

Abstract
Background
Mitral valve prolapse (MVP) is one of the most common forms of cardiac valve disease and affects 2% to 3% of the population. Previous imaging reports have indicated that myocardial fibrosis is common in MVP and described its association with sudden cardiac death. These data combined with evidence for postrepair ventricular dysfunction in surgical patients with MVP support a link between fibrosis and MVP.
Methods and Results
We performed histopathologic analysis of left ventricular (LV) biopsies from peripapillary regions, inferobasal LV wall and apex on surgical patients with MVP, as well as in a mouse model of human MVP (Dzip1S14R /+ ). Tension‐dependent molecular pathways were subsequently assessed using both computational modeling and cyclical stretch of primary human cardiac fibroblasts in vitro. Histopathology of LV biopsies revealed regionalized fibrosis in the peripapillary myocardium that correlated with increased macrophages and myofibroblasts. The MVP mouse model exhibited similar regional increases in collagen deposition that progress over time. As observed in the patient biopsies, increased macrophages and myofibroblasts were observed in fibrotic areas within the murine heart. Computational modeling revealed tension‐dependent profibrotic cellular and molecular responses consistent with fibrosis locations related to valve‐induced stress. These simulations also identified mechanosensing primary cilia as involved in profibrotic pathways, which was validated in vitro and in human biopsies. Finally, in vitro stretching of primary human cardiac fibroblasts showed that stretch directly activates profibrotic pathways and increases extracellular matrix protein production.
Conclusions
The presence of prominent regional LV fibrosis in patients and mice with MVP supports a relationship between MVP and progressive damaging effects on LV structure before overt alterations in cardiac function. The regionalized molecular and cellular changes suggest a reactive response of the papillary and inferobasal myocardium to increased chordal tension from a prolapsing valve. These studies raise the question whether surgical intervention on patients with MVP should occur earlier than indicated by current guidelines to prevent advanced LV fibrosis and potentially reduce residual risk of LV dysfunction and sudden cardiac death.
Keywords: computational modeling, DZIP1, fibrosis, mitral valve prolapse, primary cilia
Subject Categories: Animal Models of Human Disease, Basic Science Research, Fibrosis, Inflammation, Mechanisms
Nonstandard Abbreviations and Acronyms
- α‐SMA
α‐smooth muscle actin
- CF
cardiac fibroblast
- ECM
extracellular matrix
- HCF
human cardiac fibroblast
- KI
knock‐in
- PM
papillary muscle
- WT
wild‐type
Clinical Perspective
What Is New?
This is a study on the role of pathologic mechanical tension on the papillary muscle from a prolapsing valve and how this abnormal valve motion induces regional fibrosis.
Computational modeling paired with molecular, immunologic, and genetic studies indicate mechanosensory nodes that are abnormally stimulated in response to increased mechanical stress in fibrotic development.
What Are the Clinical Implications?
These studies raise the question whether surgical intervention on patients with mitral valve prolapse should occur earlier than indicated by current guidelines to prevent advanced left ventricular fibrosis and potentially reduce residual risk of left ventricular dysfunction and sudden cardiac death.
More research is needed that includes the marriage of enhanced imaging techniques with genetic and cellular mechanosensing mechanisms to understand initiating and sustaining signals driving regional fibrosis in patients with mitral valve prolapse.
Mitral valve prolapse (MVP) is a common degenerative disease of the left‐heart inlet valve defined by billowing of one or both leaflets above the annulus. It affects 1 in 40 individuals and carries the risk of serious secondary complications such as heart failure, arrhythmias, and sudden cardiac death. 1 , 2 Histologically, MVP is characterized by myxomatous degeneration of the mitral leaflets, defined as loss of the normal stratified extracellular matrix layers with increased proteoglycans throughout the valve, and collagen and elastin fragmentation. Disruption of these extracellular matrix (ECM) components, combined with valve interstitial cell hyperplasia, results in tissue enlargement and mechanical incompetence with leaflet billowing, malcoaptation, and chordal stretching and rupture. 3
There is growing recognition that pathologic changes in MVP are not limited to the valve but commonly include myocardial fibrosis, which is associated with arrhythmias, sudden cardiac death, and potential for postrepair ventricular dysfunction. 4 , 5 , 6 Lethal left ventricular (LV) arrhythmias have been reported in association with inferobasal LV and papillary muscle (PM) fibrosis. 2 , 7 , 8 These findings were further supported by studies that found increased fibrosis in patients with ventricular arrhythmias, 8 , 9 , 10 and cardiac magnetic resonance imaging demonstrates regional LV fibrosis is more prevalent in MVP than patients without MVP with mitral regurgitation. 8 , 10 , 11 Furthermore, Basso et al and other groups have described fibrosis in patients with MVP as a potential arrhythmogenic substrate and plausible cause for sudden cardiac death in a subset of these patients. 2 , 12 The location of fibrosis and PM involvement suggest a relationship to localized LV stresses exerted by abnormal valve motion. How and if these stresses are translated into molecular and cellular changes within the LV wall to drive reactive fibrotic responses is unknown but would likely invoke mechanosensors on various cell types within the myocardial wall. Of potential relevance, genetic studies identified mechanosensing primary cilia as prominently involved in human MVP 13 , 14 ; however, whether these cellular appendages can transmit altered mechanical stresses from a prolapsing valve to reactive changes in the LV remains to be determined.
To date, LV histopathology in patients with MVP as well as application of appropriate models to study mechanisms underlying potential regional LV fibrosis have been lacking. In this study we provide histopathological, cellular, and molecular evidence for profound regionalized LV fibrosis in a sample of patients with MVP undergoing surgical repair. To establish a platform for studying fibrosis mechanisms and potential therapies in MVP, a genetically and phenotypically accurate murine model of MVP (Dzip1S14R/+ ) was evaluated. 13 In this model, we tested the hypothesis that myocardial fibrosis in MVP is progressive and has comparable location as well as histopathologic and molecular characteristics similar to patients undergoing mitral valve surgery. Computational models and validation in vitro revealed mechanosensory molecular nodes that promote fibrosis proportional to stress, consistent with the valve‐related location of myocardial fibrosis. Our data demonstrate that regionalized LV fibrosis is a conserved process across species and involves a localized mechanomolecular response to sense and increased chordal tension from a prolapsing mitral valve.
METHODS
Materials Disclosures
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Patient Consent and Recruitment
All procedures performed in the studies involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study protocol was approved by the local ethics committee (study protocol number 450/18‐ek), and informed written consent was obtained from each patient before study enrollment. Patients (n=6) with severe mitral regurgitation secondary to MVP and indications for mitral valve repair 15 were enrolled in the study. Exclusion criteria consisted of other possible causes of myocardial fibrosis including coronary artery disease, nonvalvular cardiomyopathy, aortic stenosis, and previous cardiac surgery. Patients underwent transthoracic and transesophageal echocardiography to confirm the cause of the MVP and assess LV function before surgery (Table S1). Cardiac magnetic resonance imaging, using T1 mapping before and after gadolinium contrast to calculate extracellular volume fraction and assess regional tissue abnormalities, as well as late gadolinium enhancement, was performed in all patients. Extracellular volume values for each patient were as follows: patient 1: 28.1%, patient 2: 31.8%, patient 3: 28.0%, patient 5: 25.9%, patient 6: 33%. Patient 4 stopped his CMR study before meaningful images could be obtained. During mitral valve repair surgery, biopsies were obtained from the inferobasal myocardium between the PMs in all patients (n=6), with biopsies of the interventricular septum (n=3) or LV apex (n=3) to serve as within‐patient controls. Biopsies were obtained with a scalpel with a mean size of 84±71 mg SD. All samples were processed for pathology and immunohistochemistry.
Mouse Model of Nonsyndromic MVP
A single missense mutation in the DZIP1 gene was identified as disease‐causing in a large family with nonsyndromic MVP. This mutation was introduced into mice through CRISPR (clustered regularly interspaced short palindromic repeats)‐Cas9 (CRISPR‐associated protein‐9) (designated Dzip1S14R/+ ), and the knock‐in (KI) mutant mouse was validated as having myxomatous mitral leaflets and functional MVP by echocardiography at 6 months of age, as described previously. 13 All animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committees at the Medical University of South Carolina (Protocol No. 170092). Before cardiac resection, mice were anesthetized by a 1‐time inhaled dose of 10 mL isofluorane (Piramal) for 1 minute in a closed chamber. Toe pinch confirmed deep anesthesia, and the method of euthanasia was cervical dislocation in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication No. 85–23, revised 1996). Hearts were removed following cervical dislocation.
Histopathology and Immunohistochemistry
Masson’s trichrome stain was performed on 5‐μm paraffin‐embedded sections from surgical biopsies of patients with MVP (n=6). Sections were deparaffinized and rehydrated through a graded series of 100%, 95%, and 70% EtOH and washed in distilled water. Samples were refixed in Bouin’s solution for 1 hour at 56 °C and rinsed in running water. Sections were then stained in Weigert’s iron hematoxylin solution (10 minutes), washed in water, stained in Biebrich scarlet‐acid fuchsin (15 minutes), and washed in water. Samples were placed in phosphomolybdic‐phosphotungstic solution (15 minutes) and transferred to aniline blue solution (10 minutes). Samples were briefly washed in water and placed in 1% acetic acid solution (5 minutes) and washed, followed by dehydration (95% and 100% EtOH) and cleared in xylene before mounting.
