

Abstract
A zirconium 1,4-dicarboxybenzene metal–organic framework (UiO-66 MOF) was successfully used as a template to enhance the distribution and activity of nanoscale zero-valent iron (NZVI). MOF-NZVI showed good anti-interference ability to co-existing ions (Ca2+, Mn2+, Cu2+, H2PO4− and SO42−) and organic acids (oxalic acid and citric acid). SEM and TEM analyses indicated that the MOF as a support efficiently prevent NZVI from aggregation for quick and effective removal of As(iii). Through the non-linear least-squares (NLLS) adjustment, As(iii) removal by MOF-NZVI could be well fitted by pseudo first and second order reaction kinetics, as well as the Freundlich isotherm. FTIR, XRD and XPS results verified that NZVI and iron oxyhydroxides (Fe3O4, γ-Fe2O3, γ-FeOOH and α-FeOOH) might be responsible for the effective removal of As(iii) and its oxidized product As(v) with an adsorption capacity of 360.6 mg As per g NZVI through chemical oxidation and physical adsorption. This work indicates that MOF-NZVI with good reusability and high efficiency is promising for application in As(iii)-polluted wastewater treatment.
A zirconium 1,4-dicarboxybenzene metal–organic framework (UiO-66 MOF) was successfully used as a template to enhance the distribution and activity of nanoscale zero-valent iron (NZVI).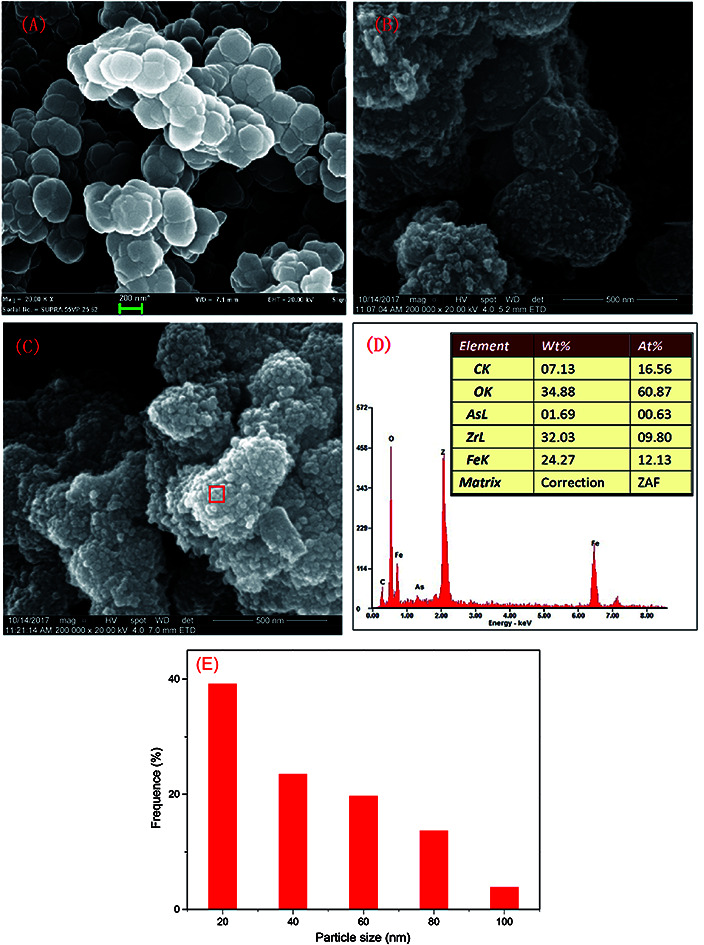
Introduction
Arsenic(iii) is a serious health and environmental hazard.1 Due to the high affinity of arsenic(iii) to vicinal sulfhydryl groups through reacting with various proteins, arsenic(iii) can inhibit the activity of proteins.2 The long-term ingestion of arsenic(iii) polluted groundwater can cause many serious diseases and even cancer.3 The World Health Organization (WHO) recommended that As(iii) in drinking water should be less than 10 μg L−1 to protect against health impacts.4 To meet these stringent guidelines for drinking-water quality, particularly in developing nations where technologies and resources are limited and most communities are at risk, there is a need to develop efficient, inexpensive and practical technologies to remove As(iii) from contaminated water.
Nanoscale zero-valent iron (NZVI) is known to efficiently remove toxic As(iii) from aqueous solutions.5 Unexpectedly, the aggregation and instability of NZVI frequently happened, consequently restricting the efficient removal of contaminants from effluents.6 To address these issues, surfactants and noble metals have been used to improve the stability and dispersion of NZVI in the aqueous solution.7,8 More recently, many porous materials were also used as a support to enhance their dispersion and reactivity.9,10
Metal–organic frameworks (MOFs) with the exceptionally high surface areas and porous networks may bring an inspiring breakthrough for wastewater treatment.11 MOFs have been modified by other functional materials to improve their properties and performances in various advanced fields, such as gas separations, sensors, drug transport and environmental remediation.12,13 MOFs can be used as a promising platform or support for other nanomaterials to develop novel catalytic composites.11,14 Recently, MOFs with single-molecule traps (SMTs) prevented enzyme from the aggregation through encapsulating each enzyme molecule in a SMT.15
Several MOFs have been designed to remove heavy metals from aqueous solution by adsorption.16 MOF based on Cu-benzene tricarboxylates has already been proven to effectively adsorb Cr(vi) from aqueous solution.17 Zeolite–imidazolate frameworks (ZIF) could remove heavy metals with high concentrations.18 Enhanced ZIF-8 nanocomposites offered a highly efficient reduction of Cr(vi) from aqueous solution.19 The zinc(ii) metal–organic frameworks showed effective adsorption of Cd(ii), Co(ii), Cr(iii), Cu(ii) and Pb(ii) in trace amounts.20 Moreover, MOFs showed excellent performance for the removal of As(iii) at ppb levels from aqueous solution.21 However, As(iii) adsorption onto ZIFs through surface complexation occurred within 2 h, which was a comparatively slow process,21 as well as metals adsorption on Cu-BTC.17 New designs that improve the stability and decontamination performance will overcome the current limitations of MOF composites.
Inspiringly, NZVI modified by MOFs was successfully prepared for solid phase extraction of N- and S-containing polycyclic aromatic hydrocarbons.22 Fe-based MOFs were used as a chemical precursor to synthesize NZVI on porous carbon to efficiently remove Cr(vi).23 NZVI was distributed uniformly in the carbon support, leading to prevent the agglomeration of these nanoparticles.5 MOFs could also be employed as templates for the fabrication of magnetic iron oxide composites for the adsorption of organic dyes from wastewater.24 UiO-66 with high surface area and stability has become one of the most popular MOFs,25 leading to its potential application as a support for nanoparticles. Furthermore, UiO-66 is effective to capture heavy metals from aqueous solution, such as As(v) and U(vi).26,27 Herein, our central hypothesis is that UiO-66 MOFs as the template for supporting NZVI can substantially enhance the removal efficiency of As(iii) from aqueous solutions.
