

Abstract
Background
Post‐resuscitation syndrome, involves a severe inflammatory response following successful cardiopulmonary resuscitation. The potential mechanism of Vitamin C (VitC) after cardiopulmonary resuscitation on myocardial and cerebral function, duration of survival is undefined.
Methods and Results
A first set of experiments were done in 18 male Sprague‐Dawley rats for the investigation of short‐term follow‐up, randomized into 3 groups: (1) sham; (2) controls; (3) VitC. Ventricular fibrillation was electrically induced and untreated for 6 minutes. Cardiopulmonary resuscitation including chest compression and mechanical ventilation were then initiated and continued for 8 minutes followed by defibrillation. At 5 minutes after return of spontaneous circulation, either VitC (200 mg/kg) or placebo was administered by intravenous infusion with a syringe pump for half an hour. There were significant improvements in myocardial function and buccal microcirculation in rats treated with VitC after return of spontaneous circulation 4 hours compared with controls. VitC inhibited proinflammatory cytokines (interleukin‐6 and tumor necrosis factor‐α), SDC‐1 (Syndecan‐1), and hyaluronic acid in plasma compared with controls (P<0.01). VitC decreased reactive oxygen species production and inhibited p38/MAPK (mitogen‐activated protein kinase) pathway phosphorylation. A second set with 20 animals was used for assessing the neurological deficit score after return of spontaneous circulation 72 hours, randomized into 2 groups: 1) controls; 2) VitC. The survival rate and neurological deficit score after return of spontaneous circulation 72 hours were improved in VitC‐treated animals compared with those of the control group.
Conclusions
VitC reduces the severity of post‐resuscitation myocardial and cerebral dysfunction and improves the survival. The mechanisms may involve inhibiting transcription of inflammatory cytokines and oxidative stress, thus protecting the integrity of the vascular endothelium. Meanwhile VitC reduces shedding of SDC‐1 and alters p38/MAPK phosphorylation and microcirculation.
Keywords: cardiac arrest, inflammatory cytokines, p38/MAPK pathway, Syndecan‐1, Vitamin C
Subject Categories: Cardiopulmonary Resuscitation and Emergency Cardiac Care
Nonstandard Abbreviations and Acronyms
- CA
cardiac arrest
- CO
cardiac output
- cTnI
cardiac troponin I
- ET
ejection time
- HA
hyaluronic acid
- I/R
ischemia‐reperfusion
- ROSC
return of spontaneous circulation
- SDC‐1
syndecan‐1
- VitC
vitamin C
Clinical Perspective
What Is New?
To the best of our knowledge, this is the first study to evaluate the therapeutic strategy of vitamin C on improving the outcomes of cardiopulmonary resuscitation by reducing the severity of post‐resuscitation myocardial and cerebral dysfunction.
Vitamin C improved myocardial and cerebral dysfunction after cardiopulmonary resuscitation which mechanisms may involve inhibiting transcription of inflammatory cytokines, thus protecting the integrity of the vascular endothelium via reducing shedding of SDC‐1 (Syndecan‐1) which could regulate p38/MAPK (mitogen‐activated protein kinase) phosphorylation and microcirculation.
What Are the Clinical Implications?
Our research supports and highlights the perspective on protection of the vascular endothelium with its glycocalyx is especially vulnerable to post‐resuscitation after cardiac arrest induced by the initiation of oxidative stress and inflammatory reactions.
This research is also translational that the conception of protecting the integrity of the vascular endothelium via reducing shedding of SDC‐1 provides a new, and effective option for improving the outcomes of cardiopulmonary resuscitation.
Cardiac arrest (CA) is a major public health problem causing substantial morbidity and mortality. Approximately 350 000 adults in the United States experienced out‐of‐hospital CA. 1 Only half of these patients arrived at the hospital alive. Of these survivors, 50% patients die in the hospital or remain severely disabled because of post‐CA syndrome. 2
Post‐resuscitation syndrome, involves a severe inflammatory response following successful cardiopulmonary resuscitation (CPR). This has been described as to be similar to sepsis and sepsis‐like syndromes. The syndrome can be initiated either by the initial ischemia/reperfusion(I/R) injury during CA and subsequent resuscitation or by a prolonged low reperfusion state as a result of persistent cardiogenic dysfunction after initial successful resuscitation. 3 Systemic inflammation with consecutive macro‐ and micro‐circulatory dysfunction further aggravates organ dysfunction adjusted as well by direct inflammatory cytokines transmitted by I/R tissue injury. 4 Geppert et al. postulate the presence of a “systemic inflammatory response syndrome” in 66% of their patients after CA and return of spontaneous circulation (ROSC). 5 The vascular endothelium with its glycocalyx is especially vulnerable to initial ischemic damage and may play a harmful role in the initiation of post‐ischemic inflammatory reactions. It is not known to date if early damage to the endothelial glycocalyx, detected by on‐the‐scene blood sampling and measurement of soluble components (hyaluronan and SDC‐1 [Syndecan‐1), precedes and predicts survival after ROSC. 6
Vitamin C (VitC) is a key circulating antioxidant with anti‐inflammatory and immune‐supporting effects and a cofactor for important mono‐ and dioxygenase enzymes. Endothelial dysfunction is mediated by oxidative stress that VitC is able to attenuate and counteract, making it a serious candidate for adjunctive treatments in sepsis and other I/R injury. The aim of the present study was to investigate the potential protective effects of VitC as a therapeutic agent for CA. We explore whether VitC attenuates global I/R injury in our rat model of CA and investigates the effects on reducing shedding of SDC‐1 and altering p38/MAPK (mitogen‐activated protein kinase) phosphorylation.
