

Abstract
Pulmonary hypertension (PH) is a debilitating condition characterized by increased pulmonary arterial pressures and remodeling of pulmonary arteries, leading to right heart failure. Women have a higher prevalence of PH, whereas men have more severe disease and poorer outcomes. Animal models also show female-predominant disease. Despite the known sex differences in PH, little is known about how pathogenesis differs between the sexes. There is growing evidence of mitochondrial dysfunction, as well as altered mitophagy in PH. We hypothesized that sexual dimorphism contributes to mitochondrial dysfunction and altered mitophagy in PH. Using mouse lung endothelial cells, we exposed both wild-type and Parkin−/− cells to hypoxia and measured the effects on mitochondrial function and mitophagy-associated proteins. Our results show that females have more Parkin expression at baseline as well as increased mitochondrial respiratory capacity when exposed to oxidative stress. Inhibition of Parkin increased metabolic activity but reduced cell proliferation but to different degrees depending on sex, with results differing by sex. Our findings demonstrate sexual dimorphism in mitophagy-associated proteins and in mitochondrial respiration, which may help shed light on how the pathogenesis of PH may differ between the sexes.
Keywords: endothelial cells, mitochondrial dysfunction, mitophagy, pulmonary hypertension, sexual dimorphism
INTRODUCTION
Pulmonary hypertension (PH) is a progressive and often fatal disease. PH is characterized by vascular remodeling of pulmonary arteries and elevated pulmonary artery pressure, which can lead to progressive dyspnea and right heart failure. Despite advancements in therapies, the 3-yr survival is still estimated to be between 58% and 70% (1, 2). The pathogenesis of PH is complicated and not well understood. Pathogenic triggers occur and result in pulmonary vasoconstriction, endothelial cell proliferation, and remodeling of pulmonary arterioles (3). Currently available therapies target pulmonary vasoconstriction, which have been shown to improve functional status and/or hemodynamics (4–6), but fail to show a mortality benefit or reversal of vascular remodeling.
Several epidemiological studies have reported a female predominance in the prevalence of PH, as high as a 4:1 female to male ratio (7–9). Despite a higher prevalence in women, men who develop PH are more likely to have more severe hemodynamics and poorer outcomes (10, 11). Women have a better 5-yr survival rate at 74% compared with 53% in men (12). There are also differing responses to available medical therapy (12, 13). It is not known what underlying pathogenic mechanism results in these differences.
Sexual dimorphism in PH has also been seen in animal models. Both chronic hypoxia and monocrotaline-induced PH models have shown more severe PH in male than female rats (14–16). There are also animal models that have demonstrated female susceptibility to PH. Mouse models including those that overexpress a human serotonin transporter (17), overexpress S100A (18), or those that are treated with dexfenfluramine (19) show more severe PH in females. These data are conflicting but ultimately highlight that sexual dimorphism exists, and studies are needed to identify the mechanism of these differences.
At a cellular level, there is growing evidence that mitochondrial dysfunction may play a role in the pathogenesis of PH (20). Endothelial cells, smooth muscle cells, and fibroblasts all show a shift toward more anaerobic respiration, a response that is usually transient in the setting of exposure to hypoxia, but in PH continues chronically and results in vasoconstriction and ultimately vascular remodeling (21, 22). In addition to this glycolytic shift, cells also show a disruption in calcium homeostasis that contributes to vasoconstriction and proliferation. The mitochondrial membrane is also hyperpolarized in pulmonary artery smooth muscle cells (23, 24), which prevents release of proapoptotic chemicals and leads to an apoptosis-resistant phenotype that results in proliferation and remodeling (25).
Removal of damaged mitochondria occurs through a process called mitophagy. The most well studied pathway of mitophagy begins with depolarization of the mitochondrial membrane and accumulation of PTEN-induced kinase 1 (PINK1) on the mitochondrial membrane. PINK1 recruits cytosolic Parkin, and Parkin causes ubiquination of mitochondrial surface proteins resulting in formation of an autophagosome around the damaged mitochondria and subsequent degradation of the autophagosome in the lysosome. Mitophagy can also occur in a Parkin-independent manner, via mitochondrial receptors FUN14 Domain Containing 1, which is constitutively present on the outer mitochondrial membrane, or Nix, which upregulates BCL2 interacting protein 3 (BNIP3) to induce mitophagy in the presence of hypoxia. Although defective mitophagy has been implicated in cancer and other pulmonary diseases (26, 27), the role of mitophagy in the pathogenesis of PH is still unclear. Dynamin-related protein-1 (DRP1), a member of the dynamin superfamily of proteins, is an essential protein in mitochondrial quality control and helps to maintain cellular homeostasis. DRP1 finalizes the fission of the mitochondria by pinching off the membrane, after which mitophagy can begin to remove the damaged mitochondria. DRP1 depends on the phosphorylation of Ser616 and the dephosphorylation of Ser 637 for translocation to the mitochondria (28, 29). There are increased levels of active DRP1 in human lungs of patients with PH, and inhibition of DRP1 in a chronic hypoxia rat model attenuated the development of PH (30). There are more fragmented mitochondria, suggesting altered mitochondrial dynamics (31).
Our previous work suggested that mitochondrial dysfunction contributes to hypoxia-induced PH (32). In this model, we identified increased mitophagy and increased PINK1 and Parkin, and inhibition of mitophagy through silencing of PINK1 reversed the consequences of mitochondrial dysfunction. Together, this data suggested that mitophagy was a mediator of mitochondrial-induced dysfunction in PH (32).
