

Keywords: 5/6 nephrectomy, autoregulation, blood pressure, chronic kidney disease, renin-angiotensin-aldosterone system
Abstract
The 5/6 nephrectomy rat remnant kidney model is commonly used to study chronic kidney disease (CKD). This model requires the removal of one whole kidney and two-thirds of the other kidney. The two most common ways of producing the remnant kidney are surgical resection of poles, known as the polectomy model, or ligation of superior and inferior segmental renal arteries, resulting in pole infarction. These models have much in common, but also major phenotypic differences, and thus respectively model unique aspects of human CKD. The purpose of this review is to summarize phenotypic similarities and differences between these two models and their relation to human CKD while emphasizing their vascular phenotype. In this article, we review studies that have evaluated arterial blood pressure, the renin-angiotensin-aldosterone-system, autoregulation, nitric oxide, single-nephron physiology, angiogenic and antiangiogenic factors, and capillary rarefaction in these two models. In terms of phenotypic similarities, both models spontaneously develop hallmarks of human CKD including uremia, fibrosis, capillary rarefaction, and progressive renal function decline. They both undergo whole organ hypertrophy, hyperfiltration of functional nephrons, reduced renal expression of vascular endothelial growth factor, increased renal expression of antiangiogenic thrombospondin-1, impaired renal autoregulation, and abnormal vascular nitric oxide physiology. In terms of key phenotypic differences, the infarction model develops rapid-onset, moderate to severe systemic hypertension and the polectomy model develops early normotension followed by mild to moderate hypertension. Rats subjected to the infarction model have a markedly more active renin-angiotensin-aldosterone system. Comparison of these two models facilitates understanding of how they can be used for studying CKD pathophysiology.
INTRODUCTION
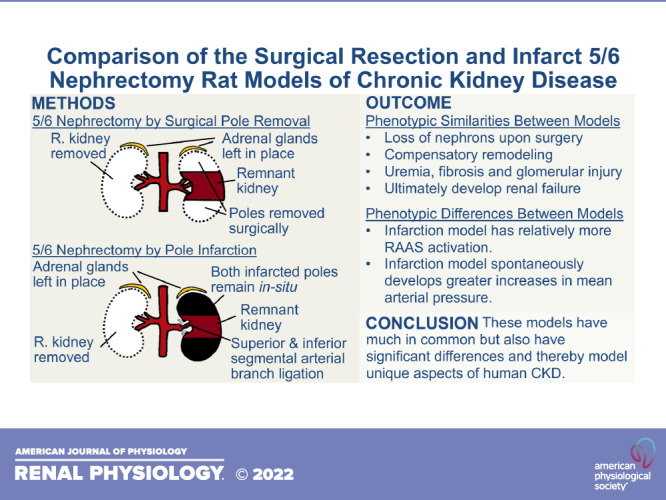
Chronic kidney disease (CKD) is the loss of kidney function, often irreversible, that occurs over a time of months or years. CKD may be characterized by reduced glomerular filtration rate (GFR), uremia, proteinuria, renal fibrosis, capillary rarefaction, metabolic acidosis, and skeletal myopathy and ultimately leads to organ failure (1–3). Approximately 15% of United States adults have CKD (4). The rat remnant kidney model of CKD is widely used experimentally due to its relative ease of generation and recapitulation of many human CKD characteristics. The most common rat remnant kidney model is the 5/6 nephrectomy (5/6Nx), in which one whole kidney is removed and the poles of the remaining kidney are ablated. Pole ablation is most commonly performed in one of two ways: surgically (e.g., removal by scalpel) (1, 5, 6) or infarction by ligation of the inferior and superior segmental arteries (Fig. 1, A and B) (6). The sham procedure includes laparotomy and renal pedicle manipulation (Fig. 1C). Although infarction and polectomy rats are both models of chronic renal insufficiency, they have important phenotypic differences and thereby model unique aspects of human CKD. Although the primary renal insult that initiates CKD in infarction and polectomy rats differ from typical human CKD, both rat models exhibit similar pathological progression from that point on. This progression includes compensatory adaptation of the remaining nephrons that, over time, leads to further damage, thus establishing a progressive cycle that may culminate with organ failure (Fig. 1D).
Figure 1.

Schematic summary of 5/6 nephrectomy (5/6Nx) by surgical resection (A), 5/6Nx by infarction (B), and sham-operated nephrectomy (C). All three procedures include kidney access by laparotomy. D: although the mechanism of primary renal insult is variable in human chronic kidney disease and different from both the infarction and polectomy 5/6Nx models, progressive decline of renal function is common to all. R, right.
The infarction and polectomy models recapitulate hallmarks of human CKD. Both models spontaneously develop elevated serum creatinine (5–12). Importantly, several studies have reported similar serum creatinine levels between infarction and polectomy rats 3 days following surgery (6, 13–15), suggesting a similar initial reduction of nephron number. Both models become uremic (16–20) and develop proteinuria (5, 6, 17, 21). Direct comparison studies have indicated that infarction rats have more severe proteinuria than polectomy rats through 6 wk postsurgery (Table 1) (6, 14, 15). For example, Ibrahim and Hostetter (15), at 2 wk postsurgery, found 154 mg/24 h of protein in polectomy rat urine compared with 249 mg/24 h in infarction rat urine. At 6 wk postsurgery, Griffin et al. (6) reported ∼10 mg/24 h of protein in polectomy rat urine compared with ∼50 mg/24 h in infarction rat urine. Studies that compared glomerular injury between infarction and polectomy rats have reported no difference (8, 15) or significantly more in infarction rats (6, 14). Both models develop some degree of tubulointerstitial injury and fibrosis (17, 22–26). Fibrosis, glomerular injury, and tubulointerstitial injury are virtually universal outcomes in human CKD (3, 27–29).
Table 1.
Proteinuria values for infarction and polectomy rats subjected to 5/6 nephrectomy from studies that compared these two models directly in a prospective manner
| Week Postsurgery | Average Proteinuria, mg/24 h |
References | |
|---|---|---|---|
| Infarction Model | Polectomy Model | ||
| 2 | 249 ± 124 (±SD) | 154 ± 73 (±SD)* | Ibrahim and Hostetter (15) |
| 4 | 225 ± 93 (±SD) | 135 ± 67 (±SD)* | |
| 3 | 11 ± 6 (±SE) | 6 ± 0.6 (±SE)* | Griffin et al. (14) |
| 4 | ∼35 ± 8 (±SE) | ∼10 ± 3 (±SE)* | Griffin et al. (6)† |
| 6 | ∼50 ± 10 (±SE) | ∼10 ± 3 (±SE)* | |
These studies were performed in Sprague–Dawley rats. SD, standard deviation; SE, standard error; ∼, estimated values.
Significantly different than infarction rats from the corresponding time point; †proteinuria values for this table were estimated from a graphical figure in the referenced study.
Although these two ubiquitous rat models have much in common and both ultimately develop renal failure, they also have major phenotypic differences [e.g., degree of renin-angiotensin-aldosterone system (RAAS) activation, as detailed elsewhere] and therefore model unique aspects of human CKD. These differences may be underappreciated by the scientific community. Many published 5/6Nx rat studies 1) do not specify which 5/6Nx model they used, 2) lack rationale for model selection, and 3) omit discussion of their respective model’s unique pathophysiology. The aforementioned omissions may contribute to misinterpretation of results and inappropriate 5/6Nx rat publication referencing. Therefore, the goal of this review article is to raise awareness of the similarities and differences of these respective model phenotypes and aid investigators in model selection. Given that the kidney’s central function is blood filtration and the prominence of renal vasculature pathology in renal disease, we emphasize comparison of the infarction and polectomy rat renal vascular phenotypes. Because this topic is too large to comprehensively review, we compiled “mini-reviews,” each focused on a specific facet of vascular pathology. These include arterial blood pressure, the RAAS, autoregulation, nitric oxide (NO), single-nephron physiology, angiogenic and antiangiogenic factors, and capillary rarefaction.
In this review, we focus on assessing the variable of surgical approach (infarction vs. polectomy) upon multiple phenotypes. All studies referenced here used Sprague–Dawley or Wistar rat strains. Because the overwhelming majority of published studies used male rats, the majority of data we present are for male rats. However, recent studies have identified a number of phenotypic differences between males and females. We address these in sex differences in the 5/6nx model. In addition, the 5/6Nx surgery is sometimes performed as two separate surgeries. For example, a uniphrectomy performed at one surgery, and then sometime later, the poles of the remaining kidney are removed at a second surgery. The two-surgical step approach has been used for both polectomy (30–32) and infarction rats (12, 33). Although it is clear that both the one-step and two-step models manifest classical markers of CKD, including elevated phosphocreatine, fibrosis, proteinuria, etc., it is difficult to clearly determine differences between one-step and two-step surgeries for other phenotypes of interest. For example, in some reports, the first surgical step is pole ablation and the contralateral kidney is removed as a second surgery at a later time (32, 34, 35); other times, the reverse is used (36, 37). Within the body of two-surgical step 5/6Nx rat studies, there is variation in the amount of time elapsed between surgical steps (30, 32, 34–39), and there is variation in the surgical order. The effects of these variables upon the CKD phenotype are difficult to assess. Therefore, this review will focus on one-surgical step infarction and polectomy rats. Although we focus on reviewing the one-surgical step model, some two-surgical step publications are referenced. For these studies, we explicitly communicate, in text, that they are two-surgical step publications.
ARTERIAL BLOOD PRESSURE
Elevated blood pressure, as most recently defined by the American College of Cardiology and American Heart Association, is defined as a systolic blood pressure (SBP) of 120–129 mmHg and diastolic blood pressure (DBP) of <80 mmHg, stage 1 hypertension as a SBP of 130–139 mmHg or DBP of 80–89 mmHg, and stage 2 hypertension as a SBP of ≥140 mmHg or DBP of ≥90 mmHg (40). Hypertension affects ∼30% of United States adults (41). People with CKD are disproportionately hypertensive (42). Although there is no doubt that hypertension can contribute to CKD pathogenesis and progression, treatment of hypertension in people with CKD slows progression without abrogating it (43, 44), and not all people with CKD are hypertensive (42, 45). Thus, hypertensive and nonhypertensive CKD models are necessary to reflect different clinical manifestations of CKD.
Infarction and polectomy rats have markedly different arterial blood pressure phenotypes. The infarction model is characterized by rapid and robustly increased SBP (6, 8, 14, 15, 46, 47), whereas the polectomy model, in general, is characterized by early normotension, followed by slowly developing (over months) mild to moderate hypertension. For example, Griffin et al. (6), using telemetry-based measurements, found an infarction rat SBP of ∼160 mmHg with 3–5 days of surgery when both polectomy and sham-operated rats were normotensive. Many studies have reported later-onset (>6 wk postsurgery) hypertension in polectomy rats, typically 125–150 mmHg of SBP (5, 8, 17, 48, 49). In infarction rats, SBPs of >180 mmHg are common (20, 21, 25, 34, 50–54). Infarction and polectomy rat arterial blood pressures respond to angiotensin-converting enzyme (ACE) inhibitors and Ca2+ channel blockers (9, 35, 47–50, 52, 55–57). Significant changes in arterial blood pressure in response to experimental modulation of diet salt content have been reported in polectomy (58–60) and infarction rats (20, 21). In contrast, in infarction rats, Daniels and Hostetter (61) reported no statistically significant change in mean arterial blood pressure in high, low, and deficient (no salt) diet-fed relative to normal diet-fed controls. Table 2 shows SBP data for polectomy and infarction rats.
Table 2.
