

Abstract
Background
Cerebrovascular autoregulation (CA) regulates cerebral vascular tone to maintain near‐constant cerebral blood flow during fluctuations in cerebral perfusion pressure (CPP). Preclinical and clinical research has challenged the classic triphasic pressure‐flow relationship, leaving the normal pressure‐flow relationship unclear.
Methods and Results
We used in vivo imaging of the hemodynamic response in pial arterioles to study CA in a porcine closed cranial window model during nonpharmacological blood pressure manipulation. Red blood cell flux was determined in 52 pial arterioles during 10 hypotension and 10 hypertension experiments to describe the pressure‐flow relationship. We found a quadriphasic pressure‐flow relationship with 4 distinct physiological phases. Smaller arterioles demonstrated greater vasodilation during low CPP when compared with large arterioles (P<0.01), whereas vasoconstrictive capacity during high CPP was not significantly different between arterioles (P>0.9). The upper limit of CA was defined by 2 breakpoints. Increases in CPP lead to a point of maximal vasoconstriction of the smallest pial arterioles (upper limit of autoregulation [ULA] 1). Beyond ULA1, only larger arterioles maintain a limited additional vasoconstrictive capacity, extending the buffer for high CPP. Beyond ULA2, vasoconstrictive capacity is exhausted, and all pial arterioles passively dilate. There was substantial intersubject variability, with ranges of 29.2, 47.3, and 50.9 mm Hg for the lower limit, ULA1, and ULA2, respectively.
Conclusions
We provide new insights into the quadriphasic physiology of CA, differentiating between truly active CA and an extended capacity to buffer increased CPP with progressive failure of CA. In this experimental model, the limits of CA widely varied between subjects.
Keywords: cerebral blood flow, cerebral perfusion pressure, cerebrovascular autoregulation
Subject Categories: Animal Models of Human Disease, Basic Science Research, High Blood Pressure, Imaging, Cerebrovascular Disease/Stroke
Nonstandard Abbreviations and Acronyms
- ABP
arterial blood pressure
- CA
cerebrovascular autoregulation
- CBF
cerebral blood flow
- CPP
cerebral perfusion pressure
- CVR
cerebrovascular resistance
- LLA
lower limit of autoregulation
- ULA
upper limit of autoregulation
Clinical Perspective
What Is New?
Instead of the classic triphasic cerebrovascular autoregulation relationship between cerebral perfusion pressure and cerebral blood flow (passive‐active‐passive autoregulation), we demonstrated a quadriphasic relationship (passive‐active‐active and passive‐passive autoregulation) in a porcine model using in vivo imaging of the hemodynamic response in pial arterioles.
The range of entirely active autoregulation is more restricted than classically assumed.
Toward high cerebral perfusion pressures, the cerebral pial arterioles provide an extended capacity to buffer increased perfusion pressure with progressive failure of autoregulation; important intersubject variability in the active regions of autoregulation was found.
What Are the Clinical Implications?
Improved understanding of cerebrovascular autoregulation can lead to better clinical autoregulation monitoring tools that have the potential to define individualized blood pressure targets for adequate cerebral perfusion pressure to improve patient outcomes during general anesthesia, and many cerebral and cardiovascular diseases.
Given the potential large intersubject variability in the limits of autoregulation, the clinical use of fixed threshold numbers from grouped analyses could easily overestimate or underestimate the true limits of autoregulation in individual patients, highlighting the need for individualized clinical autoregulation monitoring tools.
The structural and functional integrity of the brain is highly dependent on the homeostasis of cerebral blood flow (CBF). 1 Cerebrovascular autoregulation (CA) protects the brain against fluctuations in cerebral perfusion pressure (CPP) by actively adjusting the cerebrovascular resistance (CVR) to ensure a near‐constant CBF. The role and implications of impaired CA are increasingly being recognized in the pathophysiology of acute brain injuries, such as stroke, 2 traumatic brain injury, 3 subarachnoid hemorrhage, 4 and prematurity‐related intracranial hemorrhage, 5 , 6 but also in chronic neurological conditions, such as vascular dementia and Alzheimer disease. 7 , 8 The principle site of CVR changes is believed to be small pial arteries and arterioles on the surface of the brain. 9 , 10 Larger arteries of the brain and neck also contribute to modulate CVR. 11 , 12 , 13 CVR is regulated by changes in luminal diameter through vasodilation and vasoconstriction, producing large changes in flow according to the Poiseuille law. 9 , 14 , 15
In 1959, Lassen established the concept of static CA in humans in terms of a triphasic curve consisting of a plateau of steady CBF defined by the upper limit of autoregulation (ULA) and lower limit of autoregulation (LLA) of CA. In Lassen’s review of 7 studies involving 11 different patient groups, CBF appeared to be stable across a wide range of blood pressures, ranging from 60 to 150 mm Hg. 10 CBF was measured per individual at one single mean arterial pressure value and was not tested throughout a range in arterial pressures. Subjects had different pathological conditions and/or were receiving pharmaceutical agents. Lassen’s CA concept does not necessarily represent the pressure‐flow relationship within an individual. 16 , 17 , 18 In healthy individuals, the study of CA is limited by the need for pharmacological blood pressure manipulations with associated confounding effects on cerebrovascular tone and/or indirect effects from alterations of arterial gasses. 19 Only few studies have corrected for the effects of CO2. 20 The validity of transcranial Doppler ultrasonography as a noninvasive measurement method depends on the assumption of a constant diameter of the insonated vessel and remains questionable. 19 , 20 , 21 , 22 Another limitation in human studies is the diversity in measurement protocols between studies.
In the face of these limitations, available data suggest that Lassen’s curve might not be the optimal representation of the pressure‐flow relationship within an individual. Several studies in humans and large animal models have demonstrated autoregulatory capacity. However, important disagreements exist: plateau regions are either wide (>40 mm Hg) or narrow (10–15 mm Hg) and efficacy of CA in maintaining CBF could depend on the direction of change of CPP above or below baseline. 20 , 21 , 23 , 24 , 25 , 26 In a meta‐analysis of human studies, Numan et al pooled the within‐subject responses across 40 studies and found little evidence of a plateau region. 20 Interestingly, this meta‐analysis supported the concept of CA asymmetry, with more efficient mechanisms to buffer increases in mean arterial pressure, compared with decreases. 27 , 28
A definitive within‐subject assessment of CBF across a range of nonpharmacologically altered blood pressures with maintained CO2 has not been completed. 19 The exact role of disturbed CA in pathological conditions and its therapeutic potential to steer hemodynamic management can only be established by unraveling the mechanisms of normal CA first.
The purpose of this study was to investigate CBF regulation in response to CPP changes in pial arterioles of different sizes and to characterize the normal cerebral pressure‐flow relationship.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Animals and Anesthesia
The methods and validation of CA assessment by direct visualization of pial arteriolar blood flow in the piglet brain have been described earlier. 29 Twenty 6‐week‐old male piglets (domestic swine; Zootechnical Center, KU Leuven University) were anesthetized by continuous intravenous infusion of propofol (2–4 mg/kg per hour), midazolam (B. Braun) (0.3–0.7 mg/kg per hour), fentanyl (2 µg/kg per hour, adjusted to response to painful stimuli up to 20 µg/kg per hour), and pancuronium (Inresa) (0.3 mg/kg per hour). Only male animals were studied to avoid the potential confounding role of sex‐related differences. 30 No inhalation anesthetics were used. Mechanical ventilation was adjusted to maintain an end‐tidal CO2 tension of 40 mm Hg, verified by arterial blood gas sampling.
