

Abstract
Concentric pulmonary vascular wall thickening due partially to increased pulmonary artery (PA) smooth muscle cell (PASMC) proliferation contributes to elevating pulmonary vascular resistance (PVR) in patients with pulmonary hypertension (PH). Although pulmonary vasoconstriction may be an early contributor to increasing PVR, the transition of contractile PASMCs to proliferative PASMCs may play an important role in the development and progression of pulmonary vascular remodeling in PH. A rise in cytosolic Ca2+ concentration ([Ca2+]cyt) is a trigger for PASMC contraction and proliferation. Here, we report that upregulation of Piezo1, a mechanosensitive cation channel, is involved in the contractile-to-proliferative phenotypic transition of PASMCs and potential development of pulmonary vascular remodeling. By comparing freshly isolated PA (contractile PASMCs) and primary cultured PASMCs (from the same rat) in a growth medium (proliferative PASMCs), we found that Piezo1, Notch2/3, and CaSR protein levels were significantly higher in proliferative PASMCs than in contractile PASMCs. Upregulated Piezo1 was associated with an increase in expression of PCNA, a marker for cell proliferation, whereas downregulation (with siRNA) or inhibition (with GsMTx4) of Piezo1 attenuated PASMC proliferation. Furthermore, Piezo1 in the remodeled PA from rats with experimental PH was upregulated compared with PA from control rats. These data indicate that PASMC contractile-to-proliferative phenotypic transition is associated with the transition or adaptation of membrane channels and receptors. Upregulated Piezo1 may play a critical role in PASMC phenotypic transition and PASMC proliferation. Upregulation of Piezo1 in proliferative PASMCs may likely be required to provide sufficient Ca2+ to assure nuclear/cell division and PASMC proliferation, contributing to the development and progression of pulmonary vascular remodeling in PH.
Keywords: mechanosensitive channel, phenotypic transition, Piezo1, pulmonary hypertension, smooth muscle cell
INTRODUCTION
The transition or switch of smooth muscle cells (SMCs) from a contractile phenotype to a synthetic or proliferative phenotype in blood vessels is associated with abnormal vascular remodeling and vascular lesions observed in multiple vascular diseases (1–5). Sustained pulmonary vasoconstriction, concentric pulmonary vascular remodeling, and occlusive vascular lesions are the major causes for the elevated pulmonary vascular resistance (PVR) and pulmonary arterial pressure (PAP) in patients with pulmonary arterial hypertension (PAH) and animals with experimental pulmonary hypertension (PH). Pulmonary vasoconstriction due to pulmonary artery (PA) smooth muscle cell (PASMC) contraction is potentially an early cause for the elevated PVR and PAP in patients with PAH, whereas concentric pulmonary vascular wall thickening and arteriole muscularization due to, at least in part, PASMC proliferation (and migration) become predominant pathogenic mechanisms in the development and progression of severe PH. In some patients with PAH, obliterative vascular lesions (e.g., plexiform and neointimal lesions) result in distal and proximal pulmonary artery (PA) occlusion further contributing to the increased PVR and PAP (6–9).
PASMC is the major cell type in the media of proximal and distal PA. PASMC contraction with precisely controlled excitation-contraction coupling is the major cause for sustained pulmonary vasoconstriction in patients with PAH and animals with experimental PH. Abnormal PASMC proliferation is known to be one of the major contributors to the concentric PA wall thickening or medial hypertrophy (10–13), pulmonary arteriole, and precapillary muscularization (14–17), and concentric laminar intimal lesions and plexiform lesions (9). In patients with PAH with occlusive vascular lesions, histological and pathological examination also found ample PASMCs in the obliterative lesions in the proximal and distal PAs (7, 9, 12, 18) indicating that highly proliferative PASMCs also contribute to the formation and progression of occlusive lesions. It has been demonstrated that SMCs undergo phenotypical transitions during vascular development, vascular injury repair, and pathological vascular remodeling in patients with cardiovascular diseases (5, 19–22). We (23, 24) and others (3) reported that the contractile-to-proliferative phenotypical transition in vascular SMCs was associated with significant changes in the expression and function of membrane receptors and ion channels. It is possible that the contractile-to-proliferative phenotypical transition of vascular SMCs or PASMCs is also a process of membrane receptor and ion channel “remodeling” or adaptation. The reorganization or rearrangement of different ion channels and membrane receptors is part of the transition that enables vascular SMCs and PASMCs to proliferate in response to serum and growth factors.
Piezo1 is a mechanosensitive cation channel responsible for sensing physical forces including stretch, touch, flow, and pressure; its role in mechanical sensing in various tissues and cells has been well described (25–28). During SMC proliferation, the proliferating cells in the cell cycle undergo many physical changes including mitosis and division in which the force applied to the plasma membrane varies dramatically (29). It is, however, unknown whether Piezo1 (25), along with other receptors and channels, is required for or associated with the PASMC contractile-to-proliferative phenotypic transition and whether physical stress or stretch reinforced during cell mitosis and division serves as a trigger for activating Piezo1 to bring in more Ca2+ to assure progression of the cell mitosis and division. In this study, we aimed to further identify the cation channels and membrane receptors that undergo “remodeling” or adaptation (e.g., transcriptional and/or translational upregulation and/or downregulation) during PASMC contractile-to-proliferative phenotypic transition and define whether mechanosensitive channels (e.g., Piezo1) are involved in the transition and in the development and progression of experimental PH.
MATERIALS AND METHODS
Animal Experiments
All experimental protocols on animals were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, San Diego (UCSD). For the generation of monocrotaline (MCT)-induced PH in rats, we used 6- to 8-wk-old Sprague–Dawley rats (males) that were randomly divided into two groups: 1) control group and 2) MCT-injected group. For the MCT group, the rats were injected subcutaneously with a single dose of MCT (60 mg/kg, Sigma, Cat. No. C2401). For the control group, the rats were injected with a single dose of vehicle (dissolving solution) subcutaneously. MCT was dissolved in 0.5 N HCl into a concentration of 200 mg/mL. It was then neutralized with 0.5 N NaOH to a pH of 7.4. Finally, it was diluted with sterilized water to generate a final concentration of 10 mg/mL. All animals resided in the UCSD Vivarium. Food and water were provided at will. The rats were utilized for hemodynamic measurement and phenotypic characteristic studies 4 wk after being injected with MCT. For pulmonary hemodynamics measurement in rats, the catheter (SPR-513, 2F, ADInstruments) was inserted into the right ventricle (RV) through the external right jugular vein for observing the right ventricular pressure (RVP) that was recorded and analyzed by the PowerLab data acquisition system (ADInstruments) and the LabChart software (ADInstruments) (30, 31). After measuring the hemodynamics, the entire heart was dissected and weighed to calculate the Fulton index.
For the development of PH in mice induced by Nω-nitro-l-arginine methyl ester (l-NAME) (a cell-permeable nitric oxide synthase inhibitor) (32–35) and Yoda1 (a selective activator of Piezo1 channels) (28, 36–38), the 8-wk-old mice (males) were randomly divided into four groups: 1) control group; 2) Yoda1 group; 3) l-NAME group; and 4) l-NAME and Yoda1 group. For the Yoda1 group, the mice were injected intraperitoneally (ip) with Yoda1 (1 mg/kg, everyday; Sigma, Cat. No. SML1558). For the l-NAME group, the mice were administered with l-NAME that was dissolved in drinking water (0.1%, Cayman Chemical, Cat. No. 80210) and injected intraperitoneally with vehicle. For the l-NAME and Yoda1 group, the mice were administered with l-NAME in drinking water (0.1%) and injected intraperitoneally with Yoda1 (1 mg/kg, everyday). For the control group, the mice were injected intraperitoneally (ip) with vehicle (DMSO). All animals were housed in the UCSD Vivarium. The mice were utilized for hemodynamic measurement and phenotypic characteristic studies 6 wk after being injected with Yoda1 and administered with l-NAME. For pulmonary hemodynamics measurement in mice, the catheter (PVR1030, 1F, ADInstruments) was used as described in the previous section. The mean pulmonary arterial pressure (mPAP) was calculated using the following formula: mPAP = 0.61 × right ventricular systolic pressure (RVSP) + 2 (in mmHg) (39, 40). The mean systemic arterial pressure (SAP) was calculated using the following formula: mean SAP = (systolic SAP + diastolic SAP × 2) ÷ 3 (in mmHg).
To assess RV hypertrophy in rats and mice, we measured the Fulton Index using the isolated heart after hemodynamic measurement. First, the heart was dissected from the animal. Next, both the left and the right atria were separated. The RV and the rest of the heart tissues, including the left ventricle (LV) and the septum (S) were carefully separated and weighed immediately. The ratio of the weight of the RV to the weight of the LV and S [RV/(LV + S)] was then calculated and utilized to access RV hypertrophy, which also referred to Fulton Index.
Ex Vivo Lung Angiogram
Lung angiogram was utilized to evaluate the pulmonary vascular density and remodeling as described previously (41). First, heparin was injected into the RV to prevent congelation in the pulmonary vessel immediately after right heart catheterization. Then, PBS was perfused through the polyethylene tube inserted into the main PA to wash out vestigial blood within the pulmonary vessel. Next, the casting polymer (MV-122, Flow-Tech, Inc., Carver, MA) was perfused into the PA using a pump (NE-1000, New Era Pump Systems, Inc., Farmingdale, NY) at a speed of 0.05 mL/min for 1–2 min until the casting polymer was infiltrated into all the branches of the pulmonary arteries. The casting polymer-filled lungs were stored at 4°C overnight and then immersed in ethanol (50%, 70%, 80%, 90%, and 100% for 1 h in succession) to dehydrate the lung tissues. The dehydrated lungs were then immersed in methyl salicylate solution (Sigma, Cat. No. M6752) placed on a shaker overnight to hyalinization of the surrounding tissues around the Microfil-casted pulmonary vasculature to display the vascular structure. Finally, the transparent and dehydrated lung filled with polymers was photographed with a digital camera (MU1000, FMA050, Amscope, CA) through a microscope (WILD M651, Leica, Switzerland) with a ×45 objective. The pixel size is 3,584 width × 2,748 height. The apical, middle, and basal regions of the peripheral area of the lung vascular images were selected with Photoshop software and converted to binary images using National Institutes of Health (NIH) ImageJ software for quantification of the total length of the vascular branches of the lungs (mm/mm2), the number of vascular branches (mm−2), and the number of vascular branch junctions (mm−2) at a given area (mm2). The initial lung images from different groups of animals were collected and processed on the same day and under the same conditions.