Summary of arterial blood pressure phenotype from infarction and polectomy rats subjected to 5/6 nephrectomy
| Method of Measurement | Animal Sex | Animal Strain | Time Postsurgery | Average Systolic Blood Pressure, mmHg |
References | |
|---|---|---|---|---|---|---|
| Infarction | Polectomy | |||||
| Radiotelemetry of the abdominal aorta | Male | Sprague–Dawley | 1 wk | ∼160 ± 5 (±SE) | ∼125 ± 3 (±SE) | Griffin et al. (6)* |
| 3 wk | ∼155 ± 5 (±SE) | ∼120 ± 3 (±SE) | ||||
| 6 wk | ∼155 ± 5 (±SE) | ∼125 ± 3 (±SE) | ||||
| Tail-cuff sphygmomanometer | Male | Sprague–Dawley | 2 wk | 169 ± 8 (±SE) | 123 ± 6 (±SE) | Garber et al. (8) |
| 6 wk | 169 ± 7 (±SE) | 129 ± 9 (±SE) | ||||
| Radiotelemetry of the abdominal aorta | Male | Sprague–Dawley | 3 days | ∼160 ± 10 (±SE) | ∼130 ± 5 (±SE) | Griffin et al. (14)* |
| 1 wk | ∼170 ± 10 (±SE) | ∼120 ± 5 (±SE) | ||||
| 3 wk | ∼170 ± 10 (±SE) | ∼120 ± 5 (±SE) | ||||
| Tail-cuff sphygmomanometer | Male | Sprague–Dawley | 2 wk | 166 ± 31 (±SD) | 120 ± 16 (±SD) | Ibrahim and Hostetter (15) |
| 4 wk | 161 ± 28 (±SD) | 120 ± 12 (±SD) | ||||
| Carotid artery using a pressure transducer catheter | Male | Sprague–Dawley | 2 wk | 126 ± 9 (±SE) | Chuppa et al. (5) | |
| 4 wk | 121.7 ± 7 (±SE) | |||||
| 5 wk | 139.6 ± 4 (±SE) | |||||
| 7 wk | 135 ± 5 (±SE) | |||||
| Tail-cuff sphygmomanometer | Male | Sprague–Dawley | Baseline | 95.4 ± 10.8 (±SD) | Kang et al. (17) | |
| 2 wk | 100.8 ± 15.4 (±SD) | |||||
| 4 wk | 104.3 ± 15.4 (±SD) | |||||
| 8 wk | 145.3 ± 25.8 (±SD) | |||||
| Radiotelemetry of the abdominal aorta | Male | Sprague–Dawley | 14-15 wk | 128.9 ± 2.3 (±SE) | Griffin et al. (48) | |
| Radiotelemetry of the abdominal aorta | Male | Sprague–Dawley | 1 wk | 160 ± 5 (±SE) | Griffin et al. (9)* | |
| 3 wk | 160 ± 5 (±SE) | |||||
| 6 wk | 185 ± 5 (±SE) | |||||
| Tail-cuff sphygmomanometer | Male | Sprague–Dawley | Baseline | ∼110 ± 5 (±SE) | Sun et al. (25)* | |
| 4 wk | ∼180 ± 10 (±SE) | |||||
| 8 wk | ∼190 ± 10 (±SE) | |||||
| Radiotelemetry of the abdominal aorta | Male | Sprague–Dawley | 1 wk | ∼165 ± 4 (±SE) | Griffin et al. (13)* | |
| 2 wk | ∼160 ± 4 (±SE) | |||||
| 3 wk | ∼160 ± 4 (±SE) | |||||
| 4 wk | ∼162 ± 4 (±SE) | |||||
| 5 wk | ∼170 ± 8 (±SE) | |||||
| 6 wk | ∼180 ± 8 (±SE) | |||||
| 7 wk | ∼182 ± 8 (±SE) | |||||
| Carotid artery using a pressure transducer catheter | Male | Wistar | 3 wk | 115 ± 0.5 (±SE) | Juncos et al. (62) | |
| 4 wk | 116 ± 1.5 (±SE) | |||||
| Tail-cuff sphygmomanometer | Male | Wistar | 2 wk | 139 ± 4 (±SE) | Sampaio-Maia et al. (63) | |
| Tail-cuff sphygmomanometer | Male | Wistar | 3 wk | ∼122 ± 3 (±SE) | Bai et al. (64)*† | |
| 5 wk | ∼138 ± 4 (±SE) | |||||
| 9 wk | ∼140 ± 5 (±SE) | |||||
| 11 wk | ∼139 ± 4 (±SE) | |||||
| Tail-cuff sphygmomanometer | Male | Wistar | 9 wk (60 days) | 191.3 ± 4.2 (±SD) | Correa et al. (16) | |
| Tail-cuff sphygmomanometer | Male | Wistar | 2 wk | ∼190 ± 10 (±SE) | Lax et al. (21)* | |
| 4 wk | ∼195 ± 10 (±SE) | |||||
| 6 wk | ∼200 ± 10 (±SE) | |||||
| 8 wk | ∼220 ± 10 (±SE) | |||||
| Tail-cuff sphygmomanometer | Male | Wistar | Baseline | 124 ± 3 (±SE) | Vavrinec et al. (46) | |
| 12 wk | 176 ± 12 (±SE) | |||||
| Tail-cuff sphygmomanometer | Male | Wistar | Baseline | ∼125 ± 5 (±SE) | Vettoretti et al. (47)* | |
| 6 wk | ∼165 ± 5 (±SE) | |||||
| 15 wk | ∼170 ± 5 (±SE) | |||||
| Tail-cuff sphygmomanometer | Male | Wistar | 2 wk | ∼175 ± 5 (±SE) | Anderson et al. (50)* | |
| 4 wk | ∼160 ± 10 (±SE) | |||||
| 6 wk | ∼170 ± 5 (±SE) | |||||
| 8 wk | ∼190 ± 5 (±SE) | |||||
| 10 wk | ∼185 ± 5 (±SE) | |||||
| 12 wk | ∼210 ± 5 (±SE) | |||||
| Tail-cuff sphygmomanometer | Male | Wistar | 3 wk | ∼175 ± 5 (±SE) | Mackenzie et al. (52)* | |
| 6 wk | ∼185 ± 5 (±SE) | |||||
| 9 wk | ∼210 ± 5 (±SE) | |||||
| 12 wk | 220 ± 15 (±SE) | |||||
| Tail-cuff sphygmomanometer | Male | Wistar | 2 wk | ∼170 ± 5 (±SE) | Anderson et al. (55)* | |
| 3 wk | ∼160 ± 10 (±SE) | |||||
| 4 wk | ∼170 ± 15 (±SE) | |||||
| 5 wk | ∼175 ± 10 (±SE) | |||||
| 6 wk | ∼175 ± 10 (±SE) | |||||
| 7 wk | ∼170 ± 15 (±SE) | |||||
| 8 wk | ∼180 ± 5 (±SE) | |||||
| Tail-cuff sphygmomanometer | Male | Sprague–Dawley | 3–4 wk | 170.6 ± 7.9 (±SE) | Bidani et al. (65) | |
| 6–8 wk | 158.9 ± 8.7 (±SE) | |||||
| Tail-cuff sphygmomanometer | Male | Wistar | 2 wk | ∼175 ± 10 (±SD) | Tapia et al. (26)* | |
| 4 wk | ∼185 ± 15 (±SD) | |||||
| Tail-cuff sphygmomanometer | Male | Sprague–Dawley | 2 wk | 144 ± 3.2 (±SE) | Bidani et al. (66) | |
| 4 wk | 134 ± 3.9 (±SE) | |||||
| Tail-cuff sphygmomanometer | Female | Wistar | 9 wk | 158 ± 4 (±SE) | Tanaka et al. (67) | |
SD, standard deviation; SE, standard error; ∼, estimated values.
Systolic blood pressure values for this table were estimated from a graphical figure in the referenced study; †5/6 nephrectomy surgery was performed over two surgical steps performed at different times.
People with chronic renal failure exhibit sympathetic overactivity (68, 69), which likely contributes to hypertension in this population (68, 69). There is evidence of increased sympathetic tone in both polectomy (58, 70) and infarction rats (51, 71). For example, Yuhara et al. (70) found that polectomy rats had plasma norepinephrine, a biomarker of sympathetic tone, to be on average 536 pg/mL compared with 230 pg/mL in sham-operated controls 4 wk postsurgery. In these animals, there was a significant, positive, linear correlation between mean arterial pressure (MAP) and plasma norepinephrine (70), consistent with the idea that sympathetic activity contributes to elevations of arterial blood pressure. Similarly, in polectomy rats, Cao et al. (58) found plasma norepinephrine to be, on average, 671 pg/mL in 5/6Nx rats compared with 335 pg/mL in sham-operated controls 10 wk postsurgery. We were unable to find reports of plasma norepinephrine in the infarction rat model; however, Paterno et al. (71) measured elevated renal sympathetic nerve activity in infarction rats relative to normal controls, and Augustyniak et al. (51) found that “neonatal sympathectomy” (neonatal guanethidine injection) reduced arterial blood pressure and proteinuria in infarction rats. The RAAS affects arterial blood pressure and sympathetic tone will be discussed in the renin angiotensin aldosterone system.
Arterial blood pressure is a prominent phenotypic difference between infarction and polectomy rats. Reduced nephron count per se is not responsible for these differences as both models likely have a similarly reduced nephron count following surgery. Although the infarction rat spontaneously develops greater increases in blood pressure than the polectomy rat, blood pressure and blood pressure-related injury can remain a prominent component of pathology in the polectomy rat. Moreover, although the polectomy rat does not spontaneously develop hypertension as the infarction rat does, polectomy rat blood pressure may increase in response to a variety of stimuli, such as a high-salt diet, for example. Investigators may need to carefully consider the arterial blood pressure phenotype when deciding on the use of the infarction or polectomy model to achieve their study aims.
THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM
The RAAS is differentially activated in infarction and polectomy rats. Multiple publications that included prospectively obtained infarction and polectomy 5/6Nx rats have shown significant differences in specific elements of the RAAS (e.g., renin content). These values are shown in Table 3. A study by Ibrahim and Hostetter (15) found that at 2 wk postsurgery, infarction rats, relative to polectomy rats, had significantly greater plasma renin activity, plasma aldosterone, total kidney renin content, SBP, and proteinuria. Ibrahim and Hostetter (15) observed an elevated infarction rat kidney scar (infarct) region renin content relative to the nonscar region. In contrast, polectomy rats had the same renin content at the resection edge as away from it, and both regions had less than the infarction rat scar region (Fig. 2) (15). Others have shown a threefold increase in infarction plasma renin content relative to polectomy rats 3 days following surgery with a correlation between plasma renin content and arterial blood pressure (14). These observations suggest that increases in arterial blood pressure by day 3 in infarction rats are caused in part by RAAS activation. Moreover, this rapid RAAS activation likely contributes to augmented glomerular pathology in infarction rats compared with polectomy rats in the weeks immediately following surgery, as previously reported in these specific studies (6, 14). The findings that similar remnant kidney hypertrophic responses occur between models following surgery despite differential RAAS activation suggests that elements of the RAAS (e.g., ANG II) are not primarily responsible for this hypertrophic remodeling. RAAS activation is associated with elevations in sympathetic activity. For example, increased plasma renin activity is associated with elevations in arterial blood pressure and plasma norepinephrine concentration in a variety of settings (72–75).
Table 3.
Measurements of renin-angiotensin-aldosterone system elements as reported in publications that included direct, prospectively obtained comparison of infarction and polectomy rats subjected to 5/6 nephrectomy
| Time Postsurgery | Measurement | Value |
Reference | |
|---|---|---|---|---|
| Infarction | Polectomy | |||
| 2 wk | Plasma renin content, ng ANG I/mL/h | 10.2 ± 8.9 (±SD) | 2.5 ± 1.8 (±SD)* | Ibrahim et al. (15) |
| Plasma aldosterone, pg/mL | 672 ± 591 (±SD) | 194 ± 107 (±SD)* | ||
| Total kidney renin content, µg ANG I/h/kidney | 127 ± 115 (±SD) | 26 ± 18 (±SD)* | ||
| Systolic blood pressure, mmHg | 166 ± 31 (±SD) | 120 ± 16 (±SD) | ||
| 3 days | Plasma renin content, ng ANG I/mL/h | 59.9 ± 9.6 (±SE) | 12.8 ± 1.5 (±SE)* | Griffin et al. (14) |
| Systolic blood pressure, mmHg | ∼160 ± 10 (±SE)† | ∼130 ± 5 (±SE)† | ||
| 3 wk | Plasma renin content, ng ANG I/mL/h | 18.6 ± 3.5 (±SE) | 9.3 ± 1.3 (±SE)* | |
| Systolic blood pressure, mmHg | ∼170 ± 10 (±SE)† | ∼120 ± 5 (±SE)† | ||
Both studies were performed in Sprague–Dawley rats. SD, standard deviation; SE, standard error.
Significantly different than infarction rats from the corresponding time point; †proteinuria values for this table were estimated from a graphical figure in the referenced study.
Figure 2.
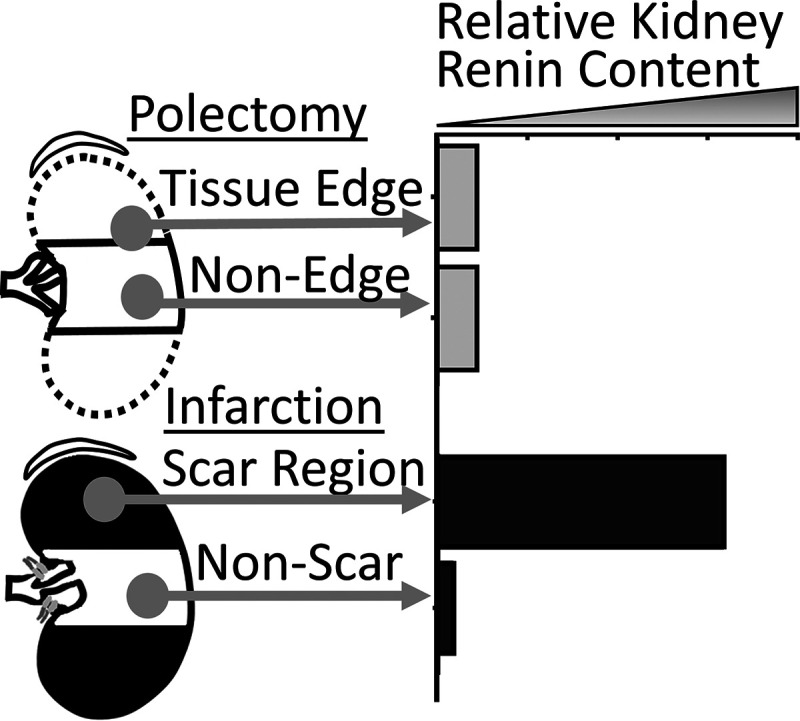
Tissue renin content in the infarction rat scar (infarcted poles) regions is highly elevated relative to nonscar tissue and is increased relative to the polectomy rat tissue edge and nonedge renin content. Graphical data were recreated from tabular data (Table 2) from Ibrahim et al. (15).
Interestingly, Griffin et al. (14) observed that by 3 wk postsurgery, polectomy plasma renin dropped to significantly below sham levels (9.3 ± 1.3 vs. 17.9 ± 0.9 ng ANG I/mL/h, average ± SE) and that despite maintenance of hypertension (SBP: 185 mmHg), infarction rat plasma renin content dropped to approximate sham levels (18.6 ± 3.5 ng ANG I/mL/h). This temporal pattern of plasma renin reduction is supported by a Daniels et al. study (50) of infarction rats. Moreover, Ismail et al. (76) observed polectomy rats (two-step surgery with left kidney surgical polectomy performed followed 1 wk later by right kidney removal) to have ∼30% of sham rat plasma ANG II levels 8–10 wk postsurgery. Jackson et al. (77) observed infarction rats to have twofold sham levels of ANG II 1 wk postsurgery but exactly sham levels of ANG II 4 wk postsurgery. Thus, it is possible that elevated RAAS activity contributes to early infarction rat arterial blood pressure increases, but that over time, following volume retention and increased blood pressure, physiological feedback mechanisms enable the maintenance of elevated arterial blood pressure in infarction rats even with ANG II and plasma renin levels reduced relative to their previous peak. Infarction-to-polectomy rat transition experiments in which infarcted poles are later surgically removed may inform upon short- versus long-term contributions of this tissue to RAAS activity. In addition, the presence of the infarcted tissue itself may elicit an immune and/or inflammatory reaction in a manner that polectomy rats would lack due to pole tissue removal. Although markers of renal and plasma inflammation are elevated in both models (24, 53, 58, 78–81), we were unable to find investigations that detailed the specific contribution of the infarcted tissue to an inflammatory phenotype. However, interactions between inflammatory processes and the RAAS are well established. For example, ANG II has known proinflammatory effects such as increasing vascular permeability and expression of proinflammatory cytokines within specific settings (82–84). Thus, the infarcted tissue may exhibit a primary inflammatory reaction as well as a reaction derived from elevated local RAAS activity.
RAAS involvement in infarction rats, polectomy rats, and humans with CKD is demonstrated by the efficacy of RAAS targeting treatments, such as ACE inhibitors and angiotensin receptor blockers (43, 44, 48–50, 52, 55, 57, 85). Part of the mechanism by which RAAS inhibitors function in people with chronic renal failure is through a reduction of sympathetic tone (86). RAAS blockade is a standard treatment for people with CKD and is associated with mitigated progression of proteinuria, increased serum creatinine doubling time, reductions in blood pressure, and delays in reaching end-stage renal disease (43, 44, 87–89).