Surgical Procedure
The femoral artery was cannulated for placement of a pressure and blood gas monitoring line. In the hypertensive group, the contralateral femoral artery was cannulated for placement of a 5F balloon occlusion catheter (LeMaitre embolectomy catheter; 1601‐54; LeMaitre Vascular) into the thoracic aorta. In the hypotensive group, a 5F balloon catheter was placed in the femoral vein and advanced to the inferior vena cava. Heparin intravenous infusion was started (50 IU/kg per hour). An intraparenchymal probe was placed in the right hemisphere for continuous measurement of intracranial pressure and brain temperature (Neurovent‐PTO; Raumedic AG, Muenchenberg, Germany). A 3.5‐mm diameter laser Doppler flow probe was placed in contact with the dura (Moor VMS‐LDF1 with VP14‐CBF probe; Moor Instruments, Devon, UK), hereby avoiding large dural or pial vessels (not used in the current analysis). A cranial window was placed anterior to the coronal suture using a high‐speed drill, continuously cooled with cold saline, according to the technique described by Levasseur et al and Busija et al. 31 , 32 The dura was elevated, cut, and removed under optical magnification to avoid damaging underlying structures. The cranial window consists of a stainless‐steel ring with 3 injection ports and a central 15‐mm diameter opening sealed with a coverslip glass. The space under window was filled with artificial cerebrospinal fluid (NaCl 132 mmol/L, KCl 3.0 mmol/L, MgCl2 1.5 mmol/L, CaCl2 1.5 mmol/L, urea 6.6 mmol/L, glucose 3.7 mmol/L, NaHCO3 24.6 mmol/L at 37°C and equilibrated with 6% O2 and 6% CO2 in N2 to a pH 7.35–7.45, partial pressure of CO2 40–42 mm Hg, and partial pressure of O2 42–50 mm Hg). Piglets were allowed to recover for 2 hours.
Physiological Monitoring
Arterial blood pressure (ABP) and intracranial pressure signals were continuously sampled at 250 Hz. Blood oxygen level was monitored using a pulse oximeter and kept at 99% to 100%. Inspired and expired concentrations of CO2 and oxygen were monitored with a gas analyzer (Phillips M1026B; Philips Medical Systems, the Netherlands). Arterial blood Paco 2 was sampled for verification of continuously monitored end‐tidal CO2. pH was kept between 7.35 and 7.45, Pao 2 at 200 mm Hg, and Paco 2 at 38 mm Hg. Rectal temperature was maintained at 38 to 39°C by a warming mattress and blankets. Signals were stored using ICM+ software (Cambridge University, Cambridge, UK). CPP was calculated as the difference between ABP and intracranial pressure.
Functional CA Challenge
Two series of experiments were performed, a hypertensive and a hypotensive group, with 10 animals per group. A sample size of 10 per group will yield statistically significant results at the P=0.05 level with a power of 0.9 (1‐tailed paired t‐test). Progressive hypertension or hypotension was induced by gradual inflation of a balloon catheter in the aorta or inferior vena cava, respectively. The balloon catheter was gradually inflated over 2 to 3 hours. Total duration of the experimental procedures ranged from 7 to 9 hours.
In Vivo Imaging
Red blood cells (RBCs) were labeled with carboxyfluorescein diacetate succinimidyl ester (Invitrogen/Molecular Probes), as described earlier. 29 Carboxyfluorescein diacetate succinimidyl ester is a membrane‐permeable esterified cytoplasmatic dye commonly used for staining of viable cells without compromising their functional properties. 33 Pial vessels were observed using an epifluorescence microscope (SMZ18; Nikon) with green fluorescent filter (GFP‐B Filter Cube 470–535 nm; Nikon), and captured with a high‐speed digital CMOS camera (Orca Flash 4.0V2; Hamamatsu) controlled by NIS‐Elements software (Nikon). Images were acquired at 170 to 200 frames per second. RBC velocity was determined by threshold pixel intensity detection of fluorescent RBCs to create a binary object. An individual movement track was created, and the average path speed was calculated for each track. Per arteriole, an overall average of RBC path speeds was calculated for each time point. Diameter measurements were performed for each time point by pixel intensity thresholding between background and arteriole in the same region of interest per arteriole.
Under the assumption of steady‐state laminar flow in a cylindrical vessel, RBC velocity and lumen diameter measured from a single vessel can be used to define the average volumetric flux F⃗, by:
where V is velocity, A is the luminal cross‐sectional area, r is the vessel radius, and D is the vessel diameter. 34 , 35 We randomly selected up to 3 arterioles per cranial window experiment, ranging from 20 to 140 μm. Data from 52 arterioles (diameter range, 16–123 µm) were obtained in 10 hypotensive (26 arterioles) and 10 hypertensive (26 arterioles) experiments. Arterioles were separated into 3 categories: small, <40 μm (n=20); medium, 40 to 70 μm (n=19); and large, >70 μm (n=13).
Ethical Considerations
All animal care and procedures were approved by the Independent Ethics Committee for Animal Experimentation at the KU Leuven University (P105‐2015) in compliance with the Belgian Royal Decree (May 29, 2013) and European Directive 2010/63/EU on the protection of animals used for scientific purposes. All animal procedures were conducted under veterinarian supervision, according to the guidelines imposed by the Ethical Committee, and reported in compliance with the Animal Research: Reporting of In Vivo Experiments guidelines.
Statistical Analysis
Analysis was performed using R statistical software (R Core Team 2016; URL: https://www.R‐project.org/) using following packages: ggplot2, mgcv, dplyr, and segmented. A generalized additive model smoothing function was used to visualize the global estimate of the physiological variables in response to changes in CPP. Segmental linear regression was used to define breakpoints in the relation between pial arteriolar lumen diameter, RBC velocity, and RBC flux measurements and CPP. Breakpoints correspond to the LLA and ULA. Baseline was determined as the mean value during 60 minutes before the start of invasive blood pressure manipulation. Tests for normal distribution were performed using Shapiro‐Wilk test at the level of significance 0.05. In case of a normal distribution, the mean and SD are given; otherwise, median and interquartile ranges (IQRs) are provided. Differences between 2 variables were evaluated using a Wilcoxon‐Mann‐Whitney ranks sum test. Comparisons between pial arteriolar diameter groups were performed with 1‐way ANOVA. All P values of <0.05 were considered significant.
Results
Pial Arteriolar Response to Changes in CPP
Briefly, a closed cranial window was placed over the right cerebral hemisphere in 20 piglets to evaluate the pial arteriolar hemodynamic response during gradual manipulation of ABP by progressive inflation of a balloon catheter in the thoracic aorta (induced hypertension; n=10 experiments) or inferior vena cava (induced hypotension; n=10 experiments) over a period of 2 to 3 hours. Figure 1 demonstrates the experimental setup, cranial window, microscopic imaging of pial arterioles and fluorescent RBCs, and experimental timeline. 29 Baseline measurements performed during 1 hour before the start of invasive ABP manipulation are described in Table 1. Balloon inflation allowed for a median decrease in CPP of ≈84% in the hypotensive experiments and a median increase of ≈95% in the hypertensive experiments (Table 2). End‐tidal CO2 was kept relatively constant (Table 2). Experimental timeline per arteriole, demonstrating the changes over time of CPP, arteriolar diameter, RBC velocity, and RBC flux per arteriole (n=52), is shown in Figure S1. Intracranial pressure remained within normal limits (Figure S2).
