

Abstract
Background
Myocardial dysfunction is the leading cause of early death following successful cardiopulmonary resuscitation (CPR) in people with cardiac arrest (CA), which is potentially driven by cell pyroptosis mediated by NOD‐like receptor pyrin domain 3 (NLRP3) inflammasome. Recently, histone deacetylase 6 (HDAC6) inhibition was shown to exert effective myocardial protection against regional ischemia/reperfusion injury. In this study, we investigated whether tubastatin A, a specific histone deacetylase 6 inhibitor, could improve postresuscitation myocardial dysfunction through the inhibition of NLRP3‐mediated cell pyroptosis and its modulation mechanism.
Methods and Results
Healthy male white domestic swine were used to establish the model of CA/CPR in vivo, and the H9c2 cardiomyocyte hypoxia/reoxygenation model was used to simulate the CA/CPR process in vitro. Consequently, tubastatin A inhibited NLRP3 inflammasome activation, decreased proinflammatory cytokines production and cell pyroptosis, and increased cell survival after hypoxia/reoxygenation in H9c2 cardiomyocytes in vitro. In addition, tubastatin A increased the acetylated levels of transcription factor EB and its translocation to the nucleus, and its protective effect above was partly abrogated by transcription factor EB short interfering RNA after hypoxia/reoxygenation in H9c2 cardiomyocytes. Similarly, tubastatin A promoted cardiac transcription factor EB nuclear translocation, inhibited NLRP3‐mediated cell pyroptosis, and mitigated myocardial dysfunction after CA/CPR in swine.
Conclusions
The inhibition of histone deacetylase 6 activity by tubastatin A limited NLRP3 inflammasome activation and cell pyroptosis probably through the enhancement of transcription factor EB signaling, and therefore improved myocardial dysfunction after CA/CPR.
Keywords: cardiopulmonary resuscitation, cell pyroptosis, myocardial dysfunction, transcription factor EB, tubastatin A
Subject Categories: Cardiopulmonary Resuscitation and Emergency Cardiac Care
Nonstandard Abbreviations and Acronyms
- CA
cardiac arrest
- H/R
hypoxia/reoxygenation
- HDAC6
histone deacetylase 6
- I/R
ischemia/reperfusion
- NC
normal control
- NLRP3
NOD‐like receptor protein 3
- PI
propidium iodide
- ROS
reactive oxygen species
- TFEB
transcription factor EB
Clinical Perspective
What Is New?
The inhibition of histone deacetylase 6 activity by tubastatin A can alleviate cell damage after hypoxia/reoxygenation in H9c2 cardiomyocytes and improve myocardial dysfunction after cardiac arrest and resuscitation in swine, in which the protective effect is related to the inhibition of cell pyroptosis induced by NOD‐like receptor pyrin domain 3 inflammasome activation.
Furthermore, transcription factor EB, a potential regulator of NOD‐like receptor pyrin domain 3 inflammasome, mediates the protective role of tubastatin A probably through its acetylation and nuclear translocation.
What Are the Clinical Implications?
The inflammation, which is potentially controlled by cell pyroptosis through NOD‐like receptor pyrin domain 3 inflammasome activation, has been proven to be a key contributing factor to cardiomyocyte death after cardiac arrest and resuscitation.
Our findings indicate that histone deacetylase 6 and transcription factor EB are the key upstream regulators for NOD‐like receptor pyrin domain 3 inflammasome activation and cell pyroptosis, which may become effective therapeutic targets for postresuscitation myocardial protection through the regulation of inflammatory pathways in the clinical setting.
Sudden cardiac arrest (CA) is always a major public health problem. In China, it is estimated that 550 000 individuals annually have CA events, and their survival rate is <1%. 1 Following successful cardiopulmonary resuscitation (CPR), the ensuing myocardial dysfunction will result in systemic circulatory failure and early death. 2 , 3 Although several therapeutic interventions including remote ischemic postconditioning, polyethylene glycol‐20k, and aldehyde dehydrogenase 2 have been shown to provide effective myocardial protection after resuscitation in animal models 4 , 5 , 6 ; however, almost none of them have been successfully translated into clinical practice.
Cell pyroptosis, one novel form of programmed cell death characterized by plasma membrane rupture and inflammatory cytokine release, plays a key role in the development of regional ischemia/reperfusion (I/R) injury of various organs. 7 , 8 , 9 Studies have confirmed that cell pyroptosis is mainly induced by the NOD‐like receptor protein 3 (NLRP3)–caspase‐1 pathway, in which the NLRP3 inflammasome promotes the activation of pro‐caspase‐1 into caspase‐1, then cleaves the proinflammatory cytokines interleukin‐1β and interleukin‐18 and the pyroptotic substrate gasdermin D, and finally results in cell death and excessive release of interleukin‐1β and interleukin‐18. 10 In the setting of CA/CPR, one investigation has preliminarily demonstrated that cell pyroptosis induced by NLRP3 inflammasome was involved in the development of myocardial dysfunction after resuscitation. 11
Recently, there has been increasing evidence that histone deacetylase inhibitors are a promising strategy for the treatment of I/R injuries. 12 In particular, tubastatin A, a specific inhibitor of histone deacetylase 6 (HDAC6), has been proven to alleviate regional cardiac and cerebral I/R injury through its prevention of target protein deacetylation. 13 , 14 Currently, another 2 studies have demonstrated that the inhibition of HDAC6 by tubastatin A protected the macrophage and neuron against nicotine‐ and 6‐hydroxydopamine–induced injury, respectively, in which the mechanism was related to the decrease of cell pyroptosis and proinflammatory cytokines through inhibiting NLRP3 inflammasome activation. 15 , 16 However, it is unknown whether tubastatin A alleviates regional organ I/R injury by targeting NLRP3‐mediated pyroptosis. In addition, whether tubastatin A protects the heart against global I/R injury triggered by CA/CPR and its potential mechanism require further investigations.
In the present study, we investigated the influence of HDAC6 inhibition by tubastatin A on NLRP3‐mediated pyroptosis in an H9c2 cardiomyocyte hypoxia/reoxygenation (H/R) model and its effect on postresuscitation myocardial dysfunction in a swine CA/CPR model. We demonstrated that tubastatin A blocked NLRP3–caspase‐1 pathway activation, and then decreased cell pyroptosis after H/R in H9c2 cardiomyocytes. Likewise, tubastatin A inhibited myocardial NLRP3 inflammasome activation and cell pyroptosis and therefore improved its dysfunction after CA/CPR in swine. In addition, we identified that tubastatin A promoted the acetylation and nuclear translocation of transcription factor EB (TFEB), which is an essential regulator for the autophagy‐lysosome pathway and can negatively regulate the activation of NLRP3 inflammasome through autophagy induction. 17 , 18 We further demonstrated that TFEB siRNA partly abrogated the protective effect of tubastatin A after H/R in H9c2 cardiomyocytes. Thus, our study suggested that tubastatin A enhanced the TFEB signaling to inhibit NLRP3 inflammasome activation and cell pyroptosis, and further improved postresuscitation myocardial dysfunction after CA/CPR.
Methods
The data, analytic methods, and study materials in this study will be made available from the corresponding author on reasonable request to other researchers for purposes of reproducing the results or replicating the procedure.
Cell Culture Studies
The H9c2 cardiomyocyte cell line was purchased from Meixuan Biotechnology Inc. (Shanghai, China). The cells were normally cultured in DMEM supplemented with 10% fetal bovine serum and kept at 37 °C in a humidified 95% air/5% CO2 incubator. The procedure of H/R was performed by initially exposing the cells in a multigas incubator (Mco‐5m, Panasonic, Tokyo, Japan) containing 94% N2, 5% CO2, and 1% O2 at 37 °C with glucose‐deprived media for 2 hours and then culturing cells under normal condition with complete media for another 24 hours.
Initially, the safety of tubastatin A was evaluated in H9c2 cardiomyocytes under normal conditions, in which the cells were treated with tubastatin A at concentrations of 0.5, 1, 2.5, 5, 10, 20, 40, 80, and 160 μmol/L. Subsequently, 6 safe concentrations of tubastatin A were chosen to observe its protective role in H9c2 cardiomyocytes after H/R. After that, the optimal concentration of tubastatin A was chosen to use for the remaining experiments.