AUTOREGULATION
The ability of the kidney to maintain a relatively constant GFR, despite changes in systemic blood pressure, is referred to as autoregulation. People with normally autoregulated renal blood flow and essential hypertension are thought to typically exhibit slow developing glomerulosclerosis, because blood pressure elevations elicit reactionary elevations in renal arteriole resistance, thus reducing glomerular pressure and consequent barotrauma (90–92). If blood pressure elevates beyond the autoregulatory range, malignant nephrosclerosis can develop rapidly (92, 93). Parving et al. (94) measured autoregulation in people with nephropathy by quantifying GFR changes in response to acute arterial blood pressure reduction by clonidine treatment. They observed significantly more GFR reduction in people with nephropathy relative to controls, including some individuals that exhibited pressure passivity [ΔMAP (%) = ΔGFR (%)], indicative of highly impaired autoregulation (94–96). Non-nephrotic controls had an ∼2% GFR reduction despite an 18-mmHg MAP reduction, indicating normally autoregulated renal blood flow (96). A recent study by Bidani et al. (56) in healthy conscious rats demonstrated that spontaneous changes in systemic blood pressure are normalized at the renal level within 5–10 s by autoregulation but that normalization is significantly slower in polectomy rats. Thus, even without sustained hypertension, transient blood pressure elevations could cause glomerular barotrauma by slow-acting autoregulation (48). This may be one reason why CKD progresses in normotension, albeit more slowly.
Griffin and Bidani (97) have identified the autoregulation range in healthy rats to be ∼110–210 mmHg and have identified autoregulation impairments in infarction and polectomy rats (6, 9, 56, 65, 98, 99). For example, Griffin et al. (6) demonstrated infarction and polectomy rats to have similarly impaired “static” autoregulation within 2 wk of nephrectomy surgery. This static autoregulation measure is referred to as the autoregulation index and was produced by assessing changes in renal artery blood flow in response to large step changes in renal perfusion pressure, from ∼100 to 140 mmHg (6). An autoregulation index of 0 represents “perfect” autoregulation (no change in renal blood flow despite a renal perfusion pressure change), whereas an autoregulation index of 1 indicates a lack of autoregulation (vessels acting as passive conduits) (6). A lack of difference between infarction and polectomy rats (6) is consistent with the idea that autoregulation is abnormal due to hyperfiltration consequent to reduced nephron count rather than hypertension of the infarction model; the polectomy rats in this study were normotensive and the infarction rats were hypertensive (6). In a different study, Griffin et al. (9) demonstrated that the impaired autoregulation of infarction rats was further impaired by treatment with the Ca2+ channel blocker nifedipine, resulting in little glomerular protection, despite reduced blood pressure. In contrast, treatment of infarction rats with the ACE inhibitor enalapril resulted in reduced blood pressure, no change in autoregulation, and significant reductions in glomerular injury (9). The differential effects of these drugs likely occur, in part, by differential autoregulation modulation.
Lin et al. (100) demonstrated impaired autoregulation in infarction and polectomy rats 3 and 4 wk postsurgery. They assessed juxtamedullary glomeruli afferent arteriole diameter at several set perfusion pressures in freshly excised and perfused remnant kidneys (100). Infarction and polectomy rat arterioles exhibited blunted diameter reductions, relative to sham controls, in response to increased perfusion pressure, including virtually no diameter change between perfusion pressures of 100 and 140 mmHg (100). Dilation in response to abluminal acetylcholine application resulted in similar dilation between polectomy and sham rat but a highly blunted effect in infarction rat afferent arterioles, suggesting impairment of endothelium-dependent vasodilation (100). This finding could represent a significant difference between models. That both nephrectomy models had similarly blunted autoregulation despite large differences in the acetylcholine response suggests that acetylcholine-dependent vasodilation has little impact on autoregulation, at least as it is assayed here.
Infarction and polectomy rats spontaneously develop autoregulation impairments. This may be more consequential in infarction rats than in polectomy rats because they spontaneously develop elevated arterial blood pressure relative to polectomy rats, although even transient blood pressure elevations amid normotension could impart renal damage with impaired autoregulation.
NITRIC OXIDE
NO is produced from the oxidation of l-arginine to l-citrulline by NO synthases (NOSs). Although NO has many effects on renal physiology, it is most well known as a vasorelaxation agent. People with chronic renal failure have reduced NO production (101, 102), reduced plasma l-arginine (103–106), and increased plasma concentration of the NOS inhibitor asymmetric dimethylarginine (ADMA) (101, 107, 108). Higher ADMA plasma concentrations are associated with worse disease (101, 108, 109) and are a strong, independent predictor of adverse outcomes in people with end-stage renal disease (109, 110). These observations illustrate the importance of NO handling in renal health maintenance.
Albeit with exceptions, the infarction and polectomy rat models are both associated with decreased NO bioavailability (23, 81, 111, 112). Infarction and polectomy rat studies using urine total nitrite (NO2) and nitrate (NO3), stable NO metabolites, as an index of NO production, have reported decreases at a variety of time points (19, 39, 81, 111, 113). Aiello et al. (113) observed a reduction in renal NO production in week 1 postsurgery infarction rats and a near absence 90 days postsurgery, as evidenced by the ex vivo conversion of l-arginine to l-citrulline by renal tissue explants. Ni et al. (23), in 12-wk infarction rats, found significantly reduced kidney tissue NO. Similar to people with CKD, increased plasma ADMA has been reported in polectomy rats (19, 114) and in two-surgical step infarction rats (33). We were unable to find plasma ADMA reported for one-surgical step infarction rats. Moreover, the ADMA concentration has a significant, negative correlation with peritubular (19), and glomerular capillary density (114) in polectomy rats. In infarction rats, renal endothelial NOS (eNOS) levels have been reported as increased (115), not different (111), and decreased (23) relative to sham rats. Similarly, in polectomy rats, renal eNOS levels have been reported as increased (62), not different (116), and decreased (39); the latter two references performed 5/6Nx surgery as a two-step procedure with left kidney polectomy as a first surgery and right kidney removal as a second surgery. Renal neuronal NOS expression has been reported as decreased in infarction rats by Erdely (111) and Szabo et al. (115) and decreased in two-surgical step polectomy rats by Tain et al. (116). Multiple studies have reported elevations in renal eNOS expression in renal biopsy tissue from people with diabetic nephropathy relative to normal controls (117, 118). NO physiology is clearly abnormal in people with CKD. Despite this, establishing a cause-and-effect relationship between CKD pathology and NOS function in people can be difficult. Doubtless, in some cases, intrinsic impairments of NOS function helped precipitate CKD. In fact, certain eNOS polymorphisms are associated with CKD (119, 120). In other cases, the onset of renal disease drives changes in NOS function that contribute to impaired NO production. In infarction rats, Reyes et al. (121) reported that treatment with l-arginine, the oxidation of which NOS produces NO, improves outcomes. Treatment with NOS inhibitors (e.g., N-nitro-l-arginine methyl ester) has been reported to exacerbate pathology in polectomy and infarction rats (22, 33, 111, 121, 122). Studies of l-arginine supplementation in people with CKD have reported little to no apparent effect (123, 124), whereas others have reported modest improvement (125, 126).
Experimental modulation of NO has been used to identify NO-dependent, pathogenic mechanisms endemic to the remnant kidney model, although interstudy methodological differences can complicate interpretation. For instance, Vaziri et al. (39) observed a blunted MAP reduction in 6-wk postsurgery, two-surgical step, polectomy rats relative to sham rats in response to intravenous injection of l-arginine, a blunted pressor response to N-monomethyl-l-arginine (l-MMA; NOS inhibitor, 10 mg/kg iv), and no difference in MAP reduction to sodium nitroprusside, an NO donor. This result suggests impaired NO production in polectomy rats rather than an impaired reaction to adequate NO. In contrast, Griffin et al. (127) reported, in 3–4 wk postpolectomy rats, that l-NMMA administration (50 mg/kg) resulted in nearly identical drops in MAP between polectomy and sham rats. The disparate results of these two studies in the polectomy rat response to l-NMMA treatment, while compatible with each other, could be due to differences in dose (10 vs. 50 mg/kg) or study time points (3–4 vs. 6 wk). They may also be due to differences in baseline arterial blood pressure: despite both being polectomy rats, the rats in Griffin et al. study (127) were normotensive, whereas those in the Vaziri et al. study (39) were mildly hypertensive.
SINGLE-NEPHRON PHYSIOLOGY ASSESSED BY MICROPUNCTURE ANALYSIS
In humans, a GFR below 60 mL/min/1.73 m2 for 3 mo is one diagnostic criterion for CKD and below 15 mL/min/1.73 m2 is kidney failure. Whole kidney GFR and renal blood flow are initially reduced in infarction and polectomy remnant kidneys (6, 14, 38, 66, 98), but single-nephron GFR and single-nephron plasma flow are elevated in both models 3–4 wk postsurgery (14, 26, 50, 98), indicating compensatory hyperfiltration.
The micropuncture technique enables measurement of individual nephron flows and pressures. Here, we summarize a Griffin et al. study conducted on rats 3–4 wk postsurgery. They reported a single-nephron GFR of 38 ± 18 nL/min in sham rats, 78 ± 9.7 nL/min in polectomy rats, and 73 ± 9.0 nL/min in infarction rats (means ± SE) (14). The glomerular plasma flow was markedly elevated in polectomy (427 ± 39 nL/min) and infarction rats (440 ± 64 nL/min) compared with sham controls (183 ± 18 nL/min) (14). These elevated single-nephron flow rates are consistent with other micropuncture studies (50, 61, 81, 98). Both models exhibited reductions in resistance of glomerular afferent arteries (sham: 1.6 ± 0.2 dyn·s·cm−5, polectomy: 0.7 ± 0.15 dyn·s·cm−5, and infarction: 1.0 ± 0.16 dyn·s·cm−5) and efferent arteries (sham: 1.0 ± 0.1 dyn·s·cm−5, polectomy: 0.5 ± 0.08 dyn·s·cm−5, and infarction: 0.6 ± 0.09 dyn·s·cm−5) (14). Importantly, polectomy rats exhibited elevated glomerular capillary pressures (PGC; 50 ± 0.9 mmHg) compared with sham rats (46 ± 0.8 mmHg), and infarction rat PGC (56 ± 1.1 mmHg) was significantly elevated compared with polectomy and sham rats (14). Polectomy and sham rat MAP, at the time of micropuncture, were both 104 mmHg, whereas for infarction rats, MAP was 136 mmHg (14). This finding suggests that not only is arterial blood pressure different between models but perhaps also glomerular pressure. This finding may represent a significant phenotype difference between models. Anderson et al. (50) reported that treatment of infarction rats with the ACE inhibitor enalapril normalized their systemic blood pressure and PGC with associated decreases in proteinuria and glomerular injury. In contrast, triple drug therapy (reserpine, hydralazine, and hydrochlorothiazide) normalized systemic arterial blood pressure, but not PGC, resulting in, over time, a similar proteinuria and glomerulosclerosis rate to untreated infarction rats (50). Taken together, these observations suggest that elevated PGC in infarction rats relative to polectomy rats contributes to their exacerbated glomerular injury as shown in these specific studies. These observations also suggest that the elevated PGC in infarction relative to polectomy rats is due to transmission of systemic blood pressure to glomeruli and not hyperfiltration per se, as both groups’ single-nephron GFR were similar (14).
Reduced glomerular resistance, as measured by micropuncture in 5/6Nx rats, suggests arteriole dilation. Kimura et al. (128) measured afferent and efferent glomerular arteriole diameter using microvascular casts. In 2 wk postsurgery, two-surgical step, polectomy rats, they reported significantly increased glomerular afferent arteriole diameter (polectomy: 17.9 ± 0.8 µm and sham: 13.8 ± 0.5 µm, P < 0.05) and glomerular efferent arterial diameter (polectomy: 13.7 ± 0.5 µm and sham: 11.5 ± 0.4 µm, P < 0.05) (128). Lin et al. (100) found greatly dilated afferent juxtaglomerular arterioles in both models 3–4 wk postsurgery. In people with hypertension with proteinuria and/or elevated serum creatinine, Hill et al. (129) reported significantly increased glomerular afferent arterioles in renal biopsy samples relative to age-matched, normotensive controls. Like the 5/6Nx rat models, increased afferent arteriole diameter in these individuals was associated with glomerular hypertrophy and sclerosis (129). Collectively, these studies highlight a complex interplay between nephron count, single-nephron GFR, glomerular arteriole diameters, transmission of systemic blood pressure to the glomeruli, and CKD progression.
GLOMERULAR AND PERITUBULAR CAPILLARY RAREFACTION
Capillary loss impairs oxygen delivery, nutrient delivery, and waste removal while promoting tissue ischemia and scar formation (130). Scarring further impairs oxygen delivery, establishing a progressive cycle. Glomerular capillary rarefaction occurs spontaneously in both models (17, 23, 67, 131–133). Interestingly, in polectomy rats, Kang et al. reported an increase in proliferating glomerular endothelial cells, as measured by proliferating cell nuclear antigen (PCNA)-positive cells 2 wk postsurgery. This is followed by a significant reduction by week 8 to below sham levels with a concomitant reduction in glomerular capillary loop number (17). In infarction rats, Floege et al. (134) also found an approximately fivefold increase in glomerular PCNA-positive cells 2 wk postsurgery relative to sham controls, and in 2–10 wk postsurgery, glomerular PCNA gradually reduced until at week 10 it was approximately threefold sham levels. Glomerular capillary loss and glomerulosclerosis are human CKD hallmarks (3, 135, 136). Hohenstein et al. (135), in human glomerulonephritis (of various etiology) biopsy samples, observed elevated glomerular capillary rarefaction relative to normal controls and a linear correlation between this rarefaction and glomerulosclerosis, GFR, and serum creatinine.
