

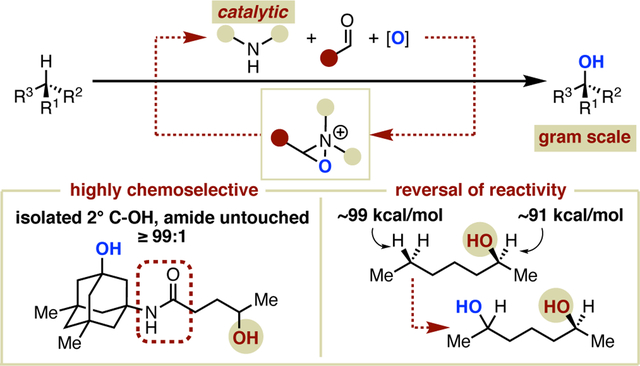
Abstract
We introduce an organocatalytic approach for oxaziridinium-mediated C–H hydroxylation that employs secondary amines as catalysts. We also demonstrate the advantages of this operationally simple catalytic strategy for achieving high yielding and highly selective remote hydroxylation of compounds bearing oxidation-sensitive functional groups such as alcohols, ethers, carbamates, and amides. By employing hexafluoroisopropanol as the solvent in the absence of water, a proposed hydrogen bonding effect leads to, among other advantages, as high as ≥99:1 chemoselectivity for remote aliphatic hydroxylation of 2° alcohols, an otherwise unsolved synthetic challenge normally complicated by substantial amounts of alcohol oxidation. Initial studies of the reaction mechanism indicate the formation of an oxaziridinium salt as the active oxidant, and a C–H oxidation step that proceeds in a stereospecific manner via concerted insertion or hydrogen atom transfer/radical rebound. Furthermore, preliminary results indicate that site selectivity can be affected by amine catalyst structure. In the long term, we anticipate that this will enable new strategies for catalyst control of selectivity based on the abundance of catalytic scaffolds that have proliferated over the last twenty years as a result of Nobel Prize-winning discoveries.
Keywords: organocatalysis, hydroxylation, amine, chemoselective, C-H functionalization
Graphical Abstract
INTRODUCTION
Selective hydroxylation of inert C–H bonds continues to receive increasing attention from the organic chemistry community due to the promise of dramatic improvements in synthetic efficiency and rapid optimization of drugs and materials via late-stage functionalization.1 Therefore, the development of new strategies and methods to accomplish such reactions, particularly those that enable progress toward the goal of catalyst-controlled selectivity, is highly valued. In this context, catalytic atom-transfer C(sp3)–H oxidation, in which a metal-oxo or an organic oxidant, such as an oxaziridine,2 is generated from an appropriate catalyst has been actively pursued and has shown considerable promise for this purpose. Early examples of metal-catalyzed processes from several groups focused on the use of bioinspired Fe and later Mn complexes, or on catalytic access to other high-valent metal-oxo species known to hydroxylate C–H bonds when used as stoichiometric oxidants.3 Concurrently, organocatalytic methods using benzoxathiazine catalysts such as 1a to access oxaziridines capable of C–H hydroxylation were developed by Du Bois (Figure 1).4 In comparison to metal-catalyzed methods, organocatalysis is underexplored, despite its potential complementarity and advantages in certain scenarios, such as late-stage oxidation in pharmaceutical synthesis, where even trace amounts of metal impurities can be problematic.5 We have established a research program focused on identifying new organocatalytic methods for atom-transfer C–H functionalization,6 and as part of these efforts have developed the first examples of ketone-catalyzed7 and iminium-catalyzed8 C–H hydroxylation, which generate dioxiranes and oxaziridinium salts, respectively, as the active oxidant. We anticipate that these platforms could be complementary to metal-catalyzed and enzymatic methods in addressing longstanding challenges in catalyst control of site selectivity,9 enantioselectivity,10 and chemoselectivity.11
Figure 1.
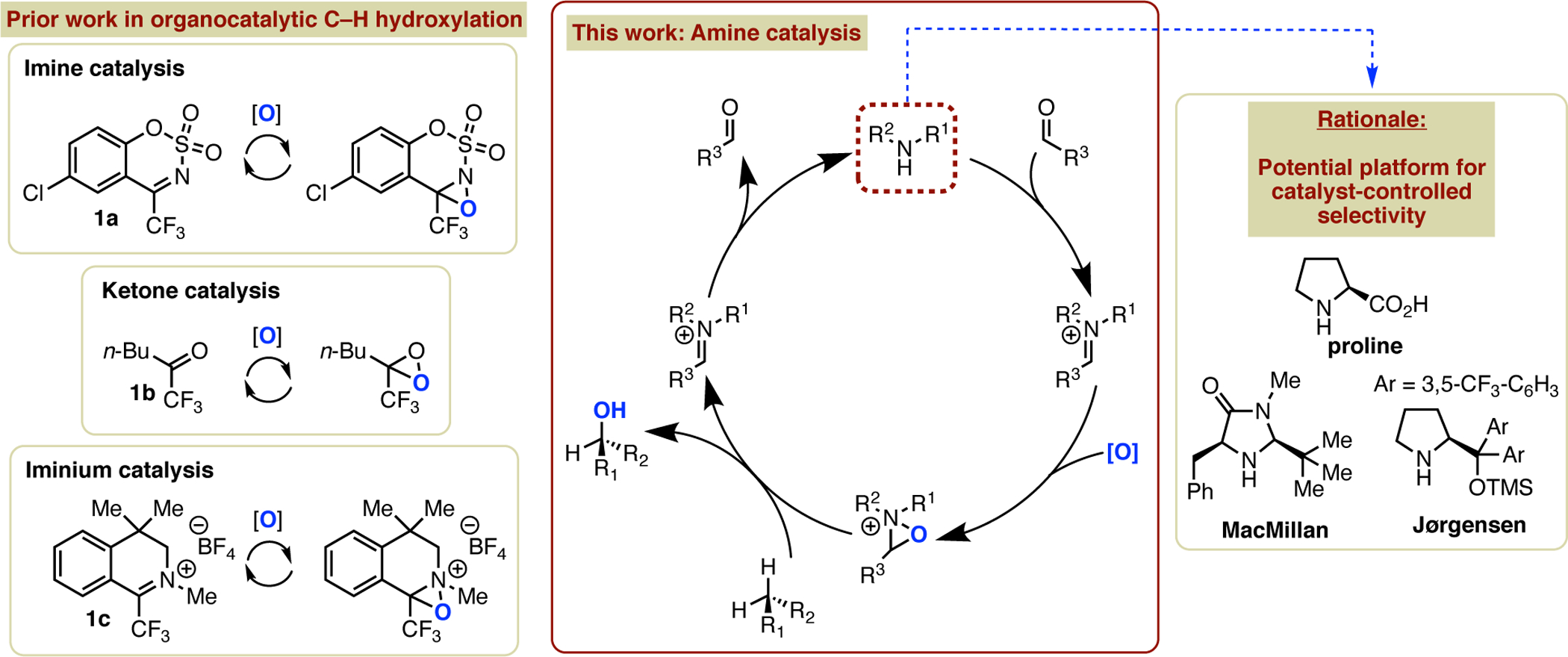
Atom-transfer organocatalysis of C–H oxidation.