Immunohistochemical and fluorescence stains were performed on 5‐μm paraffin‐embedded sections from surgical biopsies of patients with MVP (n=6) as well as Dzip1S14R/+ KI MVP mice 13 and control wild‐type (WT) littermates (Dzip1+/+ ) at 2 months (KI: n=3, WT: n=5), 4 months (KI: n=4, WT: n=4), 5 to 6 months (KI: n=4, WT: n=3), and ≥7 months (KI: n=5, WT=3) of age, as previously described. 13 , 16 , 17 , 18 , 19 , 20 Primary antibodies and their dilutions included: acetylated tubulin (Sigma; No. T6793, 1:500), α‐smooth muscle actin (α‐SMA) (Sigma; No. A2547, 1:500), cluster of differentiation 206 (CD206) (R&D; No. AF2535, 1:100), cluster of differentiation 163 (Abcam; No. ab199402, 1:100), ADP ribosylation factor like GTPase 13B (ARL13B) (Protein Tech; No. 17711‐1‐AP, 1:500), and Collagen Telo (a generous gift from Dr. Stanley Hoffman; 1:250). Secondary antibodies (Invitrogen), used at a 1:100 dilution, included fluorophores 488, 568, and Cy5. Nuclei were stained with Hoechst (Life Technologies; No. H3569, 1:10 000). Slides were cover‐slipped using SlowFade Gold Antifade Reagent (Invitrogen; No. S36936). Images were captured using a Leica TCS SP5 AOBS Confocal Microscope System and LAS AF version 2.6.3 build 8173 acquisition and analysis software, Zeiss Axioscope M2, or Olympus BH‐2 brightfield microscope.
Statistical Analysis
Quantification of collagen content on immunohistochemical stains was performed in Adobe Photoshop CS6. For human biopsies, total pixel counts for collagen stains were obtained. Images from each of the experimental groups and anatomical locations were generated, and percent collagen content in each region was calculated. For the human biopsies, 6 equal surface area measurements were obtained for collagen expression per patient. Percentages were calculated based on collagen pixel intensity divided by total pixel intensity. These 6 measurements were averaged to generate percent SEM. A similar approach was performed for mouse collagen fraction with minor modification. Papillary, peripapillary, and apex (control) regions were selected, and pixel intensity for collagen was obtained and divided by total pixels within each field. This generated a percent collagen fraction, which is graphically depicted by showing total percent collagen (fibrosis) within the papillary region and apex control tissue across multiple mice at varying ages (control WT littermates [Dzip1+/+ ] at 2 months [KI: n=3, WT: n=5], 4 months [KI: n=4, WT: n=4], 5 to 6 months [KI: n=4, WT: n=3], and ≥7 months [KI: n=5, WT: n=3]). To assess normality of the data, a Shapiro‐Wilk test was performed for each group of samples. For data that were normally distributed, a parametric test was used; when data were not normally distributed, a nonparametric test was used. For individual patient comparisons, a nonparametric Mann‐Whitney U test was used to test for statistical changes in percent collagen fraction between either septum and papillary biopsies or apical and papillary biopsies, depending on which samples were available per patient. To compare the average percent collagen fraction of apical and septal biopsies with PM biopsies across multiple patients, a Student t test was used, because data were determined to be parametric. To compare the average percent collagen fraction for remote myocardium, which includes apical and septal samples depending on what was available per patient (n=6), compared with papillary myocardium (n=6), a Student t test was used because the data were normally distributed. For mouse analyses, to compare percent collagen fraction across different time points, a 1‐way ANOVA was used with post hoc analysis using Dunn multiple comparisons test with the mean of the 2‐month time point used as a referent control. To compare collagen fractions between control and Dzip1 KI mice over time, we used a 2‐way ANOVA, with post hoc testing using Bonferroni multiple comparisons test. For all analyses, an * indicates P<0.05, whereas ** and *** indicate P<0.01 and 0.001, respectively. Having 6 collagen measurements of each biopsy type (remote, papillary) per patient provided 80% power to detect large within‐patient differences in collagen fractions between biopsy types (eg, Cohen d effect sizes equivalent to 1.8 SD units), as well as large differences (d=1.4) in average collagen fractions between biopsy types across all 6 patients, assuming 2‐sided hypothesis testing and α=0.05. Only large differences (d=3–4, depending on whether data from 5 or 6 patients were available) were detectable between the patient subgroups with 80% power.
Quantification of α‐SMA was performed by comparing pixel intensity within 6 representative fields. Quantification of CD206 was generated by counting total number of positive cells within 6 representative fields. Data are shown as fold‐change in CD206 cell numbers between experimental and controls and percent of CD206 cells within particular regions. Total cells analyzed are presented in the figures. To detect statistically significant differences between test groups with 2‐sided α=0.05, a Student t test was used.
Computational Modeling
To help identify potential signaling connections between mechanical tension and regional fibrosis, we adapted a previously published computational model of cardiac fibroblast (CF) signaling pathways. 21 , 22 This model captured the activity levels of 109 signaling molecules and 174 reactions connecting biochemical and mechanical input stimuli (mechanical tension, transforming growth factor beta 1, angiotensin II, interleuken 1, interleuken 6, platelet‐derived growth factor, tumor necrosis factor alpha, endothelin 1, norepinephrine, and natriuretic peptide) to predict fibrosis‐related outputs (collagen I, collagen III, fibronectin, periostin, α‐SMA, matrix metalloproteinases, tissue inhibitors of metalloproteinases, and others). In the present study, we integrated an additional 27 new signaling molecules and 50 new network reactions related to cilia signaling that were manually curated from existing literature reports (Tables S2 and S3). Reactions were added if 2 or more prior experimental results were found that confirmed each particular reaction in CFs or similar cell types. The word “reaction” here is a general term used in reference to all reactions between species in the network. For example, TGFβ ligand interacting with its receptor is a reaction within the network. Our modeling approach simulated each reaction as a logic‐based ordinary differential equation, wherein each molecule’s activity is represented as a fractional value between 0 (fully off) and 1 (fully saturated), calculated from upstream input nodes using Hill‐type sigmoidal ordinary differential equations and the associated reaction logic (ie, activation, inhibition, coactivation). Simulations of the CF signaling network and construction of ordinary differential equations were completed using Netflux software in MATLAB (MathWorks), as previously described. 22 In our past modeling studies, this model has successfully predicted ≈82% (96/118) of previously reported signaling responses by CFs under a wide variety of biomechanical and biochemical stimulation conditions. 21 , 22 Importantly, these validation agreements are based on >100 independent experimental studies, not including the ≈300 experimental studies used to build the model. In the current study, we investigated the connection between mechanical tension, cilia signaling, and downstream collagen production. For the tension dose‐response simulations, tension stimulation was applied at increasing intervals of 10% activation from 0% to 100% until species steady‐state activation levels were achieved (11 total simulations). Biochemical input nodes were initialized at 10% activation to replicate basal activation of the CF. All other noninput nodes were initialized at 0% activation. The condensed network reconstruction and species heatmap were based on the steady‐state species activation levels derived from these simulations.
Cell Culture and Western Analyses
Human CFs (HCFs) (Cell Applications) were cultured on substrates of increasing elastic moduli to interrogate the effects of ECM stiffness on HCF primary cilia. CytoSoft (Advance BioMatrix, Cat. No. 5142‐5) collagen‐coated silicone hydrogels were used with elastic moduli resembling normal working myocardium (8 kPa) compared with pathological conditions (16, 32, and 64 kPa). 23 HCFs were seeded at a density of 3×105 in 6‐well CytoSoft plates in triplicate and allowed to reach near confluence over 48 hours. HCF‐specific growth media were used for all plates (Cell Applications; 315‐500). At 48 hours, HCFs were released from the silicone hydrogels using trypsin‐EDTA, and protein lysate was collected. Western blot analysis of this protein lysate was conducted in triplicate and probed for the ciliary axoneme marker Arl13b (Proteintech; Cat No. 17711‐1‐AP). Goat‐HRP secondary antibodies were used to detect Arl13b followed by West‐Femto chemiluminescence detection reagent (ThermoFisher). Band intensities were calculated as described above and normalized against total protein loading via Ponceau S stain.
In addition, human cardiac fibroblasts were cultured in a 16‐well silicone plate coated with 150 µL of collagen type 1 (3.37 mg/mL; Corning) at a seeding density of 2.5×104 per well. These cells were then subjected to either 10% mechanical strain (frequency 1 Hz) or 0% strain (static control) using a MechanoCulture FX plate stretcher (CellScale) for 24 hours. Following stretch, cells were lysed using RIPA buffer containing 1× Halt protease and phosphatase inhibitors cocktail (ThermoFisher) to extract proteins. To detect statistically significant differences between test groups with 2‐sided α=0.05, a Student t test was used.
RNA Sequencing
Total RNA was isolated from the cells using the RNeasy Mini Kit (Qiagen), and RNA quality was determined using a nanodrop spectrophotometer (ThermoFisher). For each sample, 2 µg of total RNA were used in Illumina’s TruSeq Stranded mRNA Library Kit (Cat No. 20020594). Libraries were sequenced on Illumina NextSeq 500 as paired‐end 42‐nt reads. Sequence reads were analyzed with the spliced transcripts alignment to a reference alignment–DESeq2 software pipeline. For read mapping, the paired‐end 42 bp sequencing reads generated by Illumina sequencing were mapped to the genome using the spliced transcripts alignment to a reference algorithm with default settings. Alignment information for each read is stored in the binary alignment map format. For fragment assignment, the number of fragments overlapping predefined genomic features of interest (eg, genes) were counted. Only read pairs that have both ends aligned were counted. Read pairs that have their 2 ends mapping to different chromosomes or mapping to the same chromosome but on different strands were discarded. The gene annotations used were obtained from the Subread package. These annotations were originally from the National Center for Biotechnology Information RefSeq database and then adapted by merging overlapping exons from the same gene to form a set of disjoint exons for each gene. Genes with the same Entrez gene identifiers were also merged into 1 gene. For differential analysis, after obtaining the gene table containing the fragment counts of genes, differential analyses to identify statistically significant differential genes using DESeq2 were performed. The following lists the preprocessing steps before differential calling: (1) Data normalization: DESeq2 expects unnormalized count matrix of sequencing fragments. The DESeq2 model internally corrects for library size using their median‐of‐ratios method. The gene table obtained from analysis step 2 is used as input to perform the DESeq2’s differential test. (2) Filtering before multiple testing adjustment: After a differential test has been applied to each gene except the ones with zero counts, the P value of each gene is calculated and adjusted to control the number of false positives among all discoveries at a proper level. During this process, DESeq2 by default filters out statistical tests (ie, genes) that have low counts by a statistical technique called independent filtering. It uses the average counts of each gene (ie, base mean), across all samples, as its filter criterion, and it omits all genes with average normalized counts below a filtering threshold from multiple testing adjustment. This filtering threshold is automatically determined to maximize detection power (ie, maximize the number of differential genes detected) at a specified false discovery rate. (3) Differential calling: Differential genes are detected by DESeq2 at 0.1 (or 10%) false discovery rate (ie, adjusted P value). (4) Gene set enrichment analysis: Using DESeq2 normalized gene counts, we performed gene set enrichment analysis with default settings to determine whether members of an a priori defined gene set based on biological knowledge (eg, genes sharing the same gene ontology category) are enriched. Before running the gene set enrichment analysis, we add a small pseudocount to the normalized counts to avoid dividing by0 errors. Our standard gene set enrichment analysis uses MSigDB’s C5 (gene ontology gene set) collection. AdvaitaBio was also used as an efficient tool to prioritize differentially expressed gene pathways.