Similarly, peritubular capillary loss is a human CKD hallmark and correlates with renal health biomarkers (3, 28, 137, 138). Infarction and polectomy rats exhibit peritubular capillary rarefaction (17, 67, 80, 131, 132). Reminiscent of glomerular capillaries, Kang et al. (17) observed an increase in peritubular capillary proliferating endothelial cells at week 1 but then a rapid decline to below sham levels by week 2 and near absence at week 8. This was accompanied by significant peritubular capillary rarefaction by week 2, with further rarefaction by week 8 (17). Interestingly, the timing of this marked rarefaction increase coincides temporally with increases in proximal tubular damage including dilation, apical blebbing, and brush border loss, as previously described by Chuppa et al. (5) and Adam et al. (139) in polectomy rats. In infarction rats, Advani et al. (131) described loss of peritubular capillaries with the endothelial cell marker JG12 as well as three-dimensional fluorescent microangiography imaging, and Ni et al. (23) described at 12 wk with concomitant tubular dilation and a strong linear correlation with interstitial fibrosis. Similar to human CKD biopsy studies, associations and direct correlations between renal capillary rarefaction and fibrosis have been identified in numerous infarction and polectomy rat studies (17, 23, 131, 133, 140). Glomerular and peritubular capillaries interact with renal tubules in complex ways. Although there is clearly an association between capillary loss with functional loss of tubules, the timing and exact causal relationship between the two and other factors varies with the specific pathological setting. For instance, capillary loss may drive tubular loss and vice versa.
ANGIOGENIC AND ANTIANGIOGENIC FACTORS
Imbalances of proangiogenic and antiangiogenic have been identified in 5/6Nx rats of both models. Although many factors affect endothelial function and growth, we focus here on thrombospondin-1 (TSP-1) and vascular endothelial growth factor (VEGF) because of the extent of their characterization and effect magnitude in progressive CKD. TSP-1, a glycoprotein, facilitates fibroblast activation and inhibits endothelial cell proliferation, among other functions (133, 141, 142). VEGF exerts potent mitogenic activity on endothelial cells and in health is constitutively expressed primarily in the outer medulla, medullary rays, and podocytes (143–145).
Renal VEGF expression has been reported as decreased in infarction and polectomy rats (17, 23, 133). Kang et al. (17) reported significant reductions in polectomy rat tubular VEGF expression by week 4, with additional progressive loss through week 8. These VEGF reductions occurred with reduced peritubular endothelial cell proliferation and increased peritubular capillary rarefaction (17). The antiangiogenic factor TSP-1 was significantly elevated in tubules and glomeruli by week 2 and remained elevated through 8 wk (17). Administration of VEGF to polectomy rats reduced capillary rarefaction, increased the number of proliferating endothelial cells, and improved renal function compared with untreated polectomy controls (132), demonstrating that VEGF can independently alter CKD progression. In a longitudinal study of infarction rats, Hugo et al. (146) observed increases in renal expression of TSP-1 within 1 wk of surgery and steady increases through 10 wk. Collectively, these observations suggest important roles for VEGF and TSP-1 in CKD progression.
Renal expression of VEGF in human CKD is complex and varies with disease and cell type (137, 147–149). For example, Bailey et al. (147) found significantly less VEGF mRNA in glomerular cells in diabetic nephropathy human biopsy samples than controls, but more in people with minimal nephropathy. Namikoshi et al. (148) found that VEGF expression was elevated in biopsy tissue from people with IgA nephropathy with mild/moderate interstitial injury compared with samples with little interstitial injury. However, VEGF levels in advanced interstitial injury were below those with very minimal interstitial injury, despite the highest degree of peritubular capillary rarefaction (148). Analysis of human CKD biopsy samples, of various etiology, consistently demonstrate increased levels of renal TSP-1 (150–152).
SEX DIFFERENCES IN THE 5/6Nx MODEL
Many sex differences in the etiology, progression, and end points of human CKD have been reported (153–164). Women have higher rates of CKD, but men are more likely to progress to end-stage renal disease (156, 165, 166). Despite women having a higher age-adjusted CKD prevalence than men (167), several studies have suggested females are protected from developing kidney disease due to a cardiovascular benefit of gonadal hormones (168–182). However, it has been noted that apparent protection from CKD progression in premenopausal females is lost if the data are adjusted for baseline proteinuria, MAP, and high-density lipoprotein-cholesterol (156). In addition, because of the intimate connection between the cardiovascular system and kidney disease, sex differences in blood pressure and response to chronic hemodynamic stress likely differentially effects renal disease progression between sexes (183, 184).
A unique aspect of 5/6Nx model utilization for exploration of sex differences in CKD is that the initial induction of renal insufficiency is common to both sexes; the renal mass reduction surgery can be replicated between sexes. In contrast, in other models, the nature of the renal insult is itself sex dependent (e.g., hypertension). Thus, use of the 5/6Nx model allows researchers to study sex-specific consequences of renal insufficiency without upstream sources of experimental variation. Despite this, to date, there are relatively few 5/6Nx studies that have included male and female rats. Indeed, the published medical literature as a whole focuses on male subjects disproportionately (185, 186). Within the 5/6Nx female rat literature, most investigations used the polectomy model; we were only able to find four published studies using the infarction model (67, 121, 187, 188). Moreover, very few female 5/6Nx rat studies made assessment of the renal vasculature a point of investigation. For example, we were unable to find any publications that assessed renal autoregulation or capillary rarefaction in female 5/6Nx rats. For the purposes of this review, the paucity of female 5/6Nx publications, especially those with vascular-focused data, greatly hindered our ability to adequately assess sex differences in 5/6Nx rat renovascular pathology. This reality represents an opportunity for investigators to fill these knowledge gaps.
Within published studies that included both male and female 5/6Nx polectomy rats, assessment of basic renal pathology measures such as creatinine clearance and proteinuria in some studies suggested worse pathology in males (18) or worse pathology in females (11, 189). All of the studies referenced in the previous sentence used the two-surgical step polectomy model except for Paterson et al. (11). We were unable to find any published studies that included male and female infarction model rats. Blood urea nitrogen and serum creatinine comparisons across sexes can be difficult to interpret given their dependence upon sex-dependent variables such as muscle mass (190, 191). Clearance measurements (e.g., creatinine clearance) may facilitate comparison across sexes as they normalize for some sex-dependent factors. Some studies used exclusively female rats because they investigated female specific physiology. For example, Antus et al. (168) performed a hormone replacement study in ovariectomized 5/6Nx rats, and Deng and Baylis (187) used female rats to investigate CKD in pregnancy. In contrast, a number of publications used exclusively female 5/6Nx rats (all of these happened to be two-surgical step polectomy models) without providing a clear rationale for the selection of sex (192–195). Similarly, most publications that used exclusively male 5/6Nx rats also lacked an explanation for the selection of sex.
In 5/6Nx polectomy rats assessed 7 wk postsurgery, we found evidence of exacerbated renal pathology in females relative to males (11). For example, creatinine clearance was more than twofold lower in females than in males (11). In addition, through chronic conscious telemetric blood pressure recordings, we found females to have a MAP of 161 ± 7 mmHg (means ± SE) at 7 wk postsurgery compared with 126 ± 5 mmHg in males. Matched sham-operated female and male MAP averaged 114 ± 7 and 102 ± 2 mmHg, respectively (11). Interestingly, despite sex differences in arterial blood pressure, renal fibrosis, as assessed by histological trichrome staining and gene expression, was not different between sexes (11). Furthermore, we found that ovariectomy in 5/6Nx polectomy rats improved creatinine clearance (11). This is likely due to the loss of progesterone rather than the loss of estrogens because it has been shown that estradiol replacement in ovariectomized 5/6Nx rats attenuated indexes of declining renal function, whereas combined treatment of estradiol and progesterone did not (168).
CONCLUSIONS
Infarction and polectomy rats have value in modeling a wide range of CKD types due to shared pathology between these models and people with CKD. Both models spontaneously develop uremia, impaired autoregulation, fibrosis, altered NO physiology, capillary rarefaction, imbalances in pro/antiangiogenic factors, and ultimately renal failure. These two models also exhibit clear phenotypic differences from each other. Infarction rats have augmented RAAS activity. The infarction rat is characterized by rapid-onset, moderate to severe hypertension, whereas the polectomy rat is characterized by slow-developing, mild to moderate hypertension. Differences in blood pressure likely contribute to the apparent faster onset of glomerular injury and proteinuria in infarction rats than polectomy rats. Ca2+ channel blockers further impair autoregulation in people with diabetic nephropathy and infarction rats (9, 196). Infarction and polectomy rat models both exhibit increased renal TSP-1, consistent with human CKD of various etiology. Infarction and polectomy rat kidneys consistently have reduced VEGF expression, but VEGF expression is variable in people with CKD. To date, there have been very few 5/6Nx studies that have used female rats for understanding sex differences in CKD. This knowledge gap represents an area of opportunity for discovery. In conclusion, infarction and polectomy rats, despite having much in common, also have major phenotypic differences and thereby model unique aspects of human CKD.
GRANTS
This work was supported by National Institutes of Health Grants R01HL128332 and T32HL134643 and by the Medical College of Wisconsin Cardiovascular Center’s A.O. Smith Fellowship Scholars Program (to R.J.A.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.J.A. and A.J.K. conceived and designed research; R.J.A. prepared figures; R.J.A., A.C.W., and A.J.K. drafted manuscript; R.J.A., A.C.W., and A.J.K. edited and revised manuscript; R.J.A., A.C.W., and A.J.K. approved final version of manuscript.
REFERENCES
- 1.Kim K, Anderson EM, Thome T, Lu G, Salyers ZR, Cort TA, O’Malley KA, Scali ST, Ryan TE. Skeletal myopathy in CKD: a comparison of adenine-induced nephropathy and 5/6 nephrectomy models in mice. Am J Physiol Renal Physiol 321: F106–F119, 2021. doi: 10.1152/ajprenal.00117.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mannon EC, O'Connor PM. Alkali supplementation as a therapeutic in chronic kidney disease: what mediates protection? Am J Physiol Renal Physiol 319: F1090–F1104, 2020. doi: 10.1152/ajprenal.00343.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet 389: 1238–1252, 2017. doi: 10.1016/S0140-6736(16)32064-5. [DOI] [PubMed] [Google Scholar]
- 4.Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, Van Lente F, Levey AS. Prevalence of chronic kidney disease in the United States. JAMA 298: 2038–2047, 2007. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 5.Chuppa S, Liang M, Liu P, Liu Y, Casati MC, Cowley AW, Patullo L, Kriegel AJ. MicroRNA-21 regulates peroxisome proliferator-activated receptor α, a molecular mechanism of cardiac pathology in cardiorenal syndrome type 4. Kidney Int 93: 375–389, 2018. doi: 10.1016/j.kint.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffin KA, Picken M, Bidani AK. Method of renal mass reduction is a critical modulator of subsequent hypertension and glomerular injury. J Am Soc Nephrol 4: 2023–2031, 1994. doi: 10.1681/ASN.V4122023. [DOI] [PubMed] [Google Scholar]
- 7.Benetti A, Martins FL, Sene LB, Shimizu MHM, Seguro AC, Luchi WM, Girardi ACC. Urinary DPP4 correlates with renal dysfunction, and DPP4 inhibition protects against the reduction in megalin and podocin expression in experimental CKD. Am J Physiol Renal Physiol 320: F285–F296, 2021. doi: 10.1152/ajprenal.00288.2020. [DOI] [PubMed] [Google Scholar]