Notwithstanding the promise of ketone and iminium catalysis, we envisioned a next-generation organocatalytic C–H hydroxylation method based on the concept of amine organocatalysis.12 Although the use of secondary amines as catalysts for LUMO or HOMO activation of aldehydes and ketones via iminium or enamine formation is well established, there is potential synergy with iminium organocatalysis of atom-transfer reactions that is underexplored. Employing a combination of an amine catalyst and an aldehyde or ketone in the presence of an oxidant can result in three-component formation of an oxaziridinium salt (Figure 1). In the only prior example of this concept, Yang has demonstrated proof of principle of this idea for oxaziridinium-mediated epoxidation13,14 To the best of our knowledge, there are no examples of the application of this concept, nor of amine organocatalysis more broadly, to atom-transfer hydroxylation of unactivated C–H bonds. As part of our ongoing interests in this area, we have initiated an effort to explore the utility of amine catalysis of C–H oxidation in both intermolecular and intramolecular contexts. One appealing advantage of this approach compared to prior oxaziridinium-mediated oxidations is the abundance of privileged amine catalyst scaffolds and other commercially available secondary amines, which could facilitate a more rapid and diverse exploration of catalyst control of selectivity compared to dihydroisoquinolinium catalysts based on 1c (Figure 1).
Herein, we describe the first examples of amine catalyzed atom-transfer C–H hydroxylation. In addition to establishing the feasibility of this catalytic approach, we also demonstrate that this method enables dramatic increases in chemoselectivity, as well as substantial increases in yield, in the substrate-limited remote hydroxylation of isolated secondary and primary alcohols. The remote hydroxylation of methylene C–H bonds selectively to 2° alcohol products is also possible. In a broader sense, these results also establish a novel platform for investigation of catalyst control of selectivity in aliphatic C–H oxidation that is poised to benefit from a wealth of well-studied approaches to amine catalysis.
RESULTS AND DISCUSSION
Remote Hydroylation of Alcohols.
A secondary reason for our interest in amine catalysis of oxaziridinium-mediated reactions was to explore its potential for remote hydroxylation of alcohols. We had previously reported, in an oxaziridinium-mediated hydroxylation using catalyst 1c, the first examples of chemoselective, remote hydroxylation of unprotected alcohols, which are typically incompatible with aliphatic hydroxylation due to the lower bond dissociation energies of C–H bonds α to hydroxyl groups. Evidence based on our report8 and subsequent studies by others15 suggests that this selectivity is mediated by the reaction solvent, hexafluoroisopropanol (HFIP), which, as a strong hydrogen bond donor, can deactivate potential reactive sites proximal to nitrogen or oxygen and thus promote remote hydroxylation. Even though this solvent effect could in principle be generalized to other catalyst platforms, to date this has not occurred; very few catalysts have been shown to be compatible. In addition to our iminium catalyst, chemoselective remote hydroxylation of alcohols has only been reported using Mn(TIPSmcp).15a Secondary alcohols pose a greater chemoselectivity challenge than primary alcohols, which has only been addressed for polyhydroxylated substrates, particularly 1,2-diols, wherein H-bonding effects are presumably enhanced by the presence of an additional acceptor.16 For mono-ols, prior reports of remote oxidation of the relatively simple 6-methylheptan-2-ol and related substrates proceed with low yields and/or selectivities, or employed the oxidant as the limiting reagent.15a High-yielding, highly site- and chemoselective remote hydroxylation of isolated secondary alcohols remains an unsolved challenge.
Catalyst Screening and Reaction Development.
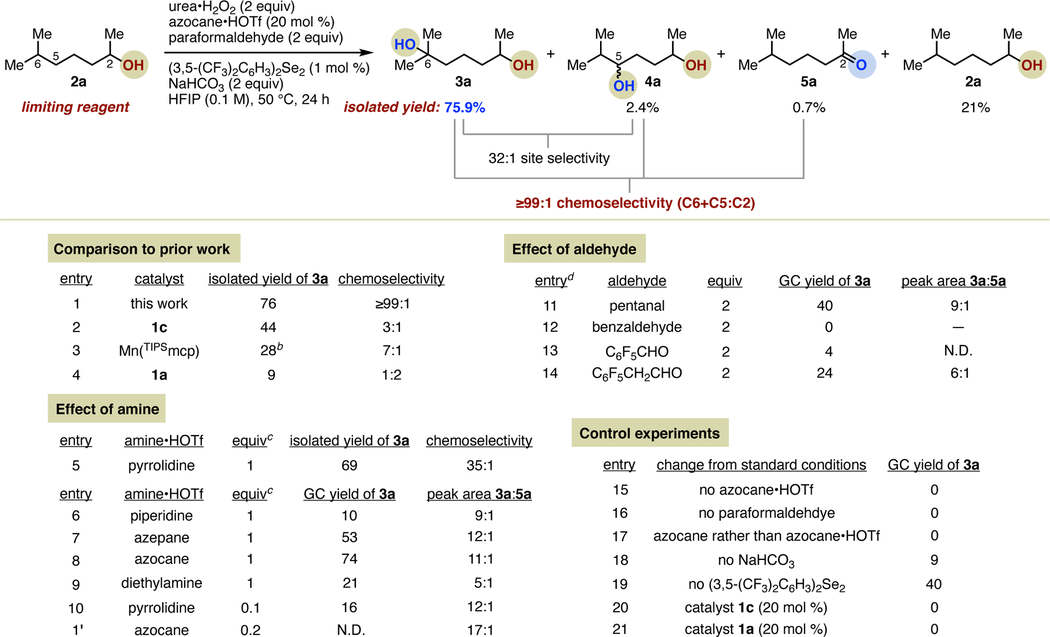
Accordingly, we initiated our studies by investigating the hydroxylation of 6-methylheptan-2-ol (2a) as a model example of a challenging remote oxidation of a 2° alcohol, using a combination of commercially available secondary amines and aldehydes (Table 1). At the outset of our investigation, we hypothesized that the HFIP-mediated chemoselectivity observed in previous reports was likely counteracted by aqueous oxidants (H2O2) and other reagents (acetic acid) introducing co-solvent amounts of water into the reaction conditions, partially disrupting any beneficial hydrogen bonding effects. Accordingly, we focused on the use of water-free oxidants such as urea•H2O2 (UHP), which proved well suited to this purpose. Initial studies identified pyrrolidine (as its triflate salt), paraformaldehyde, and UHP as a promising combination, providing 69% yield of 3a and, remarkably, 35:1 chemoselectivity, based on isolated yields, for remote hydroxylation over oxidation of the C2-OH to a ketone (entry 5). Also identified during these studies was the advantageous use of 1,2-bis(3,5-bis(trifluoromethyl)phenyl)diselane17 in catalytic amounts to promote the activation of the peroxide (entry 19). Regarding the aldehyde, advantages of steric size and electrophilicity offered by formaldehyde are apparent. The use of aliphatic or aromatic aldehydes (entries 11–12) led to reduced yields, although a beneficial influence of electron-with-drawing groups was noted (entries 13–14).