RESULTS
Surgical Patients With MVP Display Regional Fibrosis
Clinical information on the 6 patients with MVP can be found in Table S1. Sample preoperative transesophageal echocardiography (Videos S1 and S2) and cardiac magnetic resonance videos (Videos S3 and S4) are shown in supplemental files. Still images of 3‐dimensional and 2‐dimensional echocardiography show prominent posterior leaflet prolapse (Figure 1A and 1B). Patients typically had evidence of gross myocardial fibrosis in the PMs and surrounding inferobasal myocardium as determined by late gadolinium enhancement cardiac magnetic resonance (Figure 1C) and upon intraoperative visual inspection, with less/absent fibrosis in the interventricular septum and apex (Video S5). Successful mitral valve repair was performed via a minithoracotomy approach in all patients without any significant perioperative complications.
Figure 1. Echocardiographic visualization of mitral valve prolapse (MVP) and associated left ventricular (LV) fibrosis.
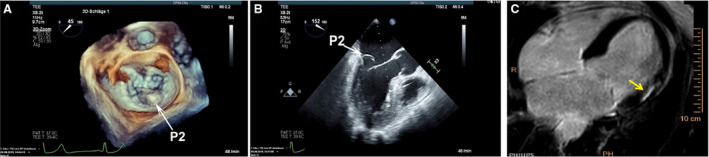
A and B, Three‐dimensional and 2‐dimensional transesophageal echocardiography MVP showing prolapse of the P2 segment (white arrows). C, Cardiac magnetic resonance with late gadolinium enhancement showing evidence of inferobasal LV fibrosis underlying the papillary muscles (yellow arrows).
Biopsies of the 6 patients were obtained from the inferobasilar (peripapillary) region, where gross fibrosis was typically observed during surgery (Video S5). Additional samples from each patient were harvested from either the interventricular septum or the apex of the heart to serve as within‐person controls. Masson’s trichrome stain revealed pronounced replacement fibrosis within the inferobasal myocardium, with little evidence of fibrosis within the apex or interventricular septum (Figure 2 and Figure S1). Immunohistochemistry (IHC) confirmed a statistically significant increase in collagen type I protein in these fibrotic regions (Figure 2). Within the scar regions, myocyte loss was evident by both Masson’s trichrome and IHC stains, and myocytes bordering the fibrotic zone displayed qualitative changes in cell size and presence of disorganized sarcomeres by IHC (Figure 2 and Figure S2). Those patients who displayed more pronounced fibrosis by IHC and Masson’s trichrome stains (patients 2 and 6) also had more diffuse prolapse (P1, P2, P3 segments in patient 2 and bileaflet involvement in patient 6) (Figure S1). Although all biopsied patients showed substantial localized LV fibrosis, echocardiographic data did not reveal overt LV dysfunction (Table S1). Taken together, these histological and molecular data confirm regional LV fibrosis at the time of surgery in patients with MVP with nonischemic disease.
Figure 2. Regional left ventricular (LV) fibrosis in human patients with mitral valve prolapse (MVP).
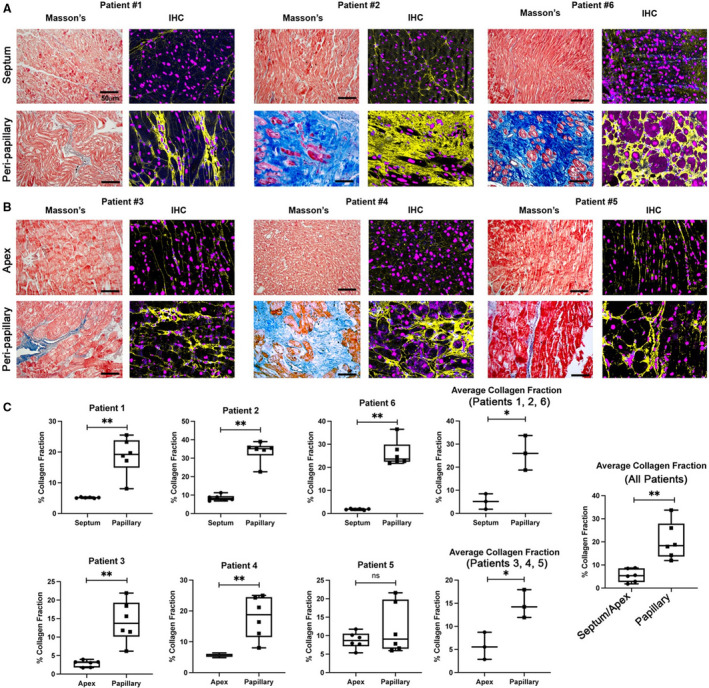
Masson’s trichrome and immunohistochemistry (IHC) for collagen (yellow) shows prominent LV fibrosis in the peripapillary region of surgical mitral valve repair patients compared with either septal (A) or apex (B) biopsies. C, Quantification of fibrosis shows significant elevation of collagen I protein in peripapillary regions compared with either septal or apex in‐person control tissue. Amounts are shown as percent collagen fraction (positive pixel staining/total pixels). Zones of myocyte loss are evident, indicating replacement fibrosis. Scale bars=50 µm. Blue=collagen histological (Masson) stain, red=myocytes histological (Masson) stain, purple=nuclei (Hoechst). *P<0.05, **P<0.01.
Regional LV Fibrosis Correlates With Myofibroblasts and Inflammation
Myofibroblast activity and inflammation have previously been correlated with fibrosis in numerous disease states. Thus, IHC was used to quantify whether molecular markers that demarcate these cell types correlated with the regional fibrosis observed in the LV surgical biopsies. High magnification of the fibrotic region in the peripapillary zone revealed numerous cells that stained for α‐SMA, a marker for activated myofibroblasts (Figure 3). α‐SMA–positive cells were also observed within the apex of the myocardium but were restricted, as expected, to small blood vessels and therefore do not represent activated fibroblasts (arrowheads in Figure 3A). Quantification of pixel density revealed a statistically significant increase in α‐SMA within the fibrotic zone compared with remote myocardium (Figure 3B). As previous reports have demonstrated a role for innate immune cells in both initiating and contributing to fibrotic events in the heart, 23 , 24 , 25 , 26 , 27 we assayed whether macrophage markers were also uniquely present within these collagen‐rich regions. As shown in Figure 3C and Figure S3, CD206‐ and cluster of differentiation 163‐positive macrophages were abundant within the fibrotic zones. Quantification of CD206+ cells revealed a 3‐fold increase in macrophages when comparing peripapillary region to apex and a 5‐fold increase when compared with septal myocardium (Figure 3D). The total average percentage of CD206+ cells within the peripapillary region was ≈25% (N=826 cells) compared with ≈5% within the septum (N=586 cells) or apex (N=502 cells). These data demonstrate that activated myofibroblasts and inflammation are associated with regional LV fibrosis in patients with MVP.
Figure 3. Fibrosis in patients with mitral valve repair correlates with activated cell types specific to peripapillary regions.
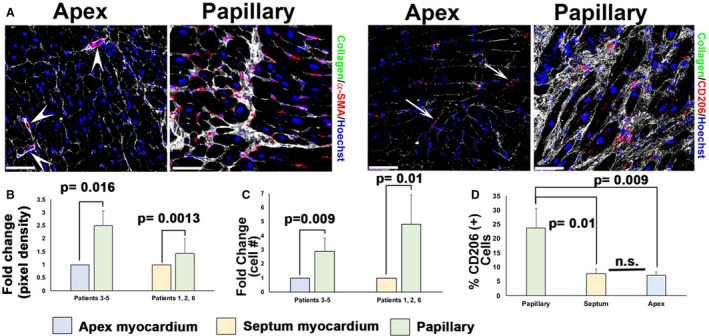
A, Immunohistochemistry (IHC) (white arrows, purple staining) shows increased α‐smooth muscle actin (α‐SMA) and cluster of differentiation 206 positive (CD206+) macrophages within fibrotic areas (white, collagen) localized to peripapillary regions (arrows) of the left ventricle compared with in‐person apex tissue. Scant α‐SMA+ cells within capillaries are observed in apex tissue (arrowheads in A), and few macrophages are present within the apex (arrows). B through D, Quantification of IHC data shows 2‐ to 3‐fold increase in α‐SMA expression, a 3‐ to 5‐fold increase in cluster of differentiation 206 (CD206) macrophages, and ≈25% total CD206+ cells within the fibrotic peripapillary region compared with apex or septal in‐person control tissues. Scale bars=100 µm.
Regional LV Fibrosis Is Conserved Across Species
In our previously reported mouse Dzip1S14R/+ model for nonsyndromic MVP, 100% of mice with the human mutation developed myxomatous valves and functional MVP by around 6 months of age. 13 To determine whether Dzip1S14R/+ mice also develop regionalized fibrosis, we conducted similar stains as in the human biopsies. For consistency of analysis, we performed IHC initially on the exact Dzip1S14R/+ and control mice that were previously reported as having myxomatous valves and functional MVP at 6 months of age. 13 As shown in Figure 4, Dzip1S14R/+ mice show significant elevation of collagen I in both the inferobasal wall (Figure 4A through 4C) and PM (Figure 4D and 4E) compared with age‐matched WT control animals. We also observed robust increases in both α‐SMA and CD206 staining, demonstrating activated myofibroblasts and macrophage presence within the fibrotic papillary and inferobasal LV. These activated cell types were largely absent from the same regions in control animals. Consistent with our human data, these increases in collagen content, activated cell types, and macrophages were uniquely observed within the peripapillary and inferobasal myocardium and absent within myocardial apex tissue (Figure 4F).
