

Abstract
Brugada syndrome (BrS) is an inherited and rare cardiac arrhythmogenic disease associated with an increased risk of ventricular fibrillation and sudden cardiac death. Different genes have been linked to BrS. The majority of mutations are located in the SCN5A gene, and the typical abnormal ECG is an elevation of the ST segment in the right precordial leads V1 to V3. The pathophysiological mechanisms of BrS were studied in different models, including animal models, heterologous expression systems, and human‐induced pluripotent stem cell–derived cardiomyocyte models. Currently, only a few BrS studies have used human‐induced pluripotent stem cell–derived cardiomyocytes, most of which have focused on genotype–phenotype correlations and drug screening. The combination of new technologies, such as clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 (CRISPR associated protein 9)‐mediated genome editing and 3‐dimensional engineered heart tissues, has provided novel insights into the pathophysiological mechanisms of the disease and could offer opportunities to improve the diagnosis and treatment of patients with BrS. This review aimed to compare different models of BrS for a better understanding of the roles of human‐induced pluripotent stem cell–derived cardiomyocytes in current BrS research and personalized medicine at a later stage.
Keywords: Brugada syndrome, human‐induced pluripotent stem cell‐derived cardiomyocytes, model systems, precision medicine
Subject Categories: Developmental biology, Ion Channels/Membrane Transport, Physiology, Arrhythmias, Sudden Cardiac Death
Nonstandard Abbreviations and Acronyms
- BrS
Brugada syndrome
- HEK
human embryonic kidney
- hiPSC‐CMs
human‐induced pluripotent stem cell–derived cardiomyocytes
- INa
sodium current
- Ito
transient outward potassium current
Brugada syndrome (BrS) is an inherited arrhythmic disorder that is characterized by an increased risk of sudden cardiac death. It was first described in 1992 by research on 8 patients who were resuscitated from sudden cardiac death because of ventricular fibrillation and/or suffered from atrial fibrillation. 1 Patients can have symptoms of nocturnal breathing, syncope, seizures, and in worse cases ventricular fibrillation. 2 Fever can increase the risk of ventricular fibrillation 3 . In a 2012 expert consensus report, 2 types of ECG patterns of BrS were described, type 1 (coved type) and type 2 (saddleback type). 4 However, only the type 1 ECG pattern is regarded as the diagnostic criterion for BrS, and type 2 is only suggestive of BrS. The typical type 1 ECG pattern of BrS is an ST‐segment elevation ≥2 mm in 1 or more leads among right precordial leads V1 to V3 2 (Figure 1A). To date, the definition of diagnosis for BrS is mainly based on its typical ECG changes or the application of different sodium channel blockers as described in 2015 by the European Society of Cardiology guidelines. 5 Although the incidence of BrS appears to be low, only 0.12% to 0.8%, it is responsible for 4% to 12% of sudden cardiac deaths in the general population at a young age. According to a recent study, men are more likely to suffer from BrS than women. 6
Figure 1. Main characteristics and possible mechanism of Brugada syndrome (BrS).
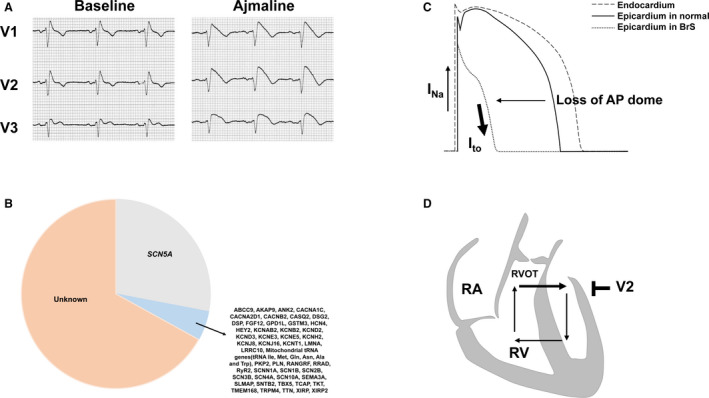
A, The Ajmaline test unmasked the typical cover‐type ST‐segment elevation in a BrS patient. B, All reported genes related to Brugada syndrome. C, Repolarization hypothesis of the BrS. D, Depolarization hypothesis of the BrS. INa indicates sodium current; Ito, transient outward potassium current; RA, right atrium; RV, right ventricle; and RVOT, right ventricular outflow tract.
Approximately 11% to 28% of patients with BrS were found to be carriers of genetic mutations, the majority of which are located in the SCN5A gene. 7 This gene encodes the α subunit of the cardiac sodium channel Nav1.5, which plays a significant role in phase 0 of the action potential in cardiomyocytes. In addition, >50 genes that were also reported to be associated with BrS were responsible for 2% to 5% of diagnosed cases 8 (Figure 1B). However, a recent study indicated that most of the putative genes associated with BrS had no sufficient evidence for causality in BrS, which indicated that BrS is likely polygenic rather than being influenced by mutations of a single gene. 9 To date, thousands of mutations have been reported to be related or possibly related to BrS, but only a small proportion of them have been experimentally investigated.
Most pathogenic or likely pathogenic variants were detected in the SCN5A gene, such as E1784K, p.S1812X, and p.Val1429Met. 10 , 11 , 12 In other rare genes, few pathogenic and benign variants as well as variants with unknown significance were identified. 13 In the study by Chen, 14 SCN5A accounted for 87.5% of pathogenic and likely pathogenic variants, whereas others accounted for only 12.5%. Despite advances in understanding the genotype association with the clinical phenotype, there is a reappraisal of reported genes with an evidence‐based evaluation of gene validity for BrS.
Although loss of function of the Nav1.5 channel in BrS is not generally controversial, 2 main hypotheses about the pathophysiological mechanism of BrS are disputed. The repolarization hypothesis indicated that the stronger expression of transient outward potassium current (Ito) in the epicardium led to the transmural dispersion of repolarization in the right ventricle, which could cause ST‐segment elevation in BrS 15 (Figure 1C); the depolarization hypothesis suggested that a right ventricular outflow tract conduction delay caused an intercellular current from the right ventricle to right ventricular outflow tract and a back current from the right ventricular outflow tract to the right ventricle, which caused ST‐segment elevation in right precordial leads in BrS 16 (Figure 1D). In recent years, conventional ECG analysis has been commonly applied to explore the features and mechanisms of BrS, but it could provide limited information for researchers. Because of the development of next‐generation sequencing, the identification of pathogenic genes of BrS has also become possible and is performed in clinical practice. However, these technologies are not able to study the detailed molecular mechanisms of the syndrome in vivo. Therefore, experimental models are commonly used in BrS studies. Although the human heart is the best choice for building models, animal and cellular models have been chosen by most researchers because of the difficulty in obtaining human heart tissues and ethical limitations. Several models have been used to simulate the disease, including transgenic mice and porcine, canine, and rabbit heart preparations, and various cellular models with expression of mutant SCN5A and human‐induced pluripotent stem cell–derived cardiomyocytes (hiPSC‐CMs) 17 (Table 1). Another difficulty is the current technological inability to study the detailed impulse propagation mechanisms of the syndrome in vivo. In this regard, mapping analysis could be helpful. In the study by Calvo et al, 18 the 3‐dimensional (3D) electroanatomic mapping system, which can create 3D endocardial reconstructions, were used to study the mechanism of ventricular tachycardia and early ventricular fibrillation in patients with BrS. 3D mapping showed that SCN5A‐positive patients with BrS exhibited a larger epicardial arrhythmogenic substrate area. In addition, more prolonged electrograms and a higher frequency of noninvasive late potentials were detected. The analysis showed a predictable sequence in the frequency‐phase domain, which is consistent with the anatomic location of the arrhythmogenic substrate. 19
Table 1.
Experimental Models for BrS
| Model | Methods | Advantages | Disadvantages |
|---|---|---|---|
| Animal | Scn5a+/− mouse; Scn5a(1798insD/+) mouse |
Easier to feed than other animals Allows investigators to study BrS in physiological environment and different developmental stages |
Different electrophysiological properties and cardiac markers between human and mouse heart Hard to evaluate the typical ECG pattern of BrS in the model |
| Canine |
Similar electrophysiology properties and profile of ion channel expression compared with the human heart Allows investigation in epicardium and endocardium with intact structural organization in the heart |
Transgenic model is difficult to obtain and drug could be required for inducing the phenotype Not easier to feed and relatively expensive |
|
| Porcine |
Similar electrophysiology properties compared with the human heart Allows investigation in epicardium and endocardium with intact structural organization in the heart |
High price and long reproductive cycles of cells Transgenic model is difficult to obtain and a drug may be required for inducing the phenotype Did not display the typical ECG pattern of BrS |
|
| Rabbit |
Similar electrophysiology properties and profile of ion channel expression compared with the human heart Allows investigation in epicardium and endocardium with intact structural organization in the heart |
Transgenic model is difficult to obtain and a drug could be required for inducing the phenotype | |
| Heterologous expression | HEK‐293 cells; Xenopus oocytes; tsA201 cells; CHO cells |
Easier to feed than animal models Allows investigation by more detailed experiments in cellular mechanism of the disease |
Lack of cardiac markers and electrophysiological properties of cardiomyocytes Results will be various because of the different cell lines |
| Native human cardiomyocytes | Native human cardiomyocytes from right atrial tissue | Carries patient’s exact genetic background, cardiac markers and electrophysiological properties of cardiomyocytes |
Difficult to obtain cardiomyocytes from ventricular tissue Ethical limitations |
| hiPSC‐CMs | hiPSCs derived from human somatic cells |
Carries patient’s exact genetic background; expression profile is similar to cardiomyocytes Relatively easier to feed and combine with other advanced technologies |
Immature phenotype of cells Composed of a mixture of ventricular‐, atrial‐, and nodal‐like cells Hard to conduct studies in a physiological environment |
BrS indicates Brugada syndrome; CHO cells, Chinese hamster ovary; HEK, human embryonic kidney; hiPSCs, human‐induced pluripotent stem cells; and hiPSC‐CMs, human‐induced pluripotent stem cell–derived cardiomyocytes.
Compared with other models, hiPSC‐CMs from patients who carry the desired genetic profile have unique advantages. These are relatively easy to obtain from somatic cells, including peripheral blood mononuclear cells or fibroblasts of patients. 20 Although animal models can allow integral studies from whole animals, organs, and tissues, there are some fundamental differences in physiology between humans and animals. Patient‐specific hiPSC‐CMs carry the exact genetic background of patients. Recent studies have indicated that hiPSC‐CMs can effectively recapitulate the electrophysiological pattern in several cardiac disorders, including BrS, 21 , 22 long‐QT syndrome, 23 arrhythmogenic right ventricular cardiomyopathy, 24 catecholaminergic polymorphic ventricular tachycardia, 25 Timothy syndrome, 26 short‐QT syndrome, 27 dilated cardiomyopathy, 28 and Noonan syndrome, 29 at the cellular level. Other cellular models of BrS, such as human embryonic kidney (HEK) cells or Xenopus oocytes, can easily explore the impact of mutations on ion channels, but these cells lack many cardiac proteins. Previous studies 30 , 31 suggested that hiPSC‐CMs possess basic features of human cardiomyocytes and express a variety of cardiac‐specific genes, including those that can encode ion channels in cardiomyocytes. 32 This circumstance makes it possible for investigators to use hiPSC‐CMs combined with other techniques to recapitulate disease phenotypes in vitro and conduct various studies to explore the underlying mechanism of the disease or new therapeutic strategies (Figure 2).
Figure 2. Applications of human‐induced pluripotent stem cell–derived cardiomyocytes (hiPSC‐CMs) in disease research by using different technologies.
