

Abstract
Background
Transient receptor potential canonical (TRPC) channels play a role in angiogenesis. However, the involvement of TRPC1 in myocardial infarction (MI) remains unclear. The present study was aimed at investigating whether TRPC1 can improve the recovery of cardiac function via prompting angiogenesis following MI.
Methods and Results
In vitro, coronary artery endothelial cells from floxed TRPC1 mice and endothelial cell‐specific TRPC1 channel knockout mice were cultured to access EC angiogenesis. Both EC tube formation and migration were significantly suppressed in mouse coronary artery endothelial cells from endothelial cell‐specific TRPC1 channel knockout mice. In vivo, coronary artery endothelial cells from floxed TRPC1 and endothelial cell‐specific TRPC1 channel knockout mice were subjected to MI, then echocardiography, triphenyltetrazolium chloride staining and immunofluorescence were performed to assess cardiac repair on day 28. Endothelial cell‐specific TRPC1 channel knockout mice had higher ejection fraction change, larger myocardial infarct size, and reduced capillary density in the infarct area compared with coronary artery endothelial cells from floxed TRPC1 mice. Furthermore, we found underlying regulation by HIF‐1α (hypoxic inducible factor‐1α) and MEK‐ERK (mitogen‐activated protein kinase/extracellular signal‐regulated kinase) that could be the mechanism for the angiogenetic action of TRPC1. Significantly, treatment with dimethyloxaloylglycine, an activator of HIF‐1α, induced cardiac improvement via the HIF‐1α‐TRPC1‐MEK/ERK pathway in MI mice.
Conclusions
Our study demonstrated TRPC1 improves cardiac function after MI by increasing angiogenesis via the upstream regulator HIF‐1α and downstream MEK/ERK, and dimethyloxaloylglycine treatment has protective effect on MI through the HIF‐1α‐TRPC1‐MEK/ERK pathway.
Keywords: angiogenesis, hypoxia‐inducible factor, myocardial infarction, transient receptor potential channels, vascular biology
Subject Categories: Angiogenesis, Cardiovascular Disease, Myocardial Infarction
Nonstandard Abbreviations and Acronyms
- MCAECs
mouse coronary artery endothelial cells
- TRPC1EC ‐/‐
endothelial cell‐specific TRPC1 channel knockout
Clinical Perspective
What Is New?
Transient receptor potential canonical 1 plays a role in recovery of cardiac function via prompting angiogenesis of coronary artery endothelial cells following myocardial infarction.
HIF‐1α (hypoxic inducible factor‐1α) and MEK‐ERK (mitogen‐activated protein kinase/extracellular signal‐regulated kinase) affected the angiogenetic action of transient receptor potential canonical 1.
Dimethyloxaloylglycine treatment has a protective effect on myocardial ischemia through the HIF‐1α‐transient receptor potential canonical 1‐MEK/ERK pathway.
What Are the Clinical Implications?
This finding provides potential therapeutics targeting endothelial TRPC1 to treat myocardial infarction.
Among the cardiovascular diseases, myocardial infarction (MI) is the leading cause of mortality and disability worldwide. 1 It damages the structure and function of the heart, and ultimately leads to cardiac arrhythmia and heart failure. Nevertheless, the effective formation of coronary collaterals in the infarcted zone is crucial for the maintenance of cardiac function after MI. 2 In this regard, enhanced angiogenesis could be a promising approach to improve the post‐MI prognosis. 3 , 4
Angiogenesis refers to the budding of new vessels from original vessels by migration and proliferation, with subsequent modification and maturation. Through sprouting of capillary networks via the migration and proliferation of previously differentiated endothelial cells (ECs), angiogenesis is essential to extend the vascular bed initially formed by vasculogenesis. A variety of angiogenic growth factors have been used in therapy, including vascular endothelial growth factor (VEGF), fibroblast growth factor, hepatocyte growth factor, platelet‐derived growth factor, angiotension‐1, and insulin‐like growth factor 1. 5 , 6 , 7 , 8 However, considering the advantages and disadvantages of each agent, the ideal angiogenic growth factor for therapeutic angiogenesis application has not been found so far. 9 Consequently, it is critical to search for other crucial angiogenic factors with potential to treat ischemic heart disease.
Endothelial transient receptor potential canonical (TRPC) channels have long been associated to angiogenesis and vascular remodeling because of their ability to engage intracellular signaling pathways associated to endothelial cell proliferation, migration, adhesion, tubulogenesis, and permeability. 10 , 11 , 12 Endothelial TRPC channels affect the angiogenic process by maintaining the calcium ion response to pro‐angiogenic factors, including VEGF, basic fibroblast growth factor, and/or by detecting delicate variations in the composition of the local microenvironment. 13 , 14 TRPC1 regulates angiogenesis following VEGF‐, basic fibroblast growth factor‐, insulin‐like growth factor‐, and thrombin‐induced stimulation. 15 TRPC4 is responsive to oxidized low‐density lipoprotein, epidermal growth factor, and hypoxia. 16 TRPC3 is sensitive to VEGF activation and erythropoietin, similar to TRPC6, which is mediated by thrombin, oxidized low‐density lipoprotein, and 14,15‐epoxyeicosatrienoic acid. 17 TRPC5 responds to oxidized low‐density lipoprotein‐dependent stimulation, hypoxia, and riluzole. 18 , 19 Although the angiogenic effect of TRPC channels is generally understood, their involvement in MI is still shrouded in mystery. The aim of this study was to explore whether TRPC channels mediate post‐MI angiogenesis and which TRPC channel provides an appropriate therapeutic option for ischemic heart disease.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animal Sources and Cell Cultures
Six‐ to eight‐week‐old C57BL/6 male mice were purchased from the Shanghai SLAC Laboratory Animal Co., Ltd. Coronary artery endothelial cells from floxed TRPC1 (TRPC1fl/fl) and Tek‐Cre mice were from GemPharmatech (Jiangsu, China). EC‐specific TRPC1 knockout mice (TRPC1fl/fl‐Tek‐Cre; TRPC1EC ‐/‐) were generated by breeding TRPC1fl/fl mice with Tek‐Cre mice. Littermates without Tek–Cre were used as controls. Genotyping was performed by PCR (Figure S1) with the use of the F1‐R1 and F2‐R2 primer sets listed below: F1, 5’‐CGAGTGATGAGGTTCGCAAG‐3’; R1, 5’‐TGAGTGAACGAACCTGGT‐3’; and F2, 5’‐CTTCTCCTTCCAGTTTCCACTCTG‐3’; R2, 5’‐CTACTTGATCTACCAGGAGCTGGC‐3’. TRPC1 mRNA level was reduced in heart, mesentery and aorta of TRPC1EC‐/‐ mice (Figure S2A) and almost not expressed in endothelial cells (Figure S2B). Immunohistochemical staining confirmed TRPC1 deficiency in endothelial layer of artery of TRPC1EC‐/‐ mice (Figure S2C). All mice were raised in a specific pathogen free—rated environment. Each group had 5 mice. All animal experiments were approved by the Animal Experimentation Ethics Committee of Jiangnan University and performed in compliance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication, 8th edition, updated 2011).
Primary mouse coronary artery endothelial cells (MCAECs) were isolated as described previously. 20 Briefly, the freshly taken out heart tissues were collected in PBS, after removing the surrounding excess tissues and blood, the hearts were cut to pieces until there were no obvious large tissue fragments. Then, the tissues were digested with collagenase type IA (Sigma, USA) for 30 to 35 minutes at 37 °C. The collagenase solution was harvested and after centrifugation at 1200g for 5 minutes, EC pellet was resuspended in 1 mL of endothelial cell growth medium (ScienCell, USA) with 5% of fetal bovine serum. Cultures were maintained at 37 °C in humidified CO2 incubator (Thermo Scientific Pierce, Rockford, IL, USA). Medium were changed after the endothelial cells adhered to the plates for 2 hours and then were changed regularly according to the cell growth situation.