- 8.Garber SL, Mirochnik Y, Brecklin C, Slobodskoy L, Arruda JA, Dunea G. Effect of relaxin in two models of renal mass reduction. Am J Nephrol 23: 8–12, 2003. doi: 10.1159/000066302. [DOI] [PubMed] [Google Scholar]
- 9.Griffin KA, Picken MM, Bidani AK. Deleterious effects of calcium channel blockade on pressure transmission and glomerular injury in rat remnant kidneys. J Clin Invest 96: 793–800, 1995. doi: 10.1172/JCI118125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mannon EC, Sartain CL, Wilkes TC, Sun J, Polichnowski AJ, O'Connor PM. Renal mass reduction increases the response to exogenous insulin independent of acid-base status or plasma insulin levels in rats. Am J Physiol Renal Physiol 321: F494–F504, 2021. doi: 10.1152/ajprenal.00679.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paterson MR, Geurts AM, Kriegel AJ. miR-146b-5p has a sex-specific role in renal and cardiac pathology in a rat model of chronic kidney disease. Kidney Int 96: 1332–1345, 2019. doi: 10.1016/j.kint.2019.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida T, Kakizawa S, Totsuka Y, Sugimoto M, Miura S, Kumagai H. Effect of endurance training and branched-chain amino acids on the signaling for muscle protein synthesis in CKD model rats fed a low-protein diet. Am J Physiol Renal Physiol 313: F805–F814, 2017. doi: 10.1152/ajprenal.00592.2015. [DOI] [PubMed] [Google Scholar]
- 13.Griffin KA, Picken M, Giobbie-Hurder A, Bidani AK. Low protein diet mediated renoprotection in remnant kidneys: renal autoregulatory versus hypertrophic mechanisms. Kidney Int 63: 607–616, 2003. doi: 10.1046/j.1523-1755.2003.00759.x. [DOI] [PubMed] [Google Scholar]
- 14.Griffin KA, Picken MM, Churchill M, Churchill P, Bidani AK. Functional and structural correlates of glomerulosclerosis after renal mass reduction in the rat. J Am Soc Nephrol 11: 497–506, 2000. doi: 10.1681/ASN.V113497. [DOI] [PubMed] [Google Scholar]
- 15.Ibrahim HN, Hostetter TH. The renin-aldosterone axis in two models of reduced renal mass in the rat. J Am Soc Nephrol 9: 72–76, 1998. doi: 10.1681/ASN.V9172. [DOI] [PubMed] [Google Scholar]
- 16.Correa F, Buelna-Chontal M, Hernandez-Resendiz S, Garcia-Nino WR, Roldan FJ, Soto V, Silva-Palacios A, Amador A, Pedraza-Chaverri J, Tapia E, Zazueta C. Curcumin maintains cardiac and mitochondrial function in chronic kidney disease. Free Radic Biol Med 61: 119–129, 2013. doi: 10.1016/j.freeradbiomed.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 17.Kang DH, Joly AH, Oh SW, Hugo C, Kerjaschki D, Gordon KL, Mazzali M, Jefferson JA, Hughes J, Madsen KM, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J Am Soc Nephrol 12: 1434–1447, 2001. doi: 10.1681/ASN.V1271434. [DOI] [PubMed] [Google Scholar]
- 18.Lu H, Lei X, Klaassen C. Gender differences in renal nuclear receptors and aryl hydrocarbon receptor in 5/6 nephrectomized rats. Kidney Int 70: 1920–1928, 2006. doi: 10.1038/sj.ki.5001880. [DOI] [PubMed] [Google Scholar]
- 19.Matsuguma K, Ueda S, Yamagishi S, Matsumoto Y, Kaneyuki U, Shibata R, Fujimura T, Matsuoka H, Kimoto M, Kato S, Imaizumi T, Okuda S. Molecular mechanism for elevation of asymmetric dimethylarginine and its role for hypertension in chronic kidney disease. J Am Soc Nephrol 17: 2176–2183, 2006. doi: 10.1681/ASN.2005121379. [DOI] [PubMed] [Google Scholar]
- 20.Purkerson ML, Hoffsten PE, Klahr S. Pathogenesis of the glomerulopathy associated with renal infarction in rats. Kidney Int 9: 407–417, 1976. doi: 10.1038/ki.1976.50. [DOI] [PubMed] [Google Scholar]
- 21.Lax DS, Benstein JA, Tolbert E, Dworkin LD. Effects of salt restriction on renal growth and glomerular injury in rats with remnant kidneys. Kidney Int 41: 1527–1534, 1992. doi: 10.1038/ki.1992.222. [DOI] [PubMed] [Google Scholar]
- 22.Kang DH, Nakagawa T, Feng L, Johnson RJ. Nitric oxide modulates vascular disease in the remnant kidney model. Am J Pathol 161: 239–248, 2002. doi: 10.1016/S0002-9440(10)64175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ni H, Chen J, Pan M, Zhang M, Zhang J, Chen P, Liu B. FTY720 prevents progression of renal fibrosis by inhibiting renal microvasculature endothelial dysfunction in a rat model of chronic kidney disease. J Mol Histol 44: 693–703, 2013. doi: 10.1007/s10735-013-9521-8. [DOI] [PubMed] [Google Scholar]
- 24.Souza MK, Neves RVP, Rosa TS, Cenedeze MA, Arias SCA, Fujihara CK, Bacurau RFP, Camara NOS, Moraes MR, Pacheco ESFA. Resistance training attenuates inflammation and the progression of renal fibrosis in chronic renal disease. Life Sci 206: 93–97, 2018. doi: 10.1016/j.lfs.2018.05.034. [DOI] [PubMed] [Google Scholar]
- 25.Sun L, Zhang D, Liu F, Xiang X, Ling G, Xiao L, Liu Y, Zhu X, Zhan M, Yang Y, Kondeti VK, Kanwar YS. Low-dose paclitaxel ameliorates fibrosis in the remnant kidney model by down-regulating miR-192. J Pathol 225: 364–377, 2011. doi: 10.1002/path.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tapia E, Soto V, Ortiz-Vega KM, Zarco-Márquez G, Molina-Jijón E, Cristóbal-García M, Santamaría J, García-Niño WR, Correa F, Zazueta C, Pedraza-Chaverri J. Curcumin induces Nrf2 nuclear translocation and prevents glomerular hypertension, hyperfiltration, oxidant stress, and the decrease in antioxidant enzymes in 5/6 nephrectomized rats. Oxid Med Cell Longev 2012: 269039, 2012. doi: 10.1155/2012/269039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bitzer M, Sterzel RB, Bottinger EP. Transforming growth factor-β in renal disease. Kidney Blood Press Res 21: 1–12, 1998. doi: 10.1159/000025837. [DOI] [PubMed] [Google Scholar]
- 28.Bohle A, Mackensen-Haen S, Wehrmann M. Significance of postglomerular capillaries in the pathogenesis of chronic renal failure. Kidney Blood Press Res 19: 191–195, 1996. doi: 10.1159/000174072. [DOI] [PubMed] [Google Scholar]
- 29.Schainuck LI, Striker GE, Cutler RE, Benditt EP. Structural-functional correlations in renal disease. II. The correlations. Hum Pathol 1: 631–641, 1970. doi: 10.1016/s0046-8177(70)80061-2. [DOI] [PubMed] [Google Scholar]
- 30.Fleck C, Scholle T, Schwertfeger M, Appenroth D, Stein G. Determination of renal porphyrin handling in rats suffering from different kinds of chronic renal failure (CRF): uranyl nitrate (UN) induced fibrosis or 5/6-nephrectomy (5/6NX). Exp Toxicol Pathol 54: 393–399, 2003. doi: 10.1078/0940-2993-00276. [DOI] [PubMed] [Google Scholar]
- 31.Rangarajan S, Rezonzew G, Chumley P, Fatima H, Golovko MY, Feng W, Hua P, Jaimes EA. COX-2-derived prostaglandins as mediators of the deleterious effects of nicotine in chronic kidney disease. Am J Physiol Renal Physiol 318: F475–F485, 2020. doi: 10.1152/ajprenal.00407.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rezonzew G, Chumley P, Feng W, Hua P, Siegal GP, Jaimes EA. Nicotine exposure and the progression of chronic kidney disease: role of the α7-nicotinic acetylcholine receptor. Am J Physiol Renal Physiol 303: F304–F312, 2012. doi: 10.1152/ajprenal.00661.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamamizu K, Shinozaki K, Ayajiki K, Gemba M, Okamura T. Oral administration of both tetrahydrobiopterin and L-arginine prevents endothelial dysfunction in rats with chronic renal failure. J Cardiovasc Pharmacol 49: 131–139, 2007. doi: 10.1097/FJC.0b013e31802f9923. [DOI] [PubMed] [Google Scholar]
- 34.Faraj AH, Morley AR. Remnant kidney pathology after five-sixth nephrectomy in rat. I. A biochemical and morphological study. APMIS 100: 1097–1105, 1992. doi: 10.1111/j.1699-0463.1992.tb04046.x. [DOI] [PubMed] [Google Scholar]
- 35.Yatsu T, Sanagi M, Fujimori A, Tomura Y, Hayashi K, Tanahashi M, Inagaki O. Progression of renal failure with anaemia and multiple effects of angiotensin-converting enzyme inhibitor in rats with renal mass reduction. Pharmacol Res 47: 243–252, 2003. doi: 10.1016/s1043-6618(02)00321-3. [DOI] [PubMed] [Google Scholar]
- 36.Adamczak M, Gross ML, Amann K, Ritz E. Reversal of glomerular lesions involves coordinated restructuring of glomerular microvasculature. J Am Soc Nephrol 15: 3063–3072, 2004. doi: 10.1097/01.ASN.0000146121.72699.86. [DOI] [PubMed] [Google Scholar]
- 37.Jacobi J, Porst M, Cordasic N, Namer B, Schmieder RE, Eckardt KU, Hilgers KF. Subtotal nephrectomy impairs ischemia-induced angiogenesis and hindlimb re-perfusion in rats. Kidney Int 69: 2013–2021, 2006. doi: 10.1038/sj.ki.5000448. [DOI] [PubMed] [Google Scholar]
- 38.Allison ME, Lipham EM, Lassiter WE, Gottschalk CW. The acutely reduced kidney. Kidney Int 3: 354–363, 1973. doi: 10.1038/ki.1973.57. [DOI] [PubMed] [Google Scholar]
- 39.Vaziri ND, Ni Z, Wang XQ, Oveisi F, Zhou XJ. Downregulation of nitric oxide synthase in chronic renal insufficiency: role of excess PTH. Am J Physiol Renal Physiol 274: F642–F649, 1998. doi: 10.1152/ajprenal.1998.274.4.F642. [DOI] [PubMed] [Google Scholar]
- 40.Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, MacLaughlin EJ, Muntner P, Ovbiagele B, Smith SC Jr, Spencer CC, Stafford RS, Taler SJ, Thomas RJ, Williams KA Sr, Williamson JD, Wright JT Jr.. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 71: 1269–1324, 2018. [Erratum in Hypertension 71: e136–e139, 2018 and in Hypertension 72: e33, 2018]. doi: 10.1161/HYP.0000000000000066. [DOI] [PubMed] [Google Scholar]
- 41.Egan BM, Zhao Y, Axon RN. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA 303: 2043–2050, 2010. doi: 10.1001/jama.2010.650. [DOI] [PubMed] [Google Scholar]
- 42.Muntner P, Anderson A, Charleston J, Chen Z, Ford V, Makos G, O'Connor A, Perumal K, Rahman M, Steigerwalt S, Teal V, Townsend R, Weir M, Wright JT Jr;. Chronic Renal Insufficiency Cohort Study Investigators. Hypertension awareness, treatment, and control in adults with CKD: results from the Chronic Renal Insufficiency Cohort (CRIC) study. Am J Kidney Dis 55: 441–451, 2010. doi: 10.1053/j.ajkd.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The collaborative study group. N Engl J Med 329: 1456–1462, 1993. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 44.Maschio G, Alberti D, Janin G, Locatelli F, Mann JF, Motolese M, Ponticelli C, Ritz E, Zucchelli P. Effect of the angiotensin-converting-enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting-Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med 334: 939–945, 1996. doi: 10.1056/NEJM199604113341502. [DOI] [PubMed] [Google Scholar]
- 45.James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, Lackland DT, LeFevre ML, MacKenzie TD, Ogedegbe O, Smith SC Jr, Svetkey LP, Taler SJ, Townsend RR, Wright JT Jr, Narva AS, Ortiz E. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 311: 507–520, 2014. [Erratum in JAMA 311: 1809, 2014]. doi: 10.1001/jama.2013.284427. [DOI] [PubMed] [Google Scholar]
- 46.Vavrinec P, Henning RH, Goris M, Vavrincova-Yaghi D, Buikema H, van Dokkum RP. Vascular smooth muscle function of renal glomerular and interlobar arteries predicts renal damage in rats. Am J Physiol Renal Physiol 303: F1187–F1195, 2012. doi: 10.1152/ajprenal.00653.2011. [DOI] [PubMed] [Google Scholar]
- 47.Vettoretti S, Vavrinec P, Ochodnicky P, Deelman LE, De Zeeuw D, Henning RH, Buikema H. Renal endothelial function is associated with the anti-proteinuric effect of ACE inhibition in 5/6 nephrectomized rats. Am J Physiol Renal Physiol 310: F1047–F1053, 2016. doi: 10.1152/ajprenal.00325.2015. [DOI] [PubMed] [Google Scholar]
- 48.Griffin KA, Picken MM, Bidani AK. Blood pressure lability and glomerulosclerosis after normotensive 5/6 renal mass reduction in the rat. Kidney Int 65: 209–218, 2004. doi: 10.1111/j.1523-1755.2004.00356.x. [DOI] [PubMed] [Google Scholar]
- 49.Meggs LG, Garrick R, Chander P, Ben-Ari J, Gammon D, Goodman AI. Amelioration of systemic hypertension by converting enzyme inhibition in the renal ablation model. Am J Hypertens 1: 190–192, 1988. doi: 10.1093/ajh/1.2.190. [DOI] [PubMed] [Google Scholar]
- 50.Anderson S, Rennke HG, Brenner BM. Therapeutic advantage of converting enzyme inhibitors in arresting progressive renal disease associated with systemic hypertension in the rat. J Clin Invest 77: 1993–2000, 1986. doi: 10.1172/JCI112528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Augustyniak RA, Picken MM, Leonard D, Zhou XJ, Zhang W, Victor RG. Sympathetic nerves and the progression of chronic kidney disease during 5/6 nephrectomy: studies in sympathectomized rats. Clin Exp Pharmacol Physiol 37: 12–18, 2010. doi: 10.1111/j.1440-1681.2009.05253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mackenzie HS, Troy JL, Rennke HG, Brenner BM. TCV 116 prevents progressive renal injury in rats with extensive renal mass ablation. J Hypertens Suppl 12: S11–S16, 1994. [PubMed] [Google Scholar]
