

Abstract
Background
Pediatric dilated cardiomyopathy (DCM) is a well‐known clinical entity; however, phenotype–genotype correlations are inadequately described. Our objective was to provide genotype associations with life‐threatening cardiac outcomes in pediatric DCM probands.
Methods and Results
We performed a retrospective review of children with DCM at a large pediatric referral center (2007–2016), excluding syndromic, chemotherapy‐induced, and congenital heart disease causes. Genetic variants were adjudicated by an expert panel and an independent clinical laboratory. In a cohort of 109 pediatric DCM cases with a mean age at diagnosis of 4.2 years (SD 5.9), life‐threatening cardiac outcomes occurred in 47% (42% heart transplant, 5% death). One or more pathogenic/likely pathogenic variants were present in 40/109 (37%), and 36/44 (82%) of pathogenic/likely pathogenic variants occurred in sarcomeric genes. The frequency of pathogenic/likely pathogenic variants was not different in patients with familial cardiomyopathy (15/33 with family history versus 25/76 with no family history, P=0.21). TTN truncating variants occurred in a higher percentage of children diagnosed as teenagers (26% teenagers versus 6% younger children, P=0.01), but life‐threatening cardiac outcomes occurred in both infants and teenagers with these TTN variants. DCM with left ventricular noncompaction features occurred in 6/6 patients with MYH7 variants between amino acids 1 and 600.
Conclusions
Sarcomeric variants were common in pediatric DCM. We demonstrated genotype‐specific associations with age of diagnosis and cardiac outcomes. In particular, MYH7 had domain‐specific association with DCM with left ventricular noncompaction features. Family history did not predict pathogenic/likely pathogenic variants, reinforcing that genetic testing should be considered in all children with idiopathic DCM.
Keywords: dilated cardiomyopathy, genotype, pediatrics, transplant
Subject Categories: Cardiomyopathy, Heart Failure, Genetics, Arrhythmias, Mortality/Survival
Nonstandard Abbreviations and Acronyms
- DCM
dilated cardiomyopathy
- M‐LVNC
dilated cardiomyopathy with left ventricular noncompaction features
- P/LP
pathogenic/likely pathogenic
- VUS
variants of uncertain significance
Clinical Perspective
What Is New?
Life‐threatening cardiac outcomes occurred in approximately half of patients with pediatric dilated cardiomyopathy.
Pathogenic/likely pathogenic variants occurred in one third of patients and there was a high prevalence of variants in sarcomeric genes (>80%).
Dilated cardiomyopathy in the setting of TTN truncating variants was associated with life‐threatening cardiac outcomes in infants and older children.
What Are the Clinical Implications?
Family history did not predict pathogenic/likely pathogenic variants, reinforcing that genetic testing should be considered in all children with idiopathic dilated cardiomyopathy.
TTN truncating variants were associated with severe outcomes at all ages and should be considered as potentially pathogenic even in infants and young children.
Dilated cardiomyopathy (DCM) is a heart muscle disease characterized by ventricular dilation associated with systolic dysfunction. 1 Between one third and one half of children with DCM progress to heart failure, death, or transplantation within 2 years of diagnosis. DCM is the indication for 45% of heart transplants in the pediatric population, primarily associated with chronic symptomatic heart failure. 2 Sudden death has an annual incidence of 2% to 3% in pediatric patients with DCM. 3 , 4
Between 25% and 50% of DCM cases are familial, underscoring the importance of genetic contribution. 5 , 6 The pathogenesis of genetically mediated DCM has been associated with genes that encode sarcomeric proteins, components of the cytoskeleton, nuclear envelope proteins, and ion channels. 7 Even among DCM associated with nongenetic causes such as hypertension, valve disease, inflammatory/infectious causes, and toxins, genetic background may influence clinical phenotype and outcome. 5
Genetic Variation Linked to Age of Onset and Clinical Course
For certain genes, age of onset and genetic DCM have been associated. For example, some RAF‐1 variants have been associated with childhood‐onset DCM. 8 In Becker muscular dystrophy, region‐specific dystrophin deletions are associated with earlier onset of cardiomyopathy. 9 Adults with nonsense mutations in the BAG3 gene had early‐onset DCM with arrhythmias. 10 In addition, there is evidence that some genetic variants predict disease severity. LMNA variants in DCM can present with conduction system abnormalities and arrhythmia before ventricular dilation. 11 Adult studies have also demonstrated that patients with LMNA variants have high rates of heart transplantation and those with RBM20 variants were transplanted at a younger age. 12 Specific variants in PLN and TPM1 have also been identified as particularly severe in certain populations. 13 , 14
Clinical Implications in Children
Although studies have examined outcomes of genetic testing in adults with DCM, there is a paucity of data on the yield, mutations, and clinical correlates with genetic testing in the pediatric DCM population. To address this, we collected information on children who presented with DCM and had undergone clinical genetic testing using gene panels.