Isolation of Intrapulmonary Arteries and Preparation of Primary Cultured PASMCs
Isolation of rat PAs was performed as previously described (23). The isolated lungs and heart were submerged in Hanks’ balanced salt solution (HBSS). First, the fat and connective tissues were removed, and intrapulmonary arterial branches were isolated under a magnified microscope. Then, the adventitia and the endothelium were cautiously removed from the PA with fine forceps under the microscope. The freshly isolated PA was used as contractile PASMCs for extracting protein. The PA smooth muscle tissues from the same rat were used to perform primary cultured PASMCs. The PA smooth muscle segment was dissolved in HBSS supplemented with 1,750 U/mL of Type I collagenase (Sigma, Cat. No. C-0130), 9.5 U/mL of papain (Sigma, Cat. No. P-4762), 2 mg/mL bovine serum albumin (BSA, Sigma, Cat. No. A-2058), and 0.14 mg/mL dithiothreitol (DTT, Sigma, Cat. No. D-9779) at 37°C for 20 min followed by gentle blowing with a Pasteur pipette to disconnect the cells. The digestion was then stopped by adding 10 mL of smooth muscle growth medium (Lifeline Cell Technologies, Cat. No. LL-0014) with 10% fetal bovine serum (FBS). The cell suspension was centrifuged, and the supernatant was discarded. The pellet was resuspended in an SMC growth medium containing 10% FBS and transferred onto cell culture dish. The cells were cultured in 5% CO2 incubator at 37°C. The primary cultured PASMCs, which were initially prepared from the same rat from which we obtained freshly isolated PA (as contractile PASMCs), were used as proliferative PASMCs at days 3–5.
Culture of Human PASMCs
Normal human PASMCs were purchased from Lonza (Cat. No. CC-2581, Alps, Switzerland). The 4th-5th passages of cells were first cultured in 10% FBS medium (Cat. No. LL-0014, Lifeline Cell Technology) in 10-cm Petri dishes in a 37°C-incubator equilibrated with 5% CO2. The cells were subcultured by trypsinization with 0.05% trypsin EDTA (Thermo Fisher Scientific) when 80%–90% confluence was reached and then plated in 6-mm Petri dishes or 6-well Petri dishes. The medium was changed 24 h after the initial plating and then every other day.
Transfection of Small Interfering RNA
The human PASMCs were transfected with control small interfering RNA (siRNA) (siNT) or siRNA for Piezo1 (siPiezo1, Cat. No. sc-93227, Santa Cruz Biotechnology, TX) using the RNAiMax transfection reagent (Cat. No. 13778075, Invitrogen) for 72 h. Then, the cells transfected with siNT or siPiezo1 were incubated in 10% FBS medium for 48 h before Western blot and immunocytochemistry experiments were performed to determine changes in PCNA, YES-associated protein (YAP), AKT kinase (or protein kinase B), and mammalian target of rapamycin (mTOR), and the nuclear translocation of YAP.
Western Blot Experiment
Rat whole lung tissue, freshly isolated PA tissue, primary cultured rat PASMCs, and cultured human PASMC were used for Western blot experiments using standard protocol (23, 24). Briefly, the tissue and cell were lysed with cold radio-immunoprecipitation cell lysis (RIPA) lysis buffer (Thermo Fisher Scientific, Cat. No. 89901) supplemented with protease inhibitor cocktail (Roche, Cat. No. 11836153001) and centrifugated at 4°C (12,000 g for 15 min). The supernatant was collected and used to measure protein concentration. Protein samples were loaded to SDS-PAGE and transferred onto 0.45-µm nitrocellulose membranes. Then the membranes were blocked in 0.1% Tween 20 in 1× Tris-buffered saline (TBS) (TBST) with 5% BSA for 1 h at 24°C followed by the incubation with primary antibodies in 5% BSA-TBST on the rocker for 12 h at 4°C. Membranes were then washed and incubated with secondary antibody for 1 h at 24°C in TBST. Membranes were washed again, and the blots were developed with enhanced chemiluminescence substrate (Cat. No. RPN2232, Amersham, Cytiva). The primary antibodies used in this study included anti-SMα2-actin (ACTA2, 1:1,000, Cat. No. ab7817, Abcam), anti-myosin heavy chain 11 (MHC11, (1:1,000, Cat. No. sc-376157, Santa Cruz Biotechnology), anti-transgelin (TAGLN, 1:1,000, Cat. No. ab14106, Abcam), anti-PCNA (1:1,000, Cat. No. sc-25280, Santa Cruz Biotechnology), anti-Piezo1 (1:1,000, Cat. No. 15939-1-AP, Proteintech), anti-CaSR (1:1,000, Cat. No. ab137408, Abcam), anti-Notch2 (1:1,000, Cat. No. 5732, Cell Signaling Technology), anti-Notch3 (1:1,000, Cat. No. 2889, Cell Signaling Technology), anti-F-actin (1:1,000, Cat. No. ab205, Abcam), anti- budding uninhibited by benzimidazole-related 1 (BUBR1, 1:1,000, Cat. No. ab28193, Abcam), anti-Calpain1 (1:1,000, Cat. No. 2556, Cell Signaling Technology), anti-Calpain2 (1:1,000, Cat. No. 2539, Cell Signaling Technology), anti-pYAP (1:1,000, Cat. No. 4911, Cell Signaling Technology), anti-YAP (1:1,000, Cat. No. 4912, Cell Signaling Technology), anti-pAKT (1:1,000, Cat. No. 4060, Cell Signaling Technology), anti-AKT (1:1,000, Cat. No. 4691, Cell Signaling Technology), anti-pmTOR (1:1,000, Cat. No. 2971, Cell Signaling Tech), anti-mTOR (1:1,000, Cat. No. 2983, Cell Signaling Technology, Inc.), anti-Caveolin1 (1:1,000, Cat. No. sc-70516, Santa Cruz Biotechnology), anti-GAPDH (1:1,000, Cat. No. ab8245, Abcam), and anti-Actin (1:1,000, Cat. No. 4968, Cell Signaling Technology). The secondary antibodies used for the experiments included anti-mouse (1:2,000, Cat. No. 7076S, Cell Signaling Technology) or anti-rabbit (1:2,000, Cat. No. 7074S, Cell Signaling). The band intensity was quantified with ImageJ and normalized to pan-actin or GAPDH as a loading control. For quantifying the target protein expression or protein level, we first measured the band intensity of the target protein and then divided it by the band intensity of loading control (e.g., Aβ-actin or GAPDH); the ratio is defined as the level of the target protein (as arbitrary unit). Then, we further normalized the values (the ratio of the target band intensity to the loading control band intensity) in different experimental groups by the values in the control group; these data are presented in the figures as “normalized” values to controls. Most of the antibodies used in this study, for example, anti-PCNA, anti-CaSR, anti-Notch2, anti-Notch3, anti-Calpain1, anti-Calpain2, anti-pYAP, anti-YAP, anti-pAKT, anti-AKT, anti-pmTOR, anti-mTOR, and anti-Caveolin1 antibodies, were validated by the experiments showing that the siRNA knockdown decreased the band intensity. The anti-Piezo1 antibody was validated by comparing Piezo1 protein level in control human embryonic kidney (HEK) cells and Piezo1-transfected HEK cells.
Immunocytochemistry Experiment
Primary cultured rat and human PASMCs grown on the glass bottom cell culture dish (Cat. No. 801002, Pipette) were fixed in 4% paraformaldehyde at +4°C for 15 min and then washed three times with PBS and permeabilized with 0.1% Triton X-100 in PBS for 15 min. Unspecific binding was reduced by blocking with 5% BSA (Cat. No. A3059, Sigma) in PBS for 60 min. The PASMCs were then incubated with primary antibody at +4°C. Twenty-four hours later, the cells were incubated with secondary antibody at 24°C for 2 h. Nuclei were stained with DAPI. Fluorescent images were acquired using a confocal microscope (Laser Scanning Microscopes 880, Zeiss, Oberkochen, Germany) with a T-PMT confocal detector and a ×63/1.40 oil Microscope objective (Plan-Apochromat, ZEISS). The image size was 135 μm × 135 μm. The pixel size was 0.13 μm. The excitation and emission wavelengths were 405 nm and 460 nm, respectively, for DAPI fluorescence measurement. The primary antibodies used in this study included anti-ACTA2 (or anti-α-SMA) antibody (1:100, Cat. No. ab7817, Abcam) and anti-YAP antibody (1:100, Cat. No. 4912, Cell Signaling Technology). The secondary antibodies used in this study included anti-mouse (1:500, Alexa Fluor 488, Cat. No. 4408S, Cell Signaling Technology) and anti-rabbit (1:500, Alexa Flour 488, Cat. No. 4412S, Cell Signaling Technology) antibodies. The excitation and emission wavelengths were 488 nm and 519 nm, respectively, for Alexa 488 fluorescence measurement. We used ImageJ to first measure the averaged fluorescence intensity of the region of interest (ROI) (e.g., the cytoplasmic or nuclear area) of each cell. Then, we calculated the ratio of the fluorescence intensity of the nuclear area to the fluorescence intensity of the cytoplasm to indicate the nuclear translocation of the target proteins. We used similar methods for quantitating the ratio of the nuclear level to the cytoplasmic level that are previously published by other investigators (42–47). The initial fluorescent images from cells cultured in 0.3% FBS and 10% FBS media with siNT and siPiezo1 were collected and processed at the same time under the same conditions.