Methods
The data, analytic methods, and study materials that support the findings of this study are available from the corresponding author upon reasonable request to other researchers for purposes of reproducing the results or replicating the procedure. This study was performed under a protocol (AD10001396, specific aim 23) approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University. All animals were handled in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Animal Preparation
Animal preparation was performed according to our previous studies. 7 , 8 In brief male Sprague‐Dawley rats, 6 to 10 months of age weighing between 450 and 550 g, were anesthetized by an intraperitoneal injection of pentobarbital (45 mg/kg). The trachea was orally intubated. End‐tidal CO2 and ECG were monitored. Arterial and right atrial pressures were recorded using PE‐50 catheters. A thermocouple microprobe was inserted into the left femoral to measure core temperature. A guide‐wire was advanced through the right external jugular vein and into the right ventricle to induce ventricular fibrillation (VF).
Experimental Protocol
A first set of experiments were done in 18 male Sprague‐Dawley rats for the investigation of short‐term follow‐up, randomized into 3 groups (each group n=6): Group 1: sham; Group 2: controls; Group 3: VitC. Fifteen minutes before induction of VF, baseline measurements and echocardiography were obtained. Mechanical ventilation was established at a tidal volume of 0.60 mL/100 g of body weight, a frequency of 100 breaths/min, and an inspired O2 fraction of 0.21. VF was then induced through a guide wire advanced from the right jugular vein into the right ventricle. A progressive increase in 60‐Hz current to a maximum of 3.5 mA was then delivered to the right ventricular endocardium. The current flow was continued for 3 minutes to prevent spontaneous defibrillation. Mechanical ventilation was discontinued after onset of VF. After 6 minutes of untreated VF, precordial compressions, together with mechanical ventilation (tidal volume 0.60 mL/100 g body weight, frequency 100 breaths/min, inspired O2 fraction of 1.0), were initiated using a pneumatically driven mechanical chest compressor. Precordial chest compressions were maintained at a rate of 200/min and synchronized to provide a compression/ventilation ratio of 2:1 with equal compression‐relaxation for a duration of 8 minutes. Defibrillation was attempted with up to 3 4‐J counter shocks. ROSC was defined as the return of supraventricular rhythm with a mean aortic pressure >50 mm Hg for 5 minutes. If ROSC was not achieved after the first defibrillation attempt, a 30‐second interval of CPR was performed before the next defibrillation attempt (up to 3 attempts). After ROSC, an inspired O2 fraction of 1.0 was continued for 1 hour, adjusted to 0.5 for the second hour and 0.21 for the last 2 hours. To observe survival time and assess neurological deficit score (NDS) 72 hours after ROSC, 20 additional rats were randomized into 2 groups: 1) controls; 2) VitC.
At 5 minutes after ROSC, either VitC (200 mg/kg) 9 or vehicle (0.9% NaCl solution) was administered by continuous intravenous infusion with a syringe pump (Genie Touch, Kent Scientific Corporation USA) for half an hour by the same volume of 1 mL. In the present study, rats (except the sham group) were not injected with additional pentobarbital since they were in a coma within 4 hours following ROSC (Figure S1).
Measurements
Hemodynamics and Other Parameters
ECG, aortic and right atrial pressures, blood temperature, and end‐tidal CO2 values were continuously recorded on a PC‐based data‐acquisition system supported by WINDAQ software (DATAQ, Akron, OH, USA). Coronary perfusion pressure was calculated as the difference between aortic and time‐coincident right atrial diastolic pressures measured at the end of each minute of precordial compression.
At baseline, 1, 2, 3, and 4 hours after ROSC, ejection fraction (EF), cardiac output (CO), and myocardial performance index (MPI) were measured by echocardiography (HD11XE; Philips Medical Systems, Eindhoven, Netherlands, USA) with a 12.5 Hz transducer. From the long‐axis view, left ventricular end‐systolic volume, left ventricular end‐diastolic volume, and left ventricular outflow tract dimension were obtained. From pulse‐Doppler imaging, aortic velocity time integral, isovolumic contraction time, isovolumic relaxation time, ejection time were obtained. Heart rate was obtained by ECG. The parameters CO, EF, and MPI were calculated and confirmed separately by 2 investigators. CO and EF are adopted to estimate the myocardial contractility; MPI was adapted to estimate the left ventricular diastolic function. EF= (left ventricular end‐diastolic volume ‐ left ventricular end‐systolic volume)/ left ventricular end‐diastolic volume×100%. CO= velocity time integra×π (left ventricular outflow tract D/2)2×heart rate. MPI=isovolumic contraction time + isovolumic relaxation time)/ ejection time.
Buccal microcirculation was measured at baseline, 2 and 4 hours after ROSC with the aid of a sidestream dark‐field imaging device (Micro Scan; Micro Vision Medical Inc., Amsterdam, the Netherlands) that has a 5x imaging objective, resulting in an on‐screen magnification of 276x. Three discrete fields for each were captured with the intention to minimize motion artifacts. Microvascular images were recorded on a DVD recorder (DMR‐EZ47V; Panasonic AVC Networks, Dalian, China). The image was divided into 4 quadrants, and the predominant flow type was assessed in the small vessels (<20 µm) of each quadrant. The microvascular flow index score represents the average values of the 4 quadrants. Perfused vessel density was measured based on the method of De Backer et al. 10
ELISA
Cardiac troponin I (cTnI), interleukin 6 (IL‐6), tumor necrosis factor‐α (TNF‐α), SDC‐1, and hyaluronic acid (HA) serum levels were measured with ELISA kits in accordance with the manufacturer’s instructions (category NO. LS‐F23616, LSBio; category NO. R6000B, R&D SYSTEM; category NO. RTA00, R&D SYSTEM; category NO. LS‐F 5983; category NO. LS‐F13033, LSBio; respectively). Enzymatic activity was determined spectrophotometrically at an absorbance of 450 nm using a 96‐well plate reader. The detection limit for cTnI, IL‐6, TNF‐α, SDC‐1, and HA was 31.25, 62.5, 15.6, 1.563, and 6.25 pg/mL, respectively.
