

Abstract
Decoding the epigenomic landscapes in diverse tissues and cell types is fundamental to understanding molecular mechanisms underlying many essential cellular processes and human diseases. Recent advances in artificial intelligence provide new methods and strategies for imputing unknown epigenomes based on existing data, yet how to reveal the predictive relationships among epigenetic marks remains largely unexplored. Here we present a machine learning approach for epigenomic imputation and interpretation. Through dissection of the spatial contributions from six histone marks, we reveal the prevalent and asymmetric cross-prediction relationships among these marks. Meanwhile, our approach achieved high predictive performance on held-out prospective epigenomes and outperformed the state-of-the-art. To facilitate future research, we further applied this approach to impute a total of 527 and 2,455 unavailable genome-wide histone modification signal tracks for the ENCODE3 and Roadmap datasets, respectively.
Keywords: Epigenome, Machine Learning, Histone Modification
Introduction
Biomolecules cooperatively orchestrate a variety of cellular processes through decoding genetic information from the human genome, epigenetically modifying DNAs and chromosomes, and chemophysically interacting with each other to catalyze metabolic reactions 1–4. These cellular processes underlie the molecular mechanisms of tissue-specific gene expression and complex human diseases 5–8. To understand the genome-wide signals associated with diverse tissues and cell types, the Encyclopedia of DNA Elements (ENCODE) 9,10 and Roadmap 11 Epigenomics Consortiums have systematically characterized the in vivo biochemical signatures including histone modification, chromatin accessibility, and DNA methylation. These data collections have provided us invaluable insights into the functions and regulations of epigenetic marks under different tissue and cellular conditions.
However, our available information is far from complete and even for these two comprehensive datasets, only a subset of all possible mark-cell type combinations were measured experimentally owing to resource and sample availability. An alternative is building in silico models to impute unknown epigenomic profiles based on existing ones. Epigenomic imputation methods, e.g. ChromImpute 12, PREDICTD 13, and Avocado 14, have been created to address this problem, aiming at accurate and precise imputation of missing data. In addition to pursuing higher predictive performance on epigenomic imputation, a major challenge is how to understand an algorithm and reveal the underlying biological insights from a new computational perspective. Approaches have been developed to define chromatin states associated with cell-type specific biological activities 15–17. Yet the modulatory relationships and interplays among a multitude of epigenetic marks in computational models remain largely unexplored, especially given the ever-growing epigenomics datasets.
In this work, we present an interpretable machine learning approach, Ocelot (Optimized Complementation of Epigenomes by neuraL netwOrk and Tree-based modeling), for predicting the epigenomes across cell types. Ocelot leverages both the cutting-edge tree-based and deep learning models to exploit available data. It integrates information from mark-specific signals across cell types, cell type-specific signals across epigenetic marks, and DNA sequence to impute epigenomic signals. Meanwhile, the neighboring information around the center of interest is considered to improve the prediction accuracy. This approach ranked first in the recent ENCODE Imputation Challenge, in which computational methods were developed and comprehensively evaluated on a large testing dataset of 51 held-out prospective epigenomes. Moreover, we investigated the cross-histone modulation patterns in Ocelot based on game theory analysis, and revealed the asymmetric predictive relationships among six representative histone marks. Finally, to facilitate potential research that requires complete epigenomes, we further applied our approach to impute 527 (51.31%) missing entries in the ENCODE3 dataset of 13 histone marks in 79 cell types and 2,455 (71.60%) missing entries in the Roadmap Epigenomics dataset of 27 histone marks in 127 cell types, corresponding to a total of 361 billion predicted values.
Results
Overview of experimental design
Key challenges in epigenome imputation include how to exploit existing data to improve imputation accuracy and reveal the predictive relationships among epigenetic marks. We address this problem by developing tree-based lightGBM 18 and deep learning models that integrate signal tracks of multiple epigenetic marks across tissues and cell types (Fig. 1a). For each mark-cell type combination, e.g. CiMj (cell type Ci and mark Mj in Fig. 1b), we leverage available information from other cell types of the same mark (row Ci), and other marks in the same cell type (column Mj). To understand the regulatory relationships across marks in Ocelot, we calculate the spatial contribution of each mark to predicting every other mark, where the upstream and downstream neighboring genomic regions are considered as well (Fig. 1c). Then the “predicting” and “being predicted” strengths of each pair of marks are summarized to reveal the cross-prediction patterns among marks (Fig. 1d). Finally, to evaluate the predictive performance, our in silico imputations are compared with held-out in vivo measurements (Fig. 1e). Multiple scoring metrics are used to comprehensively evaluate the quality of imputation, including global correlations and mean squared errors (MSEs), and local MSEs and overlapping for the top 1% − 5% regions (Fig. 1f).
Figure 1: Overview of experimental design.
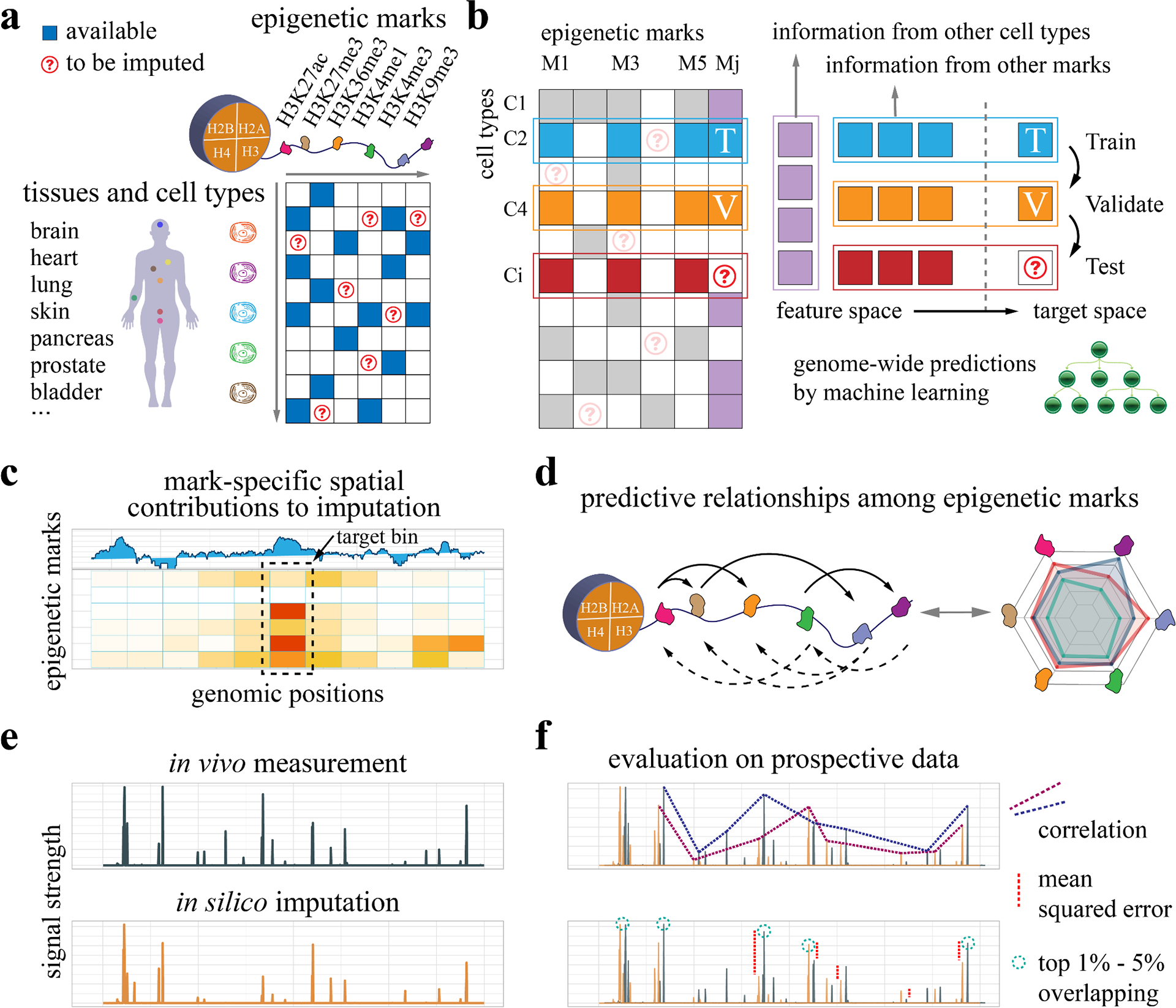
a, We develop machine learning models to impute genome-wide epigenetic signal tracks across cell types and investigate the regulatory relationships among epigenetic marks based on available data. b, For each cell type-mark combination to be imputed, we consider both the information from other cell types of the same mark and the information from other marks in the same cell type. The available data are partitioned into the training and validation sets to build machine learning models for epigenome imputation. c, To investigate the cross-prediction relationships among epigenetic marks, we dissect the machine learning models and extract the spatial contribution of each feature epigenetic mark to a target mark. d, The pairwise modulatory relationships among marks are summarized. The relationships are directional and asymmetric, where the solid and dashed arrows represent predicting others and being predicted respectively. e, The in silico imputation from our approach is compared with held-out in vivo measurement, which is collected prospectively to avoid information leakage or overfitting. f, The imputed data are compared with observed data based on multiple evaluation metrics, including correlations, mean squared errors (MSEs), and overlapping for the top 1% - 5% regions.