Western Blot
Cells and tissues were lysed for 2 hours in RIPA lysis solution freshly prepared with a protease inhibitor (Roche). The concentration of extracted proteins was determined with a bicinchoninic acid protein determination kit (enhanced) (P0010, Beyotime, Shanghai, China). The extracted proteins were then separated by molecular size on SDS‐PAGE gels, followed by transfer to polyvinylidene fluoride membranes, which were incubated at 4 °C overnight with primary antibodies (1:200–1:1000) dissolved in Tris‐buffered saline containing Tween 20 containing 5% skimmed milk powder. The antibodies were as follows: anti‐TRPC1 (sc‐133076, Santa Cruz Biotechnology), anti‐HIF‐1α (abs130612, Absin), anti‐MEK (2352S, Cell Signaling Technology), anti‐p‐MEK (ab96379, Abcam), anti‐Erk (ab32537, Abcam), anti‐p‐Erk (4370S, Cell signaling technology), and anti‐GAPDH (21612, Signalway Antibody). Afterwards, the membrane was washed with Tris‐buffered saline containing Tween 20 and incubated with the corresponding secondary antibodies for 2 hours at room temperature. All the secondary antibodies were from Abcam. All blots were detected with an ECL substrate (180‐5001, Tanon, Shanghai, China), and images were captured with Image Lab Software (Bio‐Rad).
Tube‐Formation Assay
Matrigel matrix (354234, BD Biosciences, Oxford, UK) with reduced growth factors was thawed overnight at 4 °C. Ninety‐six well plates and tips were initially cooled at 4 °C for at least 2 hours, then 50 µL Matrigel was added and incubated at 37 °C for 30 to 45 minutes. ECs (4×104) were added to the plates after the Matrigel had solidified, then the plates were incubated at 37 °C for 12 hours. A hypoxic environment was established in a humidified multi‐gas incubator containing 1% O2, 5% CO2, and 94% N2 (4040TS, Thermo, MA, USA). Images of networks were captured with a microscope‐mounted camera (Nikon Coolpix 54, Tokyo, Japan). Five randomly selected regions were used to quantify the results of tube formation.
Transwell
Cells were digested and then re‐suspended in serum‐free medium at 4×104 cells/chamber. Fetal bovine serum containing medium (500 µL) was added to the 24‐well plate in advance, then 200 µL of the cell suspension was added to the chamber. After 24 hours, cells were fixed with 4% paraformaldehyde, then stained with 0.1% crystal violet. The numbers of migrated cells were counted in 9 to 12 randomly selected fields.
Transfection of siRNA
The cells were inoculated on a 6‐well plate, and the cell density reached 70% to 80% confluence before transfection with siRNA. The sequences of siRNAs are shown in Table S1. Lipofectamine 2000 (cat. no. 11668027; Invitrogen; Thermo Fisher Scientific, Inc.), Opti‐MEM (cat. no. 31985070; Gibco; Thermo Fisher Scientific, Inc.), and siRNA were mixed and slowly dripped into the 6‐well plate. During transfection, the 6‐well plate medium was changed to serum‐free medium (1 mL per well). After culture at 37 °C and 5% CO2 for 5 to 6 hours, the mixed transfection reagents were replaced with complete endothelial cell growth medium (without antibiotics), and experiments were performed after 48 hours of transfection. 21
Reverse Transcriptase‐Quantitative Polymerase Chain Reaction
Total RNA was extracted using TRIzol (Ambion, 262301, USA) and reverse‐transcribed from RNA to cDNA using the corresponding kit (RR036A, TAKARA, Japan). The sequences of the primers used to amplify the cDNA are listed in Table S2. Reverse transcriptase‐quantitative polymerase chain reaction used the SYBR Green kit (RR420A, TAKARA). The thermocycling procedure was as follows: pre‐denaturation at 95 °C for 30 seconds, followed by 40 cycles of 95 °C for 5 seconds, and 60 °C for 30 seconds using a real‐time PCR cycler (Roche Light Cycler 480 II, Basel, Switzerland).
Mouse Model of MI
C57BL/6 male mice were anesthetized by intraperitoneal. injection of pentobarbital (10 mg·kg−1 body weight), while artificial ventilation (SAR‐1000 Small Animal Ventilator, CWE, Inc.) was used to provide life support. After exposing the heart, one third of the left anterior descending artery was ligated. Iodophor was used to disinfect the incision; 30 000 U of penicillin was injected intramuscularly; and tidal volume was adjusted appropriately. 22 Echocardiography was taken at days 3 and 28 after left anterior descending artery ligation, and left ventricle (LV) dimensions were quantified by digitally recorded two‐dimensional clips and M‐mode images from the mid‐LV just below the papillary muscles to allow for consistent measurements from the same anatomical location in different mice. Corresponding index like ejection fraction, fractional shortening, diastolic LV internal dimensions and systolic LV internal dimensions were measured to analyze heart function. After taking echocardiogram, mice were euthanized immediately, their hearts were harvested and reserved for next experiments. Mice received dimethyloxaloylglycine at 20 mg/kg per day intraperitoneally for 28 days after surgery.
Triphenyltetrazolium Chloride Staining
Three mice with MI were randomly selected from each group, euthanized, and the heart quickly removed. Heart tissue was quickly frozen in a –20 °C refrigerator for 20 minutes. The tissue was cut into slices perpendicular to the long axis and placed in 2% triphenyltetrazolium chloride staining solution (T7788, Sigma‐Aldrich) for 25 to 30 minutes, protected from light, and the container was gently shaken at 5‐minute intervals. After that, images of the slices were captured.
Immunofluorescence Analysis
In brief, the frozen sections were placed in an oven at 60 °C for 30 minutes or 2 hours at room temperature, blocked in 5% bovine serum albumin in PBS for 30 minutes, then 0.1% Triton X‑100 was added and incubated for 10 to 20 minutes. All these steps were performed at room temperature. The samples were incubated overnight at 4 °C with CD31 antibody (1:100; AF3628, R&D Systems) to stain microvascular endothelium. Then, the sections were incubated with fluorescent secondary antibodies (1:500; 2045332, Invitrogen) for 2 hours at room temperature. After that, 4',6‐diamidino‐2‐phenylindole diluted to 1:1000 with PBS was incubated for 8 minutes at room temperature. Images were captured on a confocal laser scanning microscope (Leica TCS SP8, Wetzlar, Germany). To assess micro‐vessel density, the numbers of vessels were counted in 5 random locations on every fourth section (from each sample) and recorded as CD31+ vessels/mm2.
Luciferase Assay
Coronary endothelial cells were transiently co‐transfected using lipofectamine TM2000 (116680119, Invitrogen, USA) with 1 μg TRPC plasmids plus 0.5 μL HIF‐1α siRNA (siHIF‐1α) (10 nmol/L) or negative control. There was change to cell growth medium 4 hours after transfection. After 36 hours of transfection, samples were collected for dual fluorescence detection. Then firefly and Renilla luciferase experiments were measured consecutively with Dual‐Luciferase Reporter Assay System (E5331, Promega, USA), according to the manufacturer’s instructions and normalized for transfection efficiency by the control vector pGL3‐basic (E1751, Promega, USA) that contained Renilla luciferase. All the experiments were repeated 3 times.
Statistical Analysis
Comparisons among groups were made using ANOVA followed by Dunnett or Tukey multiple comparison tests, with P<0.05 as the threshold for a significant difference.
Results
TRPC1 Plays a Role in Coronary Artery Angiogenesis
To study the contribution of TRPC channels in the angiogenesis of MCAECs, we first evaluated their role by EC tube formation and migration assays. The area of tube formation was decreased after knockdown of TRPC1, TRPC3, TRPC4, TRPC5, and TRPC6 (Figures 1A and 1B, S3). Among these, tube formation of MCAECs was reduced by 25% (P<0.01) and 28% (P<0.01) upon siRNA‐mediated knockdown of TRPC1 and TRPC5, which resulted in greater impairment of the tube formation than other groups. However, there was no significant difference of tube formation between knockdown of TRPC1 and TRPC5. To clarify which one predominated in post‐MI angiogenesis, we next used a mouse model of MI to investigate the involvement of TRPC1 and TRPC5. As shown in Figure 1C, TRPC1 protein expression significantly increased in hearts from MI‐model mice compared with sham‐model mice (P<0.01). The protein expression of TRPC5 was low in heart tissues and there was no difference between MI‐ and sham‐model mice (Figure 1D). Thus, TRPC1 may be involved in post‐MI angiogenesis rather than TRPC5. Because few previous studies have been concerned with the role of endothelial TRPC1 in post‐MI angiogenesis, we decided to confirm the involvement of TRPC1 using primary MCAECs from TRPC1fl/fl mice and endothelial cell‐specific TRPC1 channel knockout (TRPC1EC ‐/‐) mice by tube formation and migration assays (Figure 1E and 1F). Both EC tube formation and migration were significantly suppressed in MCAECs from TRPC1EC ‐/‐ mice by (P<0.01), which also indicated a role for TRPC1 in angiogenesis.