The main objectives are to: (1) test the stable UiO-66 MOFs as the template to support and distribute NZVI, (2) examine removal efficiencies of As(iii) at different conditions using MOF-NZVI and (3) reveal the removal mechanism of As(iii) using MOF-NZVI by electron microscopic and spectroscopic analysis.
Experimental
UiO-66 MOFs (C48H24O30Zr6, BET surface area > 1100 m2 g−1) were provided by Strem Chemicals, Inc. (Massachusetts, United States). 1 g L−1 Na3AsO3 solution was purchased from National Centre for Certified Reference Materials (Beijing, China). BH4− and Fe(ii) stock solutions were freshly prepared using sodium borohydride (NaBH4) and ferrous sulfate tetrahydrate (FeSO4·4H2O) (Sigma-Aldrich Co., USA), respectively. All other chemicals were analytical reagent grade.
MOF-NZVI was in situ synthesized using UiO-66 MOFs as a support model according to a previous procedure.28 After ferrous ions (0.1 g of FeSO4·4H2O) was dissolved into 100 mL of alcohol–water solution (8 : 1 v/v) in a three-necked open flask, commercial MOFs (0.125 g) was added into the flask. After the magnetic stirring at 65 °C for an hour, a freshly prepared solution through the dissolution of 0.95 g NaBH4 in 100 mL deionized water was introduced into the above mixture drop by drop under constant stir of 180 rpm. The freshly prepared MOF-NZVI was put quietly for 24 h, and then washed using deionized water for three times. The MOF-NZVI was stored in a brown bottle for further use. The theoretical mass fraction of NZVI via the reduction of FeSO4·4H2O by NaBH4 on the surface of the MOF-NZVI was about 16.7% and MOF-NZVI had a SBET of 82.2 m2 g−1. The whole process was carried out in a nitrogen atmosphere.
Effects of different experimental factors on the removal efficiencies and mechanism of As(iii) using MOF-NZVI were conducted according to the following procedure. Effects of pH on the removal efficiency of As(iii) were investigated and the pH was adjusted to 3.0, 5.0, 7.0, 9.0 and 12.0 using 0.1 mol L−1 HCl and NaOH, respectively. The concentration of As(iii) was also chosen as a variable in five levels at 20, 40, 60, 80 and 100 mg L−1. Effects of temperatures on the removal efficiency of As(iii) were conducted at 15, 20, 25 and 30 °C. MOF-NZVI (0.15 g) was mixed with the prepared As(iii) wastewater (100 mL) in brown bottles, which were put in a water-bathing constant temperature vibrator to achieve the stable reaction temperature and uniform mixing. Then, 5 mL dispensable syringes and 0.45 μm filters were used to withdraw and filter samples for further analysis. All experiments were performed in duplicate.
Kinetics of As(iii) removal by MOF-NZVI were described using the pseudo-first-order and pseudo-second-order reaction.29 According to a previously procedure,6 the nature of adsorption process was also evaluated using Langmuir and Freundlich isotherm models, as well as the thermodynamic parameters such as enthalpy change (ΔH0), Gibbs free energy change (ΔG0) and entropy change (ΔS0).
Recyclability investigation of MOF-NZVI to remove As(iii) was according to the previous procedure.5 MOFs-NVZI (mMOF-NZVI = 0.15 g, mass fraction of NZVI = 16.7%) was added to 20 mg L−1 As(iii) solution (100 mL) and the mixture was left at room temperature with slightly stirring. After 1 h, MOF-NZVI was collected and washed by 0.01 mol L−1 HCl solution for 3 times. Then, these MOF-NZVI was soaked in deionized–deoxygenated water for 24 h. The MOF-NZVI was used to remove As(iii) for 5 times in succession to test the efficiencies of recyclability under the identical experimental conditions.
The morphological structure of MOF-NZVI was evaluated using scanning electron microscope (SEM) (SEM, FEI Nova NanoSEM 230) at a accelerating voltage of 10–20 kV, as well as transmission electron microscope (TEM, FEI Tecnai G2 F30). Inductively coupled plasma-mass spectrometry (ICP-MS, Elan-9000, PE) was used to determine the concentrations of arsenic and Fe ions. A Nexus fourier transform infrared (FTIR) spectroscopy was used to offer FTIR spectra for MOF-NZVI before and after their reaction to As(iii). X-ray diffraction (XRD) using an X-ray diffractometer (IPDSII T, STOE Company, Germany) with a copper target was conducted to characterize the MOF-NZVI materials at 40 kV and 40 mA. The valence change of arsenic and Fe on the surface of MOF-NZVI was investigated using the X-ray photoelectron spectroscope (XPS, PHI 5000 Versa Probe) at an energy spectrum range of 40–48 eV for As and 700–730 eV for Fe. The specific surface area (SBET) of MOF-NZVI was also estimated with Brunauer–Emmett–Teller (BET) N2 method.
Results and discussion
The SEM characterization is shown in Fig. 1. The particle size of MOFs (UiO-66) was 0.20–0.36 μm and the particles had clearly visible facets (Fig. 1(A)). The Zr–O bonds between the cluster and carboxylate ligands were known to increase the stability of Zr-based MOFs.30 The strong Zr–O bonds and the stable Zr6-cluster core also contributed to the special stability of UiO-66.31 After NZVI was supported on MOFs (UiO-66), the surfaces of MOFs were rough, however, the structure of MOFs was completely preserved (Fig. 1(B)). The stable structure of MOFs (UiO-66) providing a steady support for NZVI was contributed to the great stability of MOF-NZVI. There were many crystallized products formed on the surfaces of MOF-NZVI after their reactions with As(iii) (Fig. 1(C)). Weight percentage of arsenic was about 1.69% on the surface of MOF-NZVI (Fig. 1(D)). Other report showed that weight percentage of arsenic was in the range of 2.48% to 2.97% on the FeOOH surface, which was much less than the thin layer of arsenic between the Fe(0) core and iron oxide shell of NZVI.32 These crystallized products may cause the microstructure changes of MOF-NZVI, which will be characterized by TEM as following. The size distribution for a random sample of 295 particles. The particles are nearly uniform in size with a mean diameter of 36.6 nm and more than 82.4% are below 60 nm (Fig. 1(E)).
Fig. 1. SEM images of (A) MOFs (UiO-66), (B) fresh MOFs-NZVI and (C) MOFs-NZVI reacted with As(iii), (D) EDS of main elements in the region of (C) in the red frame and (E) histogram showing the size distribution of NZVI on MOFs-NZVI (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7.0, T = 25 °C).