[Ca2+]Cyt Measurement
Digital imaging fluorescence microscopy and Fura-2, a ratiometric Ca2+ indicator, were utilized to determine [Ca2+]cyt in human and rat PASMCs grown on glass coverslips. Cells were first loaded with Fura-2 by incubation in media containing 5 µM Fura-2-acetoxymethyl ester (Fura-2/AM, Cat. No. F1201, Invitrogen) at 37°C for 40 min. Then, the cells were superfused with HEPES-buffered solution for 10–30 min to wash out extracellular Fura-2/AM and allow intracellular esterase to cleave acetoxymethyl ester from Fura-2/AM. The Fura-2-loaded cells were then placed in the perfusion chamber on the stage of an inverted fluorescence microscope (DMi8, Leica, Germany) with a microscope objective lens [Cat. No. 506365, HC PL FLUOTAR (340) ×40/1,30 OIL, Leica, Germany] and a filter cube set for Fura-2 (Cat. No. 11525348) that includes the excitation filter (bandpass: 340–380 nm) and the emission filter (bandpass: HQ 540/50 m) and illuminated at 340- and 380-nm wavelengths with a Mercury lamp (X-Cite 200 DC, Excelitas Canada Inc., QC, Canada). The emitted fluorescence at 510 nm was captured by a fluorescent camera (Leica DFC9000 GT, Germany) and the ratio of fluorescence intensity (F340 and F380) was recorded every 5 s using the LAS X Synapse software. The ratio (F340/F380) was then used to calculate [Ca2+]cyt (48). PASMCs were first superfused with HEPES-buffered solution for 5 min, and then perfused with HEPES-buffered solution containing 5-µm Yoda1 for 10–15 min. All the experiments for measuring [Ca2+]cyt were performed at room temperature (22°C–24°C).
Cell Proliferation Assay
Change in number of viable cells before (0 h) and after (48 h) incubation of cells in different media was measured to determine PASMC proliferation. First, human PASMCs (at passages of 4–5) were evenly distributed in 96-well plates containing 10% fetal bovine serum (FBS) media with growth factors (growth medium). When the cells grew to ∼60%–70% confluence, the growth medium containing 10% FBS was changed to 0.3% FBS medium (for 24 h) to synchronize the cells in the G0/G1 phases (49). The synchronized cells were then split into six groups: 0.3% FBS group, 0.3% FBS + Yoda1 (0.3 μM) group, 0.3% FBS + Yoda1 (0.3 μM) + EGTA (2 mM) group, 5% FBS group, 5% FBS + Yoda1 (0.3 μM) group, and 5% FBS + Yoda1 (0.3 μM) + EGTA (2 mM) group. The cells were treated for 48 h with the media mentioned earlier. Then, 10 µL of CCK8 solution (Cell Counting Kit-8, Cat. No. K1018, ApexBio, Houston, TX) was added into each well of the two groups of cells incubated for 2 h as basal controls. The absorbance at 450 nm was measured with a microplate reader (iMark, Bio-Rad Laboratories, Hercules, CA) for 2 h to illustrate the basal number of viable cells. Furthermore, a new set of primary cultured rat PASMCs from PA were evenly distributed in 96-well plates and incubated for 12 h to enable cells to attach to the plates. Then, the cells were split into three groups: 5% FBS group, 5% FBS with Yoda1 (0.3 μM)-treated group, and 5% FBS with GsMTx4 (1 μM)-treated group. The cells were treated for 48 h. We examined cell proliferation by measuring the number of viable cells using Cell Counting Kit 8 (CCK8) and the DNA synthesis or BrdU DNA incorporation. To measure the viable cell number, CCK8 solution was added to each well (for 2 h) and then absorbance at 450 nm or optical density at 450 nm (OD450) was measured with a microplate reader (iMark, Bio-Rad Laboratories, Hercules, CA). To measure BrdU incorporation, we used the BrdU Cell Proliferation ELISA Kit (Cat. No. ab126556, Abcam). BrdU reagent (20 µL/well) was first added into 96-well plates at the end of 4-h incubation. Fixing solution (200 µL/well, for 30 min) was used to fix the cells and denature the DNA at room temperature. Then, the anti-BrdU monoclonal detector antibody (100 µL/well, for 1 h) was added to the cells, followed by addition of the Peroxidase Goat Anti-Mouse IgG Conjugate (100 μL/well, for 30 min) and incubation of the cells with the 3,3′,5,5′-tetramethylbenzidine (TMB) peroxidase substrate (100 µL/well, for 30 min) in the dark. Stop solution (100 µL/well) was used to stop the reaction. The optical density at 450 nm or the absorbance at 450 nm was then measured with a microplate reader (iMark, Bio-Rad Laboratories, Hercules, CA).
Drugs and Chemicals
Yoda1 (Sigma, Cat. No. SML1558), a selective activator of Piezo1 channels (28, 36, 38), and GsMTx4 (MedChemExpress, Cat. No. HY-P1410A), a blocker of Piezo1 channels (50–53), were dissolved into DMSO and sterilized water, respectively, to make stock solutions. The stock solutions were then aliquoted and kept frozen at −80°C. Aliquots of the stock solutions were then used to make the final solutions to treat cells at the time when the experiments were conducted.
Statistical Analysis
Data are expressed as means ± standard deviation (SD) with the number (n) of individual biological replicates. Statistical significance was analyzed by Student’s t test (paired or unpaired as applicable) between two groups or by one-way ANOVA (Holm–Sidak method) between many groups using the software SigmaPlot. A value of P < 0.05 was considered as statistically significant. Significant difference is indicated in the figures or figure legends as *P < 0.05, **P < 0.01, and ***P < 0.001 or #P < 0.05, ##P < 0.01, and ###P < 0.001.
RESULTS
Upregulation of Piezo1 during the Contractile-to-Proliferative Phenotypic Transition of PASMC
We used freshly isolated PA, in which the endothelium was functionally removed, as the contractile PASMC, and primary cultured PASMC incubated in growth media containing 10% FBS and growth factors as synthetic or proliferative PASMC (Fig. 1A). First, we measured the protein level of the markers for contractile SMCs [e.g., MHC11, ACTA2, and TAGLN) and the marker for cell proliferation, PCNA and Piezo1 in freshly isolated PA (contractile PASMCs) and primary cultured PASMCs (proliferative PASMCs). As shown in Fig. 1, B and C, freshly isolated PA exhibited significantly higher expression levels of MHC11, ACTA2, and TAGLN than in primary cultured PASMCs; whereas primary cultured PASMCs exhibited significantly higher expression of PCNA. More importantly, the protein level of Piezo1 in primary cultured PASMCs (proliferative PASMCs) was significantly higher than in freshly isolated PA (contractile PASMCs). The upregulated Piezo1 was positively associated with the increased PCNA in proliferating PASMCs and inversely associated with the protein levels of the contractile markers (e.g., MHC11, ACTA2, and TAGLN) in contractile PASMCs (Fig. 1, B and C). In addition to upregulated Piezo1 channels in synthetic or proliferative PASMCs compared with contractile PASMCs (freshly isolated PA), we also observed upregulation of CaSR and Notch2/3 in proliferative PASMCs (Fig. 1, D and E). Since the primary cultured PASMCs were initially prepared from freshly isolated PA of the same rats, these data indicate that Piezo1, CaSR, and Notch2/3 are upregulated during the contractile-to-proliferative phenotypic transition of PASMCs.
Figure 1.