Figure 1. Illustration of the in vivo measurement of pial arteriolar red blood cell (RBC) flux.
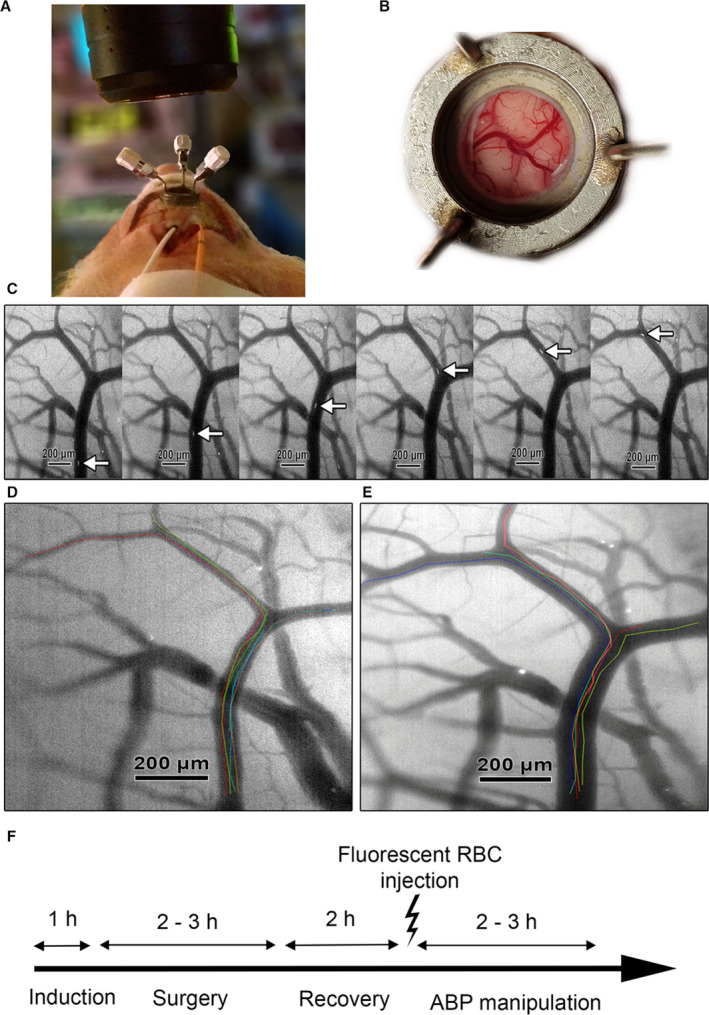
A, Microscope positioned over the closed cranial window. Intraparenchymal intracranial pressure‐probe (orange) and cortical laser Doppler flow probe (white; not used in current analysis) placed ipsilateral immediately behind the cranial window. B, Overview of cortical vessels as seen through the closed cranial window. C, Fluorescent‐labeled RBCs moving through a pial arteriole at 200 frames per second. D, Baseline visualization of pial arterioles and individual RBC tracks. Individual RBC tracks are superimposed on the original image in different colors. E, Vasodilation of pial arterioles and individual RBC tracks during induced hypotension. Individual RBC tracks are superimposed on the original image in different colors. F, Experimental timeline: induction 1 h, surgery 2 to 3 h, recovery 2 h, gradual arterial blood pressure (ABP) manipulation 2 to 3 h. Adapted from Klein et al under CC BY 4.0. 29
Table 1.
Baseline Measurements of Pial Arteriolar Diameter, RBC Velocity, RBC Flux, CPP, and ETCO2 as Well as Number of Arterioles Studied
| Variable | No. of arterioles |
Diameter, median (minimum‐maximum), µm |
RBC velocity, median (minimum‐maximum), mm/s |
RBC flux, median (minimum‐maximum), nL/s |
CPP, median (IQR), mm Hg |
ETCO2, median (IQR), mm Hg |
|---|---|---|---|---|---|---|
| Hypotensive experiments | 26 |
42 (18–105) |
6.7 (4.9–9.3) |
10.3 (1.4–54.9) |
63.7 (5.7)* |
39.9 (1.1) |
| Hypertensive experiments | 26 |
49 (16–123) |
7.0 (4.5–13.6) |
14.2 (1.7–71.9) |
70.8 (8.9)* |
38.4 (3.1) |
| Overall | 52 |
46 (16–123) |
6.8 (4.5–13.6) |
13.1 (1.4–71.9) |
68.2 (6.3) |
39.9 (2.7) |
CPP indicates cerebral perfusion pressure; ETCO2, end‐tidal CO2; and RBC, red blood cell.
*P<0.05, significant differences determined by a Mann‐Whitney U test comparing values from hypotension and hypertension experiments.
Table 2.
Maximal Percentage Change From Baseline in CPP, Pial Arteriole Diameter, RBC Velocity, RBC Flux, and ETCO2 During Arterial Blood Pressure Manipulations
| Variable | CPP, median (IQR), % change | Diameter, % change, median (IQR) | RBC velocity, % change, median (IQR) | RBC flux, % change, median (IQR) | ETCO2, % change, median (IQR) |
|---|---|---|---|---|---|
| Hypotensive experiments |
−84.6 (13.6) |
56.3 (43.3) |
−86.5 (8.7) |
−81.5 (14.5) |
−5.0 (4.8) |
| Hypertensive experiments |
95.3 (19.3) |
−16.4 (13.6) |
123.7 (84.1) |
130.7 (14.2) |
4.3 (7.1) |
CPP indicates cerebral perfusion pressure; ETCO2, end‐tidal CO2; IQR, interquartile range; and RBC, red blood cell.
Relative changes in pial arteriolar diameter, RBC velocity, and RBC flux per arteriole as a function of CPP and stratified according to arteriolar size category to demonstrate the differential role of baseline arteriolar caliber are shown in Figure 2. A combined overview of net changes in pial arteriole diameter, RBC velocity, and RBC flux as a function of CPP during hypotension and hypertension experiments combined is visualized in Figure 3. Median changes in pial arteriolar lumen demonstrated ≈56% vasodilation from baseline in the hypotensive experiments versus ≈16% vasoconstriction from baseline in the hypertensive experiments. Maximal RBC velocity and RBC flux changes are described in Table 2. Differential size‐based response in pial arteriolar diameter to changes in CPP is illustrated in Figure 4. Maximal vasodilation compared with baseline was median 77.8% (IQR, 20.7%) at CPP of 30.4 mm Hg (IQR, 4.5 mm Hg), 52.5% (IQR, 33.5%) at CPP 36.7 mm Hg (IQR, 6.4 mm Hg), 35.6% (IQR, 19.6%) at CPP 40.5 mm Hg (IQR, 14.1 mm Hg), for small, medium, and large arterioles, respectively. Size‐based differences in maximal vasodilation in response to low CPP were significant (Figure 4A). Maximal vasoconstriction compared with baseline was median −16.5% (IQR, 11.3%) at CPP of 86.0 mm Hg (IQR, 35.8 mm Hg), −18.8% (IQR, 12.0%) at CPP 87.3 mm Hg (IQR, 11.8 mm Hg), −16.0% (IQR, 8.6%) at CPP 106.0 mm Hg (IQR, 27.2 mm Hg), for small, medium, and large arterioles, respectively. Size‐based differences in maximal vasoconstriction in response to high CPP were not significant (Figure 4B). Relative differences compared with baseline in pial arteriolar RBC velocity and RBC flux to changes in CPP according to size category are shown in Figure S3.