Figure 4. Dzip1S14R/+ mitral valve prolapse mice have regionalized left ventricular (LV) fibrosis similar to human patients.
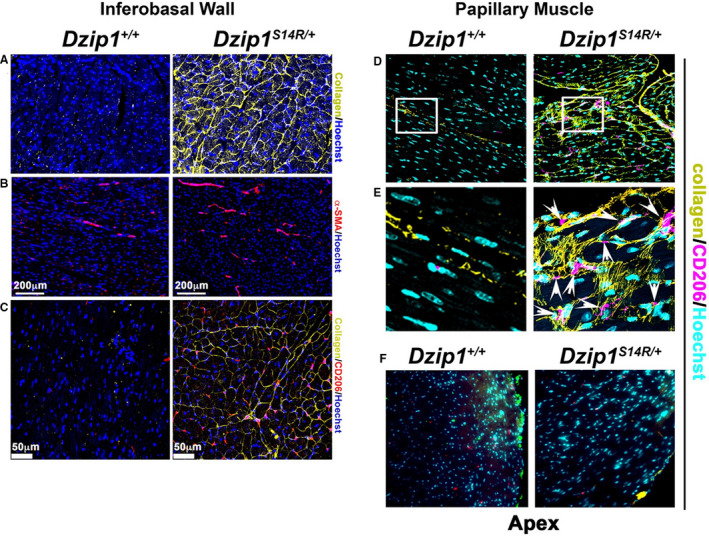
Immunohistochemistry at 6 months showing regionalized LV fibrosis (collagen, yellow) within the inferobasal wall (A and C) and papillary muscle (D) that correlates with increased presence of α‐smooth muscle actin (α‐SMA+) (B) and cluster of differentiation 206 positive (CD206+) (C and D, arrow heads in E) cells compared with wild‐type Dzip1 mice (Dzip1+/+ ). Higher magnification of boxed regions in (D) are shown in (E). Activated cells were specific to fibrotic regions in Dzip1S14R/+ mice, because apex tissue from control and mutant animals did not reveal evidence of increased α‐SMA or cluster of differentiation 206 (CD206) cells (F). Nuclei=blue (A through C), turquoise (D through F). Scale bar sizes are noted in the figure.
LV Fibrosis in MVP Is Progressive
As our data demonstrated significant regional LV fibrosis in a validated nonsyndromic MVP mouse model, follow‐up studies tested whether fibrosis with collagen deposition progresses in Dzip1S14R/+ mice at 2‐, 4‐, 5 to 6‐, and ≥7‐month time points. As shown in Figure 5, IHC shows that 2‐month Dzip1S14R/+ mice have low‐level collagen expression that is comparable to control animals (Dzip1+/+ ) (Figure 5A and 5E). By 4 months of age, collagen deposition becomes elevated within the PM and adjacent LV myocardium of Dzip1S14R/+ mice compared with controls (Figure 5B and 5F). By 6 months of age, abundant collagen is observed throughout the PMs of the Dzip1S14R/+ mice compared with controls (Figure 5C and 5G). At ≥7 months of age, control animals still have little detectable collagen (Figure 5D), whereas Dzip1S14R/+ mice display pronounced fibrosis with globular zones of collagen accretion (Figure 5H). The highest level of collagen expression begins primarily at the PM tip at 4‐months and then extends through the PM belly in a gradient fashion as the animals age. Quantification of collagen content by IHC revealed no statistically significant increase in collagen I protein expression in WT (Dzip1+/+ ) control tissues as a function of age (Figure 5I). However, in the Dzip1S14R/+ mice, significant elevation of expression is observed with time (1‐way ANOVA P=0.0004) (Figure 5J). From 2 to 6 months of age, there was a trend toward increased collagen as seen in the IHC; however, this did not reach significance. Following 6 months of age, a time point in which we previously detected MVP in the Dzip1S14R/+ mouse, 13 we observed a significant increase in collagen deposition (Figure 5J). Expression of collagen continued to increase after this time point and was statistically significant from all previous time points studied. Comparison between the control and Dzip1S14R/+ mice for collagen within the posterior–medial papillary showed that Dzip1S14R/+ mice develop progressive fibrosis over time compared with control mice (2‐way ANOVA P for interaction=0.004). Statistically significant increases for collagen for time points >6 months of age were identified (Bonferroni P<0.0001 at the 7+‐month time point) (Figure 5K). These data highlight a regionalized and progressive accumulation of fibrosis within the PMs and inferobasal LV, and is consistent with MVP‐driven mechanical tension as contributing to a progressive, regionalized fibrotic phenotype.
Figure 5. Regionalized fibrosis progression is observed in Dzip1S14R/+ mice.
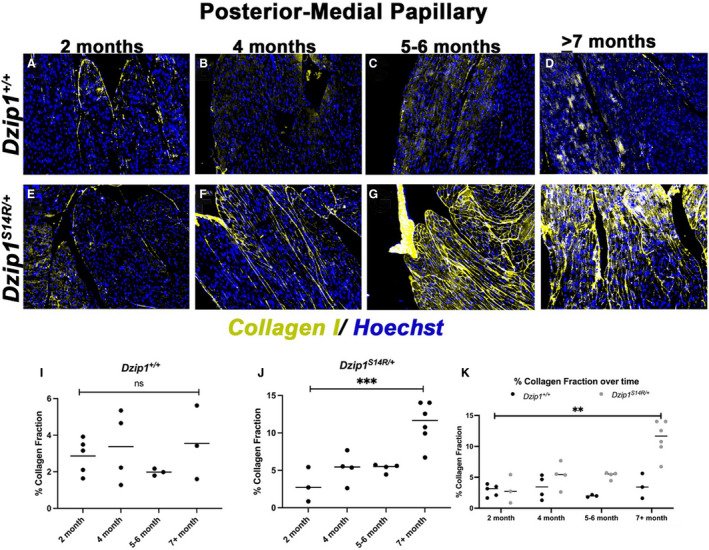
A through D, Immunohistochemistry for collagen I (yellow) over time shows no discernible increase of collagen production within the posterior–medial papillary muscle of control mice (Dzip1+/+ ). E through H, Dzip1S14R/+ mitral valve prolapse mice show increased collagen I staining (yellow) over time within the papillary muscle. I, Quantification of percent collagen fraction of tissue within the control papillary muscle shows no significant change in expression over time. One‐way ANOVA resulted in a P=0.55. J, Compared with 2 months of age, the Dzip1S14R/+ posterior–medial papillary muscle shows a trend toward increased fibrosis by 5 to 6 months of age and ≈5‐fold increase by ≥7 months. ***One‐way ANOVA resulted in a P=0.0004. K, Compared with control animals, significant differences in collagen within the papillary muscle is observed in Dzip1S14R/+ mice (2‐way ANOVA P for genotype=0.0005, P for time point<0.0001, P for interaction=0.004). Post hoc comparison of individual time points using Bonferroni multiple comparisons test found that significant differences in collagen are present by ≥7 months of age (P<0.0001), whereas a trend for increased collagen is observed by 6 months of age (P=0.12). ns, not significant; **P<0.01; ***P<0.001.
Computational Modeling Indicates Mechanical Tension as a Driver of Fibrosis
To probe a link between MVP and an LV fibrotic phenotype, we hypothesized that increased chordal tension from the prolapsing valve can promote CF activation in regions of increased stress. We applied a validated in silico model for how the CF responds to tension. 21 , 22 This model is based on our previously established computational model of the CF and uses a logic‐based, ordinary differential equation modeling approach to provide insight into CF dynamics. 21 , 22 This model was expanded to include a signaling network related to primary cilia activity and to integrate cilia signaling with mechanosensing signaling nodes. We included primary cilia in our analyses, because recent data have suggested a role for these mechanosensing cellular antennae in both MVP and cardiac fibrosis. 13 , 14 , 17 , 20 , 28 , 29 In addition, primary cilia have been previously shown to function in signal transduction pathways that are linked to collagen synthesis and deposition, including hedgehog, WNT/β‐catenin, TGF‐β, and cytoskeletal organization cascades. 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 Activation of these various signaling cascades were interrogated by assessing node activation patterns for well‐characterized downstream effectors of these pathways (Figure S4). A condensed form of the CF network is shown with nodes relevant to fibrotic pathways (Figure 6A). CFs, in response to high‐tension stimulation (tension activation=90%), showed a robust activation of profibrotic signaling pathways (Figure 6B). Next, we evaluated the activation of species directly related to the development of fibrosis. Expected increases in canonical and noncanonical TGF‐β signaling as well as fibrosis‐related transcription factors (myocardin related transcription factor, extracellular related kinase, serum response factor, mothers against decapentaplegic homolog 3, yes‐associated protein) are shown and are associated with increased collagen‐1 and α‐SMA activity (Figure 6B and 6C). Furthermore, a positive dose‐response relationship is evident between the CF profibrotic response and tension stimulation. Additionally, we identified primary cilia as most responsive in areas of low tension, with a dose‐dependent loss of these structures as tension increases.
Figure 6. Predictive computational model of cardiac fibroblasts in response to tension indicates mechanosensory responses.