Methods
We performed a retrospective review of patients with DCM at a single tertiary pediatric referral center. Cases were included if they presented in the inpatient or outpatient setting from 2007 to 2016, received a clinical evaluation by a cardiomyopathy specialist, and had at least 1 commercial genetic panel that included cardiomyopathy genes (Table S1). The Institutional Review Board at Ann & Robert H. Lurie Children’s Hospital provided human subjects protection. The data that support the findings of this study are available in the Supplemental Material or can be obtained from the corresponding author upon reasonable request.
Inclusion and Exclusion Criteria
Clinical inclusion was determined using echocardiographic criteria for DCM established by the PCMR (Pediatric Cardiomyopathy Registry). 15 These criteria require 1 or more of the following: (1) left ventricular fractional shortening or ejection fraction (EF) >2 SDs below the normal mean for age, (2) end systolic left ventricular posterior wall thickness >2 SDs below the normal mean for body‐surface area, or (3) left ventricular end‐diastolic dimension or volume >2 SDs above the normal mean for body‐surface area. Three patients with DCM received a heart transplant elsewhere and detailed echocardiographic measurements from the explanted heart were not available for review.
Clinical exclusion criteria included having a known genetic syndrome with definitive extracardiac manifestations including neuromuscular disease; a complex congenital heart defect leading to ventricular dysfunction; significant iatrogenic exposures associated with the development of cardiomyopathy (eg, anthracycline therapy); or a minimal genetic evaluation (eg, specific testing for only 1 gene rather than a panel approach). To minimize selection bias, we included patients with a diagnosis of myocarditis if they also met the primary PCMR inclusion criteria.
Data Collection
Study data were collected and managed using REDCap (Research Electronic Data Capture) hosted at Northwestern University Clinical and Translational Sciences Institute. 16 Baseline information included demographics, presentation data (age at diagnosis, length of stay, and presence of significant arrhythmias), family history data, and cardiac diagnostic data. Report of race was obtained by retrospective chart review and classified according to the nomenclature available in our electronic medical record. The primary outcome measure was a life‐threatening cardiac outcome, defined as 1 or more of the following: resuscitated cardiac arrest, mechanical circulatory support, heart transplant, or death secondary to a cardiac cause.
All available data in the echocardiographic database were reviewed and data from the echocardiogram with the lowest EF (or fractional shortening if EF was not available) were used for primary analysis. Mitral regurgitation was based on a 7‐point ordinal scale ranging from “Absent” to “Severe,” based on the echocardiographer’s written interpretation. Unclear interpretations were evaluated by direct image review. Rhythm abnormalities were tabulated exclusively from pretransplant hearts and included sustained atrial, junctional, or ventricular arrhythmias or nonsustained ventricular arrhythmias (≥4 beats).
A patient was classified with familial cardiomyopathy if any relative(s) had cardiomyopathy. The subgroup with familial cardiomyopathy was divided into those who had first‐degree relatives with cardiomyopathy and those with only higher‐degree relatives with cardiomyopathy.
During the study dates, the following commercial panels were used: the Pan Cardiomyopathy Panel and the DCM Panels A/B by Laboratory for Molecular Medicine (Harvard Partners), the Comprehensive Cardiomyopathy Panel and the Combined Cardiac Panel (GeneDx), and the Familion DCM Test (Transgenomic).
Variant Classification
All variants were clinically adjudicated using American College of Medical Genetics Standards and Guidelines interpreted by a committee of adult and pediatric cardiomyopathy and arrhythmia practitioners, and a genetic counselor with expertise in cardiomyopathy (authors R.K., E.M, E.P., L.D‐C., and G.W.), all blinded to the identity of the patients, but aware of the clinical phenotype and variant segregation within the family. This clinical adjudication was used for the primary analysis. ACTN2, MYBPC3, MYH7, MYL2, TNNC1, TNNT2, TPM1, and TTN were classified as genes encoding sarcomeric proteins. Missense variants in TTN were excluded from analysis. To minimize potential classification bias, we also submitted all variants for independent classification in a Clinical Laboratory Improvement Amendments (CLIA)‐certified laboratory according to the American College of Medical Genetics Standards and Guidelines. 17 The independent laboratory received the standard phenotype details that would be available during clinical variant analysis. The classification from the original report at the time of patient care and both reclassifications are tabulated in Table S2.