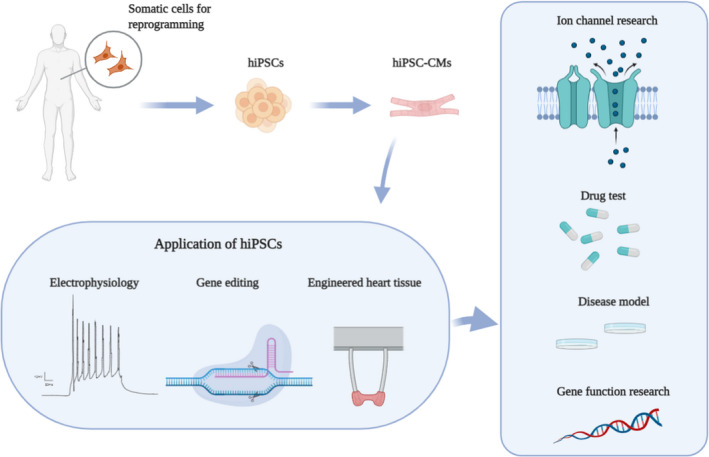
hiPSCs were generated by somatic reprogramming from Brugada syndrome patients and then were differentiated into patient‐specific hiPSC‐CMs. hiPSC‐CMs combined with other advanced technologies have been used for disease modeling of inherited cardiomyopathies and channelopathies, drug testing, and studying gene function. Created with http://BioRender.com/.
This review aimed to summarize studies that use different BrS models and focuses on the application of hiPSC‐CMs in BrS studies.
Experimental Models of BrS
Animal Models
Several animal models have often been used for BrS research, including murine models, porcine models, canine models, and rabbit models. Each model has its own unique advantages and limitations. The murine model has been commonly used because mice are easier to feed than other animals, which can allow researchers to study BrS in its physiological environment and different developmental stages. However, there are significant differences in the electrophysiology and profile of ion channel expression between the human heart and murine heart. Therefore, this type of model is not perfect for BrS studies. Alternatively, porcine, canine, and rabbit models show electrophysiological properties and profiles of ion channel expression similar to those of the human heart, and these models allow investigation of cells in the epicardium and endocardium with intact structural organization in the heart. Nevertheless, they still have their own limitations. Porcine and canine animals are expensive and have long reproductive cycles. The canine and rabbit models of BrS were induced by ion channel blocking agents, 33 , 34 which could be different from the disease caused by mutations in ion channel genes.
Intact Animal Models
Two methods are commonly used in murine models of BrS: Scn5a +/‐ mice 35 and Scn5a1798insD mice. 36 In 2002, Papadatos and colleagues generated a mouse model by targeted disruption of the cardiac sodium channel gene Scn5a. Homozygous knockout mice showed severe defects in ventricular morphogenesis and eventually died. However, heterozygous knockout mice survived and exhibited similar cardiac morphology compared with the wild type. Furthermore, whole‐cell patch clamp analyses of ventricular myocytes showed an ≈50% reduction in sodium conductance and impaired conduction compared with the wild type. 35 Subsequent studies used this model to investigate the drug response and underlying mechanisms of the phenotypes observed before. Several studies recapitulated the effect of pro‐ and antiarrhythmic drugs, including flecainide and quinidine, in a Scn5a +/‐ mouse model. 37 , 38 Nav1.5 messenger RNA and protein expression levels in the mouse model were found to be lower than those in the wild type, which was further related to the presence of fibrosis. 39 , 40 In 2006, Remme and colleagues generated a transgenic model of mice containing the equivalent mutation 1798insD. 36 Scn5a(1798insD/+) heterozygous mice showed lower heart rates and abnormal ECG patterns than wild‐type mice. Whole‐cell patch‐clamp analysis exhibited action potential prolongation, a ≈39% reduction in peak sodium current density, and a reduction in action potential upstroke velocity. This mutation could cause common symptoms of long‐QT syndrome type 3 or BrS or progressive cardiac conduction defects. 41 A recent study showed that conduction defects were related to the strain in a Scn5a(1795insD/+) mouse model. 42 However, the 12‐lead ECG morphology in mice is quite different from humans, so it is hard to evaluate whether the typical ECG pattern of BrS was present in the mouse model.
In 2015, Park and collaborators 43 generated a porcine model by introducing a nonsense mutation into the orthologous position (E558X) in the pig SCN5A gene. SCN5A(E558X/+) pigs displayed conduction abnormalities with no structural abnormalities. However, the porcine model did not display a BrS‐type ECG. Although the electrophysiology of porcine hearts is similar to that of humans, few studies of BrS use this model because of its high price.
In addition, although the majority of genetic mutations of BrS are located in the SCN5A gene, other factors or genetic mutations can also cause BrS. These animal models contained only SCN5A mutations and only displayed a part of BrS’s features, mainly the loss of function of sodium current and conduction slowing, rather than the characteristic ECG pattern of BrS. This may reflect the difference between humans and animals on cardiac structure and electrophysiology, and also an important limitation of animal models.
Tissue Models
The canine model first used for BrS was in the study conducted by Yan and Antzelevitch in 1996, which explored the underlying mechanism of the J wave. 44 In the canine model, arterially perfused wedges in the left or right ventricles of dogs and drugs were used to induce the BrS phenotype. Several drugs were used for the canine model, including sodium channel blockers, calcium channel blockers, and potassium channel agonists. 45 , 46 , 47 ST‐segment elevation in drug‐induced BrS in a canine model was caused by a transmural gradient across the right ventricular wall, which was generated by loss of the epicardial action potential dome. 44 Further studies showed that ventricular repolarization abnormalities that were located in the right ventricular outflow tract could lead to ST‐segment elevation. 48 In a recent study, radiofrequency catheter ablation technology was used in a canine model of BrS, and the results showed that radiofrequency catheter ablation could eliminate abnormal repolarization sites and the substrate for ventricular tachyarrhythmias. 49 Chen and colleagues 34 generated a rabbit model for J‐wave syndrome by using cyclohexyl‐[2‐(3,5‐dimethyl‐pyrazol‐1‐yl)‐6‐methyl‐pyrimidin‐4‐yl]‐amine, which decreased sodium current (INa) and action potential upstroke as well as intracellular calcium transients. At the same time, cyclohexyl‐[2‐(3,5‐dimethyl‐pyrazol‐1‐yl)‐6‐methyl‐pyrimidin‐4‐yl]‐amine triggered a significant J‐wave elevation and frequent spontaneous ventricular fibrillation.
Heterologous Expression Systems
Transfecting plasmids with mutant genes identified in patients with BrS into HEK cells, SV40 transformed (tsA201), or Chinese hamster ovary cells or other cells could help to understand the underlying molecular mechanisms and explore the biophysical properties of associated mutations in BrS. This type of model is relatively easy to use compared with animal models, and it is more suitable for studying the effect of mutations on ion channels. Casini and colleagues 50 used HEK‐293 cells to study the effect of a missense SCN5A mutation called G1319V and found that the G1319V mutation could decrease voltage‐dependent activation and channel availability and hence reduce the peak amplitude of INa. In a recent study, HEK‐293 cells were also used to identify the relationship between BrS and the SCN1B variant A197V. The results showed that this mutation could cause a reduction in the activation velocity and current density. 51 In addition, Xenopus laevis oocytes are also commonly used for heterologous expression of ion channels. In a recent study, SCN5A variants of 13 families were functionally characterized in Xenopus laevis oocytes by the 2‐electrode voltage‐clamp technique. The results showed that patients carrying variants S216L, K480N, A572D, F816Y, and G983D were more likely to develop long‐QT syndrome, and variants R222stop and R2012H were new mutations for BrS genetic causes. 52 In the study by Watanabe and colleagues, 53 Chinese hamster ovaries or tsA201 cells with the D1275N SCN5A mutation did not show a significant difference in sodium current amplitudes and gating, whereas the mouse model with the same variant displayed sodium channel dysfunction.
Although expression systems are better methods to investigate the molecular mechanism of associated mutations in BrS than animal models, the results of these experiments could differ because of the variety of cell types. In a study by Dumaine et al, transfection of the Thr1620Met mutation into a mammalian cell line showed that Thr1620Met current decay kinetics were faster, and that the recovery from inactivation was slower than that of the wild type at 32 °C, whereas the electrophysiological data failed to explain the electrocardiographic phenotype when the mutant was heterologously expressed in frog oocytes. 54 A novel mutation T1620M in the SCN5A gene also displayed different phenotypes when expressed in Xenopus oocytes and mammalian tsA201 cells. 55 The mutation led to a faster recovery from inactivation and a shift of steady‐state inactivation to more positive voltages when expressed in Xenopus oocytes, whereas it showed a slower recovery from inactivation and no effect on steady‐state inactivation when expressed in tsA201 cells. Although all of the cell models express sodium channels, the difference in Nav1.5 morphology and function among these models could cause the discrepant results. Nevertheless, heterologous expression systems are still suitable for investigating the effects of individual mutations.
Native Human Cardiomyocytes
A recent study investigated the functional roles of a mutation in SNTB2 (β‐2‐syntrophin) (SNTB2‐N167K) in native cardiomyocytes obtained from right atrial tissue of a patient with BrS. 17 Cardiomyocytes with the SNTB2‐N167K mutation showed reduced peak INa and late INa and shortened action potential duration. The results were consistent with findings in the coexpression of Nav1.5 with WT SNTB2 and SNTB2‐N167K or with SNTB2‐N167K alone in Xenopus oocytes, which showed a significant reduction in INa in both homozygous and heterozygous conditions. Although cardiomyocytes were obtained from atrial tissue because of the difficulty of obtaining them from ventricular tissue, these results can be extrapolated to ventricular electrophysiology because of the similar electrophysiological properties between atrial and ventricular tissues.
hiPSC‐CM Model
Human‐induced Pluripotent Stem Cell–Derived Cardiomyocytes
Embryonic stem cells are a type of cells that have properties of self‐renewal and pluripotency. They were first shown in the study by Evans and Kaufman 56 in 1981 and were derived from the inner cell mass of murine blastocysts. Human embryonic stem cells were successfully derived from human blastocysts in 1998. 57 Although embryonic stem cells have a unique advantage for studying cell fate and tissue development, destroying early‐stage embryos in the process of embryonic stem cell preparation makes this technology ethically controversial. Yamanaka’s research group improved the situation by the production of induced pluripotent stem cells. They found that somatic cells from mice 58 and humans 59 could be induced to a pluripotent state through the introduction of 4 reprogramming factors (Octamer‐binding transcription factor 4, Sex‐determining region Y‐box 2, Kruppel‐like factor‐4, and Cellular‐myelocytomatosis viral oncogene). The induced cells could also maintain the properties of unlimited self‐renewal and pluripotency characteristics, and they avoid the ethical issues caused by embryo destruction. Currently, induced pluripotent stem cells that can differentiate into specific cell types are commonly used in extensive studies with different differentiation methods. Induced pluripotent stem cells have considerable advantages in disease modeling and personalized medicine. To date, clustered regularly interspaced short palindromic repeats (CRISPR) gene‐editing technologies combined with induced pluripotent stem cells can create patient‐specific isogenic controls. 60 Using this method, a raised limitation of comparing diseased hiPSC‐CMs with independent healthy cells with their individual variability could be overcome.