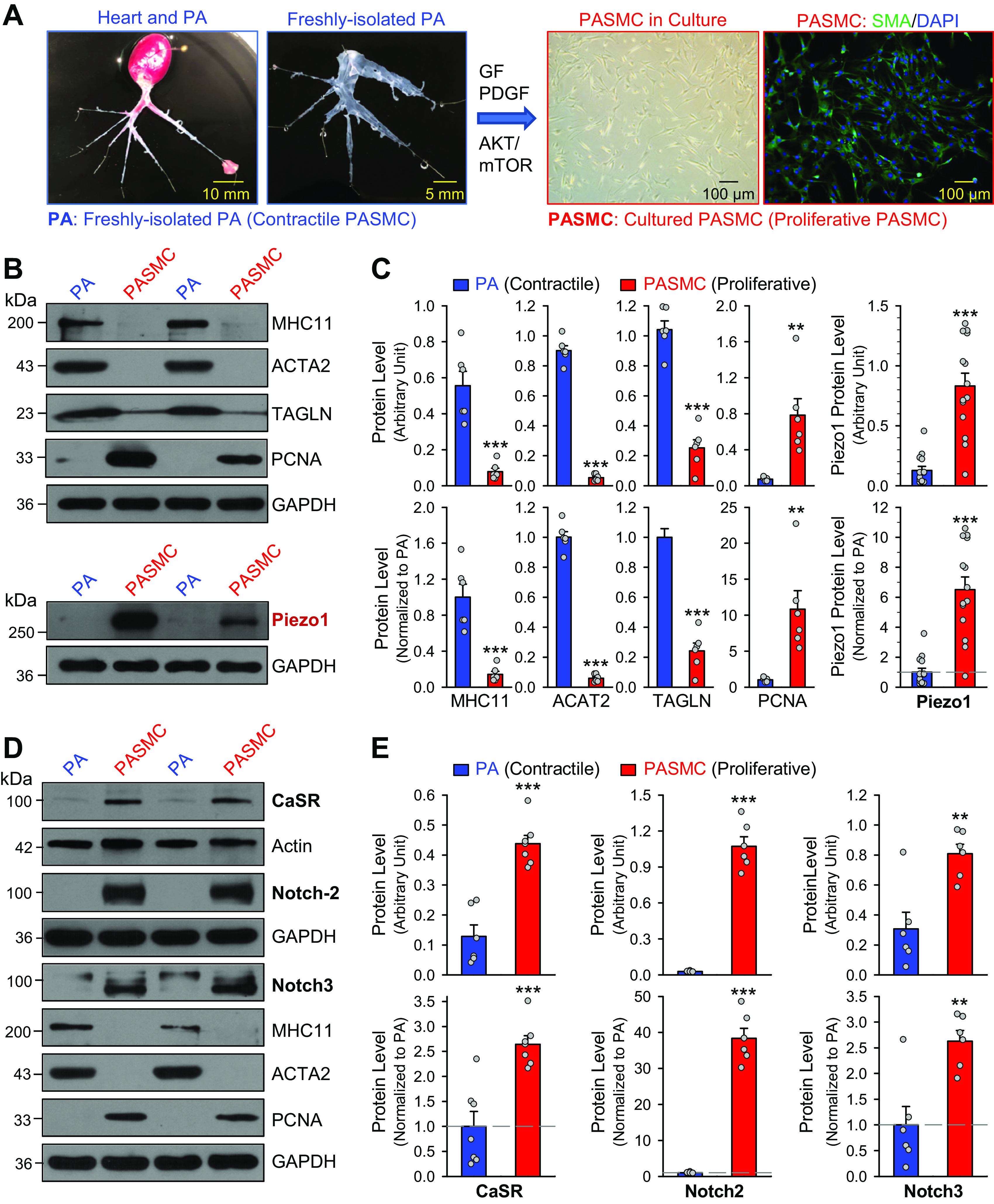
Upregulation of the Piezo1 channel and CaSR and Notch receptors in proliferative pulmonary artery smooth muscle cells (PASMCs) during the contractile-to-proliferative phenotypical transition. A: freshly isolated pulmonary artery (PA) branches from the rat lung (left) and primary cultured PASMCs derived from the same rat in medium containing serum and growth factors (right). A phase-contrast image and a fluorescent image showing DAPI (4′,6-diamidino-2-phenylindole, in blue) and α-smooth muscle actin (SMA, in green) in primary cultured PASMCs are shown. B: Western blot analyses on the contractile smooth muscle cell (SMC) markers, myosin heavy chain 11 (MHC11), SMα2-actin (ACTA2) and SM22-α or transgelin (TAGLN), and the cell proliferation marker, proliferation cell nuclear antigen (PCNA), as well as Piezo1 in freshly isolated PA (mainly containing contractile PASMC) and primary cultured PASMCs (mainly containing proliferative PASMCs). C: summarized data (means ± SE, n = 6–14 in each group) showing protein levels of MHC11, ACTA2, TAGLN, and PCNA (left) and Piezo1 (right) in freshly isolated PA [PA (contractile PASMCs)] and primary cultured PASMCs [PASMCs (proliferative PASMCs)]. Data are shown as arbitrary units (top) and values normalized to PA tissue (bottom). The horizontal dotted line in the lower panel of the Piezo1 graph depicts the level of protein in the PA. **P < 0.01, ***P < 0.001 vs. PA (contractile PASMCs) (unpaired t test). D: Western blot analyses on CaSR, Notch-2, and Notch3, as well as MHC11, ACTA2, and PCNA in freshly isolated PA (mainly containing contractile PASMCs) and primary cultured PASMCs (mainly containing proliferative PASMCs). E: summarized data (means ± SE) showing protein expression level of CaSR (n = 7), Notch2 (n = 6), and Notch3 (n = 6) in the PA and in the PASMCs. Data are presented as arbitrary units (top) or values normalized to PA tissue (bottom). The horizontal broken lines indicate the level of the CaSR, Notch2, and Notch3 proteins in the PA. **P < 0.01, ***P < 0.001 vs. PA (contractile PASMC) (unpaired t test).
Upregulation of Proteins Associated with Cell Proliferation during the Contractile-to-Proliferative Phenotypic Transition of PASMC
The next set of experiments was designed to examine and compare the protein levels of F-actin, a filamentous actin that is part of a linear polymer microfilament essential for mobility and contraction of cells during cell division (54, 55), BUBR1 (budding uninhibited by benzimidazole-related 1), a kinase of the cell cycle serving as the mitotic checkpoint for spindle assembly (56), calpain, a Ca2+-dependent nonlysosomal cysteine protease involved in regulating cell differentiation and proliferation (57), and YAP, a transcriptional coactivator involved in promoting cell proliferation (58–60) in contractile and proliferating PASMCs. Using the same ex vivo model, we found that F-actin, BUBR1, and calpain-1/-2 were all upregulated in proliferating PASMCs compared with contractile PASMCs (Fig. 2, A and B). Since YAP is reported to be involved in mechanosensitive signaling pathways involved in PH (58–60), we also examined and compared the level of YAP in freshly isolated PA and primary cultured PASMCs. The protein level of YAP, along with its phosphorylated form (pYAP), was significantly greater in synthetic or proliferative PASMCs than in contractile PASMCs (Fig. 2, C and D). The contractile-to-proliferative phenotypic transition of PASMC seems to be associated with an upregulated F-actin, calpain1/2, and BUBR1, as well as phosphorylated YAP and YAP. These observations suggest that, due to upregulated Piezo1 channels and, potentially, enhanced Ca2+ influx through membrane stretch-activated Piezo1 channels, the Ca2+-sensitive and/or mechanosensitive signaling pathways are more active in proliferative PASMCs than in contractile PASMCs.
Figure 2.
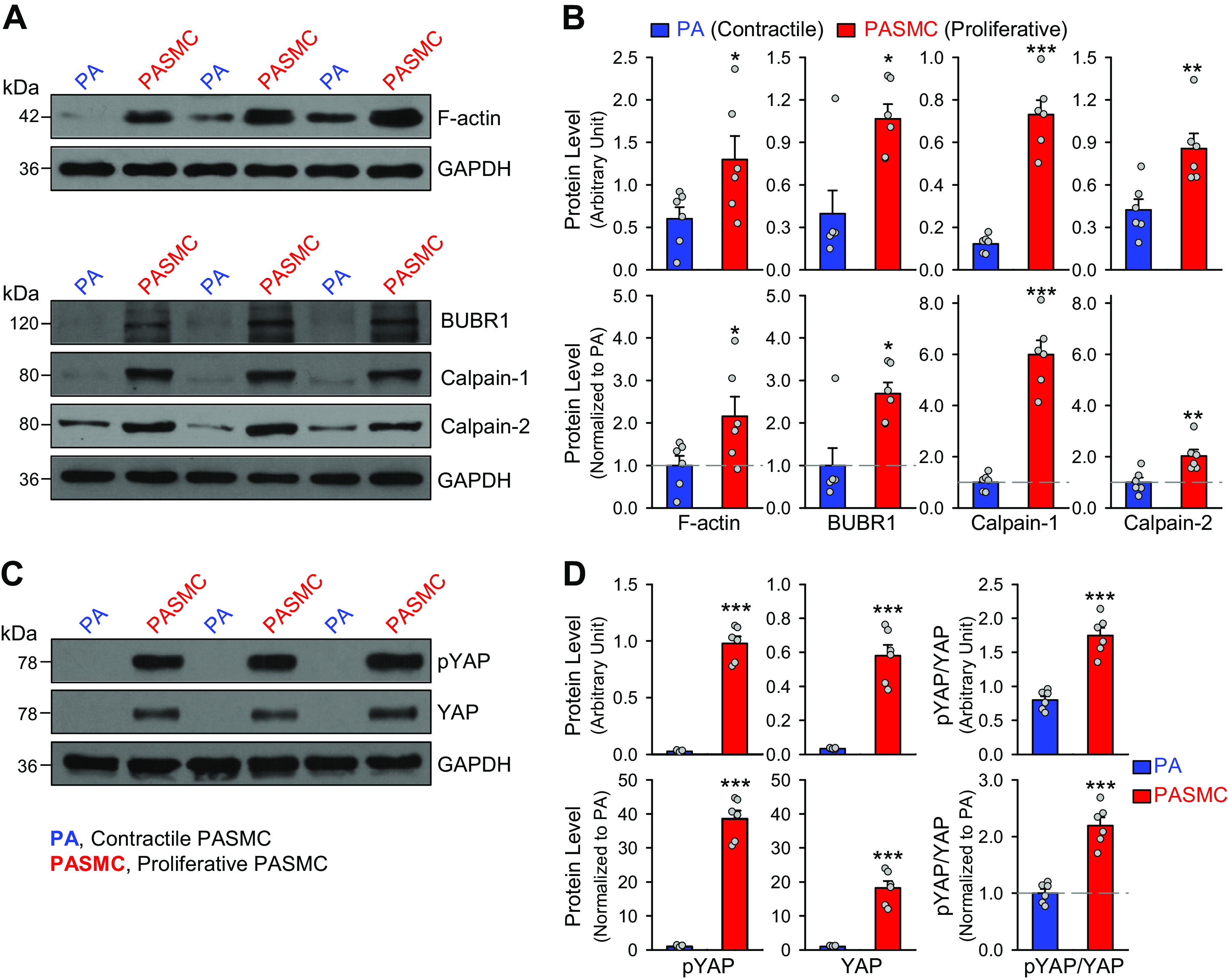
Upregulation of F-actin, budding uninhibited by benzimidazole-related 1 (BUBR1), calpains, and YES-associated protein (YAP) in proliferative pulmonary artery smooth muscle cells (PASMCs) during the contractile-to-proliferative phenotypical transition. A: representative Western blot analyses on F-actin, BUBR1, calpain-1, and calpain-2 in freshly isolated pulmonary artery (PA) (mainly containing contractile PASMCs) and primary cultured PASMCs (mainly containing proliferative PASMCs). GAPDH was used as a loading control. B: summarized data (means ± SE, n = 6) showing protein levels of F-actin, BURBR1, calpain-1, and calpain-2 in freshly isolated PA and primary cultured PASMCs. Data are presented as arbitrary units (top) and values normalized to PA tissue (bottom). The horizontal broken lines in the bottom panels indicate the level of the proteins in the freshly isolated PA. C: representative Western blot analysis on phosphorylated (p) YES-associated protein 1 (pYAP1) and total YAP1 in freshly isolated PA (mainly containing contractile PASMCs) and primary cultured PASMCs (mainly containing proliferative PASMC). GAPDH was used as a loading control. D: summarized data (means ± SE, n = 6) showing protein expression levels of pYAP1, YAP1, and the ratio of pYAP/YAP in the PA and in the PASMCs. Data are shown as arbitrary units (top) and values normalized to PA tissue (bottom). The horizontal broken lines indicate the level of the proteins in the PA. *P < 0.05, **P < 0.01, ***P < 0.001 vs. PA (unpaired t test).