Reactive Oxygen Species Assays in Heart and Brain Tissue
Heart and brain tissue were harvested, flash frozen, and stored at −80°C until assessment. Heart and brain sample reactive oxygen species (ROS) levels were measured using Reactive Oxygen Species Fluorometric Assay Kit (Catalog No. E‐BC‐K138‐F, Elabscience), according to the manufacturer’s protocol. Briefly, tissues from the heart and the brain were homogenized in RIPA buffer at a weight: volume ratio of 1:10. The homogenate was centrifuged at 1600g for 10 minutes at 4°C. Dichlorofuorescin is oxidized to dichlorofluorescein which is a strong green fluorescent substance that cannot penetrate the cell membrane. The intensity of dichlorofluorescein is proportional to the level of intracellular ROS. ROS content was assessed by fluorimetric method at a wavelength of 530 nm.
Western Blotting
Heart and brain tissue were homogenized in a RIPA buffer consisting of protease inhibitor cocktail (Roche) and phosphatase inhibitor PhosSTOP. The homogenate was centrifuged at 16 000g for 15 minutes at 4°C, and the supernatant was extracted for further protein quantitation. Thirty micrograms of protein for each sample were separated on 4% to 20% SDS/PAGE gel electrophoresis and transferred to a polyvinylidene fluoride membrane. The selected membrane was blocked with 1×Tris Buffer Saline with 1% casein for 1 hour at room temperature with 120 bpm shaking. Subsequently, the membrane was incubated with primary antibodies at a dilution 1:1000 to 1:2000 overnight at 4°C. Primary antibodies used were listed: p38 MAPK (CST#9212, Cell Signaling), p‐ p38 MAPK (CST#9211, Cell Signaling), SDC‐1 (Ab#128936, Abcam), TNF‐α (Ab#205587, Abcam), IL‐6 (Ab#9324, Abcam). In addition, GAPDH (Ab#9482, Abcam) was performed as an internal control. After washing with 1× tris buffer saline tween, the membrane was incubated with secondary anti‐rabbit/mouse immunoglobulin G for 1 hour at room temperature. Protein quantitative analysis was done using ImageJ software.
Assessment of Functional NDS and Survival
For the 72‐hour survival study, all catheters were removed and wounds were surgically sutured at 6 hours following ROSC. Rats received a subcutaneous injection of Buprenorphine (1 mg/kg) to relieve pain when they were returned to their cages and rats were observed every 24 hours after ROSC. Levels of consciousness, brain stem function, and overall performance were evaluated according to the method of NDS (normal=0, death=500). 11 NDS was examined and scored by 2 investigators unaware of group identities at 24, 48, and 72 hours after ROSC in all groups. Animals were then euthanized by Euthasol (150 mg/kg).
Statistical Analysis
Statistical analysis was performed with Prism (Version 8.00; GraphPad Software Inc.). All continuous variables are presented as mean±SD after confirming for normality of distribution by Shapiro‐Wilk test. Group comparisons were performed by 1‐way or 2‐way ANOVA with Tukey post‐hoc test. Survival analyses were conducted based on Kaplan‒Meier plots and log‐rank tests. Statistical significance was assumed at P<0.05.
RESULTS
Baseline Characteristics of 2 Groups
Forty‐two rats were used for this study and 4 rats were not resuscitated (1 rat, technical failure; 3 rats, resuscitation failure). There were no significant differences in body weight, heart rate, calculated coronary perfusion pressure, end‐tidal CO2, and temperature of rats among groups (P>0.05) (Table). There were no significant differences in baseline blood pressure and (CO, EF, MPI), or buccal microcirculation among the groups.
Table .
Baseline Characteristics in Body Weight, Heart Rate, End‐Tidal CO2, Coronary Perfusion Pressure, and Temperature
| Sham (n=6) | Control (n=16) | VitC (n=16) | |
|---|---|---|---|
| Weight (g) | 483±17.6 | 480±20.1 | 485±16.9 |
| HR (bpm) | 361±24 | 369±16 | 366±18 |
| ETCO2 (mm Hg) | 36.7±1.7 | 35.2±2.8 | 35.9±3.1 |
| CPP (mm Hg) | 39.1±4.3 | 36.8±6.8 | 36.2±5.1 |
| Temperature (°C) | 36.6±0.23 | 36.8±0.19 | 36.7±0.21 |
CPP indicates coronary perfusion pressure; ETCO2, end‐tidal CO2; HR, heart rate; and VitC, vitamin C.
VitC Ameliorate Hemodynamic and Cardiac Function
CA and I/R injury significantly impaired myocardial function, as indicated by EF, CO, and MPI. However, myocardial function was all significantly better in the VitC group at ROSC3H and ROSC4H when compared with the control group (Figure 1A through 1C and Table S1). Plasma cTnI in control group versus sham group was significantly elevated after ROSC4H (1169±109.29 versus 188.1±11.76, P<0.001). However, VitC significantly reduced the release of cTnI compared with control group (699.6±57.27 versus 1169±109.29, P<0.001) (Figure 1D and Table S1).
Figure 1. Vitamin C effects on post‐resuscitation hemodynamics and myocardial function.