In previous studies, several signaling pathways, such as bone morphogenetic protein (BMP), wingless/integrated (Wnt) and Activin/Nodal/transforming growth factor‐β (TGF‐β), have been indicated to play important roles in cardiovascular development. 61 , 62 , 63 Protocols of hiPSC‐CM differentiation aim to simulate these developmental signaling cues and generate cardiomyocytes that are highly similar to cells in the human heart. Therefore, different methods for hiPSC‐CM differentiation have been developed. Initially, hiPSC‐CMs were obtained from embryoid bodies with serum‐containing media, but the differentiation efficiency of this method was low, only 5% to 10%. 64 To date, several endogenous growth factors and small molecules of related signaling pathways have been used in different hiPSC‐CM differentiation protocols to improve reproducibility and efficiency. 30 By various methods for purification of cardiomyocytes, hiPSC‐CMs could be further purified, and more beating cardiomyocytes expressing human cardiac‐specific proteins could be obtained. 65 , 66 , 67 The expression of specific genes in several stages of normal cardiac development was also detected in hiPSC‐CMs, including genes for mesoderm formation (BRY and MIXL1), cardiogenic mesoderm (MESP1 and ISL1), cardiac‐specific progenitors (NKX2.5, GATA4, and TBX5), and mature cardiomyocyte structure (MYL2, MYH7, and TNNT2). 68
Recently, atrial‐specific differentiation has been reported after recently published techniques for the differentiation of human‐induced pluripotent stem cells into hiPSC‐CMs generated a mixture of cell types with a higher presence of ventricular cardiomyocytes. 69 , 70 Fluorescent activated cell sorting could be a useful method to distinguish subtype‐specific hiPSC‐CM populations by specific markers, but the cells would be exposed to additional influencing factors that could affect their functions. Previous studies showed that pluripotent stem cells could be directed into atrial‐like or ventricular‐like cardiomyocytes by modulating retinoic acid and Wnt signaling pathways. 71 , 72 In more recent years, Liu and colleagues directed hiPSC‐CMs into sinoatrial node‐like cells by combined modulation of the BMP, fibroblast growth factor, and retinoic acid signaling pathways. 73 In the study by Cyganek, 74 a protocol that can direct hiPSC‐CMs into either atrial‐like or ventricular‐like cardiomyocytes was provided. These methods will be useful in further BrS studies to explore the underlying mechanisms of the disease by the hiPSC‐CM model and to improve our understanding of lineage‐specific development or chamber‐specific remodeling of some heart diseases.
hiPSC‐CM Model for Precision Medicine
Use of hiPSC‐CMs for Studying the Pathogenicity of Gene Variants
Precision medicine is an emerging approach for disease treatment and prevention that accounts for individual variability in genes, environment, and lifestyle for each person. 75 In this regard, there is no doubt that hiPSCs can provide a good model for studying BrS. The key point for studying BrS by hiPSC‐CMs with respect to precision medicine is to identify personal‐specific mutations and their effect on the phenotypes of diseases. The main accomplishments of using hiPSC‐CMs in the BrS study are shown in Table 2. 11 , 21 , 22 , 31 , 76 , 77 , 78 , 79 To date, hundreds of mutations in SCN5A have been reported, including missense mutations, nonsense mutations, nucleotide insertion/deletions, and splice site mutations, which can cause loss‐of‐function in sodium channels by different mechanisms, such as reduced expression of sodium channel proteins, expression of channels with no function, and changed gating properties of sodium channels. 80 tion in hiPSC‐CMs was conducted by Davis and collaborators in 2012. 41 They used both hiPSC‐CMs derived from SCN5A(1795insD/+) and Scn5a(1798insD/+) heterozygous mice to investigate the electrophysiological properties of sodium channel mutations, and the results demonstrated that hiPSC‐CMs are suitable for use as models of cardiac sodium channel disease. In 2014, Cerrone et al showed that missense mutations in plakophilin‐2 (PKP2) could cause sodium current deficits, including decreased INa and Nav1.5 at the site of cell contact, and the deficit could be restored by transfection of wild‐type PKP2, which indicated that PKP2 mutations could be a molecular substrate of BrS. 78 Later, Liang and colleagues conducted another study of the hiPSC‐CM model for BrS. 31 Two patients with BrS carrying 2 variants in the SCN5A gene were recruited, and patient‐specific hiPSC‐CMs showed a reduction in INa and maximal upstroke velocity (Vmax) of the action potential compared with healthy control hiPSC‐CMs. Increased triggered activity and abnormal Ca2+ handling were also observed in patient‐derived hiPSC‐CMs. Furthermore, genomic correction of the causative variant in hiPSC‐CMs from 1 patient with BrS by CRISPR/Cas9 (CRISPR associated protein 9)‐mediated genome editing technology showed a rescue of triggered activity and a reversion of abnormal Ca2+ transients.
Table 2.
Current Accomplishments of Using hiPSC‐CMs in BrS
| Mechanism study | Drug testing | |||
|---|---|---|---|---|
| Mutations | Variants | Major findings | Drugs | Major findings |
| SCN5A | p.S1812X 11 | Reduction of INa density and NaV1.5 expression, impaired localization of NaV1.5 and connexin 43 at the cell surface, reduced action potential upstroke velocity and conduction slowing in BrS‐CMs | Carbachol 21 | Increasing arrhythmia events and the beating frequency in BrS |
| p.R620H & p.R811H 31 | BrS‐CMs displayed reductions in INa density, reduced maximal upstroke velocity of action potential, increased burden of triggered activity, abnormal calcium transients and beating interval variation | |||
| p.A226V & p.R1629X 76 | BrS‐CMs displayed reductions in INa density, and reduced maximal upstroke velocity and amplitude of action potential | |||
| SCN1B | p.L210P & p.P213T 21 | BrS‐CMs displayed significantly reduced peak and late sodium channel current as well as reduced amplitude and upstroke velocity of action potentials | Ajmaline 11 , 21 , 77 | Reduced amplitude and Vmax of action potential; blocking effect on both depolarization and repolarization in hiPSC‐CMs of BrS and control |
| SCN10A | p.Arg1250Gln & p.Arg1268Gln 22 | BrS‐CMs displayed significantly reduced INa as well as reduced amplitude and upstroke velocity of action potentials | Milrinone and cilostazol 11 | Inhibited Ito and alleviated the arrhythmic activity in BrS |
| PKP2 | p.D26N 78 | Loss of PKP2 caused decreased INa and NaV1.5 at the site of cell contact | ||
| RRAD | p.R211H 79 | BrS‐CMs displayed reduced action potential upstroke velocity, prolonged action potentials and increased incidence of early after depolarizations, with decreased Na+ peak current amplitude and increased Na+ persistent current amplitude as well as abnormal distribution of actin and fewer focal adhesions | ||
BrS indicates Brugada syndrome; BrS‐CMs, cardiomyocytes form Brugada syndrome‐derived human‐induced pluripotent stem cell; hiPSC‐CMs, human‐induced pluripotent stem cell–derived cardiomyocytes; INa, sodium current; Ito, transient outward potassium current; PKP2, plakophilin‐2; and Vmax, maximal upstroke velocity.
In addition to SCN5A, other genes that encoded subunits of the sodium channel have also been reported to be associated with sodium channel dysfunction in BrS. For example, 3 genes (SCN1B, SCN2B, and SCN3B) that encoded the β subunits of sodium channels, which can modify the channel function, have been discovered to cause abnormal electrical features, as in BrS. Recently, hiPSC‐CM models of BrS with double variants in SCN1B (c.629T > C and c.637C > A) and SCN10A (c.3803G>A and c.3749G>A) were established by our research group. 21 , 22 Cardiomyocytes from induced pluripotent stem cell from patients with BrS showed a significantly reduced peak INa and late INa. The action potential amplitude and Vmax were also reduced, which was consistent with the reduced peak INa.
The loss of function of sodium channels can cause conduction defects, which are important phenotypic features in BrS. To date, many studies have displayed conduction slowing in several models with a reduction in INa. In a study of BrS using the Scn5a(1798insD/+) mouse model, the hearts of mice showed prolonged atrioventricular conduction 81 and lower expression of genes that encoded proteins for conduction in the right ventricular outflow tract during cardiac development. 82 Pigs with mutations in SCN5A showed similar results, and HEK‐293 cells transfected with the SCN5A gene mutation (p.1493delK) recapitulated the loss of function of sodium channels. 43 , 83 Several studies of the hiPSC‐CM model for BrS showed that mutations in genes associated with sodium channels could reduce Vmax and peak INa amplitude with prolonged action potentials. 11 , 79 However, the conduction‐slowing effect of mutated sodium channels has not been investigated in hiPSC‐CMs.
BrS‐associated genes were also found in calcium channels and potassium channels. The calcium channel is composed of the α su84 bunit and accessory proteins. Mutations in α1 (CACNA1C), β2 (CACNB2), and α2/δ (CACNA2D1) subunit genes of cardiac long‐lasting (L)‐type channels can cause shortened action potential duration (representing shorter QT intervals) in patients with BrS 84 , 85 . A previous study showed that Chinese hamster ovary K1 Chinese hamster ovary K1 cells transfected with mutations of these genes could cause a reduction in Calcium current. 86 Because mutations in calcium channels commonly occur in patients with overlapping phenotypes of BrS and short‐QT syndrome, and it is rare to see patients with BrS without a short QT interval, functional analysis of mutant L‐type calcium channels is limited. Gain‐of‐function mutations in genes encoding potassium channels have also been reported in a few BrS studies. Mutations in KCND3 and KCNE3 identified in patients with BrS increased transient outward potassium current and loss of the action potential dome. 87 , 88 Genes that encoded adenosine triphosphate‐sensitive potassium channels, including KCNJ8 and ABCC9, also showed gain‐of‐function mutations in patients with BrS. 89 , 90 However, these mutations have not been investigated in the hiPSC‐CM model. It is meaningful to explore the effect of these mutations on the electrophysiological properties of BrS with an hiPSC‐CM model in the future.
Use of hiPSC‐CMs for Drug Screening
To date, limited numbers of drugs, including quinidine, 91 disopyramide, 92 β‐adrenergic agonists, 93 , 94 phosphodiesterase inhibitors, 95 and bepridil, 96 have been studied for pharmacological therapy of BrS 97 . Some of them exhibited beneficial effects in preventing arrhythmic events in BrS by augmenting INa and ICa or blocking Ito. However, some drugs also showed ineffective effects in BrS studies, such as amiodarone, 98 β‐blockers, calcium channel blockers, 99 and some contraindicated drugs 92 , 98 , 100 (ajmaline, procainamide, flecainide, propafenone, and pilsicainide). All of the aforementioned animal and cellular models were used to explore the effects of those drugs on BrS* (Table 3). To date, with the development of hiPSC‐CM models, several research groups have already tested the effects of some drugs on BrS patient‐specific hiPSC‐CMs. Phosphodiesterase inhibitors (cilostazol and milrinone) are phosphodiesterase III inhibitors that can increase ICa and suppress Ito by increasing the heart rate. Previous studies showed that cilostazol had a beneficial effect on preventing ventricular fibrillation storms in some patients with BrS. 96 , 101 In recent studies, cilostazol and milrinone also reduced ST elevation and arrhythmogenesis in a canine model. 46 , 102 In the study by Li and colleagues, 11 hiPSC‐CMs from patients with BrS with the SCN5A mutation p. S1812X showed a reduction in INa and the expression of sodium channels. At the same time, a significant increase in Ito was detected, and cilostazol and milrinone dramatically inhibited Ito and alleviated arrhythmic activity, which indicated the therapeutic potential of phosphodiesterase inhibitors for patients with BrS.
Table 3.
Drug Research in BrS Experimental Models
| Model | Drugs | Major findings | Reference |
|---|---|---|---|
| Murine | Quinidine | Increased regional VERPs and increased corresponding APD(90)s | 35 |
| Fewer ventricular arrhythmias, but variable effects on ST segments and worsened conduction abnormalities | 36 | ||
| Flecainide | Increased regional VERPs and decreased corresponding APD(90)s | 35 | |
| Accentuated ventricular arrhythmias, ST elevation, and conduction disorders | 36 | ||
| Ajmaline | Prolonged QRS interval and conduction defects | 103 | |
| Canine | Wenxin Keli, quinidine | Suppressed P2R and pVT | 104 |
| Isoproterenol | Spontaneous VF following premature ventricular beats was induced by vagal nerve stimulation | 105 | |
| Milrinone, cilostazol | Restored the epicardial AP dome, reduced dispersion, and abolished phase 2 reentry–induced extrasystoles and ventricular tachycardia | 101 | |
| Acacetin | Reduced Ito density, AP notch, and J‐wave area and totally suppressed the electrocardiographic and arrhythmic manifestation | 106 | |
| Ajmaline | Accentuation of epicardial AP notch and ECG J waves resulting in characteristic BrS | 106 | |
| Increased maximal J‐wave area, AP notch area, and interval between the peak | 49 | ||
| Generating polymorphic VT when combined with verapamil | 107 | ||
| Verapamil, NS5806 | Accentuation of epicardial AP notch and ECG J waves resulting in characteristic BrS | 106 | |
| Increased maximal J‐wave area, AP notch area, and interval between the peak and the end of the T wave | 49 | ||
| Terfenadine | Loss of the epicardial AP dome and resulting ST‐segment elevation | 107 | |
| Flecainide, procainamide | Generating polymorphic VT when combined with verapamil | 107 | |
| Pilsicainide, pinacidil | Coved‐type ST elevation in the ECG and longer APD in the epicardium than in the endocardium | 108 | |
| Porcine | Ajmaline | Total conduction blocked | 109 |
| Rabbit | CyPPA | Significant J‐wave elevation, frequent spontaneous ventricular fibrillation, and conduction delay | 34 |
| Heterologous expression | Quinidine | Normalized the QT interval and prevented stimulation‐induced ventricular tachycardia | 86 |
| Lidocaine | More negative shift of steady‐state inactivation | 110 | |
| hiPSC‐CMs | Ajmaline | More reduced APA and Vmax than healthy control cells | 11, 21 |
| Blocking effect on both depolarization and repolarization in hiPSC‐CMs in BrS and control | 77 | ||
| Milrinone, cilostazol | Inhibited Ito and alleviated the arrhythmic activity | 11 |
AP indicates action potential; APD, action potential duration; APD(90)s, action potential duration at 90% repolarization; APA, action potential amplitude; BrS, Brugada syndrome; CyPPA, cyclohexyl‐[2‐(3,5‐dimethyl‐pyrazol‐1‐yl)‐6‐methyl‐pyrimidin‐4‐yl]‐amine; hiPSC‐CMs, human‐induced pluripotent stem cell–derived cardiomyocytes; Ito, transient outward potassium current; P2R, phase 2 reentry; pVT, polymorphic ventricular tachycardia; VERPs, ventricular effective refractory periods; VF, ventricular fibrillation; VT, ventricular tachycardia and Vmax, maximal upstroke velocity.