In the tree-based lightGBM model, we consider information from both epigenomic tracks and DNA sequences (Fig. 2a). Specifically, for each 25bp target bin under consideration, we extract information from the neighboring 275bp = 11 bins × 25bp regions. We condense the information and define the “MMMN” features, which are the (1) Maximum, (2) Minimum, (3) Mean of each 25bp bin, and (4) the Number of unique values within each 25bp bin in the epigenomic tracks. To reduce the potential effect of sequencing biases in a specific cell type, we further define the “ΔMMMN” features, which are calculated from the Δsignal track (the difference between this track and the average signal across cell types). When Ntrack is the total number of cell type-specific and mark-specific feature tracks, the total number of epigenetic features will be 11 bins × 8 MMMN-ΔMMMN features × Ntrack. In addition to epigenomic tracks, we also considered the DNA sequence by one-hot encoding three 25bp bins, resulting in another 300 features = 3 bins × 25bp × 4 nucleotides. Finally, these two types of features are used to train a lightGBM model, which predicts the one target value for the target bin.
Figure 2: Schematic illustration of the tree-based model and neural network model design.
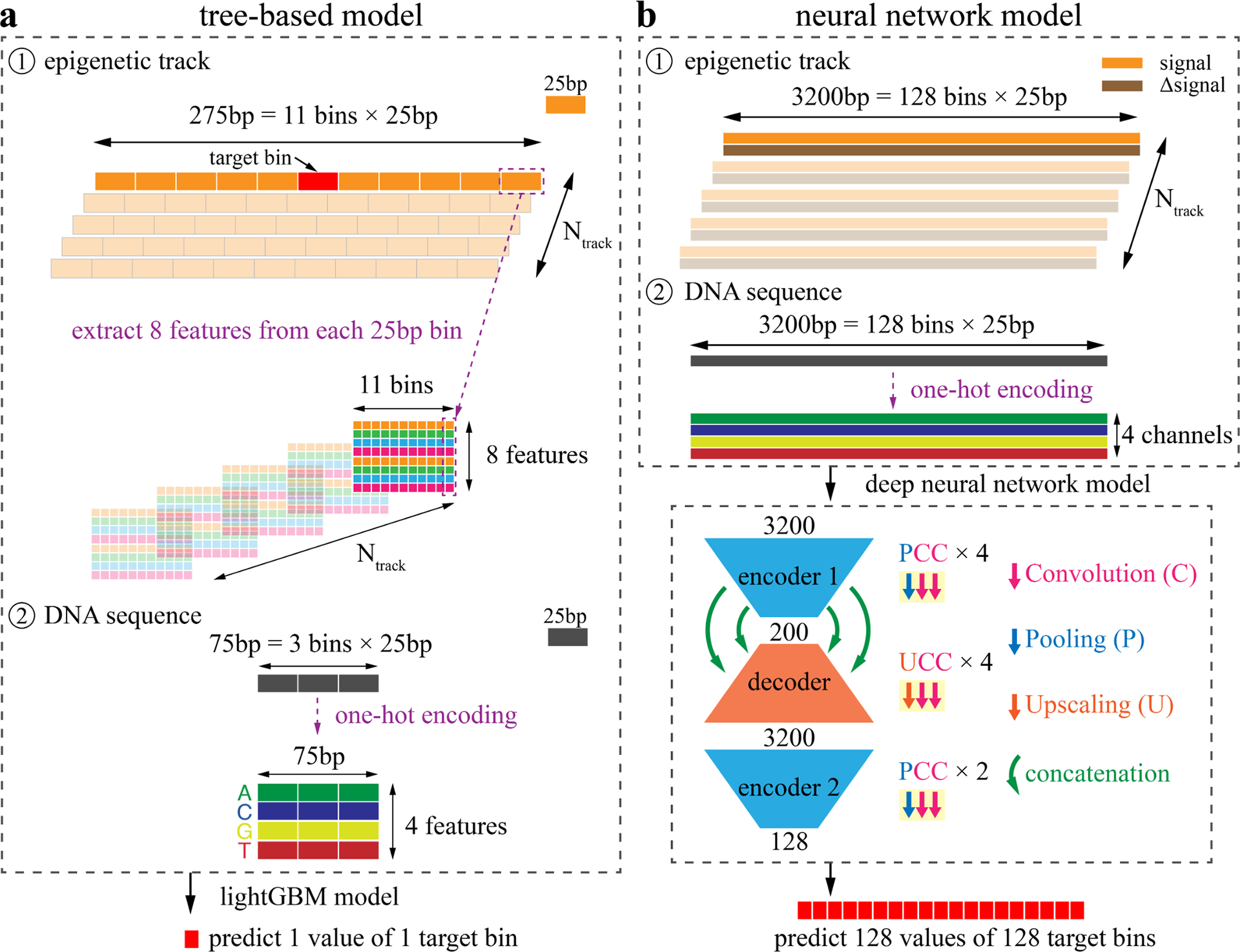
a, We first build a tree-based model through extracting features from both epigenomic tracks and DNA sequences. For each 25bp bin to be predicted, we consider the 5 upstream and 5 downstream bins to extract the neighboring information. For each 25bp bin, we calculate the (1) Maximum, (2) Minimum, (3) Mean, and (4) the Number of unique values as the “MMMN” features. Then for these 4 features, we further calculate the difference between this track and the average values across cell types, resulting in another 4 features - the “ΔMMMN” features. When Ntrack is the number of entries that are treated as feature entries, a total of 11×8×Ntrack feature values are extracted from the epigenetic tracks. In addition, the DNA sequences from the three 25bp bins are one-hot encoded into another 300 = 3×25×4 features. Then all these features are used to build a lightGBM model to predict one value of the target bin. b, In the neural network model, the signal tracks are directly used as inputs without feature extraction. Specifically, for each epigenomic entry treated as a feature entry, both the signal and Δsignal (the difference between this track and the average track across cell types) tracks are considered as two channels. The input length is 3200bp = 128 bins × 25bp. Then the DNA sequence is one-hot encoded into 4 nucleotide channels. When Ntrack tracks are considered as feature entries, the number of channels is (2×Ntrack + 4). Then we build a deep convolutional neural network with two encoders and one decoder. The building block of the encoders is Pooling-Convolution-Convolution (PCC) layers. There are four and two PCC blocks in encoder 1 and encoder 2, respectively. The building block of the decoder is Upscaling-Convolution-Convolution (UCC) layers and four UCC blocks are used in the decoder. The encoder 1 and decoder are further connected with concatenation layers to reduce information decay. Finally, 128 values are predicted simultaneously, corresponding to the 128 bins of the input.
Regarding the neural network, a commonly used design in functional genomics and epigenomics is the end-to-end model, which accepts a genomic region of multiple positions as the input and predicts one target value. This design works well when the number of input channels is small (e.g. models using DNA sequence only) or the depth of the network structure is relatively shallow. However, when the neural network becomes deep and complex (e.g. millions of parameters) with large numbers of input channels, the end-to-end model will become extremely time-consuming, especially for tasks of genome-wide predictions. A time-efficient alternative with high accuracy is the many-to-many model widely used in image segmentation, which simultaneously outputs multiple predictions 19. For this epigenomic imputation task, another challenge is the resolution shift from the input 1bp resolution to the output 25bp resolution. We therefore design a special many-to-many deep neural network model, which automatically makes predictions for multiple bins at 25bp resolution. This model considers long-range upstream and downstream information, with the input length of 3200bp = 128 bins × 25bp (Fig. 2b). We directly use the signal tracks as inputs without any feature extraction. Similar to the tree-based model, we also use Δsignal tracks as extra channels. Moreover, the DNA sequence is one-hot encoded into another four channels, corresponding to four types of nucleotides. The total number of channels will be 2 × Ntrack + 4, where Ntrack is the number of cell type-specific and mark-specific feature tracks. Based on these inputs, we build a deep convolutional neural network model that has two encoders and one decoder. We first define two types of building blocks: (1) the encoder block of Pooling-Convolution-Convolution (PCC) layers, and (2) the decoder block of Upscaling-Convolution-Convolution (UCC) layers. The encoder gradually decreases the length of the input and increases the number of channels, whereas the decoder works in an opposite way. Four and two PCC blocks are used in the encoder 1 and encoder 2, respectively. And four UCC blocks are used in the decoder. To alleviate the information decay issue of deep neural networks, we further added concatenation layers between the encoder 1 and the decoder. Finally, this neural network simultaneously outputs 128 values, corresponding to the input 128 bins.