Table 1.
Selected Optimization for Amine-Catalyzed Remote C–H Hydroxylationa
|
Unless otherwise noted, reactions were conducted on 0.2 mmol scale of 2a at 50 °C, using amounts of reagents shown and 2 mL of HFIP. Yields and selectivities determined by gas chromatography, with the exceptions of entries 1 and 5, which are based on isolated yields of products and 2a. Entry 1 performed on 1 mmol scale of 2a; isolated yield reported. Entry 2 data is from reference 8.
Entry 3 data is from reference 15a. Hydroxylation of 2a in reference 15a was calculated as 56% based on the limiting reagent (H2O2, 0.5 equiv). To compare more accurately with substrate-limited methods, we have recalculated the yield based on 2a he as the limiting reagent.
For entries employing 1 equivalent of amine salt, 3 equiv of urea•H2O2 and 5 equiv of NaHCO3 were used.
For entries 11–14, experiments were performed using 1 equiv of pyrrolidine•HOTf, 3 equiv of urea•H2O2, and 5 equiv of NaHCO3. See Supporting Information for additional details.
Despite this initial success, we found that the use of catalytic amounts of pyrrolidine failed to provide synthetically useful yields of 3a (entry 10). The evaluation of additional commercially available cyclic and acyclic amines (entries 6–9) led to the identification of azocane•HOTf as an alternative amine candidate, providing 3a in 74% yield with chemoselectivity for C6 hydroxylation over secondary alcohol oxidation of 12:1 (as measured by uncorrected GC peak areas) when used in stoichiometric amounts (entry 8). Despite the lack of notable improvement over the use of pyrrolidine under the same conditions, the further investigation of azocane as a potential catalyst proved consequential. When the loading of azocane•HOTf was reduced to 20 mol %, the isolated yield of 3a remained high at 75.9%, but the undesired C2 alcohol oxidation was advantageously suppressed, resulting in nearly complete chemoselectivity (≥99:1) for remote hydroxylation (Table 1 scheme and entry 1). In this 1 mmol reaction, the remaining mass balance was completely accounted for, allowing for an extremely accurate assessment of selectivity. In addition to unreacted starting material (21%) and C2 oxidation product 6-methylheptan-2-one (5a, 0.7%), a mixture of stereoisomers arising from 2° C–H hydroxylation at C5 (4a, 2.4%) was isolated. Thus, the site selectivity for the C–H bond most remote from the polar group observed for this reaction (32:1) is also markedly higher than what is typically observed for aliphatic hydroxylation. A strength of this method also made clear by the isolation of 4a is a preference for 2° alcohol products of methylene oxidation, presumably driven by the suppression of overoxidation via H-bonding. Importantly, the remarkable chemoselectivity observed is not solely a function of the optimized reaction conditions but also requires the simultaneous advance of amine catalysis; neither iminium catalyst 1c nor benzoxathiazine catalyst 1a promote any conversion to products under the optimized conditions (entries 20–21). Finally, all reagents can be obtained at low cost from commercial suppliers, except for the diselenide, which is easily prepared in one step on gram scale.17
Substrate Scope.
Using the optimized reaction conditions, we investigated the scope of substrate-limited remote hydroxylation of alcohols and other compounds bearing functional groups potentially susceptible to HFIP deactivation (Figure 2). Continuing our investigation of 2° alcohols (Figure 2a), we identified early on that the use of 10 mol % of pyrrolidine•HOTf (catalyst B) was sufficient for high-yielding remote 3° hydroxylation of functionalized adamantanes bearing 2° alcohols or ethers despite its reduced performance in other cases (3b-3e). We interpret this as suggesting a beneficial effect of stereoelectronic stabilization of the reaction center by adjacent C–C bonds in these rigid scaffolds. For these same examples, high chemoselectivity, as high as 85:1 or greater (3b) based on combined isolated yield of product and unreacted starting material, was observed. For 2-adamantanol hydroxylation product 3e, 31:1 chemoselectivity for C1 hydroxylation was accompanied by 34:1 diastereoselectivity over the competing C7 hydroxylation. For less reactive C–H bonds, as we had found previously for the hydroxylation of 2a, azocane•HOTf was the more effective catalyst. In the hydroxylation of menthol (products 3f and 3f′) and cis-4-methylcyclohexanol (product 3g), selectivity for remote hydroxylation was maintained, although both yields and chemoselectivities were lower in comparison to the hydroxylation of 2a. We attribute this to the closer proximity of 3° C–H bonds in these molecules to the unprotected alcohol group, leading to more substantial deactivation of these centers by propagation of the H-bonding effect, reducing the rate of aliphatic hydroxylation compared to the competing alcohol oxidation. Proximity to the alcohol can also be used to explain the approximately 1:1 site selectivity observed for the remote hydroxylation of menthol, in which the two oxidized 3° C–H bonds in 3f and 3f′ are equidistant from C1.
Figure 2.
