

Abstract
Background
Platelet‐derived growth factor is a major regulator of the vascular remodeling associated with pulmonary arterial hypertension. We previously showed that protein widely 1 (PW1+) vascular progenitor cells participate in early vessel neomuscularization during experimental pulmonary hypertension (PH) and we addressed the role of the platelet‐derived growth factor receptor type α (PDGFRα) pathway in progenitor cell‐dependent vascular remodeling and in PH development.
Methods and Results
Remodeled pulmonary arteries from patients with idiopathic pulmonary arterial hypertension showed an increased number of perivascular and vascular PW1+ cells expressing PDGFRα. PW1nLacZ reporter mice were used to follow the fate of pulmonary PW1+ progenitor cells in a model of chronic hypoxia–induced PH development. Under chronic hypoxia, PDGFRα inhibition prevented the increase in PW1+ progenitor cell proliferation and differentiation into vascular smooth muscle cells and reduced pulmonary vessel neomuscularization, but did not prevent an increased right ventricular systolic pressure or the development of right ventricular hypertrophy. Conversely, constitutive PDGFRα activation led to neomuscularization via PW1+ progenitor cell differentiation into new smooth muscle cells and to PH development in male mice without fibrosis. In vitro, PW1+ progenitor cell proliferation, but not differentiation, was dependent on PDGFRα activity.
Conclusions
These results demonstrate a major role of PDGFRα signaling in progenitor cell–dependent lung vessel neomuscularization and vascular remodeling contributing to PH development, including in idiopathic pulmonary arterial hypertension patients. Our findings suggest that PDGFRα blockers may offer a therapeutic add‐on strategy to combine with current pulmonary arterial hypertension treatments to reduce vascular remodeling. Furthermore, our study highlights constitutive PDGFRα activation as a novel experimental PH model.
Keywords: fibrosis, pulmonary hypertension, platelet‐derived growth factor receptor alpha, stem cells, vascular remodeling
Subject Categories: Vascular Disease, Pulmonary Hypertension, Stem Cells, Smooth Muscle Proliferation and Differentiation, Animal Models of Human Disease
Nonstandard Abbreviations and Acronyms
- α‐SMA
alpha smooth muscle actin
- CH
chronic hypoxia
- iPAH
idiopathic pulmonary arterial hypertension
- PAH
pulmonary arterial hypertension
- PDGFR
platelet‐derived growth factor receptor
- PH
pulmonary hypertension
- PW1
protein widely 1
- RVSP
right ventricular systolic pressure
- SMC
smooth muscle cell
Clinical Perspective
What Is New?
Platelet‐derived growth factor receptor type α (PDGFRα)+ vascular progenitor cells accumulate in the perivascular area in lungs of patients with idiopathic pulmonary arterial hypertension.
The PDGFRα pathway regulates progenitor cells proliferation and their participation in pulmonary vessels neomuscularization in a pulmonary hypertension experimental mouse model.
Activating the PDGFRα pathway leads to pulmonary vessels neomuscularization but to moderate pulmonary hypertension only in male mice.
What Are the Clinical Implications?
PDGFRα activation of PDGFRα+ progenitor cells may participate in pulmonary vascular remodeling during pulmonary arterial hypertension development.
Blocking PDGFRα could help reduce pulmonary vessel muscularization in patients with idiopathic pulmonary arterial hypertension.
Pulmonary arterial hypertension (PAH), a severe rare disease, is histologically defined by pulmonary vascular remodeling characterized by endothelial cell proliferation, media thickening, neointima formation, and neomuscularization of small pulmonary vessels. This vascular remodeling leads to increased pulmonary vascular resistance and to PAH, subsequently causing right ventricular hypertrophy and ultimately right heart failure and death. During the development of PAH, pulmonary arterioles, which are predominantly nonmuscularized, become covered by smooth muscle cells (SMCs). This neomuscularization, observed in patients with PAH and rodent models of PAH, is driven by both SMC proliferation and the recruitment of pericytes and other SMC progenitor cells. 1 , 2 , 3 , 4 We have recently identified new perivascular pulmonary progenitor cells characterized by the expression of stem cell markers PW1 (protein widely 1), platelet‐derived growth factor receptor type α (PDGFRα), and CD34 in rodent and human lungs. 3 During the development of early chronic hypoxia (CH) in an experimental pulmonary hypertension (PH) model, these PW1+ progenitor cells proliferate and differentiate into new SMCs, contributing to small pulmonary vessel neomuscularization. We demonstrated that their differentiation into SMCs was under the control of the C‐X‐C chemokine receptor type 4/C‐X‐C motif chemokine 12 pathway, but the specific factors regulating their proliferation remained unknown.
Studies from the past 25 years have highlighted the platelet‐derived growth factor (PDGF) pathway as a major regulator of pulmonary vascular remodeling and PAH development. 5 , 6 , 7 PDGF receptors (PDGFRs) are transmembrane tyrosine kinase receptors with 2 isoforms, PDGFRα and PDGFRβ, which assemble into homo‐ or heterodimers. Five PDGF ligand dimers (PDGF‐AA, ‐BB, ‐AB, ‐CC, ‐DD) differentially activate these receptors and play an important paracrine role during embryonic and vascular development (for review see Andrae et al 8 ). All PDGFR forms can activate phosphoinositide 3‐kinase, phospholipase Cγ, and Ras–mitogen‐activated protein kinase signaling pathways but trigger distinct cellular responses. Expression of PDGF‐A and ‐B ligands and PDGFRα and β was found to be significantly increased in the lungs of patients with PAH compared with control lungs. 5 Imatinib (STI 571), which targets both the PDGFRα and β subtypes as well as Bcr/Abl and c‐kit, protects against PH development in rodent experimental models. 9 These protective effects were attributed to the PDGFRβ pathway as constitutively activating or inhibiting PDGFRβ signaling, respectively, increased or reduced CH‐induced PH development in mice. 10 , 11 In contrast, it remains unknown whether PDGFRα also plays a role during PAH development. This receptor is a marker for mesenchymal stem cells in various organs (heart, skeletal muscle, adipose tissue, and bone marrow) and of pulmonary vascular progenitor cells. 3 , 12 The PDGF‐A/PDGFRα pathway controls stem cell proliferation and differentiation 13 , 14 , 15 , 16 and is involved in myofibroblast formation during lung development in particular. 17
Here, we provide evidence that PW1+/PDGFRα+ progenitor cells are actively recruited in human idiopathic pulmonary arterial hypertension (iPAH) remodeled arteries. We establish that PDGFRα activation regulates pulmonary vascular remodeling during experimental chronic hypoxia‐induced PH. We demonstrate in vitro and in vivo that this receptor controls the proliferation of PW1+/PDGFRα+ pulmonary vascular progenitor cells that form new SMCs during CH. Furthermore, we highlight that PDGFRα activation leads to PH development in both a constitutive PDGFRα activation mouse model or after chronic PDGF‐AA treatment.
Methods
The data, protocols, and materials that support the findings of this study are available from the corresponding author upon reasonable request. Detailed methods are provided in Data S1 and Tables S1 and S2.
Studies with patients complied with the Declaration of Helsinki and are part of the French Network on Pulmonary Hypertension, a program approved by our institutional Ethics Committee (2018‐A01252‐53). Written informed consent was received from participants before inclusion in the study. Animal experiments for the project were approved by our institutional review board (APAFIS#3566bis‐2016022617318129 v8).
Pw1IRESnLacZ transgenic mice (Pw1nlacZ/+) bear a nuclear operon lactose gene expressed under the control of the Pw1 gene locus. 18 The stability (perdurance) of the β‐galactosidase reporter facilitates short‐term lineage tracing of cells derived from PW1+/β‐galactosidase+ cells. These mice were crossed with RosaCreERT2xPDGFRα+/(S)K mice, which, when administered tamoxifen, express a constitutively activated form of PDGFRα (D842V) in PDGFRα‐expressing cells. 19 Mice (7‐ to 10‐week‐old male and female littermates) were exposed to normobaric room air (normoxia) or chronic normobaric hypoxia (10% O2) for 4 or 21 days. PDGFRα activity was inhibited in vivo using either imatinib or the specific blocking antibody, APA5. Imatinib was injected daily (50 mg/kg of body weight intraperitoneally), and the blocking antibody or a control IgG was injected intraperitoneally once (500 µg/mouse) 3 days before starting CH, then every 3 to 4 days throughout the CH periods (100 µg/mouse). PDGF‐AA (or vehicle) was injected daily (150 ng/mouse, intraperitoneally) for 3 weeks in Pw1nlacZ/+mice. Following the different treatments, mice were anesthetized to measure hemodynamic parameters, right ventricular hypertrophy, and to collect lung samples for tissue analysis. Fluorescence‐activated cell sorting was used to isolate PDGFRα+/PW1+ progenitor cell populations from Pw1nlacZ/+mice based on their β‐galactosidase activity and a combination of progenitor cell markers, to study them in culture. Fluorescence‐activated cell sorting–isolated CD45‐/CD34+/β‐galactosidase+/c‐kit‐ cells were cultured for 5 days in the presence of PDGF ligands (20 ng/mL), control IgG (150 ng/mL), or APA5 blocking antibody (150 ng/mL) before immunofluorescence analysis.
Results
Perivascular PW1+/PDGFRα+ Cells Are Increased in the Lungs of Patients With iPAH
Activation of the PDGFRα pathway in human iPAH has been suggested by the increased expression of PDGF‐A in patient lungs. 5 We studied the presence and regulation of PW1+ and PW1+/PDGFRα+ progenitor cells adjacent to or within pulmonary arteries of control patients and patients with iPAH. Patient characteristics are indicated in Table S3. Immunofluorescence studies (Figure 1A) confirmed the presence of PW1+ cells organized in clusters in the perivascular zone and their absence in the media of control patient lungs, as seen previously. 3 Importantly, these PW1+ cells were positive for PDGFRα expression. In patients with iPAH, perivascular PW1+/PDGFRα+/alpha smooth muscle actin (α‐SMA‐) cells were found to be significantly more numerous around remodeled arteries (P=0.016; Figure 1A and 1B), as observed in murine experimental models. 3 The number of PW1+/PDGFRα+ cells within the media and neointima of small or large arteries was equivalent in control and patients with iPAH, whereas PDGFRα+/PW1+/α‐SMA+ cells were significantly increased (P=0.008; Figure 1C). Complex lesions did not contain PW1+ cells (as illustrated in Figure S1) but showed numerous PDGFRα+ cells. We then used the mouse chronic hypoxia PH model to study the regulation of these PW1+/PDGFRα+ progenitor cells during pulmonary vascular remodeling and to decipher their role.