A recent study by Miller et al 77 showed that hiPSC‐CMs from patients with BrS with no identified pathogenic mutations and control hiPSC‐CMs had similar action potential parameters. Although ajmaline generated blocking effects on both depolarization and repolarization in hiPSC‐CMs, there was no difference between BrS and control cells. Furthermore, hiPSC‐CMs from patients with BrS with variants in the SCN1B and SCN10A proteins, which are subunits of the sodium channels, showed reduced INa, action potential amplitude and Vmax, and disease cells displayed a greater reduction in action potential amplitude and Vmax than healthy control cells after the application of ajmaline. 21 , 22
In the study by Kosmidis and colleagues, 111 hiPSC‐CMs were used to investigate the effect of several compounds, such as gentamicin and PTC124, which could increase the translational readthrough of premature stop codons in patients who carry loss‐of‐function SCN5A gene mutations. Although the hiPSC‐CMs derived from patients who carry the SCN5A mutations R1638X and W156X displayed reduced INa and action potential upstroke velocities, gentamicin and PTC124 showed no significant influence on reversing the effect of either mutation. This study not only used BrS patient‐specific hiPSC‐CMs as an experimental model but also used them to investigate novel therapeutic tools for specific mutations associated with BrS.
Latest Advances in Applications of hiPSC‐CMs
Currently, an increasing number of investigators have applied CRISPR/Cas9‐mediated genome editing technology combined with hiPSC techniques to target specific genes that cause disease to study pathophysiology, conduct drug screening, and improve cell therapeutic potential. 112
Gene editing technology is used as a tool that can modify the genome, including correcting, writing, or removing genetic information in specific DNA sequences. The CRISPR/Cas9 system, acting as the latest tool for gene editing, has shown unprecedented ease and precision compared with previous technologies. It is a system of adaptive immunity to viruses and plasmids in bacteria and archaea. Three types of CRISPR/Cas9 exist, whereas type 2 is the best known in genome editing. 113 The CRISPR/Cas9 system can target many genomic sequences through gene knock‐out or knock‐in, gene interference or activation, and other applications related to chromosomes with unchanged genetic backgrounds except for the editing part. The combination of CRISPR/Cas9 technology and hiPSC‐CMs has been widely used in recent years. It can explore the effect of variants with unknown significance mutations or variants in cardiac channelopathies by inserting the variant into wild‐type cell lines or correcting the variant in patient‐specific cell lines. 114 Furthermore, these advanced technologies have also contributed to drug screening and repurposing, which showed potential effects for precision medicine. 115 In most gene‐editing studies of BrS, the CRISPR/Cas9 system was used to investigate the effect of a mutation on the electrophysiological properties of disease after genomic correction in hiPSC‐CMs. 31 , 79 There is no doubt that the CRISPR/Cas9 system together with hiPSC‐CMs will be widely used in studies of BrS for different purposes in the future, including studying specific gene functions, identifying new associated gene mutations and drug screening.
Although most of the hiPSC‐CM models reported to date of human arrhythmias have focused on single‐cell phenotypes, studying arrhythmias at the 2‐dimensional cell culture or tissue level would be a better way to understand the mechanism of disease. In recent studies, hiPSC‐CMs were cultured by the 2‐dimensional method and provided reliable drug risk stratification in drug screening applications. Furthermore, the inclusion of conduction velocity studies improved the ability of prediction models to test proarrhythmia risk. 116 The 2‐dimensional culture of hiPSC‐CMs was also applied in another study, which indicated that mutation of MYPBC3 played a critical role in inherited hypertrophic cardiomyopathy. 117
To date, tissue engineering systems have been developed to enhance electrical coupling, promote physiological maturation, and make it possible to establish cardiac tissues with hiPSC‐CMs together with multiple cell types. 118 The main purpose of tissue engineering is to create an environment that is closely similar to that in vivo. In 1997, a type of 3D model heart tissue was generated that could directly measure isometric contractile force, and collagen played an important role in hydrogels for 3D cardiomyocyte culture. 119 In a study by Zimmermann and colleagues, 120 3D engineered heart tissue from rat cardiac myocytes in vitro improved tissue formation and inhibited dedifferentiation and overgrowth of noncardiomyocytes. These results indicated that engineered heart tissue retained many physiological characteristics of rat cardiac tissue and allowed efficient gene transfer with subsequent force measurement. This technology could also be used for drug discovery. Although previous single‐cell studies are useful for initial screening and toxicity testing, they could not organize differentiated cells into the organ‐like structures that are necessary for high‐content analysis of candidate drugs. The combination of 3D (engineered heart tissue) and 2D models using hiPSC‐CMs has significant potential for advancing preclinical drug screening and has also promoted the development of cardiovascular tissue engineering in the field of precision medicine. A particular microphysiological system using hiPSC‐CMs has been established to test the effect of pharmacological agents, including isoproterenol (β‐adrenergic agonist), E‐4031 (human ether‐a‐go‐go‐related gene blocker), verapamil (multi‐ion channel blocker), and metoprolol (β‐adrenergic antagonist). The beat rate and mechanical motion were used as metrics of cardiac tissue function. 121 Advanced technologies combined with hiPSC‐CMs could be used in BrS studies to further investigate the molecular and cellular mechanisms that underlie the disease, develop new potential target drugs, and test drug toxicity.
Limitations of hiPSC‐CMs
Although hiPSC‐CMs have great strengths in current BrS studies compared with other systems, there are several limitations for research. First, the main limitation of hiPSC‐CMs is the immature phenotype. This property leads to different expression profiles in ion channels and sarcomeric proteins, such as less organized sarcomeric structures and calcium‐handling machinery. 122 However, multiple efforts to overcome this limitation have been used recently, 123 , 124 , 125 , 126 , 127 , 128 , 129 , 130 , 131 , 132 , 133 , 134 , 135 , 136 , 137 , 138 , 139 , 140 , 141 including biochemical stimulation, electrical stimulation, mechanical stimulation, surface topography, substrate stiffness modification, 3D culture, long‐term culture of hiPSC‐CMs, and extracellular matrix culture (Table 4). All of these methods have been reported to improve hiPSC‐CM maturation.
Table 4.
Maturation Methods for hiPSC‐CMs
| Methods | Major findings | Reference | |
|---|---|---|---|
| Biochemical stimulation | Tri‐iodo‐l‐thyronine treatment | Increased cardiomyocyte size, anisotropy, and sarcomere length | 123 |
| Fatty acid treatment | Higher myofibril density and alignment, enhanced contractility and improved calcium handling | 125 | |
| Centrosome reduction | Increased postmitotic transitions and aspects of cardiomyocyte maturation | 126 | |
| Polyinosinic‐polycytidylic acid | Increased cell size, greater contractility, faster electrical upstrokes, increased oxidative metabolism, and more mature sarcomeric structure and composition | 127 | |
| Electrical stimulation | Biowire | Enhanced degree of structural and electrophysiological maturation | 128 |
| Silicon nanowire | Enhanced contractility and expression of contractile protein α‐SA and cTnI | 129 | |
| Chronic electrical stimulation | Increased connexin‐43 abundance and sarcomere ultrastructure | 130 | |
| Mechanical stimulation | Static stress | Increased contractility, construct alignment, cell size, and expression of RYR2 and SERCA2 | 131 |
| Uniaxial stress | Enhanced alignment cells/matrix fiber, myofibril genesis, and sarcomeric banding | 132 | |
| Cyclic stress | Increased expression of β‐myosin heavy chain and cardiac troponin T, and the tissue showed enhanced calcium dynamics and force production | 133 | |
| Surface topography | Bioinspired onion epithelium‐like structure | High level of genes relating to sarcomere proteins, ion channels, and calcium handling proteins | 134 |
| Substrate stiffness | Softest fibronectin‐coated PDMS surface | Increased expression of key mature sarcolemmal (SCN5A, Kir2.1, and connexin43) and myofilament markers (cTnI) | 135 |
| Undiluted Matrigel surface | More rod‐shape morphology, increased sarcomere length, cTnI and Vmax | 136 | |
| Carbon nanotube and pericardial matrix | Improved contraction amplitude, cellular alignment, connexin 43 expression and sarcomere organization | 137 | |
| 3D culture | 3D CMTs with biochemical factors | Higher fidelity of adult cardiac phenotype, including sarcoplasmic reticulum function and contractile properties | 138 |
| Scaffold‐based tissue engineering with electrical stimulation | Enhanced contractility and expression of contractile protein α‐SA and cTnI | 129 | |
| EHT with electrical stimulation | Increased connexin‐43 abundance and sarcomere ultrastructure | 130 | |
| hiPSC‐CMs spheroids with cyclic, uniaxial stretch in PDMS channels | Enhanced protein expressions of cTnI, MLC2v, and MLC2a, along with improved ultrastructure, fibril alignment, and fiber number | 139 | |
| Microfluidic platform with surface topography | Increased sarcomeric striations, highly synchronous contractions, and upregulation of several maturation genes | 140 | |
| Long‐term culture of hiPSC‐CMs | 200 d | Upregulation of cellular metabolism and increased cell contractility | 141 |
| ECM culture | Human perinatal stem cell derived ECM | More rod‐shape morphology, highly organized sarcomeres, elevated cTnI expression, mitochondrial and electrophysiological function | 142 |
3D indicates 3‐dimensional; α‐SA, α‐sarcomeric actinin; CMTs, cardiac microtissues; cTnI, cardiac troponin I; ECM, extracellular matrix; EHT, engineered heart tissue; hiPSC‐CMs, human‐induced pluripotent stem cell–derived cardiomyocytes; Kir2.1, the dominant subunit of I(K1) channel in ventricle; MLC2a, myosin light chain 2a; MLC2v, myosin regulatory light chain; PDMS, polydimethylsiloxane; RYR2, ryanodine receptor 2; SCN5A, sodium voltage‐gated channel alpha subunit 5; SERCA2, Sarco/Endoplasmic Reticulum Calcium ATPase 2 and Vmax, maximal upstroke velocity.
Second, differentiated hiPSC‐CMs can be composed of a mixture of ventricular‐, atrial‐, and nodal‐like cells, each of which showing different expression profiles of cardiac proteins and electrophysiological properties. 142 Fortunately, many studies have shown effective methods to direct hiPSC‐CMs into subtype‐specific cardiomyocytes.