Upregulated Piezo1 and Ca2+ Influx through Piezo1 Channels Are Involved in PASMC Proliferation
Compared with freshly isolated PA, the primary cultured PASMC or proliferative PASMCs exhibited a significantly higher expression level of Piezo1, along with the mechanosensitive signaling protein YAP. When the proliferative PASMCs were growth-arrested by incubating in 0.3% FBS media without growth factors, the expression level of Piezo1 was significantly reduced compared with the proliferative PASMCs cultured in 10% FBS medium (Fig. 3, A and B). The growth arrest-associated downregulation in Piezo1 was associated with a significant decrease in protein levels of PCNA, a cell proliferation marker, and YAP (Fig. 3, A and B). The growth arrest-associated decrease of YAP was correlated with the decrease of pYAP, although phosphorylation of YAP inhibits nuclear translocation of YAP to transcriptionally activate YAP/YAZ-sensitive genes associated with cell proliferation and differentiation (60–62). These data suggest that Piezo1 expression is positively associated with PASMC proliferation determined by upregulation of PCNA and YAP. The correlated decrease in pYAP in growth-arrested PASMCs (incubated in 0.3% FBS medium) or increase in pYAP in proliferating PASMCs (incubated in 10% FBS medium) are possibly related to a compensatory mechanism resulting in phosphorylation to retain YAP in the cytoplasm during PASMC proliferation. Nonetheless, a large amount of YAP could still be translocated into the nucleus in proliferating PASMCs (Fig. 3, A and B).
Figure 3.
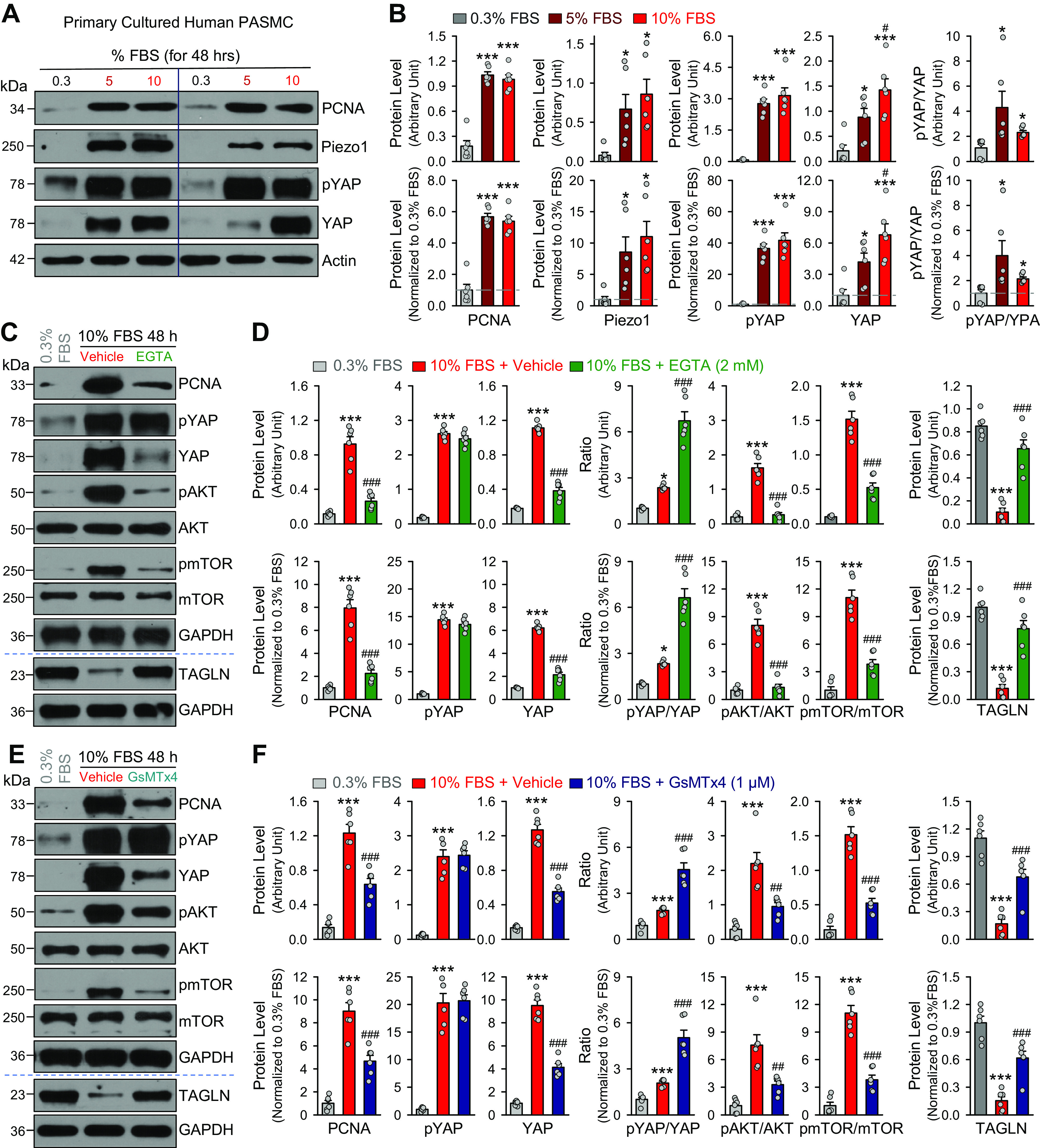
Serum starvation inhibits pulmonary artery smooth muscle cell (PASMC) proliferation, downregulates Piezo1 and decreases YES-associated protein (YAP) level in proliferative PASMCs and Ca2+ influx through Piezo1 channels is required for growth medium-induced PASMC proliferation and increases in YAP signaling and AKT/mTOR signaling. A: Western blot analyses on the cell proliferation marker, proliferation cell nuclear antigen (PCNA), phosphorylated (pYAP1), and total YAP (YAP) in primary cultured PASMCs incubated (for 48 h) in 0.3%, 5%, and 10% fetal bovine serum (FBS)-containing media. Actin was used as a loading control. B: summarized data (means ± SE, n = 6 experiments) showing protein levels of PCNA, pYAP1, YAP1, and Piezo1, along with the ratio of pYAP1/YAP1, in primary cultured PASMCs incubated in culture media containing 0.3%, 5%, and 10% FBS. Data are presented as arbitrary units (top) and values normalized to pulmonary artery (PA) tissue (bottom). *P < 0.05, ***P < 0.001 vs. 0.3% FBS; #P < 0.05 vs. 5% FBS. C: Western blot analyses on PCNA, pYAP, YAP, pAKT, AKT, pmTOR, mTOR, and TAGLN (transgelin) in quiescent PASMCs (0.3% FBS) and proliferating PASMCs incubated (for 48 h) in 10% FBS media with vehicle or EGTA (2 mM), a Ca2+ chelator. GAPDH was used as a loading control. D: summarized data (means ± SE, n = 6 experiments) showing protein levels of PCNA, pYAP, and YAP, along with the ratio of pYAP/YAP, pAKT/AKT, pmTOR/mTOR, and TAGLN in primary cultured PASMCs incubated in 0.3% FBS media and 10% FBS media with vehicle or EGTA. Data are presented as arbitrary units (top) and values normalized to 0.3% FBS media (bottom). *P < 0.05, ***P < 0.001 vs. 0.3% FBS; #P < 0.05 vs. 10%% FBS + vehicle (one-way ANOVA/Holm–Sidak). E: Western blot analyses on PCNA, pYAP, YAP, pAKT, AKT, pmTOR, mTOR, and TAGLN in quiescent PASMCs incubated in 0.3% FBS media and proliferating PASMCs incubated (for 48 h) in 10% FBS media with vehicle or GsmTx4 (1 µM), a Piezo1 channel blocker. F: summarized data (means ± SE, n = 6 experiments) showing protein levels of PCNA, pYAP, and YAP, along with the ratio of pYAP/YAP, pAKT/AKT, pmTOR/mTOR, and TAGLN in PASMCs incubated in 0.3% FBS media and 10% FBS media with vehicle or GsMTx4. Data are presented as arbitrary units (top) and values normalized to 0.3% FBS media (bottom). ***P < 0.001 vs. 0.3% FBS; ##P < 0.01, ###P < 0.001 vs. 10%% FBS + vehicle (one-way ANOVA/Holm–Sidak).