There is evidence for sexual dimorphism in metabolism and mitochondrial function. Differences have been identified in the ways that each sex responds to stress, including oxidative stress (33), exercise (34), starvation (35), injury to specific cell types including neuronal cells (36), and cardiac myocytes (37). These conditions demonstrate a protective effect of female sex hormones, a better adaptive response in females, or just highlight differing pathogenesis of protective mechanisms between the sexes. We aimed to further elucidate the effect of hypoxia on mitophagy-associated proteins and identify whether there are sex differences in mitophagy and mitochondrial function that could inform the sexual dimorphism in the pathogenesis of PH.
MATERIALS AND METHODS
Cell Culture
Primary mouse lung endothelial cells (MLECs) were isolated from C57Bl6 wild-type (WT) mice and from Parkin−/− mice between 5 and 6 wk of age as we have previously reported (38) and separated by sex. Authentication of cell type previously confirmed via CD31 staining and flow cytometry (39). Three mice were combined per isolation, and each experiment represents a different group of mice. Cells underwent immunoselection with CD31 and intercellular adhesion molecule 1 antibodies twice to ensure isolation of MLECs. Cells were between passages 5 and 9 at time of experiments. Cells were maintained at 37°C-5% CO2 in F12:DMEM containing 100 IU/mL penicillin, 100 µg/mL streptomycin, and 20% FBS (Cat. No. SA-12306C). After checking cell morphology under the microscope, chemical hypoxia was used to simulate hypoxia in vitro with 50 µM of CoCl2 (40–42). Separating by sex, CoCl2 was added to cells for 3 days total of chronic chemical hypoxia, with media changed each day to prevent nutrient deprivation. Room air control cells were subjected to the same media changes but without CoCl2. The cells were harvested on ice on day 3 and protein lysates collected as previously reported (43). The research protocols were approved by the Institutional Animal Care and Use Committee of Yale University.
Hypoxia in Cell Culture
Male and female WT MLEC (primary endothelial cells from the lungs of C57BL/6J, mice were purchased from Jackson Laboratory) were seeded on 60-mm culture dishes and located in the hypoxia chamber. The chamber was attached to hypoxia tank containing a 1% O2 gas mixture of (1% O2, 5% CO2, balanced with nitrogen) and all oxygen present in the chamber was removed by flushing chamber with 1% O2 gas mixture for at least 2 min. After removing all present oxygen, chamber was closed and incubated at 37°C for 24 h. Cells were collected for Western blot analysis.
Western Blot Analysis
Protein concentrations of lysates were determined by BCA Protein Assay (Thermo Scientific). Samples were electrophoresed in a 4%–20% ready-made Tris-HCl gel (Bio-Rad Laboratories) and electrophoretically transferred onto a PVDF membrane using a Trans Blot Turbo transfer system. Membranes then were incubated overnight at 4°C with primary antibodies at 1:5,000 dilution (Parkin, Cat. No. SC-32282; BNIP3, Cat. No. ab-109362; DRP1, Cat. No. ab-56788; p-DRP1 Cell Signaling, Cat. No. 4867, B-actin, Cat. No. sc-47778). Antibodies were validated elsewhere (44–47). The membranes were incubated with HRP-conjugated secondary antibodies (at 1:1,000 dilution) followed by the detection of signal with a Clarity Western ECL Substrate (Bio-Rad Laboratories).
Seahorse Assay
Cellular metabolism was measured with the Seahorse XFe96 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA) for real-time analysis of extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) according to the manufacturer’s protocol. Briefly, following treatment with chemical hypoxia or room air for 3 days, cells were detached with 0.05% trypsin/EDTA, counted, and ∼10,000 cells per well were seeded onto Seahorse XF Cell Culture Microplates. The chemical hypoxia group cells were then treated with CoCl2 for 24 h. The assay was performed with the Seahorse XF Cell Mito Stress Test with oligomycin (1.0 μM), carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) (1.0 μM), and rotenone/antimycin A (0.5 μM). Each measurement cycle included mixing for 5 min and measuring for 5 min; results were analyzed using Wave Desktop. Following the assay, cells were recounted and data were normalized to cell number.
WST Assay
The Abcam Quick Cell Proliferation Assay Kit was used, with water-soluble tetrazolium salt-1 as the reagent, and cells were cultured according to their protocol. Cells were incubated for 72 h, and absorbance was measured with a microtiter plate reader at 420–480 nm.
BrdU Assay
WT and Parkin−/− MLEC were isolated from mice and seeded on 96-well plate (cell density: 2 × 104 cells/well). The next day, cells were incubated with BrdU solution for 24 h at 37°C incubator. The amount of BrdU incorporated cells was determined according to manufacturer’s instructions (BrdU Cell Proliferation Assay Kit, Cell Signaling, Cat. No. 6813).
Statistics
Data are represented as means ± SD. Mean values were normalized with the male-room air group used as the control. Comparison of multiple groups was done with GraphPad Prism, version 8.0, using two-way ANOVA with Sidak’s multiple comparisons test. Mean values were considered significant at P < 0.05.
RESULTS
Effect of Sex and Hypoxia on Mitophagic Markers
We sought to determine if mitochondrial function differed between sexes in MLECs from WT C57BL/6 mice. We measured Parkin (Fig. 1), BNIP3 (Fig. 2), and DRP1 (Fig. 3) expression in MLECs exposed to either room air versus chemical hypoxia for 3 days. We found that in room air, Parkin and DRP1 levels were higher in females than in males, whereas BNIP3 expression was not statistically different between the sexes. Following exposure to chemical hypoxia, Parkin and total DRP1 levels decreased by 50% whereas BNIP3 levels significantly increased in both sexes. These findings suggest that at baseline, females have more Parkin activity.