Figure 1. Transient receptor potential canonical 1 (TRPC1) promotes tube formation and migration in mouse coronary artery endothelial cells.
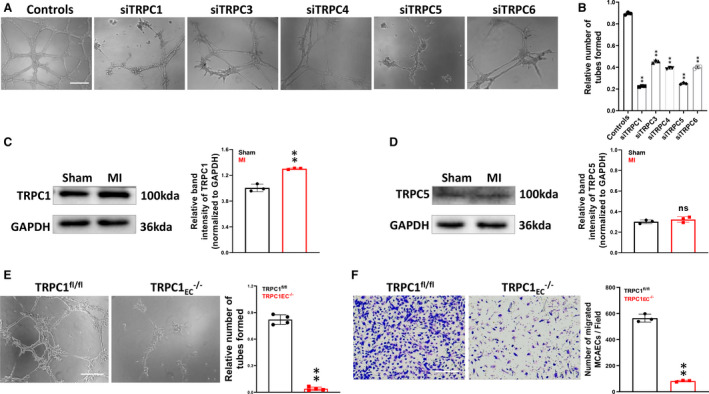
A and B, Representative images of tube formation (measured after 12 hours) and scratch wound assays in primary mouse coronary artery endothelial cells pretreated with control or TRPC siRNA (siTRPC) (n=3; ** P<0.01 vs ctl, 1‐way ANOVA and Dunnett multiple comparison test). C and D, Protein expression levels of TRPC1 and TRPC5 in sham and post‐infarct heart tissues (n=3; ** P<0.01 vs sham group, unpaired t test). E, Representative images and quantification of tube formation in primary mouse coronary artery endothelial cells from floxed TRPC1 (TRPC1fl/fl) and endothelial cell‐specific TRPC1 channel knockout (TRPC1EC ‐/‐) mice (n=4; ** P<0.01 vs TRPC1fl/fl, unpaired t test). F, Representative images and quantification of migrated TRPC1fl/fl and TRPC1EC ‐/‐ mouse coronary artery endothelial cells (n=3; ** P<0.01 vs TRPC1fl/fl, unpaired t test; scale bar, 50 μm). The data are presented as the mean ± SD. MI indicates myocardial infarction; TRPC1, transient receptor potential canonical 1; TRPC1EC ‐/‐, endothelial cell‐specific TRPC1 channel knockout; and TRPC1fl/fl; coronary artery endothelial cells from floxed TRPC1.
TRPC1 Is Involved in Recovery From MI
To validate these in vitro observations, we investigated the angiogenic role of TRPC1 in TRPC1fl/fl and TRPC1EC ‐/‐ MI‐model mice. Echocardiography was used to quantify the effect of TRPC1 on cardiac function. Based on the analysis of echocardiography (Figure 2A through 2E), on day 28 after left anterior descending artery ligation, TRPC1EC ‐/‐ mice had more significantly reduced ejection fraction (P<0.01) and fractional shortening (P<0.05), more significantly increased systolic left ventricular internal dimensions (P<0.01) and diastolic left ventricular internal dimensions (P<0.01) than TRPC1fl/fl mice. Triphenyltetrazolium chloride staining revealed a larger myocardial infarct in TRPC1EC ‐/‐ mice than in TRPC1fl/fl mice (P<0.05) (Figure 2F and 2G). Using an anti‐CD31 antibody to identify ECs, we found a significant decrease of capillary density in the infarct area of hearts from TRPC1EC ‐/‐ mice compared with TRPC1fl/fl mice (P<0.01) (Figure 2H and 2I). These data revealed that TRPC1 contributes to the functional recovery of the post‐ischemic heart and is a potential target for treating ischemic heart disease.
Figure 2. Effects of transient receptor potential canonical 1 (TRPC1) on angiogenesis in myocardial infarction‐model mice.
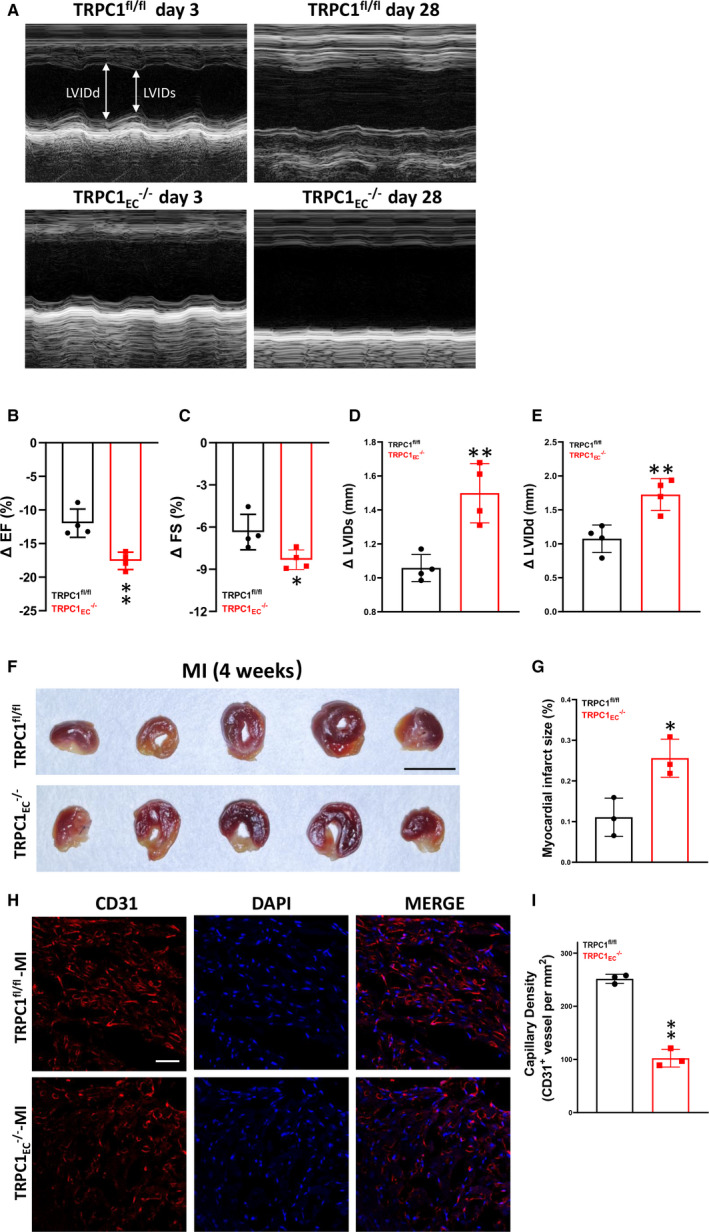
A, Representative echocardiogram of coronary artery endothelial cells from floxed TRPC1 (TRPC1fl/fl) and TRPC1EC ‐/‐ mice at 3 days and 28 days post‐myocardial infarction. B, Change in ejection fraction from 3 days to 28 days after left anterior descending artery ligation in TRPC1fl/fl and TRPC1EC ‐/‐ mice. C, Quantitative analysis of fractional shortening. D, Quantitative analysis of systolic left ventricular internal dimensions. E, Quantitative analysis of diastolic left ventricular internal dimensions. (n=4; ** P<0.01 vs TRPC1fl/fl, unpaired t test). F, Infarct size in TRPC1fl/fl and TRPC1EC ‐/‐ groups measured by triphenyltetrazolium chloride staining (scale bar, 0.5 cm). G, Statistics of the relative infarct size (n=3; * P<0.05 vs TRPC1fl/fl, unpaired t test). H, Representative confocal laser scanning microscope images showing capillary density in hearts from TRPC1fl/fl and TRPC1EC ‐/‐ myocardial infarction‐model mice (red, cardiac CD31; blue, DAPI; scale bar, 20 μm). I, Quantitation of capillary density in remote zone of left ventricle from TRPC1fl/fl and TRPC1EC ‐/‐ myocardial infarction‐model mice (n=3; * P<0.05 vs TRPC1fl/fl, unpaired t test). The data are presented as mean ± SD. DAPI indicates 4’,6‐diamidino‐2‐phenylindole; EF, ejection fraction; FS, fractional shortening; LVIDd, diastolic left ventricular internal dimensions; LVIDs, systolic left ventricular internal dimensions; MI, myocardial infarction; TRPC1EC ‐/‐, endothelial cell‐specific TRPC1 channel knockout; TRPC1fl/fl, coronary artery endothelial cells from floxed TRPC1; and siTRPC, TRPC siRNA.