NZVI was generally well dispersed on MOFs (Fig. 2(A)). The shape of NZVI became indistinct after the removal of As(iii) using MOF-NZVI (Fig. 2(D)), indicating that new products were formed on the surface of NZVI in the process. Comparing Fig. 2(B) with Fig. 2(E), it could be obviously seen that evident core–shell structures appeared on MOF-NZVI after the exposure to As(iii) rather than fresh MOF-NZVI, meaning that MOFs as a stable support could prevent NZVI from the oxidation at a certain extent. NZVI had a face-centered cubic structure (fcc) in the space group O.32Fig. 2(C) showed that the structure of NZVI was predominant along (111) orientation in fresh MOF-NZVI. Other researchers found that the interatomic spacing along this direction was 1.70 Å, which was in good agreement with the calculated interatomic spacing along 〈110〉 direction.33Fig. 2(F) showed that the structure of NZVI had changed to a body-centered cubic structure (bcc), which was predominant along (111) orientation in the consumed MOF-NZVI. The similar results had been achieved by using NZVI to enrich silver from wastewater and the interatomic spacing along this direction was measured at 2.36 Å.34 The structural transformation of NZVI may be due to adsorption of As on the surface of NZVI, which is agreement with the change of morphology and structure of MOF-NZVI shown in Fig. 1.
Fig. 2. TEM images of (A)–(C) of fresh MOFs-NZVI and (D)–(F) of MOFs-NZVI reacted with As(iii) (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7.0, T = 25 °C).

HADDF image of MOF-NZVI and elemental mapping of Fe showed that NZVI was dispersedly distributed on the surface of MOF-NZVI (Fig. 3(A)–(C)), which proved again that MOFs could be used as a stable support to successfully prevent NZVI from aggregation. Elemental mapping of As showed that As was evenly scattered on the surface of MOF-NZVI (Fig. 3(D)), indicating that As was adsorbed by both MOFs and NZVI on the surface of MOF-NZVI. It was interesting that As aggregation did not happen although the slight aggregation of NZVI on the edge of MOF-NZVI (Fig. 3(C)). According to a single NZVI particle, arsenic was mainly enriched in the thin layer between the Fe(0) core and iron oxide shell.33 Elemental mapping of Zr (Fig. 3(E)) indicated that Zr was stable at MOFs structure, which played an important for supporting NZVI. The results proved again that As(iii) could be effectively removed by MOF-NZVI, leading to the change of morphology and structure of MOF-NZVI shown in Fig. 1 and 2, which could help to demonstrate the removal mechanism.
Fig. 3. (A) TEM image and (B) HADDF image of MOFs-NZVI reacted with As(iii), elemental mappings of (C) Fe, (D) As and (E) Zr (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7.0, T = 25 °C).

The effect of NZVI amounts on As(iii) removal using MOF-NZVI is shown in Fig. 4(A). The removal (%) rates were more than 98.6% at different NZVI dosages within 1 min, whereas there was less than 10% of As(iii) and As(v) adsorbed by only MOFs (Fig. 4(A)). MOF-NZVI was efficient and rapid to remove As(iii) from wastewater and NZVI had a critical role in achieving the excellent performance. MOFs with a high BET surface area provided a support for uniform distribution of NZVI, promoting full contact between As(iii) and NZVI active sites.23 The MOFs appeared to prevent the agglomeration of NZVI, increasing the efficiency of As(iii) removal. The similar phenomena were also found in the removal of mercury(ii), chromium(vi) and lead(ii) by other NZVI systems.28,35 Unexpectedly, the removal (%) decreased slightly as the NZVI dosage increased from 0.025 to 0.2 g (Fig. 4(A)). Other reports showed that the removal (%) and the rate constants (Kobs) of As(iii) were enhanced with an increase in NZVI dosages.5 Obviously, an increase of NZVI amount could offer more active sites for contaminants.36 However, a given amount of MOFs in our study had a certain surface area and the slight agglomeration of NZVI might happen when the amount of NZVI reached a certain value. According to the situation, the removal rate (%) was not enhanced all the time as an increase of the NZVI dosage in MOF-NZVI. The tendency toward the agglomeration of NZVI at certain thresholds might be a critical factor, leading to a slight decrease of the removal (%) as an increase of the NZVI dosage. Therefore, MOF-NZVI compounds containing 0.025 g NZVI and 0.125 g MOFs were used in all the following experiments.
Fig. 4. (A) The effect of NZVI dosages on the removal (%) of As(iii)/As(v), (B) the effect of pH values on the removal (%) of As(iii) and inset: adsorption kinetics plots fitted with pseudo-first-order model, (C) zeta-potential as a function of solution pH, (D) the effect of initial concentrations of As(iii) on the removal (%) of As(iii) and inset: the effect of initial concentrations of As(iii) on the adsorption capacity in 60 min, (E) the effect of temperatures on the removal (%) of As(iii) and inset: adsorption kinetics plots fitted with pseudo-second-order model and (F) Freundlich and Langmuir adsorption isotherms of As(iii) onto MOFs-NZVI.

The removal (%) of As(iii) were all more than 98.2% at pH between 3.0 and 12.0 and the removal (%) were highest at pH = 7.0 (Fig. 4(B)). ZVI corrosion at pH < 7.0 would produce a great number of iron oxides/hydroxides, which showed good adsorption effect on As(iii).37 In order to better understand and discuss the phenomenon, zeta potential of MOF-NZVI is shown in Fig. 4(C). MOF-NZVI was positively charged at pH < 8.1 and negatively charged at pH > 8.1, indicating that the isoelectric point (IEP) of MOF-NZVI was 8.1. As pH value increases up to 9.2, H2AsO3− became the main specie5 and the surface of MOF-NZVI was negative charge (Fig. 4(C)). Consequently, the potential repulsion force between MOF-NZVI and H2AsO3− would be formed, which hinder the contact reaction process, contributing to declining As removal efficiency.
The pseudo-first-order reaction kinetics data has been adjusted following the non-linear least-squares (NLLS) regression according to previous procedures38 and the results are shown in Fig. 4(B) and inset. High correlation coefficients (R2) (>0.98) stated clearly that the experimental data could be well fitted to pseudo-first-order reaction kinetics for adsorption of As(iii) on MOF-NZVI (Fig. 4(B) and Table 1). The calculated adsorption capacity (qe) was 360.6 mg As per g NZVI, which was very close to the experimental value (qexp) 360.8 mg As per g NZVI (Table 1). The similar performance has also been reported in other systems using hydrogel to remove Cr(vi).38 The structure of MOFs was complex and stable for dispersedly supporting NZVI that benefited contact reaction between NZVI active sites and As(iii),39 contributing to the high removal (%) of As(iii) by MOF-NZVI in a wide pH range. It is apparent that MOF-NZVI is an effective and fast composite for removing As(iii) from wastewater.