- 53.Saud A, Luiz RS, Leite APO, Muller CR, Visona I, Reinecke N, Silva WH, Gloria MA, Razvickas CV, Casarini DE, Schor N. Resistance exercise training ameliorates chronic kidney disease outcomes in a 5/6 nephrectomy model. Life Sci 275: 119362, 2021. doi: 10.1016/j.lfs.2021.119362. [DOI] [PubMed] [Google Scholar]
- 54.Uchida L, Tanaka T, Saito H, Sugahara M, Wakashima T, Fukui K, Nangaku M. Effects of a prolyl hydroxylase inhibitor on kidney and cardiovascular complications in a rat model of chronic kidney disease. Am J Physiol Renal Physiol 318: F388–F401, 2020. doi: 10.1152/ajprenal.00419.2019. [DOI] [PubMed] [Google Scholar]
- 55.Anderson S, Meyer TW, Rennke HG, Brenner BM. Control of glomerular hypertension limits glomerular injury in rats with reduced renal mass. J Clin Invest 76: 612–619, 1985. doi: 10.1172/JCI112013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bidani AK, Polichnowski AJ, Licea-Vargas H, Long J, Kliethermes S, Williamson GA, Griffin KA. BP fluctuations and the real-time dynamics of renal blood flow responses in conscious rats. J Am Soc Nephrol 31: 324–336, 2020. doi: 10.1681/ASN.2019070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Noda M, Matsuo T, Fukuda R, Ohta M, Nagano H, Shibouta Y, Naka T, Nishikawa K, Imura Y. Effect of candesartan cilexetil (TCV-116) in rats with chronic renal failure. Kidney Int 56: 898–909, 1999. doi: 10.1046/j.1523-1755.1999.00614.x. [DOI] [PubMed] [Google Scholar]
- 58.Cao W, Li A, Wang L, Zhou Z, Su Z, Bin W, Wilcox CS, Hou FF. A salt-induced reno-cerebral reflex activates renin-angiotensin systems and promotes CKD progression. J Am Soc Nephrol 26: 1619–1633, 2015. doi: 10.1681/ASN.2014050518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hinojosa-Laborde C, Frohlich BH, Cowley AW Jr.. Whole body autoregulation in reduced renal mass hypertension. Hypertension 20: 659–665, 1992. doi: 10.1161/01.hyp.20.5.659. [DOI] [PubMed] [Google Scholar]
- 60.Ylitalo P, Hepp R, Möhring J, Gross F. Effects of varying sodium intake on blood pressure and renin-angiotensin system in subtotally nephrectomized rats. J Lab Clin Med 88: 807–816, 1976. [PubMed] [Google Scholar]
- 61.Daniels BS, Hostetter TH. Adverse effects of growth in the glomerular microcirculation. Am J Physiol Renal Physiol 258: F1409–F1416, 1990. doi: 10.1152/ajprenal.1990.258.5.F1409. [DOI] [PubMed] [Google Scholar]
- 62.Juncos LI, Martin FL, Baigorria ST, Pasqualini ME, Fiore MC, Eynard AR, Juncos LA, Garcia NH. Atorvastatin improves sodium handling and decreases blood pressure in salt-loaded rats with chronic renal insufficiency. Nutrition 28: e23–e28, 2012. doi: 10.1016/j.nut.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 63.Sampaio-Maia B, Serrao P, Moura M, Pestana M. Jejunal dopamine and Na,K-ATPase activity in early chronic renal insufficiency. Nephrology (Carlton) 11: 63–67, 2006. doi: 10.1111/j.1440-1797.2006.00533.x. [DOI] [PubMed] [Google Scholar]
- 64.Bai F, Makino T, Ono T, Mizukami H. Anti-hypertensive effects of shichimotsukokato in 5/6 nephrectomized Wistar rats mediated by the DDAH-ADMA-NO pathway. J Nat Med 66: 583–590, 2012. doi: 10.1007/s11418-011-0625-8. [DOI] [PubMed] [Google Scholar]
- 65.Bidani AK, Schwartz MM, Lewis EJ. Renal autoregulation and vulnerability to hypertensive injury in remnant kidney. Am J Physiol Renal Physiol 252: F1003–F1010, 1987. doi: 10.1152/ajprenal.1987.252.6.F1003. [DOI] [PubMed] [Google Scholar]
- 66.Bidani AK, Griffin KA, Picken M, Lansky DM. Continuous telemetric blood pressure monitoring and glomerular injury in the rat remnant kidney model. Am J Physiol Renal Physiol 265: F391–F398, 1993. doi: 10.1152/ajprenal.1993.265.3.F391. [DOI] [PubMed] [Google Scholar]
- 67.Tanaka T, Kojima I, Ohse T, Ingelfinger JR, Adler S, Fujita T, Nangaku M. Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab Invest 85: 1292–1307, 2005. doi: 10.1038/labinvest.3700328. [DOI] [PubMed] [Google Scholar]
- 68.Converse RL Jr, Jacobsen TN, Toto RD, Jost CM, Cosentino F, Fouad-Tarazi F, Victor RG. Sympathetic overactivity in patients with chronic renal failure. N Engl J Med 327: 1912–1918, 1992. doi: 10.1056/NEJM199212313272704. [DOI] [PubMed] [Google Scholar]
- 69.Ligtenberg G, Blankestijn PJ, Oey PL, Klein IH, Dijkhorst-Oei LT, Boomsma F, Wieneke GH, van Huffelen AC, Koomans HA. Reduction of sympathetic hyperactivity by enalapril in patients with chronic renal failure. N Engl J Med 340: 1321–1328, 1999. doi: 10.1056/NEJM199904293401704. [DOI] [PubMed] [Google Scholar]
- 70.Yuhara M, Ikeda T, Toya Y, Sakurai J, Gomi T, Ikeda T. Participation of the sympathetic nervous system in hypertension in rats with subtotal renal ablation. J Hypertens 7: 443–446, 1989. doi: 10.1097/00004872-198906000-00002. [DOI] [PubMed] [Google Scholar]
- 71.Paterno JC, Bergamaschi CT, Campos RR, Higa EM, Soares MF, Schor N, Freire AO, Teixeira VP. Electroacupuncture and moxibustion decrease renal sympathetic nerve activity and retard progression of renal disease in rats. Kidney Blood Press Res 35: 355–364, 2012. doi: 10.1159/000336095. [DOI] [PubMed] [Google Scholar]
- 72.Hubbard JW, Buchholz RA, Keeton TK, Nathan MA. Parabrachial lesions increase plasma norepinephrine concentration, plasma renin activity and enhance baroreflex sensitivity in the conscious rat. Brain Res 421: 226–234, 1987. doi: 10.1016/0006-8993(87)91292-3. [DOI] [PubMed] [Google Scholar]
- 73.Jensen LW, Pedersen EB. Nocturnal blood pressure and relation to vasoactive hormones and renal function in hypertension and chronic renal failure. Blood Press 6: 332–342, 1997. doi: 10.3109/08037059709062092. [DOI] [PubMed] [Google Scholar]
- 74.Levine TB, Francis GS, Goldsmith SR, Simon AB, Cohn JN. Activity of the sympathetic nervous system and renin-angiotensin system assessed by plasma hormone levels and their relation to hemodynamic abnormalities in congestive heart failure. Am J Cardiol 49: 1659–1666, 1982. doi: 10.1016/0002-9149(82)90243-0. [DOI] [PubMed] [Google Scholar]
- 75.Robertson D, Frolich JC, Carr RK, Watson JT, Hollifield JW, Shand DG, Oates JA. Effects of caffeine on plasma renin activity, catecholamines and blood pressure. N Engl J Med 298: 181–186, 1978. doi: 10.1056/NEJM197801262980403. [DOI] [PubMed] [Google Scholar]
- 76.Ismail B, deKemp RA, Hadizad T, Mackasey K, Beanlands RS, DaSilva JN. Decreased renal AT1 receptor binding in rats after subtotal nephrectomy: PET study with [18F]FPyKYNE-losartan. EJNMMI Res 6: 55, 2016. doi: 10.1186/s13550-016-0209-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jackson B, Hodsman P, Johnston CI. Changes in the renin-angiotensin system, exchangeable body sodium, and plasma and atrial content of atrial natriuretic factor during evolution of chronic renal failure in the rat. Am J Hypertens 1: 298–300, 1988. doi: 10.1093/ajh/1.3.298. [DOI] [PubMed] [Google Scholar]
- 78.Mu W, Long DA, Ouyang X, Agarwal A, Cruz PE, Roncal CA, Nakagawa T, Yu X, Hauswirth WW, Johnson RJ. Angiostatin overexpression is associated with an improvement in chronic kidney injury by an anti-inflammatory mechanism. Am J Physiol Renal Physiol 296: F145–F152, 2009. doi: 10.1152/ajprenal.90430.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mu W, Ouyang X, Agarwal A, Zhang L, Long DA, Cruz PE, Roncal CA, Glushakova OY, Chiodo VA, Atkinson MA, Hauswirth WW, Flotte TR, Rodriguez-Iturbe B, Johnson RJ. IL-10 suppresses chemokines, inflammation, and fibrosis in a model of chronic renal disease. J Am Soc Nephrol 16: 3651–3660, 2005. doi: 10.1681/ASN.2005030297. [DOI] [PubMed] [Google Scholar]
- 80.Rempel LCT, Faustino VD, Foresto-Neto O, Fanelli C, Arias SCA, Moreira G, Nascimento TF, Avila VF, Malheiros D, Camara NOS, Fujihara CK, Zatz R. Chronic exposure to hypoxia attenuates renal injury and innate immunity activation in the remnant kidney model. Am J Physiol Renal Physiol 317: F1285–F1292, 2019. doi: 10.1152/ajprenal.00367.2018. [DOI] [PubMed] [Google Scholar]
- 81.Tapia E, Sanchez-Lozada LG, Soto V, Manrique AM, Ortiz-Vega KM, Santamaría J, Medina-Campos ON, Cristóbal M, Avila-Casado C, Pedraza-Chaverri J, Rodríguez-Iturbe B, Franco M. Sildenafil treatment prevents glomerular hypertension and hyperfiltration in rats with renal ablation. Kidney Blood Press Res 35: 273–280, 2012. doi: 10.1159/000334952. [DOI] [PubMed] [Google Scholar]
- 82.Huang H, Fan Y, Gao Z, Wang W, Shao N, Zhang L, Yang Y, Zhu W, Chen Z, Hu J, Ding G. HIF-1α contributes to Ang II-induced inflammatory cytokine production in podocytes. BMC Pharmacol Toxicol 20: 59, 2019. doi: 10.1186/s40360-019-0340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol 35: 881–900, 2003. doi: 10.1016/s1357-2725(02)00271-6. [DOI] [PubMed] [Google Scholar]
- 84.Williams B, Baker AQ, Gallacher B, Lodwick D. Angiotensin II increases vascular permeability factor gene expression by human vascular smooth muscle cells. Hypertension 25: 913–917, 1995. doi: 10.1161/01.hyp.25.5.913. [DOI] [PubMed] [Google Scholar]
- 85.Adamczak M, Gross ML, Krtil J, Koch A, Tyralla K, Amann K, Ritz E. Reversal of glomerulosclerosis after high-dose enalapril treatment in subtotally nephrectomized rats. J Am Soc Nephrol 14: 2833–2842, 2003. doi: 10.1097/01.asn.0000095248.91994.d3. [DOI] [PubMed] [Google Scholar]
- 86.Klein IH, Ligtenberg G, Oey PL, Koomans HA, Blankestijn PJ. Enalapril and losartan reduce sympathetic hyperactivity in patients with chronic renal failure. J Am Soc Nephrol 14: 425–430, 2003. doi: 10.1097/01.asn.0000045049.72965.b7. [DOI] [PubMed] [Google Scholar]
- 87.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S, Investigators RS. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345: 861–869, 2001. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 88.Hou FF, Xie D, Zhang X, Chen PY, Zhang WR, Liang M, Guo ZJ, Jiang JP. Renoprotection of Optimal Antiproteinuric Doses (ROAD) study: a randomized controlled study of benazepril and losartan in chronic renal insufficiency. J Am Soc Nephrol 18: 1889–1898, 2007. doi: 10.1681/ASN.2006121372. [DOI] [PubMed] [Google Scholar]
- 89.Hou FF, Zhang X, Zhang GH, Xie D, Chen PY, Zhang WR, Jiang JP, Liang M, Wang GB, Liu ZR, Geng RW. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N Engl J Med 354: 131–140, 2006. doi: 10.1056/NEJMoa053107. [DOI] [PubMed] [Google Scholar]
- 90.Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension 44: 595–601, 2004. doi: 10.1161/01.HYP.0000145180.38707.84. [DOI] [PubMed] [Google Scholar]
- 91.Bidani AK, Griffin KA, Williamson G, Wang X, Loutzenhiser R. Protective importance of the myogenic response in the renal circulation. Hypertension 54: 393–398, 2009. doi: 10.1161/HYPERTENSIONAHA.109.133777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Griffin KA. Hypertensive kidney injury and the progression of chronic kidney disease. Hypertension 70: 687–694, 2017. doi: 10.1161/HYPERTENSIONAHA.117.08314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bidani AK, Polichnowski AJ, Loutzenhiser R, Griffin KA. Renal microvascular dysfunction, hypertension and CKD progression. Curr Opin Nephrol Hypertens 22: 1–9, 2013. doi: 10.1097/MNH.0b013e32835b36c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Parving HH, Kastrup H, Smidt UM, Andersen AR, Feldt-Rasmussen B, Christiansen JS. Impaired autoregulation of glomerular filtration rate in type 1 (insulin-dependent) diabetic patients with nephropathy. Diabetologia 27: 547–552, 1984. doi: 10.1007/BF00276965. [DOI] [PubMed] [Google Scholar]
- 95.Christensen PK, Hansen HP, Parving HH. Impaired autoregulation of GFR in hypertensive non-insulin dependent diabetic patients. Kidney Int 52: 1369–1374, 1997. doi: 10.1038/ki.1997.463. [DOI] [PubMed] [Google Scholar]
- 96.Christensen PK, Hommel EE, Clausen P, Feldt-Rasmussen B, Parving HH. Impaired autoregulation of the glomerular filtration rate in patients with nondiabetic nephropathies. Kidney Int 56: 1517–1523, 1999. doi: 10.1046/j.1523-1755.1999.00676.x. [DOI] [PubMed] [Google Scholar]
- 97.Griffin KA, Bidani AK. Progression of renal disease: renoprotective specificity of renin-angiotensin system blockade. Clin J Am Soc Nephrol 1: 1054–1065, 2006. doi: 10.2215/CJN.02231205. [DOI] [PubMed] [Google Scholar]
- 98.Bidani AK, Mitchell KD, Schwartz MM, Navar LG, Lewis EJ. Absence of glomerular injury or nephron loss in a normotensive rat remnant kidney model. Kidney Int 38: 28–38, 1990. doi: 10.1038/ki.1990.163. [DOI] [PubMed] [Google Scholar]
- 99.Griffin K, Polichnowski A, Licea-Vargas H, Picken M, Long J, Williamson G, Bidani A. Large BP-dependent and -independent differences in susceptibility to nephropathy after nitric oxide inhibition in Sprague-Dawley rats from two major suppliers. Am J Physiol Renal Physiol 302: F173–F182, 2012. doi: 10.1152/ajprenal.00070.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lin JJ, Tonshoff B, Bouriquet N, Casellas D, Kaskel FJ, Moore LC. Insulin-like growth factor-I restores microvascular autoregulation in experimental chronic renal failure. Kidney Int Suppl 67: S195–S198, 1998. doi: 10.1046/j.1523-1755.1998.06745.x. [DOI] [PubMed] [Google Scholar]