Ocelot reveals spatial regulatory relationships among epigenetic marks
To investigate the predictive relationships among histone marks in Ocelot, for each histone mark pair (mark A and mark B), we calculated the pairwise Shapley values to interpret predictions 20,21. Specifically, we analyzed two types of models: (1) using “predictor” mark A as a feature to predict “target” mark B, and (2) using mark B to predict mark A. The pairwise absolute SHAP values of six representative histone marks (H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me3, H3K9me3) are shown as heatmaps in Fig. 3a, where the predictor marks are listed vertically on the left and the target marks are listed horizontally on the top. These heatmaps were created from 10 bootstraps and the original SHAP values of each bootstrap are shown in Supplementary Table 1 (see details in Methods – SHAP analysis). In each model, we extracted features from the neighboring genomic regions around the center bin of interest: every non-overlapping 25bp bin from upstream −125bp to downstream 125bp. These 11 bins are shown along the x-axis in each heatmap, representing the spatial distribution of contributions. In general, the high absolute SHAP value (red) of a feature indicates its relatively large contribution to the prediction. For most predictor-target pairs, the strongest feature resides in the center and the importance gradually decreases in the distal intervals. This indicates that in addition to the immediate genomic interval of interest, the epigenetic signals in neighboring bins also contribute to epigenomic imputation. For example, considering the H3K27ac-H3K4me3 pair, there are multiple contributing features distributed in the neighboring region in addition to the center bin.
Figure 3: Ocelot reveals the asymmetric and spatial cross-regulation of multiple histone modifications in epigenome imputation.
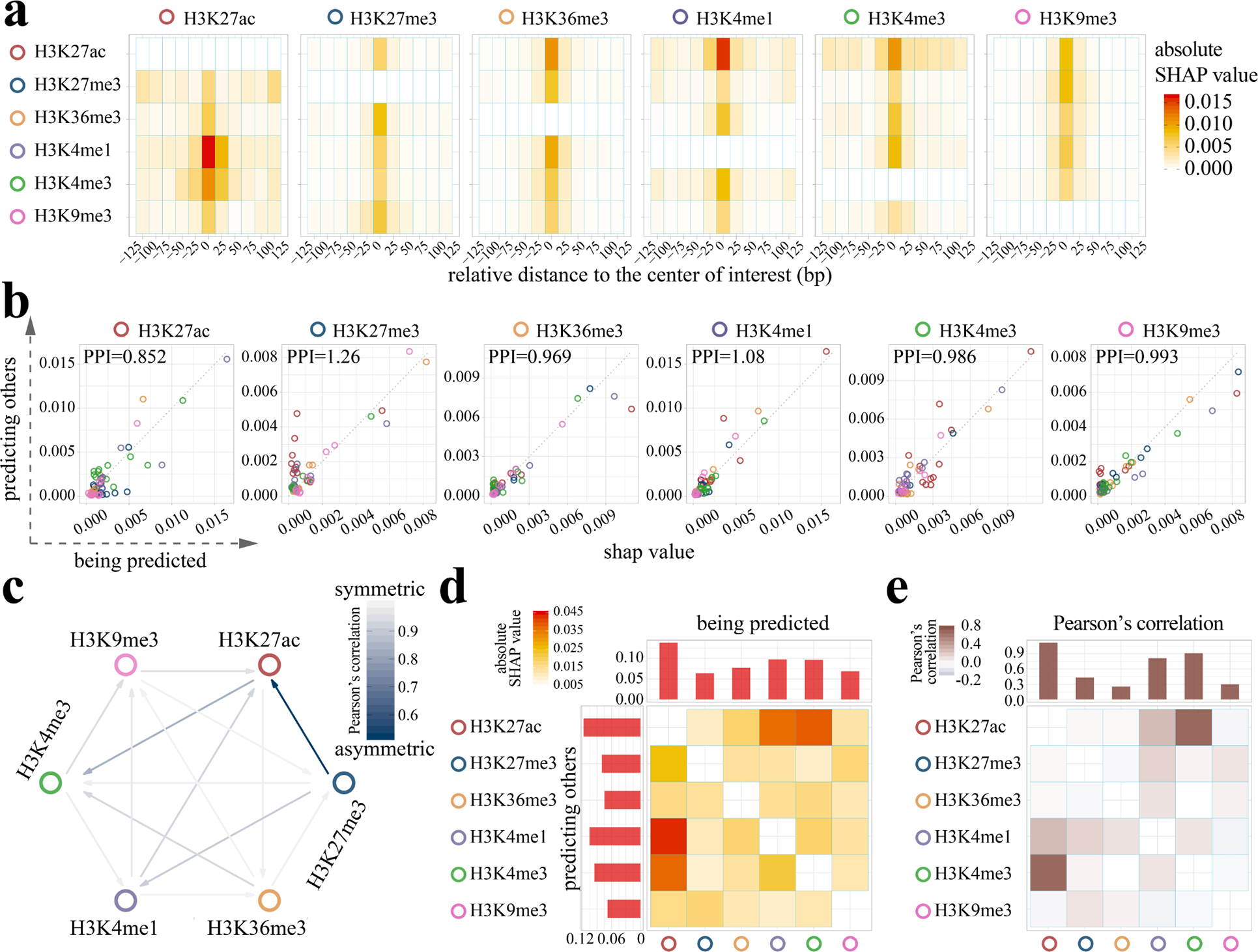
a, The SHAP analysis was performed on Ocelot models to reveal the pairwise cross-regulation between six histone modifications represented as six heatmaps. The histone marks along the six heatmap rows serve as predictors to predict other marks, whereas the marks along the column are the target to be predicted. Each row in a heatmap has 11 positions, covering the upstream −125bp to downstream +125bp around the center of the target 25bp bin to be predicted. High SHAP values are shown in red. For example, in the first heatmap the H3K4me1 row has a high SHAP value (the red block) in the center, indicating that H3K4me1 largely contributes to the prediction of H3K27ac at the center position. b, The pairwise comparison of SHAP values between two scenarios: (1) using mark A to predict mark B and (2) using mark B to predict mark A. For each histone mark, we compare it with the other 5 marks and represent these SHAP values as circles. The colors represent the other marks. For each color, there are 11 circles, corresponding to the 25bp bins around the center bin of interest in panel a. For example, in the first scatter plot, if a circle is above the diagonal dashed line, it indicates that H3K27ac has larger predictive power (higher SHAP values) as features in predicting the other marks. We define an indicator, Predictive Power Index (PPI), which is the ratio of the average SHAP value when this mark predicts others over the average SHAP value when other marks predict this mark. c, We further calculate Pearson’s correlation of SHAP values between (1) using mark A to predict mark B and (2) using mark B to predict mark A in all histone mark pairs. Lower correlation (dark blue) indicates higher level of asymmetry in cross-regulation and the direction of stronger prediction power is represented by the arrow. d, In addition to analysis at the 25bp bin level, the SHAP values of 11 bins in panel a are summed to obtain the matrix at the histone mark level. Each row and column of this matrix are also summed to obtain the accumulated SHAP values of predicting other marks (the bar plot on the top) and being predicted by other marks (the bar plot on the left). This matrix is asymmetric and directional. e, We also calculated the pairwise correlation among histone marks based on the average signal tracks from all training cell types. The accumulated correlations are shown as bars on the top. This correlation matrix is symmetric and undirected.
We further analyze the overall prediction contribution of a specific mark against all other marks (Fig. 3b). For each mark under consideration, we present all the pairwise SHAP values in Fig. 3a as colored circles when treating it as the predictor (y-axis) or the target (x-axis). If a circle is above the diagonal dashed line, it indicates that this specific mark has larger predictive power (higher absolute SHAP values) as features in predicting the other target mark. If the predictor-target relationship is symmetric, the circles should distribute along the diagonal. In fact, many predictor-target pairs are off the diagonal, indicating the prevalent asymmetric cross-prediction relationships among these histone modifications.
To quantitatively characterize this, we define a statistics, Predictive Power Index (PPI), which is the ratio of the average predictor SHAP value over the average target SHAP value. If PPI of a histone modification is higher than one, it means this histone has higher contributions as a predictor than being a target predicted by others. We find that H3K27me3 participates more as a predictor with a PPI value of 1.26, whereas H3K27ac is more likely being a target with a PPI value of 0.852. The other four histone marks are more balanced serving as both a predictor and a target, with a PPI value ranging from 0.969 and 1.08. We further calculated Pearson’s correlation of SHAP values between (1) using mark A to predict mark B and (2) using mark B to predict mark A in all histone mark pairs (Fig. 3c). Lower correlation (dark blue) indicates higher level of asymmetry in cross-regulation and the direction of stronger predictive power is represented by the arrow. We observe a strong asymmetric relationship between H3K27me3 and H3K27ac, possibly due to the fact that the trimethylation and acetylation can not coincide on the same lysine. In fact, dynamic and reciprocal changes of H3K27me3 and H3K27ac have been observed in the promoter regions of related genes during decidualization 22. The ratio of dimethylation and trimethylation of H3K27 is associated with its acetylation level in embryonic stem cells 23. Compared with these experimental observations, we provide a new computational approach to investigate the complete pairwise relationships among marks with spatial distributions.