Comparison of pseudo first and second order kinetics model constants (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 100 mg L−1, VAs(iii) = 100 mL, pH = 7.0, T = 25 °C).
| q exp (mg As per g NZVI) | Pseudo-first-order kinetics | Pseudo-second-order kinetics | ||||
|---|---|---|---|---|---|---|
| k 1 (min−1) | q e (mg As per g NZVI) | R 2 | k 2 (g (mg min)−1) | q e (mg As per g NZVI) | R 2 | |
| 360.8 | 0.63 | 360.6 | 0.98 | 0.16 | 360.7 | 0.99 |
Initial concentrations of As(iii) showed an obvious effect on removal efficiencies of As(iii).40 With an increase of the initial As(iii) concentration, the removal (%) of As(iii) using MOF-NZVI declined gradually in 10 min, however, these rates were almost all the same after 10 min (Fig. 4(D)). As the initial As(iii) concentration increased from 20 to 100 mg L−1, the adsorbed amount was enhanced from 72.6 to 360.6 mg As per g NZVI (Fig. 4(D) and inset), explaining that MOF-NZVI had a good adsorption capacity to As(iii) at different initial concentrations of As(iii). The higher adsorption capacity to As(iii) by MOF-NZVI than those by other NZVI, ZIF-8 nanoparticles and Fe3O4@TiO2 nanoparticles might be due to the highly dispersed distribution of NZVI in the stable matrix structure of MOFs,5,21,41 which provided effectively active sites for As(iii) adsorption (Table 2).
The comparison of adsorption capacity of As(iii) from wastewater by different adsorbent.
| Type of adsorbent | Adsorption conditions | Adsorption capacity (mg g−1) | References |
|---|---|---|---|
| NZVI | C NZVI = 0.5 g L−1, CAs(iii) = 100 mg L−1, VAs(iii) = 20 mL, pH = 6.5, t = 45 min | 3.5 | 5 |
| ZIF-8 nanoparticles | C ZIF-8 = 0.2 g L−1, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7, t = 24 h | 49.49 | 33 |
| Fe3O4@TiO2 nanoparticles | C Fe3O4@TiO2 = 0.32 g L−1, CAs(iii) = 0.91 mg L−1, VAs(iii) = 1000 mL, pH = 7, t = 180 min | 30.96 | 50 |
| MOFs-NZVI | m MOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7.0, t = 60 min | 360.6 | a |
The result of our study.
The effects of co-existing cations, anions and organic acids are pivotal factors in evaluating the adsorption capacity to As(iii) using MOF-NZVI. The presence of PO43− and CO32− inhibited the removal of both pentavalent and trivalent arsenic by ZIF-8 nanoparticles.21 Magnesium and calcium played a stimulative role in arsenate rather than arsenite adsorption onto a goethite-based material at higher pH values.42 The arsenite remediation using Fe(ii)–KMnO4 was enhanced by Ca2+ at pH 7.0–9.0.43 Mainly through competitive adsorption, humic acid (HA), phosphate and sulfate decreased As(iii) adsorption onto Fe(ii)–KMnO4.44 The removal of arsenic was adversely affected by phosphates when their concentrations were higher than 200 μg L−1.45 PO43− showed certain interference on the adsorption of As(iii) when the concentration of PO43− was much lower than that of As(iii).46
The effects of co-existing cations, anions and organic acids on As(iii) removal by MOF-NZVI are shown in Table 3. However, these co-existing cations (Ca2+, Mg2+ and Cu2+) and anions (PO43− and SO42−) had no noticeable inhibitory effect on As(iii) removal by MOF-NZVI in a pH range of 3.0–12.0 (Table 3). The similar results were also reported in other systems. The presence of SO42− showed no significant interference on the adsorption of As(iii) by ZIF-8 nanoparticles and GO-ZrO(OH)2 nanocomposites.21,46 Mg2+, Ca2+, PO43− and SO42− had few effects on As(iii) removal using cement modified by iron oxide.47 Oxalate had scarcely any effect on nitrate removal by ZVI through surface complexation.48 Nevertheless, the removal of heavy metals ions using Na-montmorillonite relied heavily on oxalic acid and citric acid.49 Hg(ii) adsorption onto kaolinite could be hindered by citric acid at pH values of 6.0 and 8.0.50 Oxalic acid could inhibit Hg(ii) adsorption at high concentrations.51 However, it was surprising that oxalic acid and citric acid did not inhibit As(iii) adsorption onto MOF-NZVI in this study (Table 3). Overall, MOF-NZVI showed good anti-interference ability to co-existing cations, anions and organic acids at a wide range of pH.
The effect of co-existing cations and anions on the removal (%) of As(iii) at different pH values (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 3.0, 5.0, 7.0, 9.0 and 12.0, T = 25 °C).
| pH | Co-existing ions | ||||||
|---|---|---|---|---|---|---|---|
| Ca2+ (100 mg L−1) | Mn2+ (100 mg L−1) | Cu2+ (20 mg L−1) | H2PO4− (20 mg L−1) | SO42−(20 mg L−1) | Oxalic acid (20 mg L−1) | Citric acid (20 mg L−1) | |
| 3.0 | 99.7 | 99.6 | 99.5 | 99.4 | 99.5 | 99.2 | 99.2 |
| 5.0 | 99.5 | 99.3 | 99.2 | 99.6 | 99.2 | 99.1 | 99.0 |
| 7.0 | 99.5 | 99.3 | 99.0 | 99.2 | 99.0 | 99.1 | 99.0 |
| 9.0 | 99.4 | 99.2 | 98.7 | 99.0 | 98.7 | 99.3 | 98.5 |
| 12.0 | 99.4 | 99.3 | 99.3 | 98.5 | 99.3 | 97.3 | 96.8 |
The effects of temperatures on removal (%) rates are shown in Fig. 4(E). The pseudo-second-order reaction kinetics data has been adjusted following the non-linear least-squares (NLLS) regression according to previous procedures38 and the results are shown in Fig. 4(E) and Table 1. Removal (%) are >99.0% at temperatures ranging from 15 to 35 °C (Fig. 4(E)), displaying that MOF-NZVI is effective to remove As(iii). Furthermore, R2 is more than 0.99 and the calculated adsorption capacity (qe) 360.7 mg As per g NZVI was very close to the experimental value (qexp) 360.8 mg As per g NZVI (Fig. 4(E) inset and Table 1), indicating that the experimental data of As(iii) adsorption onto MOF-NZVI could be well fitted to a pseudo-second-order kinetic expression. Pseudo-second order model describe well the adsorption system, implying that chemical sorption dominates the removal process of As(iii) by MOF-NZVI through electronic exchange or transfer between sorbent and sorbate.52
Freundlich and Langmuir equations to As(iii) adsorption by MOF-NZVI at different temperature are adjusted with NLLS and the results are shown in Table 4 and Fig. 4(F). Nonlinearized Freundlich and Langmuir equations was also used to treat the equilibrium data in order to obtain more isotherm constants (Table 4), which could characterize the nature of As(iii) adsorption onto MOF-NZVI. By comparing the correlation coefficients between the isotherm models, As(iii) adsorption on MOF-NZVI better followed Freundlich isotherm (Table 4). The strong correlation (R2 > 0.98) with Freundlich isotherm (Table 4) meant that As(iii) adsorption on MOF-NZVI was via a heterogeneous system, which might contain physical adsorption of As(iii) by MOFs and NZVI, quick oxidization of As(iii) to As(v) by NZVI and its corrosion products (iron oxyhydroxides), and then physical adsorption of As(v) by MOFs, NZVI and iron oxyhydroxides. All of the exponent n was larger than 3.6 (Table 4), indicating that the adsorption of As(iii) on MOF-NZVI was a favorable adsorption process.53 The increasing K values as an increase in temperatures predicated that the higher temperature was more favorable to As(iii) adsorption. Nearly similar result has been reported for Langmuir and Freundlich models in other literature.9 It was obvious that qe of As(iii) adsorption by MOF-NZVI increased with an increase of Ce. Moreover, Freundlich adsorption isotherm of As(iii) onto MOF-NZVI showed a proper fit with the non-linear regression model (Fig. 4(F)), indicating that As(iii) adsorption on MOF-NZVI occurred in a heterogeneous system. The phenomenon is in accordance with the kinetics behavior previously (Fig. 4(B) and (E)). The similar phenomenon was also reported in other research.54 The analyses hence suggested that MOF-NZVI with a heterogeneous surface was rapid and effective to remove As(iii), which might also help us explain the removal behavior previously shown in Fig. 4(A) and (B).