Figure 1.

A: Western blot analyses of Parkin/β-actin expression in male and female wild-type (WT) MLECs at room air and following 3 days of chemical hypoxia (CoCl2) exposure. Data represented a means ± SD of four independent experiments, normalized to the male room air group. Two-way ANOVA used with Sidak’s multiple comparisons test. *P < 0.05. B: representative Western blot also shown. MLECs, mouse lung endothelial cells.
Figure 2.

A: Western blot analyses of BNIP3//β-actin expression in male and female wild-type (WT) MLECs at room air and following 3 days of chemical hypoxia exposure. Data represented a means ± SD of four independent experiments, normalized to the male room air group. Two-way ANOVA used with Sidak’s multiple comparisons test. **P < 0.01. B: representative Western blot also shown. MLECs, mouse lung endothelial cells.
Figure 3.

A: Western blot analyses of total DRP1 (tDRP1)/β-actin expression in male and female wild-type (WT) MLECs at room air and following 3 days of chemical hypoxia exposure. Data represented as means ± SD of three independent experiments, normalized to the male room air group. Two-way ANOVA used with Sidak’s multiple comparisons test. *P < 0.05, **P < 0.01. B: representative Western blot also shown. MLECs, mouse lung endothelial cells.
We also exposed MLEC to 1% O2 and found that 1% O2-induced hypoxia decreased Parkin expression in females and increased BNIP3 expression in both female and male MLEC (Fig. 4, A and B). These data are consistent with what we found in CoCl2-induced chemical hypoxia. Next, we measured p-DRP1 (Ser637) expression to determine DRP1 activity (Fig. 4C). P-DRP1 (Ser637) increased in male MLECs after 24 h of 1% O2-induced hypoxia. Given that phosphorylation of DRP1 on Ser637 residue inhibits translocation of this protein to the mitochondrial outer membrane (28), these results suggest that not only total DRP1 but also its activity were decreased in hypoxia. This may be due to increased mitophagy, although mitophagic flux was not directly measured. Increased Parkin activity in females strengthens the hypothesis of sexual dimorphism in mitochondrial function. Following chemical hypoxia, both male and female cells appear to decrease Parkin activity and increase BNIP3 activity, suggesting a possible switch to BNIP3-mediated mitophagy.
Figure 4.
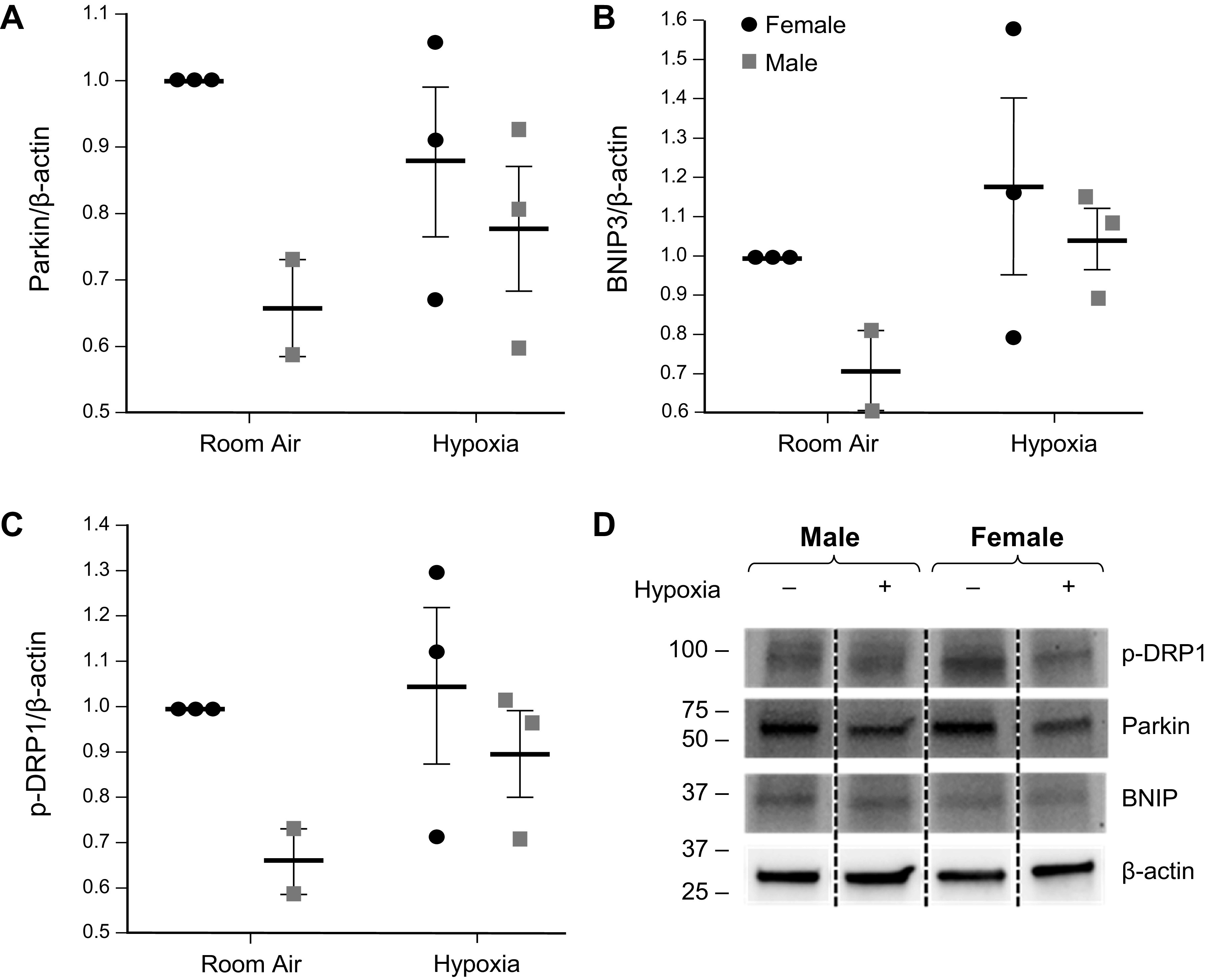
Protein expression of Parkin (A), BNIP3 (B), and p-DRP1 (C) in 1% O2-exposed female and male MLECs. Cells were exposed to 1% O2 for 24 h in a hypoxia chamber. Protein expressions were determined by western blot analyses. Data are represented as means ± SD and each dot represents independent experiments (N = 3 for groups except N = 2 for RA Male group). There were no statistically significant differences observed between groups (two-way ANOVA used with Sidak’s multiple comparisons test). D: representative Western blot also shown. The lanes for the controls and hypoxic samples for each sex were run on the same gel with nonessential lanes removed for clarity. MLECs, mouse lung endothelial cells.