Figure 1. Perivascular PW1+/PDGFRα+ cells are increased in the perivascular zone and within the vascular wall of remodeled arteries in patients with iPAH.
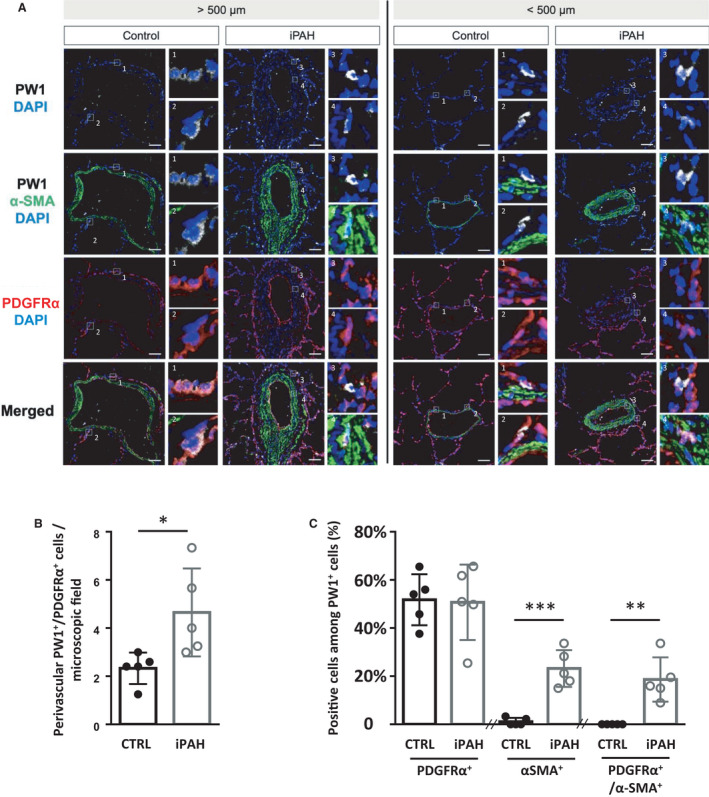
Lung sections from control (CTRL) or patients with idiopathic pulmonary arterial hypertension (iPAH) were labeled for PW1 (white), PDGFRα (red), and α‐SMA (green). A, Representative confocal images of pulmonary vessels (>500 µm diameter, left panels; <500 µm diameter, right panels) from control patients (n=5) and patients with iPAH (n=5). Panels 1, 2, and 3 show details of perivascular PW1+/PDFGFRα+ cells. Panel 4 shows details of PW1+/PDFGFRα+/α‐SMA+ cells found within arteries in patients with iPAH. B, quantification of perivascular PW1+/PDGFRα+ cells. C, quantification of PDGFRα+ or α‐SMA+ or PDGFRα+/α‐SMA+ cells among perivascular and vascular PW1+. Scale bar, 50 µm. Bars represent means and whiskers represent SD. *P<0.05, **P<0.01, ***P<0.001; ns indicates not significant (2‐tailed Mann‐Whitney); PDGFRα indicates platelet‐derived growth factor receptor type α; PW1, protein widely 1; and SMA, smooth muscle actin.
Chronic Hypoxia Induces the PDGFRα Pathway and Increases the Number of Perivascular PDGFRα+/PW1+ Progenitor Cells in Mouse Lungs
We reported previously that early neomuscularization of pulmonary arteries observed in the chronic hypoxic PH experimental model relies on the recruitment of pulmonary resident CD34+/PDGFRα+/PW1+ progenitor cells. 3 To study the role of the PDGFRα pathway in the activation of these progenitors and associated vascular remodeling, we first analyzed the expression of the 4 PDGF ligands and the 2 PDGFR isoforms in total mouse lung samples extracted during the first 4 days of CH with reverse transcriptase polymerase chain reaction (Figure S2A). PDGF‐A, ‐B, and ‐D and PDGFRα and β demonstrated increased expression (×1.3 to ×2, with P=0.000, P=0.002, P=0.001, P=0.012, and P=0.002, respectively) after 1 day of CHs. It is noteworthy that PDGF‐A, a specific PDGFRα ligand, remained upregulated after 4 days of CH compared with normoxia. PDGFRα protein expression also showed a tendency to increase after 1 day of CH (Figure S2B). Concomitantly with the increased expression of PDGFRα ligands, we observed an early and significant increase in the number of pulmonary vessels associated with perivascular PDGFRα+/PW1+ progenitor cells (P=0.024; Figure S3A and B). On the contrary, there was no difference in the number of medial PDGFRα+/α‐SMA+ SMC during CH (Figure S4A and S4B).
PDGFRα Inhibition During Chronic Hypoxia Inhibits PW1+ Progenitor Cells Proliferation and Differentiation as Well as Pulmonary Vascular Remodeling In Vivo
To explore whether PDGFRα influences PW1+ progenitor cell participation in vascular remodeling during CH, we inhibited PDGFRα using 2 approaches: (1) intraperitoneal injection of imatinib, a pharmacological inhibitor that also targets PDGFRβ, c‐kit, and c‐abl, and has been used in PH models 9 , 11 ; and (2) intraperitoneal injection of the PDFGRα‐specific blocking antibody APA5. 20
Experiments were performed in the PW1nLacz transgenic mouse model, in which the innate stability of the β‐galactosidase reporter enables short‐term lineage tracing of PW1+‐derived cells. As such, PW1+ progenitor cell proliferation and their differentiation into SMCs can be assessed. Mice were maintained under normoxia or CH for 4 days and treated daily with DMSO or imatinib. Pulmonary vessel neomuscularization (the total percentage of partially and fully muscularized vessels), as observed by α‐SMA and vWF immunofluorescence colabeling (Figure 2A), was increased by CH (83%±4.7% of muscularized arterioles versus 36%±6.7% in normoxia, P<0.0001; Figure 2B). As previously shown, 3 this neomuscularization was at least partially attributable to an increased number of β‐galactosidase+ vascular SMC derived from PW1+ progenitor cells (72%±4% per vessel versus 42%±7% in normoxia, P<0.0001; Figure 2C and 2D) associated with a greater number of pulmonary PW1+ progenitor cells (19%±2% of parenchymal cells versus 8%±1% in normoxia, P<0.0001; Figure 2E and 2F). Imatinib treatment had no effect on these parameters under normoxia but significantly reduced CH‐induced neomuscularization by 40% (from 83%±4.7% in CH+DMSO to 65%±4.5% in CH+IMA, P<0.0001; Figure 2B). Imatinib completely abolished PW1+ progenitor cell participation in neomuscularization under CH, reducing the number of PW1+ progenitor‐derived SMCs to basal levels (from 72%±4% per vessel in CH+DMSO to 47%±8% in CH+IMA; P<0.0001 versus DMSO; Figure 2D). PW1+ cell numbers also returned to baseline with imatinib treatment (from 19%±2% of parenchymal cells in CH+DMSO to 10%±1% in CH+IMA, P<0.0001; Figure 2F). Imatinib treatment did not modify PDGFRα mRNA or protein expression (Figure S5). As we demonstrated previously, 3 PW1+ progenitor cells involved in early CH‐induced neomuscularization are PDGFRα+, suggesting that PDGFRα activation could be involved in increased levels of PW1+ cells and their differentiation into SMCs.
Figure 2. Imatinib treatment prevents early chronic hypoxia (CH)‐induced neomuscularization and PW1+ progenitor cell recruitment and differentiation in SMCs.
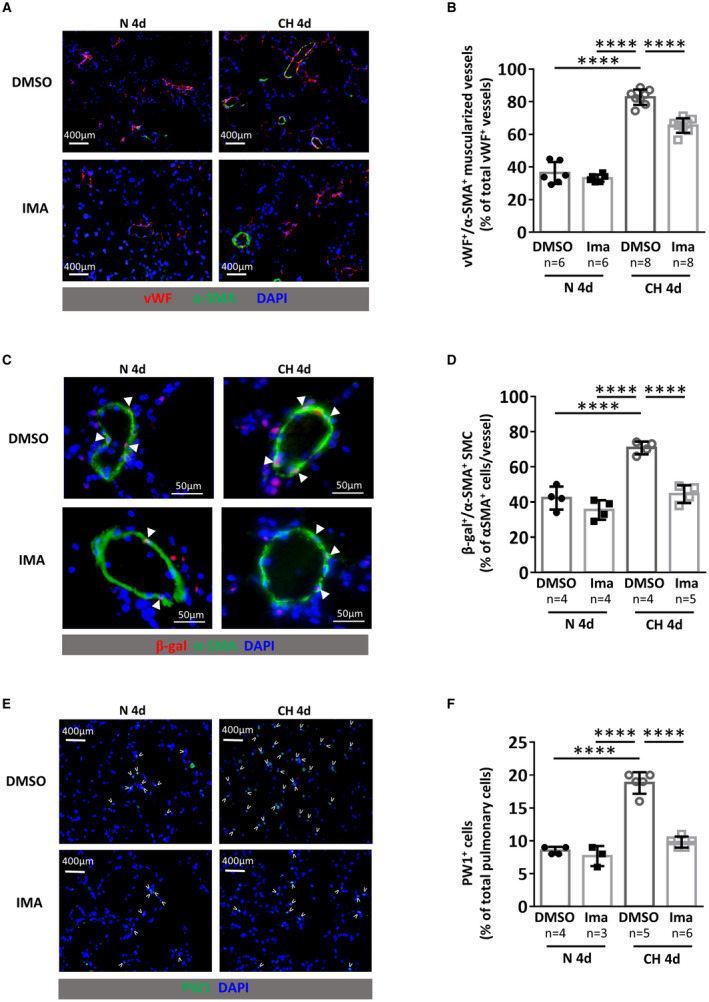
PW1nLacz mice were maintained under normoxia (N 4d) or chronic hypoxia (CH 4d) for 4 days and treated daily with DMSO or imatinib (IMA). A, Representative images of von Willebrand factor (vWF, red) and α‐smooth muscle actin (α‐SMA, green) staining in lungs. B, Quantification of muscularized vessels (fully+partially) (n=6–8 mice/group). C Representative images of pulmonary muscularized vessel stained with α‐SMA (green) and β‐galactosidase (red) in lungs (double positive are marked by arrowheads). D, Quantification of lung β‐galactosidase (β‐gal)‐expressing SMC in fully‐muscularized vessels (n=4–5 mice/group). E, Representative images of lung parenchyma stained for PW1 (PW1 in green, positive cells are marked by arrowheads). F, Quantification of PW1+ cells in lung parenchyma (n=3–6 mice/group). Bars represent means and whiskers represent SD. ****P<0.0001, 2‐way ANOVA and Tukey. PW1 indicates protein widely 1; and SMCs, smooth muscle cells.