We also conducted a series of pharmacological experiments to examine whether Ca2+ influx through the upregulated Piezo1 was involved in the enhanced PASMC proliferation through increased YAP and enhanced AKT/mTOR signaling. Chelation of extracellular free Ca2+ with 2 mM EGTA, an aminopolycarboxylic acid with a higher affinity for Ca2+ than Mg2+ (63), significantly inhibited 10% FBS media-mediated increases in PCNA, a marker for cells in early G1 phase and S phase of the cell cycle, as well as the ratios of pAKT/AKT and pmTOR/mTOR in human PASMCs (Fig. 3, C and D). Inhibition of Ca2+ influx by chelating extracellular free Ca2+ also inhibited the 10% FBS-mediated increase in YAP but had little effect on the 10% FBS-mediated increase in phosphorylated YAP (pYAP) (Fig. 3, C and D). The EGTA-mediated increase in the ratio of pYAP/YAP in PASMCs incubated in 10% FBS was associated with a significant decrease in the ratio of pAKT/AKT and pmTOR/mTOR (Fig. 3, C and D). These data imply that extracellular free Ca2+ or Ca2+ influx through cation channels in the plasma membrane is required for the growth media-mediated PASMC proliferation (increased PCNA) via YAP and AKT/mTOR signaling pathways.
GsMTx4 is a spider venom peptide (34 aa) that blocks mechanosensitive cation channels such as Piezo1 (50, 52). Treatment of PASMCs incubated in 10% FBS media with GsMTx4 significantly inhibited the growth media-induced increases in PCNA and YAP, as well as the ratios of pAKT/AKT and pmTOR/mTOR (Fig. 3, E and F). Block of Piezo1 channels by GsMTx4 (1 µM) also inhibited the 10% FBS-mediated decrease in the ratio of pYAP/YAP, or further enhanced the ratio of pYAP/YAP (Fig. 3, E and F). The GsMTX4-induced increase in the ratio of pYAP/YAP in PASMCs incubated in 10% FBS was associated with a significant decrease in the ratio of pAKT/AKT and pmTOR/mTOR (Fig. 3, E and F). These observations provide compelling evidence that Ca2+ influx through upregulated Piezo1 channels in the plasma membrane of PASMCs is required for the contractile-to-proliferative phenotypic transition and/or growth media-mediated PASMC proliferation.
siRNA-Mediated Piezo1 Downregulation Inhibits Cell Proliferation and Attenuates YAP Nuclear Translocation in Proliferative PASMCs
To further confirm the correlation of the results shown earlier to Piezo1, we used siRNA to decrease Piezo1 expression level in proliferative PASMCs and examined the changes of PCNA, YAP, and AKT/mTOR. As shown in Fig. 4, downregulation of Piezo1 with siRNA (siPiezo1) (Fig. 4, A and B) resulted in a similar inhibitory effect as the blocker GsMTx4 of Piezo1 on the 10% FBS growth media-induced increases in PCNA and the ratios of pAKT/AKT and pmTOR/mTOR (Fig. 4D). The siPiezo1-mediated downregulation of Piezo1 in proliferating PASMCs significantly decreased YAP level, negligibly affected pYAP level, and thus significantly enhanced the ratio of pYAP/YAP (Fig. 4C). The 10% FBS media-mediated increases in YAP protein level were associated with a significant enhancement of the nuclear translocation of YAP, whereas the enhanced YAP nuclear translocation in proliferating PASMCs was inhibited by siPiezo1 (Fig. 4, E and F). Furthermore, siRNA-mediated downregulation of Piezo1 in proliferative PASMCs significantly inhibited proliferation of PASMCs incubated in 10% FBS media. These observations suggest that Piezo1 or upregulated Piezo1 is required for and/or correlated with increasing YAP expression and YAP nuclear translocation in proliferating PASMCs.
Figure 4.

Downregulation of Piezo1 channels with siRNA inhibits growth medium-induced pulmonary artery smooth muscle cell (PASMC) proliferation and increases in YES-associated protein (YAP) signaling and AKT/mTOR signaling. A: Western blot analyses on Piezo1, proliferation cell nuclear antigen (PCNA), phosphorylated (pYAP) and total (YAP) YAP, phosphorylated (pAKT) and total (AKT) AKT, and phosphorylated (pmTOR) and total (mTOR) mTOR, and TAGLN in quiescent PASMCs incubated in fetal bovine serum (FBS)-containing media and proliferating PASMCs incubated (for 48 h) in 10% FBS media containing control siRNA (siNT) or siRNA for Piezo1 (siPiezo1). B: summarized data (means ± SE, n = 6 experiments) showing protein levels of Piezo1, PCNA, pYAP, and YAP, and TAGLN in quiescent PASMCs incubated in 0.3% FBS media and proliferating PASMCs incubated in 10% FBS media with siNT or siPiezo1. Data are presented as arbitrary units (top) and values normalized to 0.3% FBS media (bottom). **P < 0.01, ***P < 0.001 vs. 0.3% FBS; ##P < 0.01, ###P < 0.001 vs. 10%% FBS + siNT (one-way ANOVA/Holm–Sidak). Summarized data (means ± SE, n = 6 experiments) showing the ratio of pYAP/YAP (C), pAKT/AKT and pmTOR/mTOR (D) in quiescent PASMCs incubated in 0.3% FBS media and proliferating PASMCs incubated in 10% FBS media with siNT or siPiezo1. Data are presented as arbitrary units (top) and values normalized to 0.3% FBS media (bottom). **P < 0.01, ***P < 0.001 vs. 0.3% FBS; #P < 0.05, ###P < 0.001 vs. 10%% FBS + siNT (one-way ANOVA/Holm–Sidak). E: representative fluorescence imaging showing YAP (green) and DAPI (blue) in quiescent PASMCs incubated in 0.3% FBS media (left) and proliferating PASMCs incubated in 10% FBS media (right) containing siNT or siPiezo1. The overlay images in the bottom showing nuclear staining of YAP. F: summarized data (means ± SE, n = 111–137 cells from 5 independent experiments) showing the ration of nuclear staining intensity of YAP to cytoplasmic staining intensity (nuclear/cytosolic) in quiescent PASMCs incubated in 0.3% FBS media (gray and black bars) and proliferating PASMCs incubated in 10% FBS media (red and dark red bars) containing siNT or siPiezo1. ***P < 0.001 vs. 0.3% FBS + siNT; ###P < 0.001 vs. 10% FBS + siPiezo1 (one-way ANOVA/Holm–Sidak).
Upregulation of Piezo1 in Freshly Isolated PA from Animals with Experimental PH
To define whether Piezo1 upregulation is associated with the development of PH due to potential contractile-to-proliferative phenotypical transition and enhanced PASMC proliferation, we examined and compared protein expression of Piezo1 in freshly isolated PA from control rats and rats with experimental PH. In these experiments, we used the MCT-induced PH rat model (Fig. 5, A–C). As shown in Fig. 5, injection of MCT resulted in significant increases in RVSP and mean PAP (Fig. 5, A and B), and RV hypertrophy (Fig. 5B, inset). The increased pulmonary hemodynamics and RV hypertrophy were associated with pulmonary vascular remodeling defined by significant decreases in the total number of lung vascular branches (branch length), the number of lung vascular branches (branch No.), and the number of lung vascular branch junctions (junction No.) shown in ex vivo pulmonary angiogram (Fig. 5C). By comparison of the protein levels in freshly isolated PA tissues from control rats (Cont) and rats with MCT-induced PH (MCT), we found that Piezo1, along with calpain-1/2 and caveolin-1 (Cav-1), was significantly upregulated in MCT-PH rats (Fig. 5, D and E). The upregulation of Piezo1 in freshly isolated PA from MCT-PH rats was associated with a significant increase in pAKT, AKT, pmTOR, and mTOR (Fig. 5F) as well as the ratio of pAKT/AKT and pmTOR/mTOR (Fig. 5G). The upregulation of Piezo1 was also detected in whole lung tissues from MCT-PH rats compared with control rats (Fig. 5H). These data imply that Piezo1 is upregulated in isolated PA from PH animals because more PASMCs are undergoing contractile-to-proliferative phenotypic transition and/or because PASMC proliferation is enhanced, which contributes to pulmonary vascular remodeling in experimental PH.
Figure 5.