Langmuir and Freundlich isotherm constants at different temperatures (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7.0, T = 15, 20, 25 and 35 °C).
| T (K) | Langmuir isotherm parameters | Freundlich isotherm parameters | ||||
|---|---|---|---|---|---|---|
| q m (mg g−1) | b | R 2 | n | K | R 2 | |
| 288 | 360.6 | 2.08 | 0.79 | 3.66 | 198.8 | 0.98 |
| 293 | 360.8 | 2.13 | 0.88 | 3.72 | 198.6 | 0.99 |
| 298 | 361.2 | 2.16 | 0.86 | 3.81 | 199.4 | 0.99 |
| 308 | 361.3 | 2.02 | 0.89 | 3.96 | 199.6 | 0.99 |
At the reaction temperature of 288–303 K, Gibbs free energy change (ΔG0) was negative and decreased with an increase in temperature (Table 5), indicating that the process of As(iii) removal by MOF-NZVI was spontaneous and more favorable at higher temperature. Enthalpy change (ΔH0) was positive (Table 5), suggesting that As(iii) removal by MOF-NZVI was an endothermic process. Entropy change (ΔS0) was positive (Table 5), which reflected the good affinity between As(iii) and MOF-NZVI. The similar phenomena were also found in the other NZVI system.6
Thermodynamic parameters for adsorption of As(iii) onto MOFs-NZVI (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7.0, T = 15, 20, 25 and 35 °C).
| Temperature (K) | ΔH0 (kJ mol−1) | ΔG0 (kJ mol−1) | ΔS0 (J K−1 mol−1) |
|---|---|---|---|
| 288 K | 56.2 | −8.62 | 1.83 |
| 293 K | −9.96 | ||
| 298 K | −11.67 | ||
| 308 K | −13.56 |
In order to reveal the removal mechanism of As(iii) by MOF-NZVI, XRD patterns of MOF-NZVI were investigated (Fig. 5). According to Valenzano et al.,31 the characteristic peaks of MOFs centered at 7.2° and 8.4°, which did not disappear or diminish in intensity in Fig. 5, indicating that MOFs structure was very stable as the support to enhance the dispersity and stability of NZVI. Three NZVI characteristic peaks of fresh MOF-NZVI centered at 45.06°, 65.0° and 82.62° according to Fe0,55 indicating that the Fe0 was the main structural composition of NZVI samples in fresh MOF-NZVI (Fig. 5). As the reaction between MOF-NZVI and As(iii) proceeded, NZVI characteristic peaks at 45.06° and 65.0° gradually disappeared and new peaks appeared at 35.6° assigned to γ-Fe2O3, 43.6° and 56.8° to Fe3O4, and 14.9°, 26.7°, 31.2°, 50.7° and 63.6° to γ-FeOOH (Fig. 5).56 Consequently, iron oxyhydroxides (γ-Fe2O3, Fe3O4 and γ-FeOOH) were formed on the surface of NZVI, indicating that iron oxyhydroxides played a critical role in As(iii) removal using MOF-NZVI through multiple mechanisms containing oxidation, adsorption, precipitation and coprecipitation.57 Similar corrosion products were found by other researchers using NZVI to remove As(iii).5
Fig. 5. X-ray diffraction patterns of fresh MOFs-NZVI, MOFs-NZVI reacted with As(iii) after 10 min and 60 min. Peaks are due to NZVI (Fe0), magnetite (Fe3O4) (MN), maghemite (γ-Fe2O3) (MH), lepidocrocite (γ-FeOOH) (LE), respectively (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7.0, T = 25 °C).

The FTIR spectra between 4000 to 390 cm−1 of fresh MOF-NZVI and MOF-NZVI reacted with As(iii) after 60 min are shown in Fig. 6. The peak at 3428 cm−1 and the new one at 1633 cm−1 after the reaction with As(iii) was ascribed to the O–H stretching vibration of surface-adsorbed water (Fig. 6), partly due to the O–H stretching vibration of γ-FeOOH,58 indicating that γ-FeOOH was formed as one the corrosion products playing an important role in the removal of As(iii). The peaks at 1165 and 1060 cm−1 according to γ-FeOOH weakened (Fig. 6), clearly stating the binding of arsenic onto γ-FeOOH. Because of the bands at 3000 and 885 cm−1 assigned to hydroxyl groups in the goethite structure,59 the rising peaks at 3000 and 885 cm−1 proved the binding of arsenic onto α-FeOOH (Fig. 6). Bands at 570 cm−1 and 630 cm−1 assigned to the Fe–O stretching models on magnetite (Fe3O4) and maghemite (γ-Fe2O3),60 respectively, are also found in Fig. 6 and inset. Moreover, the peak values at 455 and 589 cm−1 represented the Fe–O bonds.60 As a result, magnetite (Fe3O4) and maghemite (γ-Fe2O3) were also involved in the removal of As(iii). The peak at 812 cm−1 corresponds to the Zr–O–As band,61 identifying the binding of arsenic onto MOFs. This is in agree with the results of Fig. 5. As(iii) could be effectively removed by adsorption on CS-P-NZVI in a very short time through quick adsorption and oxidation.5 Through the electrostatic attraction between the negatively charged As–OH and the positively charged Zr–OH, monodentate and bidentate complexes were formed on GO-ZrO(OH)2.40
Fig. 6. FTIR of fresh MOFs-NZVI (A) and MOFs-NZVI (B) reacted with As(iii) after 60 min. Inset: the spectra from 395–1200 cm−1 is enlarged (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7.0, T = 25 °C).