To understand the role of PDGFRα in the proliferation and differentiation of PW1+ cells during CH, PW1nLacz transgenic mice were maintained under CH for 4 days and treated with control IgG or the PDGFRα‐blocking antibody APA5. Specific inhibition of PDGFRα during CH reproduced the results obtained with imatinib: Early CH‐induced pulmonary arterial neomuscularization was significantly reduced from 86%±2.3% to 63%±4% (P=0.0006; Figure 3A). In parallel, the CH‐induced increase in PW1+ progenitor‐derived β‐galactosidase+ vascular SMC was significantly inhibited (from 80.1%±4.2% to 50.3%±2.4%, P=0.0006; Figure 3B). We previously established that the increase in PW1+ progenitor cells observed during early CH was explained by induced proliferation. 3 The proportion of proliferating ethynyl‐deoxyuridine+ PW1+ cells was reduced by 66% from 46.0%±7.0% to 15.6%±9.1% (P=0.016; Figure 3C and 3D), whereas pulmonary cell apoptosis, as measured by terminal deoxynucleotidyl transferase dUTP nick end labeling, was very low and did not differ between control IgG‐ and APA5‐treated mice (Figure 3E).
Figure 3. PDGFRα inhibition with the specific blocking antibody APA5 prevents early chronic hypoxia (CH)‐induced neomuscularization and PW1+ progenitor cell proliferation and differentiation in SMCs.
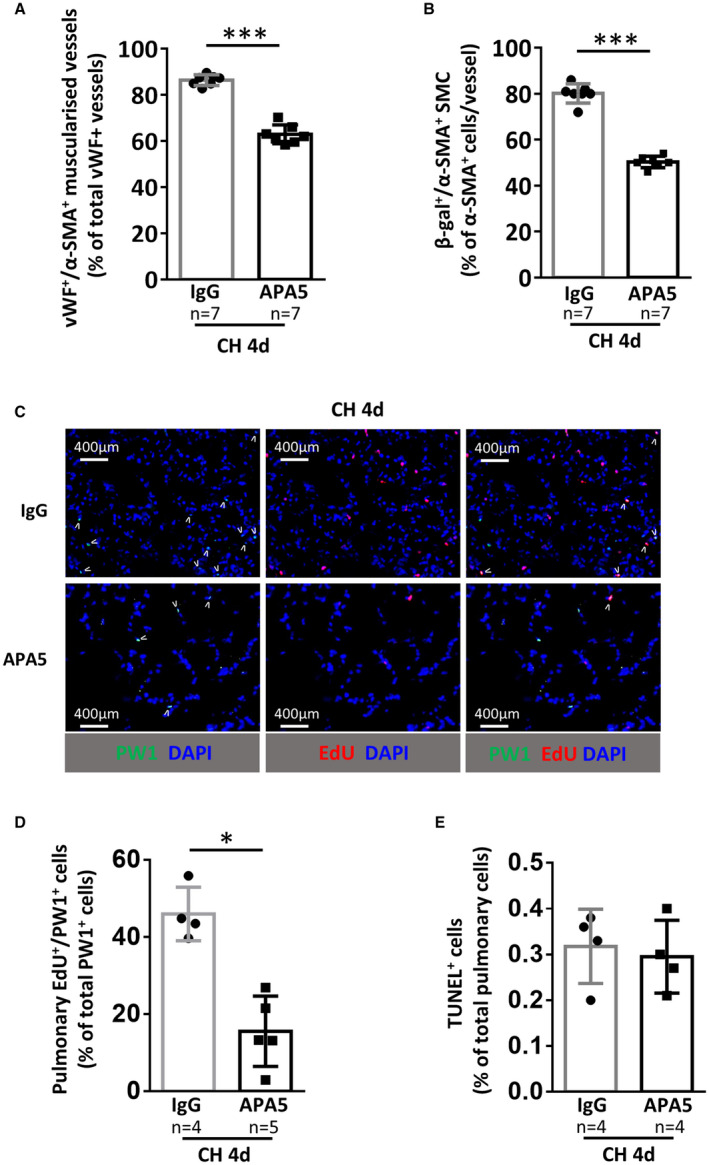
PW1nLacz mice were maintained under CH for 4 days and treated with control antibody (IgG) or PDGFRα blocking antibody (APA5). A, Quantification of pulmonary muscularized vessels (fully+partially). For each animal (n=7 mice/group), at least 100 vWF+ vessels (<100 µm) were analyzed for muscularization (α‐SMA+). B, Quantification of lung β‐galactosidase (β‐gal)‐expressing PW1‐derived SMCs in fully‐muscularized vessels (<100 µm) (n=7 mice/group). C, Representative images of lung parenchyma from CH mice treated with control IgG or APA5 and stained for PW1 (green) and EdU (red), double‐positive cells are marked by arrowheads. D, Quantification of EdU+/PW1+ cells in lung parenchyma (n=4–5 mice/group). E, Quantification of pulmonary apoptotic cells. The percentage of TUNEL+ cells in lung parenchyma was determined by immunofluorescence (n=4 mice/group). Bars represent means and whiskers represent SD. *P<0.05, **P<0.01, ***P<0.001. 2‐tailed Mann‐Whitney. Scale bar 400 µm. EdU indicates ethynyl‐deoxyuridine; PDGFRα, platelet‐derived growth factor receptor type α; PW1, protein widely 1; SMA, smooth muscle actin; SMC, smooth muscle cell; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; and vWF, von Willebrand factor.
Together, these data show that PDGFRα activation is essential for CH‐induced proliferation and differentiation of PW1+ progenitor cells into vascular SMCs, and that PDGFRα participates in the induction of early pulmonary arterial neomuscularization during CH.
PDGFRα Activation Elicits Neomuscularization of Pulmonary Arterioles Through PW1+ Progenitor Cell Differentiation Into New Smooth Muscle Cells Under Normoxic Conditions In Vivo
We developed a novel PW1nLacz/+/PDGFRα+/(S)K/Rosa‐CREERT2 mouse model to directly examine the role of PDGFRα in the regulation of pulmonary progenitor cells and vascular remodeling. We crossed PDGFRα+/(S)K mice with (1) Rosa‐CREERT2 mice for tamoxifen induction and generation of an inducible model of a mutated constitutively‐activated form of PDGFRα 19 and (2) with PW1nLacz mice to follow PW1+ progenitor cell fates (Figure 4A). Two weeks after the final tamoxifen injection, normoxic male and female PW1nLacz/+/PDGFRα+/(S)K/Rosa‐CREERT2 mice demonstrated significant neomuscularization with a 50% augmentation in the number of muscularized vessels (67%±13.9% of muscularized arteries in induced mice versus 35.5%±10.3% in noninduced littermates, P=0.004; Figure 4B). This was associated with increased numbers of PW1+ progenitor‐derived vascular SMC (65.8%±11.3% per vessel versus 48.8%±9.8% in noninduced littermates, P=0.026; Figure 4C). Both the vascular remodeling and increased number of PW1+ cells was similar to observations after 4 days of chronic hypoxia 3 and was not associated with PH development at this early time point (Figure S6).
Figure 4. Constitutive PDGFRα activation leads to pulmonary vessel neomuscularization because of increased numbers of PW1+ progenitor‐derived SMCs.
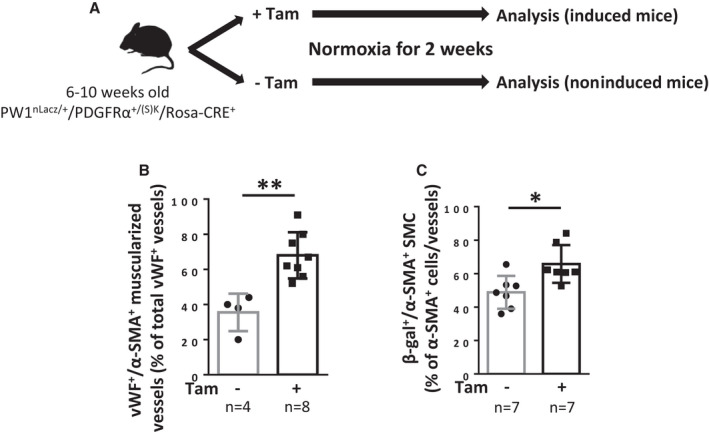
A, Timeline of tamoxifen (Tam) induction and analysis of PW1nLacz/+/PDGFRα+/(S)K/Rosa‐CRE+ mice (males + females). B and C, Analysis of mice lungs 2 weeks after tamoxifen induction. B, Quantification of muscularized vessels in lungs from control or tamoxifen‐treated mice. Pulmonary vessel muscularization was determined by immunofluorescence using anti–α‐smooth muscle actin (α‐SMA) and anti‐von Willebrand factor (vWF) antibodies (n=4–8). For each animal, ≈100 vWF+ vessels (<100 µm) were analyzed for muscularization (α‐SMA+). C, Quantification of lung β‐galactosidase (β‐gal)‐expressing SMC in fully‐muscularized pulmonary vessels. The percentage of lung β‐gal+/α‐SMA+ cells among α‐SMA+ cells in fully‐muscularized vessels (<100 µm) was determined by immunofluorescence (n=7 for each condition). Bars represent means and whiskers represent SD. *P<0.05, **P<0.01 (2‐tailed Mann‐Whitney). PDGFRα indicates platelet‐derived growth factor receptor type α; PW1, protein widely 1; SMA, smooth muscle actin; and SMC, smooth muscle cell.