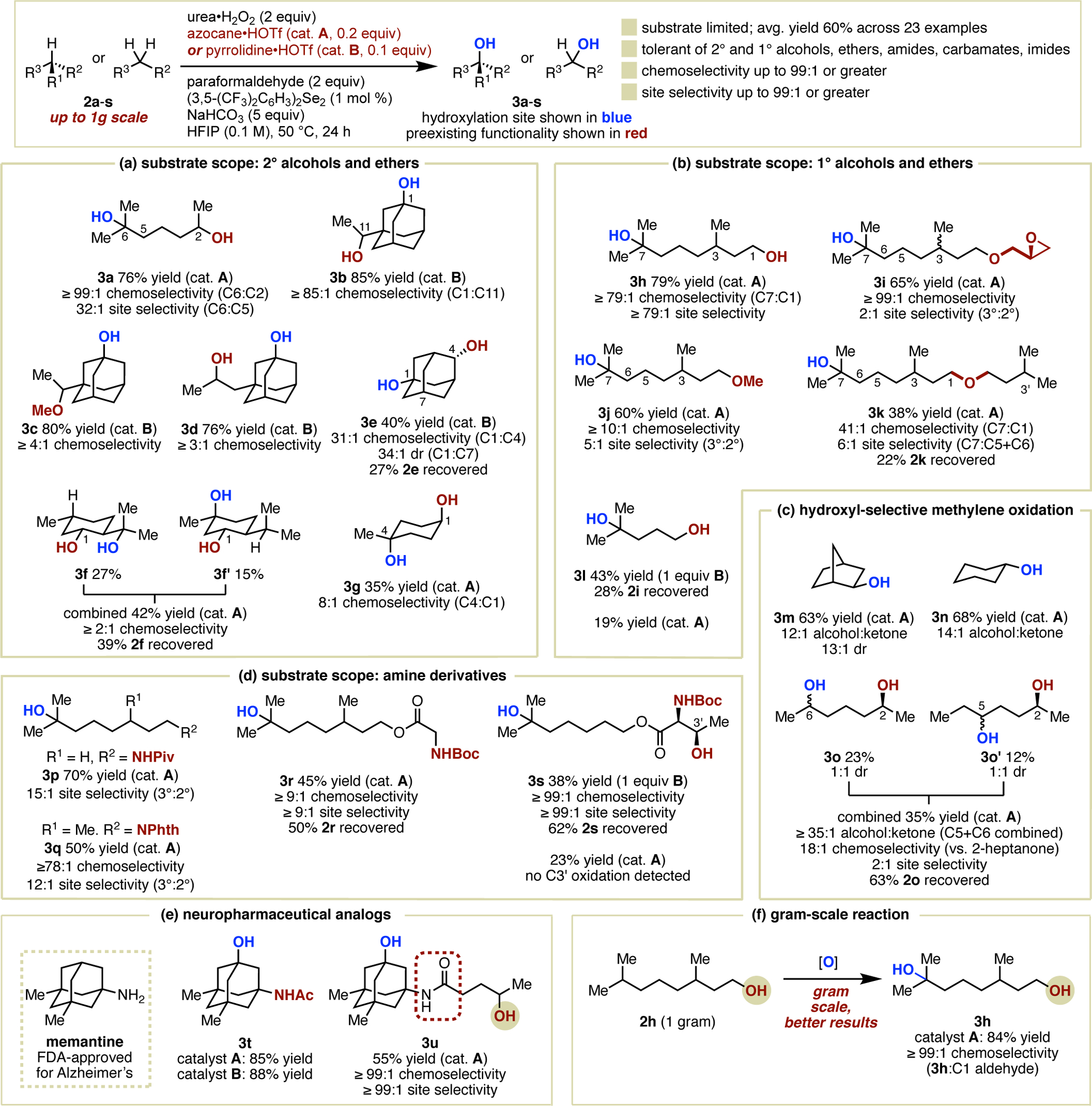
Scope of amine-catalyzed remote hydroxylation. Isolated yields are provided apart from 3n, which was assayed by gas chromatrography using dodecane as an internal standard. Selectivities that are reported as precise ratios based on isolated yields of the relevant major and minor products, are based on NMR analysis of inseparable product mixture, or are based on GC analysis of the crude reaction mixture with the use of an internal standard and comparison to authentic samples. Selectivities that are reported as minimum ratios (e.g. 3b at ≥ 85:1 chemoselectivity) are provided when the relevant products (i.e. alcohol oxidation products or alternative oxidation sites) were not observed, and are provided as ratios of the major product to the unrecovered mass balance of the reaction. See Supporting Information for details.
We also evaluated the oxidation of 3,7-dimethyloctanol and several derivatives as representative examples of remote hydroxylation of primary alcohols and their corresponding ethers (Figure 2b). Oxidation of the parent alcohol occurred selectively at C7 (product 3h) in 79% yield, with no other aliphatic hydroxylation products (including C3 hydroxylation) present in identifiable amounts by gas chromatography. Both chemoselectivity and site selectivity for this reaction was determined to be ≥79:1 based on a recovered starting material yield of 20%. Trace (±)-dihydrocitronellal was detected by GC. Regarding ethers, we observed that the chemoselectivity for remote alcohol oxidation also extends to this class of compounds (products 3i-3k, as well as 3c). This includes epoxides (3i), which are less sensitive to oxidative conditions but much more sensitive to acidic and basic environments, highlighting the mild nature of the reaction conditions. The degree of chemoselectivity for remote hydroxylation compared to hydroxylation of αC–H bonds of the ethers remained high, as determined for 3k, which produced a small but detectable amount of (±)-dihydrocitronellal as a side product. In contrast, we noted a trend of dramatically reduced site selectivity for oxidation of the ethers versus 3,7-dimethyloctanol (e.g. 3j vs 3h). However, the minor products were not C3 3° hydroxylation products, as would normally be expected,2c but rather mixtures of 2° hydroxylation products at C5 and C6. We attribute the diminished selectivity to a reduced tendency of these compounds to participate in multiple hydrogen bonds and extended H-bond networks, potentially influenced by both the lack of a hydrogen bond donor and steric restrictions. This would explain the retention of high chemoselectivity, mediated by a short-range inductive effect, which should be less sensitive to diminished polarization compared to the longer-range effects thought to influence site selectivity. This also accounts for the lack of C3 hydroxylation, if one presumes that the inductive deactivation is sufficient to overcome the intrinsic reactivity advantage of the 3° C3 C–H versus 2° hydroxylation at more remote sites. Pertinent to this latter point is the substantially reduced reaction performance observed for the remote hydroxylation of isohexanol (3l), which required the use of stoichiometric pyrrolidine•HOTf to achieve 43% isolated yield of 3° hydroxylation at a site with similar electron density to the proximal C3 and C4 sites in 3h; only 19% yield was observed for 3l using catalytic azocane.
Site-selective hydroxylation of methylenic sites to 2° alcohol products rather than ketones, which typically arise from overoxidation of the more reactive intermediate alcohol in the absence of substrate-controlled factors, is an unsolved problem.18 In addition to saving a synthetic step when alcohol products are desired, progress in this direction might ultimately enable new general methods for enantioselective alcohol synthesis. In this context, we observe preliminary success for hydroxyl-selective methylene oxidation, as shown by the examples in Figure 2c. Hydroxylation of alkanes norbornane and cyclohexane occurred in good yield, providing 3m and 3n with synthetically useful selectivities of 12:1 and 13:1 over the corresponding ketones, respectively, highlighting a reversal of the relative rates of alcohol versus aliphatic oxidation. Hydroxylation of 2-heptanol, bearing four methylenic sites in addition to a secondary alcohol, occurred site-selectively at the two most remote positions (3o and 3o′). No influence of the distant C2 stereocenter on diastereoselectivity was observed. The chemoselectivity of 18:1 over alcohol oxidation for this transformation is notable given the greater challenge associated with remote hydroxylation of a stronger 2° C–H bond relative to the prior 3° hydroxylation examples.