Because our prior work suggested excessive PINK1-Parkin-mediated mitophagy in mouse PH (32), and there was reduced Parkin in male MLECs at room air and following chemical hypoxia, we examined MLECs from Parkin−/− mice and performed the same experiment. Following 3 days of CoCl2 exposure, BNIP3 levels increased in both male and female MLECs. There was 50% less BNIP3 expression in males following chemical hypoxia, but this difference was not statistically different from room air (Fig. 5). Interestingly, DRP1 levels did not change following chemical hypoxia exposure but were statistically different between the sexes both at room and following chemical hypoxia exposure, with males having higher levels of DRP1 (Fig. 6). This suggests that inhibition of Parkin results in sexually dimorphic activity of DRP1, with reduction of these mitophagic markers in females and increased activity in males.
Figure 5.

A: Western blot analyses of BNIP/β-actin expression in male and female Parkin−/− MLECs at room air and following 3 days of chemical hypoxia exposure. Data are represented as means ± SD of three independent experiments, normalized to the male room air group. Two-way ANOVA used with Sidak’s multiple comparisons test. **P < 0.01. B: representative Western blot also shown. MLECs, mouse lung endothelial cells.
Figure 6.

A: Western blot analyses of DRP1/β-actin expression in male and female Parkin−/− MLECs at room air and following 3 days of chemical hypoxia exposure. Data are represented as means ± SD of three independent experiments, normalized to the male room air group. Two-way ANOVA used with Sidak’s multiple comparisons test. **P < 0.01. B: representative Western blot also shown. MLECs, mouse lung endothelial cells.
Better Mitochondrial Function in Female MLECs
Given the room air sex difference seen in our Western blot analyses, we wanted to further compare the mitochondrial function of each sex, with and without chemical hypoxia exposure using the Seahorse XFe96 Extracellular Flux Analyzer. We found that although females trended toward having a higher basal respiration both at room and following chemical hypoxia exposure, this difference was not statistically different (Fig. 7). At maximal respiration, more differences were seen (Fig. 8). Both sexes had a decrease in maximal respiration following exposure to chemical hypoxia, as expected. However, we also found that females had a higher oxygen consumption rate as compared with males both at room air and following chemical hypoxia exposure. To be sure that this difference was not due to the initial trend toward higher basal respiration in females, we looked at the spare respiratory capacity (Fig. 9), which is the maximal respiration minus the basal respiration. Here, we still found that respiratory capacity decreased following chemical hypoxia exposure, and females under both conditions had a higher respiratory capacity, suggesting a higher tolerance to oxidative stress. A summary of these findings is depicted in Fig. 10.
Figure 7.

Basal oxygen consumption rate (OCR) of male and female wild-type (WT) MLECs at room air versus 3 days of chemical hypoxia exposure. Data are represented as means ± SD of three independent experiments (24 technical replicates per biologic replicate), normalized to cell number, and then normalized to the male room air group. Two-way ANOVA used with Sidak’s multiple comparisons test. MLECs, mouse lung endothelial cells.
Figure 8.

Maximal oxygen consumption rate (OCR) of male and female wild-type (WT) MLECs at room air versus 3 days of chemical hypoxia exposure. Data are represented as means ± SD of three independent experiments (24 technical replicates per biologic replicate), normalized to cell number, and then normalized to the male room air group. Two-way ANOVA used with Sidak’s multiple comparisons test. **P < 0.01, ***P < 0.001, and ****P < 0.0001. MLECs, mouse lung endothelial cells; OCR, oxygen consumption rate.
Figure 9.

Spare respiratory capacity as determined by the mean maximal OCR minus the mean basal OCR of male and female wild-type (WT) MLECs at room air versus 3 days of chemical hypoxia exposure. Data are represented as means ± SD of three independent experiments (24 technical replicates per biologic replicate), normalized to cell number, and then normalized to the male room air group. Two-way ANOVA used with Sidak’s multiple comparisons test. **P < 0.01, ***P < 0.001, and ****P < 0.0001. MLECs, mouse lung endothelial cells; OCR, oxygen consumption rate.