Statistical Analysis
Chi‐squared or Fisher exact analyses (Stata 15.1, College Station, TX) were used to compare categorical variables, including univariate analysis of variant pathogenicity and clinical/demographic features. A Spearman correlation coefficient was used to assess mitral regurgitation against continuous variables and a Student t test was used for left ventricular end diastolic Z‐scores, EF, and age at presentation. A value of P<0.05 was accepted as statistically significant. The clinical variant adjudication was used for analysis; however, analyses were repeated using the independent classification from the independent laboratory to ensure that the classification schema did not change the statistical significance.
Results
Patient Characteristics
We identified 299 children with DCM from January 1, 2007 to December 31, 2016, without syndromic features, congenital heart disease, or chemotherapy exposure. Of those, 109 of 299 (36%) were tested with a cardiomyopathy genetic panel. Patients who received a genetic panel had a more severe clinical presentation, with a lower mean left ventricular EF at diagnosis (29.4% versus 50.1%, P<0.001) and a lower EF on their most severely depressed echocardiogram (24.1% versus 39.9%, P<0.001), when compared with nongenotyped patients.
Among the 109 genotyped patients retained for further analysis, the mean age at diagnosis was 4.2 years (±5.9), with a range of 0 to 17.6 years. Life‐threatening cardiac outcomes (resuscitated cardiac arrest, mechanical circulatory support, heart transplant, or death) occurred in 51 patients (47%), of whom 46 underwent heart transplant. Figure 1 shows the time from diagnosis to the first life‐threatening cardiac outcome. Two patients died without having undergone heart transplant and 6 died after heart transplant.
Figure 1. Kaplan–Meier analysis, time from diagnosis to life‐threatening cardiac outcome (years).
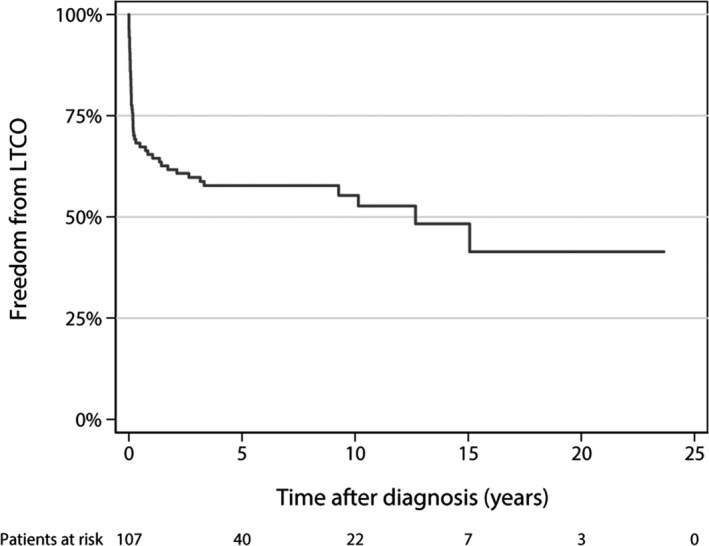
A life‐threatening cardiac outcome (LTCO) was 1 or more of the following: resuscitated cardiac arrest, mechanical circulatory support, heart transplant, or death secondary to a cardiac cause.
A more concise end point of death or transplant was also considered. Death or transplant occurred in 48/51 patients with a life‐threatening cardiac outcome. The other 3 patients had a cardiac arrest or sustained ventricular tachycardia with syncope. A sensitivity analysis for the population with only death or transplant was unchanged with respect to genotype prevalence.
Table tabulates clinical and echocardiographic data. There was no meaningful latency between presentation with symptoms and initial imaging diagnosis. An imaging diagnosis was obtained on the same day as clinical presentation in 91/96 patients (95%); the other 5 received definitive imaging within 10 days of presentation. Moderate or higher‐grade mitral regurgitation was present at diagnosis in 31% of hearts (13 severe or moderate‐to‐severe, 15 moderate, 31 mild or mild‐to‐moderate, and 30 trivial or none). As expected, severity of mitral regurgitation at presentation correlated with left ventricular end‐diastolic diameter (r=0.47; P<0.001).
Table 1.