Interaction of the HIF‐1α and MEK/ERK Pathway in TRPC1‐Regulated Angiogenesis
To elucidate the mechanism of TRPC1‐mediated post‐infarct angiogenesis, we used reverse transcriptase‐quantitative polymerase chain reaction to assess the expression of TRPC1 and several TRPC1‐interacting factors in hearts from sham and MI mice. As shown in Figure 3A, the hearts from the MI group showed higher HIF‐1α (hypoxic inducible factor‐1α) mRNA level than hearts from the sham group (P <0.01). However, no remarkable changes were found in the expression of HNF‐4α, COX‐2, NF‐kBp65, FOXO1, Smad3, and Mef2c. Intriguingly, HIF‐1α has been documented to improve cardiac function and ameliorate MI 23 , 24 ; it elicits therapeutic angiogenesis, restricts infarct size, and ameliorates myocardial function after acute coronary occlusion. Its reported role as a potent pro‐angiogenic factor was in accordance with our findings for the roles of TRPC1 in the MI model (Figure 2). Therefore, we speculated about the relationship between TRPC1 and HIF‐1α in modulating post‐infarct angiogenesis. Then, we investigated whether HIF‐1α could transcriptionally regulate TRPC1 in MCAECs. To validate our hypothesis, a series of TRPC1 promoter truncation mutants was generated targeting the potential binding sites of HIF‐1α on TRPC1 promoter predicted by Jaspar (Table S3 and S4). The results of the luciferase assay after co‐transfected with siHIF‐1α showed that the regulatory region may be located between −1344 and −1016 bp (Figure 3B). Knocking down HIF‐1α by siRNA in primary MCAECs (Figure 3C) resulted in decreased protein expression of TRPC1 (P<0.05). Therefore, we proposed that HIF‐1α could directly target TRPC1 in MCAECs. Next, we evaluated the effect of HIF‐1α in EC tube formation and migration (Figure 3D and 3E). We found that siRNA‐mediated knockdown of HIF‐1α in MCAECs led to the inhibition of tube formation and migration (P<0.01), which was slightly stronger than that in MCAECs after siRNA‐mediated knockdown of TRPC1. These findings suggested a relationship between TRPC1 and HIF‐1α in the regulation of angiogenesis.
Figure 3. Analysis of interplay between transient receptor potential canonical 1 (TRPC1), HIF‐1α (hypoxic inducible factor‐1α), and MEK‐ERK (mitogen‐activated protein kinase/extracellular signal‐regulated kinase) in mouse coronary artery endothelial cells (MCAECs).
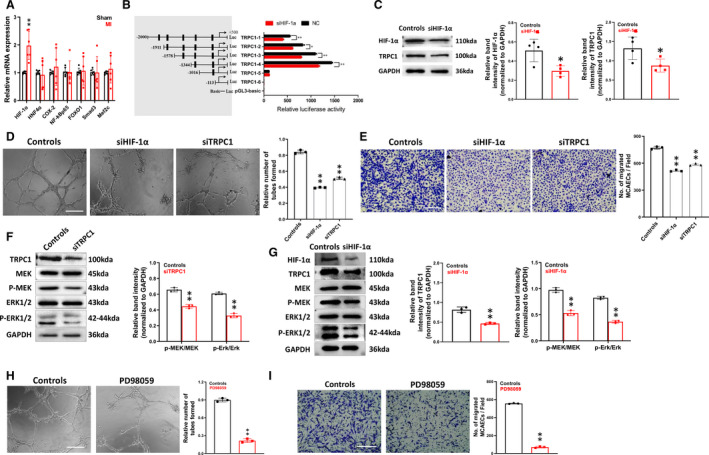
A, Relative mRNA levels of HNF‐4α, COX‐2, NF‐kBp65, FOXO1, Smad3, and Mef2c in hearts from sham and myocardial infarction‐model mice (n=6; ** P<0.01 vs sham, paired t test). B, Predicted consequential pairing of TRPC1 region and HIF‐1α by Dual‐Luciferase Assay System in MCAECs. pLUC‐TRPC1 vector was co‐transfected with HIF‐1α siRNA (siHIF‐1α) or negative control. TRPC1‐1‐6 represents individual mutations at 1‐6 sites, respectively. (n=3 independent experiments performed in triplicate. *P<0.05 and **P<0.01 by unpaired t test). C, Protein expression levels of HIF‐1α and TRPC1 in MCAECs from control and si‐HIF‐1α groups (n=4; * P<0.05 vs ctl, unpaired t test). D, Representative images and quantification of tube formation in primary MCAECs from wild type primary MCAECs treated with scrambled siRNA (control) or siHIF‐1α (n=3; ** P<0.01 vs ctrl, one‐way ANOVA and Dunnett multiple comparisons test; scale bar, 50 μm). E, Representative images and quantification of cell migration in primary MCAECs from control and siHIF‐1α groups (n=3, ** P<0.01 vs ctl, one‐way ANOVA and Dunnett’s multiple comparisons test; scale bar, 50 μm). F, Protein expression levels of pMEK and pERK in MCAECs from control and TRPC1 siRNA (siTRPC1) groups (n=3; ** P<0.01 vs ctl, unpaired t test). G, Protein expression levels of TRPC1, pMEK and pERK in MCAECs from control and siHIF‐1α groups (n=3; ** P<0.01 vs ctl, unpaired t test). H, Representative images and quantification of tube formation in primary MCAECs from wild type primary MCAECs in the absence and presence of 50 μmol/L PD98059 for 24 hours (n=3; ** P<0.01 vs ctrl, one‐way ANOVA and Dunnett multiple comparisons test; scale bar, 50 μm). I, Representative images and quantification of cell migration in primary MCAECs from wild type primary MCAECs in the absence and presence of 50 μmol/L PD98059 for 24 hours (n=3, ** P<0.01 vs control, 1‐way ANOVA and Dunnett multiple comparisons test; scale bar, 50 μm). The data are presented as mean ± SD. ERK indicates extracellular signal‐regulated kinase; HIF‐1α, hypoxic inducible factor‐1α; MAPK, mitogen‐activated protein kinase; and TRPC1, transient receptor potential canonical 1.
The mitogen‐activated protein kinase/extracellular signal‐regulated kinase (MEK‐ERK) signaling pathway has been demonstrated to regulate different biological processes, including proliferation, migration and differentiation, in numerous different cells. 25 , 26 Modulation of the MEK/ERK pathway (for example, by therapeutic strategies involving insulin‐like growth factor, statins, or ischemic postconditioning) is a universally recognized and classical mechanism for the treatment of myocardial ischemia injury. 27 , 28 Previous reports show that ERK signaling is activated by hypoxia and that HIF‐1α is involved in an ERK‐dependent mechanism. 29 , 30 Thus we determined to evaluate the possible impact of HIF‐1α and TRPC1 on MEK/ERK signaling pathway.
Knocking down TRPC1 and HIF‐1α by siRNA in primary MCAECs (Figure 3F and 3G) led to decreased phosphorylation of MEK and ERK (P<0.01), whereas the total MEK and ERK protein levels remained unaltered. We also found that treatment of MCAECs with the MEK inhibitor, PD98059, resulted in the inhibition of tube formation and migration (Figure 3H and 3I), which mimicked the angiogenic defects caused by TRPC1 knockdown. Taken together, these findings indicated that HIF‐1α and TRPC1 is required for MEK/ERK phosphorylation and suggest that disruption of MEK/ERK signaling by TRPC1 knockdown may account for TRPC1 deficiency‐induced angiogenic defects.