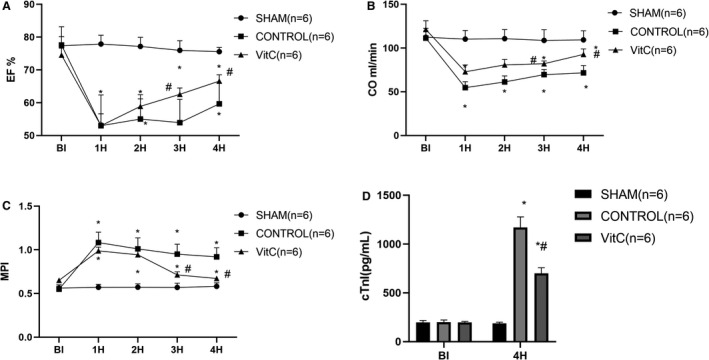
A, Ejection fraction among groups; B, Cardiac output among groups; C, Myocardial performance index among group; D, plasma cardiac troponin I concentration. Data are presented as mean±SD. CO indicates cardiac output; cTnI, cardiac troponin I; EF, ejection fraction; and MPI, myocardial performance index. *P<0.05 vs sham group, #P<0.05 vs control group.
In the control group, buccal microcirculatory blood flow presented at a low density and inadequate perfused state following resuscitation, which was also observed in the VitC group. However, in the VitC group there was significant improvements in perfused vessel density and microvascular flow index compared with the control group at post‐resuscitation 4 hours (P<0.0001, Figure 2 and Table S2).
Figure 2. Vitamin C effects on post‐resuscitation buccal microcirculation.
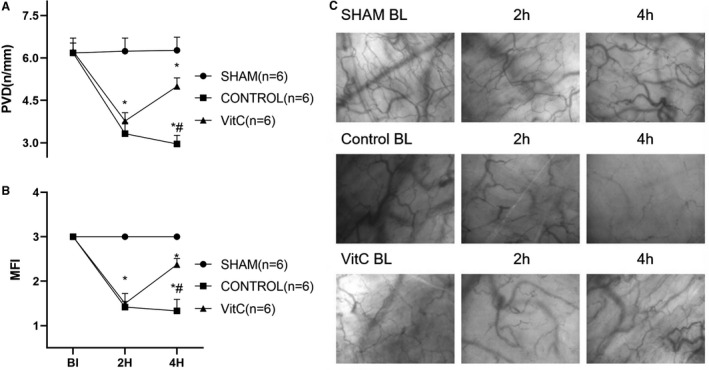
A, Perfused vessel density; B, Microcirculatory flow index; C, Digital photomicrographs of buccal microcirculation. Data are presented as mean±SD. MFI indicates microcirculatory flow index; and PVD, perfused vessel density. *P<0.05 vs sham group, #P<0.05 vs control group.
Plasma IL‐6 and TNF‐α levels were significantly increased after ROSC 4 hours in control and VitC (P<0.0001, Figure 3A and 3B and Table S3). However, VitC significantly reduced systemic inflammatory response compared with control which was also observed in the brain and heart tissue (Figure 3D and 3E and Table S3). ROS production had the same tendency in both heart and brain. VitC significantly decreased ROS level compared with controls (Figure 3F and Table S3).
Figure 3. Vitamin C effects on post‐resuscitation proinflammatory cytokines oxidative damage in heart and brain tissue.
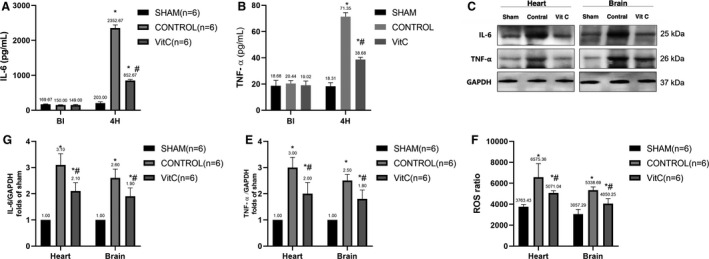
A, Levels of Interleukin‐6 levels in plasma, B, Tumor necrosis factor‐alpha levels in plasma. C, Representative protein bands of tumor necrosis factor‐alpha, Interleukin‐6, and GAPDH. D, Quantification of the expression levels of Interleukin‐6 in the heart and brain. E, Quantification of the expression levels of tumor necrosis factor‐alpha in the heart and brain. F, Reactive oxygen species levels in heart and brain. Data are presented as mean±SD. IL‐6 indicates interleukin 6; ROS, reactive oxygen species; and TNF‐α, tumor necrosis factor‐alpha. *P<0.05 vs sham group, #P<0.05 vs control group.
Plasma SDC‐1 and HA were significantly increased after ROSC 4 hours in controls and VitC (Figure 4B and 4D and Table S4). Meanwhile VitC retrieved vascular endothelial injury compared with controls. As expected, there was the opposite SDC‐1 expression in the heart and brain (Figure 4C and Table S4). Phosphorylation of p38 in controls was significantly increased compared with sham, while VitC significantly reduced p‐p38 expression (Figure 4E and Table S4).
Figure 4. Vitamin C effects on post‐resuscitation of SDC‐1 (Syndecan‐1), p38/MAPK (mitogen‐activated protein kinase) protein expression in heart and brain tissue.
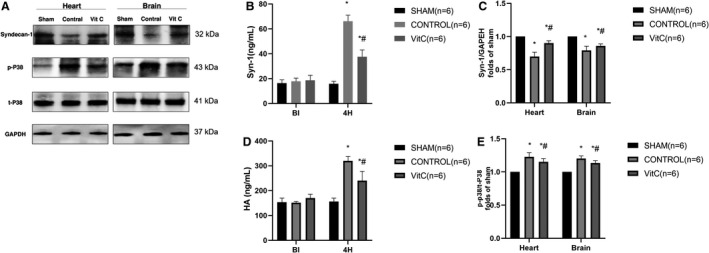
A, Representative protein bands of SDC‐1, p‐p38, t‐p38, and GAPDH. B, Quantification of the expression levels of SDC‐1 in plasma. C, Quantification of the expression levels of SDC‐1 in the heart and brain. D, Quantification of the expression levels of hyaluronan in plasma; E, Quantification of the expression levels of p‐p38/t‐p38 in the heart and brain. Data are presented as mean±SD. HA indicates hyaluronic acid; SDC‐1, Syndecan‐1; and VitC, vitamin C. *P<0.05 vs sham group, #P<0.05 vs control group.