Clinical Characteristics of Genotyped Pediatric DCM Cohort
| All patients (N=109) | Patients with P/LP variants (N=40) |
Patients with no P/LP variants (N=69) |
P value | |
|---|---|---|---|---|
| Age at diagnosis, y | 4.2 (5.9) | 4.2 (6.1) | 4.3 (5.9) | 0.96 |
| Sex | ||||
| Male | 59 | 19 | 40 | 0.32 |
| Female | 50 | 21 | 29 | |
| Race or ethnicity | ||||
| Black | 25 | 7 | 18 | 0.52 |
| Asian | 7 | 3 | 4 | |
| Hispanic | 24 | 7 | 17 | |
| White | 46 | 21 | 25 | |
| Other | 7 | 2 | 5 | |
| Life‐threatening cardiac outcome* | ||||
| Yes | 51 | 20 | 31 | 0.61 |
| No | 58 | 20 | 38 | |
| Family history | ||||
| Yes | 33 | 15 | 18 | 0.21 |
| No | 76 | 25 | 51 | |
| Myocarditis history | ||||
| Yes | 16 | 3 | 13 | 0.11 |
| No | 93 | 37 | 56 | |
| Any arrhythmia | 0.40 | |||
| Yes | 41 | 13 | 28 | |
| No | 68 | 27 | 41 | |
| Ventricular arrhythmias | ||||
| Yes | 27 | 11 | 16 | 0.62 |
| No | 82 | 29 | 53 | |
| EF at diagnosis | 29.4 (16.7) | 29.3 (15.8) | 29.5 (16.7) | 0.95 |
| LVEDD Z‐score at diagnosis | 4.3 (3.3) | 4.0 (3.1) | 4.6 (3.5) | 0.44 |
| Lowest EF | 24.1 (13.2) | 21.3 (12.3) | 26.2 (13.6) | 0.10 |
| Largest LVEDD Z‐score | 5.1 (3.3) | 5.2 (3.8) | 5.0 (3.0) | 0.81 |
Numbers in parentheses are SD in normally distributed variables. DCM indicates dilated cardiomyopathy; EF, ejection fraction; LVEDD, left ventricular end‐diastolic dimension; and P/LP, pathogenic/likely pathogenic.
“Life‐threatening cardiac outcome” was 1 or more of the following: resuscitated cardiac arrest, mechanical circulatory support, heart transplantation, or death secondary to a cardiac cause.
Pathogenic Gene Variants
Forty children (37%) had at least 1 likely pathogenic/likely pathogenic (P/LP) variant in a gene associated with DCM and only a single P/LP variant was present in 37 of these 40 children. A life‐threatening cardiac outcome occurred in half of patients with a P/LP variant (20/40; 19 transplants and 1 mechanical support). The 3 children with >1 P/LP variant had varying severity of clinical courses from mildly affected to arrest and transplant (Cases 12, 16, and 37 in Table S4).
In total, there were 44 P/LP variants in the cohort, and 36/44 (82%) occurred in genes encoding sarcomeric proteins (Figure 2). One or more P/LP sarcomeric variants occurred in 29 of the 40 patients with a P/LP variant (73%). MYH7 was the gene with the greatest number of P/LP variants; 11 patients had a P/LP variant in MYH7.
Figure 2. Distribution of genes for pathogenic and likely pathogenic variants.

Pie chart displaying the distribution of all genes containing pathogenic or likely pathogenic (P/LP) variants, weighted by the number of P/LP variants in each gene.
TTN Variants in Teenage Patients
Premature termination codons, insertion/deletions, and splice‐site variants in TTN were considered P/LP since they result in TTN truncations (Table S3). TTN truncation variants were the second most common gene mutation observed, occurring in 10 patients, with a median age of 9.7 years. Patients diagnosed with DCM as teenagers (between age 13 and 18 years) were >4 times as likely to have TTN truncations as those diagnosed before age 13 years (5/19 versus 5/90, P=0.01). Nonetheless, life‐threatening cardiac outcomes did not exclusively occur in teenage presentations. Life‐threatening cardiac outcomes occurred in 6 of the 10 patients with P/LP TTN variants, including 2 patients who presented before 1 year of age.
Variants of Uncertain Significance
At least 1 variants of uncertain significance (VUS) was identified in 43/109 patients (39%), of whom 30 had no P/LP variants. In contrast to the 82% of P/LP variants that were in genes encoding sarcomeric proteins, only 19% of VUS were in genes encoding sarcomeric proteins (16/85 VUS versus 36/44 P/LP, P<0.01). Neither the presence of a P/LP variant in a sarcomeric gene nor the presence of any P/LP/VUS variant in a sarcomeric gene was associated with life‐threatening cardiac outcome (P=0.85 and 0.83, respectively).
DCM With Left Ventricular Noncompaction Features in Domain‐Specific MYH7
All 6 children with MYH7 variants encoding changes within amino acid residues 1 and 600 presented with a dilated cardiomyopathy with left ventricular noncompaction features (DCM‐LVNC) phenotype. However, none of the 6 patients with DCM with MYH7 variants encoding changes in amino acids 601 through 1935 had a DCM‐LVNC phenotype (Figure 3).