In addition to analysis at the bin level, we further summed the SHAP values of all 11 bins and obtained a heatmap matrix at the histone mark level (Fig. 3d). The accumulated values for each row (predicting others) and column (being predicted) are shown as bars on the left and top. Meanwhile, for each histone mark pair, we calculated Pearson’s correlation between the average signals of all available cell types (Fig. 3e). In both matrices, H3K27ac has higher SHAP values or correlations with H3K4me1 and H3K4me3. However, the correlation is symmetric and undirected for a pair of marks A and B, whereas SHAP captures the asymmetric and directional predictive importance, and the feature importance of A predicting B is not equal to the feature importance of B predicting A. For example, the first rows of two matrices in Fig. 3d (SHAP values of H3K27ac predicting other marks) and Fig. 3e (H3K27ac’s correlations with other marks) have a similar pattern and trend. Yet the first columns of two matrices have different patterns - the largest contribution to H3K27ac’s predictions comes from H3K4me1, whereas H3K27ac has the largest correlation with H3K4me3 instead of H3K4me1. Moreover, the SHAP analysis of machine learning models reveal the spatial distributions of contributions, while correlations only reflect the global trends between two signals.
Ocelot achieved high predictive performance on held-out prospective data
To stringently and systematically evaluate the predictive performance of Ocelot, we participated in the recent ENCODE Imputation Challenge, where a large-scale dataset of 363 epigenomes of 35 marks in 51 cell types were used to train and benchmark different methods. Here we first compared Ocelot with a recent deep learning method, Avocado 14, which reported lower imputation MSEs than PREDICTD 13 and ChromImpute 12. Nine scoring metrics were used to thoroughly compare the differences between the imputed and observed data globally and locally on specific peaks or functional regions. The first three metrics are (1) Pearson’s correlation, (2) mean squared error (MSEglobal), and (3) Spearman’s correlation between the predicted and observed values across the entire human genome. The next three metrics are MSEs across genomic regions that are annotated as (4) promoters (MSEProm), (5) genes (MSEGene), and (6) enhancers (MSEEnh). The last three metrics are (7) MSE weighted by the cross-cell-type variance (MSEvar), (8) MSE across genomic regions with top 1% observed values (MSE1obs) and (9) MSE across genomic regions with top 1% predicted values (MSE1imp).
The ENCODE Imputation Challenge blind testing dataset consists of a total of 51 prospective epigenomes covering 8 assays (H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me3, H3K9me3, DNase-seq, and ATAC-seq) in 12 cell types. These 51 testing epigenomes were newly acquired and only released for one-shot evaluation after predictions were made, so that potential information leakage or overfitting was completely avoided. For each testing mark-cell type pair, bootstrap subsampling from the human genome was performed 10 times to calculate scores for 10 subsampled region sets. At the mark level, the comparison of global Pearson’s correlations is shown in Supplementary Fig. 1. The top panel is the scatter plot of Ocelot (y-axis) and Avocado (x-axis). Each square represents a bootstrap subsampled region for a testing cell type. The bottom panel is the density distribution of Ocelot (solid line) and Avocado (dashed line). Since each mark contains multiple testing cell types, there are multiple peaks in the density distribution plot as we expected. For different types of marks, we observe varied correlation scores, reflecting different imputation difficulties. For example, H3K4me3 is relatively easy to predict with the highest Pearson’s correlations of 0.807 and 0.660 for Ocelot and Avocado, respectively. For marks related to the open chromatin, Ocelot also achieved relatively high correlations of 0.683 and 0.623 for ATAC-seq and DNase-seq, respectively. Our predictions for H3K27ac and H3K36me3 have medium correlations around 0.5, whereas we only achieved relatively low correlations for the other three histone modifications (H3K27me3, H3K4me1 and H3K9me3). Similarly, we compared Ocelot and Avocado using the other 8 scoring metrics in Supplementary Fig. 2 – 9. The paired Wilcoxon signed-rank test was used to determine the statistical significance. Overall, at the mark level, Ocelot significantly outperformed Avocado in 62 out of 72 scores (86.1%; 72 scores = 8 marks × 9 scoring metrics). Similarly, at the mark-cell type combination level, Ocelot also significantly outperformed Avocado in 376 out of 459 scores (81.9%; 459 scores = 51 combinations × 9 scoring metrics; Supplementary Fig. 10 – 18). These results demonstrate that Ocelot has considerably advanced the imputation accuracy over Avocado when evaluated on completely unseen prospective data.
In addition to the MSE based evaluation metrics emphasized in Avocado 14, we further considered more non-MSE based metrics proposed in ChromImpute 12. As a pioneering approach, ChromImpute has shown better performance than Avocado on multiple non-MSE based metrics in epigenomics imputation 14. We included three non-MSE based metrics that focus on peak regions: (1) the overlap between the top 1% observed and imputed signals (Match1), (2) the overlap between the top 1% observed and 5% imputed signals (Catch1obs), and (3) the overlap between the top 5% observed and 1% imputed signals (Catch1imp). Now we have 5 non-MSE based metrics and 7 MSE based metrics to compare different methods. We trained ChromImpute on the challenge training data and benchmarked the performance of Ocelot, ChromImpute and Avocado on the challenge held-out testing set of 51 mark-cell type pairs using 12 evaluation metrics. For each testing pair, we calculated genome-wide evaluation scores through concatenating signals of 23 chromosomes. For each metric, we performed paired one-sided Wilcoxon signed-rank tests across 51 testing mark-cell type pairs. The significantly different ones are labeled by asterisks (* p-value < 0.01 and ** p-value < 0.001). In terms of the 5 non-MSE based metrics, Ocelot significantly outperformed ChromImpute in 2 correlation metrics and achieved comparable performance without statistical differences in the other three overlapping metrics (Fig. 4a and Supplementary Table 2). In terms of the 7 MSE based metrics, Ocelot significantly outperformed ChromImpute in 6 metrics except for MSE1obs. We compared Ocelot with Avocado using these metrics as well. Ocelot significantly outperformed Avocado in 9 out of 12 metrics, including 2 correlation metrics, Catch1obs and 6 MSE based metrics (Fig. 4b and Supplementary Table 2). In general, genomic regions of cell type-specific signals have larger cross-cell-type variance than those of constitutive signals. In addition to the global MSE that considers both cell type-specific and constitutive regions, the MSEvar metric (MSE weighted by the cross-cell-type variance) emphasizes more on cell type-specific regions through assigning larger weights to them. In terms of MSEvar, Ocelot significantly outperformed both ChromImpute (Fig. 4a) and Avocado (Fig. 4b).
Figure 4: Predictive performance comparisons between Ocelot, ChromImpute and Avocado.