To observe the role of TFEB in cardiomyocyte protection provided by tubastatin A after H/R, the cells were transfected with TFEB siRNA for 48 hours and then used to establish the H/R model. Initially, 4 different RNAi duplexes were obtained from Meixuan Biotechnology Inc. (Shanghai, China), and their sequences were as follows: TFEB siRNA1 forward CGGACUCAGUUUCUCCUUAUGCACATT, reverse UGUGCAUAAGGAGAAACUGAGUCCGTT; TFEB siRNA2 forward GAUGAGUACUUCAUGGACCUGUCUUTT, reverse AAGACAGGUCCAUGAAGUACUCAUCTT; TFEB siRNA3 forward GUCCUGUUUCAAUGUCGGUUUCACCTT, reverse UGAUGUUGAACCUGCGUCUUCUCUCTT; control siRNA forward GUCCUGUUUCAAUGUCGGUUUCACCTT, reverse GGUGAAACCGACAUUGAAACAGGACTT. Subsequently, H9c2 cardiomyocytes were seeded into plates and then transfected by short interfering RNA (siRNA) using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA). Finally, the transfection efficiency was examined by reverse transcriptase polymerase chain reaction and western blot. The TFEB siRNA with the best knockdown efficiency was used in the studies.
Swine Study
This animal study was reviewed and approved by the Institutional Animal Care and Use Committee of the Second Affiliated Hospital, Zhejiang University School of Medicine. All the experimental procedures were in accordance with institutional guidelines. Healthy male white domestic swine, aged 4 to 6 months, weighing 36±2 kg, were purchased from Shanghai Jiagan Biotechnology Inc. (Shanghai, China). The animals were sedated by intramuscular tiletamine/zolazepam (5 mg/kg) and xylazine (1 mg/kg), and then anesthetized by intravenous propofol (2 mg/kg) followed by a continuous infusion of propofol (4 mg/kg per hour). Subsequently, the animals were intubated and ventilated with a ventilator (Monnal T75, Air Liquide Medical Systems, Antony Cedex, France) with a tidal volume of 10 mL/kg, the peak flow of 40 L/min and fraction of inspired oxygen of 0.21. End‐tidal CO2 was monitored with a monitor/defibrillator (M Series, ZOLL Medical Corporation, Chelmsford, MA). For the measurements of aortic and right atrial pressures and the collection of arterial and venous blood samples, 2 fluid‐filled 7 Fr thermodilution‐tipped catheters were advanced from the right femoral artery and vein into the thoracic aorta and right atrium, respectively. For the measurements of stroke volume and global ejection fraction, a 4 Fr thermistor‐tipped arterial catheter was inserted into the left femoral artery, another 7 Fr central venous catheter was inserted into the right internal jugular vein, and then both were connected to the PiCCO system (PiCCOplus, Pulsion Medical Systems, Munich, Germany). For the induction of ventricular fibrillation, a 5 F pacing catheter was inserted from the right external jugular vein into the right ventricle. Heart rate and mean arterial pressure were continuously recorded by a patient monitoring system (iM60, Edan, Shenzhen, China). Baseline arterial pH and lactate concentrations were measured with a blood gas analyzer (i15, Edan, Shenzhen, China).
After the surgical preparation was completed, the animals were then randomized with the sealed envelope method into 1 of the 3 groups: sham group (n=6), CPR group (n=8), and CPR+tubastatin A group (n=8). A dose of 4.5 mg/kg of tubastatin A was intravenously administered at 5 minutes after successful resuscitation in the CPR+tubastatin A group, and the same volume of vehicle was similarly given in the sham and CPR groups. Sham animals only underwent the surgical preparation without experiencing the CA/CPR procedure. In the other 2 groups, ventricular fibrillation was induced by 1 mA alternating current delivered to the right ventricular endocardium. After 9 minutes of untreated ventricular fibrillation, CPR was manually performed by a ratio of 30:2 of compression to ventilation. The compression quality was monitored by a CPR feedback device (PlamCPR, Sunlife, Shanghai, China). The changes of coronary perfusion pressure during CPR were dynamically calculated according to the difference between decompression diastolic aortic and time‐coincident right atrial pressures. After 2 minutes of CPR, a dose of 20 µg/kg of epinephrine was given followed by the same administration at an interval of 3 minutes. After 6 minutes of CPR, a single 150‐J biphasic electrical shock was delivered by the monitor/defibrillator. If an organized rhythm with a mean arterial pressure >50 mm Hg persisted for ≥5 minutes, the animal was regarded as having return of spontaneous circulation. If not, CPR was immediately resumed for 2 minutes before another electrical shock. This protocol was repeated until successful resuscitation or for a total of 16 minutes. Following successful resuscitation, the animals were monitored for 4 hours and then sent back to their cages for an additional 20 hours of observation.
Cell Viability and Lactate Dehydrogenase Release Assay
Cell viability was evaluated using a CCK‐8 assay kit (Biyuntian Biotechnology, Shanghai, China) in a 96‐well plate. First, 10 μL of CCK‐8 reagent was added to each well and then incubated for 3 hours at 37 °C. Subsequently, the absorbance was measured at 450 nm using a microplate reader (Multiskan MK3, Thermo Fisher Scientific, Waltham, MA). Lactate dehydrogenase (LDH) release analysis was performed using an LDH assay kit (Abcam, Cambridge, MA). Briefly, the cell supernatant was collected, then mixed with the LDH assay buffer, and finally incubated at 37 °C for 1 hour. The mixture was measured at 450 nm using the microplate reader above.
Cell Apoptosis Detection
Cell apoptosis was measured using an annexin V‐fluorescein isothiocyanate/propidium iodide (PI) apoptosis kit (Solarbio, Beijing, China). Briefly, the cells were harvested and resuspended, and then incubated with 5 μL of annexin V–fluorescein isothiocyanate and 5 μL of PI for 20 minutes in the dark at room temperature. Subsequently, cell apoptosis was detected by flow cytometry (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (FlowJo LLC, Ashland, OR).
Reactive Oxygen Species Assay
The production of reactive oxygen species (ROS) was measured using an ROS assay kit (Solarbio, Beijing, China). First, the cells were washed with PBS, and then incubated with 10 μmol/L of 2′,7′‐dichloroflurorescein diacetate in a serum‐free medium for 30 minutes at 37 °C. Subsequently, the cells were washed again, and then their fluorescent intensity was immediately measured by flow cytometry (BD Biosciences) and analyzed using FlowJo software (FlowJo LLC).
Measurement of Myocardial Function and Cardiac Injury
The changes of stroke volume and global ejection fraction indicating myocardial function were measured with the PiCCO system at baseline and 0.5, 1, 2, and 4 hours after resuscitation. The serum levels of cardiac troponin I indicating cardiac injury were measured with ELISA kits (Meixuan Biotechnology Inc.) at baseline and 1, 2, 4, and 24 hours after resuscitation.
Immunoblotting Analysis
Protein samples were harvested from H9c2 cardiomyocytes after H/R and the myocardium at 24 hours after resuscitation. The separation of cytosolic and nuclear fractions was performed by centrifugation of samples at 16 000g. Subsequently, the samples were separated by SDS‐PAGE, then transferred to a polyvinylidene fluoride membrane, and finally blocked with 5% nonfat milk. After that, the membranes were incubated with primary anti‐NLRP3 (1:1000, Proteintech, Rosemount, IL), anticleaved caspase‐1 (1:1000, Cell Signaling Technology Inc., Danvers, MA), anti–gasdermin D (1:1000, Proteintech), anti‐HDAC6 (1:1000, Proteintech), anti‐GAPDH (1:5000, BBI Life Science Corporation, Shanghai, China), antiacetylated α‐tubulin (1:1000, Proteintech), anti–α‐tubulin (1:1000, Proteintech), anti‐TFEB (1:1000, Proteintech), antiacetylated lysine (1:1000, Cell Signaling Technology Inc.), antihistone H3 (1:1000, Abcam) at 4 °C for 24 hours, then rinsed with TBST solution, and finally incubated with the secondary antibody (1:5000, BBI Life Science Corporation) at room temperature for 1 hour. The protein was visualized with enhanced chemiluminescence substrates and analyzed by Image J software (National Institutes of Health, Bethesda, MD).
Measurement of Proinflammatory Cytokines
In the cell study, the cell supernatant was collected and then the contents of interleukin‐1β and interleukin‐18 were measured with ELISA kits (Meixuan Biotechnology Inc.) according to the manufacturer’s instructions. In the animal study, cardiac tissue specimens were harvested at 24 hours after resuscitation, then homogenized with normal saline on ice and centrifuged at 2500g at 4 °C for 15 minutes, and finally, the supernatants were collected for measuring the contents of interleukin‐1β and interleukin‐18 according to the same method above.
PI Staining
Pyroptotic cells were detected using PI staining. First, the cells were washed with PBS, then stained with 5 µmol/L of PI in each well at 37 °C for 15 minutes, and finally stained with DAPI for 5 minutes. After that, the images of the cells were immediately observed at ×200 magnification under a fluorescence microscope (Olympus, Tokyo, Japan). The average fluorescence intensity and the percentage of positive cells were assessed with Image J software (National Institutes of Health).