Altogether, these results demonstrate that PDGFRα activation leads to pulmonary vessel neomuscularization through PW1+ progenitor cell proliferation.
PDGFRα Controls PW1+ Progenitor Cell Proliferation In Vitro
To confirm the direct role of the PDGFRα pathway on PW1+ progenitor cell proliferation or differentiation, we studied fluorescence‐activated cell sorting–purified pulmonary CD34+/PDGFRα+/PW1+ progenitor cells, which are amplified after chronic hypoxia. 3 These cells can be separated into 2 subpopulations according to PDGF‐dependent c‐kit expression. 3 As c‐kit+ cells could not be cultured at low serum concentrations, we were unable to determine whether PDFG‐AA activation affected this cell population. We therefore focused on the role of the PDGFRα pathway in the c‐kit‐ cell population, which undergoes spontaneous differentiation into α‐SMA‐expressing SMCs under normal culture conditions. 3 CD34+/PDGFRα+/β‐galactosidase+/c‐kit‐ cells were sorted and cultured using the strategy previously published 3 (Figure S7). Incubation with PDGFRα‐blocking antibody APA5 significantly inhibited their proliferation by 50% as compared with control IgG (P=0.016, Figure 5A) and reduced their differentiation by 40% (P=0.016, Figure 5B) without inducing apoptosis as measured by the terminal deoxynucleotidyl transferase dUTP nick end labeling assay (Figure 5C). While treatment of these progenitor cells with PDGF‐AA (a specific PDGFRα ligand) did not induce their differentiation into α‐SMA+ SMC (Figure 5D), it significantly increased their proliferation by 2‐fold (P=0.0625, Figure 5E). This effect was also observed with PDGF‐AB and PDGF‐BB, which bind both PDGFRα and β. These data demonstrate that PDGFRα activation directly controls the proliferation of PW1+/PDGFRα+/ckit‐ progenitor cells.
Figure 5. PDGFRα regulates PW1+ progenitor cell proliferation in vitro.
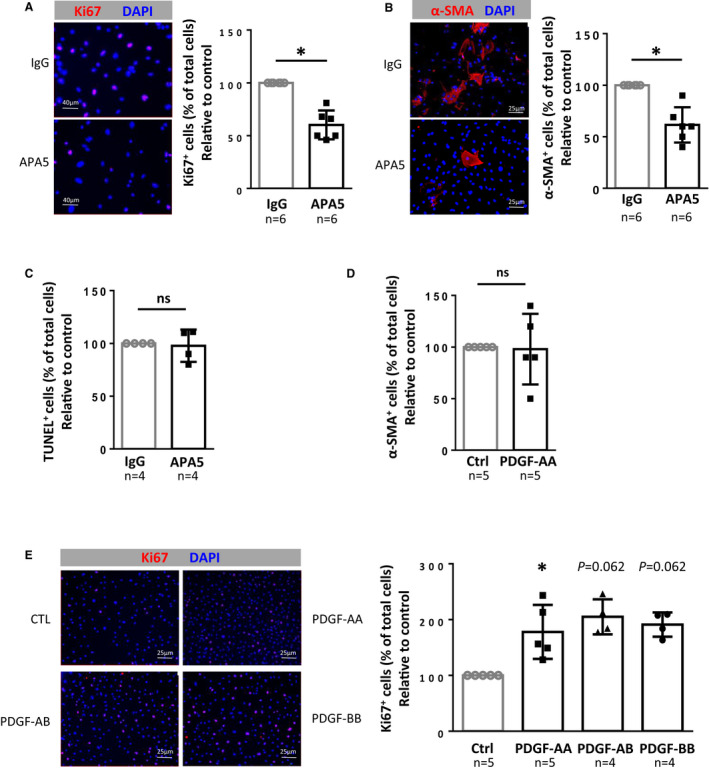
A, Effect of PDGFRα inhibition on PW1+ progenitor cell proliferation. Pulmonary PW1+ progenitor cells were sorted by FACS, incubated for 5 days with IgG control or APA5 and proliferative cells were then labeled with anti‐Ki67 antibody (red). (Left) Representative immunofluorescence images and (right) quantification of PW1+ progenitor cell proliferation as a ratio of Ki67+ cells over total cells reported to control (n=6). B, Effect of PDGFRα inhibition on PW1+ progenitor cell differentiation into SMC. FACS‐sorted pulmonary PW1+ progenitor cells were incubated for 5 days with IgG control or APA5 and then labeled with anti‐α‐SMA antibody (red). (Left) Representative immunofluorescence images and (right) quantification of PW1+ progenitor cell differentiation as a ratio of α‐SMA+ cells over total cells reported to control (n=6). C, Effect of PDGFRα inhibition on PW1+ progenitor cell apoptosis. FACS‐sorted pulmonary PW1+ progenitor cells were incubated for 5 days with IgG control or APA5 and then labeled following the TUNEL method. The ratio of TUNEL+ cells over total cells was determined and reported to control (n=4). D, Effect of PDGFRα activation on PW1+ progenitor cell differentiation into SMCs. FACS‐sorted pulmonary PW1+ progenitor cells were incubated for 5 days with IgG control or PDGF‐AA, ‐AB, or ‐BB (20 ng/mL) and labeled with anti–α‐SMA antibody (red). The ratio of α‐SMA+ cells over total cells was reported to control (n=5). E, Effect of PDGFRα activation on PW1+ progenitor cell proliferation. FACS‐sorted pulmonary PW1+ progenitor cells were incubated for 5 days with either vehicle, PDGF‐AA, PDGF‐AB, or PDGF‐BB (20 ng/mL) and then labeled with anti‐Ki67 antibody (red). (Left) Representative immunofluorescence images and (right) quantification of PW1+ progenitor cell proliferation as a ratio of Ki67+ cells over total cells reported to control (n=4–5). Bars represent means and whiskers represent SD. *P<0.05, ns indicates not significant (1‐tailed Wilcoxon signed‐ranked test). FACS indicates fluorescence‐activated cell sorting; PDGF, platelet‐derived growth factor; PDGFRα, platelet‐derived growth factor receptor type α; PW1, protein widely 1; SMA, smooth muscle actin; and SMC, smooth muscle cell; and TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; and vWF, von Willebrand factor.
PDGFRα Inhibition Limits Vascular Remodeling After Long‐Term Chronic Hypoxia but Does Not Reduce Right Ventricular Systolic Pressure, Whereas PDGFRα Activation Induces PH
Our in vitro and in vivo data show that the PDGFRα pathway controls pulmonary neomuscularization by regulating PW1+/PDGFRα+ progenitor cell proliferation. We therefore investigated the long‐term effects of PDGFRα inhibition or activation on PH development.
PDGFRα was inhibited during CH by treating mice (7 males and 1 female per group) with APA5 blocking antibody. Chronic hypoxia was confirmed by increased blood hemoglobin concentration (Figure S8). After 21 days of CH, PDGFRα inhibition significantly reduced the number of fully muscularized vessels (27.2%±9.7% with APA5 treatment versus 39.8%±6.2% with IgG treatment; P=0.039) and increased the number of partially muscularized vessels (53.8%±8% with APA5 treatment versus 39.7%±7.1% with IgG treatment; P=0.008) as compared with IgG‐treated control mice (Figure 6A). However, APA5 treatment did not significantly modify right ventricular systolic pressure (RVSP) or the Fulton index (Figure 6B and 6C) as compared with IgG‐treated control mice, and both values were increased as compared with normoxic mice. This result demonstrates that PDGFRα inhibition for 21 days reduces CH‐induced pulmonary vessel neomuscularization but does not prevent either increased RVSP or right ventricular hypertrophy.
Figure 6. PDGFRα inhibition with APA5 limits vascular neomuscularization but does not prevent chronic hypoxia (CH)‐induced PH.
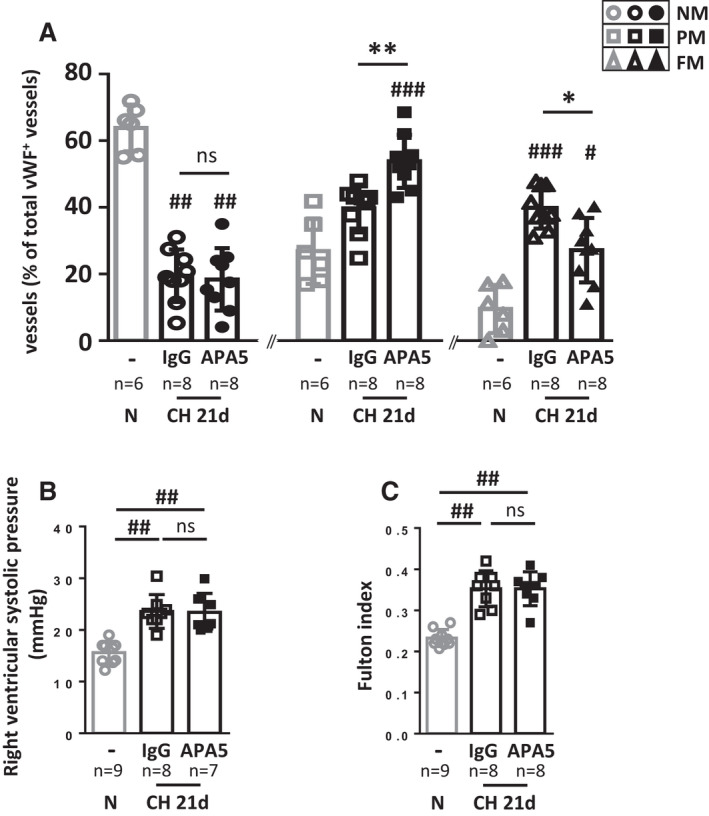
PW1nLacz mice were maintained untreated under normoxia (N) or under CH for 21 days and treated with control IgG or PDGFRα blocking antibody APA5. A, Pulmonary vessel muscularization was determined by immunofluorescence in lungs of control untreated normoxic mice (n=6) or of IgG‐ and APA5‐treated mice after 21 days of chronic hypoxia (CH 21d, n=9) using anti–α‐smooth muscle actin (α‐SMA) and anti–von Willebrand factor (vWF) antibodies. For each animal, >100 vWF+ vessels (<100 μm) were analyzed for muscularization (α‐SMA+). Vessels were identified as nonmuscularized (NM), partially muscularized (PM) or fully muscularized (FM). B, Right ventricular systolic pressure (RVSP, mm Hg) was measured in untreated normoxic mice (n=9) and in IgG‐ and APA5‐treated mice after 21 days of CH (CH 21d, n=7–8). C, Right ventricle hypertrophy was measured as the Fulton index in normoxic untreated mice (n=9) and in IgG‐ and APA5‐treated mice in CH (CH 21d, n=8). Bars represent means and whiskers represent SD. *P<0.05, **P<0.01, ns indicates not significant for APA5 vs IgG within CH 21d group; # P<0.05, ## P<0.01 vs normoxic; ns indicates not significant (Kruskal‐Wallis and Dunn). PDGFRα, platelet‐derived growth factor receptor type α; PH, pulmonary hypertension; PW1, protein widely 1; and SMA, smooth muscle actin.