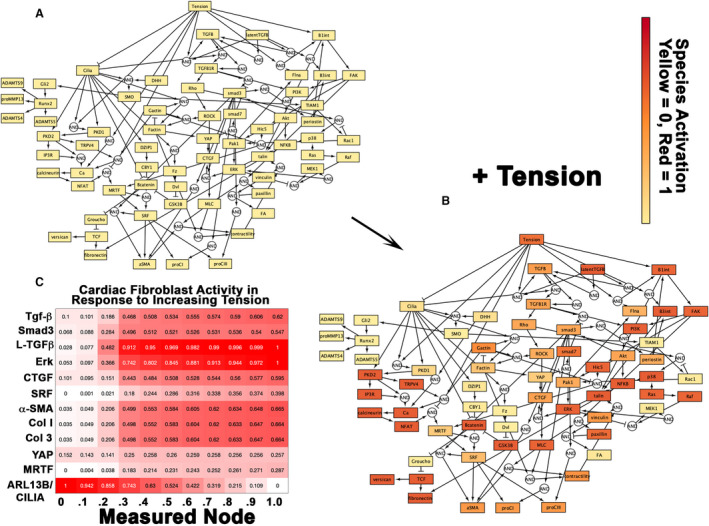
For the tension dose‐response simulations, tension stimulation was applied at graduating intervals of 10% activation from 0% (static, A) to 100% (B) until species steady‐state activation levels were achieved. The condensed network reconstruction (A and B) and species heatmap (C) were based on the steady‐state species activation levels derived from these simulations. Cardiac fibroblasts, in response to high tension stimulation (tension activation=90%), showed a robust activation of profibrotic signaling pathways (B and C). Tension stimulation was integrated into the cardiac fibroblast (CF) signaling network dynamics through the primary cilia and β‐integrin nodes. Expected increases in canonical and noncanonical TGFβ signaling as well as fibrosis‐related transcription factors (myocardin related transcription factor [MRTF], connective tissue growth factor [CTGF], yes‐associated protein [YAP], serum response factor [SRF], mothers against decapentaplegic homolog 3 [Smad3], extracellular related kinase [Erk]) are shown and are associated with increased collagen‐1, 3, and α‐smooth muscle actin (α‐SMA) activity (C). A positive dose‐response relationship is evident between the CF profibrotic response and tension stimulation but inversely related to primary cilia and ADP ribosylation factor like GTPase 13B (ARL13B) (C).
To determine if there is a correlation between primary cilia and tension in vivo, we analyzed papillary and apex biopsies from surgical patients with MVP. As shown in Figure 7A and 7B, in apical myocardium, under low/steady‐state levels of mechanical tension, prominent ciliary axonemes are observed. However, in the peripapillary regions that show prominent replacement fibrosis, reduced axoneme staining is observed. Quantification of pixel intensities for acetylated tubulin revealed ≈6‐fold reduction in staining in the fibrotic compared with nonfibrotic regions (Figure 7C). We further tested whether substrate stiffness impacts expression levels of ARL13B, a surrogate for axonemal biogenesis. As shown in Figure 7D and 7E, substrates with low elastic moduli (8 kPa), which represent the mechanical environment of normal working myocardium, show robust expression of ARL13B. However, as the substrate increased in stiffness, as occurs during fibrosis, expression of ARL13B was significantly downregulated (Figure 7D and 7E). These in vivo and in vitro results validate our computational model prediction that increased mechanical tension can simultaneously lead to decreased cilia and increased fibrosis.
Figure 7. Primary cilia are deficient within human peripapillary fibrotic regions in patients with mitral valve prolapse.
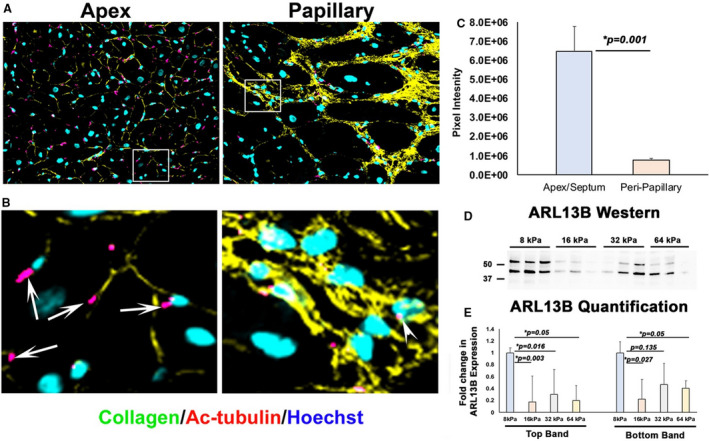
A and B, Within apex myocardium of patients with mitral valve repair, primary cilia axonemes (pink) are evident (arrows). Within fibrotic zones of the peripapillary regions, axonemes are either absent or deficient as quantified by immunohistochemistry and pixel intensity (C). Western analyses of human cardiac fibroblasts plated on matrices of increasing stiffness showed a significant downregulation of ADP ribosylation factor like GTPase 13B (ARL13B) protein, a surrogate for primary ciliogenesis (D and E). Scale bars sizes are denoted in panels.
RNA Sequencing Identifies Tension‐Driven Profibrotic Pathways
Our human, mouse, and in silico modeling data demonstrated fibrosis in areas of myocardium that experienced increased mechanical tension and suggested that tension drives fibrogenesis in MVP. To test this hypothesis, we performed mechanical stretch experiments on human cardiac fibroblasts. Following 24 hours of cyclical longitudinal strain (10%, 1 Hz), RNA sequencing identified 1761 transcripts that were nominally significant (adjusted P<0.1), of which 232 transcripts were statistically significant after correction for multiple testing (adjusted P<1.6×10−6; Figure 8A, 8B and Figure S5). These 232 transcripts included collagens 1α1, 1α2, 4α1, 5α1, 5α3, 7α1, as well as proteins involved in translation initiation (eukaryotic translation initiation factor 4A2), posttranslational collagen assembly, trafficking, and processing, (prolyl 4‐hydroxylase subunit alpha 1, prolyl 4‐hydroxylase subunit alpha 2, prolyl 4‐hydroxylase subunit Alpha 3, prolyl 3‐hydroxylase family member 4, procollagen‐lysine, 2‐oxoglutarate 5‐sioxygenase 2, and lysyl oxidase, and serpin family H member 1), and markers of fibroblast activation (actin alpha 2, smooth muscle). Conversely, proteolytic enzymes (matrix metalloproteinase 1 and a disintegrin and metalloproteinase with thrombospondin motifs 15) were downregulated (Figure 8B). Gene ontology analyses of cellular components using all nominally significant transcripts revealed 75 pathways that were statistically enriched following mechanical stretch. The most significantly different pathways corresponded to the extracellular space, secretory pathways such as vesicles and endosomes, and pathways relating to the cell membrane and cell–cell and cell–matrix adhesion (Figure 8C). To validate our computational model and RNA sequencing data sets, Western analyses were conducted on cell extract and media from stretched cells. Following 24 hours of mechanical stretch (10%, 1 Hz), proteins extracted from cell lysates showed increased production of eukaryotic translation initiation factor 4A2, an initiation factor in protein biosynthesis, and collagen type IV (Figure 8D and 8E). An increase in secreted collagen type I was observed in the media from stretched cells compared with static (Figure 8D and 8E). These data confirmed that mechanical stretch of human ventricular cardiac fibroblasts enhances a fibrogenic response in vitro.
Figure 8. Cyclical stretch of human cardiac fibroblasts (HCFs) reveals tension responsive changes in extracellular matrix gene expression and protein production.
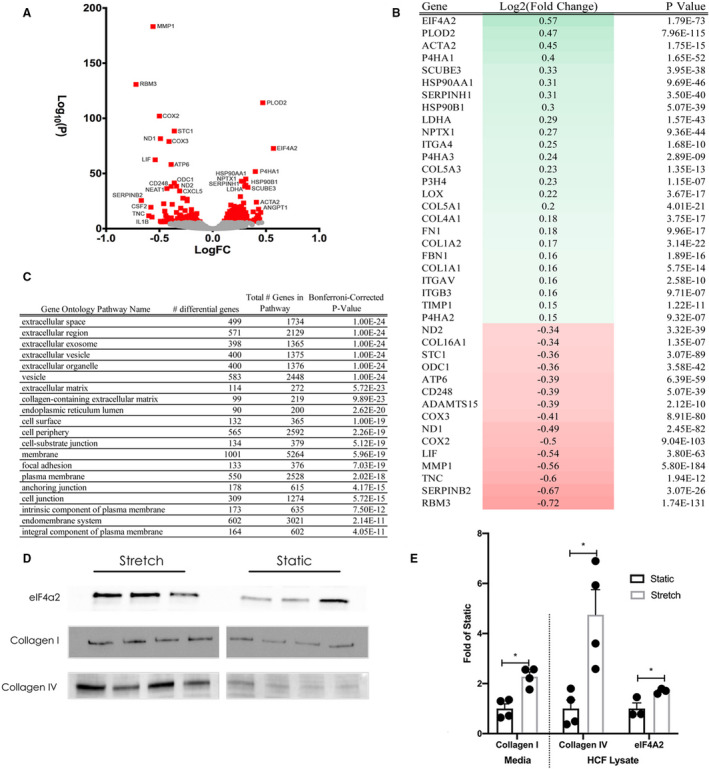
A, Volcano plot showing global changes in gene expression between stretched (10% longitudinal strain, 24 hours) vs static (0% longitudinal strain, 24 hours) HCFs. B, Heatmap of the top differentially expressed genes. C, Gene ontology analysis revealed that pathways corresponding to the extracellular space were enriched by mechanical stretch. D and E, Western blots performed on cell lysates and media from stretched and static HCF showed differences in collagen 1 protein in the media, and collagen IV and eukaryotic translation initiation factor 4A2 (eIF4A2) protein in cell lysates. FC indicates fold change. *P<0.05
DISCUSSION
Our results show histological and molecular evidence for regional LV myocardial fibrosis in patients with MVP. Cross‐species analyses in murine models validates a similarly localized, evolutionarily conserved fibrotic process observed in patients with MVP. Myofibroblast presence, inflammation, and fibrosis progression are predominantly localized to the inferobasal wall and attached PM that are physically and mechanically linked to the prolapsing leaflets. These data suggest an effect of valve‐induced mechanical stresses through the chordae tendineae on interstitial cell biology and tissue‐level responses. Lending additional support to this hypothesis, our studies show that profibrotic molecular changes can be reproduced in a validated computational model of fibroblast activation, as well as in in vitro models that apply biomechanical tension to cells. A colocalized presence of myofibroblasts and inflammation is consistent with previous reports of cardiac fibrosis, mostly in ischemic settings. 23 , 24 , 25 , 26 , 27 In that context, lack of blood flow induces cell death, a known potent nidus for early and prolonged inflammatory processes that initiate and propagate scar formation to prevent catastrophic cardiac rupture. Our studies show an inflammatory and activated cell phenotype response in the absence of ischemia in human and mouse MVP, and support the notion that changes in the mechanical environment are sufficient to engender reactive phenotypic and molecular responses that culminate in fibrosis.