Immunofluorescence Staining
The distribution of TFEB in H9c2 cardiomyocytes after H/R and the myocardium at 24 hours after resuscitation were detected using immunofluorescence staining. Briefly, the specimens were fixed with 4% paraformaldehyde, then permeated with 0.3% Triton X‐100, and finally blocked by 10% goat serum. Subsequently, the specimens were incubated with anti‐TFEB (1:200, Proteintech) at 4 °C for 24 hours, then incubated with secondary antibody for 1 hour, and finally stained with DAPI for 10 minutes. After that, the fluorescence of TFEB protein adducts and DAPI were observed with the fluorescence microscope.
Statistical Analysis
The statistical analysis was performed with SPSS 20.0 statistical software (IBM, Armonk, NY). Continuous variables were presented as mean±SD when the data were confirmed with the Kolmogorov‐Smirnov test to be normally distributed, and then the comparisons between 2 groups were performed using the Student t test, and ≥2 groups were performed by 1‐way ANOVA. Nonnormally distributed data were presented as a median (25th–75th percentiles) and then compared with the Kruskal‐Wallis test. Comparisons between time‐based measurements within each group were performed with repeated‐measurement ANOVA. If there was a significant difference in the overall comparison of groups, comparisons between any other 2 groups were made by the Bonferroni test. Categorical variables were compared with Fisher’s exact test. A value of P<0.05 was considered statistically significant.
Results
Tubastatin A Treatment Reduced H/R‐Induced Cell Damage in H9c2 Cardiomyocytes
To investigate the effect of tubastatin A on cell damage after H/R in H9c2 cardiomyocytes, we established a H9c2 cardiomyocyte H/R model by 2 hours of hypoxia and 24 hours of reoxygenation. First, the safety of tubastatin A in H9c2 cardiomyocytes was evaluated, in which cell viability was significantly decreased when the concentration of tubastatin A was ≥80 μmol/L (Figure 1A). Subsequently, H9c2 cardiomyocytes were treated with 6 safe concentrations of tubastatin A 3 hours before cell hypoxia. We observed that cell viability was significantly decreased in all groups experiencing H/R when compared with the normal control (NC) group; however, it was gradually increased in a concentration‐dependent manner in all tubastatin A–treated groups, in which the viability was significantly higher in H9c2 cardiomyocytes receiving tubastatin A treatment at a concentration of ≥2.5 μmol/L when compared with the H/R group (Figure 1B). Thereafter, a concentration of 40 μmol/L of tubastatin A was chosen to investigate its effect on cell LDH release, apoptosis, and ROS after H/R in H9c2 cardiomyocytes. The level of LDH was detected by an LDH assay kit, and the ratio of cell apoptosis and ROS production were evaluated by flow cytometric analysis. Consequently, LDH level, apoptosis ratio, and ROS production in H9c2 cardiomyocytes were significantly increased in the H/R and H/R+tubastatin A groups compared with the NC group; however, all of them were significantly decreased in the H/R+tubastatin A group compared with the H/R group (Figure 1C through 1E). These data indicated that tubastatin A treatment markedly reduced H/R‐induced H9c2 cardiomyocyte damage by increasing cell viability and decreasing LDH release, apoptosis, and ROS production.
Figure 1. The effect of tubastatin A (TubA) on cell damage after hypoxia/reoxygenation (H/R) in H9c2 cardiomyocytes.
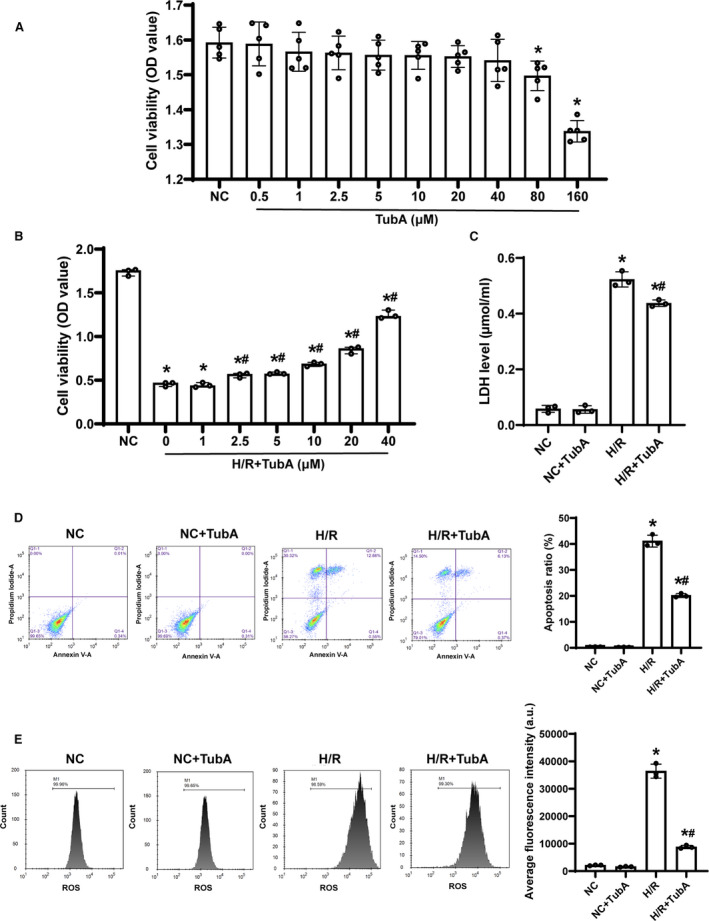
A and B, Cell viability evaluated by cell counting kit‐8 assay. C, The level of lactate dehydrogenase (LDH) detected by an LDH assay kit. D, Flow cytometric analysis and quantification of cell apoptosis. E, Flow cytometric analysis and quantification of reactive oxygen species (ROS) production. A and B, The cells were treated with different concentrations of tubastatin A. C through E, The cells were treated with tubastatin A (40 μmol/L). A, Each group contained 5 samples. B through E, Each group contained 3 samples. Data were presented as mean±SD. Group comparisons were performed by 1‐way ANOVA with Bonferroni test. *P<0.05 vs normal control (NC) group, # P<0.05 vs H/R group. OD indicates optical density.
Tubastatin A Treatment Inhibited H/R‐Induced NLRP3 Inflammasome Activation, Proinflammatory Cytokine Production, and Cell Pyroptosis in H9c2 Cardiomyocytes
To investigate the effect of tubastatin A on cell pyroptosis induced by NLRP3 inflammasome after H/R in H9c2 cardiomyocytes, a concentration of 40 μmol/L of tubastatin A was similarly administered in the same H/R model. The expression of NLRP3 inflammasome‐ and pyroptosis‐related proteins, the contents of proinflammatory cytokines, and cell pyroptosis were examined after H/R. Consequently, the protein levels of NLRP3, cleaved caspase‐1, and gasdermin D were significantly increased accompanied by significantly higher contents of interleukin‐1β and interleukin‐18 in H9c2 cardiomyocytes in the H/R and H/R+tubastatin A groups compared with the NC group; however, all of them were significantly decreased in H9c2 cardiomyocytes treated with tubastatin A compared with the H/R group (Figure 2A and 2B). In addition, the rate of positive cells measured by PI staining was significantly increased in the 2 groups compared with the NC group; nevertheless, tubastatin A treatment significantly decreased cell pyroptosis compared with the H/R group (Figure 2C). These results indicated that the protective role of tubastatin A was related to the inhibition of NLRP3 inflammasome activation, inflammatory response, and cell pyroptosis after H/R in H9c2 cardiomyocytes.
Figure 2. The effect of tubastatin A (TubA) on cell pyroptosis induced by NOD‐like receptor pyrin domain 3 (NLRP3) inflammasome after hypoxia/reoxygenation (H/R) in H9c2 cardiomyocytes.