Finally, the capability of amine catalysis for the remote hydroxylation of amine derivatives was evaluated. Prior successful strategies to overcome N-oxidation and oxidation of C(α)–H bonds of amines and their derivatives, such as amides, rely on temporary blocking of reactive sites via amine or imidate salt formation, respectively.11 As shown in Figure 2d, amine- catalyzed hydroxylation in HFIP removes the need for covalent modification of amides in order to achieve remote hydroxylation (product 3p).15a Imides are also tolerated with similarly high site selectivity for remote 3° hydroxylation (3q). The mild reaction conditions of this method, along with the lack of requirement for strong acids or electrophiles for functional group deactivation, are highlighted by the tolerance for the popular but acid sensitive Boc protecting group (products 3r and 3s). The modest yield observed for hydroxylation of a (–)-threonine derivative (3s), along with the low yield observed under the catalytic conditions, belies the challenge associated with simultaneously avoiding alcohol oxidation and hydroxylation adjacent to the carbamate. In this example, chemoselectivity for remote aliphatic hydroxylation in the presence of both the carbamate and 2° alcohol was ≥99:1 based on combined isolated yields of all products when 1 equivalent of pyrrolidine•HOTf was used.19
Amides having direct pharmaceutical relevance were also evaluated as preliminary examples of late-stage API functionalization (Figure 2e). Using either azocane•HOTf or pyrrolidine•HOTf as the catalyst, hydroxylation of an N-acetyl derivative of memantine, used as a treatment for dementia,20 occurred chemoselectively at the 3° C–H bond and in high yield, providing 3t. The high chemoselectivity over 2° alcohol oxidation also extends to the memantine system, as shown by product 3u. In this case, the combined yield of 3u and recovered starting material was 100%, indicating that the reaction proceeded with ≥99:1 chemoselectivity and site selectivity. Finally, the scalability of any new reaction is a key determinant of potential applications in synthesis. On the highest scale evaluated (1 gram), we were delighted to observe that reaction performance in fact improves, with remote hydroxylation of 2h proceeding in higher yield (84%) and with effectively complete chemoselectivity for remote hydroxylation (≥99:1, Figure 2f).
Catalytic Cycle and Preliminary Mechanistic Studies.
Two highlights of this method are its operational simplicity and superior results for remote alcohol oxidation. With proof of concept for this catalytic approach established, there is also long-term potential for development of catalyst-controlled selectivity based on structural modifications to the amine. In support of this potential, we have obtained preliminary evidence that site selectivity is dependent on amine structure. As shown in Figure 2d, hydroxylation of 2q using 20 mol % azocane•HOTf provides 3° alcohol 3q in 50% yield. The other products isolated from this reaction are an inseparable mixture of 2° alcohols 3q′ and unreacted starting material in 4.3% and 45% yield, respectively (Scheme 1). Thus, the site selectivity for 3° over 2° hydroxylation can be reasonably determined to be 12:1 based on these isolated yields, with only 0.7% of the reaction mixture unaccounted for. Using this combined yield of unknowns (0.7%) along with the combined yields of 3q and 3q′, chemoselectivity for aliphatic hydroxylation over any other possible reactions is calculated to be ≥78:1. When using an equivalent amount of pyrrolidine•HOTf instead, but maintaining reaction conditions otherwise the same, the site selectivity based on isolated yields of 3q (30.3%) and 3q′ (8.4%) is 3-fold lower at 3.6:1, with only 2.3% of the material unaccounted for. This difference in site selectivity is also consistent with spectroscopic analysis of the unpurified reaction mixtures. Therefore, we can conclude that changes in amine structure alone are responsible for the differences in hydroxylation selectivity.
Scheme 1.
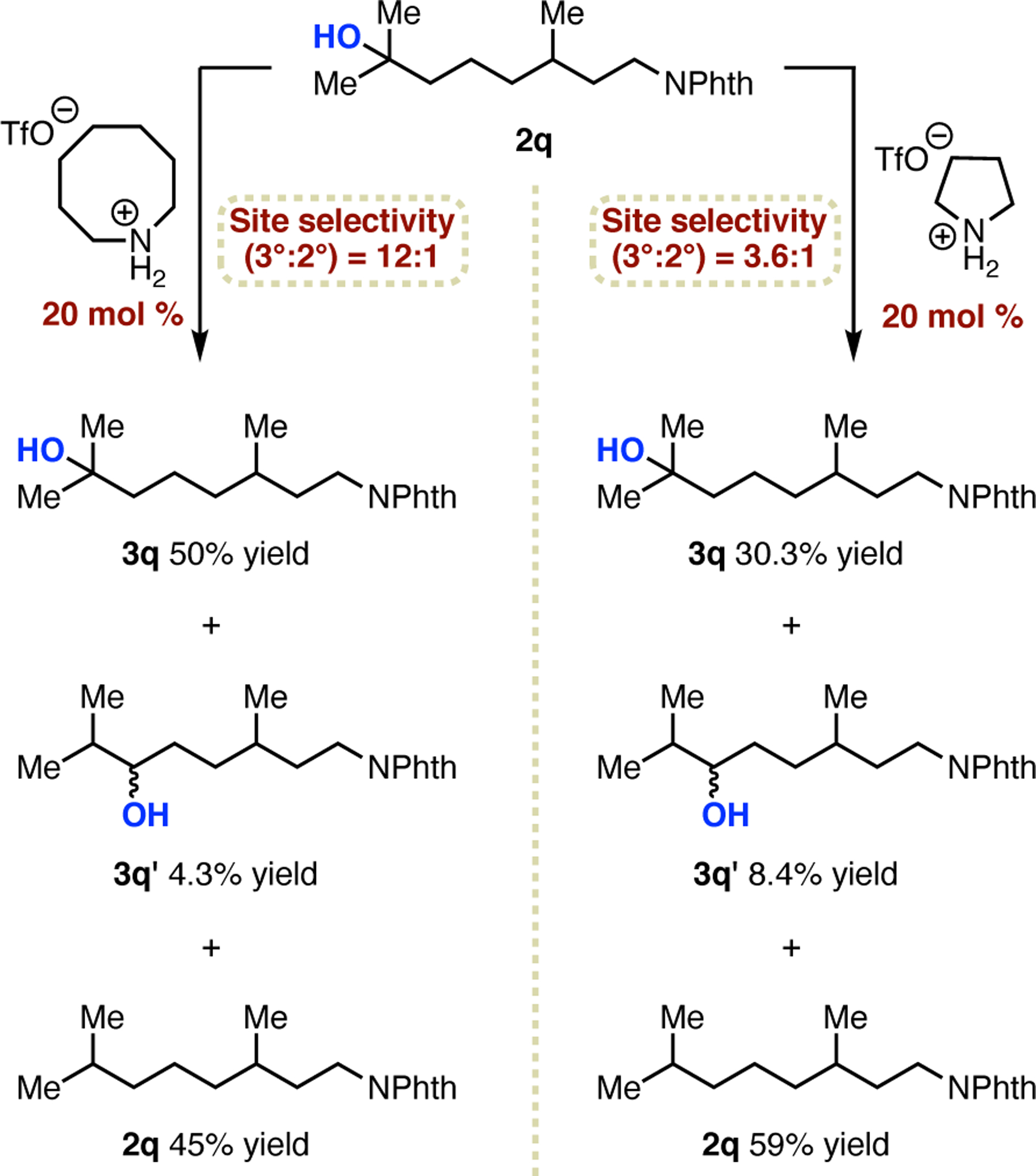
Dependence of site selectivity on amine structure
To aid in the future design of amine catalysts to build on these results, we have developed an initial proposal for the operative catalytic cycle, as shown in Figure 3A. The control experiments shown in Table 1 indicate that both the amine (as its triflate salt) and paraformaldehyde are required to observe any product formation. As previously noted, the diselenide is not strictly required for the reaction to proceed, but its presence is necessary to observe the highest possible yields. Consistent with these observations, we presuppose the formation of iminium salt I from the amine and aldehyde. In the presence of H2O2, seleninic acid III is known to form rapidly from the diselenide and undergo further oxidation to perseleninic acid IV, which we propose oxidizes the iminium salt to oxaziridinium II,4a,21 performing better in this regard than H2O2 alone. From there, the oxaziridinium is reduced back to iminium I upon O-atom transfer to the substrate. Iminium I is a potential catalyst resting state that can be re-oxidized to II, but can also regenerate the amine salt upon hydrolysis, as has been clearly established in the organocatalysis literature.12a,b
Figure 3.