Figure 10.

Representative schematic of mitochondrial respiration in MLECs at room air and after chemical hypoxia, depicting increased basal, maximal, and spare respiratory capacity in females, both at room air and following exposure to chemical hypoxia. Dashed lines denote injection of mitochondrial inhibitors: oligomycin, an ATP synthase inhibitor (A); FCCP, a protonophore that uncouples ATS synthesis from oxygen consumption (B); rotenone/antimycin A that inhibits Complex I and III of the electron transport chain, respectively (C). Time before A signifies basal respiration, time between A and B signifies proton leak, time between B and C signifies maximal respiration, and time following C signifies nonmitochondrial oxygen consumption. Spare respiratory capacity is the maximal respiration minus the basal respiration. Data from a single experiment (12 replicates) are represented as means ± SD of nonnormalized OCR value. MLECs, mouse lung endothelial cells; OCR, oxygen consumption rate.
Sex Differences in Cell Growth with Parkin Is Inhibited
As we found differences in mitochondrial function between the sexes, we wanted to see how this would affect mitochondrial metabolic activities and cell proliferation (Figs. 11 and 12). We found that there was no difference in metabolic activity between the sexes in WT MLECs. However, in Parkin−/− MLECs, the males showed significantly increased metabolic activity, whereas the female Parkin−/− MLECs matched the WT cells. Next, we determined cell proliferation by BrdU analysis. In Parkin−/− MLECs, males showed more proliferation than female. However, Parkin−/− MLECs showed less proliferation than WT MLECs. These results suggest that loss of Parkin may result in increased mitochondrial activity but decreased in cell proliferation especially in males.
Figure 11.

WST assay of wild-type (WT) and Parkin−/− MLECs after 3 days of cell growth. Data are represented as means ± SD of two independent experiments, each with n = 6. One-way ANOVA with Tukey’s multiple comparisons test. ****P < 0.0001. MLECs, mouse lung endothelial cells.
Figure 12.

BrdU assay in WT and Parkin−/− MLECs. Cells (2 × 104 cells) were incubated with BrdU staining solution for 24 h and BrdU incorporated cells were determined. Data are represented as means ± SD of six independent experiments. Two-way ANOVA are used for statistical analysis. *P < 0.05, ***P < 0.001. MLECs, mouse lung endothelial cells; WT, wild type.
DISCUSSION
There are known sex differences between the incidence, hemodynamics, and mortality of human PH. There is accumulating evidence for mitochondrial dysfunction in PH, as well differences in mitochondrial function and stress responses between the sexes. Though prior studies have suggested altered mitophagy in PH, it is not clear whether there is excessive, insufficient, or defective mitophagy in PH and which pathway is used. In the current study, we found decreased Parkin and DRP1 activity in WT MLECs but increased BNIP3 expression following chemical hypoxia, suggesting that the BNIP3 may be more important in the pathogenesis of PH. In addition, we found higher Parkin and DRP1 in WT female MLECs in room air, strengthening the argument that sexual dimorphism exists in mitochondrial function. Higher Parkin levels in females MLEC could be deleterious, making them more susceptible to injury and remodeling or could be protective and contribute to the mortality benefit seen in females. Interestingly, inhibition of Parkin resulted in higher DRP1 levels in males rather than females, which suggests that Parkin or Parkin-mediated mitophagy may play a larger role or a different role depending on sex. There is evidence of estrogen and progesterone effects on mitochondrial fission and fusion. These hormones have been shown to increase cell number, proliferation, and increase transcription of fission and fusion genes in females. When exposed to the same hormones, there was no change in male cells (48). Decreased Parkin levels may suggest loss of hormonal benefit and a resultant decrease in DRP1-mediated fission in females. Of note, we have only performed in vitro experiments, which lacks the hormonal interaction that can contribute to sexual dimorphism; however, we believe sex differences persist due to sex-dependent gene expression, as has been described by others (49). Epigenetic sex differences in both methylation and histone modification have also been described (50, 51).
In terms of the respiratory capacity of MLECs, female cells had higher oxygen consumption rates than males, both at room air and following exposure to chemical hypoxia. The spare respiratory capacity is a measure of the cell’s ability to respond to increased energy demands or stress. Our findings suggest that female cells may have a better ability to handle stress, which could contribute to how they respond to the initial stress or injury and the fact that women have milder PH than men.
A surprising finding was the difference in cell growth between sexes when Parkin was inhibited. As noted above, inhibiting Parkin resulted in a change from higher DRP1 in females to higher DRP1 in males. Parkin inhibition also resulted in a significant increase in cellular metabolic activity in males with a decrease in newly synthesized DNA in both female and male cells. Female cellular metabolic activity was unchanged compared with WT cells. It is possible that the increased DRP1 levels found in Parkin-inhibited male MLECs is due to increased cell turnover, which results in more DRP1-mediated fission (52, 53). Hyperactive metabolism via increased mitochondrial biogenesis as a result of decreased proliferation has been described (54). Collectively, reduced proliferation from loss of Parkin could have resulted in increased mitochondrial biogenesis, thus accounting for hyperactive metabolism. Parkin is a downstream target of transcription factor p53. Other studies suggest that Parkin may be an important mediator for p53, particularly in its role in glucose metabolism. Inhibition of Parkin or p53 resulted in increased glucose uptake, rate of glycolysis, and lactate production, though not as much as when both were inhibited together (55). Increased rates of glycolysis may explain the hyperactive metabolism seen in our Parkin-inhibited MLECs. Sex-dependent differences in metabolic regulation would explain the lack of a similar finding in the Parkin-inhibited females. There is evidence for differing epigenetic effects between the sexes, such as methylation patterns, on the p53 pathway (50). It is possible that somehow these epigenetic sex differences are altering the cell’s response to Parkin inhibition. These findings again strengthen the idea that mitochondrial sexual dimorphism is biologically relevant and suggests that inhibiting Parkin may be more advantageous in males, and/or more deleterious in females.