Third, the human heart consists of various cell types, including smooth muscle cells, endothelial cells, leukocytes, and fibroblasts. Cardiomyocytes account for only ≈30% of the heart cell numbers. Therefore, a single‐cell model of hiPSC‐CMs cannot fully reflect the physiological and pathological conditions of the heart. The 3D tissue engineering platforms established in recent years have improved this situation. Tissue engineering systems can also promote the maturation of hiPSC‐CMs and make it possible for investigators to conduct studies of hiPSC‐CMs from patients with BrS at a complex organ level.
Future Perspectives
In recent years, hiPSC‐CMs have been widely applied in disease models, drug screening, studying pathological mechanisms, and developing novel treatments. With the development of precision medicine, hiPSCs have received increasing attention from researchers in this field. The main purpose of precision medicine is to provide an individual patient or a group of patients with more effective treatment or preventive actions. 75 hiPSCs with individual patient genetic profiles are ideal tools for precision medicine. They can differentiate into specific cell types and be used in related studies to better understand the underlying mechanisms of disease, improve risk stratification, and develop a more personalized treatment. To date, cardiomyocytes derived from hiPSCs have been used to investigate the pathogenicity of gene variants and drug screening in BrS studies, which has made large contributions to mechanistic studies and personalized medicine. With the identification of genetic variants, it is possible to more specifically identify genotype–phenotype links of BrS to generate more precise risk stratification and to fulfill more exact clinical management for patients with BrS. The results of drug screening could also help to evaluate the response of specific genetic variants to specific pharmacological treatments. Furthermore, in combination with novel technologies, such as genome editing technology and tissue engineering systems, hiPSC‐CMs would play a more important role in the study of BrS in the future.
Conclusions
BrS is a heterogeneous disease, and its clinical course cannot be predicted. Numerous studies have been conducted to explore its underlying mechanism, but the exact pathophysiological mechanism of BrS remains disputed. Different experimental models possess different advantages and disadvantages, leading to different relevance for BrS study. Animal models can model arrhythmias and some ECG changes but are different from humans in gene background. Heterologous expression in cells is ideal for examining the effects of mutations or variants on ion channel functions but cannot simulate action potential changes and arrhythmias. HiPSC‐CMs can overcome the shortcomings of animal and heterologous expression models but cannot model the ECG phenotype of BrS. Nevertheless, HiPSCs possess their own advantages in BrS studies, especially in the identification of pathogenicity of gene variants and drug screening.
The hiPSC‐CM model derived from human somatic cells shows unique advantages, which makes it possible for investigators to conduct studies of BrS in cell models with desired genetic profiles. The immaturity of hiPSC‐CMs is the main limitation of this model. Fortunately, different efforts have been made to solve this problem, and most of them have been shown to be effective in promoting the maturation of hiPSC‐CMs. Furthermore, several advanced technologies, including CRISPR/Cas9‐mediated genome editing and tissue engineering technology, have been combined with hiPSC‐CMs, which can improve mechanistic and therapeutic studies on BrS.
From the available research data in the field of iPSC studies, we believe that studies using patient‐specific hiPSC‐CMs with unique advantages will provide novel insights into the mechanisms and pathophysiology of the disease and could offer opportunities to improve the diagnosis and treatment of patients with BrS.
Sources of Funding
The work was supported by the Hector‐Foundation (MED1814).
Disclosures
None.
For Sources of Funding and Disclosures, see page 13.
Footnotes
References
- 1. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391–1396. doi: 10.1016/0735-1097(92)90253-J [DOI] [PubMed] [Google Scholar]
- 2. Brugada J, Campuzano O, Arbelo E, Sarquella‐Brugada G, Brugada R. Present status of Brugada syndrome: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2018;72:1046–1059. doi: 10.1016/j.jacc.2018.06.037 [DOI] [PubMed] [Google Scholar]
- 3. Roterberg G, El‐Battrawy I, Veith M, Liebe V, Ansari U, Lang S, Zhou X, Akin I, Borggrefe M. Arrhythmic events in Brugada syndrome patients induced by fever. Ann Noninvasive Electrocardiol. 2020;25:e12723. doi: 10.1111/anec.12723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bayés de Luna A, Brugada J, Baranchuk A, Borggrefe M, Breithardt G, Goldwasser D, Lambiase P, Riera AP, Garcia‐Niebla J, Pastore C, et al. Current electrocardiographic criteria for diagnosis of Brugada pattern: a consensus report. J Electrocardiol. 2012;45:433–442. doi: 10.1016/j.jelectrocard.2012.06.004 [DOI] [PubMed] [Google Scholar]
- 5. Priori SG, Blomstrom‐Lundqvist C. 2015 European Society of Cardiology guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death summarized by co‐chairs. Eur Heart J. 2015;36:2757–2759. doi: 10.1093/eurheartj/ehv445 [DOI] [PubMed] [Google Scholar]
- 6. Quan XQ, Li S, Liu R, Zheng K, Wu XF, Tang Q. A meta‐analytic review of prevalence for Brugada ECG patterns and the risk for death. Medicine (Baltimore). 2016;95:e5643. doi: 10.1097/MD.0000000000005643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris‐Kerr C, et al. An international compendium of mutations in the SCN5A‐encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7:33–46. doi: 10.1016/j.hrthm.2009.09.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Campuzano O, Sarquella‐Brugada G, Cesar S, Arbelo E, Brugada J, Brugada R. Update on genetic basis of Brugada syndrome: monogenic, polygenic or oligogenic? Int J Mol Sci. 2020;21:7155. doi: 10.3390/ijms21197155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hosseini SM, Kim R, Udupa S, Costain G, Jobling R, Liston E, Jamal SM, Szybowska M, Morel CF, Bowdin S, et al. Reappraisal of reported genes for sudden arrhythmic death: evidence‐based evaluation of gene validity for Brugada syndrome. Circulation. 2018;138:1195–1205. doi: 10.1161/CIRCULATIONAHA.118.035070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Monasky MM, Micaglio E, Ciconte G, Borrelli V, Giannelli L, Vicedomini G, Ghiroldi A, Anastasia L, Locati ET, Benedetti S, et al. Novel SCN5A p. V1429M variant segregation in a family with Brugada syndrome. Int J Mol Sci. 2020;21:5902. doi: 10.3390/ijms21165902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li W, Stauske M, Luo X, Wagner S, Vollrath M, Mehnert CS, Schubert M, Cyganek L, Chen S, Hasheminasab S‐M, et al. Disease phenotypes and mechanisms of iPSC‐derived cardiomyocytes from Brugada syndrome patients with a loss‐of‐function SCN5A mutation. Front Cell Dev Biol. 2020;8:592893. doi: 10.3389/fcell.2020.592893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wijeyeratne YD, Tanck MW, Mizusawa Y, Batchvarov V, Barc J, Crotti L, Bos JM, Tester DJ, Muir A, Veltmann C, et al. SCN5A mutation type and a genetic risk score associate variably With Brugada syndrome phenotype in SCN5A families. Circ Genom Precis Med. 2020;13:e002911. doi: 10.1161/CIRCGEN.120.002911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Campuzano O, Sarquella‐Brugada G, Fernandez‐Falgueras A, Cesar S, Coll M, Mates J, Arbelo E, Perez‐Serra A, del Olmo B, Jordá P, et al. Genetic interpretation and clinical translation of minor genes related to Brugada syndrome. Hum Mutat. 2019;40:749–764. doi: 10.1002/humu.23730 [DOI] [PubMed] [Google Scholar]
- 14. Chen C‐Y, Lu T‐P, Lin L‐Y, Liu Y‐B, Ho L‐T, Huang H‐C, Lai L‐P, Hwang J‐J, Yeh S‐F, Wu C‐K, et al. Impact of ancestral differences and reassessment of the classification of previously reported pathogenic variants in patients with Brugada syndrome in the genomic era: a SADS‐TW BrS Registry. Front Genet. 2018;9:680. doi: 10.3389/fgene.2018.00680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST‐segment elevation. Circulation. 1999;100:1660–1666. doi: 10.1161/01.CIR.100.15.1660 [DOI] [PubMed] [Google Scholar]
- 16. Meregalli PG, Wilde AA, Tan HL. Pathophysiological mechanisms of Brugada syndrome: depolarization disorder, repolarization disorder, or more? Cardiovasc Res. 2005;67:367–378. doi: 10.1016/j.cardiores.2005.03.005 [DOI] [PubMed] [Google Scholar]
- 17. Schmidt C, Wiedmann F, El‐Battrawy I, Fritz M, Ratte A, Beller CJ, Lang S, Rudic B, Schimpf R, Akin I, et al. Reduced Na(+) current in native cardiomyocytes of a Brugada syndrome patient associated with beta‐2‐syntrophin mutation. Circ Genom Precis Med. 2018;11:e002263. doi: 10.1161/CIRCGEN.118.002263 [DOI] [PubMed] [Google Scholar]
- 18. Calvo D, Atienza F, Saiz J, Martínez L, Ávila P, Rubín J, Herreros B, Arenal Á, García‐Fernández J, Ferrer A, et al. Ventricular tachycardia and early fibrillation in patients with Brugada syndrome and ischemic cardiomyopathy show predictable frequency‐phase properties on the precordial ECG consistent with the respective arrhythmogenic substrate. Circ Arrhythm Electrophysiol. 2015;8:1133–1143. doi: 10.1161/CIRCEP.114.002717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishikawa T, Kimoto H, Mishima H, Yamagata K, Ogata S, Aizawa Y, Hayashi K, Morita H, Nakajima T, Nakano Y, et al. Functionally validated SCN5A variants allow interpretation of pathogenicity and prediction of lethal events in Brugada syndrome. Eur Heart J. 2021;42:2854–2863. doi: 10.1093/eurheartj/ehab254 [DOI] [PubMed] [Google Scholar]
- 20. Ross SB, Fraser ST, Nowak N, Semsarian C. Generation of induced pluripotent stem cells (iPSCs) from a hypertrophic cardiomyopathy patient with the pathogenic variant p.Val698Ala in beta‐myosin heavy chain (MYH7) gene. Stem Cell Res. 2017;20:88–90. doi: 10.1016/j.scr.2017.02.015 [DOI] [PubMed] [Google Scholar]
- 21. El‐Battrawy I, Müller J, Zhao Z, Cyganek L, Zhong R, Zhang F, Kleinsorge M, Lan H, Li X, Xu Q, et al. Studying Brugada syndrome with an SCN1B variants in human‐induced pluripotent stem cell‐derived cardiomyocytes. Front Cell Dev Biol. 2019;7:261. doi: 10.3389/fcell.2019.00261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. El‐Battrawy I, Albers S, Cyganek L, Zhao Z, Lan H, Li X, Xu Q, Kleinsorge M, Huang M, Liao Z, et al. A cellular model of Brugada syndrome with SCN10A variants using human‐induced pluripotent stem cell‐derived cardiomyocytes. Europace. 2019;21:1410–1421. doi: 10.1093/europace/euz122 [DOI] [PubMed] [Google Scholar]
- 23. Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott‐Flügel L, Dorn T, Goedel A, Höhnke C, Hofmann F, et al. Patient‐specific induced pluripotent stem‐cell models for long‐QT syndrome. N Engl J Med. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679 [DOI] [PubMed] [Google Scholar]