Figure 2. Relative changes in pial arteriolar diameter, red blood cell (RBC) velocity, and RBC flux per arteriole compared with baseline (Δ%) as a function of cerebral perfusion pressure (CPP; mm Hg).
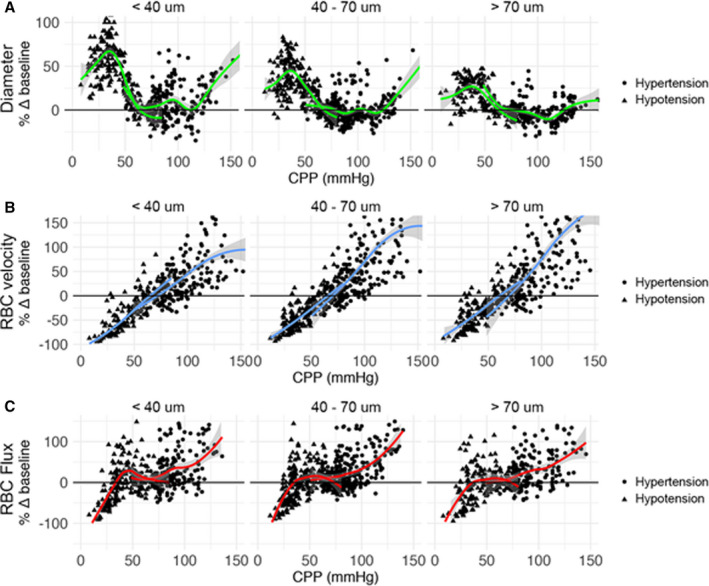
Individual data points are shown (triangle=hypotension experiment, and circle=hypertension experiment). Plots are stratified according to arteriolar size category to demonstrate the differential role of baseline arteriolar caliber. A generalized additive model smoothing function is applied separately on 2 sets of experiments (hypotension and hypertension) to visualize trends. A, Pial arteriolar diameter. B, RBC velocity. C, RBC flux.
Figure 3. Grouped illustration of relative changes in pial arteriolar vasodynamics as a function of cerebral perfusion pressure (CPP) in hypotension and hypertension experiments combined.
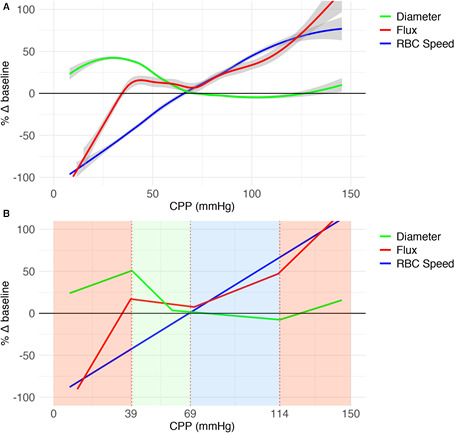
A, Grouped analysis of changes from baseline (Δ%) in red blood cell (RBC) flux, pial arteriole diameter, and RBC velocity as a function of CPP (mm Hg) for 10 hypotensive and 10 hypertensive experiments combined using a generalized additive model smoothing function. Gray shading: SE. B, Segmented regression of data in A to illustrate 4 physiological phases. Active cerebrovascular autoregulation (CA) consists of 2 phases: “active contraction/relaxation” between the lower limit of autoregulation and upper limit of autoregulation (ULA) 1 (green area) and “maximally maintained vasoconstriction” between ULA1 and ULA2 (blue area). Red areas represent passive CA.
Figure 4. Relative differences in pial arteriolar diameter adjustment to changes in cerebral perfusion pressure (CPP) according to size category.
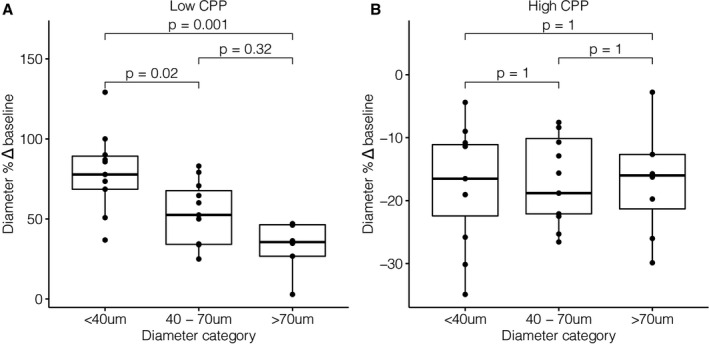
A, Maximal vasodilation per arteriole (n=26) relative to baseline (Δ%) during hypotension. Median CPP when reaching maximal vasodilation was 30.4 mm Hg (interquartile range [IQR], 4.5 mm Hg), 36.7 mm Hg (IQR, 6.4 mm Hg), and 40.5 mm Hg (IQR, 14.1 mm Hg), for small, medium, and large arterioles, respectively. B, Maximal vasoconstriction per arteriole (n=26) relative to baseline (Δ%) during hypertension. Median CPP when reaching maximal vasoconstriction was 86.0 mm Hg (IQR, 35.8 mm Hg), 87.3 mm Hg (IQR, 11.8 mm Hg), and 106.0 mm Hg (IQR, 27.2 mm Hg), for small, medium, and large arterioles, respectively. Bonferroni‐adjusted significance levels for the Wilcoxon signed‐rank test are shown above the box plots.
Phases of CBF Regulation: Quadriphasic Curve
To examine the general physiological phases of CBF regulation, we used segmented regression analysis of the CPP‐RBC flux curve of the whole cohort. A quadriphasic curve with 3 breakpoints was demonstrated (Figure 3B). The aggregated LLA based on the whole cohort breakpoint occurred at a CPP of 38.9 mm Hg (SE, 1.3 mmHg). For the aggregated ULA, 2 separate breakpoints could be observed: at 70.9 mm Hg (SE, 3.8 mm Hg) and at 113.3 mm Hg (SE, 5.5 mm Hg), termed ULA1 and ULA2, respectively. CA thus consisted of 4 phases: 2 active phases between LLA and ULA1 and ULA1 and ULA2, flanked by 2 passive phases defined by CPP below LLA or above ULA2. In the active phase between LLA and ULA1, vessel radius of the whole arteriolar bed can dilate or constrict to maintain quasi‐steady CBF (Figure 3B green area). LLA is reached before the actual maximal vasodilatory capacity is reached (Figure 3A). A second active phase between ULA1 and ULA2 consists of a differential capacity of different sizes of arterioles for further/maintained vasoconstriction at increasing CPP (Figure 3B blue area). Failure to resist increasing CPP and associated gradual passive vasodilation occurs initially at the level of the smallest arterioles (Figure 2A: hypertension experiments, circular points). Smallest pial arterioles reached maximal vasoconstriction at ULA1. CBF regulation beyond ULA1 depends on larger arterioles with additional capacity for vasoconstriction at higher CPP than ULA1, until even they fail to resist increasing CPP at ULA2 (Figure 2A: hypertension experiments, circular points). Completely passive CA is seen at CPP below LLA and above ULA2 (Figure 3B red areas).