In summary, our findings demonstrate sex differences between the mitophagy-associated protein Parkin, cellular respiration, stress-handling and growth. Although our experiments were conducted in vitro, epigenetic differences and its resultant effects on gene expression may account for the sexual dimorphism found in vivo. Although premature to generalize our findings to human PH, other studies have found parallel sex disparities in both murine and human pulmonary artery endothelial cells (56). Our goal is to bring sex considerations into basic biology in order to advance our understanding of disease pathogenesis and to develop targeted, personalized therapeutic strategies.
DATA AVAILABILITY
The data that support the findings of this study will be made available upon reasonable request from the corresponding author.
GRANTS
I.S.B. was funded by the NIH Grant T32 HL007778. M.S. was funded by the NIH/National Heart, Lung, and Blood Institute (NHLBI) Grant K08HL135402. P.J.L. was funded by the NIH/NHLBI Grant R01HL138396; Department of Defense Grant PR150809; and Veterans Affairs Office of Research and Development Grant 11858595.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.S.B. and P.J.L. conceived and designed research; I.S.B., S.-J.K., T.A.A., Y.Z., and P.S. performed experiments; I.S.B., S.-J.K., Y.Z., and P.S. analyzed data; I.S.B., S.-J.K., Y.Z., P.S., and P.J.L. interpreted results of experiments; I.S.B., S.-J.K., and P.S. prepared figures; I.S.B. drafted manuscript; I.S.B., S.-J.K., T.A.A., M.S., and P.J.L. edited and revised manuscript; P.J.L. approved final version of manuscript.
REFERENCES
- 1.Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 142: 448–456, 2012. doi: 10.1378/chest.11-1460. [DOI] [PubMed] [Google Scholar]
- 2.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaïci A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E, Cottin V, Degano B, Jaïs X, Montani D, Souza R, Simonneau G. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 122: 156–163, 2010. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 3.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43: 13S–24S, 2004. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 4.Galiè N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, Shapiro S, White RJ, Chan M, Beardsworth A, Frumkin L, Barst RJ. Tadalafil therapy for pulmonary arterial hypertension. Circulation 119: 2894–2903, 2009. doi: 10.1161/CIRCULATIONAHA.108.839274. [DOI] [PubMed] [Google Scholar]
- 5.Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 46: 903–975, 2015. [Erratum in Eur Heart J 46: 1855–1856, 2015]. doi: 10.1183/13993003.01032-2015. [DOI] [PubMed] [Google Scholar]
- 6.Galiè N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, Badesch DB, McGoon MD, McLaughlin VV, Roecker EB, Gerber MJ, Dufton C, Wiens BL, Rubin LJ; Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Studies (ARIES) Group. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 117: 3010–3019, 2008. doi: 10.1161/CIRCULATIONAHA.107.742510. [DOI] [PubMed] [Google Scholar]
- 7.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E, Simonneau G. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 173: 1023–1030, 2006. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 8.Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, Howard LS, Pepke-Zaba J, Sheares KK, Corris PA, Fisher AJ, Lordan JL, Gaine S, Coghlan JG, Wort SJ, Gatzoulis MA, Peacock AJ. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 186: 790–796, 2012. doi: 10.1164/rccm.201203-0383OC. [DOI] [PubMed] [Google Scholar]
- 9.McGoon MD, Miller DP. REVEAL: a contemporary US pulmonary arterial hypertension registry. Eur Respir Rev 21: 8–18, 2012. doi: 10.1183/09059180.00008211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, Barst RJ, Benza RL, Liou TG, Turner M, Giles S, Feldkircher K, Miller DP, McGoon MD. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest 137: 376–387, 2010. doi: 10.1378/chest.09-1140. [DOI] [PubMed] [Google Scholar]
- 11.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 122: 164–172, 2010. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 12.Kozu K, Sugimura K, Aoki T, Tatebe S, Yamamoto S, Yaoita N, Shimizu T, Nochioka K, Sato H, Konno R, Satoh K, Miyata S, Shimokawa H. Sex differences in hemodynamic responses and long-term survival to optimal medical therapy in patients with pulmonary arterial hypertension. Heart Vessels 33: 939–947, 2018. doi: 10.1007/s00380-018-1140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marra AM, Benjamin N, Eichstaedt C, Salzano A, Arcopinto M, Gargani L, D Alto M, Argiento P, Falsetti L, Di Giosia P, Isidori AM, Ferrara F, Bossone E, Cittadini A, Grünig E. Gender-related differences in pulmonary arterial hypertension targeted drugs administration. Pharmacol Res 114: 103–109, 2016. doi: 10.1016/j.phrs.2016.10.018. [DOI] [PubMed] [Google Scholar]
- 14.Bal E, Ilgin S, Atli O, Ergun B, Sirmagul B. The effects of gender difference on monocrotaline-induced pulmonary hypertension in rats. Hum Exp Toxicol 32: 766–774, 2013. doi: 10.1177/0960327113477874. [DOI] [PubMed] [Google Scholar]