The effect of PDGFRα activation was first studied in tamoxifen‐induced PW1nLacz/+/PDGFRα+/(S)K/Rosa‐CREERT2 mice (Figure 7A). After 5 weeks of constitutive PDGFRα activation, male mice displayed both a significant RVSP increase (22.5±5.2 mm Hg, Figure 7B) and right ventricular hypertrophy (Fulton index of 0.31±0.02; Figure 7C) as compared with noninduced littermates (17.1±3.5 mm Hg, P=0.0185, Figure 7B; and 0.24±0.03, P=0.0043, Figure 7C, respectively). Interestingly, female mice did not develop PH by this time point, with no demonstrable increase in RVSP and no right ventricular hypertrophy (Figure 7D and 7E), although they did present with increased neomuscularization 15 days after tamoxifen induction, similar to males (data not shown). Total weight, heart rate, and left ventricle weight were not modified in tamoxifen‐induced mice (Figure S9). These results show that PDGFRα activation first triggers vascular remodeling and eventually PH. To further confirm that PDGFRα activation induces PH, we treated male and female mice with PDGF‐AA (150 ng/mouse daily) for 5 weeks (Figure 7F). In male mice, this treatment increased RVSP (19.7±2.6 mm Hg versus 14.7±1.8 mm Hg in the control group, P=0.03; Figure 7G) but not to the same extent as observed after a similar duration of constitutive PDGFRα activation (22.5±5.2 mm Hg; Figure 7B). Five weeks of PDGF‐AA treatment also did not lead to right ventricular hypertrophy (Figure 7H). In female mice, the RVSP or Fulton index were not significantly modified by PDGF‐AA treatment in contrast to the constitutive PDGFRα activation model (Figure 7I and 7J).
Figure 7. Constitutive PDGFRα activation leads to pulmonary hypertension.
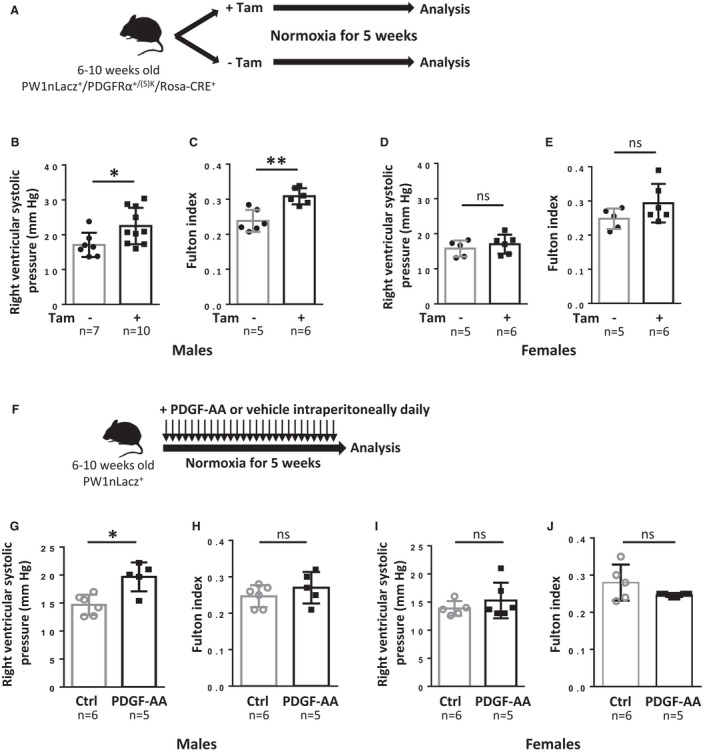
A, Timeline of tamoxifen (Tam) induction and analysis of PW1nLacz/PDGFRα+/(S)K/Rosa‐CRE+ mice. B to E, Analysis of mice 5 weeks after tamoxifen induction. B and D, Right ventricular systolic pressure (RVSP) was measured in control or tamoxifen‐treated (B), males (n=7–10) and (D), females (n=5–6). C and E, Right ventricular hypertrophy was determined using the Fulton index (right ventricular weight to left ventricular+septum weight ratio) in control or tamoxifen‐treated (C), males (n=5–6) and (E), females (n=5–6). F, Timeline of PDGF‐AA treatment and analysis of PW1nLacz+ mice. G through J, Analysis of mice after 5 weeks of PDGF‐AA treatment. G and I, Right RVSP was measured in control or PDGF‐AA‐treated (G), males (n=5–6) and (I), females (n=5–6). H and J, Right ventricular hypertrophy was determined using the Fulton index in control or PDGF‐AA–treated (H), males (n=5–6) and (J), females (n=5–6). Bars represent means and whiskers represent SD. *P<0.05, **P<0.01; ns indicates not significant (2‐tailed Mann‐Whitney); PDGF, platelet‐derived growth factor; PDGFRα, platelet‐derived growth factor receptor type α; and PW1, protein widely 1.
Altogether, these results establish that constitutive PDGFRα activation is a novel mouse model of mild pulmonary hypertension in males, recapitulating the pulmonary vascular alterations observed during chronic hypoxia.
Lung Fibrosis Is Not Induced by PDGFRα Activation nor Reduced by PDGFRα Inhibition During Chronic Hypoxia
Long‐term constitutive PDGFRα activation was reported to induce fibrosis in multiple organs including the lung. 19 We therefore sought to determine whether lung fibrosis was induced by PDGFRα activation in our model or if it was inhibited by blocking PDGFRα during CH. We measured the mRNA expression of fibrosis factors (collagens 1 and 3 and transforming growth factor‐β) and assessed changes in collagen levels by quantifying picrosirius red histological staining in lung samples from male PW1nLacz/+/PDGFRα+/(S)K/Rosa‐CREERT2 mice. After 5 weeks of constitutive PDGFRα activation, collagen 1a1 and 3a1 mRNA levels remained unchanged, but transforming growth factor‐β mRNA expression increased in lungs (Figure 8A). Histological analyses and quantification of picrosirius red‐stained lung sections did not indicate any significant differences in fibrotic area size between tamoxifen‐induced mice and noninduced littermates (Figure 8B and 8C and Figure S10). Samples did, however, demonstrate areas of thickened alveolar walls in tamoxifen‐induced mice indicating local tissue alterations (Figure S10). Constitutive PDGFRα activation for 5 to 6 weeks was therefore not sufficient to elicit pulmonary fibrosis at this stage. We also investigated the effect of PDGFRα inhibition on late CH‐induced pulmonary fibrosis with the APA5 blocking antibody 19 , 20 (7 males and 1 female per group). As expected, CH significantly increased transforming growth factor‐β and collagen 3a1 and 1a1 mRNA expression (Figure 8D). No difference was observed between control IgG and APA5‐treated mice exposed to CH. Histological analyses and quantification of fibrotic areas by picrosirius red staining (Figure 8E and 8F and Figure S11) confirmed similar CH‐induced increases in collagen‐rich areas in both control IgG‐ and APA5‐treated mice (20.1%±4% and 17.5%±5.1%, respectively) as compared with control normoxic mice (6.3%±1.7%; P=0.001 and P=0.004, respectively). Taken together, our results concerning PDGFRα inhibition under chronic hypoxia or constitutive PDGFRα activation under normoxia demonstrate that PDGFRα signaling does not affect lung fibrosis in our models.
Figure 8. Lung fibrosis is not induced by PDGFRα activation and is not reduced by PDGFRα inhibition.