Photoelectron peaks centered at 707.8 and 725.1 eV (Fig. 7(A)) to the binding energies of 2p3 of oxidized iron [Fe(ii)/Fe(iii)]. The peak at 706.6 eV to Fe0 did not appear in this study,6 which indicated that NZVI was extensively oxidized and little Fe0 remained on the surface of MOF-NZVI. Detailed XPS scans on the region of As 3d show that the photoelectron peak centers at 45.2 eV which corresponds to As(v) and 43.4 eV which corresponds to As(iii)5 (Fig. 7(B)). About 58% As(iii) was oxidized to As(v) and 42% As(iii) was therefore physically adsorbed on MOF-NZVI. The removal of As(iii) by MOF-NZVI included both oxidization and physical adsorption. The iron oxyhydroxides containing γ-FeOOH, α-FeOOH, Fe3O4 and γ-Fe2O3 were responsible for removing As(iii) and its oxidation product (As(v)). The removal mechanism of As(iii) by MOF-NZVI can be proposed that the quick capture of As(iii) by MOFs and NZVI on the surface of MOF-NZVI facilitates the reaction between NZVI and As(iii). As a result, As(iii) was quickly oxidized to As(v) by NZVI and its corrosion products (iron oxyhydroxides), which were also responsible for physically adsorbing As(iii) and its oxidation product (As(v)). Therefore, the removal mechanism of As(iii) by the dual processes of chemical oxidation and physical adsorption is shown schematically in Fig. 8, which is consistent with other research on As(iii) removal in the Fenton reaction.62 The inner sphere complex was formed through As–O linkages in the process of the As(iii) adsorption on COIMA.63 The outer-sphere complexes containing As(v) was formed through surface anion exchange and As(iii) was adsorbed in the inter-sphere complexes through physisorption by Si–O and Al–O groups bonded to the surfactant-modified bentonite.64 The As(iii) removal mechanism by Fe-exchanged natural zeolite contained oxidation of As(iii) to As(v) and adsorption of As(iii) and As(v) on the surface of the adsorbent.65
Fig. 7. High-resolution XPS survey of (A) Fe 2p3 and (B) As 3d for MOFs-NZVI reaction with As(iii) after 60 min (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7.0, T = 25 °C).

Fig. 8. Schematic diagram showing the removal mechanism of As(iii) using MOFs-NZVI.

The regeneration efficiency of MOF-NZVI is presented in Fig. 9(A). The removal (%) of As(iii) decrease markedly at the beginning of each cycle. After several cycles, NZVI might be generally dwindled away. Furthermore, some of active sites of MOF-NZVI would be generally occupied by As(iii) and As(v) through physical and chemical adsorption. As a result, the decay appeared in the recyclability after the 5th cycles. However, a high removal (%) > 95.4% after 60 min could still be achieved on the fifth run (Fig. 9(A)), indicating good reusability of MOF-NZVI. Washing MOF-NZVI before each reuse by dilute HCl to dissolve corrosion products of NZVI (iron oxyhydroxides) on the surface of MOF-NZVI appeared to be an effective at expose fresh Fe0 to As(iii).5 The results suggest that MOF-NZVI is reusable. Furthermore, the proportion of As(v) adsorbed on MOF-NZVI was gradually reduced from 58% to 39% and the proportion of As(iii) adsorbed on MOF-NZVI was gradually increased from 42% to 56.4% as MOF-NZVI was recycled for 5 times (Fig. 9(B)–(F)), indicating that oxidation capability of As(iii) by MOF-NZVI slowly decreased as regeneration of MOF-NZVI for several times. Due to the exhaustion of NZVI step by step, the concentration of Fe ions in the leaching solution after each recycle was gradually decreased (Fig. 10). However, according to the results of TEM, XRD and FTIR (Fig. 2, 5 and 6), the educated guess can be made that the adsorption capacity of As(iii) by MOF-NZVI will be gradually diminished due to the oxidation and exhaustion of NZVI. MOF-MIL-101 modified by iron nanoparticles also showed excellent reusability.66 The recirculation could improve the efficiency of NZVI and enhance the removal of heavy metals by reacted NZVI.
Fig. 9. (A) As(iii) removal efficiency in each cycle after regeneration using MOFs-NZVI, high-resolution XPS survey of As 3d for MOFs-NZVI reaction with As(iii) after 60 min at the first time (B), the second time (C), the third time (D), the fourth time (E) and the fifth time (F) (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7.0, T = 25 °C).

Fig. 10. The concentration of Fe ions in the leaching solution after each recycle (mMOFs-NZVI = 0.15 g, mass fraction of NZVI = 16.7%, CAs(iii) = 20 mg L−1, VAs(iii) = 100 mL, pH = 7.0, T = 25 °C, t = 60 min).

Conclusions
MOFs (UiO-66) was successfully used as a support model to improve the distribution and activity of NZVI. As(iii) could be quickly and effectively removed by MOF-NZVI in a very short time (minute scale) over a wide range of pH (3.0–12.0) and concentration (20–100 mg L−1) with co-existing cations (Ca2+, Mn2+ and Cu2+), anions (H2PO4− and SO42−) and organic acids (oxalic acid and citric acid). Furthermore, the experimental data of As(iii) adsorption onto MOF-NZVI could be well fitted to a pseudo first and second order kinetic expression by the non-linear least-squares (NLLS) adjustment. As(iii) adsorption on MOF-NZVI was heterogeneous adsorption, which could be well described by Freundlich isotherm. SEM characterization indicated that MOFs could prevent NZVI from aggregating in MOF-NZVI. TEM characterization showed clearly structural transformation of NZVI on the surface of MOF-NZVI. FTIR and XRD results indicated that NZVI and the iron oxyhydroxides containing γ-FeOOH, α-FeOOH, Fe3O4 and γ-Fe2O3 might play the major role (>91%) in the effective and quick removal of As(iii) through oxidation and its oxide As(v) through complexation/precipitation. At the same time, MOFs also take part in the removal of As(iii) (<9%) through physical adsorption. XPS result stated that about 58% As(iii) was oxidized to As(v) and 42% As(iii) was therefore physically adsorbed on MOF-NZVI. However, oxidation capability of As(iii) by MOF-NZVI slowly decreased as regeneration of MOF-NZVI for several times and the oxidation and chemical adsorption play the leading role. By now, it can be concluded that MOF-NZVI with good reusability and high efficiency is a suitable candidate for the remediation of arsenic.
Conflicts of interest
There is no conflicts to declare.
Supplementary Material
Acknowledgments
This work is financially supported by the Youth Scientific Research Talent Support Program of Tianjin Normal University (135202RC1706), Young and Middle-aged Backbone Innovative Talents Training Project Foundation of Tianjin (135305JF76), 131 Talent Project in the Second Level of Tianjin (135305QR06), National Natural Science Foundation of China (21307090) and Tianjin Municipal Natural Science Foundation of China (18JCYBJC96600).