- 15.Farhat MY, Chen MF, Bhatti T, Iqbal A, Cathapermal S, Ramwell PW. Protection by oestradiol against the development of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br J Pharmacol 110: 719–723, 1993. doi: 10.1111/j.1476-5381.1993.tb13871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rafikova O, Rafikov R, Meadows ML, Kangath A, Jonigk D, Black SM. The sexual dimorphism associated with pulmonary hypertension corresponds to a fibrotic phenotype. Pulm Circ 5: 184–197, 2015. doi: 10.1086/679724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17β oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res 90: 373–382, 2011. doi: 10.1093/cvr/cvq408. [DOI] [PubMed] [Google Scholar]
- 18.Dempsie Y, Nilsen M, White K, Mair KM, Loughlin L, Ambartsumian N, Rabinovitch M, MacLean MR. Development of pulmonary arterial hypertension in mice over-expressing S100A4/Mts1 is specific to females. Respir Res 12: 159, 2011. doi: 10.1186/1465-9921-12-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dempsie Y, MacRitchie NA, Morecroft I, Nilsen M, Loughlin L, MacLean MR. The effects of gender on the development of dexfenfluramine-induced pulmonary arterial hypertension in mice. Am J Respir Crit Care Med 179: A1808, 2009. doi: 10.1164/ajrccm-conference.2009.179.1_MeetingAbstracts.A1808. [DOI] [Google Scholar]
- 20.Freund-Michel V, Khoyrattee N, Savineau J-P, Muller B, Guibert C. Mitochondria: roles in pulmonary hypertension. Int J Biochem Cell Biol 55: 93–97, 2014. doi: 10.1016/j.biocel.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Schumacker PT, Gillespie MN, Nakahira K, Choi AM, Crouser ED, Piantadosi CA, Bhattacharya J. Mitochondria in lung biology and pathology: more than just a powerhouse. Am J Physiol Lung Cell Mol Physiol 306: L962–L974, 2014. doi: 10.1152/ajplung.00073.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033, 2009. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pak O, Sommer N, Hoeres T, Bakr A, Waisbrod S, Sydykov A, Haag D, Esfandiary A, Kojonazarov B, Veit F, Fuchs B, Weisel FC, Hecker M, Schermuly RT, Grimminger F, Ghofrani HA, Seeger W, Weissmann N. Mitochondrial hyperpolarization in pulmonary vascular remodeling. Mitochondrial uncoupling protein deficiency as disease model. Am J Respir Cell Mol Biol 49: 358–367, 2013. doi: 10.1165/rcmb.2012-0361OC. [DOI] [PubMed] [Google Scholar]
- 24.Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC, Michelakis ED. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med 3: 88ra55, 2011. doi: 10.1126/scitranslmed.3002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J 19: 1178–1180, 2005. doi: 10.1096/fj.04-3261fje. [DOI] [PubMed] [Google Scholar]
- 26.Aggarwal S, Mannam P, Zhang J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol 311: L433–L452, 2016. doi: 10.1152/ajplung.00128.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chourasia AH, Boland ML, Macleod KF. Mitophagy and cancer. Cancer Metab 3: 4, 2015. doi: 10.1186/s40170-015-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med 369: 2236–2251, 2013. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 29.Xie L, Shi F, Li Y, Li W, Yu X, Zhao L, Zhou M, Hu J, Luo X, Tang M, Fan J, Zhou J, Gao Q, Wu W, Zhang X, Liao W, Bode AM, Cao Y. DRP1-dependent remodeling of mitochondrial morphology triggered by EBV-LMP1 increases cisplatin resistance. Signal Transduct Target Ther 5: 56, 2020. doi: 10.1038/s41392-020-0151-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110: 1484–1497, 2012. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan J, Dasgupta A, Huston J, Chen KH, Archer SL. Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med (Berl) 93: 229–242, 2015. doi: 10.1007/s00109-015-1263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haslip M, Dostanic I, Huang Y, Zhang Y, Russell KS, Jurczak MJ, Mannam P, Giordano F, Erzurum SC, Lee PJ. Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent-hypoxia. Arterioscler Thromb Vasc Biol 35: 1166–1178, 2015. doi: 10.1161/ATVBAHA.114.304865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farhat F, Amérand A, Simon B, Guegueniat N, Moisan C. Gender-dependent differences of mitochondrial function and oxidative stress in rat skeletal muscle at rest and after exercise training. Redox Rep 22: 508–514, 2017. doi: 10.1080/13510002.2017.1296637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamont LS. Gender differences in amino acid use during endurance exercise. Nutr Rev 63: 419–422, 2005. doi: 10.1301/nr.2005.dec.419-422. [DOI] [PubMed] [Google Scholar]
- 35.Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, Kochanek PM, Jenkins LW, Ren J, Gibson G, Chu CT, Kagan VE, Clark RS. Starving neurons show sex difference in autophagy. J Biol Chem 284: 2383–2396, 2009. doi: 10.1074/jbc.M804396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodríguez-Navarro JA, Solano RM, Casarejos MJ, Gomez A, Perucho J, de Yébenes JG, Mena MA. Gender differences and estrogen effects in parkin null mice. J Neurochem 106: 2143–2157, 2008. doi: 10.1111/j.1471-4159.2008.05569.x. [DOI] [PubMed] [Google Scholar]