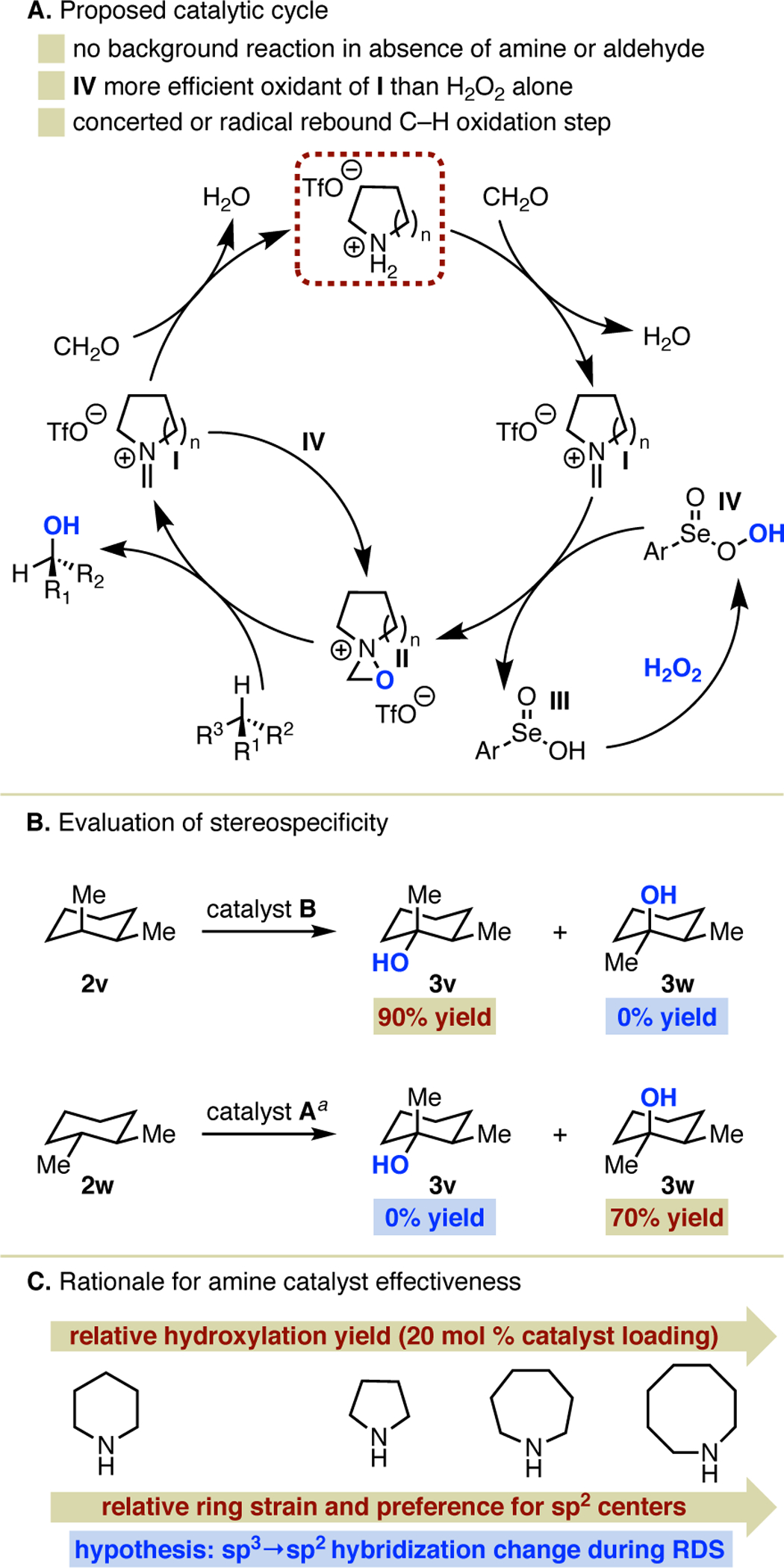
Proposed catalytic cycle and preliminary mechanistic studies. aHydroxylation of 2w using catalyst B (pyrroldine•HOTf) also provides exclusively 3w, but in lower yield. GC yield of 3v and isolated yield of 3w are reported.
To further investigate the mechanism of the C–H hydroxylation step, we performed hydroxylation reactions of cis- and trans-1,2-dimethylcyclohexane (Figure 3B) to evaluate stereo-specificity. In each case, hydroxylation occurred with complete retention of stereochemistry. This suggests the likelihood of a concerted insertion or HAT/radical rebound oxidation mechanism mediated by II, not unlike that proposed for the closely related dioxiranes.22 In addition to revealing aspects of mechanism, this fulfills an important prerequisite for pursuing enantioselective transformations.
Finally, we propose a rationalization of the relative effectiveness of cyclic amine catalysts as a function of ring size (Figure 3C). Combined yields of all products, which display an inverse relationship to starting material consumption, for hydroxylation of 2a exhibit a positive correlation with increasing ring strain of the amine (Table 1). For the ring sizes in question, ring strain itself arises from a larger contribution of torsional and transannular strain relative to angle strain, and by extension an increasing preference for sp2 relative to sp3 hybridization of at least one atom.23 This points to the potential importance of sp3 to sp2 re-hybridization during the rate-determining step, though it may also be relevant to key off-cycle processes. Regarding the former, this would be consistent with cleavage of the N–O bond of II, in the proposed HAT or concerted insertion step, occurring during the RDS. Overall, this may explain the superior performance of the eight-membered azocane, which to our knowledge has never been identified as a preferred amine catalyst for any transformation, in this reaction.