Comparison With the Classic Triphasic Cerebrovascular Pressure‐Flow Curve
To examine differences between our results and the classic concept of CA, we compared our segmented regression breakpoints and curve slopes to a theoretical classic triphasic CA curve (Figure 5). CA is classically visualized as a triphasic curve consisting of a wide plateau of steady CBF between a CPP of 50 to 150 mm Hg flanked by a passive CPP‐CBF relation below and beyond these limits (Figure 5A). The physiology of the plateau is explained by maximal changes in cerebrovascular resistance (CVR) between the LLA and ULA. The capacity to adjust CVR from baseline is thought to be equal toward both the LLA and ULA and finite at these limits. The slope of the theoretical plateau is 0, and the slope of passive CA would be 2. We demonstrated a quadriphasic curve with a relatively narrow plateau between LLA and ULA1. Between LLA and ULA1, CVR demonstrates a large capacity for adjustments and can maintain a close to zero CPP‐CBF slope of −0.3 (95% CI, −0.7 to 0.1). Below LLA, the slope of the curve is 4.0 (95% CI, 3.3–4.6). From ULA1 toward ULA2, capacity for CVR adaptation is relatively limited, resulting in an attenuated slope of 0.9 (95% CI, 0.6–1.2) in the CPP‐CBF curve. Beyond ULA2, the slope of the curve is 2.3 (95% CI, 1.3–3.6).
Figure 5. Quadriphasic curve compared with the classic triphasic cerebral perfusion pressure (CPP)–cerebral blood flow (CBF) curve.
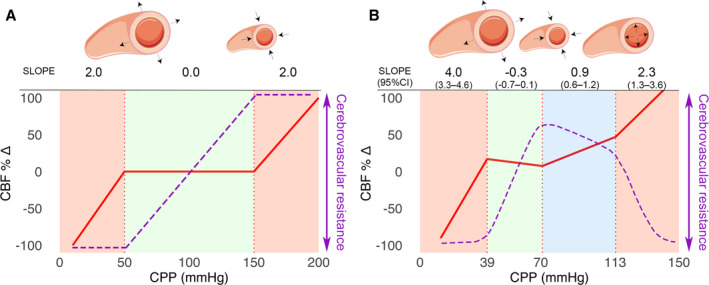
A, Cerebrovascular autoregulation (CA) is classically visualized as a triphasic curve consisting of a wide plateau of steady CBF (red curve) between a CPP of 50 and 150 mm Hg (green area). Red areas indicate absent CA with a passive CPP‐CBF relationship. Cerebrovascular resistance is represented by the purple dashed line. The underlying physiology is explained by maximal vasodilation at the lower limit of autoregulation (LLA) and maximal vasoconstriction at the upper limit of autoregulation (ULA). B, We have demonstrated a quadriphasic curve with a relatively narrow plateau between the LLA and ULA1 (green area). Between ULA1 and ULA2, there is progressive failure of CA starting at the level of the smallest arterioles and progressing toward larger arterioles (blue area) until all arterioles fail to resist increasing CPP and flow becomes completely pressure passive (right‐sided red area). During maximally maintained vasoconstriction (blue area), the CPP‐CBF relationship is sloped but attenuated.
Intersubject Variability in the Limits of CA
Our analysis has thus far focused on aggregated numbers from the total cohort to examine general physiological mechanisms. To examine intersubject variability in the limits of CA, we first determined breakpoints for each arteriole by segmented regression. For each arteriole, one breakpoint was estimated in the hypotensive experiments (LLA) and 2 breakpoints were estimated in the hypertensive experiments (ULA1 and ULA2), based on the quadriphasic curve described earlier. Breakpoint values of individual arterioles were then used to calculate one overall breakpoint per experiment. Mean LLA was 43.1 mm Hg (95% CI, 37.5–48.7 mm Hg) with an intersubject range of 29.2 mm Hg (minimum 27.9–maximum 57.1 mm Hg). Mean ULA1 was 95.4 mm Hg (95% CI, 86.1–104.7 mm Hg) with an intersubject range of 47.3 mm Hg (minimum 65.3–maximum 112.6 mm Hg). Mean ULA2 was 128.8 mm Hg (95% CI, 118.8–138.8 mm Hg) with an intersubject range of 50.9 mm Hg (minimum 97.2–maximum 148.1 mm Hg). Distribution of the intersubject limits of CA is visualized in Figure 6. Mean SD of the LLA, ULA1, and ULA2 per experiment was 1.7, 3.2, and 2.4 mm Hg, respectively, demonstrating that absolute differences in breakpoints between arterioles per experiment were relatively small. Mean baseline CPP is centered between LLA and ULA1, indicating that normal physiological CA operates between these limits.
Figure 6. Intersubject variability in the limits of cerebrovascular autoregulation (CA).
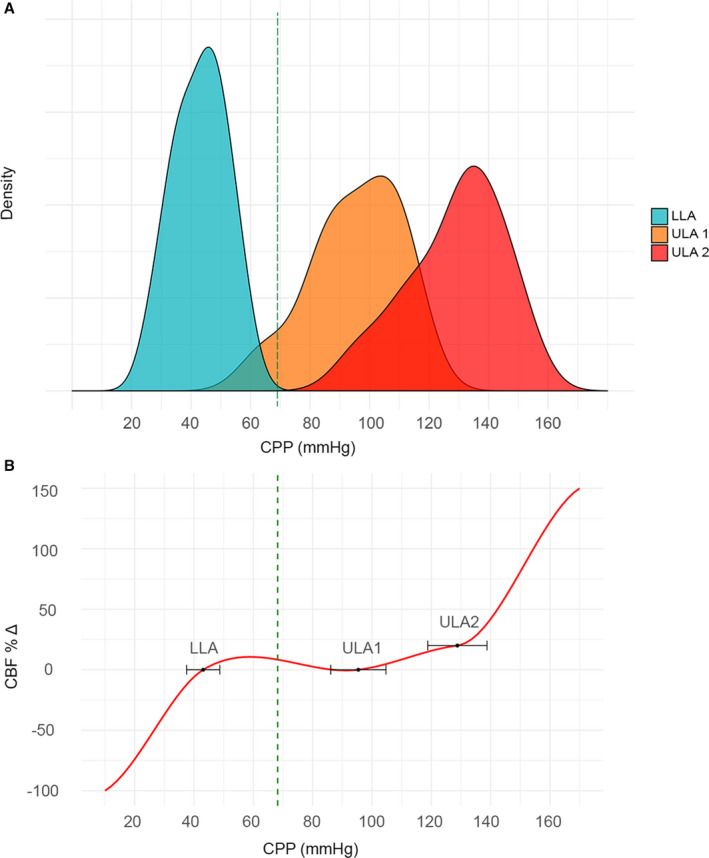
A, Density plot demonstrating the distribution of intersubject limits of CA (cerebral perfusion pressure [CPP]; in mm Hg): lower limit of autoregulation (LLA), upper limit of autoregulation (ULA) 1, and ULA2. Mean baseline CPP of 68.2 mm Hg is indicated by a green dashed vertical line. B, Illustration of the CPP–cerebral blood flow (CBF) relationship (red curve), demonstrating intersubject variability in the limits of CA (CPP in mm Hg). Black dots and horizontal error bars indicate mean LLA of 43.1 mm Hg (95% CI, 37.5–48.7 mm Hg), ULA1 of 95.4 mm Hg (95% CI, 86.1–104.7 mm Hg), and ULA2 of 128.8 mm Hg (95% CI, 118.8–138.8 mm Hg).