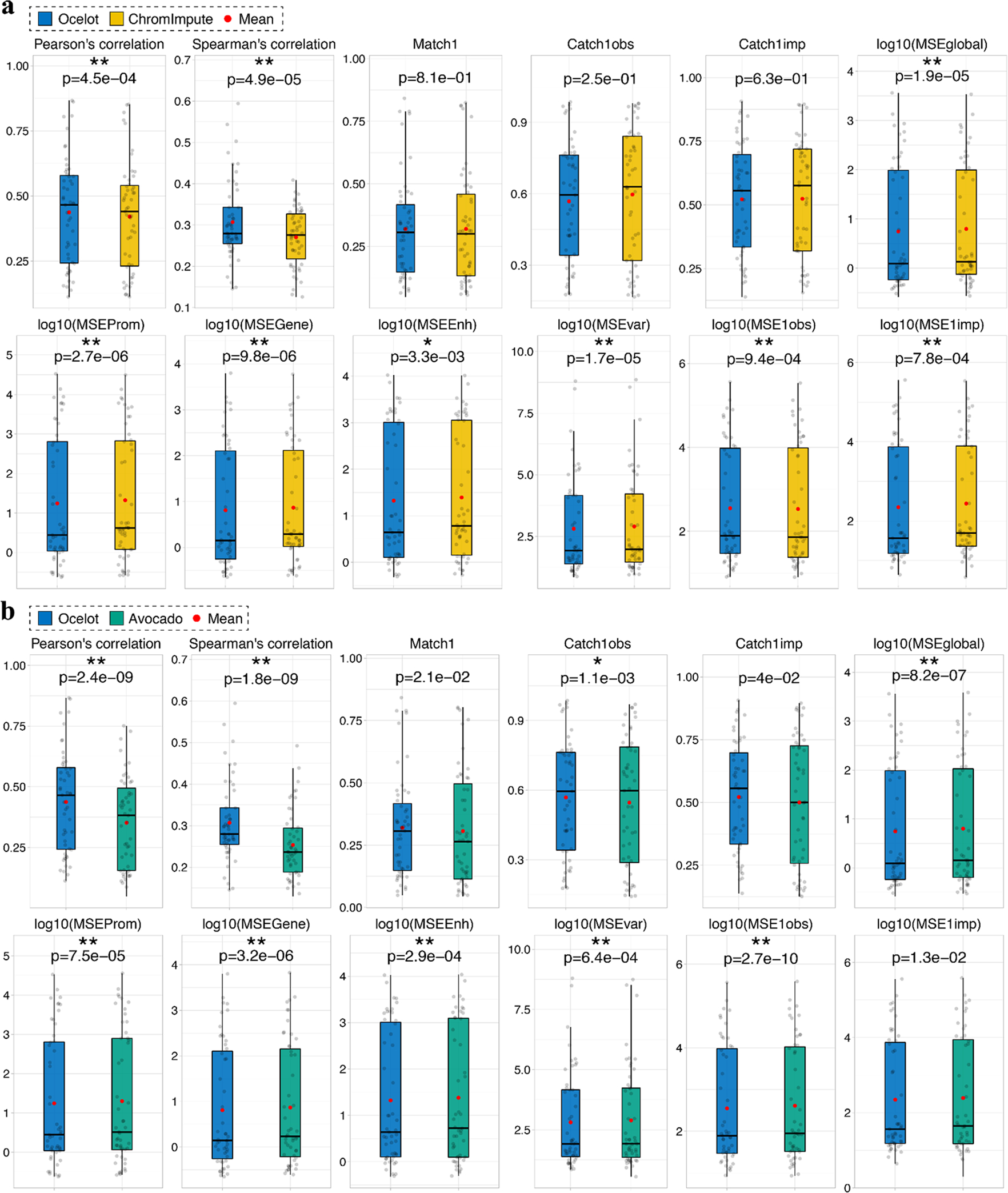
We benchmarked Ocelot with a, ChromImpute and b, Avocado, on 51 genome-wide mark profiles collected prospectively, including multiple histone modifications and chromatin accessibility (DNase-seq and ATAC-seq). Predictive performance was evaluated using 12 scoring metrics on the ENCODE Imputation Challenge testing set of 51 mark-cell type pairs. For each metric, the paired one-sided Wilcoxon signed-rank test was performed between two methods across 51 testing pairs. The significantly different ones are labeled by asterisks (* p-value < 0.01 and ** p-value < 0.001)
To visualize the imputation result together with the mark-specific and cell type-specific signals, we plot an example 200-kbp region of the H3K27ac mark in the WERI-Rb-1 cell line, which was a held-out testing entry in the ENCODE Imputation Challenge (bottom right heatmap in Fig. 5). We first compare the imputed signal and the observed ground truth (top left in Fig. 5), both of which have a high peak in the middle. In general, signals of the same mark across cell types are similar. We find similar central high peaks for this 200-kbp region in most cell types as expected (left in Fig. 5). Yet this peak is missing in several cell types (C17/H1-hESC, C18/H9, C19/HAP-1, and C34/OCI-LY7), which complicates the task of cell type-specific imputation of histone marks. We also compare the six histone marks (H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me3, and H3K9me3), which display diverse patterns (top right in Fig. 5).
Figure 5: An imputation example on the ENCODE Imputation Challenge dataset.
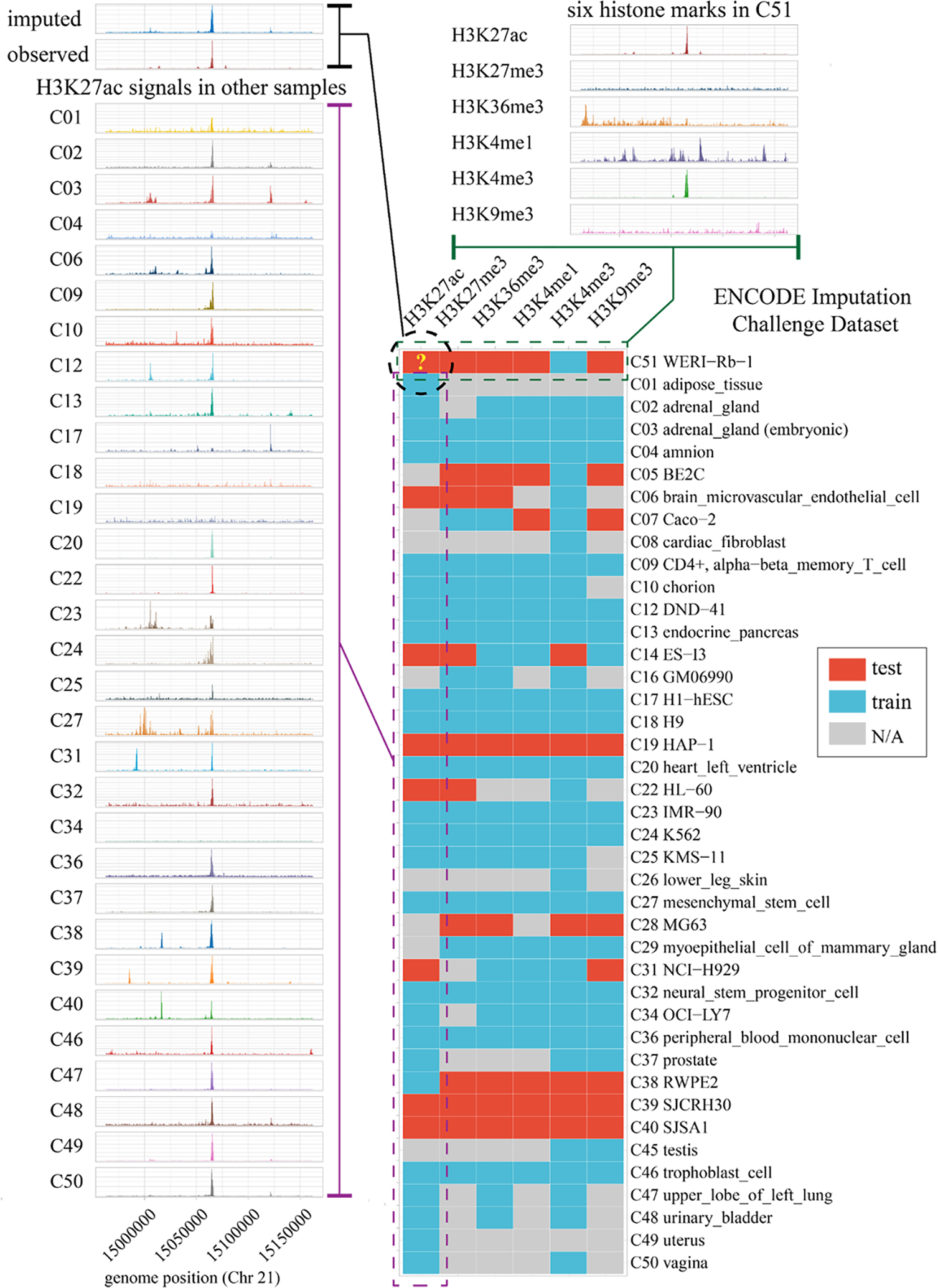
The heatmap is the matrix of the ENCODE Imputation Challenge partial dataset of six histone marks across 41 tissue and cell types that have at least one histone mark as the train or test data (bottom right). The complete challenge data matrix is shown in Supplementary Table 10. A 200-kbp region in Chr 21 of H3K27ac mark in the C51 (WERI-Rb-1) cell line is used to compare our imputation and the held-out observed ground truth (top left). For comparison, the signals of H3K27ac mark in other cell types are shown on the left, most of which are similar to the ground true as expected. In addition, all six marks of the same region in the C51 cell line are shown on top right, which are quite different from each other.
Analysis of key factors that determine predictive performance in Ocelot
To gain more insights into the epigenomic imputation problem, we further investigated factors that are potentially critical in the improved predictive performance of Ocelot. We first studied the effect of involving DNA sequences as input features. In ChromImpute and Avocado, DNA sequences are not used as features, whereas we included DNA in both lightGBM and neural network models in our challenge final submission. We re-trained models without DNA and compared the predictive performance on the same challenge test set using 12 evaluation metrics. In lightGBM, the predictive performances of 5 non-MSE based metrics are comparable between models with and without DNA sequence information (Supplementary Fig. 19 and Supplementary Table 3). Intriguingly, 3 out of 7 MSE based metrics are even better without DNA. In the neural network, only Pearson’s correlation significantly dropped without DNA, yet the change of median value is relatively small from 0.424 to 0.418 (Supplementary Fig. 20 and Supplementary Table 4). In addition, for the lightGBM model with DNA input, we calculated the absolute SHAP values that represent the contributions to predictions from three types of features: (1) DNA sequence, (2) epigenomic signals from other cell types, and (3) epigenomic signals from other marks (Supplementary Fig. 21). The average contribution (absolute SHAP value) per feature from DNA is only between 1.26% and 2.16% in six histone marks. Since the number of DNA features is smaller than the number of epigenomic features, the accumulated contribution from DNA is even smaller. Therefore, the majority (>97%) of contributions come from epigenomic signals in lightGBM models. We further re-ensembled predictions of Ocelot without DNA (Supplementary Fig. 22–23 and Supplementary Table 5) and the results are consistent with Ocelot with DNA (Fig. 4 and Supplementary Table 2). Ocelot still significantly outperformed Avocado in 9 out of 12 metrics and ChromImpute in 8 out of 12 metrics. Therefore, the contribution from DNA sequence is minimal. Ocelot is able to accurately impute epigenomes based on available epigenomic signals without DNA.