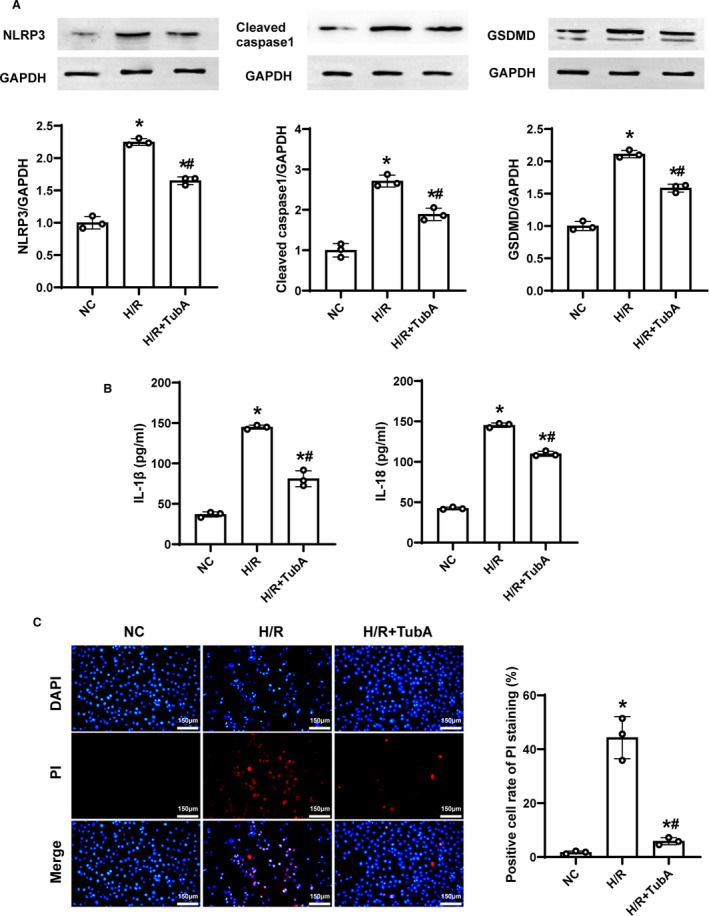
A, Representative immunoblotting and quantification of NLRP3, cleaved caspase‐1 and gasdermin D (GSDMD). B, The contents of interleukin‐1β (IL‐1β) and interleukin‐18 (IL‐18) measured by ELISA. C, Representative photographs of propidium iodide (PI) staining and quantification of positive cells (scale bar=150 µm, ×200 magnification). A through C, The cells were treated with tubastatin A (40 μmol/L). Each group contained 3 samples. Data were presented as mean±SD. Group comparisons were performed by 1‐way ANOVA with Bonferroni test. *P<0.05 vs normal control (NC) group, # P<0.05 vs H/R group.
Tubastatin A Treatment Promoted TFEB Acetylation and Nuclear Translocation After H/R in H9c2 Cardiomyocytes
To investigate the effect of tubastatin A on TFEB signaling after H/R in H9c2 cardiomyocytes, the levels of acetylated, cytoplasmic, and nuclear TFEB in H9c2 cardiomyocytes were measured after H/R. We observed that the acetylation of α‐tubulin considered as the established HDAC6 substrate was significantly decreased in the H/R and H/R+tubastatin A groups compared with the NC group; however, its acetylation was significantly restored in H9c2 cardiomyocytes receiving tubastatin A treatment compared with the H/R group (Figure 3A). Similarly, we found that the level of acetylated TFEB measured by immunoprecipitation and immunoblotting was significantly lower in the 2 groups compared with the NC group; nevertheless, tubastatin A significantly enhanced TFEB acetylation compared with the H/R group (Figure 3B). Furthermore, the levels of cytoplasmic and nuclear TFEB were measured by immunoblotting and immunofluorescence staining, in which cytoplasmic TFEB was decreased while nuclear TFEB was increased in the 2 groups experiencing H/R compared with the NC group. In particular, tubastatin A treatment further promoted cytoplasmic TFEB into the nucleus compared with the H/R group (Figure 3C through 3E). Our results indicated that tubastatin A treatment enhanced TFEB signaling after H/R in H9c2 cardiomyocytes.
Figure 3. The effect of tubastatin A (TubA) on transcription factor EB (TFEB) signaling after hypoxia/reoxygenation (H/R) in H9c2 cardiomyocytes.
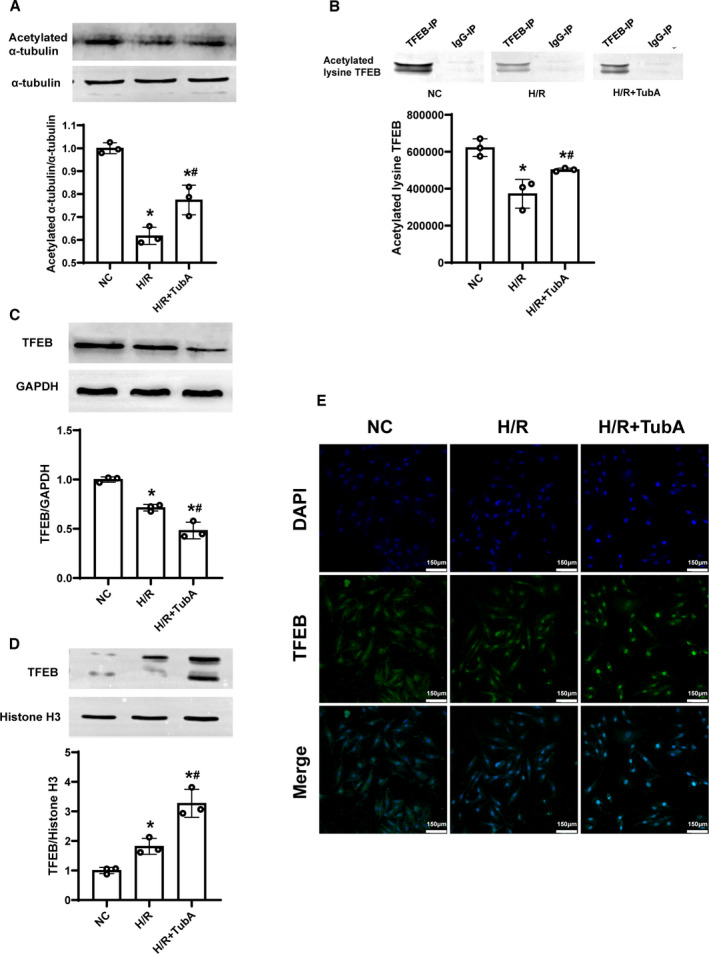
A, Representative immunoblotting and quantification of acetylated α‐tubulin. B, Immunoprecipitation of TFEB, and representative immunoblotting and quantification of acetylated lysine TFEB. C and D, Representative immunoblotting and quantification of cytoplasmic and nuclear TFEB. E, Representative photographs of immunofluorescence staining of TFEB nuclear translocation (scale bar=150 µm, ×200 magnification). A through E, The cells were treated with tubastatin A (40 μmol/L). Each group contained 3 samples. Data were presented as mean±SD. Group comparisons were performed by 1‐way ANOVA with Bonferroni test. *P<0.05 vs normal control (NC) group, # P<0.05 vs H/R group.
TFEB Mediated the Protective Role of Tubastatin A After H/R in H9c2 Cardiomyocytes
To determine if TFEB is a key regulator of tubastatin A in cardiomyocyte protection after H/R, we investigated the effect of TFEB knockdown after H/R in H9c2 cardiomyocytes. TFEB was knocked down with siRNA, which was confirmed by reverse transcriptase polymerase chain reaction and immunoblotting (Figure 4A). Similarly, the activation of NLRP3 inflammasome, the release of proinflammatory cytokines, and the occurrence of cell pyroptosis in H9c2 cardiomyocytes were observed in all groups experiencing H/R; however, all of them were significantly milder in the H/R+tubastatin A group compared with the H/R group. Most importantly, cell pyroptosis induced by NLRP3 inflammasome after H/R was significantly aggravated in the H/R+siTFEB (the knockdown of TFEB with short interfering RNA) group compared with the H/R group. In addition, cardiomyocyte protection provided by tubastatin A treatment was partly abrogated in the H/R+tubastatin A+siTFEB group compared with the H/R+tubastatin A group (Figure 4B through 4D). These results indicated that tubastatin A treatment alleviated H/R‐induced cardiomyocyte NLRP3 inflammasome activation and pyroptosis probably through the enhancement of TFEB signaling.
Figure 4. The effect of transcription factor EB (TFEB) knockdown on cardiomyocyte protection provided by tubastatin A (TubA) treatment after hypoxia/reoxygenation (H/R) in H9c2 cardiomyocytes.