Discussion
To investigate the physiology of the cerebral pressure–flow relationship, we studied the CBF response to gradual changes in CPP in single‐vessel pial arterioles by direct in vivo measurement of RBC flux in a porcine animal model. The data set led to new insights into the physiology of CA and can help to improve current uncertainties about active CBF regulation and interindividual differences in CA.
Four major findings are highlighted: (1) A quadriphasic CPP‐CBF relationship with 3 breakpoints was demonstrated instead of the classic triphasic curve with 2 breakpoints. The upper limit of CA is defined by 2 breakpoints (ULA1 and ULA2), with distinct underlying mechanisms. (2) A significantly more restricted entirely active range of CA was found between LLA and ULA1. (3) CA physiology is different depending on the direction of change in CPP. (4) Substantial intersubject variability in the active regions of CA exists.
Low and high intraluminal pressure led to distinct arteriolar responses, dependent on baseline arteriolar size. The maximal capacity of pial arterioles to dilate was a median of 3.4 times larger than the capacity to constrict. These findings are in line with earlier reports by Fog and Kontos et al. 11 , 14 , 36 The smallest pial arterioles demonstrated the largest capacity to dilate in response to hypotension. The greater increase in vessel radius of the smallest arterioles can lead to a proportionally equal increase of flow, whereas larger arterioles require a smaller relative increase in radius to achieve equal increases of flow (Hagen‐Poiseuille law). Size‐based differences in vasodilation appropriately compensated for low CPP maintaining equal flow across all arterioles, regardless of baseline diameter. In contrast, the magnitude of maximal vasoconstriction in response to increasing CPP did not differ between arterioles of different size categories. At the same time, we found differences in absolute CPP at which maximal vasoconstriction was reached. Larger arterioles demonstrated a greater capacity to maintain vasoconstriction in response to high CPP, probably explained by a thicker layer of smooth muscle cells. The capacity to increase CVR depends on smooth muscle strength and thickness of the vascular smooth muscle cell layer. 37 Toward the LLA, vessels reached a point where vasodilation was no longer capable of maintaining quasi‐steady CBF (LLA), and this occurred before maximal vasodilatation was reached. The limiting factor in the LLA seems to be CPP, not the intrinsic capacity to decrease CVR. Once the LLA was reached, CBF could not be maintained any longer by decreasing vascular resistance, leading to a clear inflection point in the CPP‐CBF relationship. This difference between the LLA and maximal vasodilation has been previously reported by MacKenzie et al. 38 Toward the upper limits of CA, 2 different vascular compensatory phases can be distinguished. Initially, CVR was increased by active global vasoconstriction until the smallest arterioles reached maximal vasoconstriction (ULA1). From the point of maximal vasoconstriction at the level of the small pial arterioles, and while further increasing CPP, a second phase with a relatively wide CPP range was seen. Larger arterioles continued to actively constrict while the smallest arterioles were forced to passively dilate. Once the maximum capacity of larger arterioles and arteries was exceeded, a third inflection point was encountered beyond which all vessels passively dilated (ULA2). The limiting factor of the upper limits is the maximal capacity to increase CVR, which is dependent on arteriolar diameter.
Our experimental findings are in line with contemporary data from healthy humans that also indicated a more restricted plateau of active CA and an asymmetric response to changes in perfusion pressure. 19 , 20 , 24 , 27 , 28 Under the assumption of rightful extrapolation from the porcine model to the human physiology, we estimate a plateau region of 52.3 mm Hg with a 95% CI of 37.4 to 67.2 mm Hg between mean values of LLA and ULA1. We emphasize that LLA and ULA were not tested in the same experiments, thereby possibly explaining the wide CI. Nevertheless, the range of active CA was more restricted than the range of 100 mm Hg between a CPP of 50 and 150 mm Hg, as often cited. 16 , 17 , 18 Baseline CPP was centered between the LLA and ULA1, indicating that normal physiological CA operates between these limits. From baseline CPP, the capacity of CA to buffer increased CPP is greater than the capacity to buffer decreased CPP. The extended capacity toward increased CPP is at the expense of an attenuated but slightly passive CPP‐CBF relationship between ULA1 and ULA2. More important, our results also confirm a large intersubject variability on the limits of CA. Several authors have raised attention to the limitations of grouped analysis, such as the Lassen curve. 19 , 21 , 39 Grouped analysis can provide general physiological insights, but cannot be used on an individual level. We have demonstrated important differences in CA limits between the grouped analysis and the individual intersubject analysis. Therefore, the clinical use of fixed numbers from such grouped analyses can easily overestimate or underestimate the true limits of CA in individual patients. Also, various pathological conditions associated with disturbed CA can lead to even greater variability. Hence, research focused on individual monitoring of CA in normal and pathological conditions remains highly relevant. Patient‐specific CA monitoring tools have the potential to determine individualized optimal blood pressure treatment strategies to improve patient outcomes across many clinical fields. 40 , 41
A porcine animal model was chosen for similarities in cardiorespiratory physiology, brain anatomy, growth, and development, and cerebrovascular anatomy and physiology to humans, improving the animal‐to‐human translation. 42 , 43 , 44 , 45 , 46 , 47 The model allowed good control of Paco 2, an important confounder during ABP manipulation. 48 The model combined vessel diameter and RBC velocity to provide a complete description of RBC flux in individual pial arterioles. 29 The use of vessel diameter or RBC velocity in isolation has been proven insufficient. 49 , 50 CPP was manipulated nonpharmacologically to avoid direct effects on cerebrovascular tone. Still, several possible limitations should be considered. First, limitations can arise from the experimental protocol. Although significant precautions were taken to avoid damage, installation of a closed cranial window may have disturbed the local physiological environment to some extent. We chose an anesthesia protocol commonly used in neurointensive care, excluding inhalation anesthetics. Nevertheless, undesired interference of anesthesia cannot be ruled out. Placement of an uninflated balloon catheter in the inferior vena cava or abdominal aorta inadvertently led to a slight change in ABP that was reflected in a limited but statistically significant difference in baseline CPP between hypertension and hypotension experiments. Second, RBC flux is not a perfect representation of whole blood flow, as it does not include plasma, white blood cells, platelets, and other blood components. Although calculated RBC flux is not exactly similar to absolute CBF, it does allow to reliably reflect relative changes. Third, larger upstream vessels were not directly studied. However, upstream effects should be indirectly reflected downstream by changes in RBC flux. Fourth, the mix of arterioles of different diameters studied per experiment was chosen on the basis of sufficient resolution under the microscope and may not have been fully representative of diameter distribution. Fifth, CPP was manipulated in one direction per experiment, limiting intrasubject interpretations of differences between the lower and upper limits of CA. We speculated that manipulation of CPP beyond the limits of CA could damage normal physiology and possibly confound experimental data from a subsequent CPP manipulation in another direction. Sixth, because of technical complexity and practical and economical restrictions in this large animal model, a randomized and blinded experimental design was not possible. The lack of randomization and blinding may have unwillingly led to the introduction of a bias. Each experimental animal served as its own control by multimodal physiological monitoring during the entire experimental duration and comparison with baseline values before and during ABP manipulation.