HIF‐1α Activation Enhances TRPC1 Expression, Which Ameliorates MI‐Induced Ischemia
Dimethyloxaloylglycine, a small molecule drug, is a proteasomal degradation inhibitor that enhances the expression of HIF‐1α in cells. 31 First, to test whether dimethyloxaloylglycine mediated cardioprotection through promoting angiogenesis by MCAECs, we assayed EC tube formation and migration in MCAECs treated with dimethyloxaloylglycine compared with saline treatment. Both EC tube formation and migration were enhanced in MCAECs after dimethyloxaloylglycine treatment (P<0.05; Figure 4A and 4B). These data point out that dimethyloxaloylglycine promotes angiogenesis by MCAECs. Western blot data showed that dimethyloxaloylglycine enhanced both HIF‐1α and TRPC1 protein expression in MCAECs (P<0.01; Figure 4C). To test whether TRPC1 participated in dimethyloxaloylglycine‐induced cardiac improvement in MI, we further elucidated the regulatory effect of dimethyloxaloylglycine on cardiac function, infarct size, and capillary density after MI in TRPC1fl/fl and TRPC1EC ‐/‐ mice. Dimethyloxaloylglycine has less significantly reduced ejection fraction (P<0.01) and fractional shortening (P<0.01), less significantly increased systolic LV internal dimensions (P<0.05) and diastolic LV internal dimensions (P<0.01) (Figure 4D), less myocardial infarct size (P<0.01; Figure 4E) and more prominent increased the capillary density (P<0.01; Figure 4F) in TRPC1fl/fl group than in TRPC1EC ‐/‐ group. In TRPC1EC ‐/‐ mice, treatment of dimethyloxaloylglycine slightly recovered the changes of ejection fraction, fractional shortening, systolic LV internal dimensions, and diastolic LV internal dimensions of infarcted heart compared with TRPC1EC ‐/‐ saline group (Figure 4D). Besides, dimethyloxaloylglycine increased both HIF‐1α and TRPC1 level of MCAECs in MI‐model TRPC1fl/fl mice. In MCAECs from TRPC1EC ‐/‐ mice, dimethyloxaloylglycine could also increase the expression of HIF‐1α, but the protein level of HIF‐1α was lower than that from TRPC1fl/fl mice (Figure 4G). These data demonstrate that dimethyloxaloylglycine treatment has a protective effect on MI via upregulating HIF‐1α and TRPC1 expressions. Immunoblots results showed that endothelial cell‐specific knockout of TRPC1 significantly reduced the phosphorylated MEK and ERK activity in MCAECs, whereas treatment of dimethyloxaloylglycine caused an apparent increase (Figure 4H). We further found that dimethyloxaloylglycine‐mediated activation of MEK‐ERK pathways was also inhibited by knockout of TRPC1 in hearts of MI‐model TRPC1fl/fl mice (Figure 4I), demonstrating the cardioprotective effects of dimethyloxaloylglycine via the TRPC1‐MEK/ERK pathway. In conclusion, we have demonstrated that TRPC1 plays a role in mediating cardiac function after MI by stimulating angiogenesis that may involve the HIF‐1α‐MEK/ERK pathway and TRPC1 might be a new therapeutic target for ischemic heart diseases.
Figure 4. Upregulation of transient receptor potential canonical 1 (TRPC1) by dimethyloxalylglycine (an HIF‐1α [hypoxic inducible factor‐1α] activator) promotes recovery from ischemia.
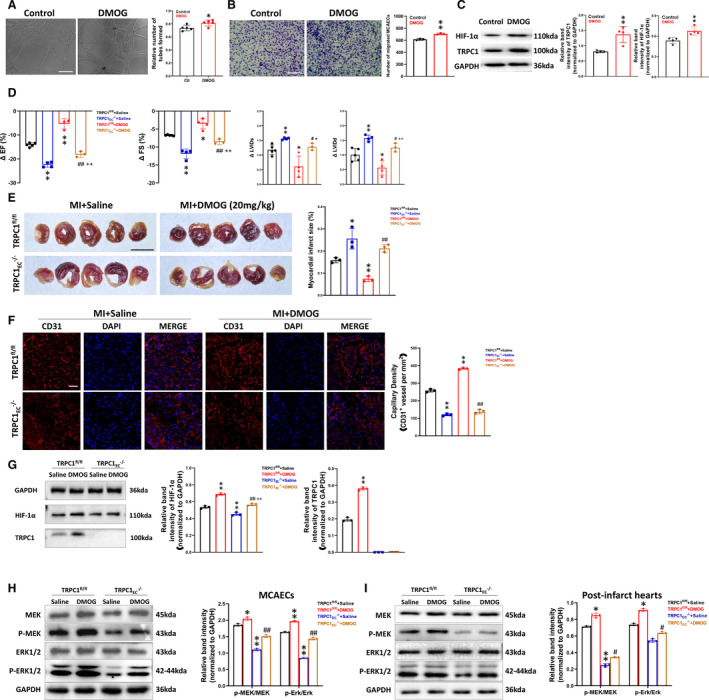
A, Representative images and quantification of tube formation in primary wild‐type mouse coronary artery endothelial cells (MCAECs) treated with saline or dimethyloxaloylglycine (200 μmol/L, 24 hours) (n=5; * P<0.05 vs control, unpaired t test; scale bar, 50 μm). B, Representative images and quantification of migrated wild‐type MCAECs treated with saline or dimethyloxaloylglycine (n=3; *P<0.01 vs control, unpaired t test; scale bar, 50 μm). C, Protein expression levels of HIF‐1α and TRPC1 in wild‐type MCAECs treated with saline or dimethyloxaloylglycine (n=4; ** P<0.01 vs control, unpaired t test). D, Change in ejection fraction, fractional shortening, systolic LV internal dimensions, and diastolic left ventricular internal dimensions from baseline to 28 days after left anterior descending artery ligation in coronary artery endothelial cells from floxed TRPC1 (TRPC1fl/fl) and TRPC1EC ‐/‐ mice treated with saline or dimethyloxaloylglycine (30 mg/kg per day intraperitoneally) (n=3–5; ** P<0.01 vs TRPC1fl/fl + saline, ## P<0.01 vs TRPC1EC ‐/‐ + saline, ++ P<0.01 vs TRPC1fl/fl + dimethyloxaloylglycine, 1‐way ANOVA and Tukey multiple comparisons test). E, Infarct size in TRPC1fl/fl and TRPC1EC ‐/‐ myocardial infarction‐model mice treated with saline or dimethyloxaloylglycine measured by triphenyltetrazolium chloride staining (n=3; * P<0.05 and ** P<0.01 vs TRPC1fl/fl + saline, ## P<0.01 vs TRPC1fl/fl + dimethyloxaloylglycine, 1‐way ANOVA and Tukey multiple comparisons test; scale bar, 0.5 cm). F, Representative confocal laser scanning microscope images and statistics for capillary density in TRPC1fl/fl and TRPC1EC ‐/‐ myocardial infarction‐model mice treated with saline or dimethyloxaloylglycine (red, cardiac CD31; blue, DAPI, scale bar, 20 μm; n=3; ** P<0.01 vs TRPC1fl/fl + saline, ## P<0.01 vs TRPC1fl/fl + dimethyloxaloylglycine, one‐way ANOVA and Tukey multiple comparisons test). G, Protein expression levels of HIF‐1α and TRPC1 in MCAECs of post‐infarct hearts from TRPC1fl/fl and TRPC1EC ‐/‐ myocardial infarction‐model mice treated with saline or dimethyloxaloylglycine (n=3; ** P<0.01 vs TRPC1fl/fl + saline, ## P<0.01 vs TRPC1EC ‐/‐ + saline, ++ P<0.01 vs TRPC1fl/fl + dimethyloxaloylglycine, 1‐way ANOVA and Tukey multiple comparisons test). H, Representative blots and quantitative analysis of Western blot of P‐MEK and P‐ERK in primary TRPC1fl/fl and TRPC1EC ‐/‐ MCAECs treated with saline or dimethyloxaloylglycine (n=3; * P<0.05 and ** P<0.01 vs TRPC1fl/fl + saline, ## P<0.01 vs TRPC1EC ‐/‐ + saline, 1‐way ANOVA and Tukey multiple comparisons test). I, The protein levels of P‐MEK and P‐ERK in post‐infarct hearts from TRPC1fl/fl and TRPC1EC ‐/‐ myocardial infarction‐model mice treated with saline or dimethyloxaloylglycine (n=3; * P<0.05 and ** P<0.01 vs TRPC1fl/fl + saline, ## P<0.01 vs TRPC1EC ‐/‐ + saline, 1‐way ANOVA and Tukey multiple comparisons test). The data are presented as mean ± SD. DAPI, 4',6‐diamidino‐2‐phenylindole; EF, ejection fraction; FS, fractional shortening; HIF‐1α, hypoxic inducible factor‐1α; LVIDd, diastolic left ventricular internal dimensions; LVIDs, systolic left ventricular internal dimensions; MI, myocardial infarction; TRPC1EC ‐/‐, endothelial cell‐specific TRPC1 channel knockout; and TRPC1fl/fl, coronary artery endothelial cells from floxed TRPC1, P‐MEK, phosphorylated mitogen‐activated protein kinase; and P‐ERK, phosphorylatedextracellular signal‐regulated kinase.