How regionalized LV inflammation is activated in MVP is unknown. Based on the localization of inflammation and fibrosis, it is possible that mechanically induced cell membrane deformation results in release of proinflammatory cytokines, ions, and/or other small molecules (ie, ATP). 39 This effect is likely felt by all cell types within the affected areas, including fibroblasts, myocytes, endothelial cells, and resident inflammatory cells. 40 , 41 If various proinflammatory or profibrotic stimuli are released from these cells, or whether these stimuli act locally and/or systemically to induce/enhance inflammation are not known. Damage of stretched cell membranes may also induce cell death, which in turn would incite an innate inflammatory response initiated by neutrophils and propagated by macrophages. Although our data do not explicitly demonstrate that membrane stretch is inducing release of inflammatory attractants, cell death, and/or autophagy, we do show evidence of myocyte loss and subsequent replacement fibrosis in both human patients with MVP and our mouse model. Thus, myocyte loss, replacement fibrosis, and an altered mechanical environment may all be potent stimuli for continued inflammatory involvement necessary for scar progression. These molecular and cellular changes are only observed within regions of the LV that are tethered to the prolapsing valve through the chordae tendineae and PM, leading us to conclude that mechanical changes induced by a prolapsing valve likely drive phenotypic responses within the LV, independent of mitral regurgitant volume overload, although that can augment the process. 11 Recent studies have shown that isolated mitral regurgitation without MVP has a much smaller effect on magnetic resonance imaging–indicated fibrosis than MVP. 11
Transmission of excess force by a prolapsing valve will likely be felt by various mechanonsensors within tissue and cells that are tethered to these excess forces within the PM and inferobasal myocardium. The primary cilia serve as cellular antennae that respond to these forces within the LV myocardium. Cell membrane tension on the primary cilia will affect all functional aspects of their mechanosensing. 42 , 43 Recent studies have invoked mechanosensing primary cilia as involved in ECM production and/or fibrotic diseases. Profibrotic molecules such as TGFβ have been shown to suppress ciliogenesis through negative regulation of the essential ciliogenic gene, Ift88, and knockdown of Ift88 enhances TGFβ‐induced collagen expression. 44 This finding of enhanced collagen synthesis during impaired ciliogenesis is consistent with recent data from multiple groups in ciliopathy patients who have mutations in critical ciliogenesis genes. 45 Human ciliopathy conditions, such as polycystic kidney disease, Bardet‐Biedl syndrome, and others, commonly have extensive fibrosis within affected organs. Knockdown of zebrafish polycystins, mechanosensitive receptors/channels localized to the primary cilium, induces substantial collagen type I overexpression. 46 Similar findings were obtained from our group in mitral and aortic valve studies showing that loss of ciliogenic genes results in a robust increase in ECM synthesis, including collagen type I. 13 , 17 , 20 , 31 Although clinical cardiovascular studies are limited relative to the function of primary cilia, one study has described loss of primary cilia correlated with increased fibrosis in biopsies from patients with atrial fibrillation. 47 In this same study, loss of primary cilia by RNAi in fibroblasts increased presence of myofibroblasts and expression of ECM genes in response to transforming growth factor beta 1. 47 Because the potential role of cilia in fibrosis is gaining increased attention, it is becoming more evident that mechanical stresses acting through these structures have an impact on fibrosis pathways. In this study, we show that primary cilia are present within normal adult working myocardium in humans but are deficient in areas of regional fibrosis in MVP biopsies. Although fibroblast primary cilia appear to be required for fibrosis suppression, our data also support a role for ECM stiffness as a suppressive ciliogenic signal as shown in Figure 7. Mechanical forces may stimulate a fibrogenic response regardless of any particular genetic mechanism or mechanosensor. Also worthy of consideration for future investigation are other stretch‐activated signaling pathways associated with inflammation. Of these, those linked to mechanosensitive Cx43 (connexin 43) hemichannels may be of interest and relevance. 48 , 49 Release of purinergic signaling molecules by hemichannels such as ATP in response to disease, injury, or infection are a known activator of the innate inflammatory response. 50 , 51 , 52 Cx43 hemichannels have also recently emerged as key pathogenic determinants of myocardial fibrosis, in diseases such as arrhythmic right ventricular cardiomyopathy 53 and Duchenne muscular dystrophy. 54 Interestingly, Cx43 hemichannels have been localized on primary cilia in auricular chondrocytes where they are thought to function as a mechanosensitive ATP‐release channel. 55
Clinical Implications and Conclusions
MVP has been associated with LV remodeling, LV dysfunction, heart failure, arrhythmogenesis, and sudden cardiac death if left untreated. 7 , 56 Mitral valve repair is a highly effective therapy for severe mitral regurgitation because of MVP but often leaves patients with residual fibrosis and LV dysfunction and arrhythmias. 57 Our mouse models demonstrate that regionalized LV fibrosis in MVP is progressive and has a similar localization and molecular and cellular signature as in the human disease. The localization of fibrosis within the myocardium suggests a role for increased mechanical stress through the chordae in MVP (Figure 9), consistent with recent reports. 58 , 59 , 60 Support for increased mechanical stress is also based on a recent study that showed primary chordal forces, and therefore the force on the attached papillary muscles, are 5‐fold higher in bileaflet prolapse compared with post–mitral valve repair. 61 Although our current findings and previous reports strongly support primary cilia defects in the cause of MVP, it is likely that MVP and MVP‐associated LV remodeling do not solely stem from defects in the cilia pathway but instead through alternate independent mechanisms. Nonetheless, our computational modeling, which in turn is validated by molecular and cellular analyses in mouse and human MVP samples, further supports induction of fibrosis through increased mechanical stress. Waiting for traditional indications for mitral valve repair (ie, symptoms, LV dilation, LV dysfunction) may be associated with reduced survival after repair, 5 and therefore, identification of other important predictors of outcome (eg, advanced fibrosis) may have the potential to affect clinical guidelines and practice. Although no studies to date have unequivocally shown that earlier surgery leads to less fibrosis and sudden death, several studies have demonstrated better long‐term survival if surgery is performed earlier in the disease process. 57 However, more and long‐term data are required to link MVP‐induced fibrosis with postoperative LV dysfunction and long‐term mortality, which are beyond the scope of this study. Regardless, uncovering mechanisms of mechanical, cellular, and molecular changes within the left ventricle of patients with MVP, as shown in this study, may provide understanding of residual contractile dysfunction and electrical instability after mitral repair.
Figure 9. Model for stress‐induced fibrosis in mitral valve prolapse (MVP).
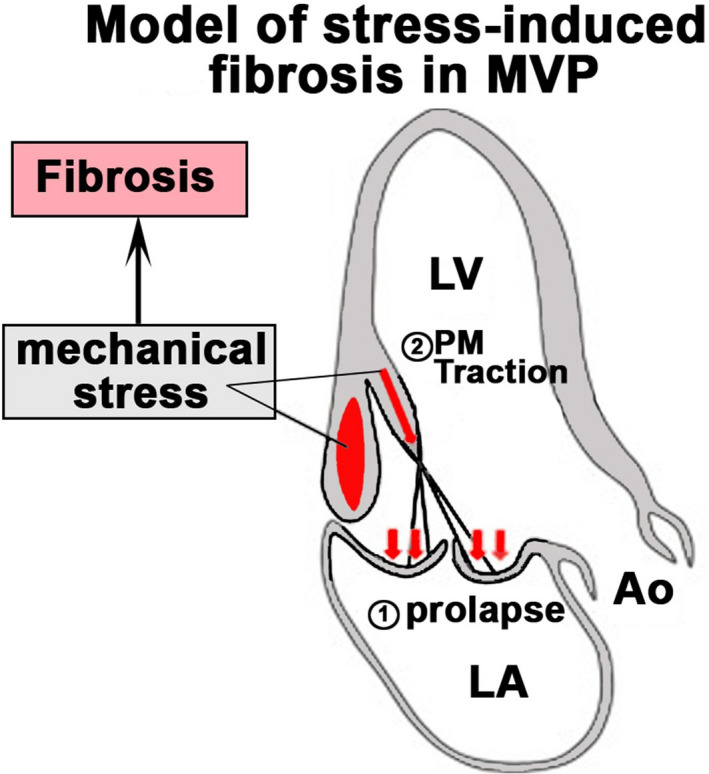
MVP (1) induces increased PM traction (2) and basal LV wall tension as fibrotic stimuli resulting in regionalized fibrosis (red). Ao indicates aorta; LA, left atrium; LV, left ventricle; and PM, papillary muscle.
Sources of Funding
This work was supported by the National Institutes of Health grant numbers: HL131546 (Norris), GM103444 (Norris), HL149696 (Norris), T32HL007260 (Beck, Gensemer, and Moore), F31152494 (Moore), T32GM132055 (Gensemer), F31HL142159 (Fulmer), GM12132 (Richardson), HL144927 (Richardson), HL141855 (Gourdie), HL56728 (Gourdie), HL128099 (Levine), and HL141917 (Levine). Grant sponsor: American Heart Association grant numbers: 19TPA34850095 (Norris), 17CSA33590067 (Norris), and 18PRE34080172 (Guo), and a contribution from the Ellison Foundation (Levine) Boston, Massachusetts. This project was supported, in part, by the National Center for Advancing Translational Sciences of the National Institutes of Health under grant number UL1 TR001450. The work at the Medical University of South Carolina was performed in a facility constructed with support from the National Institutes of Health grant number C06 RR018823 from the Extramural Research Facilities Program of the National Center for Research Resources.