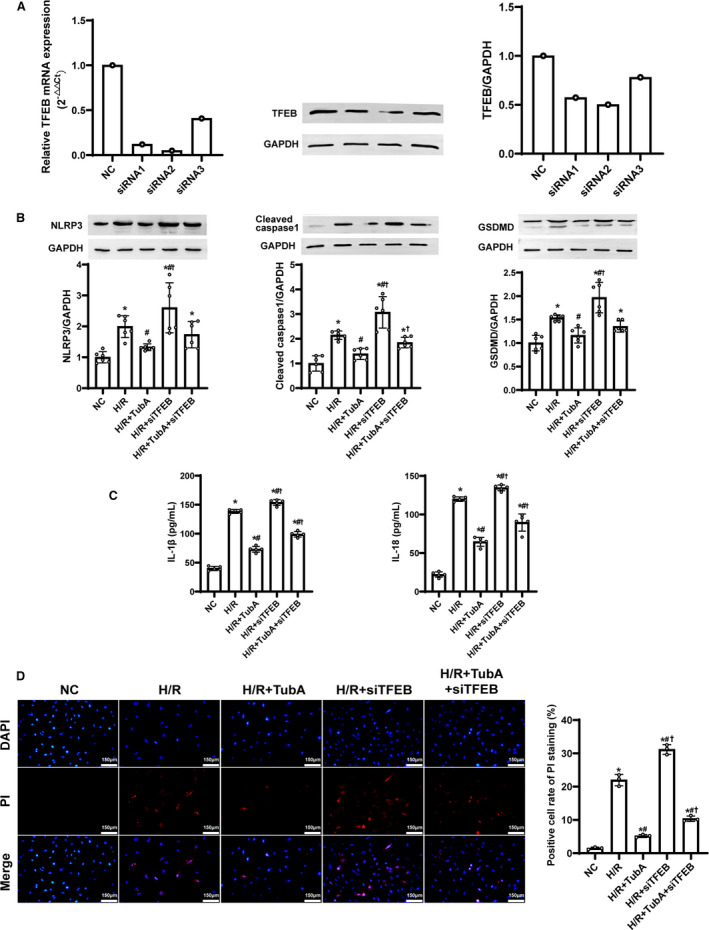
A, Knockdown of TFEB with short interfering RNA (siRNA) and its verification by reverse transcriptase polymerase chain reaction and immunoblotting. B, Representative immunoblotting and quantification of NOD‐like receptor pyrin domain 3 (NLRP3), cleaved caspase‐1 and gasdermin D (GSDMD) (n=6 samples per group). C, The contents of interleukin‐1β (IL‐1β) and interleukin‐18 (IL‐18) measured by enzyme‐linked immunosorbent assay (n=5 samples per group). D, Representative photographs of propidium iodide (PI) staining and quantification of positive cells (n=3 samples per group, scale bar=150 µm, ×200 magnification). B through D, The cells were treated with tubastatin A (40 μmol/L). Data were presented as mean±SD. Group comparisons were performed by 1‐way ANOVA with Bonferroni test. *P<0.05 vs normal control (NC) group, # P<0.05 vs H/R group, † P<0.05 vs H/R+tubastatin A group. siTFEB indicates the knockdown of TFEB with short interfering RNA.
Baseline and CPR Outcomes of the Animal Study
In total, 22 experiments were performed and completed in the animal study. There were no significant differences in baseline body weight, heart rate, mean arterial pressure, end‐tidal CO2, and arterial pH, and lactate among the 3 groups (Figure 5A through 5F). A swine model of CA/CPR was established by 9 minutes of untreated ventricular fibrillation and then 6 minutes of CPR in the CPR and CPR+tubastatin A groups. The levels of coronary perfusion pressure during CPR were maintained at an even level, and no significant difference was observed between the 2 groups (Figure 5G). Consequently, 7 of the 8 animals in the CPR group and 6 of the 8 animals in the CPR+tubastatin A group were successfully resuscitated, in which the rate of return of spontaneous circulation was not significantly different between the 2 groups (Figure 5H). In addition, the differences in duration of CPR, dosage of epinephrine, and number of defibrillations were not significant between the 2 groups (Figure 5I through 5K). These results indicated that the same baseline and procedural characteristics of CA/CPR were achieved in the CPR and CPR+tubastatin A groups.
Figure 5. Baseline characteristics and cardiopulmonary resuscitation (CPR) outcomes of the animal study.
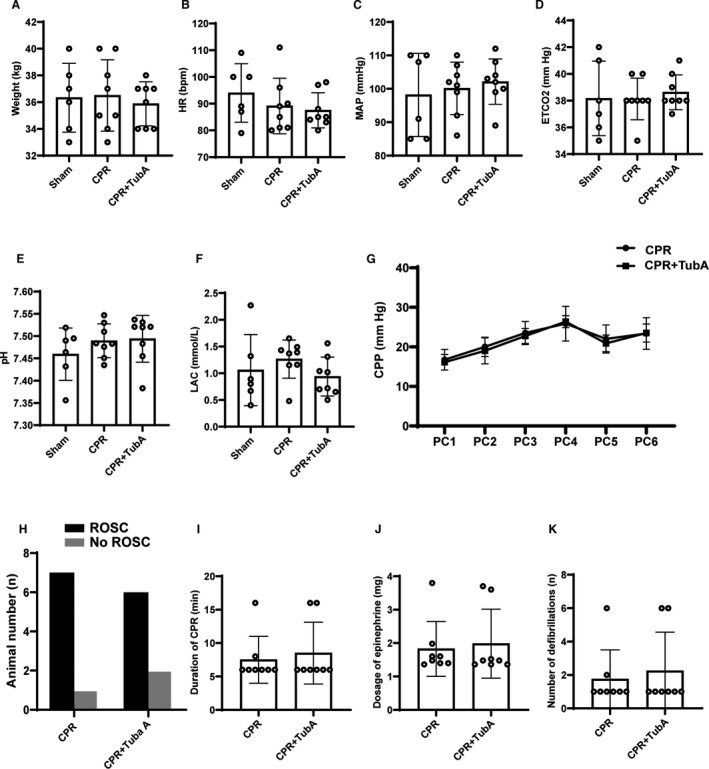
A through F, Baseline body weight (BW), heart rate (HR), mean arterial pressure (MAP), end‐tidal CO2, and arterial pH and lactate (LAC). G, The changes of coronary perfusion pressure (CPP) during CPR. PCn indicates n minutes after precordial compression. H through K, Return of spontaneous circulation (ROSC), duration of CPR, dosage of epinephrine and number of defibrillations during CPR. A through K, The sham group contained 6 swine at baseline, and the other 2 groups had 8 swine at baseline and during CPR. Data were presented as mean±SD in BW, HR, MAP, end‐tidal CO2, pH, LAC and CPP, in which group comparisons were performed by 1‐way ANOVA with Bonferroni test or Student t test. Data were presented as median (25th–75th percentiles) in duration of CPR, dosage of epinephrine, and number of defibrillations, in which group comparisons were performed by Kruskal‐Wallis test. ROSC was compared with Fisher’s exact test.
Tubastatin A Treatment Improved Myocardial Dysfunction and Alleviated Cardiac Injury After CA/CPR in Swine
To investigate the effect of tubastatin A on myocardial damage after CA/CPR in swine, a dose of 4.5 mg/kg of tubastatin A was administered at 5 minutes after resuscitation. The PiCCO system was used to measure myocardial function at baseline and during the 4 hours of postresuscitation observation. Consequently, postresuscitation myocardial function, as evaluated by the changes in stroke volume and global ejection fraction, was significantly impaired in all the resuscitated animals in the CPR and CPR+tubastatin A groups when compared with the sham group. However, myocardial dysfunction was gradually improved after resuscitation in the two groups. Most importantly, the improvement in myocardial function was significantly greater at all time points after resuscitation in the CPR+tubastatin A group than in the CPR group (Figure 6A and 6B).
Figure 6. The effect of tubastatin A (TubA) on myocardial damage after cardiac arrest (CA) and resuscitation in swine.
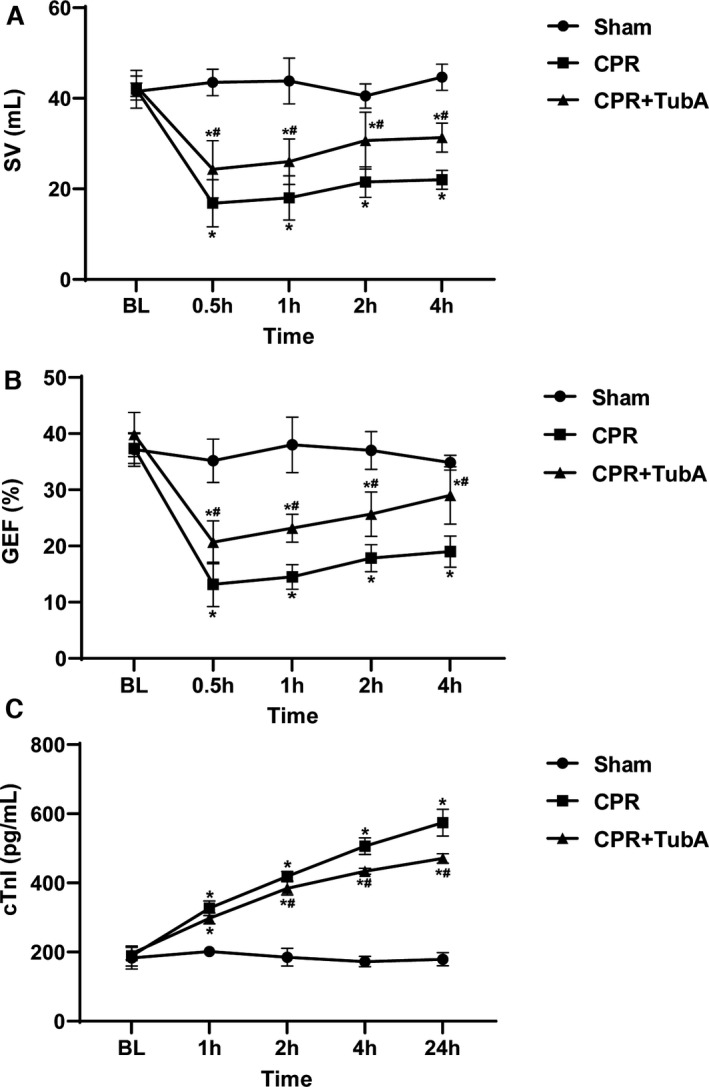
A and B, Stroke volume (SV) and global ejection fraction (GEF) evaluated by the PiCCO system at baseline and 0.5, 1, 2, and 4 hours after resuscitation. C, Serum cardiac troponin I (cTnI) evaluated by ELISA at baseline and 1, 2, 4, and 24 hours after resuscitation. A through C, Tubastatin A (4.5 mg/kg) was administered at 5 minutes after resuscitation. The sham group contained 6 swine throughout the experiment. The cardiopulmonary resuscitation (CPR) group had 8 swine at baseline, 7 swine at 1, 2, and 4 hours after resuscitation, and 6 swine at 24 hours after resuscitation. The CPR+tubastatin A group had 8 swine at baseline, and 6 swine at each time point after resuscitation. Data were presented as mean±SD. Myocardial function and injury biomarker between groups were compared by 1‐way ANOVA with Bonferroni test, and time‐based measurements within each group were compared by repeated‐measurement ANOVA. *P<0.05 vs sham group, # P<0.05 vs CPR group. BL indicates baseline.