Discussion
Intracellular Ca2+ signaling is an integral second messenger in the regulation of cell proliferation, migration, angiogenesis, differentiation, and inflammation. 32 It participates in a multi‐step process that controls angiogenesis. 33 Store‐operated Ca2+ entry is the main pathway for Ca2+ entry in miscellaneous cell types such as ECs and endothelial progenitor cells. 34 , 35 The TRPC1 channel mediating store‐operated Ca2+ entry is a Ca2+‐permeable, non‐selective cation membrane channel. Knockout of TRPC1 impairs angiogenesis by endothelial progenitor cells in vitro by suppressing the calmodulin/endothelial nitric oxide synthase signaling pathway. 36 Furthermore, the ectopic expression of TRPC1 increases the angiogenic sprouting of intersegmental vessels in zebrafish. 15 Nevertheless, it remains unknown whether TRPC1 plays a pro‐angiogenesis role in ischemic heart disease.
Based on these studies, we proposed that TRPC1 may be involved in post‐ischemic repair during MI. In this study, we demonstrated that in the TRP channel family, TRPC1 was a key regulator of coronary EC function and angiogenesis (Figure 1). Moreover, TRPC1 facilitated MI‐induced adaptive pathophysiological angiogenesis and the restoration of blood flow, providing cardioprotection in mouse model of MI in vivo (Figure 2). Collectively, these results indicate that TRPC1 plays a critical role in the process of angiogenesis and the protection of ischemic myocardium after MI. Unfortunately, there are no specific and potent agonists available to activate TRPC1, so we evaluated its upstream regulators.
Angiogenesis could be triggered by ischemia and has the potential to repair ischemic myocardium at the post‐MI stage. Also, it is of great importance for avoiding heart failure during long‐term left ventricular remodeling. 37 HIF‐1α, an essential transcription factor, regulates angiogenesis through transcription of a range of hypoxia response genes. 38 Interestingly, our study showed that the expression of TRPC1 was upregulated in MI mice and the upregulation was directly induced by HIF‐1α (Figure 3). There is evidence that MEK/ERK pathway plays a critical role in VEGF‐induced angiogenesis by promoting endothelial proliferation, migration and survival. 28 A recent study demonstrated that ERK might be engaged by store‐operated Ca2+ entry in endothelial progenitor cells to stimulate migration in vitro and neovessel formation in vivo. 39 Intriguingly, store‐operated Ca2+ entry in these cells is also contributed by TRPC1. 14 , 35 Consistent with a recent study, 40 we also found that knocking down of TRPC1 and HIF‐1α significantly inhibited phosphorylation of MEK and downstream ERK (Figure 3). Together, these results demonstrated the existence of a HIF‐1α–TRPC1–MEK/ERK signaling axis in MCAECs angiogenesis.
Dimethyloxaloylglycine inhibits the proteasomal degradation of HIF‐1α and upregulates the stability of HIF‐1α expression. 41 Several studies have suggested that ischemic preconditioning with dimethyloxaloylglycine can protect heart tissues from ischemic injury. 42 , 43 , 44 However, this is the first study that demonstrated that the benefit of dimethyloxaloylglycine treatment or the benefit of upregulation of HIF‐1α in the heart requires TRPC1 expression in coronary artery endothelial cells. In our study, upregulation of TRPC1 is a strategy for improving cardiac repair by promoting angiogenesis after MI. Results from this study demonstrated that dimethyloxaloylglycine treatment induces TRPC1 expression in MCAECs (Figure 4C).
Consistent with previous studies, 45 , 46 we found that dimethyloxaloylglycine treatment improved cardiac function, promoted angiogenesis, reduced infarct size, and continued to improve cardiac function 4 weeks after MI. Several mechanisms that may be responsible for dimethyloxaloylglycine induced ischemic preconditioning have been reported. Dimethyloxaloylglycine treatment upregulated the expression of HIF‐1α and VEGF in hearts 47 and attenuated post‐ischemic cardiac injury through induction of endoplasmic reticulum stress genes. 48 Dimethyloxaloylglycine can also inhibit PHD3 (prolyl hydroxylase domain protein 3), prevent activation of the ATR/CHK1/p53 pathway and decrease apoptosis induced by DNA damage. 49 Dimethyloxaloylglycine pretreatment significantly enhances angiogenesis by increasing circulating endothelial progenitor cells and bone marrow progenitor cells. 50 Also, dimethyloxaloylglycine treatment has anti‐inflammatory, anti‐apoptotic, and neuroprotective effects in ischemic diseases. 51 , 52 Importantly, in our study, the actions of dimethyloxaloylglycine were dependent on endothelial TRPC1 expression. Endothelial cell‐specific knockdown of TRPC1 inhibited the angiogenesis effect of dimethyloxaloylglycine and verified that dimethyloxaloylglycine worked through the HIF‐1α‐TRPC1‐MEK/ERK pathway in MCAECs (Figure 4). Notably, there was still a slight recovery in TRPC1EC ‐/‐ mice with dimethyloxaloylglycine treatment compared with those without treatment. This suggested that HIF‐1α can also act through other pathways to induce cardiac improvement, consistent with previous research. 45 In summary, dimethyloxaloylglycine could improve cardiac function through the HIF‐1α‐TRPC1‐MEK/ERK pathway in MCAECs. Nevertheless, there were several limitations: TRPC1 is not the only pathway that regulates angiogenesis after MI and we could not rule out the possibility that HIF‐1α activates other HIF‐1 target genes, such as erythropoietin or VEGF, which may contribute to the observed benefits. 43
In conclusion, to our knowledge, our study is the first to confirm that upregulation of endothelial TRPC1 is a strategy for improving cardiac function after MI by stimulating angiogenesis via the upstream regulator HIF‐1α and downstream MEK/ERK. Dimethyloxaloylglycine treatment has a protective effect on myocardial ischemia through the HIF‐1α‐TRPC1‐MEK/ERK pathway. Our study demonstrates TRPC1 might be a new therapeutic target for ischemic heart diseases.
Sources of Funding
This work was fully supported by the National Natural Science Foundation of China (81622007, 81960662, 81800430), the Chang Jiang Scholars Program (Q2015106), Fundamental Research Funds for the Central Universities (JUSRP51704A), the National First‐Class Discipline Program of Food Science and Technology (JUFSTR20180101), Postdoctoral Research funding program of Jiangsu Province (2020Z427), and Wuxi Health Commission.
Disclosures
None.
Supporting information
Tables S1–S4
Figures S1–S3
Acknowledgments
We thank Prof. Iain C. Bruce for critical reading of the article.
Supplemental Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.023678
For Sources of Funding and Disclosures, see page 11.