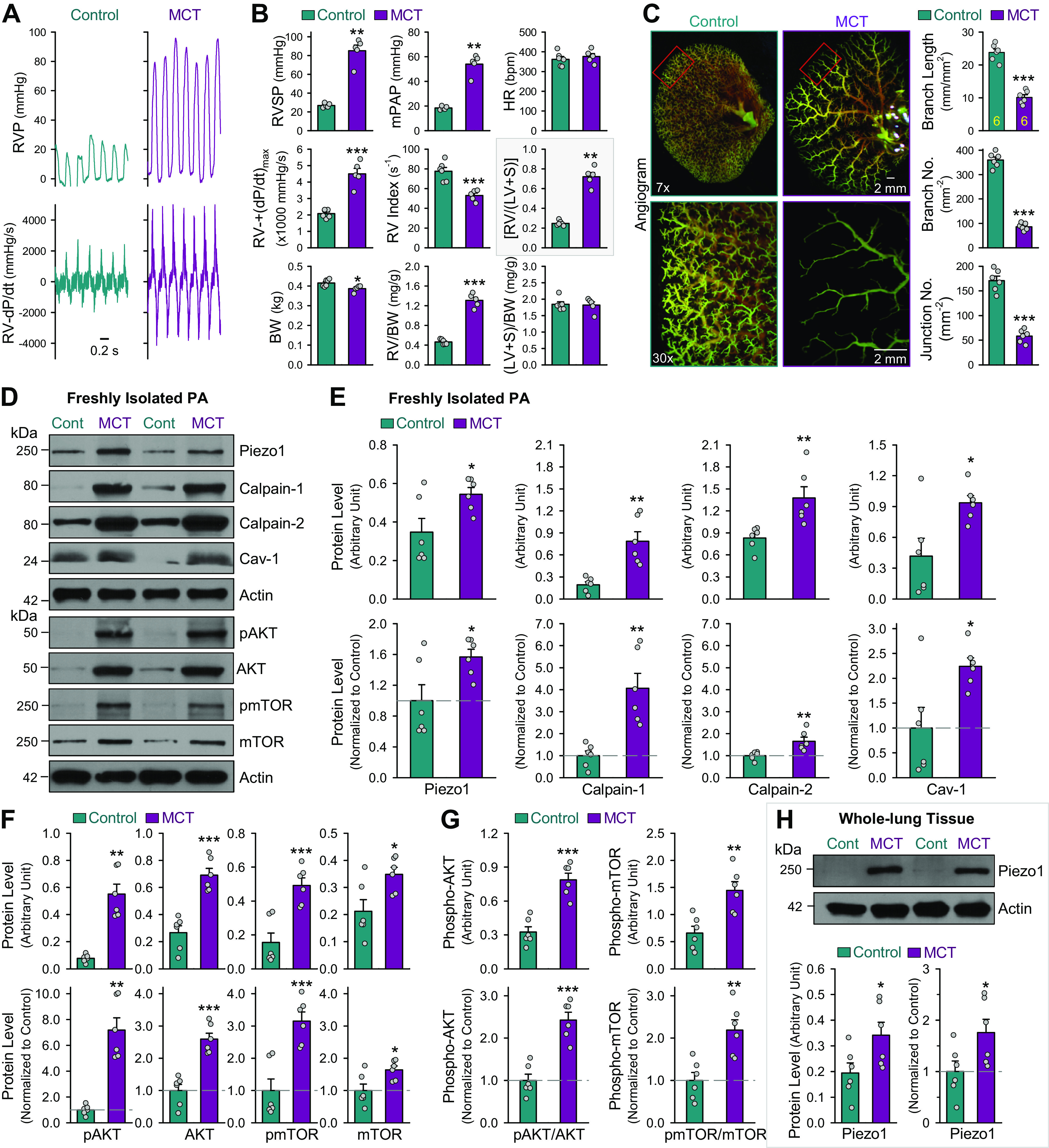
Upregulated Piezo1 in freshly isolated pulmonary artery (PA) from rats with monocrotaline (MCT)-induced pulmonary hypertension (PH). A: representative records showing right ventricular pressure (RVP) and RV contractility (RV-dP/dt) in normal control rats (Control) and rats with MCT-PH (MCT). B: summarized data (means ± SE, n = 5 for each group) showing right ventricular systolic pressure (RVSP), a surrogate measure of pulmonary arterial systolic pressure, estimated mean pulmonary arterial pressure (mPAP), heart rate (HR), RV contractility [RV- + (dP/dt)max], RV contractile index (the ratio of [RV- + (dP/dt)max)]/RVSP), Fulton Index, the ratio of the weight of RV to the weight of left ventricle (LV) and septum (S) [RV/(LV + S)], body weight (BW), RV/BW ratio, and left ventricle (LV) and septum (S) weight and BW ratio [(LV + S)/BW] in Control and MCT rats. **P < 0.01, ***P < 0.001 vs. Control (unpaired t test). C: representative angiography images of the left lung at ×7 (left) and ×30 (middle) magnification from Control and MCT rats. Images in the middle (×30) are enlarged images from the selected areas of the ×7 images in the left. Summarized data (means ± SE, right) showing the total length of lung vascular branches (branch length), the number of lung vascular branches (branch No.), and the number of vascular branch junctions (junction No.) per square millimeter of the selected lung area from Control (n = 6) and MCT (n = 6) rats. ***P < 0.001 vs. Control (unpaired t test). D: Western blot analysis on protein levels of Piezo1, Calpain-1, Calpain-2, caveolin-1 (Cav-1), phosphorylated AKT (pAKT), total AKT (AKT), phosphorylated mTOR (pmTOR), total mTOR (mTOR), and actin in freshly isolated PAs from Control (Cont) and MCT rats. E: summarized data (means ± SE, n = 6 for each group) showing protein levels of Piezo1, Calpain-1, Calpain-2, Cav-1, and actin in freshly isolated PAs from Control and MCT rats. *P < 0.05, **P < 0.01 vs. Control (unpaired t test). F: summarized data (means ± SE, n = 6 for each group) showing protein levels of pAKT, AKT, pmTOR, and mTOR in freshly isolated PAs from Control and MCT rats. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Control. G: summarized data (means ± SE, n = 6 for each group) showing the ratio of pAKT/AKT and pmTOR/mTOR in freshly isolated PAs from Control and MCT rats. **P < 0.01 and ***P < 0.001 vs. Control (Unpaired t-test). H: Western blot analysis (top) and summarized data (means ± SE, n = 6 for each group, bottom) showing the protein level of Piezo1 in whole lung tissues from Control (Cont) and MCT rats. *P < 0.05 and ***P < 0.001 vs. Control (unpaired t test). Data shown in E, F, G, and H are presented as arbitrary units and values normalized to Control (Cont). The horizontal broken lines indicate the level of the proteins in Control.
In addition, we also found upregulation of BUBR1, pYAP, and YAP, the proteins upregulated in contractile-to-proliferative phenotypical transition of PASMCs, in freshly isolated PA (Fig. 6, A and B) and whole lung tissues (Fig. 6, C and D) from MCT-PH rats compared with control rats. These observations direct us to speculate that upregulation of Piezo1 and enhancement of mechanosensitive signaling pathways (e.g., YAP signaling) are not only related to the PASMC contractile-to-proliferative phenotypical transition but also associated with the development and progression of pulmonary vascular remodeling in experimental PH.
Figure 6.
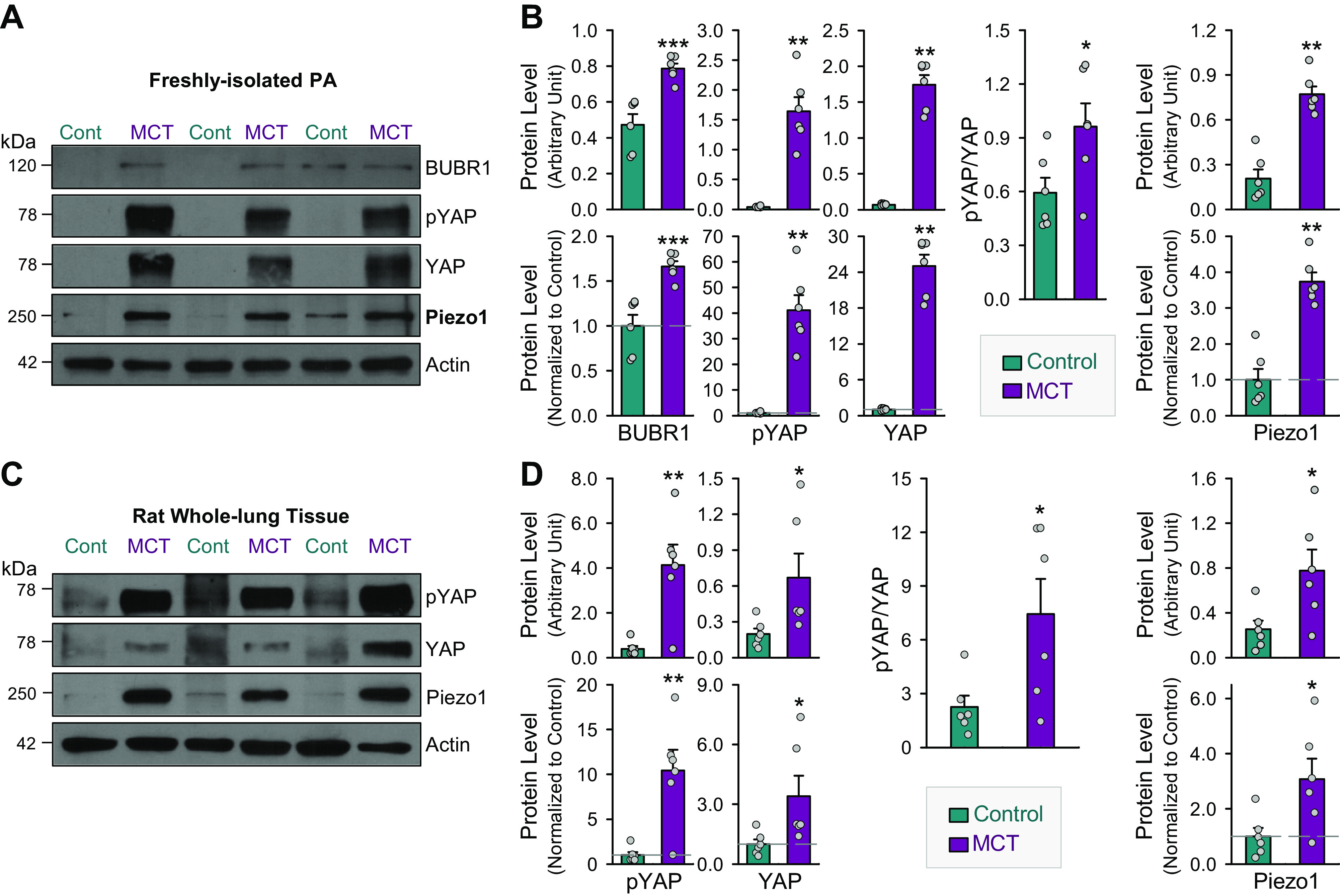
Upregulation of budding uninhibited by benzimidazole-related 1 (BUBR1) and YES-associated protein (YAP) in freshly isolated pulmonary arteries (PAs) and whole lung tissues in rats with monocrotaline (MCT)-induced pulmonary hypertension (PH). Representative Western blot images (A) and summarized data (means ± SE, n = 6 for each group) (B) showing protein levels of BUBR1, phosphorylated YAP (pYAP), and total YAP (YAP), and Piezo1, as well as the ratio (pYAP/YAP) of phosphorylated YAP (pYAP) and total YAP (YAP) in freshly isolated PAs from control rats (Cont) and rats with MCT-induced PH (MCT). *P < 0.05, **P < 0.01, ***P < 0.001 vs. Cont (unpaired t test). Representative Western blot images (C) and summarized data (means ± SE, n = 6 for each group) (D) showing protein levels of pYAP, YAP, and Piezo1, as well as the ratio (pYAP/YAP) of pYAP and YAP in whole lung tissues from Cont and MCT rats. *P < 0.05, **P < 0.01 vs. Cont (unpaired t test).