We hope the current insights will help to improve models used for measuring CA in humans and improve our general understanding of the physiology and pathophysiology of CBF regulation.
Sources of Funding
This work was supported in part by a Johnson & Johnson Research Chair in profound analysis of cerebrovascular autoregulation at KU Leuven University.
Disclosures
None.
Supporting information
Figures S1–S3
Acknowledgments
The authors would like to thank veterinarian Stéphanie De Vleeschauwer and the KU Leuven Animal Research Center for valuable technical help.
Supplemental Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.022943
For Sources of Funding and Disclosures, see page 12.
References
- 1. Hossmann KA. Pathophysiology and therapy of experimental stroke. Cell Mol Neurobiol. 2006;26:1057–1083. doi: 10.1007/s10571-006-9008-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aries MJH, Elting JW, De Keyser J, Kremer BPH, Vroomen PCAJ. Cerebral autoregulation in stroke: a review of transcranial Doppler studies. Stroke. 2010;41:2697–2704. doi: 10.1161/STROKEAHA.110.594168 [DOI] [PubMed] [Google Scholar]
- 3. Zeiler FA, Ercole A, Beqiri E, Cabeleira M, Thelin EP, Stocchetti N, Steyerberg EW, Maas AIR, Menon DK, Czosnyka M, et al. Association between cerebrovascular reactivity monitoring and mortality is preserved when adjusting for baseline admission characteristics in adult traumatic brain injury: a center‐TBI study. J Neurotrauma. 2019;1241:1233–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Budohoski KP, Czosnyka M, Kirkpatrick PJ, Smielewski P, Steiner LA, Pickard JD. Clinical relevance of cerebral autoregulation following subarachnoid haemorrhage. Nat Rev Neurol. 2013;9:152–163. doi: 10.1038/nrneurol.2013.11 [DOI] [PubMed] [Google Scholar]
- 5. O’Leary H, Gregas MC, Limperopoulos C, Zaretskaya I, Bassan H, Soul JS, Di Salvo DN, du Plessis AJ. Elevated cerebral pressure passivity is associated with prematurity‐related intracranial hemorrhage. Pediatrics. 2009;124:302–309. doi: 10.1542/peds.2008-2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vutskits L. Cerebral blood flow in the neonate. Paediatr Anaesth. 2014;24:22–29. doi: 10.1111/pan.12307 [DOI] [PubMed] [Google Scholar]
- 7. Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387 [DOI] [PubMed] [Google Scholar]
- 8. Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017;18:419–434. doi: 10.1038/nrn.2017.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fog M. The relationship between the blood pressure and the tonic regulation of the pial arteries. J Neurol Psychiatry. 1938;1:187–197. doi: 10.1136/jnnp.1.3.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lassen N. Cerebral blood flow and oxygen consumption in man. Physiol Rev. 1959;39:183–238. doi: 10.1152/physrev.1959.39.2.183 [DOI] [PubMed] [Google Scholar]
- 11. Kontos HA, Wei EP, Navari RM, Levasseur JE, Rosenblum WI, Patterson JL. Responses of cerebral arteries and arterioles to acute hypotension and hypertension. Am J Physiol. 1978;234:H371–H383. doi: 10.1152/ajpheart.1978.234.4.H371 [DOI] [PubMed] [Google Scholar]
- 12. Faraci FM, Mayhan WG, Heistad DD. Segmental vascular responses to acute hypertension in cerebrum and brain stem. Am J Physiol. 1987;252:H738–H742. doi: 10.1152/ajpheart.1987.252.4.H738 [DOI] [PubMed] [Google Scholar]
- 13. Faraci FM, Heistad DD. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ Res. 1990;66:8–17. doi: 10.1161/01.RES.66.1.8 [DOI] [PubMed] [Google Scholar]
- 14. Fog M. Cerebral circulation: the reaction of the pial arteries to a fall in blood pressure. Arch Neurol Psychiatry. 1937;37:351–364. doi: 10.1001/archneurpsyc.1937.02260140137007 [DOI] [Google Scholar]
- 15. Forbes HS, Nason MS, Wortman RCBS. Cerebral circulation. Arch Neurol Psychiatry. 1937;37:334. doi: 10.1001/archneurpsyc.1937.02260140120006 [DOI] [Google Scholar]
- 16. Dorrance AM. The effects of hypertension and stroke on the cerebral vasculature. In: Aiyagari V, Gorelick PB, eds. Hypertension and Stroke. Springer International Publishing; 2016:81–108. [Google Scholar]
- 17. Dagal A, Lam AM. Cerebral autoregulation and anesthesia. Curr Opin Anaesthesiol. 2009;22:547–552. doi: 10.1097/ACO.0b013e32833020be [DOI] [PubMed] [Google Scholar]
- 18. Walsh MP, Cole WC. The role of actin filament dynamics in the myogenic response of cerebral resistance arteries. J Cereb Blood Flow Metab. 2013;33:1–12. doi: 10.1038/jcbfm.2012.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Willie CK, Tzeng Y‐C, Fisher JA, Ainslie PN. Integrative regulation of human brain blood flow. J Physiol. 2014;592:841–859. doi: 10.1113/jphysiol.2013.268953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Numan T, Bain AR, Hoiland RL, Smirl JD, Lewis NC, Ainslie PN. Static autoregulation in humans: a review and reanalysis. Med Eng Phys. 2014;36:1487–1495. doi: 10.1016/j.medengphy.2014.08.001 [DOI] [PubMed] [Google Scholar]
- 21. Tzeng YC, Ainslie PN. Blood pressure regulation IX: cerebral autoregulation under blood pressure challenges. Eur J Appl Physiol. 2014;114:545–559. doi: 10.1007/s00421-013-2667-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zeiler FA, Donnelly J, Calviello L, Lee JK, Smielewski P, Brady K, Kim D‐J, Czosnyka M. Validation of pressure reactivity and pulse amplitude indices against the lower limit of autoregulation, part I: experimental intracranial hypertension. J Neurotrauma. 2018;35:2803–2811. doi: 10.1089/neu.2017.5603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brady KM, Easley RB, Kibler K, Kaczka DW, Andropoulos D, Fraser CD, Smielewski P, Czosnyka M, Adams GJ, Rhee CJ, et al. Positive end‐expiratory pressure oscillation facilitates brain vascular reactivity monitoring. J Appl Physiol. 2012;113:1362–1368. doi: 10.1152/japplphysiol.00853.2012 [DOI] [PubMed] [Google Scholar]