References
- 1. Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, et al. American Heart Association Council on E, Prevention Statistics C, Stroke Statistics S . Heart disease and stroke statistics‐2020 update: a report from the American Heart Association. Circulation. 2020;141:e139–e596. [DOI] [PubMed] [Google Scholar]
- 2. Seiler C, Stoller M, Pitt B, Meier P. The human coronary collateral circulation: development and clinical importance. Eur Heart J. 2013;34:2674–2682. doi: 10.1093/eurheartj/eht195 [DOI] [PubMed] [Google Scholar]
- 3. Wu W, Li X, Zuo G, Pu J, Wu X, Chen S. The role of angiogenesis in coronary artery disease: a double‐edged sword: intraplaque angiogenesis in physiopathology and therapeutic angiogenesis for treatment. Curr Pharm Des. 2018;24:451–464. doi: 10.2174/1381612824666171227220815 [DOI] [PubMed] [Google Scholar]
- 4. Chen CW, Wang LL, Zaman S, Gordon J, Arisi MF, Venkataraman CM, Chung JJ, Hung G, Gaffey AC, Spruce LA, et al. Sustained release of endothelial progenitor cell‐derived extracellular vesicles from shear‐thinning hydrogels improves angiogenesis and promotes function after myocardial infarction. Cardiovasc Res. 2018;114:1029–1040. doi: 10.1093/cvr/cvy067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu H, Jiang X, Li X, Hu M, Wan W, Wen Y, He Y, Zheng X. Intramyocardial delivery of VEGF165 via a novel biodegradable hydrogel induces angiogenesis and improves cardiac function after rat myocardial infarction. Heart Vessels. 2016;31:963–975. doi: 10.1007/s00380-015-0710-0 [DOI] [PubMed] [Google Scholar]
- 6. Cao R, Brakenhielm E, Pawliuk R, Wariaro D, Post MJ, Wahlberg E, Leboulch P, Cao Y. Angiogenic synergism, vascular stability and improvement of hind‐limb ischemia by a combination of PDGF‐BB and FGF‐2. Nat Med. 2003;9:604–613. doi: 10.1038/nm848 [DOI] [PubMed] [Google Scholar]
- 7. Luttun A, Tjwa M, Moons L, Wu Y, Angelillo‐Scherrer A, Liao F, Nagy JA, Hooper A, Priller J, De Klerck B, et al. Revascularization of ischemic tissues by PLGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti‐flt1. Nat Med. 2002;8:831–840. doi: 10.1038/nm731 [DOI] [PubMed] [Google Scholar]
- 8. Kano MR, Morishita Y, Iwata C, Iwasaka S, Watabe T, Ouchi Y, Miyazono K, Miyazawa K. VEGF‐A and FGF‐2 synergistically promote neoangiogenesis through enhancement of endogenous PDGF‐B‐PDGFRBETA signaling. J Cell Sci. 2005;118:3759–3768. [DOI] [PubMed] [Google Scholar]
- 9. Johnson T, Zhao L, Manuel G, Taylor H, Liu D. Approaches to therapeutic angiogenesis for ischemic heart disease. J Mol Med (Berl). 2019;97:141–151. doi: 10.1007/s00109-018-1729-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fantozzi I, Zhang S, Platoshyn O, Remillard CV, Cowling RT, Yuan JX. Hypoxia increases AP‐1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1233–L1245. [DOI] [PubMed] [Google Scholar]
- 11. Cheng HW, James AF, Foster RR, Hancox JC, Bates DO. VEGF activates receptor‐operated cation channels in human microvascular endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1768–1776. doi: 10.1161/01.ATV.0000231518.86795.0f [DOI] [PubMed] [Google Scholar]
- 12. Antigny F, Girardin N, Frieden M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J Biol Chem. 2012;287:5917–5927. doi: 10.1074/jbc.M111.295733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smani T, Gomez LJ, Regodon S, Woodard GE, Siegfried G, Khatib AM, Rosado JA. TRP channels in angiogenesis and other endothelial functions. Front Physiol. 2018;9:1731. doi: 10.3389/fphys.2018.01731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moccia F, Negri S, Shekha M, Faris P, Guerra G. Endothelial Ca(2+) signaling, angiogenesis and vasculogenesis: just what it takes to make a blood vessel. Int J Mol Sci. 2019;20:3962. doi: 10.3390/ijms20163962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu PC, Gu SY, Bu JW, Du JL. TRPC1 is essential for in vivo angiogenesis in zebrafish. Circ Res. 2010;106:1221–1232. doi: 10.1161/CIRCRESAHA.109.207670 [DOI] [PubMed] [Google Scholar]
- 16. Qin W, Xie W, Xia N, He Q, Sun T. Silencing of transient receptor potential channel 4 alleviates oxLDL‐induced angiogenesis in human coronary artery endothelial cells by inhibition of VEGF and NF‐KAPPAB. Med Sci Monit. 2016;22:930–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moccia F, Lucariello A, Guerra G. TRPC3‐mediated Ca(2+) signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: the role of autologous endothelial colony forming cells. J Cell Physiol. 2018;233:3901–3917. [DOI] [PubMed] [Google Scholar]
- 18. Negri S, Faris P, Berra‐Romani R, Guerra G, Moccia F. Endothelial transient receptor potential channels and vascular remodeling: extracellular Ca(2 +) entry for angiogenesis, arteriogenesis and vasculogenesis. Front Physiol. 2019;10:1618. doi: 10.3389/fphys.2019.01618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu Y, Gao M, Zhou T, Xie M, Mao A, Feng L, Yao X, Wong WT, Ma X. The TRPC5 channel regulates angiogenesis and promotes recovery from ischemic injury in mice. J Biol Chem. 2019;294:28–37. doi: 10.1074/jbc.RA118.005392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu Y, Xu M, Zhang H, Li X, Su Z, Zhang C. SEC2‐induced superantigen and antitumor activity is regulated through calcineurin. Appl Microbiol Biotechnol. 2013;97:9695–9703. doi: 10.1007/s00253-013-4764-6 [DOI] [PubMed] [Google Scholar]
- 21. Jiang H, Liu J, Fan C, Wang J, Li W. LncRNAS56464.1 as a ceRNA promotes the proliferation of fibroblastlike synoviocytes in experimental arthritis via the Wnt signaling pathway and sponges miR1523p. Int J Mol Med. 2021;47. doi: 10.3892/ijmm.2021.4850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Oliveira FCL, Roy JS, Pappas E. ACL injury, physical activity, and overweight/obesity: a vicious cycle? Knee Surg Sports Traumatol Arthrosc. 2020;28:667–669. doi: 10.1007/s00167-019-05807-6 [DOI] [PubMed] [Google Scholar]
- 23. Jianqiang P, Ping Z, Xinmin F, Zhenhua Y, Ming Z, Ying G. Expression of hypoxia‐inducible factor 1 alpha ameliorate myocardial ischemia in rat. Biochem Biophys Res Commun. 2015;465:691–695. doi: 10.1016/j.bbrc.2015.08.046 [DOI] [PubMed] [Google Scholar]
- 24. Du Y, Ge Y, Xu Z, Aa N, Gu X, Meng H, Lin Z, Zhu D, Shi J, Zhuang R, et al. Hypoxia‐inducible factor 1 alpha (HIF‐1alpha)/vascular endothelial growth factor (VEGF) pathway participates in angiogenesis of myocardial infarction in muscone‐treated mice: preliminary study. Med Sci Monit. 2018;24:8870–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zarrabi M, Afzal E, Asghari MH, Mohammad M, Es HA, Ebrahimi M. Inhibition of MEK/ERK signalling pathway promotes erythroid differentiation and reduces HSCS engraftment in ex vivo expanded haematopoietic stem cells. J Cell Mol Med. 2018;22:1464–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao X, Yi Y, Meng C, Fang N. MiRNA‐575 suppresses angiogenesis by targeting Rab5‐MEK‐ERK pathway in endothelial cells. Biosci Rep. 2019;39. doi: 10.1042/BSR20181218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Q, Wang H, Yang YJ, Dong QT, Wang TJ, Qian HY, Li N, Wang XM, Jin C. Atorvastatin treatment improves the effects of mesenchymal stem cell transplantation on acute myocardial infarction: the role of the Rhoa/ROCK/ERK pathway. Int J Cardiol. 2014;176:670–679. doi: 10.1016/j.ijcard.2014.07.071 [DOI] [PubMed] [Google Scholar]