We next examined whether tubastatin A treatment alleviated cardiac injury after CA/CPR in swine. The serum levels of cardiac troponin I used as one biomarker of cardiac injury were measured by ELISA at baseline and during the 24 hours of postresuscitation observation. Consequently, serum cardiac troponin I was significantly increased after resuscitation in the CPR and CPR+tubastatin A groups when compared with the sham group. However, the increase in cardiac troponin I was significantly slower, starting 2 hours after resuscitation in the CPR+tubastatin A group, than in the CPR group (Figure 6C). Together, our results indicated that tubastatin A treatment provided a powerful myocardial protection after CA/CPR in swine.
Tubastatin A Treatment Inhibited Cardiac NLRP3 Inflammasome‐Mediated Cell Pyroptosis and Promoted Its TFEB Nuclear Translocation After CA/CPR in Swine
To test whether the inhibition of cell pyroptosis mediated by NLRP3 Inflammasome is involved in the mechanism of myocardial protection induced by tubastatin A after resuscitation, we examined cardiac HDAC6, NLRP3 inflammasome, proinflammatory cytokines, and cell pyroptosis at 24 hours after resuscitation. Consequently, the expression levels of HDAC6, NLRP3, cleaved caspase‐1, and gasdermin D, and the contents of interleukin‐1β and interleukin‐18 in myocardium were significantly increased in all the resuscitated animals in the CPR and CPR+tubastatin A groups compared with the sham group; however, the increase in them were significantly less in the animals receiving tubastatin A treatment compared with the CPR group (Figure 7A and 7B). The results indicated that the inhibition of HDAC6 by tubastatin A treatment inhibited NLRP3 inflammasome‐mediated cell pyroptosis so as to improve myocardial dysfunction after CA/CPR in swine.
Figure 7. The effect of tubastatin A (TubA) on cardiac NOD‐like receptor pyrin domain 3 (NLRP3)‐mediated cell pyroptosis and transcription factor EB (TFEB) signaling after cardiac arrest (CA) and resuscitation in swine.
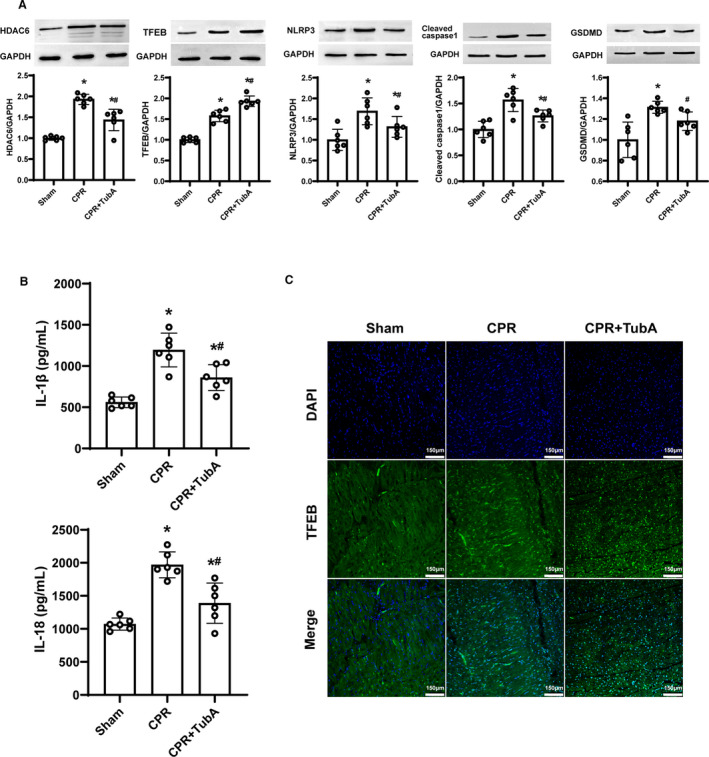
A, Representative immunoblotting and quantification of histone deacetylase 6 (HDAC6), TEFB, NLRP3, cleaved caspase‐1 and gasdermin D (GSDMD). B, The contents of interleukin‐1β (IL‐1β) and interleukin‐18 (IL‐18) measured by ELISA. C, Representative photographs of immunofluorescence staining of TFEB nuclear translocation (scale bar=150 µm, ×200 magnification). A through C, Tubastatin A (4.5 mg/kg) was administered at 5 minutes after resuscitation. The sham, cardiopulmonary resuscitation (CPR) and CPR+tubastatin A groups contained 6 swine at 24 hours after resuscitation, respectively. Myocardial tissue samples were measured at 24 hours after resuscitation. Data were presented as mean±SD. Group comparisons were performed by 1‐way ANOVA with Bonferroni test. *P<0.05 vs sham group, # P<0.05 vs CPR group.
To determine if TFEB is a downstream target of tubastatin A for myocardial protection after resuscitation, we examined the effect of tubastatin A on TFEB expression and nuclear translocation in myocardium at 24 hours after resuscitation. Consequently, the increase in total and nuclear TFEB was observed in myocardium in the CPR and CPR+tubastatin A groups compared with the sham group. However, tubastatin A treatment further enhanced TFEB expression and its nuclear translocation compared with the CPR group (Figure 7A and 7C). Together with the in vitro data in the H9c2 cardiomyocyte model, we concluded that tubastatin A treatment alleviated postresuscitation myocardial pyroptosis induced by the NLRP3 inflammasome probably through the enhancement of TFEB signaling (Figure 8).
Figure 8. The proposed molecular mechanisms of the protective effect of tubastatin A (tubastatin A) on myocardial dysfunction after cardiac arrest (CA) and resuscitation.
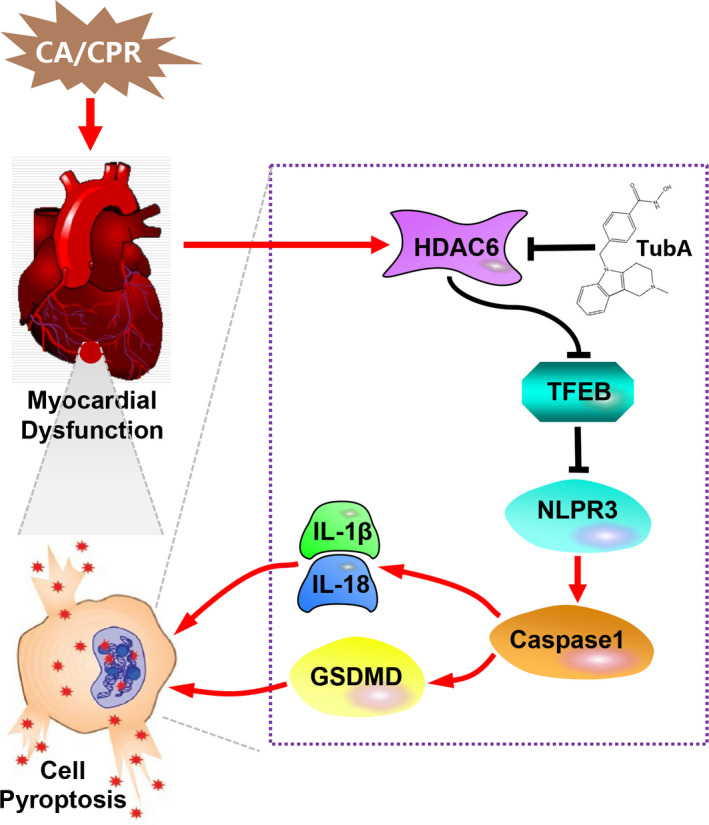
The inhibition of histone deacetylase 6 (HDAC6) activity by tubastatin A could promote transcription factor EB (TFEB) acetylation and nuclear translocation to block NOD‐like receptor pyrin domain 3 (NLPR3) inflammasome activation, and further decrease cell pyroptosis and proinflammatory cytokines production, which improve myocardial dysfunction and alleviate cardiac injury after CA and resuscitation. CPR indicates cardiopulmonary resuscitation; GSDMD, gasdermin D; IL‐1β, interleukin‐1β; and IL‐18, interleukin‐18.