- 24. Ma D, Wei H, Lu J, Ho S, Zhang G, Sun X, Oh Y, Tan SH, Ng ML, Shim W, et al. Generation of patient‐specific induced pluripotent stem cell‐derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:1122–1133. doi: 10.1093/eurheartj/ehs226 [DOI] [PubMed] [Google Scholar]
- 25. Zhang XH, Haviland S, Wei H, Saric T, Fatima A, Hescheler J, Cleemann L, Morad M. Ca2+ signaling in human induced pluripotent stem cell‐derived cardiomyocytes (iPS‐CM) from normal and catecholaminergic polymorphic ventricular tachycardia (CPVT)‐afflicted subjects. Cell Calcium. 2013;54:57–70. doi: 10.1016/j.ceca.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, Dolmetsch RE. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. El‐Battrawy I, Lan H, Cyganek L, Zhao Z, Li X, Buljubasic F, Lang S, Yücel G, Sattler K, Zimmermann W‐H, et al. Modeling short QT syndrome using human‐induced pluripotent stem cell‐derived cardiomyocytes. J Am Heart Assoc. 2018;7:e007394. doi: 10.1161/JAHA.117.007394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. El‐Battrawy I, Zhao Z, Lan H, Li X, Yücel G, Lang S, Sattler K, Schünemann J‐D, Zimmermann W‐H, Cyganek L, et al. Ion channel dysfunctions in dilated cardiomyopathy in limb‐girdle muscular dystrophy. Circ Genom Precis Med. 2018;11:e001893. doi: 10.1161/CIRCGEN.117.001893 [DOI] [PubMed] [Google Scholar]
- 29. Hanses U, Kleinsorge M, Roos L, Yigit G, Li Y, Barbarics B, El‐Battrawy I, Lan H, Tiburcy M, Hindmarsh R, et al. Intronic CRISPR repair in a preclinical model of Noonan syndrome‐associated cardiomyopathy. Circulation. 2020;142:1059–1076. doi: 10.1161/CIRCULATIONAHA.119.044794 [DOI] [PubMed] [Google Scholar]
- 30. Burridge PW, Keller G, Gold JD, Wu JC. Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell. 2012;10:16–28. doi: 10.1016/j.stem.2011.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liang P, Sallam K, Wu H, Li Y, Itzhaki I, Garg P, Zhang Y, Termglichan V, Lan F, Gu M, et al. Patient‐specific and genome‐edited induced pluripotent stem cell‐derived cardiomyocytes elucidate single‐cell phenotype of Brugada syndrome. J Am Coll Cardiol. 2016;68:2086–2096. doi: 10.1016/j.jacc.2016.07.779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao Z, Lan H, El‐Battrawy I, Li X, Buljubasic F, Sattler K, Yücel G, Lang S, Tiburcy M, Zimmermann W‐H, et al. Ion channel expression and characterization in human induced pluripotent stem cell‐derived cardiomyocytes. Stem Cells Int. 2018;2018:6067096. doi: 10.1155/2018/6067096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sendfeld F, Selga E, Scornik FS, Perez GJ, Mills NL, Brugada R. Experimental models of Brugada syndrome. Int J Mol Sci. 2019;20:2123. doi: 10.3390/ijms20092123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen MU, Xu D‐Z, Wu AZ, Guo S, Wan J, Yin D, Lin S‐F, Chen Z, Rubart‐von der Lohe M, Everett TH, et al. Concomitant SK current activation and sodium current inhibition cause J wave syndrome. JCI Insight. 2018;3:e122329. doi: 10.1172/jci.insight.122329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Papadatos GA, Wallerstein PMR, Head CEG, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AEO, Huang C L‐H, Vandenberg JI, et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA. 2002;99:6210–6215. doi: 10.1073/pnas.082121299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Remme CA, Verkerk AO, Nuyens D, van Ginneken AC, van Brunschot S, Belterman CN, van Roon MA, Tan HL, Wilde AAM, Carmeliet P, et al. Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A‐1795insD. Circulation. 2006;114:2584–2594. doi: 10.1161/CIRCULATIONAHA.106.653949 [DOI] [PubMed] [Google Scholar]
- 37. Martin CA, Grace AA, Huang CL. Refractory dispersion promotes conduction disturbance and arrhythmias in a Scn5a (+/‐) mouse model. Pflugers Arch. 2011;462:495–504. doi: 10.1007/s00424-011-0989-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Martin CA, Zhang Y, Grace AA, Huang CL. In vivo studies of Scn5a+/‐ mice modeling Brugada syndrome demonstrate both conduction and repolarization abnormalities. J Electrocardiol. 2010;43:433–439. doi: 10.1016/j.jelectrocard.2010.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martin CA, Siedlecka U, Kemmerich K, Lawrence J, Cartledge J, Guzadhur L, Brice N, Grace AA, Schwiening C, Terracciano CM, et al. Reduced Na(+) and higher K(+) channel expression and function contribute to right ventricular origin of arrhythmias in Scn5a+/‐ mice. Open Biol. 2012;2:120072. doi: 10.1098/rsob.120072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y, Guzadhur L, Jeevaratnam K, Salvage SC, Matthews GD, Lammers WJ, Lei M, Huang CL, Fraser JA. Arrhythmic substrate, slowed propagation and increased dispersion in conduction direction in the right ventricular outflow tract of murine Scn5a+/‐ hearts. Acta Physiol (Oxf). 2014;211:559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Davis RP, Casini S, van den Berg CW, Hoekstra M, Remme CA, Dambrot C, Salvatori D, Oostwaard D‐V, Wilde AAM, Bezzina CR, et al. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation. 2012;125:3079–3091. doi: 10.1161/CIRCULATIONAHA.111.066092 [DOI] [PubMed] [Google Scholar]
- 42. Stein M, van Veen TA, Remme CA, Boulaksil M, Noorman M, van Stuijvenberg L, van der Nagel R, Bezzina CR, Hauer RN, de Bakker JM, et al. Combined reduction of intercellular coupling and membrane excitability differentially affects transverse and longitudinal cardiac conduction. Cardiovasc Res. 2009;83:52–60. doi: 10.1093/cvr/cvp124 [DOI] [PubMed] [Google Scholar]
- 43. Park DS, Cerrone M, Morley G, Vasquez C, Fowler S, Liu N, Bernstein SA, Liu F‐Y, Zhang J, Rogers CS, et al. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J Clin Invest. 2015;125:403–412. doi: 10.1172/JCI76919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93:372–379. doi: 10.1161/01.CIR.93.2.372 [DOI] [PubMed] [Google Scholar]
- 45. Kimura M, Kobayashi T, Owada S, Ashikaga K, Higuma T, Sasaki S, Iwasa A, Motomura S, Okumura K. Mechanism of ST elevation and ventricular arrhythmias in an experimental Brugada syndrome model. Circulation. 2004;109:125–131. doi: 10.1161/01.CIR.0000105762.94855.46 [DOI] [PubMed] [Google Scholar]
- 46. Szel T, Koncz I, Antzelevitch C. Cellular mechanisms underlying the effects of milrinone and cilostazol to suppress arrhythmogenesis associated with Brugada syndrome. Heart Rhythm. 2013;10:1720–1727. doi: 10.1016/j.hrthm.2013.07.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Take Y, Morita H, Wu J, Nagase S, Morita S, Toh N, Nishii N, Nakamura K, Kusano KF, Ohe T, et al. Spontaneous electrocardiogram alterations predict ventricular fibrillation in Brugada syndrome. Heart Rhythm. 2011;8:1014–1021. doi: 10.1016/j.hrthm.2011.02.009 [DOI] [PubMed] [Google Scholar]
- 48. Morita H, Zipes DP, Fukushima‐Kusano K, Nagase S, Nakamura K, Morita ST, Ohe T, Wu J. Repolarization heterogeneity in the right ventricular outflow tract: correlation with ventricular arrhythmias in Brugada patients and in an in vitro canine Brugada model. Heart Rhythm. 2008;5:725–733. doi: 10.1016/j.hrthm.2008.02.028 [DOI] [PubMed] [Google Scholar]
- 49. Patocskai B, Yoon N, Antzelevitch C. Mechanisms underlying epicardial radiofrequency ablation to suppress arrhythmogenesis in experimental models of Brugada syndrome. JACC Clin Electrophysiol. 2017;3:353–363. doi: 10.1016/j.jacep.2016.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Casini S, Tan HL, Bhuiyan ZA, Bezzina CR, Barnett P, Cerbai E, Mugelli M, Wilde AAM, Veldkamp MW. Characterization of a novel SCN5A mutation associated with Brugada syndrome reveals involvement of DIIIS4‐S5 linker in slow inactivation. Cardiovasc Res. 2007;76:418–429. doi: 10.1016/j.cardiores.2007.08.005 [DOI] [PubMed] [Google Scholar]
- 51. Wang L, Han Z, Dai J, Cao K. Brugada syndrome caused by sodium channel dysfunction associated with a SCN1B variant A197V. Arch Med Res. 2020;51:245–253. doi: 10.1016/j.arcmed.2020.02.003 [DOI] [PubMed] [Google Scholar]
- 52. Ortiz‐Bonnin B, Rinné S, Moss R, Streit AK, Scharf M, Richter K, Stöber A, Pfeufer A, Seemann G, Kääb S, et al. Electrophysiological characterization of a large set of novel variants in the SCN5A‐gene: identification of novel LQTS3 and BrS mutations. Pflugers Arch. 2016;468:1375–1387. doi: 10.1007/s00424-016-1844-3 [DOI] [PubMed] [Google Scholar]
- 53. Watanabe H, Yang T, Stroud DM, Lowe JS, Harris L, Atack TC, Wang DW, Hipkens SB, Leake B, Hall L, et al. Striking In vivo phenotype of a disease‐associated human SCN5A mutation producing minimal changes in vitro. Circulation. 2011;124:1001–1011. doi: 10.1161/CIRCULATIONAHA.110.987248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, Brugada J, Brugada R, Antzelevitch C. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–809. doi: 10.1161/01.RES.85.9.803 [DOI] [PubMed] [Google Scholar]
- 55. Baroudi G, Carbonneau E, Pouliot V, Chahine M. SCN5A mutation (T1620M) causing Brugada syndrome exhibits different phenotypes when expressed in Xenopus oocytes and mammalian cells. FEBS Lett. 2000;467:12–16. [DOI] [PubMed] [Google Scholar]
- 56. Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0 [DOI] [PubMed] [Google Scholar]
- 57. Thomson JA, Itskovitz‐Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145 [DOI] [PubMed] [Google Scholar]
- 58. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 59. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- 60. Wang Y, Liang P, Lan F, Wu H, Lisowski L, Gu M, Hu S, Kay MA, Urnov FD, Shinnawi R, et al. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J Am Coll Cardiol. 2014;64:451–459. doi: 10.1016/j.jacc.2014.04.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Evans SM, Yelon D, Conlon FL, Kirby ML. Myocardial lineage development. Circ Res. 2010;107:1428–1444. doi: 10.1161/CIRCRESAHA.110.227405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. doi: 10.1126/science.1132292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Noseda M, Peterkin T, Simoes FC, Patient R, Schneider MD. Cardiopoietic factors: extracellular signals for cardiac lineage commitment. Circ Res. 2011;108:129–152. doi: 10.1161/CIRCRESAHA.110.223792 [DOI] [PubMed] [Google Scholar]
- 64. Kehat I, Kenyagin‐Karsenti D, Snir M, Segev H, Amit M, Gepstein A, Livne E, Binah O, Itskovitz‐Eldor J, Gepstein L. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407–414. doi: 10.1172/JCI200112131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Uosaki H, Fukushima H, Takeuchi A, Matsuoka S, Nakatsuji N, Yamanaka S, Yamashita JK. Efficient and scalable purification of cardiomyocytes from human embryonic and induced pluripotent stem cells by VCAM1 surface expression. PLoS One. 2011;6:e23657. doi: 10.1371/journal.pone.0023657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ban K, Wile B, Kim S, Park H‐J, Byun J, Cho K‐W, Saafir T, Song M‐K, Yu SP, Wagner M, et al. Purification of cardiomyocytes from differentiating pluripotent stem cells using molecular beacons that target cardiomyocyte‐specific mRNA. Circulation. 2013;128:1897–1909. doi: 10.1161/CIRCULATIONAHA.113.004228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, et al. Distinct metabolic flow enables large‐scale purification of mouse and human pluripotent stem cell‐derived cardiomyocytes. Cell Stem Cell. 2013;12:127–137. doi: 10.1016/j.stem.2012.09.013 [DOI] [PubMed] [Google Scholar]