CONCLUSIONS
In summary, we have reported the first examples of amine-catalyzed C–H oxidation, which relies on readily available commercial or easily-prepared reagents. This protocol is particularly well suited to the remote hydroxylation of compounds bearing oxidation-sensitive functional groups. In addition to high average yields observed for the hydroxylation of primary alcohols and amine derivatives, a considerable advantage of this process for the chemoselective remote hydroxylation of secondary alcohols (up to 99:1 or greater) was identified. Furthermore, the process allows for the selective hydroxylation of methylene C–H bonds to secondary alcohols rather than ketones, is effective for selective late-stage hydroxylation, and can be performed on gram scale. Based on our initial evaluation, the mechanism of the process likely involves in situ formation of an iminium salt that is subsequently intercepted by an oxidant to form an oxaziridinium, which then reacts with the substrate in a radical rebound or concerted insertion O-atom transfer step. We believe that a key to the success of this process may be ring strain-driven rate enhancements of C–H functionalization, which explains trends observed for different amine catalysts. In addition, we have established that the degree of site selectivity can be dependent on choice of amine catalyst. This provides a basis for future efforts in the design of amine catalysts to address unsolved challenges in catalyst control of site selectivity and stereoselectivity. Overall, we believe these combined attributes will foster interest for applications spanning academic and industrial pursuits, particularly for applications in drug discovery, where functional group tolerance and other selectivity concerns are prominent.
Supplementary Material
ACKNOWLEDGMENT
Support of this work from the National Institutes of Health (R01 GM124092) is gratefully acknowledged.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, characterization data, and spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).For recent reviews on this topic, see:; (a) Hartwig JF Evolution of C–H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc 2018, 138, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cernak T; Dykstra KD; Tyafarajan S; Vachal P; Krska SW The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug-Like Molecules. Chem. Soc. Rev 2016, 45, 546. [DOI] [PubMed] [Google Scholar]; (c) White MC; Zhao J Aliphatic C–H Oxidations for Late-Stage Functionalization. J. Am. Chem. Soc 2018, 140, 13988. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Newhouse T; Baran PS If C–H Bonds Could Talk: Selective C–H Bond Oxidation. Angewandte Chemie International Edition 2011, 50, 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Kim C; Chen K; Que L Stereospecific Alkane Hydroxylation with H2O2 Catalyzed by an Iron(II)-Tris(2-pyridylmethyl)amine Complex. J. Am. Chem. Soc 1997, 119, 5964. [Google Scholar]; (b) Lee S; Fuchs PL Chemospecific Chromium(IV) Catalyzed Oxidation of C–H Bonds at −40 °C. J. Am. Chem. Soc 2002, 124, 13978. [DOI] [PubMed] [Google Scholar]; (c) Chen MS; White MC A Predictably Selective Aliphatic C–H Oxidation Reaction for Complex Molecule Synthesis. Science 2007, 318, 783. [DOI] [PubMed] [Google Scholar]; (d) McNeill E; Du Bois J Ruthenium-Catalyzed Hydroxylation of Unactivated Tertiary C–H Bonds. J. Am. Chem. Soc 2010, 132, 10202. [DOI] [PubMed] [Google Scholar]; (e) Milan M; Bietti M; Costas M Highly Enantioselective Oxidation of Nonactivated Aliphatic C–H Bonds with Hydrogen Peroxide Catalyzed by Manganese Complexes. ACS Cent. Sci 2017, 3, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).DesMarteau DD; Donadelli A; Montanari V; Petrov VA; Resnati G Mild and Selective Oxyfunctionalization of Hydrocarbons by Perfluorodialkyloxaziridines. J. Am. Chem. Soc 1993, 115, 4897. [Google Scholar]
- (4).(a) Brodsky BH; du Bois J Oxaziridine-Mediated Catalytic Hydroxylation of Unactivated 3° C−H Bonds Using Hydrogen Peroxide. J. Am Chem. Soc 2005, 127, 15391. [DOI] [PubMed] [Google Scholar]; (d) (1) Litvinas ND; Brodsky BH; du Bois J C–H Hydroxylation Using a Heterocyclic Catalyst and Aqueous H2O2. Angew. Chem. Int. Ed 2009, 48, 4513. [DOI] [PubMed] [Google Scholar]; (c) Adams AM; du Bois J Organocatalytic C–H Hydroxylation with Oxone® Enabled by an Aqueous Fluoroalcohol Solvent System. Chem. Sci 2014, 5, 656. [Google Scholar]
- (5).MacMillan DWC The Advent and Development of Organocatalysis. Nature 2008, 455, 304. [DOI] [PubMed] [Google Scholar]
- (6).Johnson S; Combee L; Hilinski M Organocatalytic Atom-Transfer C(sp3)–H Oxidation. Synlett 2018, 29, 2331. [Google Scholar]
- (7).(a) Pierce CJ; Hilinski MK Chemoselective Hydroxylation of Aliphatic sp3 C–H Bonds Using a Ketone Catalyst and Aqueous H2O2. Org. Lett 2014, 16, 6504. [DOI] [PubMed] [Google Scholar]; (b) Shuler WG; Johnson SL; Hilinski MK Organocatalytic, Dioxirane-Mediated C–H Hydroxylation under Mild Conditions Using Oxone. Org. Lett 2017, 19, 4790. [DOI] [PubMed] [Google Scholar]
- (8).Wang D; Shuler WG; Pierce CJ; Hilinski MK An Iminium Salt Organocatalyst for Selective Aliphatic C–H Hydroxylation. Org. Lett 2016, 18, 3826. [DOI] [PubMed] [Google Scholar]
- (9).(a) Gormisky PE; White MC Catalyst-Controlled Aliphatic C–H Oxidations with a Predictive Model for Site-Selectivity. J. Am. Chem. Soc 2013, 135, 14052. [DOI] [PubMed] [Google Scholar]; (b) Font D; Canta M; Milan M; Cussó O; Ribas X; Gebbink RJMK; Costas M Readily Accessible Bulky Iron Catalysts Exhibiting Site Selectivity in the Oxidation of Steroidal Substrates. Angew. Chem. Int. Ed 2016, 55, 5776. [DOI] [PubMed] [Google Scholar]; (c) Griffin JD Vogt DB; Du Bois J; Sigman MS Mechanistic Guidance Leads to Enhanced Site-Selectivity in C–H Oxidation Reactions Catalyzed by Ruthenium bis(Bipyridine) Complexes. ACS. Catal 2021, 11, 10479. [Google Scholar]