Figure 3. Domain‐specific association with DCM‐LVNC in MYH7.
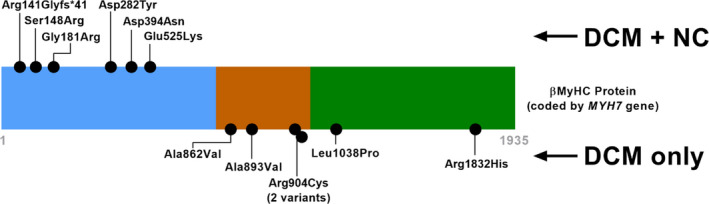
Schematic illustration of each variant positioned in the 1935 amino‐acid β‐myosin heavy chain protein. Pediatric DCM cases with variants mapping to the head region of the protein (blue) had a phenotypic presentation of DCM and DCM‐LVNC, whereas those with variants in the neck (orange) and rod (green) regions presented with DCM alone. DCM indicates dilated cardiomyopathy; DCM‐LVNC, dilated cardiomyopathy with left ventricular noncompaction; and β‐MyHC, β‐myosin heavy chain protein.
Familial Cardiomyopathy
The subject was the proband in nearly all cases (96%); the remaining 4 patients were each the sibling of a proband. Familial cardiomyopathy was present in 33 patients (30%). Among these, 25/33 (75%) had a first‐degree relative with cardiomyopathy: 14 had a parent, 14 had at least 1 sibling, and 3 had both a parent and a sibling with cardiomyopathy. Eight patients had no first‐degree relatives with cardiomyopathy, but had at least 1 higher‐degree relative with cardiomyopathy. The frequency of P/LP variants was not different in patients with familial cardiomyopathy (15/33 with family history versus 25/76 with no family history, P=0.21), but we were underpowered to detect a prevalence smaller than 26%.
Additional Patient Characteristics
Of 109 genotyped DCM cases, rhythm abnormalities occurred in 41 (38%), of which 27 had ventricular arrhythmias. Among patients with ventricular arrhythmias, 11 had P/LP variants in genes associated with pediatric DCM (MYH7, TNNT2, TPM1, and TTN). The presence of a P/LP variant was not associated with a higher rate of ventricular arrhythmia (11/27 versus 31/82, P=0.82). Two patients carried P/LP variants in TTR, a gene that has not been associated with pediatric DCM and instead is a risk factor for late‐onset adult restrictive cardiomyopathy. One patient had both MYH7 and TTR P/LP variants, and 1 patient carried only a heterozygous P/LP variant in TTR. There is insufficient evidence to link these heterozygous TTR findings to a specific phenotype, but they are included here for completeness because these findings were reported in the patients’ genetic testing.
Of the 16 patients (15%) who met criteria for DCM and had a clinical diagnosis of myocarditis, 10 also had positive viral testing. The viruses identified were typical myocarditis pathogens including enterovirus, coronavirus, parvovirus, and influenza. Biopsy was not routinely performed (1/16). There was no difference in the frequency of P/LP variants in patients with myocarditis (3/16) versus patients without a myocarditis diagnosis (37/93, P=0.11), nor was there any difference in life‐threatening cardiac outcome (8/16 with myocarditis versus 43/93 without myocarditis, P=0.78).
Cardiac magnetic resonance imaging was performed in 35 patients, of whom 34% had P/LP variants. Delayed enhancement with gadolinium contrast was present in 10 patients, but only 1 patient had both delayed enhancement and a P/LP variant.
When considering the most stringent adjudication, where the clinical adjudication and the independent classification agreed on P/LP variants, patients with 1 or more P/LP variants were more likely to have a life‐threatening cardiac outcome (15/22 with P/LP versus 36/87 without P/LP, P=0.02), perhaps suggesting that the most pathogenic variants may be associated with increased risk.
Updates in Genetic Classification
From the time when the original genetic testing was conducted, an average of 6.2 years elapsed to the time when we re‐examined and re‐classified variants as part of this study (SD 2.3 years). Among 144 variants, 115 retained their original classification (6 benign/likely benign, 82 VUS, and 27 P/LP). Among the 108 variants originally adjudicated as VUS on clinical reports, 17 were updated as P/LP (16% of variants) and 9 variants were downgraded to benign/likely benign. A full disclosure of the original report classification, clinical adjudication, and the independent laboratory adjudication is provided in Table S2.