Discussion
In the present study, we demonstrated that a specific inhibitor of HDAC6 activity, tubastatin A, significantly alleviated cell damage after H/R in H9c2 cardiomyocytes and markedly improved myocardial dysfunction after CA/CPR in swine. Furthermore, we demonstrated that the protective effect of tubastatin A was related to the inhibition of cell pyroptosis induced by the NLRP3 inflammasome in in vitro and in vivo studies. In the mechanistic studies, we revealed that tubastatin A increased TFEB acetylation and nuclear translocation, which might play an important role in the inhibition of NLRP3‐mediated cell pyroptosis.
Postresuscitation myocardial dysfunction has been confirmed to cause significant mortality during the hospital stay 2 , 3 ; however, no strategy is currently effective for myocardial protection after CPR in the clinical setting. The pathophysiology of myocardial dysfunction is mainly attributed to global I/R injury triggered by CA/CPR, which results in the increase of ROS and proinflammatory cytokines, and the activation of apoptosis, and further promotes cardiomyocyte death. 19 In theory, all these pathological injuries are mediated by histone deacetylases or lysine deacetylases, which remote the acetyl groups of histone and nonhistone proteins to alter the epigenome and gene expression. 20 , 21 Recently, histone deacetylase inhibitors have been shown to promote cell survival through the inhibition of hypoacetylation of target proteins, and thereby provide effective organ protection after I/R injury in a series of preclinical studies. 12 In particular, considering that HDAC6 has a unique structure and cellular localization, diverse substrates, and also a wider range of biological functions than other isoforms, its inhibition has been used as an effective approach for the treatment of cancers, neurodegenerative diseases, autoimmune disorders, and so on. 22 In the case of regional I/R injury, 2 investigations have demonstrated that the inhibition of HDAC6 by tubastatin A alleviated brain infarction and functional deficits through the upregulation of acetylated α‐tubulin and fibroblast growth factor‐21, and reduced cardiac infarction and dysfunction through the modulation of peroxiredoxin 1 acetylation. 13 , 14 Currently, our results showed that tubastatin A treatment significantly alleviated cell damage in H9c2 cardiomyocytes after H/R, and markedly improved myocardial dysfunction and its injury after CA/CPR in swine.
To date, the inflammation is considered as a key contributing factor to cell death in diverse I/R injuries. Recent investigation has confirmed that the inflammation is controlled by cell pyroptosis, which is triggered by NLRP3–caspase‐1 pathway activation, and then forms the holes in cell membrane and releases the proinflammatory cytokines. 23 Current studies have demonstrated that those therapeutic interventions targeting NLRP3‐mediated pyroptosis could inhibit inflammatory response so as to alleviate the damage of various target organs experiencing I/R. 7 , 8 , 9 In 2020, He et al 11 first reported that N‐acetylcysteine alleviated postresuscitation myocardial dysfunction through the inhibition of cell pyroptosis mediated by NLRP3 inflammasome activation in rats. Thus, NLRP3‐mediated pyroptosis could be a potential target for myocardial protection after resuscitation. Recently, Magupalli et al 24 demonstrated that HDAC6 was required for NLRP3 inflammasome activation in macrophage challenged with nigericin. Another 2 investigations further demonstrated that the inhibition of HDAC6 by tubastatin A protected the macrophage and neuron through the inhibition of NLRP3‐mediated pyroptosis. 15 , 16 In the present study, our results showed that tubastatin A significantly inhibited cardiac NLRP3 inflammasome activation and decreased its proinflammatory cytokine production and pyroptosis in in vitro and in vivo studies. Hence, tubastatin A could provide postresuscitation myocardial protection through the inhibition of NLRP3‐mediated pyroptosis.
TFEB, a master regulator of the autophagy‐lysosome pathway, controls the gene expression of autophagy and lysosome biogenesis in response to various stimuli. 17 Following those injury stimuli, TFEB is usually translocated from the cytoplasm to the nucleus, binds to the regions of lysosomal and autophagy promoters, and then promotes the expression of target genes. 25 In the setting of I/R injury, the enhancement of TFEB activity has emerged as a potential approach for the treatment of myocardial infarction and ischemic vascular disease. 26 , 27 , 28 Recently, Kim et al 18 demonstrated that ezetimibe promoted TFEB nuclear translocation, then induced cell autophagy, and finally blocked NLRP3 inflammasome activation in steatohepatitis. Lv et al 29 demonstrated that licochalcone A induced TFEB‐mediated autophagy activation to alleviate NLRP3‐meidated inflammatory response, and thereby improved acute liver injury. Zhou et al 30 demonstrated that TFEB overexpression reduced NLRP3 inflammasome‐mediated inflammatory response by improving autolysosome function in Alzheimer disease models. Currently, another investigation has demonstrated that HDAC6 could be a regulator of TFEB signaling, and its inhibition by tubastatin A promoted TFEB acetylation and nuclear translocation and thereby attenuated cell death in chronic kidney disease models. 31 In the present study, similar results were observed in our CA/CPR models, in which tubastatin A significantly limited NLRP3 inflammasome activation and cell pyroptosis probably through the enhancement of TFEB acetylation and nuclear translocation.
Limitations
There were several limitations in this study. First, a dose of 4.5 mg/kg of tubastatin A administered at 5 minutes after resuscitation provided effective protection for myocardial dysfunction after CA/CPR in swine; however, its optimal dosage and feasible therapeutic windows were not investigated in this animal study. Second, considering that the sex may affect the outcome of CPR according to the previous studies, 32 , 33 only male swine were used in the present study. Thus, it is unknown whether postresuscitation myocardial protection could be achieved by tubastatin A treatment in a female swine of CA and resuscitation. Third, the inhibition of HDAC6 activity by tubastatin A increased the acetylated levels of TFEB and its translocation to the nucleus in this study; however, whether the acetylation of TFEB promoted its own nuclear translocation and further its binding to the target sites of the genome remains unclear. Fourth, TFEB siRNA promoted NLRP3 inflammasome activation and subsequent cell pyroptosis after H/R in H9c2 cardiomyocytes in the present study; however, its regulatory mechanism on NLRP3 inflammasome signaling requires further investigation.
Conclusions
Our study demonstrated that tubastatin A, a specific inhibitor of HDAC6 activity, significantly reduced cell pyroptosis and further improved myocardial dysfunction after CA/CPR, in which the protective mechanism was related to the inhibition of NLRP3 inflammasome activation probably through the enhancement of TFEB signaling.
Sources of Funding
This study was supported by the Natural Science Foundation of China (82072126), the Zhejiang Provincial Key Research and Development Program of China (2021C03073, 2021C03036), and the Zhejiang Provincial Medical Science Foundation of China (2022KY958).
Disclosures
None.
Acknowledgments
The authors thank Dr Jie Zhang, Pingping zhou, Xiaochi Lu, and Yingying Kong for their contribution in the animal preparation.
For Sources of Funding and Disclosures, see page 15.