- 101.Reddy YS, Kiranmayi VS, Bitla AR, Krishna GS, Rao PV, Sivakumar V. Nitric oxide status in patients with chronic kidney disease. Indian J Nephrol 25: 287–291, 2015. doi: 10.4103/0971-4065.147376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schmidt RJ, Yokota S, Tracy TS, Sorkin MI, Baylis C. Nitric oxide production is low in end-stage renal disease patients on peritoneal dialysis. Am J Physiol Renal Physiol 276: F794–F797, 1999. doi: 10.1152/ajprenal.1999.276.5.F794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brunini TM, Mendes-Ribeiro AC, Ellory JC, Mann GE. Platelet nitric oxide synthesis in uremia and malnutrition: a role for l-arginine supplementation in vascular protection? Cardiovasc Res 73: 359–367, 2007. doi: 10.1016/j.cardiores.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 104.Brunini TM, Moss MB, Siqueira MA, Santos SF, Lugon JR, Mendes-Ribeiro AC. Nitric oxide, malnutrition and chronic renal failure. Cardiovasc Hematol Agents Med Chem 5: 155–161, 2007. doi: 10.2174/187152507780363214. [DOI] [PubMed] [Google Scholar]
- 105.da Silva CD, Brunini TM, Reis PF, Moss MB, Santos SF, Roberts NB, Ellory JC, Mann GE, Mendes-Ribeiro AC. Effects of nutritional status on the l-arginine-nitric oxide pathway in platelets from hemodialysis patients. Kidney Int 68: 2173–2179, 2005. doi: 10.1111/j.1523-1755.2005.00673.x. [DOI] [PubMed] [Google Scholar]
- 106.Mendes Ribeiro AC, Brunini TM, Ellory JC, Mann GE. Abnormalities in l-arginine transport and nitric oxide biosynthesis in chronic renal and heart failure. Cardiovasc Res 49: 697–712, 2001. doi: 10.1016/s0008-6363(00)00267-4. [DOI] [PubMed] [Google Scholar]
- 107.Schmidt RJ, Baylis C. Total nitric oxide production is low in patients with chronic renal disease. Kidney Int 58: 1261–1266, 2000. doi: 10.1046/j.1523-1755.2000.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 339: 572–575, 1992. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- 109.Oliva-Damaso E, Oliva-Damaso N, Rodriguez-Esparragon F, Payan J, Baamonde-Laborda E, Gonzalez-Cabrera F, Santana-Estupinan R, Rodriguez-Perez JC. Asymmetric (ADMA) and symmetric (SDMA) dimethylarginines in chronic kidney disease: a clinical approach. Int J Mol Sci 20: 3668, 2019. doi: 10.3390/ijms20153668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zoccali C, Bode-Boger S, Mallamaci F, Benedetto F, Tripepi G, Malatino L, Cataliotti A, Bellanuova I, Fermo I, Frolich J, Boger R. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet 358: 2113–2117, 2001. doi: 10.1016/s0140-6736(01)07217-8. [DOI] [PubMed] [Google Scholar]
- 111.Erdely A, Wagner L, Muller V, Szabo A, Baylis C. Protection of Wistar furth rats from chronic renal disease is associated with maintained renal nitric oxide synthase. J Am Soc Nephrol 14: 2526–2533, 2003. doi: 10.1097/01.asn.0000086476.48686.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Matsumoto Y, Ueda S, Yamagishi S, Matsuguma K, Shibata R, Fukami K, Matsuoka H, Imaizumi T, Okuda S. Dimethylarginine dimethylaminohydrolase prevents progression of renal dysfunction by inhibiting loss of peritubular capillaries and tubulointerstitial fibrosis in a rat model of chronic kidney disease. J Am Soc Nephrol 18: 1525–1533, 2007. doi: 10.1681/ASN.2006070696. [DOI] [PubMed] [Google Scholar]
- 113.Aiello S, Noris M, Todeschini M, Zappella S, Foglieni C, Benigni A, Corna D, Zoja C, Cavallotti D, Remuzzi G. Renal and systemic nitric oxide synthesis in rats with renal mass reduction. Kidney Int 52: 171–181, 1997. doi: 10.1038/ki.1997.317. [DOI] [PubMed] [Google Scholar]
- 114.Ueda S, Yamagishi S, Matsumoto Y, Kaida Y, Fujimi-Hayashida A, Koike K, Tanaka H, Fukami K, Okuda S. Involvement of asymmetric dimethylarginine (ADMA) in glomerular capillary loss and sclerosis in a rat model of chronic kidney disease (CKD). Life Sci 84: 853–856, 2009. doi: 10.1016/j.lfs.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 115.Szabo AJ, Wagner L, Erdely A, Lau K, Baylis C. Renal neuronal nitric oxide synthase protein expression as a marker of renal injury. Kidney Int 64: 1765–1771, 2003. doi: 10.1046/j.1523-1755.2003.00260.x. [DOI] [PubMed] [Google Scholar]
- 116.Tain YL, Freshour G, Dikalova A, Griendling K, Baylis C. Vitamin E reduces glomerulosclerosis, restores renal neuronal NOS, and suppresses oxidative stress in the 5/6 nephrectomized rat. Am J Physiol Renal Physiol 292: F1404–F1410, 2007. doi: 10.1152/ajprenal.00260.2006. [DOI] [PubMed] [Google Scholar]
- 117.Hiragushi K, Sugimoto H, Shikata K, Yamashita T, Miyatake N, Shikata Y, Wada J, Kumagai I, Fukushima M, Makino H. Nitric oxide system is involved in glomerular hyperfiltration in Japanese normo- and micro-albuminuric patients with type 2 diabetes. Diabetes Res Clin Pract 53: 149–159, 2001. doi: 10.1016/s0168-8227(01)00260-1. [DOI] [PubMed] [Google Scholar]
- 118.Hohenstein B, Hugo CP, Hausknecht B, Boehmer KP, Riess RH, Schmieder RE. Analysis of NO-synthase expression and clinical risk factors in human diabetic nephropathy. Nephrol Dial Transplant 23: 1346–1354, 2007. doi: 10.1093/ndt/gfm797. [DOI] [PubMed] [Google Scholar]
- 119.Yun Z, Yu-Ping Y, Zong-Wu T, Yang S, Fang Y, Fang S. Association of endothelial nitric oxide synthase gene polymorphisms with end-stage renal disease: a systematic review and meta-analysis. Ren Fail 36: 987–993, 2014. doi: 10.3109/0886022X.2014.900601. [DOI] [PubMed] [Google Scholar]
- 120.Zhou TB, Yin SS. Association of endothelial nitric oxide synthase Glu298Asp gene polymorphism with the risk of end-stage renal disease. Ren Fail 35: 573–578, 2013. doi: 10.3109/0886022X.2013.773834. [DOI] [PubMed] [Google Scholar]
- 121.Reyes AA, Purkerson ML, Karl I, Klahr S. Dietary supplementation with l-arginine ameliorates the progression of renal disease in rats with subtotal nephrectomy. Am J Kidney Dis 20: 168–176, 1992. doi: 10.1016/s0272-6386(12)80546-4. [DOI] [PubMed] [Google Scholar]
- 122.Ashab I, Peer G, Blum M, Wollman Y, Chernihovsky T, Hassner A, Schwartz D, Cabili S, Silverberg D, Iaina A. Oral administration of l-arginine and captopril in rats prevents chronic renal failure by nitric oxide production. Kidney Int 47: 1515–1521, 1995. doi: 10.1038/ki.1995.214. [DOI] [PubMed] [Google Scholar]
- 123.Bennett-Richards KJ, Kattenhorn M, Donald AE, Oakley GR, Varghese Z, Bruckdorfer KR, Deanfield JE, Rees L. Oral l-arginine does not improve endothelial dysfunction in children with chronic renal failure. Kidney Int 62: 1372–1378, 2002. doi: 10.1111/j.1523-1755.2002.kid555.x. [DOI] [PubMed] [Google Scholar]
- 124.De Nicola L, Bellizzi V, Minutolo R, Andreucci M, Capuano A, Garibotto G, Corso G, Andreucci VE, Cianciaruso B. Randomized, double-blind, placebo-controlled study of arginine supplementation in chronic renal failure. Kidney Int 56: 674–684, 1999. doi: 10.1046/j.1523-1755.1999.00582.x. [DOI] [PubMed] [Google Scholar]
- 125.Annavarajula SK, Dakshinamurty KV, Naidu MU, Reddy CP. The effect of l-arginine on arterial stiffness and oxidative stress in chronic kidney disease. Indian J Nephrol 22: 340–346, 2012. doi: 10.4103/0971-4065.103907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wolf SC, Erley CM, Kenner S, Berger ED, Risler T. Does l-arginine alter proteinuria and renal hemodynamics in patients with chronic glomerulonephritis and hypertension. Clin Nephrol 43: S42–S46, 1995. [PubMed] [Google Scholar]
- 127.Griffin KA, Bidani AK, Ouyang J, Ellis V, Churchill M, Churchill PC. Role of endothelium-derived nitric oxide in hemodynamic adaptations after graded renal mass reduction. Am J Physiol Regul Integr Comp Physiol 264: R1254–R1259, 1993. doi: 10.1152/ajpregu.1993.264.6.R1254. [DOI] [PubMed] [Google Scholar]
- 128.Kimura K, Tojo A, Hirata Y, Matsuoka H, Sugimoto T. Morphometric analysis of renal arterioles in subtotally nephrectomized rats. J Lab Clin Med 122: 273–283, 1993. [PubMed] [Google Scholar]
- 129.Hill GS, Heudes D, Jacquot C, Gauthier E, Bariety J. Morphometric evidence for impairment of renal autoregulation in advanced essential hypertension. Kidney Int 69: 823–831, 2006. doi: 10.1038/sj.ki.5000163. [DOI] [PubMed] [Google Scholar]
- 130.Fine LG, Orphanides C, Norman JT. Progressive renal disease: the chronic hypoxia hypothesis. Kidney Int Suppl 65: S74–S78, 1998. [PubMed] [Google Scholar]
- 131.Advani A, Connelly KA, Yuen DA, Zhang Y, Advani SL, Trogadis J, Kabir MG, Shachar E, Kuliszewski MA, Leong-Poi H, Stewart DJ, Gilbert RE. Fluorescent microangiography is a novel and widely applicable technique for delineating the renal microvasculature. PLoS One 6: e24695, 2011. doi: 10.1371/journal.pone.0024695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kang DH, Hughes J, Mazzali M, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model. II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. J Am Soc Nephrol 12: 1448–1457, 2001. doi: 10.1681/ASN.V1271448. [DOI] [PubMed] [Google Scholar]
- 133.Kang DH, Kanellis J, Hugo C, Truong L, Anderson S, Kerjaschki D, Schreiner GF, Johnson RJ. Role of the microvascular endothelium in progressive renal disease. J Am Soc Nephrol 13: 806–816, 2002. doi: 10.1681/ASN.V133806. [DOI] [PubMed] [Google Scholar]
- 134.Floege J, Burns MW, Alpers CE, Yoshimura A, Pritzl P, Gordon K, Seifert RA, Bowen-Pope DF, Couser WG, Johnson RJ. Glomerular cell proliferation and PDGF expression precede glomerulosclerosis in the remnant kidney model. Kidney Int 41: 297–309, 1992. doi: 10.1038/ki.1992.42. [DOI] [PubMed] [Google Scholar]
- 135.Hohenstein B, Colin M, Foellmer C, Amann KU, Brekken RA, Daniel C, Hugo CP. Autocrine VEGF-VEGF-R loop on podocytes during glomerulonephritis in humans. Nephrol Dial Transplant 25: 3170–3180, 2010. doi: 10.1093/ndt/gfq200. [DOI] [PubMed] [Google Scholar]
- 136.Risdon RA, Sloper JC, de Wardener HE. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet 292: 363–366, 1968. doi: 10.1016/S0140-6736(68)90589-8. [DOI] [PubMed] [Google Scholar]
- 137.Choi YJ, Chakraborty S, Nguyen V, Nguyen C, Kim BK, Shim SI, Suki WN, Truong LD. Peritubular capillary loss is associated with chronic tubulointerstitial injury in human kidney: altered expression of vascular endothelial growth factor. Hum Pathol 31: 1491–1497, 2000. doi: 10.1053/hupa.2000.20373. [DOI] [PubMed] [Google Scholar]
- 138.Kida Y, Tchao BN, Yamaguchi I. Peritubular capillary rarefaction: a new therapeutic target in chronic kidney disease. Pediatr Nephrol 29: 333–342, 2014. doi: 10.1007/s00467-013-2430-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Adam RJ, Paterson MR, Wardecke L, Hoffmann BR, Kriegel AJ. Functionally essential tubular proteins are lost to urine-excreted, large extracellular vesicles during chronic renal insufficiency. Kidney360 1: 1107–1117, 2020. doi: 10.34067/kid.0001212020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lee LK, Meyer TW, Pollock AS, Lovett DH. Endothelial cell injury initiates glomerular sclerosis in the rat remnant kidney. J Clin Invest 96: 953–964, 1995. doi: 10.1172/JCI118143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Daniel C, Wiede J, Krutzsch HC, Ribeiro SMF, Roberts DD, Murphy-Ullrich JE, Hugo C. Thrombospondin-1 is a major activator of TGF-β in fibrotic renal disease in the rat in vivo. Kidney Int 65: 459–468, 2004. doi: 10.1111/j.1523-1755.2004.00395.x. [DOI] [PubMed] [Google Scholar]
- 142.Zeisberg M, Tampe B, LeBleu V, Tampe D, Zeisberg EM, Kalluri R. Thrombospondin-1 deficiency causes a shift from fibroproliferative to inflammatory kidney disease and delays onset of renal failure. Am J Pathol 184: 2687–2698, 2014. doi: 10.1016/j.ajpath.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ferrara N. Role of vascular endothelial growth factor in the regulation of angiogenesis. Kidney Int 56: 794–814, 1999. doi: 10.1046/j.1523-1755.1999.00610.x. [DOI] [PubMed] [Google Scholar]
- 144.Kim YG, Suga SI, Kang DH, Jefferson JA, Mazzali M, Gordon KL, Matsui K, Breiteneder-Geleff S, Shankland SJ, Hughes J, Kerjaschki D, Schreiner GF, Johnson RJ. Vascular endothelial growth factor accelerates renal recovery in experimental thrombotic microangiopathy. Kidney Int 58: 2390–2399, 2000. doi: 10.1046/j.1523-1755.2000.00422.x. [DOI] [PubMed] [Google Scholar]
- 145.Masuda Y, Shimizu A, Mori T, Ishiwata T, Kitamura H, Ohashi R, Ishizaki M, Asano G, Sugisaki Y, Yamanaka N. Vascular endothelial growth factor enhances glomerular capillary repair and accelerates resolution of experimentally induced glomerulonephritis. Am J Pathol 159: 599–608, 2001. doi: 10.1016/S0002-9440(10)61731-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hugo C, Kang DH, Johnson RJ. Sustained expression of thrombospondin-1 is associated with the development of glomerular and tubulointerstitial fibrosis in the remnant kidney model. Nephron 90: 460–470, 2002. doi: 10.1159/000054735. [DOI] [PubMed] [Google Scholar]