In Ocelot, we ensemble (1) predictions from lightGBM models, (2) predictions from neural network models, and (3) the quantile normalized average signal of a mark in all training cell types. The average is a common and straightforward approach in missing data imputation, which generally captures the constituent signals across cell lines. To investigate the relative importance of different machine learning models, we performed ablation experiments by excluding lightGBM models or neural network models. Without lightGBM, the average predictive performance significantly changed for 10 out of 12 metrics, including decreases in 4 non-MSE based metrics (Spearman’s correlation, Match1, Catch1obs, and Catch1imp), and increases in 6 MSE based metrics (Supplementary Fig. 24 and Supplementary Table 6). Without neural network models, the correlations and most MSE based metrics are comparable, whereas Match1, Catch1obs and Catch1imp significantly increase (Supplementary Fig. 25 and Supplementary Table 6). This is mainly due to the relatively poor performance of the neural network in predicting H3K4me1, consistent with the ensemble weights selection and cross-validation results - we excluded neural network prediction for H3K4me1 in the challenge final submission (see details in Methods – Ensemble modeling). Another key step in Ocelot is the quantile normalization of epigenomic signals. Without quantile normalization, 3 non-MSE based metrics and 6 MSE based metrics became significantly worse in lightGBM, and only Catch1imp became better (Supplementary Fig. 26 and Supplementary Table 7). For the neural network model without quantile normalization, 11 out of 12 metrics became significantly worse and only MSEvar was comparable (Supplementary Fig. 27 and Supplementary Table 8). Therefore, quantile normalization improved the predictive performance of both lightGBM and neural network models in Ocelot.
In epigenomic imputation across multiple cell types, the similarities among cell types can potentially improve the predictive performance. A straightforward way to define similarity is using the pairwise Pearson’s correlation based on the epigenomic signal tracks in multiple cell types. For each histone mark, we calculated the pairwise correlations on the challenge training data (Supplementary Fig. 28). The correlations are weak (<0.3) for most pairs and some pairs have negative correlations close to zero, reflecting the complexity of epigenomic signals. To leverage the information of similarities among cell types, we use the pairwise correlations of the feature marks as the cell type-specific weights to re-weight predictions from multiple models. For example, when we aim to predict a cell type-mark pair CiMtarget, we train a model using CjMtarget of the same mark in another cell type Cj as the gold standard target and CjMfeature of another mark as the feature mark. The correlation between CiMfeature and CjMfeature is used as the weight for this model trained on cell type Cj. Similarly, when we have multiple training cell types and the associated cell type-specific models, we calculate the weighted predictions from multiple lightGBM models. For comparison, we also have the ensemble lightGBM prediction with a uniform weight without the similarity information (Supplementary Fig. 29 and Supplementary Table 9). Two types of weights have comparable performance in 8 metrics. Using cell type-specific weights had worse performance than using the uniform weight in 4 MSE based metrics. This is mainly because the similarity among cell types derived from feature marks can not reflect the similarity among cell types in the target mark to be imputed. These results indicate that ensembling predictions based on weights from correlation information of feature marks does not improve the predictive performance on the challenge prospective epigenome data.
Imputation of missing entries in the ENCODE3 and Roadmap datasets
The ENCODE3 and Roadmap Epigenomics Consortiums have generated two crucial epigenome datasets: (1) the ENCODE3 histone mark dataset consisting of 500 profiles of 13 epigenetic marks in 79 cell types 9, and (1) the Roadmap histone mark dataset consisting of 974 profiles of 27 epigenetic marks in 127 cell types 11. However, compared with the complete combinations of every mark in every cell type, only 49.76% and 20.34% of the epigenomes were experimentally observed in the ENCODE3 and Roadmap data matrices, respectively. To complete these two large-scale datasets, we applied Ocelot to impute missing entries. Specifically, for the ENCODE 3 data matrix, we imputed 527 (51.31%) profiles of 13 histone marks in 79 cell and tissue conditions (Fig. 6). For the Roadmap data matrix, we imputed the 2,455 (71.60%) missing entries covering 27 histone marks in 127 cell and tissue conditions (Supplementary Fig. 30). Leveraging both the conventional machine learning model and the cutting-edge deep learning technique, we provide a total of 2,982 whole-genome high-resolution (25bp) imputations, complementing the research of these two large consortiums. These imputations will further facilitate systematic studies that require complete histone mark profiles in the future.
Figure 6: Application of Ocelot to impute missing entries and complete the ENCODE3 histone mark dataset.

The ENCODE3 histone mark dataset covers 13 histone marks in 79 cell and tissue conditions, including primary tissues (n=36), primary cells (n=6), cell lines (n=28) and in vitro differentiated cells (n=9). A total of 500 (48.69%) whole-genome profiles were observed (blue blocks) and used to build machine learning models. Then we imputed the remaining 527 (51.31%) profiles (red blocks).
Discussion
Machine learning models have been widely explored and used to impute missing or unavailable data in bioinformatics. They are especially useful when experimental data are resource-intensive and hard to obtain. Similar to histone modifications, genome-wide transcription factor (TF) binding profiles are also mark- and cell type-specific. Considering the large number of TF-cell type combinations, it is infeasible to experimentally measure the genome-wide binding profile of every TF in every cell type. The number of combinations will further grow exponentially and computational predictions are necessary when we consider TF co-binding events 24,25. Conventional machine learning models were first developed to predict TF binding 26–28. Recently, deep learning models emerged and proved to be powerful 19,29,30. In this work, we leverage both the conventional tree-based model and the deep neural network model to predict histone modification marks. By exploiting the mark-specific and cell type-specific information in available observed data, we developed a method with high predictive performance and imputed missing entries in three large-scale histone modification datasets. Through dissecting the contributing elements in Ocelot models, we further revealed the cross-modulation relationships across histone modifications.
As one of the most crucial mechanisms underlying many cellular processes, epigenetic modifications together with TF bindings precisely regulate gene expression. Through quantitative analyses, it has been reported that epigenetic modifications and TF bindings are correlated and co-localized across cell types in a protein-specific manner 31,32. As a result, computational studies have shown that epigenetic modifications and TF bindings are predictive of each other 33,34. Similar to the existence of TF motifs, DNA motifs that regulate histone modifications and DNA methylation have been found in human and mouse 35,36. In this study, we mainly focused on imputation of histone marks within human tissue and cell types. Whether the regulation of histone modifications are shared or distinct across different organisms would be an interesting topic to study in the future. In fact, it has been reported that cross-species prediction of TF binding sites is feasible using neural network models 37. There are also studies about cross-species prediction of epigenomic data and regulatory sequences in gene expression 38–40. Through transfer learning on datasets of multiple organisms could be promising to improve the imputation performance. Considering the completeness and data availability, we mainly focus on six representative histone marks in this work. As more epigenomes are experimentally characterized, Ocelot can be further applied and adapted to investigate complex spatial relationships among more epigenetic marks.
Methods
Data collection
In this work, we investigated three datasets. The first one is the ENCODE Imputation Challenge dataset that contains 363 epigenomic tracks of 35 marks (33 histone modifications, DNase-seq, and ATAC-seq) in 51 cell types at 25bp resolution. During the challenge, a training set of 312 tracks were released to participants to build models, whereas the remaining 51 testing tracks were newly acquired and only released after imputations were completed by all participants (Supplementary Table 10). Therefore, potential information leakage or overfitting was avoided on the testing set. The predictive performance of our approach was comprehensively evaluated on these 51 genome-wide testing tracks. The second one is the recent ENCODE3 histone mark dataset that covers 13 histone marks in 79 cell types. The complete mark-cell type combination has a total of 1,027 entries, where 500 (48.69%) entries were experimentally observed. We applied our method by re-training models on these 500 entries and imputing the other 527 (51.31%) missing entries. The third dataset is the Roadmap histone mark dataset that covers 27 marks in 127 cell types, after excluding 4 histone marks (H3K23me2, H2AK9ac, H3T11ph, H4K12ac) that only have one or two observed whole-genome tracks. We excluded them because we could not train a solid lightGBM or neural network model with too few observed signal tracks. For this dataset, the complete mark-cell type combination has a total of 3,429 entries, where only 974 (28.40%) entries were available. We re-trained models based on the 974 entries and imputed the remaining 2,455 (71.60%) missing ones. We downloaded the standard genome-wide signal tracks that were generated by calculating the statistical significance of enrichment, −log10(p-value), where the null distribution is based on a local Poisson background estimated from the control experiment 9,11. For the ENCODE Imputation Challenge and ENCODE3 datasets, the reference genome is GRCh38. For the Roadmap dataset, the reference genome is GRCh37, which is consistent with the original release of the data.