The survival rate 72 hours after ROSC was greater in VitC compared with controls. In controls, survival rate was low (1 of 10 survival 72 hours after ROSC, Figure 5A) and the survival curves showed an initial sharp decline following ROSC (2 of 10 survival 48 hours after ROSC, Figure 5A). VitC had a higher survival rate (5 of 10 survival 72 hours after ROSC, Figure 5A) and decreased early mortality rate (6 of 10 survival 48 hours ROSC, Figure 5A). Moreover, NDS in VitC were significantly lower at 24 hours (341.50±120.53 versus 437.50±85.12; P=0.1591), 48 hours (223.57±124.89 versus 471.00±36.25; P=0.0042) and 72 hours (140±176.47 versus 462.50±53.03; P=0.0226) following ROSC compared with that in controls (Figure 5B and Table S5).
Figure 5. Vitamin C effects on survival analysis and neurological deficit score.
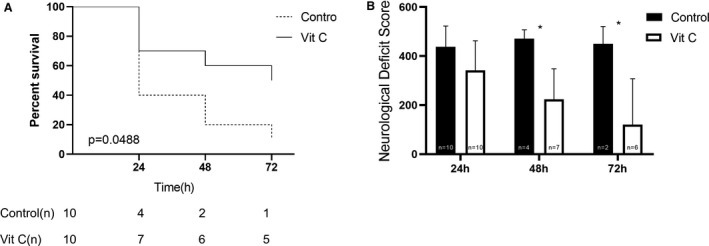
A, Survival analysis between control and vitamin C groups. B, Neurological deficit score between control and vitamin C groups at 24 hours after return of spontaneous circulation. VitC indicates vitamin C. *P<0.05 vs control group.
DISCUSSION
In the present study, we demonstrated that VitC administration following resuscitation significantly alleviated systemic inflammation, oxidative stress, microcirculation dysfunction, and shedding of SDC‐1 and HA, probably via regulating p38/MAPK phosphorylation, which may contribute to preserve cardiac and cerebral function. Finally, the effectiveness of VitC improved the 72‐hour survival and neurologic deficit in a rat model of CA and CPR was also demonstrated in the present study.
Myocardial function was impaired after ROSC. EF and CO decreased in controls compared with sham. cTnI plasma level, which indicated the severity of myocardial injury, was increased in controls. We further demonstrated that controls had high IL‐6, TNF‐α, SDC‐1, and HA plasma levels. Severity of myocardial dysfunction is closely related with elevation of ROS, TNF‐α, IL‐6, decrease of SDC‐1, and HA in the myocardium. Global I/R injury after CA and CPR is associated with post‐resuscitation myocardial dysfunction, cerebral dysfunction, and poor neurologic outcome. Perfused buccal vessel density and microcirculatory flow index at 4 hours after ROSC were impaired in controls, which is in line with the hypoperfusion in brain and cardiac regions of CA observed by Drabek et al. 12 Controls had a lower survival rate accompanied by neurologic deficits. I/R injury after successful resuscitation triggered a sterile inflammatory and oxidative stress reaction. A reduction in mean arterial pressure, elevation of inflammatory and oxidative stress response after ROSC is attributed to cerebral impairment, which contributes to a poor neurologic prognosis. 13
VitC is a key circulating antioxidant with anti‐inflammatory and immune‐supporting effects which is also a cofactor for important mono‐ and dioxygenase enzymes. VitC can suppress the oxidative stress induced by lipopolysaccharide and demonstrate increasing the activity of superoxide dismutase. 14 An increasing number of preclinical studies in trauma, I/R, and sepsis models have shown that intravenously administered VitC at pharmacological doses attenuates oxidative stress and inflammation and restores endothelial and organ function. 15 , 16 We demonstrated that VitC administration significantly decreased IL‐6, TNF‐α, and ROS, improving microcirculation and cardiac and brain function. The presence of oxidative stress is normally tested in 1 of 3 ways: direct measurement of the ROS; measurement of the resulting damage to biomolecules (eg, proteins, DNA, RNA, etc); and detection of antioxidant levels (catalase and superoxide dismutase). 17 In our research we found a huge burst of ROS was generated within minutes after the start of reperfusion. Studies have found that ROS generation can be produced by endogenous source (inflammatory cells, xanthine, and nicotinamide‐adenine dinucleotide phosphate oxidase, uncoupled eNOS or respiratory chain). 17 Fei et, al. found that after ROSC from CA a relatively “orderly” process of mitochondrial dysfuncion progressed. ROS generation was from Complex I of the mitochondrial respiratory chain. 18 The first day after CA, VitC plasma concentrations are decreased by >50% compared with healthy volunteers and after 3 days more than half the patients are insufficient. 2 The abundance of ROS, especially when insufficiently opposed by antioxidants such as VitC, leads to widespread endothelial abnormal, cellular impair, and progressive organ dysfunction. 15 ROS‐induced injury to the glycocalyx, cellular membranes, and junctions lead to increased permeability, adhesion of leukocytes and platelets with local activation of inflammation and coagulation, insensitivity of endothelial vasodilatation, and loss of the vascular response to vasoconstrictors. 19 , 20 , 21