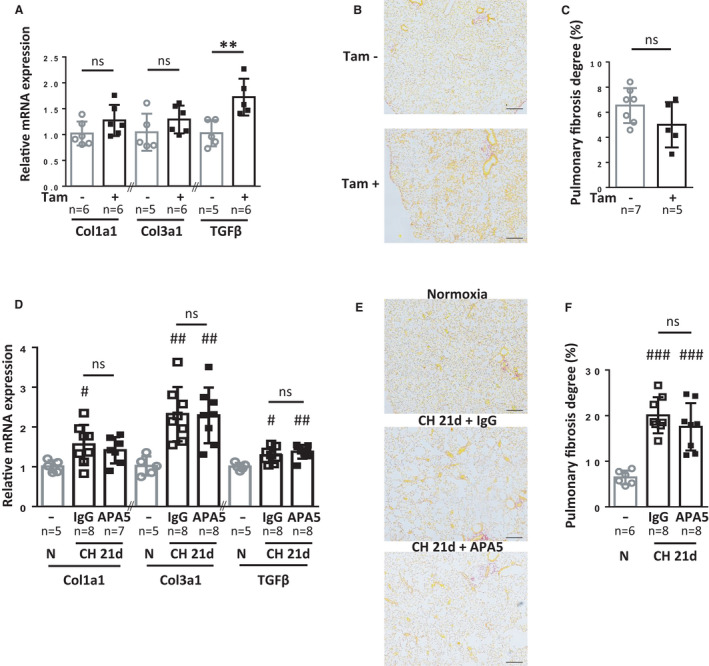
A through C, Fibrosis evaluation in noninduced (n=6–7) or tamoxifen‐induced PW1nLacz/+/PDGFRα+/(S)K/Rosa‐CRE+ mice 5 weeks after tamoxifen induction (n=5–6). D to F, Fibrosis evaluation in untreated mice under normoxia (n=5) or IgG and APA5‐treated mice after 21 days of CH (n=7–8). A and D, RT‐qPCR measurements of mRNA expression for collagen 1a1, collagen 3a1, and transforming growth factor‐β in lungs. B and E, Representative images of picrosirius red staining for collagen (red) in pulmonary parenchyma. Larger images are displayed in Figures S10 and S11. C and F, Quantification of the picrosirius red‐stained area as a measure of fibrosis using Histolab analysis. Fibrosis area (% of total area) was determined as the mean fibrotic area of four large lung sections for each animal. Bars represent means and whiskers represent SD. *P<0.05, **P<0.01 Tam+ vs Tam‐ group, 2‐tailed Mann‐Whitney; # P<0.05, ## P<0.01 vs normoxic, Kruskal‐Wallis and Dunn. CH indicates chronic hypoxia; ns, not significant for APA5 vs IgG within CH 21d group in D and F (scale bar 200 µm); PDGFRα, platelet‐derived growth factor receptor type α; PW1, protein widely 1; and RT‐qPCR, reverse transcriptase quantitative polymerase chain reaction.
Discussion
To identify new therapeutic targets for this life‐threatening pulmonary vascular disease, it is crucial that we understand the molecular mechanisms leading to new SMC production, which contributes to pulmonary vascular remodeling and ultimately PAH. In this study, we describe, for the first time, the precise role of PDGFRα in progenitor cell proliferation, in neomuscularization, and in PH development. Using a novel genetic model and a specific blocking antibody, our results provide evidence that PDGFRα is a pivotal player regulating neomuscularization, and thus participates in PH development by inducing CD34+/PDGFRα+/PW1+ vascular progenitor cell proliferation and their differentiation into SMCs. Importantly, our data suggest that this pathway intervenes during vascular remodeling of human patients with iPAH.
PDGF pathway activation has been associated with pulmonary hypertension development and related vascular remodeling in both experimental PH models and in humans. 5 , 9 Our observation of increased expression of PDGF‐A, ‐B, and ‐D after 1 day of CH suggest an early activation of both PDGFR pathways. However, most studies have examined the role of PDGFRβ signaling in experimental PH models, in particular during CH, 10 , 11 and the role of the PDGFRα pathway has never been directly investigated. In the present study, and using several experimental models, we found that this pathway induces pulmonary vascular remodeling. While PDGFRβ activation was shown to stimulate pulmonary pericyte and SMC proliferation, our data demonstrate that PDGFRα induces pulmonary vascular progenitor cell expansion, leading to their differentiation into vascular SMCs in the adult mouse lung. Related observations have been reported during pulmonary development where PDGFRα signaling is necessary to form alveolar α‐SMA+ SMCs (also known as alveolar myofibroblasts) during secondary septation. 17 , 21 Indeed, PDGFRα+ cells harvested shortly after birth express α‐SMA, but postnatal cells have lost this expression. 22 , 23 Our results (herein and in Dierick et al 3 ) are consistent with these observations and are the first—to our knowledge—to link PDGFRα activation of progenitor cells and pulmonary vascular SMC production in adult mice.
Multiple cellular sources provide new SMCs during PH development. CH‐activated SMCs have been shown to proliferate and lead to neomuscularization of downstream arterioles. 2 Pulmonary pericytes are also a source of periendothelial α‐SMA+ cells in newly muscularized vessels. 1 , 4 Our previous data 3 and our observation that CH induces an early increase in perivascular PDGFRα+/PW1+ progenitor cells argue for an important role played by PDGFRα+/PW1+ progenitor cells during PH remodeling in experimental models. We demonstrate here that PDGFRα inhibition reduces early CH‐induced neomuscularization and limits neomuscularization in late CH. Using a novel mouse model of inducible constitutive PDGFRα activation enabling PW1+ progenitor cell detection and tracing, we further establish a major role for PDGFRα signaling in inducing vascular progenitor cells to form new SMCs. Interestingly, constitutive PDGFRβ activation did not induce neomuscularization per se but slightly increased the number of fully muscularized vessels after CH. 11 These contrasting results suggest that PDGFRβ‐expressing SMCs and pericytes—which participate in CH‐induced vascular remodeling processes 1 , 2 , 24 —may first need to be activated by other pathways to become responsive to PDGFRβ activation. Strikingly, imatinib and APA5 only partially inhibited early CH‐induced neomuscularization, suggesting that pathways other than PDGF also regulate production of new SMCs. Indeed, previous studies demonstrate that factors such as interleukin‐6 and fibroblast growth factor‐2 participate in PH‐associated vascular remodeling processes. 1 , 25 These other pathways may predominate when PDGFRα+/PW1+ progenitor cell recruitment is inhibited. Further studies in other rodent PH experimental models (eg, monocrotaline or Sugen‐CH rat) will be necessary to confirm the importance of the PDGFRα pathway in vascular remodeling.
Our study establishes constitutive PDGFRα activation as a novel PH experimental model independent of CH. The pathological mechanisms leading to increased pulmonary pressure in this model are not clearly elucidated. Vessel neomuscularization might participate in disease development by inducing arteriolar stiffening and increasing the vascular resistance, thus leading to altered SMC or endothelial cell function. This result underlines the major role of PDGFRα in controlling pulmonary vascular structure and suggests that the beneficial effects observed using imatinib in PH experimental models are partly dependent on inhibiting PDGFRα activity. 9 We observed that in this transgenic model, progenitor cells differentiate into new SMCs. This observation contrasts with our in vitro results showing that PDGFRα activation alone does not induce progenitor cell differentiation. One hypothesis is that other factors are present in the local environment of these pulmonary progenitor cells in vivo that are lacking in our in vitro system. stromal cell–derived factor‐1 (C‐X‐C motif chemokine 12) could be one of these factors, as we already established that in vivo blocking of C‐X‐C chemokine receptor type 4—the stromal cell–derived factor‐1 receptor—inhibits their differentiation into vascular SMC. 3 Female mice appear to be protected against PDGFRα activation‐induced PH development, despite an induction of neomuscularization. The mechanisms leading to this sex difference are unclear but may involve estrogen‐dependent pathways, which are known to attenuate PH in several rodent models. 26 Estrogens are potent vasodilators in particular through activation of NO and prostacyclin production and inhibition of endothelin synthesis by endothelial cells. 26 One hypothesis could hence be that estrogen stimulation may favor vasodilation reducing pulmonary vascular resistance in females. Studying these protective mechanisms will help decipher the relationship between pulmonary hypertension and vascular remodeling.
PDGFRα activation has been associated with fibrosis development, where mice with a constitutively activated form of this receptor ultimately develop multiple organs fibrosis. 19 The production of collagens 1 and 3 and total collagen deposition remained unchanged in mouse lungs after 5 weeks of constitutive PDGFRα activation. Indeed, studies using this genetic model report fibrosis after 3 to 6 months. 13 , 19 Thus, in this model of constitutive PDGFRα activation, PH is induced much earlier than fibrosis development, indicating that fibrotic remodeling is not initially involved in increasing pulmonary blood pressure or pulmonary vascular neomuscularization. In addition, PDGFRα inhibition using APA5 blocking antibody did not prevent long‐term CH‐induced fibrosis, indicating that PDGFRα does not regulate CH‐associated fibrosis. Fibrocytes have been shown to contribute to CH‐induced fibrosis 27 and pericyte‐like ABCG2+ mesenchymal stem cells (PDGFRα‐) could also participate in fibrotic remodeling because of their myofibroblastic capacities. 28 Our in vitro results suggest that PDGFRα activation leads to proliferation of PDGFRα+ progenitor cells, whose fate is then controlled by other factors depending on the pathophysiological context. During early CH, these factors orientate PDGFRα+ progenitor cells toward differentiation into vascular SMCs, and our data argue against a PDGFRα‐dependent fibroblastic fate during late CH. Other factors may induce differentiation toward fibroblasts in different experimental conditions, such as aging. 19 The involvement of the PDGFRα pathway and of PDGFRα+ cells in the context of fibrosis is controversial. Several studies suggest that PDGFRα signaling is important for fibrosis development in various organs, including the lung. 13 , 16 , 19 , 23 , 29 However, Kishi et al recently showed that specific PDGFRα inhibition does not reduce bleomycin‐induced fibrosis. 30 These results, together with our data, suggest that the relationship between PDGFRα+ cells, the PDGFRα pathway, and fibrosis is more complex than simple PDGFRα activation of PDGFRα+ fibroblast precursors, and may involve other factors such as transforming growth factor‐β. 31 Interestingly, PDGF‐BB intratracheal administration was previously shown to induce SMC proliferation, confirming our results, but also perivascular and peribronchial collagen deposition. 32 These observations combined with ours suggest a complex interplay between both PDGFR receptors to induce fibrosis. In line with these results, although PDGFRα is often considered as a fibroblast marker, growing evidence now shows that a proportion of PDGFRα+ cells are progenitor or stem cells. 12 Our results confirm that the pulmonary PDGFRα+ progenitor cell population contains cells with SMC differentiation potential together with cells able to differentiate into alveolar myofibroblasts and peribronchiolar SMC, as observed during developmental alveolar septation 33 or pneumectomy‐induced realveolarization. 34 Indeed, the diversity of PDGFRα+ cells has been illustrated by single‐cell transcriptome analysis 23 ; therefore, PDGFRα activation may also target other progenitor cells because of the overlapping expression of different markers and pathways or via paracrine mechanisms.