Notes and references
- Lee Y. Um I.-H. Yoon J. Y. Environ. Sci. Technol. 2003;37:5750. doi: 10.1021/es034203+. [DOI] [PubMed] [Google Scholar]
- Aposhian H. V. Maiorino R. M. Dart R. C. Perry D. F. Clin. Pharmacol. Ther. 1989;45:520. doi: 10.1038/clpt.1989.67. [DOI] [PubMed] [Google Scholar]
- Mandal S. Sahu M. K. Patel R. K. Water Resources and Industry. 2013;4:51. doi: 10.1016/j.wri.2013.09.003. [DOI] [Google Scholar]
- WHO, Guidelines for drinking water quality, vol. 1: Recommendations, WHO, Geneva, 2nd edn, 1993 [Google Scholar]
- Liu T. Wang Z.-L. Zhao L. Yang Y. Chem. Eng. J. 2012;189–190:196. doi: 10.1016/j.cej.2012.02.056. [DOI] [Google Scholar]
- Baikousi M. Georgiou Y. Daikopoulos C. Bourlinos A. B. Filip J. Zbořil R. Deligiannakis Y. Karakassides M. A. Carbon. 2015;93:636. doi: 10.1016/j.carbon.2015.05.081. [DOI] [Google Scholar]
- He F. Zhao D. Environ. Sci. Technol. 2005;39:3314. doi: 10.1021/es048743y. [DOI] [PubMed] [Google Scholar]
- Tiraferri A. Chen K. L. Sethi R. Elimelech M. J. Colloid Interface Sci. 2008;324:71. doi: 10.1016/j.jcis.2008.04.064. [DOI] [PubMed] [Google Scholar]
- Liu T. Wang Z.-L. Sun Y. Chem. Eng. J. 2015;263:55. doi: 10.1016/j.cej.2014.11.046. [DOI] [Google Scholar]
- Teng W. Fan J. Wang W. Bai N. Liu R. Liu Y. Deng Y. Kong B. Yang J. Zhao D. Zhang W. X. J. Mater. Chem. A. 2017;5:4478. doi: 10.1039/C6TA10007D. [DOI] [Google Scholar]
- Lu G. Li S. Guo Z. Farha O. K. Hauser B. G. Qi X. Wang Y. Wang X. Han S. Liu X. DuChene J. S. Zhang H. Zhang Q. Chen X. Ma J. Loo S. C. J. Wei W. D. Yang Y. Hupp J. T. Huo F. Nat. Chem. 2012;3:310. doi: 10.1038/nchem.1272. [DOI] [PubMed] [Google Scholar]
- Li G. Kobayashi H. Taylor J. M. Ikeda R. Kubota Y. Kato K. Takata M. Yamamoto T. Toh S. Matsumura S. Kitagawa H. Nat. Mater. 2014;13:802. doi: 10.1038/nmat4030. [DOI] [PubMed] [Google Scholar]
- Xu Z. Wang Q. Li H. Meng W. Han Y. Hou H. Fan Y. Chem. Commun. 2012;48:5736. doi: 10.1039/C2CC31590D. [DOI] [PubMed] [Google Scholar]
- Wu H. B. Xia B. Y. Yu L. Yu X. Y. Lou X. W. Nat. Commun. 2015;6:6512. doi: 10.1038/ncomms7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D. Liu T. F. Su J. Bosch M. Wei Z. Wan W. Yuan D. Chen Y. P. Wang X. Wang K. Lian X. Guo Z. Y. Park J. Zou X. Zhou H. C. Nat. Commun. 2015;6:5979. doi: 10.1038/ncomms6979. [DOI] [PubMed] [Google Scholar]
- Fang Q. R. Yuan D. Q. Sculley J. Li J. R. Han Z. B. Zhou H. C. Inorg. Chem. 2010;49:11637. doi: 10.1021/ic101935f. [DOI] [PubMed] [Google Scholar]
- Maleki A. Hayati B. Naghizadeh M. Joo S. W. J. Ind. Eng. Chem. 2015;28:211. doi: 10.1016/j.jiec.2015.02.016. [DOI] [Google Scholar]
- Huang Y. Zeng X. Guo L. Lan J. Zhang L. Cao D. Sep. Purif. Technol. 2018;194:462. doi: 10.1016/j.seppur.2017.11.068. [DOI] [Google Scholar]
- Qiu J. Zhang X.-F. Zhang X. Feng Y. Li Y. Yang L. Lu H. Yao J. J. Hazard. Mater. 2018;349:234. doi: 10.1016/j.jhazmat.2018.02.009. [DOI] [PubMed] [Google Scholar]
- Tahmasebi E. Masoomi M. Y. Yamini Y. Inorg. Chem. 2015;54:425. doi: 10.1021/ic5015384. [DOI] [PubMed] [Google Scholar]
- Jian M. Liu B. Zhang G. Liu R. Zhang X. Colloids Surf., A. 2015;465:67. doi: 10.1016/j.colsurfa.2014.10.023. [DOI] [Google Scholar]
- Zhou Q. Lei M. Li J. Liu Y. Zhao K. Zhao D. Microchim. Acta. 2017;184:1029. doi: 10.1007/s00604-017-2094-6. [DOI] [Google Scholar]
- Wang Z. Yang J. Li Y. Zhuang Q. Gu J. Eur. J. Inorg. Chem. 2018;2018:23. doi: 10.1002/ejic.201701089. [DOI] [Google Scholar]
- Lee H. J. Cho W. Lim E. J. Oh M. Y. Chem. Commun. 2014;50:5476. doi: 10.1039/C4CC01914H. [DOI] [PubMed] [Google Scholar]
- Zhang X. Shen B. Zhu S. Xu H. Tian L. J. Hazard. Mater. 2016;320:556. doi: 10.1016/j.jhazmat.2016.08.039. [DOI] [PubMed] [Google Scholar]
- Wang C. Liu X. Chen J. P. Li K. Sci. Rep. 2015;5:16613. doi: 10.1038/srep16613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B.-C. Yuan L.-Y. Chai Z.-F. Shi W.-Q. Tang Q. J. Radioanal. Nucl. Chem. 2016;307:269. doi: 10.1007/s10967-015-4108-3. [DOI] [Google Scholar]
- Liu T. Wang Z.-L. Yan X. Zhang B. Chem. Eng. J. 2014;245:34. doi: 10.1016/j.cej.2014.02.011. [DOI] [Google Scholar]
- Ho Y. S. Wase D. A. J. Forster C. F. Water Res. 1995;29:1327. doi: 10.1016/0043-1354(94)00236-Z. [DOI] [Google Scholar]
- Cavka J. H. Jakobsen S. Olsbye U. Guillou N. Lamberti C. Bordiga S. Lillerud K. P. J. Am. Chem. Soc. 2008;130:13850. doi: 10.1021/ja8057953. [DOI] [PubMed] [Google Scholar]
- Valenzano L. Civalleri B. Chavan S. Bordiga S. Nilsen M. H. Jakobsen S. Lillerud K. P. Lamberti C. Chem. Mater. 2011;23:1700. doi: 10.1021/cm1022882. [DOI] [Google Scholar]
- Wang C. M. Baer D. R. Amonette J. E. Engelhard M. H. Antony J. Qiang Y. J. Am. Chem. Soc. 2009;131:8824. doi: 10.1021/ja900353f. [DOI] [PubMed] [Google Scholar]