- 28. Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a risk for cardioprotection. Heart Fail Rev. 2007;12:217–234. doi: 10.1007/s10741-007-9026-1 [DOI] [PubMed] [Google Scholar]
- 29. Liu L, Zhang H, Sun LI, Gao Y, Jin H, Liang S, Wang Y, Dong M, Shi Y, Li Z, et al. ERK/MAPK activation involves hypoxia‐induced MGr1‐Ag/37LRP expression and contributes to apoptosis resistance in gastric cancer. Int J Cancer. 2010;127:820–829. doi: 10.1002/ijc.25098 [DOI] [PubMed] [Google Scholar]
- 30. Minet E, Arnould T, Michel G, Roland I, Mottet D, Raes M, Remacle J, Michiels C. ERK activation upon hypoxia: involvement in HIF‐1 activation. FEBS Lett. 2000;468:53–58. doi: 10.1016/S0014-5793(00)01181-9 [DOI] [PubMed] [Google Scholar]
- 31. Ding H, Chen S, Song WQ, Gao YS, Guan JJ, Wang Y, Sun Y, Zhang CQ. Dimethyloxaloylglycine improves angiogenic activity of bone marrow stromal cells in the tissue‐engineered bone. Int J Biol Sci. 2014;10:746–756. doi: 10.7150/ijbs.8535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dalal PJ, Muller WA, Sullivan DP. Endothelial cell calcium signaling during barrier function and inflammation. Am J Pathol. 2020;190:535–542. doi: 10.1016/j.ajpath.2019.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Munaron L, Fiorio PA. Endothelial calcium machinery and angiogenesis: understanding physiology to interfere with pathology. Curr Med Chem. 2009;16:4691–4703. [DOI] [PubMed] [Google Scholar]
- 34. Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415 [DOI] [PubMed] [Google Scholar]
- 35. Sanchez‐Hernandez Y, Laforenza U, Bonetti E, Fontana J, Dragoni S, Russo M, Avelino‐Cruz JE, Schinelli S, Testa D, Guerra G, et al. Store‐operated Ca(2+) entry is expressed in human endothelial progenitor cells. Stem Cells Dev. 2010;19:1967–1981. [DOI] [PubMed] [Google Scholar]
- 36. Du LL, Shen Z, Li Z, Ye X, Wu M, Hong L, Zhao Y. TRPC1 deficiency impairs the endothelial progenitor cell function via inhibition of Calmodulin/eNOS pathway. J Cardiovasc Transl Res. 2018;11:339–345. doi: 10.1007/s12265-018-9798-9 [DOI] [PubMed] [Google Scholar]
- 37. van der Laan AM, Piek JJ, van Royen N. Targeting angiogenesis to restore the microcirculation after reperfused MI. Nat Rev Cardiol. 2009;6:515–523. doi: 10.1038/nrcardio.2009.103 [DOI] [PubMed] [Google Scholar]
- 38. Song W, Liang Q, Cai M, Tian Z. HIF‐1alpha‐induced up‐regulation of microRNA‐126 contributes to the effectiveness of exercise training on myocardial angiogenesis in myocardial infarction rats. J Cell Mol Med. 2020;24:12970–12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zuccolo E, Di Buduo C, Lodola F, Orecchioni S, Scarpellino G, Kheder DA, Poletto V, Guerra G, Bertolini F, Balduini A, et al. Stromal cell‐derived factor‐1alpha promotes endothelial colony‐forming cell migration through the Ca(2+)‐dependent activation of the extracellular signal‐regulated kinase 1/2 and phosphoinositide 3‐kinase/Akt pathways. Stem Cells Dev. 2018;27:23–34. [DOI] [PubMed] [Google Scholar]
- 40. Guo CX, Li ZY, Niu JB, Fan SC, Yan SY, Lu PP, Su YN, Ma LH. Qishen capsule safely boosts cardiac function and angiogenesis via the MEK/ERK pathway in a rat myocardial infarction model. J Geriatr Cardiol. 2019;16:764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sinha I, Sakthivel D, Olenchock BA, Kruse CR, Williams J, Varon DE, Smith JD, Madenci AL, Nuutila K, Wagers AJ. Prolyl hydroxylase domain‐2 inhibition improves skeletal muscle regeneration in a male murine model of obesity. Front Endocrinol (Lausanne). 2017;8:153. doi: 10.3389/fendo.2017.00153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li X, Zhao H, Wu Y, Zhang S, Zhao X, Zhang Y, Wang J, Wang J, Liu H. Up‐regulation of hypoxia‐inducible factor‐1alpha enhanced the cardioprotective effects of ischemic postconditioning in hyperlipidemic rats. Acta Biochim Biophys Sin (Shanghai). 2014;46:112–118. [DOI] [PubMed] [Google Scholar]
- 43. Poynter JA, Manukyan MC, Wang Y, Brewster BD, Herrmann JL, Weil BR, Abarbanell AM, Meldrum DR. Systemic pretreatment with dimethyloxalylglycine increases myocardial HIF‐1alpha and VEGF production and improves functional recovery after acute ischemia/reperfusion. Surgery. 2011;150:278–283. [DOI] [PubMed] [Google Scholar]
- 44. Shafighi M, Olariu R, Fathi AR, Djafarzadeh S, Jakob SM, Banic A, Constantinescu MA. Dimethyloxalylglycine stabilizes HIF‐1alpha in cultured human endothelial cells and increases random‐pattern skin flap survival in vivo. Plast Reconstr Surg. 2011;128:415–422. [DOI] [PubMed] [Google Scholar]
- 45. Mayorga M, Kiedrowski M, Shamhart P, Forudi F, Weber K, Chilian WM, Penn MS, Dong F. Early upregulation of myocardial CXCR4 expression is critical for dimethyloxalylglycine‐induced cardiac improvement in acute myocardial infarction. Am J Physiol Heart Circ Physiol. 2016;310:H20–28. doi: 10.1152/ajpheart.00449.2015 [DOI] [PubMed] [Google Scholar]
- 46. Eckle T, Kohler D, Lehmann R, El Kasmi K, Eltzschig HK. Hypoxia‐inducible factor‐1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008;118:166–175. doi: 10.1161/CIRCULATIONAHA.107.758516 [DOI] [PubMed] [Google Scholar]
- 47. Wang J, Hong Z, Zeng C, Yu Q, Wang H. NADPH oxidase 4 promotes cardiac microvascular angiogenesis after hypoxia/reoxygenation in vitro. Free Radic Biol Med. 2014;69:278–288. doi: 10.1016/j.freeradbiomed.2014.01.027 [DOI] [PubMed] [Google Scholar]
- 48. Natarajan R, Salloum FN, Fisher BJ, Smithson L, Almenara J, Fowler AA. Prolyl hydroxylase inhibition attenuates post‐ischemic cardiac injury via induction of endoplasmic reticulum stress genes. Vascul Pharmacol. 2009;51:110–118. doi: 10.1016/j.vph.2009.05.007 [DOI] [PubMed] [Google Scholar]
- 49. Xie L, Pi X, Mishra A, Fong G, Peng J, Patterson C. PHD3‐dependent hydroxylation of HCLK2 promotes the DNA damage response. J Clin Invest. 2012;122:2827–2836. doi: 10.1172/JCI62374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Takaku M, Tomita S, Kurobe H, Kihira Y, Morimoto A, Higashida M, Ikeda Y, Ushiyama A, Hashimoto I, Nakanishi H, et al. Systemic preconditioning by a prolyl hydroxylase inhibitor promotes prevention of skin flap necrosis via HIF‐1‐induced bone marrow‐derived cells. PLoS One. 2012;7:e42964. doi: 10.1371/journal.pone.0042964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang J, Liu C, Du X, Liu M, Ji X, Du H, Zhao H. Hypoxia inducible factor 1alpha plays a key role in remote ischemic preconditioning against stroke by modulating inflammatory responses in rats. J Am Heart Assoc. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nagamine Y, Tojo K, Yazawa T, Takaki S, Baba Y, Goto T, Kurahashi K. Inhibition of prolyl hydroxylase attenuates Fas ligand‐induced apoptosis and lung injury in mice. Am J Respir Cell Mol Biol. 2016;55:878–888. doi: 10.1165/rcmb.2015-0266OC [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S4
Figures S1–S3