Discussion
Genetic Landscape for Pediatric DCM Differs From Adult DCM
Clinical genetic testing with a gene panel identified a P/LP variant in 37% of children with DCM who were clinically genotyped in our center. An additional 28% had VUS, but no P/LP variant, and the final one third had only benign gene variants or no identified variants. Among the patients with P/LP variants, 73% had at least 1 P/LP variant in a sarcomeric gene and sarcomeric variants accounted for 82% of all P/LP variants (some patients had >1 P/LP variant). The high prevalence of sarcomeric P/LP variants contrasts with the adult data, where P/LP mutations have been described as being distributed more uniformly among genes encoding cytoskeletal, nucleoskeletal, mitochondrial, and calcium‐handling proteins and there is a high prevalence of private mutations, with few hotspots or recurring mutations. 18 LMNA, RBM20, and PLN are common in adults, but LMNA P/LP variants were present in only 1.8% of cases and no P/LP variants were present in RBM20 or PLN in our study population.
Overall, a TTN truncation variant was present in 9% of genotype‐positive pediatric DCM cases. Children who presented with DCM in their teenage years were 4 times more likely to harbor a P/LP TTN variant than children who were identified at a younger age. This finding is consistent with prior data in nonhypertrophic cardiomyopathy and emphasizes the age‐dependent risks associated with TTN truncating variants. 23 Others have reported TTN variants associated with DCM in childhood. 19 , 20 One important note from our study is that P/LP TTN variants determined by cardiac genetic testing in infancy may still be associated with life‐threatening cardiac outcomes.
However, these associations do not prove causality in pediatric DCM. For example, 1 patient had 2 TTN variants in cis, each predicted to result in protein truncation (Case 37). We cannot determine from these data whether 1, both, or neither variant caused TTN‐related clinical disease. Similarly, gene testing panels identified 2 cases with pathogenic TTR variants, which are typically associated with late‐onset familial amyloid cardiomyopathy (Cases 16 and 39). One TTR variant was observed in a patient who also had a pathogenic MYH7 variant. The other was seen in a toddler who presented with heart failure and ventricular arrhythmias but did not otherwise have typical features of familial amyloid cardiomyopathy. The late‐onset nature of TTR cardiomyopathy is well established, but to be comprehensive, all variants classified as pathogenic were included in the results.
Domain‐Specific MYH7 Gene Variants Are Associated With DCM‐LVNC
MYH7 has been reported in only 3% to 4% in adult‐onset DCM, whereas a P/LP variant in MYH7 occurred in 10% of patients in this pediatric cohort. 2 , 12 As an important additional finding, our data support a domain‐specific variation MYH7 associated with DCM‐LVNC. In adult DCM, MYH7 mutations associated with noncompaction have been described clustering into amino acids near the ATP binding site. 21 Recent studies have correlated DCM‐LVNC with MYH7 mutations beyond the ATPase site. 22 , 23 We found that all patients with MYH7 variants and DCM‐LVNC had variants of interest encoding the first 600 amino acids of β‐myosin heavy chain (the head region), whereas none of the patients with variants encoding the later amino acids had DCM‐LVNC.
Clinical Genetic Testing
All variants were obtained from panels ordered during clinical care. The yield of P/LP variants in this cohort supports the use of panels in children with DCM, and these data have allowed us to effectively perform cascade screening in families. In a secondary analysis, a family history of cardiomyopathy did not affect the frequency of P/LP variants. Until larger data sets are available, we continue to recommend that all children with idiopathic DCM receive genetic testing, not just those with a positive family history.
In addition, 13% of VUS were adjudicated as P/LP variants when reclassified an average of 6 years later. Families need to be educated that reclassification may change the implications for clinical care and family screening. This is consistent with data previously published in pediatric cardiology, demonstrating the long‐term value in a centralized program with access to genetic counseling and variant re‐adjudication. 24 Finally, the 2015 American College of Medical Genetics guidelines improved variant adjudication, but did not make it entirely uniform. We found differences between our clinical adjudication and the classifications made by an independent laboratory. However, the variations in classification were because of reasonable differences in application of individual American College of Medical Genetics criteria, as has been documented elsewhere. 25
Limitations
This was a single‐center retrospective review and may have selection bias toward those with worse outcomes or presentation because of the referral nature of our pediatric heart transplant center. Selection bias may impact our finding of a high prevalence of sarcomeric variants. The genes tested by the various commercial gene panels changed over time and may have biased our retrospective findings (reported in Table S1). However, these data represent a typical use of clinical genetic panels over the last 15 years. As cost and barriers to access for genetic testing continue to fall and postsequencing bioinformatics improves, a broader genotype–phenotype analysis may be possible in the future. In addition, we recognize the differences in approaches to variant classification and provide Table S4 as a full comparison of the 3 classification systems used in this article.