SDC‐1 is the core protein in heparin sulfate proteoglycan, which comprises the glycocalyx. SDC‐1 is released from the endothelium upon injury to the glycocalyx which could cause its concentration in the circulation to increase. 22 Rehm et al. reported elevated SDC‐1 and hyaluronan (HA) plasma levels as markers of glycocalyx shedding after local and regional I/R in patients who underwent vascular and cardiac surgery. 23 The glycocalyx is damaged by acute oxidative stress in CA, leading to increased SDC‐1 serum levels. 24 I/R glycocalyx damage develops at an early stage and is a strong trigger for local and systemic inflammatory mediator release. 25 Bro‐Jeppesen et al. found a significant correlation between SDC‐1 and IL‐6. 26 IL‐6 has often been suggested as a suitable inflammatory and outcome marker for post resuscitation syndrome. 25
P38/MAPKs are responsive to stress stimuli which are involved in cell differentiation, apoptosis, and autophagy, which is dependent on its phosphorylation by p38 MAPK. 27 Interestingly, ROS can activate MAPK to either stimulate or inhibit apoptosis in cardiomyocytes, depending on the isoform stimulated which could arise after CA. 28 Lei et, al 29 found that the expression of the proteoglycan SDC‐1 was increased in rats with myocardial infarction. Inhibition of pP38 MAPK has been shown to reduce infarct size and improve ventricular remodeling, 30 and p38 MAPK knockout mice show smaller infarct size, a thicker ventricular wall, and better cardiac function than wild‐type mice 7 days after myocardial infarction. 31 Therefore, some investigators believe that activation of the p38/MAPK pathway is necessary for pathological ventricular remodeling. The current study found that the pP38 MAPK was significantly increased in infarction and that SDC‐1 overexpression caused reduced expression of p38/MAPK mRNA and phosphorylated protein, resulting in inhibition of p38/MAPK pathway. Expression of SDC‐1 may inhibit this influx of inflammatory cells in ventricular remodeling. SDC‐1 overexpression clearly affects myocardial collagen synthesis and interstitial remodeling. 32 The elevation concentrations of SDC‐1 induced by degradation of the glycocalyx after CA may also be a cause for blood‐brain barrier dysfunction and cerebral edema. 33
In addition, Jaakkola et al. 34 found that p38 MAPK may in some cases activate the SDC‐1 gene, because p38/MAPK inhibitors blocked the activation of the fibroblast growth factor response element, an upstream activating element in the SDC‐1 gene. 35 p38 MAPK inhibitors were also found to reduce the shedding of the extracellular domain of the SDC‐1. The results suggest that SDC‐1 is likely to impose at least some of its actions through the p38 MAPK signal transduction pathway. 29 Morgana et, al. 36 found that VitC abolished the p38/MAPK values induced by inflammatory cytokine (TNF‐α) in mice brain. Abeer et, al. 37 found VitC decreased the p38/MAPK level induced by oxidative stress in rat liver and brain tissues, which is the same result with our study. VitC, which could mitigate the response of oxidative stress, inflammatory and ischemia, may reduce glycocalyx damage and shedding of SDC‐1 and alter p38/MAPK pathway, but the link and exact mechanism remain to be further explored. 38
There are several limitations of this study. First, only healthy animals without underlying disease were included. Second, histologic analysis to demonstrate attenuation of heart and brain injury influenced by shedding of SDC‐1 was not performed and should be validated in future studies. Thirdly, we did not measure the expression of potential sources of ROS and would perform it in further study. Finally, SDC‐1 could regulate p38/MAPK phosphorylation which needs to be established by further study in post‐resuscitation myocardial I/R.
CONCLUSIONS
VitC attenuates post‐resuscitation myocardial and cerebral dysfunction and improves the survival. The mechanisms may involve inhibiting transcription of inflammatory cytokines and oxidative stress, thus protecting the integrity of the vascular endothelium. Meanwhile VitC reduces shedding of SDC‐1 and alters p38/MAPK phosphorylation and microcirculation.
Sources of Funding
This study was funded in part by a project of the People’s Livelihood Science and Technology—Basic Research on Medical and Health Application of Soochow, Jiangsu Province, China (No. SYS20 19075) and the Zoll Foundation (2020).
Disclosures
None.
Supporting information
Tables S1–S5
Figure S1
Acknowledgments
All the work was performed at the Weil Institute of Emergency and Critical Care Research at Virginia Commonwealth University. All authors have made contributions to the research.
For Sources of Funding and Disclosures, see page 9.