- 37.Khalifa ARM, Abdel-Rahman EA, Mahmoud AM, Ali MH, Noureldin M, Saber SH, Mohsen M, Ali SS. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol Rep 5: e13125, 2017. doi: 10.14814/phy2.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Shan P, Jiang G, Cohn L, Lee PJ. Toll-like receptor 4 deficiency causes pulmonary emphysema. J Clin Invest 116: 3050–3059, 2006. doi: 10.1172/JCI28139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X, Shan P, Jiang G, Zhang SS, Otterbein LE, Fu XY, Lee PJ. Endothelial STAT3 is essential for the protective effects of HO-1 in oxidant-induced lung injury. Faseb J 20: 2156–2158, 2006. [Erratum in FASEB J 21: 630, 2007]. doi: 10.1096/fj.06-5668fje. [DOI] [PubMed] [Google Scholar]
- 40.Kim KS, Rajagopal V, Gonsalves C, Johnson C, Kalra VK. A novel role of hypoxia-inducible factor in cobalt chloride- and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J Immunol 177: 7211–7224, 2006. doi: 10.4049/jimmunol.177.10.7211. [DOI] [PubMed] [Google Scholar]
- 41.Muñoz-Sánchez J, Chánez-Cárdenas ME. The use of cobalt chloride as a chemical hypoxia model. J Appl Toxicol 39: 556–570, 2019. doi: 10.1002/jat.3749. [DOI] [PubMed] [Google Scholar]
- 42.Wu D, Yotnda P. Induction and testing of hypoxia in cell culture. J Vis Exp 54: 2899, 2011. doi: 10.3791/2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Jiang G, Sauler M, Lee PJ. Lung endothelial HO-1 targeting in vivo using lentiviral miRNA regulates apoptosis and autophagy during oxidant injury. FASEB J 27: 4041–4058, 2013. doi: 10.1096/fj.13-231225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bunnell TM, Burbach BJ, Shimizu Y, Ervasti JM. β-Actin specifically controls cell growth, migration, and the G-actin pool. Mol Biol Cell 22: 4047–4058, 2011. doi: 10.1091/mbc.E11-06-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd.. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21: 273–286, 2015. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang C, Kang X, Zhou L, Chai Z, Wu Q, Huang R, Xu H, Hu M, Sun X, Sun S, Li J, Jiao R, Zuo P, Zheng L, Yue Z, Zhou Z. Synaptotagmin-11 is a critical mediator of parkin-linked neurotoxicity and Parkinson's disease-like pathology. Nat Commun 9: 81, 2018. doi: 10.1038/s41467-017-02593-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Zhang C, Jiang X, Li L, Zhang D, Tang D, Yan T, Zhang Q, Yuan H, Jia J, Hu J, Zhang J, Huang Y. Involvement of autophagy in hypoxia-BNIP3 signaling to promote epidermal keratinocyte migration. Cell Death Dis 10: 234, 2019. [Erratum in Cell Death Dis 10: 295, 2019]. doi: 10.1038/s41419-019-1473-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Arnold S, de Araújo GW, Beyer C. Gender-specific regulation of mitochondrial fusion and fission gene transcription and viability of cortical astrocytes by steroid hormones. J Mol Endocrinol 41: 289–300, 2008. doi: 10.1677/JME-08-0085. [DOI] [PubMed] [Google Scholar]
- 49.Goring A, Sharma A, Javaheri B, Smith RC, Kanczler JM, Boyde A, Hesse E, Mahajan S, Olsen BR, Pitsillides AA, Schneider P, Oreffo RO, Clarkin CE. Regulation of the bone vascular network is sexually dimorphic. J Bone Miner Res 34: 2117–2132, 2019. doi: 10.1002/jbmr.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haupt S, Haupt Y. Cancer and tumour suppressor p53 encounters at the juncture of sex disparity. Front Genet 12: 632719, 2021. doi: 10.3389/fgene.2021.632719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai HW, Grant PA, Rissman EF. Sex differences in histone modifications in the neonatal mouse brain. Epigenetics 4: 47–53, 2009. doi: 10.4161/epi.4.1.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lutz AK, Exner N, Fett ME, Schlehe JS, Kloos K, Lämmermann K, Brunner B, Kurz-Drexler A, Vogel F, Reichert AS, Bouman L, Vogt-Weisenhorn D, Wurst W, Tatzelt J, Haass C, Winklhofer KF. Loss of parkin or PINK1 function increases DRP1-dependent mitochondrial fragmentation. J Biol Chem 284: 22938–22951, 2009. doi: 10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roy M, Itoh K, Iijima M, Sesaki H. Parkin suppresses DRP1-independent mitochondrial division. Biochem Biophys Res Commun 475: 283–288, 2016. doi: 10.1016/j.bbrc.2016.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rai Y, Pathak R, Kumari N, Sah DK, Pandey S, Kalra N, Soni R, Dwarakanath BS, Bhatt AN. Mitochondrial biogenesis and metabolic hyperactivation limits the application of MTT assay in the estimation of radiation induced growth inhibition. Sci Rep 8: 1531, 2018. doi: 10.1038/s41598-018-19930-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci USA 108: 16259–16264, 2011. doi: 10.1073/pnas.1113884108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qin S, Predescu DN, Patel M, Drazkowski P, Ganesh B, Predescu SA. Sex differences in the proliferation of pulmonary artery endothelial cells: implications for plexiform arteriopathy. J Cell Sci 133: jcs237776, 2020. doi: 10.1242/jcs.237776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study will be made available upon reasonable request from the corresponding author.