Conclusions
This pediatric DCM cohort had a high frequency of sarcomeric gene variants. TTN truncations were more common in teenagers who presented with DCM, but infants were not spared, despite TTN’s genetic profile as a late‐onset form of DCM in adults. VUS were important in this patient population, because our data suggest that many of them may be reclassified as pathogenic or likely pathogenic in the future. Finally, we provided pediatric data to support a domain‐specific link between MYH7 and a DCM‐LVNC phenotype. In summary, these data suggest genotype–phenotype correlations that emphasize the importance of clinical genetic testing in pediatric DCM.
Sources of Funding
Research reported in this publication was supported, in part, by the NIH/NHLBI, grant numbers K23HL130554, HL128075, and U01HL131914; the American Heart Association, grant numbers 17MCPRP33660457 and 19SFRN34910009. REDCap access was provided by Northwestern University Clinical and Translational Sciences Institute, funded in part by NIH UL1TR001422.
Disclosures
E.M.M. consults for Amgen, AstraZeneca, Avidity, PepGen, Pfizer, 4D Molecular Therapeutics, Janssen, Cytokinetics, Tenaya Therapeutics, and Invitae Corp. All consulting is unrelated to this article. The remaining authors have no disclosures to report.
Supporting information
Tables S1–S4
Acknowledgments
The authors thank Christina Laternser, MS, for programming assistance during analysis.
Supplemental Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.022854
For Sources of Funding and Disclosures, see page 8.
References
- 1. Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. 2010;375:752–762. doi: 10.1016/S0140-6736(09)62023-7 [DOI] [PubMed] [Google Scholar]
- 2. Rossano JW, Dipchand AI, Edwards LB, Goldfarb S, Kucheryavaya AY, Levvey BJ, Lung LH, Meiser B, Yusen RD, Stehlik J. The registry of the International Society for Heart and Lung Transplantation: nineteenth pediatric heart transplantation report‐2016; focus theme: primary diagnostic indications for transplant. J Heart Lung Transplant. 2016;35:1185–1195. doi: 10.1016/j.healun.2016.08.018 [DOI] [PubMed] [Google Scholar]
- 3. Halliday BP, Cleland JGF, Goldberger JJ, Prasad SK. Personalizing risk stratification for sudden death in dilated cardiomyopathy: the past, present, and future. Circulation. 2017;136:215–231. doi: 10.1161/CIRCULATIONAHA.116.027134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Singh RK, Canter CE, Shi L, Colan SD, Dodd DA, Everitt MD, Hsu DT, Jefferies JL, Kantor PF, Pahl E, et al. Survival without cardiac transplantation among children with dilated cardiomyopathy. J Am Coll Cardiol. 2017;70:2663–2673. doi: 10.1016/j.jacc.2017.09.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. 2017;121:731–748. doi: 10.1161/CIRCRESAHA.116.309396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rusconi P, Wilkinson JD, Sleeper LA, Lu M, Cox GF, Towbin JA, Colan SD, Webber SA, Canter CE, Ware SM, et al. Differences in presentation and outcomes between children with familial dilated cardiomyopathy and children with idiopathic dilated cardiomyopathy. Circ: Heart Fail. 2017;10(2):e002637. doi: 10.1161/CIRCHEARTFAILURE.115.002637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dellefave L, McNally EM. The genetics of dilated cardiomyopathy. Curr Opin Cardiol. 2010;25:198–204. doi: 10.1097/HCO.0b013e328337ba52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dhandapany PS, Razzaque MA, Muthusami U, Kunnoth S, Edwards JJ, Mulero‐Navarro S, Riess I, Pardo S, Sheng J, Rani DS, et al. RAF1 mutations in childhood‐onset dilated cardiomyopathy. Nat Genet. 2014;46:635–639. doi: 10.1038/ng.2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaspar RW, Allen HD, Ray WC, Alvarez CE, Kissel JT, Pestronk AP, Weiss RB, Flanigan KM, Mendell JR, Montanaro F. Analysis of dystrophin deletion mutations predicts age of cardiomyopathy onset in becker muscular dystrophy. Circ Cardiovasc Genet. 2009;2:544–551. doi: 10.1161/CIRCGENETICS.109.867242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chami N, Tadros R, Lemarbre F, Lo KS, Beaudoin M, Robb L, Labuda D, Tardif J‐C, Racine N, Talajic M, et al. Nonsense mutations in BAG3 are associated with early‐onset dilated cardiomyopathy in French Canadians. Can J Cardiol. 2014;30:1655–1661. doi: 10.1016/j.cjca.2014.09.030 [DOI] [PubMed] [Google Scholar]