Quantile normalization
To reduce potential batch effects and sequencing biases, we performed quantile normalization 41,42 on all signal tracks. Within the machine learning framework, we assume that for each histone mark, the overall distributions of epigenomic signals across different cell types are identical. This strong assumption allows for generalizability of machine learning models, though in reality the distributions of observed epigenomic signals are generally different across cell types. The quantile normalization requires a mark-specific reference. For each epigenetic mark, the reference was created by averaging the ranked original signals across all available cell types. Since the human genome is huge, we randomly subsampled signals from 0.1% of the whole genome and generated the average reference. Then for the signal track of each cell type, we created a quantile mapping function between the subsampled signal of the reference and the subsampled signal of this cell type. Then applied the quantile mapping function to normalize the whole-genome signals.
Data partition for model training, validation, and testing
To exploit available data and build high-quality models, for an entry CiMj (cell type Ci and mark Mj) to be imputed, we integrate into Ocelot both the cell type-specific information across epigenetic marks and the mark-specific information across cell types (Supplementary Fig. 31a). Specifically, we first gather all the available other marks (except mark Mj) in this cell type Ci and name it as feature set Set-Ci, representing the cell type-specific information (Supplementary Fig. 31b). Meanwhile, we gather the epigenomes for mark Mj in all other cell types (except cell type Ci), representing the mark-specific information. There are two types of mark-specific epigenomes: with or without the complete cell type-specific feature set, which are named as Set-Mj and Set-Mj’, respectively. For the Set-Mj, two cell types are randomly selected as the training and validation targets to build a model, whereas the remaining data are used as the mark-specific features. For the Set-Mj’, they can not serve as training targets owing to incompleteness of the cell type-specific features, and are therefore used as the mark-specific features.
An example case for imputing entry CiMj is shown in Fig. 1b to demonstrate how the available epigenomes are used as either features or targets to build a machine learning model. The four violet Mj entries are the common mark-specific features for model training, validation and testing. In addition, the cell type-specific features are used in model training (C2M1, C2M3, C2M5), validation (C4M1, C4M3, C4M5), and held-out testing (CiM1, CiM3, CiM5). Meanwhile, entries C2Mj and C4Mj are the targets for training and validation, respectively. We used two iterative machine learning models, convolutional neural network and tree-based lightGBM 18, where the validation-based early stopping strategy is required for hyperparameter tuning and avoiding overfitting. Of note, an epigenome in Set-Mj is used multiple times and can serve as both the feature in one model and the target in another model. Ideally, when the Set-Mj contains N entries, we can build N×(N−1) models by randomly selecting two entries as the training and validation targets. The computational cost will grow exponentially and is extremely high when N is large. To exploit the data with high efficiency during the challenge, we only built N models and each entry served as the training target once and validation target once. Then the predictions from N models are averaged to generate a single prediction.
Parameters of lightGBM model
We built regression lightGBM models using the Python module “lightgbm” (2.3.0). To avoid overfitting during model training, we used an early stopping strategy - if the validation loss did not drop for 20 boosting rounds, the training was stopped and the maximum number of boosting rounds was 500. The type of boosting is “gbdt” (gradient boosting decision tree). The maximum number of leaves within a tree is 50 with the minimum number of 20 data points with a leave. The L2 regression was used. The bagging strategy was used to introduce further randomness with the bagging frequency of 1 and bagging fraction of 0.7. In addition, we built lightGBM models in parallel with 50 boosting rounds without early stopping or bagging. Predictions from two types of lightGBM models were averaged.
Neural network architecture
We designed a special deep convolutional neural network architecture for this epigenetic imputation task. Specifically, this network contains two encoders and one decoder with multiple convolutional, max-pooling, and upscaling layers. The input layer is first connected to two convolutional layers and encoder 1. In encoder 1, there are four Pooling-Convolution-Convolution (PCC) blocks that gradually reduce the input length from 3200 to 200 and increase the number of channels. The kernel size of the max-pooling layer is 2. Then we added the decoder that has four Upscaling-Convolution-Convolution (UCC) blocks that gradually increase the length from 200 to 3200 and reduce the number of channels. Meanwhile, layers of the same length in the encoder 1 are transferred to the decoder as additional channels through four concatenation layers. Then we added the encoder 2 that has two PCC blocks and the max-pooling layers has the kernel size of 5. These two max-pooling layers reduce the length from 3200 to 128 = 3200 / 5 / 5. Finally, a last convolutional layer with one channel is added to generate the 128-by-1 output. For all the convolutional layers, the kernel size is 7 and the non-linear activation is “ReLU”. A batch normalization layer is added before each convolutional layer to accelerate the training process. We used the mean squared error loss and Adam optimizer. An epoch is defined by randomly sampling 10,000 genomic regions from the whole human genome with replacement and each sample has 3,200 bps. We first trained the neural network for 1 epoch with a relatively large learning rate of 1e-3. Then we continued to train another 2 epochs with the learning rate of 1e-4. The model was implemented using the Python module “Keras” (2.2.4) with “Tensorflow” (1.14.0) backend.
Ensemble modeling
To exploit the available training data (Supplementary Table 10) and include as many feature marks as possible, for each target mark to be imputed, we designed different combinations of feature marks for different testing cell types during the challenge. In the final submission, we ensembled predictions from (1) lightGBM, (2) neural network, and (3) the quantile normalized average signal of a mark in all training cell types. The ensemble weights are summarized in Supplementary Table 11, which were determined by cross-validation results on the training data. Specifically, for each combination of feature marks, we tested the predictive performance on five cell types (C17, C20, C24, C32, C46) and focused on three scoring metrics (1) MSEglobal, (2) Pearson’s correlation, and (3) Spearman’s correlation. Meanwhile, we tested six types of ensemble weights of lightGBM: NN: average = 4:1:1 (“l4na”), 2:1:1 (“l2na”), 1:1:1 (“lna”), 4:0:1 (“l4a”), 2:0:1 (“l2a”), and 1:0:0 (“l”). The results are shown in Supplementary Fig. 32 – 37. Each figure contains the results of one histone mark and different colors represent different ensemble weights. For example, we designed three combinations for H3K27ac shown in three rows in Supplementary Fig. 32. Three columns corresponds to three scoring metrics. For each type of weighting, we counted the number of best scores among all weightings based on 3 metrics in 5 cell lines. The weighting with the largest numbers of best scores was selected. In general, we found that the performances varied across combinations, ensemble weights, evaluation metrics and cell types, reflecting the complex nature of epigenomic imputation. There were no perfect and universal ensemble weights to optimize all different metrics in all cell types. Overall, the lightGBM model had stronger predictive power than the neural network and we assigned a larger weight for it in most combinations. The predictive power is associated with different types of histone marks as well. For example, both lightGBM and neural network models were used in predicting H3K27me3, whereas only lightGBM was used in predicting H3K4me1.
SHAP analysis
We focused on six representative histone marks (H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me3, H3K9me3) that have more than 50% available tracks within the 51 cell types from the ENCODE Imputation Challenge dataset. We randomly selected five cell types (C17, C20, C24, C32, C46) as the testing set and performed SHAP analysis of the corresponding predictions in the tree-based lightGBM models. For a pair of histone marks, A and B, we built two types of models: (1) using A as the feature mark to predict target B, and (2) using B as the feature mark to predict target A. We randomly sampled 303,104 times (0.01% of the length of the human genome) and each time we calculated the SHAP values for a 275bp bin. It corresponds to 303,104 * 275bp = 83,353,600bp, or 2.75% of the human genome. To robustly evaluate this, we further bootstrap sampled 10% of all samples from the five testing cell types for 10 times. The SHAP values from 10 bootstrapping are similar (Supplementary Table 1). We therefore used the average values from 10 bootstrap samplings and plotted the heatmap in Fig. 3a. To test the robustness of the predictive relationship pattern in different cell types, we further performed the SHAP analysis on bootstrap subsets - every 4 out of 5 cell types. The results of 5 bootstrap subsets are shown in Supplementary Fig. 38 and Supplementary Table 12. The corresponding PPI values are listed in Supplementary Table 13. The patterns and PPI values are robust across the bootstrap.
Evaluation metrics and ranking
Consistent with evaluation of the ENCODE Imputation Challenge, we used nine global and local scoring metrics to compare the observed data and imputed data of our method. These metrics includes (1) global Pearson’s correlation, (2) global MSE (MSEglobal), (3) global Spearman’s correlation across the entire human genome, three local MSEs across genomic regions that are annotated as (4) promoters (MSEProm), (5) genes (MSEGene), and (6) enhancers (MSEEnh), and (7) global MSE weighted by the cross-cell-type variance (MSEvar), (8) local MSE across genomic regions with top 1% observed values (MSE1obs), and (9) local MSE across genomic regions with top 1% predicted values (MSE1imp). The annotations of genes and promoters are based on the GENCODE annotations on GRCh38 43. The annotation of enhancers is obtained from the FANTOM5 project 44. To avoid over emphasis on MSE based metrics, we further added three non-MSE based metrics: (10) the overlap between the top 1% observed and imputed signals (Match1), (11) the overlap between the top 1% observed and 5% imputed signals (Catch1obs), and (12) the overlap between the top 5% observed and 1% imputed signals (Catch1imp). Using these five non-MSE based metrics together with seven MSE based metrics, the predictive performances of different methods and strategies are systematically and fairly compared.