We observed that PDGFRα activation by PDGF‐AA increases vascular progenitor cell proliferation but does not induce their differentiation into SMCs, indicating that other pathways could be involved. This is in agreement with previous studies showing that PDGFRα or PDGF‐AA regulate stem/progenitor cell renewal or proliferation 15 , 35 and can also maintain precursors in an immature state. 36 , 37 On the other hand, other groups have published that PDGFRα is important for stem/progenitor cell differentiation, in particular into α‐SMA‐expressing cells or fibroblasts. 13 , 38 , 39 , 40 These putative conflicting roles of PDGFRα might originate from the type of progenitor/stem cells used. We previously showed that CD34+/PDGFRα+/PW1+ cells differentiate toward terminally differentiated smooth muscle myosin heavy chain+ SMCs, 3 which might not be the case for the stem cells used in other studies. It is also possible that our in vitro experiments lack some factors that, in combination with PDGF‐AA, could induce differentiation of these cells. For example, primitive endoderm cell proliferation seems to depend on both fibroblast growth factor receptor 1 and PDGFRα, whereas cell specification seems to be only fibroblast growth factor receptor 1 dependent. 41
In keeping with our previous study, 3 PW1+ cells in control human lungs were found clustered and mostly perivascular with a large majority of these cells coexpressing PDGFRα. We observed increased numbers of perivascular PW1+/PDGFRα+ in CH mice and in patients with iPAH, suggesting similar proliferative mechanisms, in particular PDGFRα activation. PW1+/PDGFRα+/α‐SMA+ cells were absent from control arteries but were observed in the remodeled arterial wall from patients with iPAH and could be differentiating PW1+/PDGFRα+ progenitor cells participating in vascular remodeling as suggested by our data in the CH mouse (herein and in Dierick et al 3 ). However, we cannot rule out the hypothesis that a proportion of medial PW1+/PDGFRα+ cells may be derived from proliferating PW1+ SMCs reexpressing PDGFRα or from endothelial cells through endothelial‐to‐mesenchymal transition. The increased expression of PDGF and PDGFRα has been previously documented in pulmonary vessels from patients with iPAH and further strengthens our data. 5 Part of the beneficial effects observed in patients treated with imatinib could therefore be attributable to the inhibition of the PDGFRα pathway. 42 However, unlike imatinib, 11 blocking PDGFRα is not sufficient to reduce pulmonary pressure in CH mice. This suggests that the vasorelaxing effect of imatinib observed on pulmonary vessels could be attributable to PDGFRβ inhibition. 43 , 44 Therefore, a multitarget therapeutic strategy associating PDGFRα inhibition with current vasodilating therapies may provide beneficial effects by inhibiting progenitor cell recruitment and vessel neomuscularization.
In conclusion, our study highlights PDGFRα as an essential regulator of lung vessel neomuscularization via the recruitment of vascular progenitor cells and opens new avenues for pulmonary hypertension medication.
Sources of Funding
This work was supported by the Institute of Cardiometabolism and Nutrition (ANR‐10‐IAHU‐05). Drs Sassoon, Marazzi, and Harvey were supported by a grant from the Fondation Leducq (grant 13CVD01; CardioStemNet project). Drs Nadaud and Guignabert were supported by a grant from the Agence Nationale de la Recherche (ANR‐15‐CE14‐0020). Drs Humbert and C. Guignabert also acknowledge support from the Fondation pour la Recherche Médicale (DEQ20150331712), the Département Hospitalo‐Universitaire Thorax Innovation, the LabEx LERMIT (ANR‐10‐LABX‐0033), and the French PAH patient association (HTAP France). Dr Solinc was supported by the French Ministry of Research and Education. Dr Dierick was supported by the French Ministry of Research and Education and by a Canadian Institutes of Health Research project grant (to the Stephanie Lehoux laboratory). Dr Harvey also acknowledges support from the National Health and Medical Research Council of Australia (APP1118576; 1074386), the Australian Research Council Special Initiative in Stem Cell Science (SR110001002) and the New South Wales Government Department of Health.
Disclosures
Over the past 3 years, Dr Guignabert reports grants from Acceleron, Janssen, and ShouTi; and grants and personal fees from Merck, outside of the submitted work. In the past 3 years, Dr Humbert reports grants, personal fees, and nonfinancial support from Actelion, Bayer, GlaxoSmithKline, and MSD, outside of the submitted work. He is a member of the Scientific Advisory Board of Morphogen‐IX. The remaining authors have no disclosures to report.
Supporting information
Acknowledgments
We thank all participants of the French PH Network PulmoTension. The authors also thank Professor Elie Fadel, Professor Olaf Mercier, and all pathologists from the Department of Pathology at Marie Lannelongue Hospital ‐ Groupe Hospitalier Paris Saint Joseph for their expertise and support. We thank Nilay Akkoyunlu, Marta de Almeida, and Chloé Azevedo for technical help, and Professor Stephane Hatem and Dr Nadine Suffee‐Mosbah for helpful discussions We thank Doriane Forêt, Olivier Brégerie, Kim Nguyen and Yannick Martinez from the animal facility UMS28 for mice care and breeding.
Supplemental Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.023021
For Sources of Funding and Disclosures, see page 16.
References
- 1. Ricard N, Tu LY, Le Hiress M, Huertas A, Phan C, Thuillet R, Sattler C, Fadel E, Seferian A, Montani D, et al. Increased pericyte coverage mediated by endothelial‐derived fibroblast growth factor‐2 and interleukin‐6 is a source of smooth muscle‐like cells in pulmonary hypertension. Circulation. 2014;129:1586–1597. doi: 10.1161/CIRCULATIONAHA.113.007469 [DOI] [PubMed] [Google Scholar]
- 2. Sheikh AQ, Misra A, Rosas IO, Adams RH, Greif DM. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med. 2015;7:308ra159. doi: 10.1126/scitranslmed.aaa9712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dierick F, Héry T, Hoareau‐Coudert B, Mougenot N, Monceau V, Claude C, Crisan M, Besson V, Dorfmüller P, Marodon G, et al. Resident PW1+ progenitor cells participate in vascular remodeling during pulmonary arterial hypertension. Circ Res. 2016;118:822–833. doi: 10.1161/circresaha.115.307035 [DOI] [PubMed] [Google Scholar]
- 4. Bordenave J, Tu LY, Berrebeh N, Thuillet R, Cumont A, Le Vely B, Fadel E, Nadaud S, Savale L, Humbert M, et al. Lineage tracing reveals the dynamic contribution of pericytes to the blood vessel remodeling in pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2020;40:766–782. doi: 10.1161/ATVBAHA.119.313715 [DOI] [PubMed] [Google Scholar]
- 5. Perros F, Montani D, Dorfmüller P, Durand‐Gasselin I, Tcherakian C, Le Pavec J, Mazmanian M, Fadel E, Mussot S, Mercier O, et al. Platelet‐derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:81–88. doi: 10.1164/rccm.200707-1037OC [DOI] [PubMed] [Google Scholar]
- 6. Humbert M, Monti G, Fartoukh M, Magnan A, Brenot F, Rain B, Capron F, Galanaud P, Duroux P, Simonneau G, et al. Platelet‐derived growth factor expression in primary pulmonary hypertension: comparison of HIV seropositive and HIV seronegative patients. Eur Respir J. 1998;11:554–559. [PubMed] [Google Scholar]
- 7. Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, Olschewski A, Pullamsetti SS, Schermuly RT, Stenmark K, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53:1801887. doi: 10.1183/13993003.01887-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andrae J, Gallini R, Betsholtz C. Role of platelet‐derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–2821. doi: 10.1172/JCI24838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. ten Freyhaus H, Berghausen EM, Janssen W, Leuchs M, Zierden M, Murmann K, Klinke A, Vantler M, Caglayan E, Kramer T, et al. Genetic ablation of PDGF‐dependent signaling pathways abolishes vascular remodeling and experimental pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2015;35:1236–1245. doi: 10.1161/ATVBAHA.114.304864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dahal BK, Heuchel R, Pullamsetti SS, Wilhelm J, Ghofrani HA, Weissmann N, Seeger W, Grimminger F, Schermuly RT. Hypoxic pulmonary hypertension in mice with constitutively active platelet‐derived growth factor receptor‐β. Pulm Circ. 2011;1:259–268. doi: 10.4103/2045-8932.83448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Farahani RM, Xaymardan M. Platelet‐derived growth factor receptor alpha as a marker of mesenchymal stem cells in development and stem cell biology. Stem Cells Int. 2015;2015:1. doi: 10.1155/2015/362753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iwayama T, Steele C, Yao L, Dozmorov MG, Karamichos D, Wren JD, Olson LE. PDGFRα signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev. 2015;29:1106–1119. doi: 10.1101/gad.260554.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Calver AR, Hall AC, Yu WP, Walsh FS, Heath JK, Betsholtz C, Richardson WD. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron. 1998;20:869–882. doi: 10.1016/S0896-6273(00)80469-9 [DOI] [PubMed] [Google Scholar]
- 15. Rivera‐Gonzalez GC, Shook BA, Andrae J, Holtrup B, Bollag K, Betsholtz C, Rodeheffer MS, Horsley V. Skin adipocyte stem cell self‐renewal is regulated by a PDGFA/AKT‐signaling axis. Cell Stem Cell. 2016;19:738–751. doi: 10.1016/j.stem.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saito Y, Chikenji T, Ozasa Y, Fujimiya M, Yamashita T, Gingery A, Iba K. PDGFR signaling mediates hyperproliferation and fibrotic responses of subsynovial connective tissue cells in idiopathic carpal tunnel syndrome. Sci Rep. 2017;7:16192. doi: 10.1038/s41598-017-16443-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gouveia L, Betsholtz C, Andrae J. PDGF‐A signaling is required for secondary alveolar septation and controls epithelial proliferation in the developing lung. Development. 2018;145. doi: 10.1242/dev.161976 [DOI] [PubMed] [Google Scholar]