REFERENCES
- 1. Kleinman ME, Perkins GD, Bhanji F, Billi JE, Bray JE, Callaway CW, de Caen A, Finn JC, Hazinski MF, Lim SH, et al. ILCOR scientific knowledge gaps and clinical research priorities for cardiopulmonary resuscitation and emergency cardiovascular care: a consensus statement. Resuscitation. 2018;127:132–146. doi: 10.1186/2197-425X-2-11 [DOI] [PubMed] [Google Scholar]
- 2. Spoelstra‐de Man AME, Elbers PWG, Oudemans‐van Straaten HM. Making sense of early high‐dose intravenous vitamin C in ischemia/reperfusion injury. Crit Care. 2018;22:70. doi: 10.1155/2019/8071619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schroeder DC, Maul AC, Mahabir E, Koxholt I, Yan X, Padosch SA, Herff H, Bultmann‐Mellin I, Sterner‐Kock A, Annecke T, et al. Evaluation of small intestinal damage in a rat model of 6 Minutes cardiac arrest. BMC Anesthesiol. 2018;18:61. doi: 10.1016/j.resuscitation.2016.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Bottiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC, et al. Post‐cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation. 2008;118:2452‐2483. doi: 10.1161/CIRCULATIONAHA.108.190652 [DOI] [PubMed] [Google Scholar]
- 5. Geppert A, Zorn G, Karth GD, Haumer M, Gwechenberger M, Koller‐Strametz J, Heinz G, Huber K, Siostrzonek P. Soluble selectins and the systemic inflammatory response syndrome after successful cardiopulmonary resuscitation. Crit Care Med. 2000;28:2360–2365. doi: 10.1097/00003246-200007000-00030 [DOI] [PubMed] [Google Scholar]
- 6. Bogner‐Flatz V, Braunstein M, Ocker LE, Kusmenkov T, Tschoep J, Ney L, Bocker W, Annecke T. On‐the‐scene hyaluronan and syndecan‐1 serum concentrations and outcome after cardiac arrest and resuscitation. Mediators Inflamm. 2019;2019:8071619. doi: 10.1155/2019/8071619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liang L, Zhang GZ, Li H, Cheng C, Jin T, Su CL, Xiao Y, Bradley J, Peberdy MA, Ornato JP, et al. Combined therapy with polyethylene glycol‐20k and MCC950 preserves post‐resuscitated myocardial function in a rat model of cardiac arrest and cardiopulmonary resuscitation. J Am Heart Assoc. 2021;10:e019177. doi: 10.1161/JAHA.120.019177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang J, Xiao Y, Quan EY, Hu Z, Guo Q, Miao C, Bradley JL, Peberdy MA, Ornato JP, Mangino MJ, et al. Effects of polyethylene glycol‐20k on Postresuscitation myocardial and cerebral function in a rat model of cardiopulmonary resuscitation. Crit Care Med. 2018;46:e1190–e1195. doi: 10.1097/CCM.0000000000003415 [DOI] [PubMed] [Google Scholar]
- 9. Bark BP, Grande PO. The effect of vitamin C on plasma volume in the early stage of sepsis in the rat. Intensive Care Med Exp. 2014;2:11. doi: 10.1186/2197-425X-2-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Backer D, Donadello K, Cortes DO. Monitoring the microcirculation. J Clin Monit Comput. 2012;26:361–366. doi: 10.1007/s10877-012-9383-8 [DOI] [PubMed] [Google Scholar]
- 11. Benson DM, O'Neil B, Kakish E, Erpelding J, Alousi S, Mason R, Piper D, Rafols J. Open‐chest CPR improves survival and neurologic outcome following cardiac arrest. Resuscitation. 2005;64:209–217. doi: 10.1016/j.resuscitation.2003.03.001 [DOI] [PubMed] [Google Scholar]
- 12. Ishii H, Amano T, Matsubara T, Murohara T. Pharmacological intervention for prevention of left ventricular remodeling and improving prognosis in myocardial infarction. Circulation. 2008;118:2710–2718. doi: 10.1161/CIRCULATIONAHA.107.748772 [DOI] [PubMed] [Google Scholar]
- 13. He F, Zheng G, Hou J, Hu Q, Ling Q, Wu G, Zhao H, Yang J, Wang Y, Jiang L, et al. N‐acetylcysteine alleviates post‐resuscitation myocardial dysfunction and improves survival outcomes via partly inhibiting NLRP3 inflammasome induced‐pyroptosis. J Inflamm (Lond). 2020;17:25. doi: 10.1186/s12950-020-00255-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang X‐Y, Xu Z‐P, Wang W, Cao J‐B, Fu Q, Zhao W‐X, Li Y, Huo X‐L, Zhang L‐M, Li Y‐F, et al. Vitamin C alleviates LPS‐induced cognitive impairment in mice by suppressing neuroinflammation and oxidative stress. Int Immunopharmacol. 2018;65:438–447. doi: 10.1016/j.intimp.2018.10.020 [DOI] [PubMed] [Google Scholar]
- 15. Spoelstra‐de Man AME, Elbers PWG, Oudemans‐Van Straaten HM. Vitamin C: should we supplement? Curr Opin Crit Care. 2018;24:248–255. doi: 10.1097/MCC.0000000000000510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ye S, Shi K, Xu J, Wang M, Li CJ. Cholecystokinin octapeptide inhibits the inflammatory response and improves neurological outcome in a porcine model of cardiopulmonary resuscitation. Exp Ther Med. 2018;15:2583–2588. doi: 10.3892/etm.2017.5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Voronkova YS, Voronkova OS, Gorban VA, Holoborodko KK. Oxidative stress, reactive oxygen species, antioxidants: a review.Ecology and. Noospherolgoy. 2018;29:52–55. doi: 10.1186/cc11239 [DOI] [Google Scholar]
- 18. Fei H, Tong D, Natalia AR, Lance BB. Early mitochondrial dysfunction in electron transfer activity and reactive oxygen species generation following cardiac arrest. Crit Care Med 2008:36:s447–s453. doi: 10.1097/ccm.0b013e31818a8a51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burke‐Gaffney A, Evans TW. Lest we forget the endothelial glycocalyx in sepsis. Crit Care. 2012;16:121. doi: 10.1186/cc11239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van den Berg BM, Nieuwdorp M, Stroes ES, Vink H. Glycocalyx and endothelial (dys) function: from mice to men. Pharmacol Rep. 2006;58(Suppl):75–80. [PubMed] [Google Scholar]