Disclosures
Dr Gourdie is a nonremunerated member of the Scientific Advisory Board of FirstString Research, Inc., which licensed α carboxyl terminus 1 peptide. Dr Gourdie has a modest ownership interest in FirstString Research, Inc. (<3% of company stock). Dr Borger discloses that his hospital receives speakers’ honoraria and/or consulting fees on his behalf from Edwards Lifesciences, Medtronic, Abbott, and CryoLife. The remaining authors have no disclosures to report.
Supporting information
Tables S1–S3
Figures S1–S5
Videos S1
Videos S2
Videos S3
Videos S4
Videos S5
Acknowledgments
The authors acknowledge S. Hoffman for his generous contribution of the collagen antibody used for IHC in this article. Figures 8A and 9 were created with BioRender.com. We thank biostatistician Dr Nietert and the research team at the South Carolina Clinical and Translational Research Institute for assistance on all statistical aspects of this project.
Drs Norris, Levine, Borger, Richardson, Gourdie, and Poelzing, and C. Gensemer and J.E. Morningstar wrote and edited the article. C. Gensemer, J.E. Morningstar, R. Moore, Dr Fulmer, K. Watts, T.C. Beck, C. Wang, Dr Moore, Dr Guo, Dr Sieg, Dr Nagata, Dr Bertrand, Dr Spampinato, and J. Glover conducted experiments and acquired data. Dr Norris, J.E. Morningstar, C. Gensemer, Dr Levine, and Dr Borger analyzed and interpreted the data and reviewed the article.
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.022332
For Sources of Funding and Disclosures, see page 15.
REFERENCES
- 1. Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez‐Sarano M. Burden of valvular heart diseases: a population‐based study. Lancet. 2006;368:1005–1011. doi: 10.1016/S0140-6736(06)69208-8 [DOI] [PubMed] [Google Scholar]
- 2. Basso C, Perazzolo Marra M, Rizzo S, De Lazzari M, Giorgi B, Cipriani A, Frigo AC, Rigato I, Migliore F, Pilichou K, et al. Arrhythmic mitral valve prolapse and sudden cardiac death. Circulation. 2015;132:556–566. doi: 10.1161/CIRCULATIONAHA.115.016291 [DOI] [PubMed] [Google Scholar]
- 3. Levine RA, Hagége AA, Judge DP, Padala M, Dal‐Bianco JP, Aikawa E, Beaudoin J, Bischoff J, Bouatia‐Naji N, Bruneval P, et al. Mitral valve disease‐morphology and mechanisms. Nat Rev Cardiol. 2015;12:689–710. doi: 10.1038/nrcardio.2015.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miller MA, Dukkipati SR, Turagam M, Liao SL, Adams DH, Reddy VY. Arrhythmic mitral valve prolapse: JACC review topic of the week. J Am Coll Cardiol. 2018;72:2904–2914. doi: 10.1016/j.jacc.2018.09.048 [DOI] [PubMed] [Google Scholar]
- 5. Borger MA, Mansour MC, Levine RA. Atrial fibrillation and mitral valve prolapse: time to intervene? J Am Coll Cardiol. 2019;73:275–277. doi: 10.1016/j.jacc.2018.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quintana E, Suri RM, Thalji NM, Daly RC, Dearani JA, Burkhart HM, Li Z, Enriquez‐Sarano M, Schaff HV. Left ventricular dysfunction after mitral valve repair–the fallacy of "normal" preoperative myocardial function. J Thorac Cardiovasc Surg. 2014;148:2752–2760. doi: 10.1016/j.jtcvs.2014.07.029 [DOI] [PubMed] [Google Scholar]
- 7. Basso C, Iliceto S, Thiene G, Perazzolo MM. Mitral valve prolapse, ventricular arrhythmias, and sudden death. Circulation. 2019;140:952–964. doi: 10.1161/CIRCULATIONAHA.118.034075 [DOI] [PubMed] [Google Scholar]
- 8. Han H‐C, Parsons SA, Curl CL, Teh AW, Raaijmakers AJA, Koshy AN, Leong T, Burrell LM, O’Donnell D, Vohra JK, et al. Systematic quantification of histologic ventricular fibrosis in isolated mitral valve prolapse and sudden cardiac death. Heart Rhythm. 2021;18:570–576. doi: 10.1016/j.hrthm.2020.12.021 [DOI] [PubMed] [Google Scholar]
- 9. Bui AH, Roujol S, Foppa M, Kissinger KV, Goddu B, Hauser TH, Zimetbaum PJ, Ngo LH, Manning WJ, Nezafat R, et al. Diffuse myocardial fibrosis in patients with mitral valve prolapse and ventricular arrhythmia. Heart. 2017;103:204–209. doi: 10.1136/heartjnl-2016-309303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Constant Dit Beaufils A‐L, Huttin O, Jobbe‐Duval A, Senage T, Filippetti L, Piriou N, Cueff C, Venner C, Mandry D, Sellal J‐M, et al. Replacement myocardial fibrosis in patients with mitral valve prolapse: relation to mitral regurgitation, ventricular remodeling and arrhythmia. Circulation. 2021;143:1763–1774. doi: 10.1161/CIRCULATIONAHA.120.050214 [DOI] [PubMed] [Google Scholar]
- 11. Kitkungvan D, Nabi F, Kim RJ, Bonow RO, Khan MA, Xu J, Little SH, Quinones MA, Lawrie GM, Zoghbi WA, et al. Myocardial fibrosis in patients with primary mitral regurgitation with and without prolapse. J Am Coll Cardiol. 2018;72:823–834. doi: 10.1016/j.jacc.2018.06.048 [DOI] [PubMed] [Google Scholar]
- 12. Dejgaard LA, Skjølsvik ET, Lie ØH, Ribe M, Stokke MK, Hegbom F, Scheirlynck ES, Gjertsen E, Andresen K, Helle‐Valle TM, et al. The mitral annulus disjunction arrhythmic syndrome. J Am Coll Cardiol. 2018;72:1600–1609. doi: 10.1016/j.jacc.2018.07.070 [DOI] [PubMed] [Google Scholar]
- 13. Toomer KA, Yu M, Fulmer D, Guo L, Moore KS, Moore R, Drayton KD, Glover J, Peterson N, Ramos‐Ortiz S, et al. Primary cilia defects causing mitral valve prolapse. Sci Transl Med. 2019;11:eaax0290. doi: 10.1126/scitranslmed.aax0290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu M, Georges A, Tucker NR, Kyryachenko S, Toomer K, Schott JJ, Delling FN, Fernandez‐Friera L, Solis J, Ellinor PT, et al. Genome‐wide association study‐driven gene‐set analyses, genetic, and functional follow‐up suggest GLIS1 as a susceptibility gene for mitral valve prolapse. Circ Genom Precis Med. 2019;12:e002497. doi: 10.1161/CIRCGEN.119.002497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baumgartner H, Falk V, Bax JJ, De Bonis M, Hamm C, Holm PJ, Iung B, Lancellotti P, Lansac E, Rodriguez Muñoz D, et al. 2017 ESC/EACTS guidelines for the management of valvular heart disease. Eur Heart J. 2017;38:2739–2791. doi: 10.1093/eurheartj/ehx391 [DOI] [PubMed] [Google Scholar]
- 16. Durst R, Sauls K, Peal DS, deVlaming A, Toomer K, Leyne M, Salani M, Talkowski ME, Brand H, Perrocheau M, et al. Mutations in DCHS1 cause mitral valve prolapse. Nature. 2015;525:109–113. doi: 10.1038/nature14670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fulmer D, Toomer K, Guo L, Moore K, Glover J, Moore R, Stairley R, Lobo G, Zuo X, Dang Y, et al. Defects in the exocyst‐cilia machinery cause bicuspid aortic valve disease and aortic stenosis. Circulation. 2019;140:1331–1341. doi: 10.1161/CIRCULATIONAHA.119.038376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sauls K, Toomer K, Williams K, Johnson AJ, Markwald RR, Hajdu Z, Norris RA. Increased infiltration of extra‐cardiac cells in myxomatous valve disease. J Cardiovasc Dev Dis. 2015;2:200–213. doi: 10.3390/jcdd2030200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Toomer K, Sauls K, Fulmer D, Guo L, Moore K, Glover J, Stairley R, Bischoff J, Levine RA, Norris RA. Filamin‐a as a balance between Erk/Smad activities during cardiac valve development. Anat Rec (Hoboken). 2019;302:117–124. doi: 10.1002/ar.23911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Toomer KA, Fulmer D, Guo L, Drohan A, Peterson N, Swanson P, Brooks B, Mukherjee R, Body S, Lipschutz JH, et al. A role for primary cilia in aortic valve development and disease. Dev Dyn. 2017;246:625–634. doi: 10.1002/dvdy.24524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rogers JD, Richardson WJ. Fibroblast mechanotransduction network predicts targets for mechano‐adaptive infarct therapies. BioRxiv. August 14, 2020. doi: 10.1101/2020.08.13.250001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zeigler AC, Richardson WJ, Holmes JW, Saucerman JJ. A computational model of cardiac fibroblast signaling predicts context‐dependent drivers of myofibroblast differentiation. J Mol Cell Cardiol. 2016;94:72–81. doi: 10.1016/j.yjmcc.2016.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Putten S, Shafieyan Y, Hinz B. Mechanical control of cardiac myofibroblasts. J Mol Cell Cardiol. 2016;93:133–142. doi: 10.1016/j.yjmcc.2015.11.025 [DOI] [PubMed] [Google Scholar]
- 24. Milan M, Pace V, Maiullari F, Chirivì M, Baci D, Maiullari S, Madaro L, Maccari S, Stati T, Marano G, et al. Givinostat reduces adverse cardiac remodeling through regulating fibroblasts activation. Cell Death Dis. 2018;9:108. doi: 10.1038/s41419-017-0174-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schafer S, Viswanathan S, Widjaja AA, Lim W‐W, Moreno‐Moral A, DeLaughter DM, Ng B, Patone G, Chow K, Khin E, et al. IL‐11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552:110–115. doi: 10.1038/nature24676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. 2016;118:1021–1040. doi: 10.1161/CIRCRESAHA.115.306565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637–1652. doi: 10.1016/0735-1097(89)90360-4 [DOI] [PubMed] [Google Scholar]