During the challenge, the ranking of a method was determined through scoring on 10 bootstrapped regions and each bootstrap covers about 90% of the human genome. Specifically, for a bootstrap k and a testing mark-cell type pair j, the Scorejk of a method was first calculated using 9 metrics by
where Njk = 9 is the number of evaluation metrics Nsubmissions = 23, is the number of submissions and rijk is the rank among all submissions. Then the Rankjk of a method was obtained based on the Scorejk among all submissions. The Rankk of a bootstrap was calculated through averaging across 51 testing pairs by
where Npair = 51 is the number of held-out testing pairs Among the 10 bootstrap ranks, Rank1, Rank2, … Rank10, the second best rank was used as the final rank to reduce the effect of uncertainty during bootstrapping. The complete ranking results in the ENCODE Imputation Challenge for 51 testing mark-cell type pairs are available at: https://www.synapse.org/#!Synapse:syn17083203/wiki/597122. To provide an intuitive comparison between the top two methods, we summarized the median scores of 9 metrics and the percentage improvements (increase in correlations and decrease in MSEs) in Supplementary Table 14.
Benchmarking performance against ChromImpute and Avocado
We trained ChromImpute models on genome-wide signals of the challenge training data. We used default parameters during feature generation and model training. The codes and results are available at: https://guanfiles.dcmb.med.umich.edu/Ocelot/chromimpute. The Avocado imputations for the challenge final testing were directly downloaded from: http://mitra.stanford.edu/kundaje/ic/avocado/.
Supplementary Material
Acknowledgments
This work is supported by NIH/NIGMS R35GM133346 and NSF/DBI #1452656.
Footnotes
Code availability
The code of Ocelot is available on GitHub: https://github.com/GuanLab/Ocelot
Data availability
The ENCODE Imputation Challenge data were downloaded from:
(1) training data https://www.synapse.org/#!Synapse:syn18143300
(2) testing data http://mitra.stanford.edu/kundaje/ic/blind/
The ENCODE3 histone modification data were downloaded from:
https://www.encodeproject.org/ based on the accession numbers listed in Supplementary Table 15.
The Roadmap histone modification data were downloaded from:
https://egg2.wustl.edu/roadmap/data/byFileType/signal/consolidated/macs2signal/pval/
Our epigenome imputation for the missing entries in the ENCODE3 and Roadmap datasets are available at:
https://guanfiles.dcmb.med.umich.edu/Ocelot/imputation_encode3/
https://guanfiles.dcmb.med.umich.edu/Ocelot/imputation_roadmap/
References
- 1.Venter JC et al. The sequence of the human genome. Science 291, 1304–1351 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Rivera CM & Ren B Mapping human epigenomes. Cell 155, 39–55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith ZD & Meissner A DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–220 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Thiele I et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 31, 419–425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wittkopp PJ & Kalay G Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 13, 59–69 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Barrat FJ, Crow MK & Ivashkiv LB Interferon target-gene expression and epigenomic signatures in health and disease. Nat. Immunol. 20, 1574–1583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hekselman I & Yeger-Lotem E Mechanisms of tissue and cell-type specificity in heritable traits and diseases. Nat. Rev. Genet. 21, 137–150 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Lukong KE, Chang K-W, Khandjian EW & Richard S RNA-binding proteins in human genetic disease. Trends Genet. 24, 416–425 (2008). [DOI] [PubMed] [Google Scholar]
- 9.ENCODE Project Consortium et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ernst J & Kellis M Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat. Biotechnol. 33, 364–376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durham TJ, Libbrecht MW, Howbert JJ, Bilmes J & Noble WS PREDICTD PaRallel Epigenomics Data Imputation with Cloud-based Tensor Decomposition. Nat. Commun. 9, 1402 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schreiber J, Durham T, Bilmes J & Noble WS Avocado: a multi-scale deep tensor factorization method learns a latent representation of the human epigenome. Genome Biol. 21, 81 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ernst J & Kellis M Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 28, 817–825 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst J & Kellis M ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffman MM et al. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods 9, 473–476 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ke G et al. LightGBM: A Highly Efficient Gradient Boosting Decision Tree. in Advances in Neural Information Processing Systems (eds. Guyon I et al. ) vol. 30 (Curran Associates, Inc., 2017). [Google Scholar]
- 19.Li H & Guan Y Fast decoding cell type-specific transcription factor binding landscape at single-nucleotide resolution. Genome Res. 31, 721–731 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shapley LS 17. A Value for n-Person Games. in Contributions to the Theory of Games (AM-28), Volume II (eds. Kuhn HW & Tucker AW) 307–318 (Princeton University Press, 1953). [Google Scholar]
- 21.Lundberg SM et al. Explainable machine-learning predictions for the prevention of hypoxaemia during surgery. Nat Biomed Eng 2, 749–760 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katoh N et al. Reciprocal changes of H3K27ac and H3K27me3 at the promoter regions of the critical genes for endometrial decidualization. Epigenomics 10, 1243–1257 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Juan AH et al. Roles of H3K27me2 and H3K27me3 Examined during Fate Specification of Embryonic Stem Cells. Cell Rep. 17, 1369–1382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, Zhao W & Zhou X Modeling co-occupancy of transcription factors using chromatin features. Nucleic Acids Res. 44, e49 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou M, Li H, Wang X & Guan Y Evidence of widespread, independent sequence signature for transcription factor cobinding. Genome Res. (2020) doi: 10.1101/gr.267310.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghandi M, Lee D, Mohammad-Noori M & Beer MA Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput. Biol. 10, e1003711 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pique-Regi R et al. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 21, 447–455 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Quang D & Guan Y Anchor: trans-cell type prediction of transcription factor binding sites. Genome Res. 29, 281–292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou J & Troyanskaya OG Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 12, 931–934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelley DR et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 28, 739–750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Xue G, Liu J, Li Q & Wang Y Revealing transcription factor and histone modification co-localization and dynamics across cell lines by integrating ChIP-seq and RNA-seq data. BMC Genomics 19, 914 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xin B & Rohs R Relationship between histone modifications and transcription factor binding is protein family specific. Genome Res. (2018) doi: 10.1101/gr.220079.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L, Jin G & Zhou X Modeling the relationship of epigenetic modifications to transcription factor binding. Nucleic Acids Res. 43, 3873–3885 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benveniste D, Sonntag H-J, Sanguinetti G & Sproul D Transcription factor binding predicts histone modifications in human cell lines. Proc. Natl. Acad. Sci. U. S. A. 111, 13367–13372 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ngo V et al. Epigenomic analysis reveals DNA motifs regulating histone modifications in human and mouse. Proc. Natl. Acad. Sci. U. S. A. 116, 3668–3677 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang M et al. Identification of DNA motifs that regulate DNA methylation. Nucleic Acids Res. 47, 6753–6768 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cochran K et al. Domain adaptive neural networks improve cross-species prediction of transcription factor binding. bioRxiv (2021) doi: 10.1101/2021.02.13.431115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L, Fish AE & Capra JA Prediction of gene regulatory enhancers across species reveals evolutionarily conserved sequence properties. PLoS Comput. Biol. 14, e1006484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelley DR Cross-species regulatory sequence activity prediction. PLoS Comput. Biol. 16, e1008050 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schreiber J, Hegde D & Noble W Zero-shot imputations across species are enabled through joint modeling of human and mouse epigenomics. bioRxiv (2019) doi: 10.1101/801183. [DOI] [Google Scholar]
- 41.Bolstad BM, Irizarry RA, Astrand M & Speed TP A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Li H & Guan Y DeepSleep convolutional neural network allows accurate and fast detection of sleep arousal. Commun Biol 4, 18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frankish A et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lizio M et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 16, 22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ENCODE Imputation Challenge data were downloaded from:
(1) training data https://www.synapse.org/#!Synapse:syn18143300
(2) testing data http://mitra.stanford.edu/kundaje/ic/blind/
The ENCODE3 histone modification data were downloaded from:
https://www.encodeproject.org/ based on the accession numbers listed in Supplementary Table 15.
The Roadmap histone modification data were downloaded from:
https://egg2.wustl.edu/roadmap/data/byFileType/signal/consolidated/macs2signal/pval/
Our epigenome imputation for the missing entries in the ENCODE3 and Roadmap datasets are available at:
https://guanfiles.dcmb.med.umich.edu/Ocelot/imputation_encode3/
https://guanfiles.dcmb.med.umich.edu/Ocelot/imputation_roadmap/