- (10).(a) Milan M; Bietti M; Costas M Highly Enantioselective Oxidation of Nonactivated Aliphatic C–H Bonds with Hydrogen Peroxide Catalyzed by Manganese Complexes. ACS Cent. Sci 2017, 3, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qiu B; Xu D; Sun Q; Miao C; Lee Y-M; Li X-X; Nam W; Sun W Highly Enantioselective Oxidation of Spirocyclic Hydrocarbons by Bioinspired Manganese Catalysts and Hydrogen Peroxide. ACS Catalysis 2018, 8, 2479. [Google Scholar]
- (11).(a) Asensio G González-Núñez ME; Bernardini CB; Mello R; Adam W Regioselective Oxyfunctionalization of Unactivated Tertiary and Secondary C–H Bonds of Alkyla-mines by Methyl(trifluoromethyl)dioxirane in Acid Medium. J. Am. Chem. Soc 1993, 115, 7250. [Google Scholar]; (b) Lee M; Sanford MS Platinum-Catalyzed, Terminal-Selective C(sp3)–H Oxidation of Aliphatic Amines. J. Am. Chem. Soc 2015, 137, 12796. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Howell JM; Feng K; Clark JR Trzepkowski LJ; White MC Remote Oxidation of Aliphatic C–H Bonds in Nitrogen-Containing Molecules. J. Am. Chem. Soc 2015, 137, 14590. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mbofana CT; Chong E; Lawniczak J; Sanford MS Iron-Catalyzed Oxyfunctionalization of Aliphatic Amines at Remote Benzylic C–H Sites, Org, Lett 2016, 18, 4258. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lee M; Sanford MS Remote C(sp3)–H Oxygenation of Protonated Aliphatic Amines with Potassium Persul-fate. Org. Lett 2017, 19, 572. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Mack JBC; Gipson JD; Du Bois J; Sigman MS Ruthenium-Catalyzed C–H Hydroxylation in Aqueous Acid Enables Selective Functionalization of Amine Derivatives. J. Am. Chem. Soc 2017, 139, 9503. [DOI] [PubMed] [Google Scholar]; (g) Schultz DM; Lévesque F; DiRocco DA; Reibarkh M; Ji Y; Joyce LA; Dropinski JF; Sheng H; Sherry BD; Davies IW Oxyfunctionalization of the Remote C–H Bonds of Aliphatic Amines by Decatungstate Photocatalysts. Angew. Chem. Int. Ed 2017, 56, 15274. [DOI] [PubMed] [Google Scholar]; (f) Nanjo T; de Lucca EC; White MC Remote, Late-Stage Oxidation of Aliphatic C–H Bonds in Amide-Containing Molecules J. Am. Chem. Soc 2017, 139, 14586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For reviews on this topic, see:; (a) Lelais G; MacMillan DWC Modern Strategies in Organic Catalysis: The Advent and Development of Iminium Activation. Aldrichimica Acta 2006, 39, 79. [Google Scholar]; (b) Erkkilä A; Majander I; Pihko PM Iminium Catalysis. Chem. Rev 2007, 107, 5416. [DOI] [PubMed] [Google Scholar]; (c) Mukherjee S; Yang JW; Hoffmann S; List B Asymmetric Enamine Catalysis. Chem. Rev 2007, 107, 5471. [DOI] [PubMed] [Google Scholar]; (d) Bertelsen S; Jørgensen KA Organocatalysis–After the Gold Rush. Chem. Soc Rev 2009, 38, 2178. [DOI] [PubMed] [Google Scholar]
- (13).Wong M-K; Ho L-M; Zheng Y-S; Ho C-Y; Yang D Asymmetric Epoxidation of Olefins Catalyzed by Chiral Iminium Salts Generated in Situ from Amines and Aldehydes. Org. Lett 2001, 3, 2587. [DOI] [PubMed] [Google Scholar]
- (14).For an example of a related strategy applied to α-hydroxylation of β-dicarbonyl compounds using oxaziridines, see:; Cai M; Xu K; Nie Z; Zhang L; Luo S Chiral Primary Amine/Ketone Cooperative Catalysis for Asymmetric α-Hydroxylation with Hydrogen Peroxide. J. Am. Chem. Soc 2021, 143, 1078. [DOI] [PubMed] [Google Scholar]
- (15).(a) Dantignana V; Milan M; Cussó O; Company A; Bietti M; Costas M Chemoselective Aliphatic C–H Bond Oxidation Enabled by Polarity Reversal. ACS Cent. Sci 2017, 3, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun Q; Sun W Catalytic Enantioselective Methylene C(sp3)–H Hydroxylation Using a Chiral Manganese Complex/Carboxylic Acid System. Org. Lett 2020, 22, 9529. [DOI] [PubMed] [Google Scholar]
- (16).Borrell M; Gil-Caballero S; Bietti M; Costas M Site-Selective and Product Chemoselective Aliphatic C–H Bond Hydroxylation of Polyhydroxylated Substrates. ACS Catal 2020, 10, 4702. [Google Scholar]
- (17).(a) ten Brink G-D; Fernandes BCM; van Vliet MCA; Arends IWCE; Sheldon RA Seleniun Catalysed Oxidations wih Aqueous Hydrogen Peroxide. Part I: Epoxidation Reactions in Homogeneous Solution. J. Chem. Soc., Perkin Trans 1, 2001, 224. [DOI] [PubMed] [Google Scholar]; (b) Reich HJ; Renga JM; Reich IL Organoselenium Chemistry. Conversion of Ketones to Enones by Selenoxide Syn Elimination. J. Am. Chem. Soc 1975, 97, 5434. [Google Scholar]
- (18).Zhao J; Nanjo T; de Lucca EC; White MC Chemoselective Methylene Oxidation in Aromatic Molecules. Nat. Chem 2019, 11, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Based on available information, chemoselectivity was not notably different for the stoichiometric pyrrolidine and catalytic azocane conditions; for the latter unreacted starting material accounted for a large majority of the mass balance, and no C3’ ketone was detected by crude 13C NMR.
- (20).McKeage K Memantine. A Review of its Use in Moderate to Severe Alzheimer’s Disease. CNS Drugs 2009, 23, 881. [DOI] [PubMed] [Google Scholar]
- (21).Buckley BR; Elliott CE; Chan Y; Dreyfus N; Page PCB Activation of Hydrogen Peroxide by Diphenyl Diselenide for Highly Enantioselective Oxaziridinium Salt Mediated Catalytic Asymmetric Epoxidation. Synlett, 2013, 24, 2266. [Google Scholar]
- (22).Du X; Houk KN Transition States for Alkane Oxidations by Dioxiranes. J. Org. Chem 1998, 63, 6480. [Google Scholar]
- (23).(a) Brown HC; Ham G The Effect of Ring Size on the Rate of Acetolysis of the Cycloalkyl p-Toluene and p-Bromobenzenesulfonates. J. Am. Chem. Soc 1956, 78, 2735. [Google Scholar]; (b) Schneider H-J; Schmidt G; Thomas F Strain-Reactivity Relations as a Tool for the Localization of Transition States. Equilibria, Solvolysis, and Redox Reactions of Substituted Cycloalkanes. J. Am. Chem. Soc 1983, 105, 3556. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.