- 68. Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell‐derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. 2015;117:80–88. doi: 10.1161/CIRCRESAHA.117.305365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cyganek L, Tiburcy M, Sekeres K, Gerstenberg K, Bohnenberger H, Lenz C, Henze S, Stauske M, Salinas G, Zimmermann W‐H, et al. Deep phenotyping of human induced pluripotent stem cell‐derived atrial and ventricular cardiomyocytes. JCI Insight. 2018;3:e99941. doi: 10.1172/jci.insight.99941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tiburcy M, Hudson JE, Balfanz P, Schlick S, Meyer T, Chang Liao M‐L, Levent E, Raad F, Zeidler S, Wingender E, et al. Defined engineered human myocardium with advanced maturation for applications in heart failure modeling and repair. Circulation. 2017;135:1832–1847. doi: 10.1161/CIRCULATIONAHA.116.024145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Devalla HD, Schwach V, Ford JW, Milnes JT, El‐Haou S, Jackson C, Gkatzis K, Elliott DA, Chuva de Sousa Lopes SM, Mummery CL, et al. Atrial‐like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial‐selective pharmacology. EMBO Mol Med. 2015;7:394–410. doi: 10.15252/emmm.201404757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Karakikes I, Senyei GD, Hansen J, Kong C‐W, Azeloglu EU, Stillitano F, Lieu DK, Wang J, Ren L, Hulot J‐S, et al. Small molecule‐mediated directed differentiation of human embryonic stem cells toward ventricular cardiomyocytes. Stem Cells Transl Med. 2014;3:18–31. doi: 10.5966/sctm.2013-0110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu F, Fang Y, Hou X, Yan Y, Xiao H, Zuo D, Wen J, Wang L, Zhou Z, Dang X, et al. Enrichment differentiation of human induced pluripotent stem cells into sinoatrial node‐like cells by combined modulation of BMP, FGF, and RA signaling pathways. Stem Cell Res Ther. 2020;11:284. doi: 10.1186/s13287-020-01794-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kleinsorge M, Cyganek L. Subtype‐directed differentiation of human iPSCs into atrial and ventricular cardiomyocytes. STAR Protoc. 2020;1:100026. doi: 10.1016/j.xpro.2020.100026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–795. doi: 10.1056/NEJMp1500523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ma D, Liu Z, Loh LJ, Zhao Y, Li G, Liew R, Islam O, Wu J, Chung YY, Teo WS, et al. Identification of an INa‐dependent and Ito‐mediated proarrhythmic mechanism in cardiomyocytes derived from pluripotent stem cells of a Brugada syndrome patient. Sci Rep. 2018;8:11246. doi: 10.1038/s41598-018-29574-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Miller DC, Harmer SC, Poliandri A, Nobles M, Edwards EC, Ware JS, Sharp TV, McKay TR, Dunkel L, Lambiase PD, et al. Ajmaline blocks INa and IKr without eliciting differences between Brugada syndrome patient and control human pluripotent stem cell‐derived cardiac clusters. Stem Cell Res. 2017;25:233–244. doi: 10.1016/j.scr.2017.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cerrone M, Lin X, Zhang M, Agullo‐Pascual E, Pfenniger A, Chkourko Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, et al. Missense mutations in plakophilin‐2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation. 2014;129:1092–1103. doi: 10.1161/CIRCULATIONAHA.113.003077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Belbachir N, Portero V, Al Sayed ZR, Gourraud J‐B, Dilasser F, Jesel L, ` H, Wu H, Gaborit N, Guilluy C, et al. RRAD mutation causes electrical and cytoskeletal defects in cardiomyocytes derived from a familial case of Brugada syndrome. Eur Heart J. 2019;40:3081–3094. doi: 10.1093/eurheartj/ehz308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Juang JJ, Horie M. Genetics of Brugada syndrome. J Arrhythm. 2016;32:418–425. doi: 10.1016/j.joa.2016.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rivaud MR, Marchal GA, Wolswinkel R, Jansen JA, van der Made I, Beekman L, Ruiz‐Villalba A, Baartscheer A, Rajamani S, Belardinelli L, et al. Functional modulation of atrio‐ventricular conduction by enhanced late sodium current and calcium‐dependent mechanisms in Scn5a1798insD/+ mice. Europace. 2020;22:1579–1589. doi: 10.1093/europace/euaa127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Boukens BJ, Sylva M, de Gier‐de VC, Remme CA, Bezzina CR, Christoffels VM, Coronel R. Reduced sodium channel function unmasks residual embryonic slow conduction in the adult right ventricular outflow tract. Circ Res. 2013;113:137–141. doi: 10.1161/CIRCRESAHA.113.301565 [DOI] [PubMed] [Google Scholar]
- 83. Zumhagen S, Veldkamp MW, Stallmeyer B, Baartscheer A, Eckardt L, Paul M, Remme CA, Bhuiyan ZA, Bezzina CR, Schulze‐Bahr E. A heterozygous deletion mutation in the cardiac sodium channel gene SCN5A with loss‐ and gain‐of‐function characteristics manifests as isolated conduction disease, without signs of Brugada or long QT syndrome. PLoS One. 2013;8:e67963. doi: 10.1371/journal.pone.0067963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. El‐Battrawy I, Lan H, Cyganek L, Maywald L, Zhong R, Zhang F, Xu Q, Lee J, Duperrex E, Hierlemann A, et al. Deciphering the pathogenic role of a variant with uncertain significance for short QT and Brugada syndromes using gene‐edited human‐induced pluripotent stem cell‐derived cardiomyocytes and preclinical drug screening. Clin Transl Med. 2021;11:e646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fan X, Yang G, Kowitz J, Duru F, Saguner AM, Akin I, Zhou X, El‐Battrawy I. Preclinical short QT syndrome models: studying the phenotype and drug‐screening. Europace. 2021. doi: 10.1093/europace/euab214 [DOI] [PubMed] [Google Scholar]
- 86. Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, et al. Loss‐of‐function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST‐segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Delpon E, Cordeiro JM, Nunez L, Thomsen PE, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Hofman‐Bang J, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209–218. doi: 10.1161/CIRCEP.107.748103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, Albertson RM, Antzelevitch C, Schwartz PJ, Ackerman MJ. Transient outward current (I(to)) gain‐of‐function mutations in the KCND3‐encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. 2011;8:1024–1032. doi: 10.1016/j.hrthm.2011.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hu D, Barajas‐Martínez H, Terzic A, Park S, Pfeiffer R, Burashnikov E, Wu Y, Borggrefe M, Veltmann C, Schimpf R, et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol. 2014;171:431–442. doi: 10.1016/j.ijcard.2013.12.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Barajas‐Martínez H, Hu D, Ferrer T, Onetti CG, Wu Y, Burashnikov E, Boyle M, Surman T, Urrutia J, Veltmann C, et al. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm. 2012;9:548–555. doi: 10.1016/j.hrthm.2011.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Antzelevitch C. Ion channels and ventricular arrhythmias: cellular and ionic mechanisms underlying the Brugada syndrome. Curr Opin Cardiol. 1999;14:274–279. doi: 10.1097/00001573-199905000-00013 [DOI] [PubMed] [Google Scholar]
- 92. Shimizu W, Antzelevitch C, Suyama K, Kurita T, Taguchi A, Aihara N, Takaki H, Sunagawa K, Kamakura S. Effect of sodium channel blockers on ST segment, QRS duration, and corrected QT interval in patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2000;11:1320–1329. doi: 10.1046/j.1540-8167.2000.01320.x [DOI] [PubMed] [Google Scholar]
- 93. Schweizer PA, Becker R, Katus HA, Thomas D. Successful acute and long‐term management of electrical storm in Brugada syndrome using orciprenaline and quinine/quinidine. Clin Res Cardiol. 2010;99:467–470. doi: 10.1007/s00392-010-0145-7 [DOI] [PubMed] [Google Scholar]
- 94. Kyriazis K, Bahlmann E, van der Schalk H, Kuck KH. Electrical storm in Brugada syndrome successfully treated with orciprenaline; effect of low‐dose quinidine on the electrocardiogram. Europace. 2009;11:665–666. doi: 10.1093/europace/eup070 [DOI] [PubMed] [Google Scholar]
- 95. Tsuchiya T, Ashikaga K, Honda T, Arita M. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 2002;13:698–701. doi: 10.1046/j.1540-8167.2002.00698.x [DOI] [PubMed] [Google Scholar]
- 96. Kaneko Y, Horie M, Niwano S, Kusano KF, Takatsuki S, Kurita T, Mitsuhashi T, Nakajima T, Irie T, Hasegawa K, et al. Electrical storm in patients with Brugada syndrome is associated with early repolarization. Circ Arrhythm Electrophysiol. 2014;7:1122–1128. doi: 10.1161/CIRCEP.114.001806 [DOI] [PubMed] [Google Scholar]
- 97. El‐Battrawy I, Lang S, Zhou X, Akin I. Different genotypes of Brugada syndrome may present different clinical phenotypes: electrophysiology from bench to bedside. Eur Heart J. 2021;42:1270–1272. doi: 10.1093/eurheartj/ehab070 [DOI] [PubMed] [Google Scholar]
- 98. Brugada J, Brugada P, Brugada R. The syndrome of right bundle branch block ST segment elevation in V1 to V3 and sudden death–the Brugada syndrome. Europace. 1999;1:156–166. doi: 10.1053/eupc.1999.0033 [DOI] [PubMed] [Google Scholar]
- 99. Kamakura T, Wada M, Ishibashi K, Inoue YY, Miyamoto K, Okamura H, Nagase S, Noda T, Aiba T, Yasuda S, et al. Feasibility evaluation of long‐term use of beta‐blockers and calcium antagonists in patients with Brugada syndrome. Europace. 2018;20:f72–f76. doi: 10.1093/europace/eux198 [DOI] [PubMed] [Google Scholar]
- 100. Poli S, Toniolo M, Maiani M, Zanuttini D, Rebellato L, Vendramin I, Dametto E, Bernardi G, Bassi F, Napolitano C, et al. Management of untreatable ventricular arrhythmias during pharmacologic challenges with sodium channel blockers for suspected Brugada syndrome. Europace. 2018;20:234–242. doi: 10.1093/europace/eux092 [DOI] [PubMed] [Google Scholar]
- 101. Shinohara T, Ebata Y, Ayabe R, Fukui A, Okada N, Yufu K, Nakagawa M, Takahashi N. Combination therapy of cilostazol and bepridil suppresses recurrent ventricular fibrillation related to J‐wave syndromes. Heart Rhythm. 2014;11:1441–1445. doi: 10.1016/j.hrthm.2014.05.001 [DOI] [PubMed] [Google Scholar]
- 102. Szel T, Antzelevitch C. Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of Brugada syndrome. J Am Coll Cardiol. 2014;63:2037–2045. doi: 10.1016/j.jacc.2014.01.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Leoni A‐L, Gavillet B, Rougier J‐S, Marionneau C, Probst V, Le Scouarnec S, Schott J‐J, Demolombe S, Bruneval P, Huang CLH, et al. Variable Na(v)1.5 protein expression from the wild‐type allele correlates with the penetrance of cardiac conduction disease in the Scn5a(+/‐) mouse model. PLoS One. 2010;5:e9298. doi: 10.1371/journal.pone.0009298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Minoura Y, Panama BK, Nesterenko VV, Betzenhauser M, Barajas‐Martinez H, Hu D, Di Diego JM, Antzelevitch C. Effect of Wenxin Keli and quinidine to suppress arrhythmogenesis in an experimental model of Brugada syndrome. Heart Rhythm. 2013;10:1054–1062. doi: 10.1016/j.hrthm.2013.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nishida K, Fujiki A, Mizumaki K, Sakabe M, Sugao M, Tsuneda T, Inoue H. Canine model of Brugada syndrome using regional epicardial cooling of the right ventricular outflow tract. J Cardiovasc Electrophysiol. 2004;15:936–941. doi: 10.1046/j.1540-8167.2004.04041.x [DOI] [PubMed] [Google Scholar]