REFERENCES
- 1. Xu F, Zhang Y, Chen Y. Cardiopulmonary resuscitation training in China: current situation and future development. JAMA Cardiol. 2017;2:469–470. doi: 10.1001/jamacardio.2017.0035 [DOI] [PubMed] [Google Scholar]
- 2. Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Böttiger BW, Callaway C, Clark RSB, Geocadin RG, Jauch EC, et al. Post‐cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. Circulation. 2008;118:2452–2483. doi: 10.1161/CIRCULATIONAHA.108.190652 [DOI] [PubMed] [Google Scholar]
- 3. Laurent I, Monchi M, Chiche J‐D, Joly L‐M, Spaulding C, Bourgeois B, Cariou A, Rozenberg A, Carli P, Weber S, et al. Reversible myocardial dysfunction in survivors of out‐of‐hospital cardiac arrest. J Am Coll Cardiol. 2002;40:2110–2116. doi: 10.1016/s0735-1097(02)02594-9 [DOI] [PubMed] [Google Scholar]
- 4. Xu J, Sun S, Lu X, Hu X, Yang M, Tang W. Remote ischemic pre‐ and postconditioning improve postresuscitation myocardial and cerebral function in a rat model of cardiac arrest and resuscitation. Crit Care Med. 2015;43:e12–e18. doi: 10.1097/CCM.0000000000000684 [DOI] [PubMed] [Google Scholar]
- 5. Yang J, Xiao Y, Quan EY, Hu Z, Guo Q, Miao C, Bradley JL, Peberdy MA, Ornato JP, Mangino MJ, et al. Effects of polyethylene glycol‐20k on postresuscitation myocardial and cerebral function in a rat model of cardiopulmonary resuscitation. Crit Care Med. 2018;46:e1190–e1195. doi: 10.1097/CCM.0000000000003415 [DOI] [PubMed] [Google Scholar]
- 6. Zhang R, Liu B, Fan X, Wang W, Xu T, Wei S, Zheng W, Yuan Q, Gao L, Yin X, et al. Aldehyde dehydrogenase 2 protects against post‐cardiac arrest myocardial dysfunction through a novel mechanism of suppressing mitochondrial reactive oxygen species production. Front Pharmacol. 2020;11:373. doi: 10.3389/fphar.2020.00373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang J, Huang L, Shi X, Yang L, Hua F, Ma J, Zhu W, Liu X, Xuan R, Shen Y, et al. Metformin protects against myocardial ischemia‐reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging. 2020;12:24270–24287. doi: 10.18632/aging.202143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dong Z, Pan K, Pan J, Peng Q, Wang Y. The possibility and molecular mechanisms of cell pyroptosis after cerebral ischemia. Neurosci Bull. 2018;34:1131–1136. doi: 10.1007/s12264-018-0294-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ni J, Jiang L, Shen G, Xia Z, Zhang L, Xu J, Feng Q, Qu H, Xu F, Li X. Hydrogen sulfide reduces pyroptosis and alleviates ischemia‐reperfusion acute kidney injury by inhibiting NLRP3 inflammasome. Life Sci. 2021;284:119466. doi: 10.1016/j.lfs.2021.119466 [DOI] [PubMed] [Google Scholar]
- 10. Paik S, Kim JK, Silwal P, Sasakawa C, Jo EK. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol. 2021;18:1141–1160. doi: 10.1038/s41423-021-00670-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He F, Zheng G, Hou J, Hu Q, Ling Q, Wu G, Zhao H, Yang J, Wang Y, Jiang L, et al. N‐acetylcysteine alleviates post‐resuscitation myocardial dysfunction and improves survival outcomes via partly inhibiting NLRP3 inflammasome‐induced pyroptosis. J Inflamm. 2020;17:25. doi: 10.1186/s12950-020-00255-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pickell Z, Williams AM, Alam HB, Hsu CH. Histone deacetylase inhibitors: a novel strategy for neuroprotection and cardioprotection following ischemia/reperfusion injury. J Am Heart Assoc. 2020;9:e016349. doi: 10.1161/JAHA.120.016349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Z, Leng Y, Wang J, Liao HM, Bergman J, Leeds P, Kozikowski A, Chuang DM. Tubastatin A, an HDAC6 inhibitor, alleviates stroke‐induced brain infarction and functional deficits: potential roles of α‐tubulin acetylation and FGF‐21 up‐regulation. Sci Rep. 2016;6:19626. doi: 10.1038/srep19626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leng Y, Wu Y, Lei S, Zhou B, Qiu Z, Wang K, Xia Z. Inhibition of HDAC6 activity alleviates myocardial ischemia/reperfusion injury in diabetic rats: potential role of peroxiredoxin 1 acetylation and redox regulation. Oxid Med Cell Longev. 2018;2018:9494052. doi: 10.1155/2018/9494052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu S, Chen H, Ni H, Dai Q. Targeting HDAC6 attenuates nicotine‐induced macrophage pyroptosis via NF‐kappaB/NLRP3 pathway. Atherosclerosis. 2021;317:1–9. doi: 10.1016/j.atherosclerosis.2020.11.021 [DOI] [PubMed] [Google Scholar]
- 16. Yan S, Wei X, Jian W, Qin Y, Liu J, Zhu S, Jiang F, Lou H, Zhang B. Pharmacological inhibition of HDAC6 attenuates NLRP3 inflammatory response and protects dopaminergic neurons in experimental models of Parkinson’s disease. Front Aging Neurosci. 2020;12:78. doi: 10.3389/fnagi.2020.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Settembre C, Di Malta C, Polito VA, Arencibia MG, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim SH, Kim G, Han DH, Lee M, Kim I, Kim B, Kim KH, Song Y‐M, Yoo JE, Wang HJ, et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase‐TFEB‐mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy. 2017;13:1767–1781. doi: 10.1080/15548627.2017.1356977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burstein B, Jentzer JC. Comprehensive cardiac care after cardiac arrest. Crit Care Clin. 2020;36:771–786. doi: 10.1016/j.ccc.2020.07.007 [DOI] [PubMed] [Google Scholar]
- 20. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schweizer S, Meisel A, Marschenz S. Epigenetic mechanisms in cerebral ischemia. J Cereb Blood Flow Metab. 2013;33:1335–1346. doi: 10.1038/jcbfm.2013.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang X, Ma Q, Wu H, Khamis MY, Li Y, Ma L, Liu H. A review of progress in histone deacetylase 6 inhibitors research: structural specificity and functional diversity. J Med Chem. 2021;64:1362–1391. doi: 10.1021/acs.jmedchem.0c01782 [DOI] [PubMed] [Google Scholar]
- 23. Ji N, Qi Z, Wang Y, Yang X, Yan Z, Li M, Ge Q, Zhang J. Pyroptosis: a new regulating mechanism in cardiovascular disease. J Inflamm Res. 2021;14:2647–2666. doi: 10.2147/JIR.S308177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, Deng Q, Orning P, Alam HB, Maliga Z, et al. HDAC6 mediates an aggresome‐like mechanism for NLRP3 and pyrin inflammasome activation. Science. 2020;369:eaas8995. doi: 10.1126/science.aas8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Settembre C, Medina DL. TFEB and the CLEAR network. Methods Cell Biol. 2015;126:45–62. doi: 10.1016/bs.mcb.2014.11.011 [DOI] [PubMed] [Google Scholar]
- 26. Gu S, Tan J, Li Q, Liu S, Ma J, Zheng Y, Liu J, Bi W, Sha P, Li X, et al. Downregulation of LAPTM4B contributes to the impairment of the autophagic flux via unopposed activation of mTORC1 signaling during myocardial ischemia/reperfusion injury. Circ Res. 2020;127:e148–e165. doi: 10.1161/CIRCRESAHA.119.316388 [DOI] [PubMed] [Google Scholar]
- 27. Fan Y, Lu H, Liang W, Garcia‐Barrio MT, Guo Y, Zhang J, Zhu T, Hao Y, Zhang J, Chen YE, et al. Endothelial TFEB (transcription factor EB) positively regulates postischemic angiogenesis. Circ Res. 2018;122:945–957. doi: 10.1161/CIRCRESAHA.118.312672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Y, Zhang M, Zhao X, Shi K, Ye M, Tian J, Guan S, Ying W, Qu X. NAD+ administration decreases microvascular damage following cardiac ischemia/reperfusion by restoring autophagic flux. Basic Res Cardiol. 2020;115:57. doi: 10.1007/s00395-020-0817-z [DOI] [PubMed] [Google Scholar]
- 29. Lv H, Yang H, Wang Z, Feng H, Deng X, Cheng G, Ci X. Nrf2 signaling and autophagy are complementary in protecting lipopolysaccharide d‐galactosamine induced acute liver injury by licochalcone A. Cell Death Dis. 2019;10:313. doi: 10.1038/s41419-019-1543-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou W, Xiao D, Zhao Y, Tan B, Long Z, Yu L, He G. Enhanced autolysosomal function ameliorates the inflammatory response mediated by the NLRP3 inflammasome in Alzheimer’s disease. Front Aging Neurosci. 2021;13:629891. doi: 10.3389/fnagi.2021.629891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brijmohan AS, Batchu SN, Majumder S, Alghamdi TA, Thieme K, McGaugh S, Liu Y, Advani SL, Bowskill BB, Kabir MG, et al. HDAC6 inhibition promotes transcription factor EB activation and is protective in experimental kidney disease. Front Pharmacol. 2018;9:34. doi: 10.3389/fphar.2018.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Li J, Chen B, Shen Y, Wang J, Wang K, Yin C, Li Y. Interaction between gender and post resuscitation interventions on neurological outcome in an asphyxial rat model of cardiac arrest. BMC Cardiovasc Disord. 2021;21:441. doi: 10.1186/s12872-021-02262-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dietz RM, Deng G, Orfila JE, Hui X, Traystman RJ, Herson PS. Therapeutic hypothermia protects against ischemia‐induced impairment of synaptic plasticity following juvenile cardiac arrest in sex‐dependent manner. Neuroscience. 2016;325:132–141. doi: 10.1016/j.neuroscience.2016.03.052 [DOI] [PMC free article] [PubMed] [Google Scholar]