- 11. Peters S, Kumar S, Elliott P, Kalman JM, Fatkin D. Arrhythmic genotypes in familial dilated cardiomyopathy: implications for genetic testing and clinical management. Heart Lung Circ. 2019;28:31–38. doi: 10.1016/j.hlc.2018.09.010 [DOI] [PubMed] [Google Scholar]
- 12. Kayvanpour E, Sedaghat‐Hamedani F, Amr A, Lai A, Haas J, Holzer DB, Frese KS, Keller A, Jensen K, Katus HA, et al. Genotype‐phenotype associations in dilated cardiomyopathy: meta‐analysis on more than 8000 individuals. Clin Res Cardiol. 2017;106:127–139. doi: 10.1007/s00392-016-1033-6 [DOI] [PubMed] [Google Scholar]
- 13. Te Rijdt WP, van der Klooster ZJ, Hoorntje ET, Jongbloed JDH, v an der Zwaag PA , Asselbergs FW, Dooijes D, de Boer RA, van Tintelen JP, van den Berg MP, et al. Phospholamban immunostaining is a highly sensitive and specific method for diagnosing phospholamban p.Arg14del cardiomyopathy. Cardiovasc Pathol. 2017;30:23–26. doi: 10.1016/j.carpath.2017.05.004 [DOI] [PubMed] [Google Scholar]
- 14. Lakdawala NK, Dellefave L, Redwood CS, Sparks E, Cirino AL, Depalma S, Colan SD, Funke B, Zimmerman RS, Robinson P, et al. Familial dilated cardiomyopathy caused by an alpha‐tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy. J Am Coll Cardiol. 2010;55:320–329. doi: 10.1016/j.jacc.2009.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grenier MA, Osganian SK, Cox GF, Towbin JA, Colan SD, Lurie PR, Sleeper LA, Orav EJ, Lipshultz SE. Design and implementation of the North American Pediatric Cardiomyopathy Registry. Am Heart J. 2000;139:S86–S95. doi: 10.1067/mhj.2000.103933 [DOI] [PubMed] [Google Scholar]
- 16. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–381. doi: 10.1016/j.jbi.2008.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Investig. 2013;123:19–26. doi: 10.1172/JCI62862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown EE, Murray B, Vaishnav J, Tampakakis E, Barouch LA, James C, Murphy AM, Judge DP. Genetic dilated cardiomyopathy due to TTN variants without known familial disease. Circ Genom Precis Med. 2020;13:e003082. doi: 10.1161/CIRCGEN.120.003082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zaklyazminskaya E, Mikhailov V, Bukaeva A, Kotlukova N, Povolotskaya I, Kaimonov V, Dombrovskaya A, Dzemeshkevich S. Low mutation rate in the TTN gene in paediatric patients with dilated cardiomyopathy ‐ a pilot study. Sci Rep. 2019;9:16409. doi: 10.1038/s41598-019-52911-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, Greutmann M, Hürlimann D, Yegitbasi M, Pons L, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117:2893–2901. doi: 10.1161/CIRCULATIONAHA.107.746164 [DOI] [PubMed] [Google Scholar]
- 22. Miszalski‐Jamka K, Jefferies JL, Mazur W, Glowacki J, Hu J, Lazar M, Gibbs RA, Liczko J, Klys J, Venner E, et al. Novel genetic triggers and genotype‐phenotype correlations in patients with left ventricular noncompaction. Circulation. 2017;10(4):e001763. doi: 10.1161/CIRCGENETICS.117.001763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Waning JI, Caliskan K, Michels M, Schinkel AF, Hirsch A, Dalinghaus M, Hoedemaekers YM, Wessels MW, IJpma AS, Hofstra RMW, et al. Cardiac phenotypes, genetics, and risks in familial noncompaction cardiomyopathy. J Am Coll Cardiol. 2019;73:1601–1611. doi: 10.1016/j.jacc.2018.12.085 [DOI] [PubMed] [Google Scholar]
- 24. Cherny S, Olson R, Chiodo K, Balmert LC, Webster G. Changes in genetic variant results over time in pediatric cardiomyopathy and electrophysiology. J Genet Couns. 2021;30:229–236. doi: 10.1002/jgc4.1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pepin MG, Murray ML, Bailey S, Leistritz‐Kessler D, Schwarze U, Byers PH. The challenge of comprehensive and consistent sequence variant interpretation between clinical laboratories. Genet Med. 2016;18:20–24. doi: 10.1038/gim.2015.31 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S4