- Lan L. Zhang W. Environ. Sci. Technol. 2017;51:2288. doi: 10.1021/acs.est.6b06426. [DOI] [PubMed] [Google Scholar]
- Lan L. Huang X.-Y. Zhang W.-X. Adv. Mater. 2018;30:1705703. doi: 10.1002/adma.201705703. [DOI] [PubMed] [Google Scholar]
- Ponder S. M. Darab J. G. Bucher J. Caulder D. Craig I. Davis L. Edelstein N. Lukens W. Nitsche H. Rao L. Shuh D. K. Mallouk T. E. Chem. Mater. 2001;13:479. doi: 10.1021/cm000288r. [DOI] [Google Scholar]
- He D. Ma J. Collins R. N. Waite T. D. Environ. Sci. Technol. 2016;50:3820. doi: 10.1021/acs.est.5b04988. [DOI] [PubMed] [Google Scholar]
- Neumann A. Kaegi R. Voegelin A. Hussam A. Munir A. K. M. Hug S. J. Environ. Sci. Technol. 2013;47:4544. doi: 10.1021/es305176x. [DOI] [PubMed] [Google Scholar]
- Liang Q. Luo H. Geng J. Chen J. Chem. Eng. J. 2018;338:62. doi: 10.1016/j.cej.2017.12.145. [DOI] [Google Scholar]
- Abney C. W. Gilhula J. C. Lu K. Lin W. Adv. Mater. 2014;26:7993. doi: 10.1002/adma.201403428. [DOI] [PubMed] [Google Scholar]
- Han Y. S. Gallegos T. J. Demond A. H. Hayes K. F. Water Res. 2011;45:593. doi: 10.1016/j.watres.2010.09.033. [DOI] [PubMed] [Google Scholar]
- Deng M. Wu X. Zhu A. Zhuang Q. Liu Q. J. Environ. Manage. 2019;237:63. doi: 10.1016/j.jenvman.2019.02.037. [DOI] [PubMed] [Google Scholar]
- Kanematsu M. Young T. M. Fukushi K. Green P. G. Darby J. L. Geochim. Cosmochim. Acta. 2013;106:404. doi: 10.1016/j.gca.2012.09.055. [DOI] [Google Scholar]
- Guan X. Ma J. Dong H. Jiang L. Water Res. 2009;43:5119. doi: 10.1016/j.watres.2008.12.054. [DOI] [PubMed] [Google Scholar]
- Guan X. Dong H. Ma J. Jiang L. Water Res. 2009;43:3891. doi: 10.1016/j.watres.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Katsoyiannis I. A. Zouboulis A. I. Water Res. 2002;36:5141. doi: 10.1016/S0043-1354(02)00236-1. [DOI] [PubMed] [Google Scholar]
- Luo X. Wang C. Wang L. Deng F. Luo S. Tu X. Au C. Chem. Eng. J. 2013;220:98. doi: 10.1016/j.cej.2013.01.017. [DOI] [Google Scholar]
- Kunda S. Gupta A. K. Sep. Purif. Technol. 2006;51:165. doi: 10.1016/j.seppur.2006.01.007. [DOI] [Google Scholar]
- Song X. Chen Z. Wang X. Zhang S. Water Res. 2017;114:218. doi: 10.1016/j.watres.2017.02.040. [DOI] [PubMed] [Google Scholar]
- Abollino O. Aceto M. Malandrino M. Sarzanini C. Mentasti E. Water Res. 2003;37:1619. doi: 10.1016/S0043-1354(02)00524-9. [DOI] [PubMed] [Google Scholar]
- Singh J. Huang P. M. Hammer U. T. Liaw W. K. Clays Clay Miner. 1996;44:41. doi: 10.1346/CCMN.1996.0440104. [DOI] [Google Scholar]
- Jing Y. D. He Z. L. Yang X. E. Chemosphere. 2007;69:1662. doi: 10.1016/j.chemosphere.2007.05.033. [DOI] [PubMed] [Google Scholar]
- Ho Y.-S. Water Res. 2006;40:119. doi: 10.1016/j.watres.2005.10.040. [DOI] [PubMed] [Google Scholar]
- Guo T. Y. Xia Y. Q. Hao G. J. Song M. D. Zhang B. H. Biomaterials. 2004;25:5905. doi: 10.1016/j.biomaterials.2004.01.032. [DOI] [PubMed] [Google Scholar]
- Barrow N. J. Eur. J. Soil Sci. 2008;59:900. doi: 10.1111/j.1365-2389.2008.01041.x. [DOI] [Google Scholar]
- Schwertmann U. Stanjek H. Becher H.-H. Clay Miner. 2004;39:433. doi: 10.1180/0009855043940145. [DOI] [Google Scholar]
- Tang C. Huang Y. H. Zeng H. Zhang Z. Water Res. 2014;67:166. doi: 10.1016/j.watres.2014.09.016. [DOI] [PubMed] [Google Scholar]
- Appelo C. A. J. Weiden V. D. Tournassat C. Charlet L. Environ. Sci. Technol. 2002;36:3096. doi: 10.1021/es010130n. [DOI] [PubMed] [Google Scholar]
- Azzam A. M. El-Wakeel S. T. Mostafa B. B. El-Shahat M. F. J. Environ. Chem. Eng. 2016;4:2196. doi: 10.1016/j.jece.2016.03.048. [DOI] [Google Scholar]
- Liu A. Zhang W. Analyst. 2014;139:4512. doi: 10.1039/C4AN00679H. [DOI] [PubMed] [Google Scholar]
- Zhang X. Lin S. Chen Z. Megharaj M. Naidu R. Water Res. 2011;45:3481. doi: 10.1016/j.watres.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Pena M. Meng X. Korfiatis G. P. Jing C. Environ. Sci. Technol. 2006;40:1257. doi: 10.1021/es052040e. [DOI] [PubMed] [Google Scholar]
- Hug S. J. Leupin O. Environ. Sci. Technol. 2003;37:2734. doi: 10.1021/es026208x. [DOI] [PubMed] [Google Scholar]
- Pillewan P. Mukherjee S. Roychowdhury T. Das S. Bansiwal A. Rayalu S. J. Hazard. Mater. 2011;186:367. doi: 10.1016/j.jhazmat.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Su J. Huang H.-G. Jin X.-Y. Lu X.-Q. Chen Z.-L. J. Hazard. Mater. 2011;185:63. doi: 10.1016/j.jhazmat.2010.08.122. [DOI] [PubMed] [Google Scholar]
- Li Z. Jean J.-S. Jiang W.-T. Chang P.-H. Chen C.-J. Liao L. J. Hazard. Mater. 2011;187:318. doi: 10.1016/j.jhazmat.2011.01.030. [DOI] [PubMed] [Google Scholar]
- Zhou Q. Lei M. Wu Y. Yuan Y. J. Chromatogr. A. 2017;1487:22. doi: 10.1016/j.chroma.2017.01.046. [DOI] [PubMed] [Google Scholar]