- 18. Besson V, Smeriglio P, Wegener A, Relaix F, Nait Oumesmar B, Sassoon DA, Marazzi G. PW1 gene/paternally expressed gene 3 (PW1/Peg3) identifies multiple adult stem and progenitor cell populations. Proc Natl Acad Sci USA. 2011;108:11470–11475. doi: 10.1073/pnas.1103873108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Olson LE, Soriano P. Increased PDGFRalpha activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell. 2009;16:303–313. doi: 10.1016/j.devcel.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liao C‐H, Akazawa H, Tamagawa M, Ito K, Yasuda N, Kudo Y, Yamamoto R, Ozasa Y, Fujimoto M, Wang P, et al. Cardiac mast cells cause atrial fibrillation through PDGF‐A‐mediated fibrosis in pressure‐overloaded mouse hearts. J Clin Invest. 2010;120:242–253. doi: 10.1172/JCI39942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ntokou A, Klein F, Dontireddy D, Becker S, Bellusci S, Richardson WD, Szibor M, Braun T, Morty RE, Seeger W, et al. Characterization of the platelet‐derived growth factor receptor‐α‐positive cell lineage during murine late lung development. Am J Physiol Lung Cell Mol Physiol. 2015;309:L942–L958. doi: 10.1152/ajplung.00272.2014 [DOI] [PubMed] [Google Scholar]
- 22. Branchfield K, Li R, Lungova V, Verheyden JM, McCulley D, Sun X. A three‐dimensional study of alveologenesis in mouse lung. Dev Biol. 2016;409:429–441. doi: 10.1016/j.ydbio.2015.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li R, Bernau K, Sandbo N, Gu J, Preissl S, Sun X. Pdgfra marks a cellular lineage with distinct contributions to myofibroblasts in lung maturation and injury response. Elife. 2018;7. doi: 10.7554/eLife.36865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chow K, Fessel JP, Ihida‐Stansbury K, Schmidt EP, Gaskill C, Alvarez D, Graham B, Harrison DG, Wagner DH, Nozik‐Grayck E, et al. Dysfunctional resident lung mesenchymal stem cells contribute to pulmonary microvascular remodeling. Pulm Circ. 2013;3:31–49. doi: 10.4103/2045-8932.109912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Izikki M, Guignabert C, Fadel E, Humbert M, Tu LY, Zadigue P, Dartevelle P, Simonneau G, Adnot S, Maitre B, et al. Endothelial‐derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. J Clin Invest. 2009;119:512–523. doi: 10.1172/JCI35070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tofovic SP, Jackson EK. Estradiol metabolism: crossroads in pulmonary arterial hypertension. Int J Mol Sci. 2020;21:116. doi: 10.3390/ijms21010116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N, Stenmark KR. Hypoxia‐induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–669. doi: 10.2353/ajpath.2006.050599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marriott S, Baskir RS, Gaskill C, Menon S, Carrier EJ, Williams J, Talati M, Helm K, Alford CE, Kropski JA, et al. ABCG2pos lung mesenchymal stem cells are a novel pericyte subpopulation that contributes to fibrotic remodeling. Am J Physiol–Cell Physiol. 2014;307:C684–C698. doi: 10.1152/ajpcell.00114.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mueller AA, van Velthoven CT, Fukumoto KD, Cheung TH, Rando TA. Intronic polyadenylation of PDGFRα in resident stem cells attenuates muscle fibrosis. Nature. 2016;540:276–279. doi: 10.1038/nature20160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kishi M, Aono Y, Sato S, Koyama K, Azuma M, Abe S, Kawano H, Kishi J, Toyoda Y, Okazaki H, et al. Blockade of platelet‐derived growth factor receptor‐β, not receptor‐α ameliorates bleomycin‐induced pulmonary fibrosis in mice. PLoS One. 2018;13:e0209786. doi: 10.1371/journal.pone.0209786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heinzelmann K, Noskovičová N, Merl‐Pham J, Preissler G, Winter H, Lindner M, Hatz R, Hauck SM, Behr J, Eickelberg O. Surface proteome analysis identifies platelet derived growth factor receptor‐alpha as a critical mediator of transforming growth factor‐beta‐induced collagen secretion. Int J Biochem Cell Biol. 2016;74:44–59. doi: 10.1016/j.biocel.2016.02.013 [DOI] [PubMed] [Google Scholar]
- 32. Yi ES, Lee H, Yin S, Piguet P, Sarosi I, Kaufmann S, Tarpley J, Wang NS, Ulich TR. Platelet‐derived growth factor causes pulmonary cell proliferation and collagen deposition in vivo. Am J Pathol. 1996;149:539–548. [PMC free article] [PubMed] [Google Scholar]
- 33. Green J, Endale M, Auer H, Perl A‐KT. Diversity of interstitial lung fibroblasts is regulated by platelet‐derived growth factor receptor α kinase activity. Am J Respir Cell Mol Biol. 2016;54:532–545. doi: 10.1165/rcmb.2015-0095OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen L, Acciani T, Le Cras T, Lutzko C, Perl A‐KT. Dynamic regulation of platelet‐derived growth factor receptor α expression in alveolar fibroblasts during realveolarization. Am J Respir Cell Mol Biol. 2012;47:517–527. doi: 10.1165/rcmb.2012-0030OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kikuchi A, Pradhan‐Sundd T, Singh S, Nagarajan S, Loizos N, Monga SP. Platelet‐derived growth factor receptor α contributes to human hepatic stellate cell proliferation and migration. Am J Pathol. 2017;187:2273–2287. doi: 10.1016/j.ajpath.2017.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu Q, Zhao X, Zheng K, Li H, Huang H, Zhang Z, Mastracci T, Wegner M, Chen Y, Sussel L, et al. Genetic evidence that Nkx2.2 and PDGFRa are major determinants of the timing of oligodendrocyte differentiation in the developing CNS. Development. 2014;141:548–555. doi: 10.1242/dev.095323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baroti T, Zimmermann Y, Schillinger A, Liu L, Lommes P, Wegner M, Stolt CC. Transcription factors Sox5 and Sox6 exert direct and indirect influences on oligodendroglial migration in spinal cord and forebrain. Glia. 2016;64:122–138. doi: 10.1002/glia.22919 [DOI] [PubMed] [Google Scholar]
- 38. Ball SG, Shuttleworth CA, Kielty CM. Platelet‐derived growth factor receptor‐alpha is a key determinant of smooth muscle alpha‐actin filaments in bone marrow‐derived mesenchymal stem cells. Int J Biochem Cell Biol. 2007;39:379–391. doi: 10.1016/j.biocel.2006.09.005 [DOI] [PubMed] [Google Scholar]
- 39. Kim B‐J, Kim Y‐H, Lee Y‐A, Jung S‐E, Hong YH, Lee E‐J, Kim B‐G, Hwang S, Do JT, Pang M‐G, et al. Platelet‐derived growth factor receptor‐alpha positive cardiac progenitor cells derived from multipotent germline stem cells are capable of cardiomyogenesis in vitro and in vivo. Oncotarget. 2017;8:29643–29656. doi: 10.18632/oncotarget.16772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu T, Liu N, Shao Y, Huang Y, Zhu D. MiR‐218 regulated cardiomyocyte differentiation and migration in mouse embryonic stem cells by targeting PDGFRα. J Cell Biochem. 2019;120:4355–4365. doi: 10.1002/jcb.27721 [DOI] [PubMed] [Google Scholar]
- 41. Molotkov A, Soriano P. Distinct mechanisms for PDGF and FGF signaling in primitive endoderm development. Dev Biol. 2018;442:155–161. doi: 10.1016/j.ydbio.2018.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frost AE, Barst RJ, Hoeper MM, Chang H‐J, Frantz RP, Fukumoto Y, Galié N, Hassoun PM, Klose H, Matsubara H, et al. Long‐term safety and efficacy of imatinib in pulmonary arterial hypertension. J Heart Lung Transplant. 2015;34:1366–1375. doi: 10.1016/j.healun.2015.05.025 [DOI] [PubMed] [Google Scholar]
- 43. Rieg AD, Bünting NA, Cranen C, Suleiman S, Spillner JW, Schnöring H, Schröder T, von Stillfried S, Braunschweig T, Manley PW, et al. Tyrosine kinase inhibitors relax pulmonary arteries in human and murine precision‐cut lung slices. Respir Res. 2019;20:111. doi: 10.1186/s12931-019-1074-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Abe K, Toba M, Alzoubi A, Koubsky K, Ito M, Ota H, Gairhe S, Gerthoffer WT, Fagan KA, McMurtry IF, et al. Tyrosine kinase inhibitors are potent acute pulmonary vasodilators in rats. Am J Respir Cell Mol Biol. 2011;45:804–808. doi: 10.1165/rcmb.2010-0371OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takakura N, Yoshida H, Kunisada T, Nishikawa S, Nishikawa SI. Involvement of platelet‐derived growth factor receptor‐alpha in hair canal formation. J Invest Dermatol. 1996;107:770–777. doi: 10.1111/1523-1747.ep12371802 [DOI] [PubMed] [Google Scholar]
- 46. Takakura N, Yoshida H, Ogura Y, Kataoka H, Nishikawa S, Nishikawa S. PDGFR alpha expression during mouse embryogenesis: immunolocalization analyzed by whole‐mount immunohistostaining using the monoclonal anti‐mouse PDGFR alpha antibody APA5. J Histochem Cytochem. 1997;45:883–893. doi: 10.1177/002215549704500613 [DOI] [PubMed] [Google Scholar]
- 47. Dudgeon K, Rouet R, Kokmeijer I, Schofield P, Stolp J, Langley D, Stock D, Christ D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci USA. 2012;109:10879–10884. doi: 10.1073/pnas.1202866109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Asli NS, Xaymardan M, Patrick R, Farbehi N, Cornwell J, Forte E, Waardenberg AJ, Janbandhu V, Kesteven S, Chandrakanthan V, et al. PDGFRα signaling in cardiac fibroblasts modulates quiescence, metabolism and self‐renewal, and promotes anatomical and functional repair. bioRxiv. 2019;225979. doi: 10.1101/225979 [DOI] [Google Scholar]
- 49. Relaix F, Weng X, Marazzi G, Yang E, Copeland N, Jenkins N, Spence SE, Sassoon D. Pw1, a novel zinc finger gene implicated in the myogenic and neuronal lineages. Dev Biol. 1996;177:383–396. doi: 10.1006/dbio.1996.0172 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.