- 21. Rubio‐Gayosso I, Platts SH, Duling BR. Reactive oxygen species mediate modification of glycocalyx during ischemia‐reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;290:H2247–2256. doi: 10.1152/ajpheart.00796.2005 [DOI] [PubMed] [Google Scholar]
- 22. Inagawa R, Okada H, Takemura G, Suzuki K, Takada C, Yano H, Ando Y, Usui T, Hotta Y, Miyazaki N, et al. Ultrastructural alteration of pulmonary capillary endothelial glycocalyx during endotoxemia. Chest. 2018;154:317–325. doi: 10.1016/j.chest.2018.03.003 [DOI] [PubMed] [Google Scholar]
- 23. Rehm M, Bruegger D, Christ F, Conzen P, Thiel M, Jacob M, Chappell D, Stoeckelhuber M, Welsch U, Reichart B, et al. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation. 2007;116:1896–1906. doi: 10.1161/CIRCULATIONAHA.106.684852 [DOI] [PubMed] [Google Scholar]
- 24. Grundmann S, Fink K, Rabadzhieva L, Bourgeois N, Schwab T, Moser M, Bode C, Busch HJ. Perturbation of the endothelial glycocalyx in post cardiac arrest syndrome. Resuscitation. 2012;83:715–720. doi: 10.1016/j.resuscitation.2012.01.028 [DOI] [PubMed] [Google Scholar]
- 25. Callaway CW. Endothelial damage after cardiac arrest–"endotheliitis". Resuscitation. 2012;83:667–668. doi: 10.1016/j.resuscitation.2012.03.008 [DOI] [PubMed] [Google Scholar]
- 26. Bro‐Jeppesen J, Johansson PI, Hassager C, Wanscher M, Ostrowski SR, Bjerre M, Kjaergaard J. Endothelial activation/injury and associations with severity of post‐cardiac arrest syndrome and mortality after out‐of‐hospital cardiac arrest. Resuscitation. 2016;107:71–79. doi: 10.1016/j.resuscitation.2016.08.006 [DOI] [PubMed] [Google Scholar]
- 27. Gerthoffer WT, Gunst SJ. Invited review: focal adhesion and small heat shock proteins in the regulation of actin remodeling and contractility in smooth muscle. J Appl Physiol. 1985;2001(91):963–972. doi: 10.1152/jappl.2001.91.2.963 [DOI] [PubMed] [Google Scholar]
- 28. Li CJ, Lv L, Li H, Yu DM. Cardiac fibrosis and dysfunction in experimental diabetic cardiomyopathy are ameliorated by alpha‐lipoic acid. Cardiovasc Diabetol. 2012;11:73. doi: 10.1186/1475-2840-11-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lei J, Xue S, Wu W, Zhou S, Zhang Y, Yuan G, Wang J. Sdc1 overexpression inhibits the p38 MAPK pathway and lessens fibrotic ventricular remodeling in MI rats. Inflammation. 2013;36:603–615. doi: 10.1007/s10753-012-9582-y [DOI] [PubMed] [Google Scholar]
- 30. Yin H, Zhang J, Lin H, Wang R, Qiao Y, Wang B, Liu F. p38 mitogen‐activated protein kinase inhibition decreases TNFalpha secretion and protects against left ventricular remodeling in rats with myocardial ischemia. Inflammation. 2008;31:65–73. doi: 10.1007/s10753-007-9050-2 [DOI] [PubMed] [Google Scholar]
- 31. Ren J, Zhang S, Kovacs A, Wang Y, Muslin AJ. Role of p38alpha MAPK in cardiac apoptosis and remodeling after myocardial infarction. J Mol Cell Cardiol. 2005;38:617–623. doi: 10.1016/j.yjmcc.2005.01.012 [DOI] [PubMed] [Google Scholar]
- 32. Vanhoutte D, Schellings MWM, Götte M, Swinnen M, Herias V, Wild MK, Vestweber D, Chorianopoulos E, Cortés Víctor, Rigotti A, et al. Increased expression of syndecan‐1 protects against cardiac dilatation and dysfunction after myocardial infarction. Circulation. 2007;115:4754–4782. doi: 10.1161/CIRCULATIONAHA.106.644609 [DOI] [PubMed] [Google Scholar]
- 33. Zhu J, Li X, Yin J, Hu Y, Gu Y, Pan S. Glycocalyx degradation leads to blood‐brain barrier dysfunction and brain edema after asphyxia cardiac arrest in rats. J Cereb Blood Flow Metab. 2018;38:1979–1992. doi: 10.1177/0271678X17726062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jaakkola P, Kontusaari S, Kauppi T, Maata A, Jalkanen M. Wound reepithelialization activates a growth factor‐responsive enhancer in migrating keratinocytes. FASEB J. 1998;12:959–969. doi: 10.1096/fasebj.12.11.959 [DOI] [PubMed] [Google Scholar]
- 35. Popova TG, Millis B, Bradburne C, Nazarenko S, Bailey C, Chandhoke V, Popov SG. Acceleration of epithelial cell syndecan‐1 shedding by anthrax hemolytic virulence factors. BMC Microbiol. 2006;6:8. doi: 10.1186/1471-2180-6-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moretti M, Budni J, Freitas AE, Neis VB, Ribeiro CM, de Oliveira Balen G, Rieger DK, Leal RB, Rodrigues ALS. TNF‐α‐induced depressive‐like phenotype and p38MAPK activation are abolished by ascorbic acid treatment. Eur Neuropsychopharmacol. 2015;25(6):902–912. doi: 10.1016/j.euroneuro.2015.03.006 [DOI] [PubMed] [Google Scholar]
- 37. Radi AM, Mohammed ET, Abushouk AI Aleya L, Abdel‐Daim MM. The effects of abamectin on oxidative stress and gene expression in rat liver and brain tissues: modulation by sesame oil and ascorbic acid. Sci Total Environ. 2020;701:134882– 10.1016/j.scitotenv.2019.134882. doi: 10.1016/j.scitotenv.2019.134882 [DOI] [PubMed] [Google Scholar]
- 38. Burggraf D, Trinkl A, Dichgans M, Hamann GF. Doxycycline inhibits MMPs via modulation of plasminogen activators in focal cerebral ischemia. Neurobiol Dis. 2007;25:506–513. doi: 10.1016/j.nbd.2006.10.013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S5
Figure S1