- 24. Tan CO. Defining the characteristic relationship between arterial pressure and cerebral flow. J Appl Physiol. 2012;113:1194–1200. doi: 10.1152/japplphysiol.00783.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brady KM, Lee JK, Kibler KK, Easley RB, Koehler RC, Shaffner DH. Continuous measurement of autoregulation by spontaneous fluctuations in cerebral perfusion pressure comparison of 3 methods. Stroke. 2008;39:2531–2537. doi: 10.1161/STROKEAHA.108.514877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pesek M, Kibler K, Easley RB, Mytar J, Rhee C, Andropoulos D, Brady K. The upper limit of cerebral blood flow autoregulation is decreased with elevations in intracranial pressure. Neurosurgery. 2014;75:163–169. doi: 10.1227/NEU.0000000000000367 [DOI] [PubMed] [Google Scholar]
- 27. Aaslid R, Blaha M, Sviri G, Douville CM, Newell DW. Asymmetric dynamic cerebral autoregulatory response to cyclic stimuli. Stroke. 2007;38:1465–1469. doi: 10.1161/STROKEAHA.106.473462 [DOI] [PubMed] [Google Scholar]
- 28. Schmidt B, Klingelhöfer J, Perkes I, Czosnyka M. Cerebral autoregulatory response depends on the direction of change in perfusion pressure. J Neurotrauma. 2009;26:651–656. doi: 10.1089/neu.2008.0784 [DOI] [PubMed] [Google Scholar]
- 29. Klein SP, De Sloovere V, Meyfroidt G, Depreitere B. Autoregulation assessment by direct visualisation of pial arterial blood flow in the piglet brain. Sci Rep. 2019;9:13333. doi: 10.1038/s41598-019-50046-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Armstead WM, Riley J, Vavilala MS. Sex and age differences in epinephrine mechanisms and outcomes after brain injury. J Neurotrauma. 2017;34:1666–1675. doi: 10.1089/neu.2016.4770 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Levasseur JE, Wei EP, Raper AJ, Kontos HA, Patterson JL. Detailed description of a cranial window technique for acute and chronic experiments. Stroke. 1975;6:308–317. doi: 10.1161/01.STR.6.3.308 [DOI] [PubMed] [Google Scholar]
- 32. Busija DW, Leffler CW, Wagerle LC. Responses of newborn pig pial arteries to sympathetic nervous stimulation and exogenous norepinephrine. Pediatr Res. 1985;19:1210–1214. doi: 10.1203/00006450-198511000-00020 [DOI] [PubMed] [Google Scholar]
- 33. Giarratana M‐C, Kobari L, Lapillonne H, Chalmers D, Kiger L, Cynober T, Marden MC, Wajcman H, Douay L. Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. Nat Biotechnol. 2005;23:69–74. doi: 10.1038/nbt1047 [DOI] [PubMed] [Google Scholar]
- 34. Rovainen CM, Woolsey TA, Blocher NC, Wang DB, Robinson OF. Blood flow in single surface arterioles and venules on the mouse somatosensory cortex measured with videomicroscopy, fluorescent dextrans, nonoccluding fluorescent beads, and computer‐assisted image analysis. J Cereb Blood Flow Metab. 1993;13:359–371. doi: 10.1038/jcbfm.1993.49 [DOI] [PubMed] [Google Scholar]
- 35. Shih AY, Driscoll JD, Drew PJ, Nishimura N, Schaffer CB, Kleinfeld D. Two‐photon microscopy as a tool to study blood flow and neurovascular coupling in the rodent brain. J Cereb Blood Flow Metab. 2012;32:1277–1309. doi: 10.1038/jcbfm.2011.196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fog M. Cerebral circulation II: reaction of pial arteries to increase in blood pressure. Arch Neurol Psychiatry. 1939;41:260. doi: 10.1001/archneurpsyc.1939.02270140046003 [DOI] [Google Scholar]
- 37. Rhodin JAG. The ultrastructure of mammalian arterioles and precapillary sphincters. J Ultrastruct Res. 1967;18:181–223. doi: 10.1016/S0022-5320(67)80239-9 [DOI] [PubMed] [Google Scholar]
- 38. MacKenzie ET, Farrar JK, Fitch W, Graham DI, Gregory PC, Harper AM. Effects of hemorrhagic hypotension on the cerebral circulation, I: cerebral blood flow and pial arteriolar caliber. Stroke. 1979;10:711–718. doi: 10.1161/01.STR.10.6.711 [DOI] [PubMed] [Google Scholar]
- 39. Drummond JC. The lower limit of autoregulation: time to revise our thinking? Anesthesiology. 1997;86:1431–1433. doi: 10.1097/00000542-199706000-00034 [DOI] [PubMed] [Google Scholar]
- 40. Beqiri E, Smielewski P, Robba C, Czosnyka M, Cabeleira MT, Tas J, Donnelly J, Outtrim JG, Hutchinson P, Menon D, et al. Feasibility of individualised severe traumatic brain injury management using an automated assessment of optimal cerebral perfusion pressure: the COGiTATE phase II study protocol. BMJ Open. 2019;9:e030727. doi: 10.1136/bmjopen-2019-030727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pham P, Bindra J, Aneman A, Chuan A, Worthington JM, Jaeger M. Noninvasive monitoring of dynamic cerebrovascular autoregulation and “optimal blood pressure” in normal adult subjects. Neurocrit Care. 2019;30:201–206. doi: 10.1007/s12028-018-0600-2 [DOI] [PubMed] [Google Scholar]
- 42. Buckley NM. Maturation of circulatory system in three mammalian models of human development. Comp Biochem Physiol A Comp Physiol. 1986;83:1–7. doi: 10.1016/0300-9629(86)90080-0 [DOI] [PubMed] [Google Scholar]
- 43. Xiong YE, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lind NM, Moustgaard A, Jelsing J, Vajta G, Cumming P, Hansen AK. The use of pigs in neuroscience: modeling brain disorders. Neurosci Biobehav Rev. 2007;31:728–751. doi: 10.1016/j.neubiorev.2007.02.003 [DOI] [PubMed] [Google Scholar]
- 45. Sorby‐Adams AJ, Vink R, Turner RJ. Large animal models of stroke and traumatic brain injury as translational tools. Am J Physiol Integr Comp Physiol. 2018;315:R165–R190. doi: 10.1152/ajpregu.00163.2017 [DOI] [PubMed] [Google Scholar]
- 46. Duhaime AC. Large animal models of traumatic injury to the immature brain. Dev Neurosci. 2006;28:380–387. doi: 10.1159/000094164 [DOI] [PubMed] [Google Scholar]
- 47. Morales DM, Marklund N, Lebold D, Thompson HJ, Pitkanen A, Maxwell WL, Longhi L, Laurer H, Maegele M, Neugebauer E, et al. Experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience. 2005;136:971–989. doi: 10.1016/j.neuroscience.2005.08.030 [DOI] [PubMed] [Google Scholar]
- 48. Meng L, Gelb AW. Regulation of cerebral autoregulation by carbon dioxide. Anesthesiology. 2015;122:196–205. doi: 10.1097/ALN.0000000000000506 [DOI] [PubMed] [Google Scholar]
- 49. Kontos HA. Validity of cerebral arterial blood flow calculations from velocity measurements. Stroke. 1989;20:1–3. doi: 10.1161/01.STR.20.1.1 [DOI] [PubMed] [Google Scholar]
- 50. Shih AY, Friedman B, Drew PJ, Tsai PS, Lyden PD, Kleinfeld D. Active dilation of penetrating arterioles restores red blood cell flux to penumbral neocortex after focal stroke. J Cereb Blood Flow Metab. 2009;29:738–751. doi: 10.1038/jcbfm.2008.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S3