]
- 38. Hubel CA, Kagan VE, Kisin ER, McLaughlin MK, Roberts JM. Increased ascorbate radical formation and ascorbate depletion in plasma from women with preeclampsia: implications for oxidative stress. Free Radic Biol Med. 1997;23:597–609. doi: 10.1016/S0891-5849(97)00010-5 [DOI] [PubMed] [Google Scholar]
- 39. Xu A, Vita JA, Keaney JF Jr. Ascorbic acid and glutathione modulate the biological activity of S‐nitrosoglutathione. Hypertension. 2000;36:291–295. [DOI] [PubMed] [Google Scholar]
- 40. Chen X, Touyz RM, Park JB, Schiffrin EL. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke‐prone SHR. Hypertension. 2001;38:606–611. doi: 10.1161/hy09t1.094005 [DOI] [PubMed] [Google Scholar]
- 41. Groebler LK, Wang XS, Kim HB, Shanu A, Hossain F, McMahon AC, Witting PK. Cosupplementation with a synthetic, lipid‐soluble polyphenol and vitamin C inhibits oxidative damage and improves vascular function yet does not inhibit acute renal injury in an animal model of rhabdomyolysis. Free Radic Biol Med. 2012;52:1918–1928. doi: 10.1016/j.freeradbiomed.2012.02.011 [DOI] [PubMed] [Google Scholar]
- 42. Chung HS, Murray CI, Van Eyk JE. A proteomics workflow for dual labeling biotin switch assay to detect and quantify protein S‐nitroylation. Methods Mol Biol. 2018;1747:89–101. doi: 10.1007/978-1-4939-7695-9_8 [DOI] [PubMed] [Google Scholar]
- 43. Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S‐nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007;282:13977–13983. doi: 10.1074/jbc.M609684200 [DOI] [PubMed] [Google Scholar]
- 44. Chung HS, Murray CI, Venkatraman V, Crowgey EL, Rainer PP, Cole RN, Bomgarden RD, Rogers JC, Balkan W, Hare JM, et al. Dual labeling biotin switch assay to reduce bias derived from different cysteine subpopulations: a method to maximize S‐nitrosylation detection. Circ Res. 2015;117:846–857. doi: 10.1161/CIRCRESAHA.115.307336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Massy ZA, Fumeron C, Borderie D, Tuppin P, Nguyen‐Khoa T, Benoit MO, Jacquot C, Buisson C, Drueke TB, Ekindjian OG, et al. Increased plasma S‐nitrosothiol concentrations predict cardiovascular outcomes among patients with end‐stage renal disease: a prospective study. J Am Soc Nephrol: JASN. 2004;15:470–476. doi: 10.1097/01.asn.0000106716.22153.bb [DOI] [PubMed] [Google Scholar]
- 46. Raijmakers MT, Zusterzeel PL, Steegers EA, Hectors MP, Demacker PN, Peters WH. Plasma thiol status in preeclampsia. Obstet Gynecol. 2000;95:180–184. [DOI] [PubMed] [Google Scholar]
- 47. Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–626. doi: 10.1038/35054560 [DOI] [PubMed] [Google Scholar]
- 48. Hlaing KH, Clement MV. Formation of protein S‐nitrosylation by reactive oxygen species. Free Radical Res. 2014;48:996–1010. doi: 10.3109/10715762.2014.942842 [DOI] [PubMed] [Google Scholar]
- 49. Gomes SA, Rangel EB, Premer C, Dulce RA, Cao Y, Florea V, Balkan W, Rodrigues CO, Schally AV, Hare JM. S‐nitrosoglutathione reductase (GSNOR) enhances vasculogenesis by mesenchymal stem cells. Proc Natl Acad Sci USA. 2013;110:2834–2839. doi: 10.1073/pnas.1220185110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eddy AC, Bidwell GL III, George EM. Pro‐angiogenic therapeutics for preeclampsia. Biol Sex Differ. 2018;9:36. doi: 10.1186/s13293-018-0195-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cindrova‐Davies T, Sanders DA, Burton GJ, Charnock‐Jones DS. Soluble FLT1 sensitizes endothelial cells to inflammatory cytokines by antagonizing VEGF receptor‐mediated signalling. Cardiovasc Res. 2011;89:671–679. doi: 10.1093/cvr/cvq346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCance DR, Holmes VA, Maresh MJ, Patterson CC, Walker JD, Pearson DW, Young IS, Diabetes and Pre‐eclampsia Intervention Trial Study G . Vitamins C and E for prevention of pre‐eclampsia in women with type 1 diabetes (DAPIT): a randomised placebo‐controlled trial. Lancet. 2010;376:259–266. doi: 10.1016/S0140-6736(10)60630-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Poston L, Briley AL, Seed PT, Kelly FJ, Shennan AH, and Vitamins in Pre‐eclampsia Trial C . Vitamin C and vitamin E in pregnant women at risk for pre‐eclampsia (VIP trial): randomised placebo‐controlled trial. Lancet. 2006;367:1145–1154. doi: 10.1016/S0140-6736(06)68433-X [DOI] [PubMed] [Google Scholar]
- 54. Roberts JM, Myatt L, Spong CY, Thom EA, Hauth JC, Leveno KJ, Pearson GD, Wapner RJ, Varner MW, Thorp JM, et al. Vitamins C and E to prevent complications of pregnancy‐associated hypertension. New Engl J Med. 2010;362:1282–1291. doi: 10.1056/NEJMoa0908056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roberts JM. The perplexing pregnancy disorder preeclampsia: what next? Physiol Genomics. 2018;50:459–467. doi: 10.1152/physiolgenomics.00017.2018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Figures S1–S3