- 28. Dina C, Bouatia‐Naji N, Tucker N, Delling FN, Toomer K, Durst R, Perrocheau M, Fernandez‐Friera L, Solis J, Le Tourneau T, et al. Genetic association analyses highlight biological pathways underlying mitral valve prolapse. Nat Genet. 2015;47:1206–1211. doi: 10.1038/ng.3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Villalobos E, Criollo A, Schiattarella GG, Altamirano F, French KM, May HI, Jiang N, Nguyen NUN, Romero D, Roa JC, et al. Fibroblast primary cilia are required for cardiac fibrosis. Circulation. 2019;139:2342–2357. doi: 10.1161/CIRCULATIONAHA.117.028752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gerdes JM, Katsanis N. Ciliary function and Wnt signal modulation. Curr Top Dev Biol. 2008;85:175–195. doi: 10.1016/S0070-2153(08)00807-7 [DOI] [PubMed] [Google Scholar]
- 31. Fulmer D, Toomer KA, Glover J, Guo L, Moore K, Moore R, Stairley R, Gensemer C, Abrol S, Rumph MK, et al. Desert hedgehog‐primary cilia cross talk shapes mitral valve tissue by organizing smooth muscle actin. Dev Biol. 2020;463:26–38. doi: 10.1016/j.ydbio.2020.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740 [DOI] [PubMed] [Google Scholar]
- 33. Arrighi N, Lypovetska K, Moratal C, Giorgetti‐Peraldi S, Dechesne CA, Dani C, Peraldi P. The primary cilium is necessary for the differentiation and the maintenance of human adipose progenitors into myofibroblasts. Sci Rep. 2017;7:15248. doi: 10.1038/s41598-017-15649-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clement CA, Ajbro KD, Koefoed K, Vestergaard ML, Veland IR, Henriques de Jesus MP, Pedersen LB, Benmerah A, Andersen CY, Larsen LA, et al. TGF‐beta signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 2013;3:1806–1814. doi: 10.1016/j.celrep.2013.05.020 [DOI] [PubMed] [Google Scholar]
- 35. Labour MN, Riffault M, Christensen ST, Hoey DA. TGFbeta1—induced recruitment of human bone mesenchymal stem cells is mediated by the primary cilium in a SMAD3‐dependent manner. Sci Rep. 2016;6:35542. doi: 10.1038/srep35542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jones TJ, Adapala RK, Geldenhuys WJ, Bursley C, AbouAlaiwi WA, Nauli SM, Thodeti CK. Primary cilia regulates the directional migration and barrier integrity of endothelial cells through the modulation of hsp27 dependent actin cytoskeletal organization. J Cell Physiol. 2012;227:70–76. doi: 10.1002/jcp.22704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song B, Haycraft CJ, Seo HS, Yoder BK, Serra R. Development of the post‐natal growth plate requires intraflagellar transport proteins. Dev Biol. 2007;305:202–216. doi: 10.1016/j.ydbio.2007.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Z, Wann AK, Thompson CL, Hassen A, Wang W, Knight MM. IFT88 influences chondrocyte actin organization and biomechanics. Osteoarthritis Cartilage. 2016;24:544–554. doi: 10.1016/j.joca.2015.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Calder BW, Matthew Rhett J, Bainbridge H, Fann SA, Gourdie RG, Yost MJ. Inhibition of connexin 43 hemichannel‐mediated ATP release attenuates early inflammation during the foreign body response. Tissue Eng Part A. 2015;21:1752–1762. doi: 10.1089/ten.tea.2014.0651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Da Silva N, Panizzi P, van der Laan AM, et al. Differential contribution of monocytes to heart macrophages in steady‐state and after myocardial infarction. Circ Res. 2014;115:284–295. doi: 10.1161/CIRCRESAHA.115.303567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wülfers EM, Seemann G, Courties G, et al. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169:510–522.e520. doi: 10.1016/j.cell.2017.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ishikawa H, Marshall WF. Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol. 2011;12:222–234. doi: 10.1038/nrm3085 [DOI] [PubMed] [Google Scholar]
- 43. Ishikawa H, Marshall WF. Mechanobiology of ciliogenesis. Bioscience. 2014;64:1084–1091. doi: 10.1093/biosci/biu173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kawasaki M, Ezura Y, Hayata T, Notomi T, Izu Y, Noda M. TGF‐beta suppresses Ift88 expression in chondrocytic ATDC5 cells. J Cell Physiol. 2015;230:2788–2795. doi: 10.1002/jcp.25005 [DOI] [PubMed] [Google Scholar]
- 45. Seeger‐Nukpezah T, Golemis EA. The extracellular matrix and ciliary signaling. Curr Opin Cell Biol. 2012;24:652–661. doi: 10.1016/j.ceb.2012.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mangos S, Lam PY, Zhao A, Liu Y, Mudumana S, Vasilyev A, Liu A, Drummond IA. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis Model Mech. 2010;3:354–365. doi: 10.1242/dmm.003194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kawasaki M, Nariswari NA, Berg NWEVD, Neefs J, Meulendijks ER, Wesselink R, Baalman SWE, Boven WJPV, Driessen AHG, De Groot JR. The primary cilium, a sensory apparatus of cells regulates the profibrotic capacity of fibroblasts in atrial fibrillation. Eur Heart J. 2019;40:2111. doi: 10.1093/eurheartj/ehz745.0365 [DOI] [Google Scholar]
- 48. Gourdie RG, Dimmeler S, Kohl P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat Rev Drug Discov. 2016;15:620–638. doi: 10.1038/nrd.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang J, Hoagland D, Palatinus JA, He H, Iyyathurai J, Jourdan LJ, Bultynck G, Wang Z, Zhang Z, Schey K, et al. Interaction of alpha carboxyl terminus 1 peptide with the connexin 43 carboxyl terminus preserves left ventricular function after ischemia‐reperfusion injury. J Am Heart Assoc. 2019;8:e012385. doi: 10.1161/JAHA.119.012385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200 [DOI] [PubMed] [Google Scholar]
- 51. Lu D, Soleymani S, Madakshire R, Insel PA. ATP released from cardiac fibroblasts via connexin hemichannels activates profibrotic P2Y2 receptors. FASEB J. 2012;26:2580–2591. doi: 10.1096/fj.12-204677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dosch M, Zindel J, Jebbawi F, Melin N, Sanchez‐Taltavull D, Stroka D, Candinas D, Beldi G. Connexin‐43‐dependent ATP release mediates macrophage activation during sepsis. eLife. 2019;8:e42670. doi: 10.7554/eLife.42670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim JC, Perez‐Hernandez M, Alvarado FJ, Maurya SR, Montnach J, Yin Y, Zhang M, Lin X, Vasquez C, Heguy A, et al. Disruption of Ca(2+)i homeostasis and connexin 43 hemichannel function in the right ventricle precedes overt arrhythmogenic cardiomyopathy in plakophilin‐2‐deficient mice. Circulation. 2019;140:1015–1030. doi: 10.1161/CIRCULATIONAHA.119.039710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nouet J, Himelman E, Lahey KC, Zhao Q, Fraidenraich D. Connexin‐43 reduction prevents muscle defects in a mouse model of manifesting Duchenne muscular dystrophy female carriers. Sci Rep. 2020;10:5683. doi: 10.1038/s41598-020-62844-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang J, Chandrasekaran G, Li W, Kim D‐Y, Jeong IY, Lee S‐H, Liang T, Bae JY, Choi I, Kang H, et al. Wnt‐PLC‐IP3‐connexin‐Ca(2+) axis maintains ependymal motile cilia in zebrafish spinal cord. Nat Commun. 2020;11:1860. doi: 10.1038/s41467-020-15248-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ermakov S, Gulhar R, Lim L, Bibby D, Fang Q, Nah G, Abraham TP, Schiller NB, Delling FN. Left ventricular mechanical dispersion predicts arrhythmic risk in mitral valve prolapse. Heart. 2019;105:1063–1069. doi: 10.1136/heartjnl-2018-314269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. David TE, David CM, Tsang W, Lafreniere‐Roula M, Manlhiot C. Long‐term results of mitral valve repair for regurgitation due to leaflet prolapse. J Am Coll Cardiol. 2019;74:1044–1053. doi: 10.1016/j.jacc.2019.06.052 [DOI] [PubMed] [Google Scholar]
- 58. Grinberg D, Cottinet PJ, Thivolet S, Audigier D, Capsal JF, Le MQ, Obadia JF. Measuring chordae tension during transapical neochordae implantation: toward understanding objective consequences of mitral valve repair. J Thorac Cardiovasc Surg. 2019;158:746–755. doi: 10.1016/j.jtcvs.2018.10.029 [DOI] [PubMed] [Google Scholar]
- 59. Grinberg D, Le MQ, Kwon YJ, Fernandez MA, Audigier D, Ganet F, Capsal JF, Obadia JF, Cottinet PJ. Mitral valve repair based on intraoperative objective measurement. Sci Rep. 2019;9:4677. doi: 10.1038/s41598-019-41173-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Paulsen MJ, Imbrie‐Moore AM, Wang H, Bae JH, Hironaka CE, Farry JM, Lucian HJ, Thakore AD, MacArthur JW, Cutkosky MR, et al. Mitral chordae tendineae force profile characterization using a posterior ventricular anchoring neochordal repair model for mitral regurgitation in a three‐dimensional‐printed ex vivo left heart simulator. Eur J Cardiothorac Surg. 2020;57:535–544. doi: 10.1093/ejcts/ezz258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Imbrie‐Moore AM, Paulsen MJ, Zhu Y, Wang H, Lucian HJ, Farry JM, MacArthur JW, Ma M, Woo YJ. A novel cross‐species model of Barlow's disease to biomechanically analyze repair techniques in an ex vivo left heart simulator. J Thorac Cardiovasc Surg. 2021;161:1776–1783. doi: 10.1016/j.jtcvs.2020.01.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Figures S1–S5
Videos S1
Videos S2
Videos S3
Videos S4
Videos S5