- 147.Bailey E, Bottomley MJ, Westwell S, Pringle JH, Furness PN, Feehally J, Brenchley PE, Harper SJ. Vascular endothelial growth factor mRNA expression in minimal change, membranous, and diabetic nephropathy demonstrated by non-isotopic in situ hybridisation. J Clin Pathol 52: 735–738, 1999. doi: 10.1136/jcp.52.10.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Namikoshi T, Satoh M, Horike H, Fujimoto S, Arakawa S, Sasaki T, Kashihara N. Implication of peritubular capillary loss and altered expression of vascular endothelial growth factor in IgA nephropathy. Nephron Physiol 102: p9–p16, 2006. doi: 10.1159/000088405. [DOI] [PubMed] [Google Scholar]
- 149.Olszewska-Pazdrak B, Hein TW, Olszewska P, Carney DH. Chronic hypoxia attenuates VEGF signaling and angiogenic responses by downregulation of KDR in human endothelial cells. Am J Physiol Cell Physiol 296: C1162–C1170, 2009. doi: 10.1152/ajpcell.00533.2008. [DOI] [PubMed] [Google Scholar]
- 150.Hohenstein B, Daniel C, Hausknecht B, Boehmer K, Riess R, Amann KU, Hugo CP. Correlation of enhanced thrombospondin-1 expression, TGF-β signalling and proteinuria in human type-2 diabetic nephropathy. Nephrol Dial Transplant 23: 3880–3887, 2008. doi: 10.1093/ndt/gfn399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shvetsov M, Ivanov AA, Kuznetsova AV, Popova OP, Rameeva AS. [Molecular factors of angiogenesis in renal tissue of patients with chronic glomerulonephritis: association with nephrosclerosis and anemia]. Ter Arkh 81: 14–19, 2009. [PubMed] [Google Scholar]
- 152.Thompson EM, Highes J, VAN Noorden S, Sharpe J, Savill JOHN. Expression of the multifunctional extracellular matrix protein thrombospondin in crescentic glomerulonephritis. J Pathol 178: 89–94, 1996. doi:. [DOI] [PubMed] [Google Scholar]
- 153.Carrero JJ, Hecking M, Chesnaye NC, Jager KJ. Sex and gender disparities in the epidemiology and outcomes of chronic kidney disease. Nat Rev Nephrol 14: 151–164, 2018. doi: 10.1038/nrneph.2017.181. [DOI] [PubMed] [Google Scholar]
- 154.Chang PY, Chien LN, Lin YF, Wu MS, Chiu WT, Chiou HY. Risk factors of gender for renal progression in patients with early chronic kidney disease. Medicine (Baltimore) 95: e4203, 2016. doi: 10.1097/MD.0000000000004203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Clotet S, Riera M, Pascual J, Soler MJ. RAS and sex differences in diabetic nephropathy. Am J Physiol Renal Physiol 310: F945–F957, 2016. doi: 10.1152/ajprenal.00292.2015. [DOI] [PubMed] [Google Scholar]
- 156.Coggins CH, Lewis JB, Caggiula AW, Castaldo LS, Klahr S, Wang SR. Differences between women and men with chronic renal disease. Nephrol Dial Transplant 13: 1430–1437, 1998. doi: 10.1093/ndt/13.6.1430. [DOI] [PubMed] [Google Scholar]
- 157.Jafar TH, Schmid CH, Stark PC, Toto R, Remuzzi G, Ruggenenti P, Marcantoni C, Becker G, Shahinfar S, De Jong PE, De Zeeuw D, Kamper AL, Strangaard S, Levey AS. The rate of progression of renal disease may not be slower in women compared with men: a patient-level meta-analysis. Nephrol Dial Transplant 18: 2047–2053, 2003. doi: 10.1093/ndt/gfg317. [DOI] [PubMed] [Google Scholar]
- 158.Kruse NT, You Z, Moreau K, Kendrick J, Jalal D. Sex differences in endothelial function in chronic kidney disease. Am J Physiol Renal Physiol 319: F33–F40, 2020. doi: 10.1152/ajprenal.00156.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Neugarten J, Acharya A, Silbiger SR. Effect of gender on the progression of nondiabetic renal disease: a meta-analysis. J Am Soc Nephrol 11: 319–329, 2000. doi: 10.1681/ASN.V112319. [DOI] [PubMed] [Google Scholar]
- 160.O'Hare AM, Choi AI, Bertenthal D, Bacchetti P, Garg AX, Kaufman JS, Walter LC, Mehta KM, Steinman MA, Allon M, McClellan WM, Landefeld CS. Age affects outcomes in chronic kidney disease. J Am Soc Nephrol 18: 2758–2765, 2007. doi: 10.1681/ASN.2007040422. [DOI] [PubMed] [Google Scholar]
- 161.Rahman M, Xie D, Feldman HI, Go AS, He J, Kusek JW, Lash J, Miller ER 3rd, Ojo A, Pan Q, Seliger SL, Steigerwalt S, Townsend RR; CRIC Study Investigators. Association between chronic kidney disease progression and cardiovascular disease: results from the CRIC study. Am J Nephrol 40: 399–407, 2014. doi: 10.1159/000368915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Seppi T, Prajczer S, Dorler MM, Eiter O, Hekl D, Nevinny-Stickel M, Skvortsova I, Gstraunthaler G, Lukas P, Lechner J. Sex differences in renal proximal tubular cell homeostasis. J Am Soc Nephrol 27: 3051–3062, 2016. doi: 10.1681/ASN.2015080886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Silbiger SR, Neugarten J. The impact of gender on the progression of chronic renal disease. Am J Kidney Dis 25: 515–533, 1995. doi: 10.1016/0272-6386(95)90119-1. [DOI] [PubMed] [Google Scholar]
- 164.Staessen J, Bulpitt CJ, Fagard R, Lijnen P, Amery A. The influence of menopause on blood pressure. J Hum Hypertens 3: 427–433, 1989. [PubMed] [Google Scholar]
- 165.Cobo G, Hecking M, Port FK, Exner I, Lindholm B, Stenvinkel P, Carrero JJ. Sex and gender differences in chronic kidney disease: progression to end-stage renal disease and haemodialysis. Clin Sci (Lond) 130: 1147–1163, 2016. doi: 10.1042/CS20160047. [DOI] [PubMed] [Google Scholar]
- 166.Verhave JC, Hillege HL, Burgerhof JG, Navis G, de Zeeuw D, de Jong PE; PREVEND Study Group. Cardiovascular risk factors are differently associated with urinary albumin excretion in men and women. J Am Soc Nephrol 14: 1330–1335, 2003. doi: 10.1097/01.asn.0000060573.77611.73. [DOI] [PubMed] [Google Scholar]
- 167.Chino Y, Samukawa Y, Sakai S, Nakai Y, Yamaguchi J, Nakanishi T, Tamai I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm Drug Dispos 35: 391–404, 2014. doi: 10.1002/bdd.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Antus B, Hamar P, Kokeny G, Szollosi Z, Mucsi I, Nemes Z, Rosivall L. Estradiol is nephroprotective in the rat remnant kidney. Nephrol Dial Transplant 18: 54–61, 2003. doi: 10.1093/ndt/18.1.54. [DOI] [PubMed] [Google Scholar]
- 169.Gorodeski GI. Impact of the menopause on the epidemiology and risk factors of coronary artery heart disease in women. Exp Gerontol 29: 357–375, 1994. doi: 10.1016/0531-5565(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 170.Hinojosa-Laborde C, Lange DL, Haywood JR. Role of female sex hormones in the development and reversal of dahl hypertension. Hypertension 35: 484–489, 2000. doi: 10.1161/01.hyp.35.1.484. [DOI] [PubMed] [Google Scholar]
- 171.Hutchens MP, Fujiyoshi T, Komers R, Herson PS, Anderson S. Estrogen protects renal endothelial barrier function from ischemia-reperfusion in vitro and in vivo. Am J Physiol Renal Physiol 303: F377–F385, 2012. doi: 10.1152/ajprenal.00354.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kannel WB, Hjortland MC, McNamara PM, Gordon T. Menopause and risk of cardiovascular disease: the Framingham study. Ann Intern Med 85: 447–452, 1976. doi: 10.7326/0003-4819-85-4-447. [DOI] [PubMed] [Google Scholar]
- 173.Muller V, Losonczy G, Heemann U, Vannay A, Fekete A, Reusz G, Tulassay T, Szabo AJ. Sexual dimorphism in renal ischemia-reperfusion injury in rats: possible role of endothelin. Kidney Int 62: 1364–1371, 2002. doi: 10.1111/j.1523-1755.2002.kid590.x. [DOI] [PubMed] [Google Scholar]
- 174.Park YJ, Kim JM. Klotho and postmenopausal hormone replacement therapy in women with chronic kidney disease. J Menopausal Med 24: 75–80, 2018. doi: 10.6118/jmm.2018.24.2.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Reckelhoff JF, Granger JP. Role of androgens in mediating hypertension and renal injury. Clin Exp Pharmacol Physiol 26: 127–131, 1999. doi: 10.1046/j.1440-1681.1999.02996.x. [DOI] [PubMed] [Google Scholar]
- 176.Rogers JL, Mitchell AR, Maric C, Sandberg K, Myers A, Mulroney SE. Effect of sex hormones on renal estrogen and angiotensin type 1 receptors in female and male rats. Am J Physiol Regul Integr Comp Physiol 292: R794–R799, 2007. doi: 10.1152/ajpregu.00424.2006. [DOI] [PubMed] [Google Scholar]
- 177.Speroff L. Postmenopausal hormone therapy into the 21st century. Int J Gynaecol Obstet 59, Suppl 1: S3–S10, 1997. doi: 10.1016/s0020-7292(97)90193-4. [DOI] [PubMed] [Google Scholar]
- 178.Stehman-Breen CO, Gillen D, Gipson D. Prescription of hormone replacement therapy in postmenopausal women with renal failure. Kidney Int 56: 2243–2247, 1999. doi: 10.1046/j.1523-1755.1999.00793.x. [DOI] [PubMed] [Google Scholar]
- 179.Suzuki H, Kondo K. Chronic kidney disease in postmenopausal women. Hypertens Res 35: 142–147, 2012. doi: 10.1038/hr.2011.155. [DOI] [PubMed] [Google Scholar]
- 180.Szekacs B, Vajo Z, Varbiro S, Kakucs R, Vaslaki L, Acs N, Mucsi I, Brinton EA. Postmenopausal hormone replacement improves proteinuria and impaired creatinine clearance in type 2 diabetes mellitus and hypertension. BJOG 107: 1017–1021, 2000. doi: 10.1111/j.1471-0528.2000.tb10406.x. [DOI] [PubMed] [Google Scholar]
- 181.Yanes LL, Sartori-Valinotti JC, Reckelhoff JF. Sex steroids and renal disease: lessons from animal studies. Hypertension 51: 976–981, 2008. doi: 10.1161/HYPERTENSIONAHA.107.105767. [DOI] [PubMed] [Google Scholar]
- 182.Zeier M, Gafter U, Ritz E. Renal function and renal disease in males or females—vive la petite difference. Nephrol Dial Transplant 13: 2195–2198, 1998. doi: 10.1093/oxfordjournals.ndt.a027910. [DOI] [PubMed] [Google Scholar]
- 183.Melloni C, Berger JS, Wang TY, Gunes F, Stebbins A, Pieper KS, Dolor RJ, Douglas PS, Mark DB, Newby LK. Representation of women in randomized clinical trials of cardiovascular disease prevention. Circ Cardiovasc Qual Outcomes 3: 135–142, 2010. doi: 10.1161/CIRCOUTCOMES.110.868307. [DOI] [PubMed] [Google Scholar]
- 184.Sullivan JC, Gillis EE. Sex and gender differences in hypertensive kidney injury. Am J Physiol Renal Physiol 313: F1009–F1017, 2017. doi: 10.1152/ajprenal.00206.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bucholz EM, Krumholz HM. Women in clinical research: what we need for progress. Circ Cardiovasc Qual Outcomes 8: S1–S3, 2015. doi: 10.1161/CIRCOUTCOMES.115.001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mannon EC, Ray SC, Ryan MJ, Sullivan JC. Does sex matter?: an update on the implementation of sex as a biological variable in research. Am J Physiol Renal Physiol 318: F329–F331, 2020. doi: 10.1152/ajprenal.00575.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Deng A, Baylis C. Glomerular hemodynamic responses to pregnancy in rats with severe reduction of renal mass. Kidney Int 48: 39–44, 1995. doi: 10.1038/ki.1995.264. [DOI] [PubMed] [Google Scholar]
- 188.Katoh T, Takahashi K, Klahr S, Reyes AA, Badr KF. Dietary supplementation with L-arginine ameliorates glomerular hypertension in rats with subtotal nephrectomy. J Am Soc Nephrol 4: 1690–1694, 1994. doi: 10.1681/ASN.V491690. [DOI] [PubMed] [Google Scholar]
- 189.Fleck C, Appenroth D, Jonas P, Koch M, Kundt G, Nizze H, Stein G. Suitability of 5/6 nephrectomy (5/6NX) for the induction of interstitial renal fibrosis in rats–influence of sex, strain, and surgical procedure. Exp Toxicol Pathol 57: 195–205, 2006. doi: 10.1016/j.etp.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 190.Bleiler RE, Schedl HP. Creatinine excretion: variability and relationships to diet and body size. J Lab Clin Med 59: 945–955, 1962. [PubMed] [Google Scholar]
- 191.Jones CA, McQuillan GM, Kusek JW, Eberhardt MS, Herman WH, Coresh J, Salive M, Jones CP, Agodoa LY. Serum creatinine levels in the US population: third National Health and Nutrition Examination Survey. Am J Kidney Dis 32: 992–999, 1998. [Erratum in Am J Kidney Dis 35: 178, 2000]. doi: 10.1016/s0272-6386(98)70074-5. [DOI] [PubMed] [Google Scholar]
- 192.Appenroth D, Beutinger R, Lupp A, Fleck C. Effects of a therapy with losartan and quinaprilat on the progression of chronic renal failure in rats after a single dose of uranyl nitrate or 5/6 nephrectomy. Exp Toxicol Pathol 54: 359–366, 2003. doi: 10.1078/0940-2993-00271. [DOI] [PubMed] [Google Scholar]
- 193.Bai Y, Sigala W, Adams GR, Vaziri ND. Effect of exercise on cardiac tissue oxidative and inflammatory mediators in chronic kidney disease. Am J Nephrol 29: 213–221, 2009. doi: 10.1159/000156715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fleck C, Gräfe K, Kart I. Renal handling of amino acids in 5/6-nephrectomized rats: stimulation of renal amino acid reabsorption after treatment with triiodothyronine or dexamethasone under amino acid load. Amino Acids 16: 149–164, 1999. doi: 10.1007/BF01321533. [DOI] [PubMed] [Google Scholar]
- 195.Li C, Xiao Q, Gu C, Zhao Y, Liu L, Wang H. Hepatocyte growth factor inhibits renal angiotensin II expression in 5/6 nephrectomized rats. Ann Clin Lab Sci 50: 578–583, 2020. [PubMed] [Google Scholar]
- 196.Christensen PK, Akram K, Konig KB, Parving HH. Autoregulation of glomerular filtration rate in patients with type 2 diabetes during isradipine therapy. Diabetes Care 26: 156–162, 2003. doi: 10.2337/diacare.26.1.156. [DOI] [PubMed] [Google Scholar]