- 106. Di Diego JM, Patocskai B, Barajas‐Martinez H, Borbáth V, Ackerman MJ, Burashnikov A, Clatot J, Li G‐R, Robinson VM, Hu D, et al. Acacetin suppresses the electrocardiographic and arrhythmic manifestations of the J wave syndromes. PLoS One. 2020;15:e0242747. doi: 10.1371/journal.pone.0242747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fish JM, Antzelevitch C. Role of sodium and calcium channel block in unmasking the Brugada syndrome. Heart Rhythm. 2004;1:210–217. doi: 10.1016/j.hrthm.2004.03.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Morita H, Zipes DP, Morita ST, Wu J. Genotype‐phenotype correlation in tissue models of Brugada syndrome simulating patients with sodium and calcium channelopathies. Heart Rhythm. 2010;7:820–827. doi: 10.1016/j.hrthm.2010.01.039 [DOI] [PubMed] [Google Scholar]
- 109. Hoogendijk MG, Potse M, Vinet A, de Bakker JM, Coronel R. ST segment elevation by current‐to‐load mismatch: an experimental and computational study. Heart Rhythm. 2011;8:111–118. doi: 10.1016/j.hrthm.2010.09.066 [DOI] [PubMed] [Google Scholar]
- 110. Barajas‐Martínez HM, Hu D, Cordeiro JM, Wu Y, Kovacs RJ, Meltser H, Kui H, Elena B, Brugada R, Antzelevitch C, et al. Lidocaine‐induced Brugada syndrome phenotype linked to a novel double mutation in the cardiac sodium channel. Circ Res. 2008;103:396–404. doi: 10.1161/CIRCRESAHA.108.172619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kosmidis G, Veerman CC, Casini S, Verkerk AO, van de Pas S, Bellin M, Wilde AAM, Mummery CL, Bezzina CR. Readthrough‐promoting drugs gentamicin and PTC124 fail to rescue Nav1.5 function of human‐induced pluripotent stem cell‐derived cardiomyocytes carrying nonsense mutations in the sodium channel gene SCN5A. Circ Arrhythm Electrophysiol. 2016;9:e004227. doi: 10.1161/CIRCEP.116.004227 [DOI] [PubMed] [Google Scholar]
- 112. Li XF, Zhou YW, Cai PF, Fu WC, Wang JH, Chen JY, Yang Q. CRISPR/Cas9 facilitates genomic editing for large‐scale functional studies in pluripotent stem cell cultures. Hum Genet. 2019;138:1217–1225. doi: 10.1007/s00439-019-02071-z [DOI] [PubMed] [Google Scholar]
- 113. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR‐Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096 [DOI] [PubMed] [Google Scholar]
- 114. Garg P, Oikonomopoulos A, Chen H, Li Y, Lam CK, Sallam K, Perez M, Lux RL, Sanguinetti MC, Wu JC. Genome editing of induced pluripotent stem cells to decipher cardiac channelopathy variant. J Am Coll Cardiol. 2018;72:62–75. doi: 10.1016/j.jacc.2018.04.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Gnecchi M, Sala L, Schwartz PJ. Precision Medicine and cardiac channelopathies: when dreams meet reality. Eur Heart J. 2021;42:1661–1675. doi: 10.1093/eurheartj/ehab007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. da Rocha AM, Creech J, Thonn E, Mironov S, Herron TJ. Detection of drug‐induced torsades de pointes arrhythmia mechanisms using hiPSC‐CM syncytial monolayers in a high‐throughput screening voltage sensitive dye assay. Toxicol Sci. 2020;173:402–415. doi: 10.1093/toxsci/kfz235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Monteiro da Rocha A, Guerrero‐Serna G, Helms A, Luzod C, Mironov S, Russell M, Jalife J, Day SM, Smith GD, Herron TJ. Deficient cMyBP‐C protein expression during cardiomyocyte differentiation underlies human hypertrophic cardiomyopathy cellular phenotypes in disease specific human ES cell derived cardiomyocytes. J Mol Cell Cardiol. 2016;99:197–206. doi: 10.1016/j.yjmcc.2016.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ross SB, Fraser ST, Semsarian C. Induced pluripotent stem cell technology and inherited arrhythmia syndromes. Heart Rhythm. 2018;15:137–144. doi: 10.1016/j.hrthm.2017.08.013 [DOI] [PubMed] [Google Scholar]
- 119. Eschenhagen T, Fink C, Remmers U, Scholz H, Wattchow J, Weil J, Zimmermann W, Dohmen HH, Schäfer H, Bishopric N, et al. Three‐dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix: a new heart muscle model system. FASEB J. 1997;11:683–694. doi: 10.1096/fasebj.11.8.9240969 [DOI] [PubMed] [Google Scholar]
- 120. Zimmermann WH, Fink C, Kralisch D, Remmers U, Weil J, Eschenhagen T. Three‐dimensional engineered heart tissue from neonatal rat cardiac myocytes. Biotechnol Bioeng. 2000;68:106–114. doi: 10.1002/(SICI)1097-0290(20000405)68:1<106:AID-BIT13>3.0.CO;2-3 [DOI] [PubMed] [Google Scholar]
- 121. Tzatzalos E, Abilez OJ, Shukla P, Wu JC. Engineered heart tissues and induced pluripotent stem cells: macro‐ and microstructures for disease modeling, drug screening, and translational studies. Adv Drug Deliv Rev. 2016;96:234–244. doi: 10.1016/j.addr.2015.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ivashchenko CY, Pipes GC, Lozinskaya IM, Lin Z, Xiaoping XU, Needle S, Grygielko ET, Hu E, Toomey JR, Lepore JJ, et al. Human‐induced pluripotent stem cell‐derived cardiomyocytes exhibit temporal changes in phenotype. Am J Physiol Heart Circ Physiol. 2013;305:H913–H922. doi: 10.1152/ajpheart.00819.2012 [DOI] [PubMed] [Google Scholar]
- 123. Yang X, Rodriguez M, Pabon L, Fischer KA, Reinecke H, Regnier M, Sniadecki NJ, Ruohola‐Baker H, Murry CE. Tri‐iodo‐l‐thyronine promotes the maturation of human cardiomyocytes‐derived from induced pluripotent stem cells. J Mol Cell Cardiol. 2014;72:296–304. doi: 10.1016/j.yjmcc.2014.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Correia C, Koshkin A, Duarte P, Hu D, Teixeira A, Domian I, Serra M, Alves PM. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci Rep. 2017;7:8590. doi: 10.1038/s41598-017-08713-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ng DCH, Richards DK, Mills RJ, Ho UY, Perks HL, Tucker CR, Voges HK, Pagan JK, Hudson JE. Centrosome reduction promotes terminal differentiation of human cardiomyocytes. Stem Cell Reports. 2020;15:817–826. doi: 10.1016/j.stemcr.2020.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Biermann M, Cai W, Lang DI, Hermsen J, Profio L, Zhou Y, Czirok A, Isai DG, Napiwocki BN, Rodriguez AM, et al. Epigenetic priming of human pluripotent stem cell‐derived cardiac progenitor cells accelerates cardiomyocyte maturation. Stem Cells. 2019;37:910–923. doi: 10.1002/stem.3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sun X, Nunes SS. Maturation of human stem cell‐derived cardiomyocytes in biowires using electrical stimulation. J Vis Exp. 2017:55373. doi: 10.3791/55373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Tan Y, Richards D, Xu R, Stewart‐Clark S, Mani SK, Borg TK, Menick DR, Tian B, Mei Y. Silicon nanowire‐induced maturation of cardiomyocytes derived from human induced pluripotent stem cells. Nano Lett. 2015;15:2765–2772. doi: 10.1021/nl502227a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hirt MN, Boeddinghaus J, Mitchell A, Schaaf S, Börnchen C, Müller C, Schulz H, Hubner N, Stenzig J, Stoehr A, et al. Functional improvement and maturation of rat and human engineered heart tissue by chronic electrical stimulation. J Mol Cell Cardiol. 2014;74:151–161. doi: 10.1016/j.yjmcc.2014.05.009 [DOI] [PubMed] [Google Scholar]
- 130. Ruan JL, Tulloch NL, Razumova MV, Saiget M, Muskheli V, Pabon L, Reinecke H, Regnier M, Murry CE. Mechanical stress conditioning and electrical stimulation promote contractility and force maturation of induced pluripotent stem cell‐derived human cardiac tissue. Circulation. 2016;134:1557–1567. doi: 10.1161/CIRCULATIONAHA.114.014998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Tulloch NL, Muskheli V, Razumova MV, Korte FS, Regnier M, Hauch KD, Pabon L, Reinecke H, Murry CE. Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ Res. 2011;109:47–59. doi: 10.1161/CIRCRESAHA.110.237206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ruan J‐L, Tulloch NL, Saiget M, Paige SL, Razumova MV, Regnier M, Tung KC, Keller G, Pabon L, Reinecke H, et al. Mechanical stress promotes maturation of human myocardium from pluripotent stem cell‐derived progenitors. Stem Cells. 2015;33:2148–2157. doi: 10.1002/stem.2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Xu C, Wang L, Yu Y, Yin F, Zhang X, Jiang L, Qin J. Bioinspired onion epithelium‐like structure promotes the maturation of cardiomyocytes derived from human pluripotent stem cells. Biomater Sci. 2017;5:1810–1819. doi: 10.1039/C7BM00132K [DOI] [PubMed] [Google Scholar]
- 134. Herron TJ, Rocha AMD, Campbell KF, Ponce‐Balbuena D, Willis BC, Guerrero‐Serna G, Liu Q, Klos M, Musa H, Zarzoso M, et al. Extracellular matrix‐mediated maturation of human pluripotent stem cell‐derived cardiac monolayer structure and electrophysiological function. Circ Arrhythm Electrophysiol. 2016;9:e003638. doi: 10.1161/CIRCEP.113.003638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Feaster TK, Cadar AG, Wang L, Williams CH, Chun YW, Hempel JE, Bloodworth N, Merryman WD, Lim CC, Wu JC, et al. Matrigel mattress: a method for the generation of single contracting human‐induced pluripotent stem cell‐derived cardiomyocytes. Circ Res. 2015;117:995–1000. doi: 10.1161/CIRCRESAHA.115.307580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Roshanbinfar K, Mohammadi Z, Sheikh‐Mahdi Mesgar A, Dehghan MM, Oommen OP, Hilborn J, Engel FB. Carbon nanotube doped pericardial matrix derived electroconductive biohybrid hydrogel for cardiac tissue engineering. Biomater Sci. 2019;7:3906–3917. doi: 10.1039/C9BM00434C [DOI] [PubMed] [Google Scholar]
- 137. Huang CY, Peres Moreno Maia‐Joca R, Ong CS, Wilson I, DiSilvestre D, Tomaselli GF, Reich DH. Enhancement of human iPSC‐derived cardiomyocyte maturation by chemical conditioning in a 3D environment. J Mol Cell Cardiol. 2020;138:1–11. doi: 10.1016/j.yjmcc.2019.10.001 [DOI] [PubMed] [Google Scholar]
- 138. LaBarge W, Mattappally S, Kannappan R, Fast VG, Pretorius D, Berry JL, Zhang J. Maturation of three‐dimensional, hiPSC‐derived cardiomyocyte spheroids utilizing cyclic, uniaxial stretch and electrical stimulation. PLoS One. 2019;14:e0219442. doi: 10.1371/journal.pone.0219442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Veldhuizen J, Cutts J, Brafman DA, Migrino RQ, Nikkhah M. Engineering anisotropic human stem cell‐derived three‐dimensional cardiac tissue on‐a‐chip. Biomaterials. 2020;256:120195. doi: 10.1016/j.biomaterials.2020.120195 [DOI] [PubMed] [Google Scholar]
- 140. Ebert A, Joshi AU, Andorf S, Dai Y, Sampathkumar S, Chen H, Li Y, Garg P, Toischer K, Hasenfuss G, et al. Proteasome‐dependent regulation of distinct metabolic states during long‐term culture of human iPSC‐derived cardiomyocytes. Circ Res. 2019;125:90–103. doi: 10.1161/CIRCRESAHA.118.313973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Block T, Creech J, da Rocha AM, Marinkovic M, Ponce‐Balbuena D, Jimenez‐Vazquez EN, Sy G, Herron TJ. Human perinatal stem cell derived extracellular matrix enables rapid maturation of hiPSC‐CM structural and functional phenotypes. Sci Rep. 2020;10:19071. doi: 10.1038/s41598-020-76052-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Lee JH, Protze SI, Laksman Z, Backx PH, Keller GM. Human Pluripotent stem cell‐derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell. 2017;21:179–194.e4. doi: 10.1016/j.stem.2017.07.003 [DOI] [PubMed] [Google Scholar]