Yoda1-Mediated Ca2+ Influx through Piezo1 Channels Stimulates PASMC Proliferation
The next set of experiments was designed to examine whether activation of Piezo1 channels is sufficient to induce or enhance PASMC proliferation. Extracellular application of Yoda1 (0.3 µM), a well-characterized Piezo1 channel activator (36, 64, 65), resulted in a rapid increase in [Ca2+]cyt in human (Fig. 7A) and rat (Fig. 7B) PASMCs. The Yoda1-mediated Ca2+ increase due to Ca2+ influx through Piezo1 channels was sufficient to increase the number of viable PASMCs (Fig. 7C, left) and BrdU DNA incorporation (Fig. 7C, right) incubated in 0.3% FBS and 5% FBS media (Fig. 7C). Chelation of extracellular free Ca2+ with EGTA (2 mM) significantly inhibited Yoda1-mediated PASMC proliferation (Fig. 7C). We used EGTA [ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid] to reduce free [Ca2+] in the culture media since we were unable to change the ionic composition in the commercially available media. EGTA is a divalent cation chelator that has a lower affinity for Mg2+ than EDTA, which makes EGTA more selective for chelating Ca2+ than EDTA. We do not know whether EGTA, per se, has any toxic effect on PASMCs in addition to its cation-chelation effect. The in vitro experiments should be repeated in the future using low-Ca2+ or Ca2+-free culture media to exclude possible toxic effects of EGTA.
Figure 7.
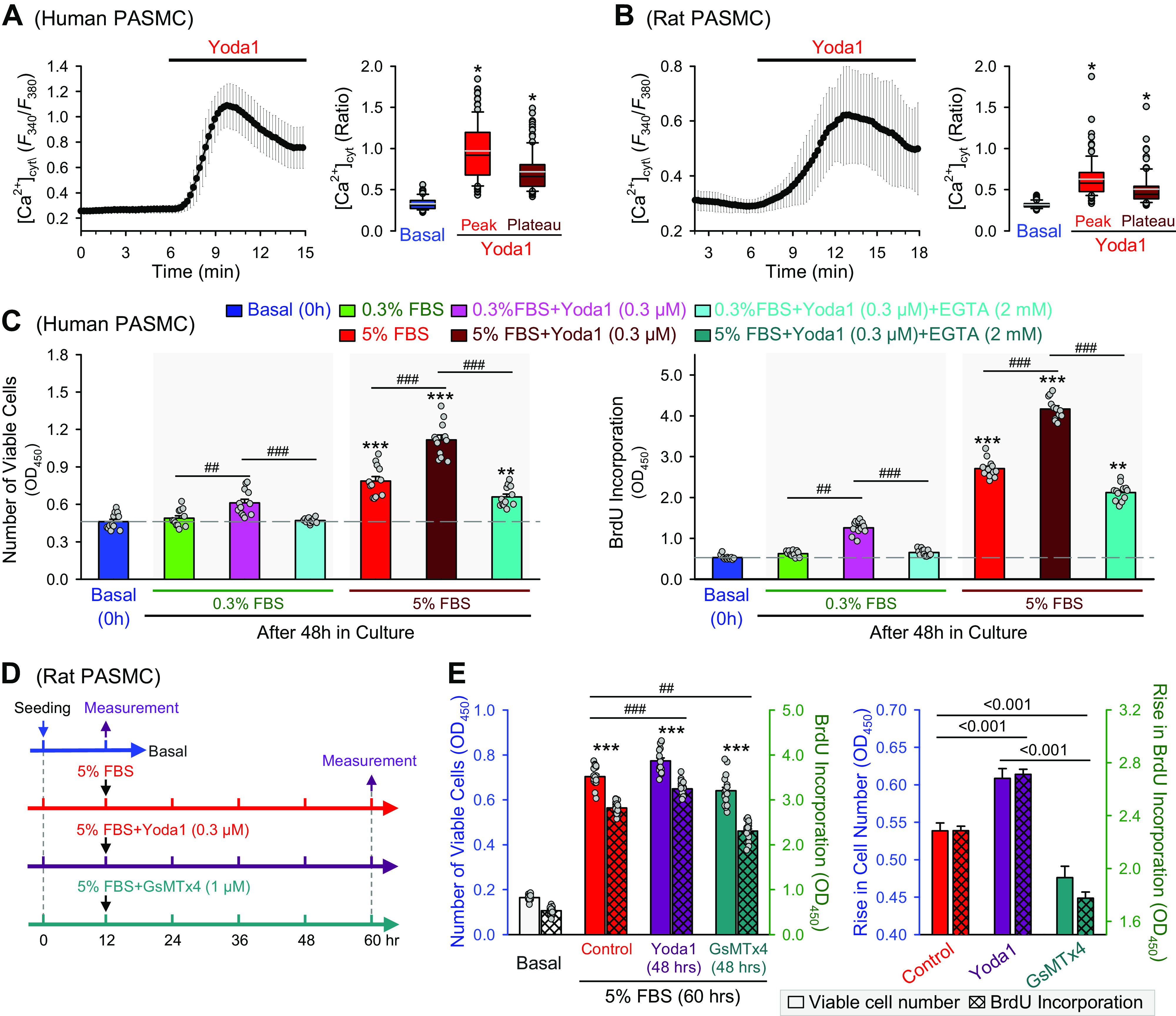
Ca2+ influx through Piezo1 channels is involved in stimulating human pulmonary artery smooth muscle cell (PASMC) proliferation. Representative record showing the change of [Ca2+]cyt (left) in human (A) and rat (B) PASMCs before and during extracellular application of Yoda 1 (5 µM). Summarized data (means ± SE, right) showing the levels of [Ca2+]cyt before (basal) and during the Yoda1 application (Peak and Plateau) in human (A, n = 117 cells) and rat (B, n = 97 cells) PASMCs. C: summarized data (means ± SE, n = 18) showing numbers of viable human PASMCs (left) and BrdU incorporation (right), determined by optical density at 450 nm (OD450), before (basal, 0 h) and after 48 h of incubation in 0.3% FBS media, 0.3% FBS with Yoda1 (0.3 μM), 0.3% FBS with Yoda1 (0.3 μM) and EGTA (2 mM), 5% FBS group, 5% FBS with Yoda1 (0.3 μM), 5% FBS with Yoda1 (0.3 μM) and EGTA (2 mM). ##P < 0.01, ###P < 0.001 between bars as indicated by the horizontal lines; *P < 0.05, **P < 0.01, ***P < 0.001 vs. 0 h (one-way ANOVA/Holm–Sidak). D: experimental protocol for the experiment using Yoda1 and GsMTx4. Primary cultured rat PASMCs were evenly distributed in 96-well plates. When cells attach to plates after 12 h of incubation, cells were split into three groups: 5% FBS, 5% FBS with Yoda1 (0.3 μM), and 5% FBS with GsMTx4 (1 μM). The cells were then treated for 48 h. CCK8 solution was used after 48 h for measurement of the number of viable cells. E: summarized data (means ± SE, n = 18, left) showing number of viable rats PASMCs (open bars) and BrdU incorporation (hatch crossed bars), determined by optical density at 450 nm (OD450), before (basal, 12 h after seeding) and after 48 h of incubation in 5% FBS media with no drugs (Control), 5% FBS with Yoda1 (0.3 μM, Yoda1), and 5% FBS with GsMTx4 (1 μM, GsMTx4). ##P < 0.01, ###P < 0.001 between bars as indicated by the horizontal lines; ***P < 0.001 vs. basal (one-way ANOVA/Holm–Sidak). Normalized data (right) showing the 5% FBS-mediated increases in the number of viable cells (open bars) and BrdU incorporation (hatch crossed bars) with no drug treatment (Control) or with 48-h treatment of Yoda1 (0.3 µM) or GsMTx4 (1 µM) are constructed from the data shown in the left. The P values between the bars are shown above the horizontal lines (one-way ANOVA/Holm–Sidak).
Furthermore, we conducted another set of experiments to examine whether rat PASMC proliferation induced by 5% FBS was sensitive to the activator (Yoda1) and blocker (GsMTx4) of Piezo1 channels. As shown in the experimental protocol (Fig. 7D), we first incubated the primary cultured rat PASMCs in 5% FBS media for 12 h and measured the basal number of viable cells. Then, the cells were further incubated in 5% FBS media in the presence of 0.3 µM Yoda1 or 1 µM GsMTx4 for 48 h before the number of viable cells was measured. Forty-eight hours of Yoda1 (0.3 µM) treatment significantly enhanced 5% FBS-mediated PASMC proliferation (determined by both the number of viable cells and BrdU DNA incorporation), whereas GsMTx4 significantly inhibited 5% FBS-mediated PASMC proliferation (Fig. 7E). The enhancement effect of Yoda1, a Piezo1 opener, and the inhibitory effect of GsMTx4, a Piezo1 blocker, on PASMC proliferation were small but statistically significant. These data indicate that, in human and rat PASMCs, activation of Piezo1 channels (with Yoda1) is sufficient to stimulate PASMC proliferation, whereas block of Ca2+ influx through Piezo1 channels (with GsMTx4) can inhibit PASMC proliferation. Piezo1 channels, which are upregulated in the PA from animals with experimental PH, are potentially an important Ca2+ channel to assure sufficient Ca2+ influx for stimulating and maintaining PASMC proliferation.
Yoda1-Mediated Activation of Piezo1 Induces PH in the Presence of l-NAME
In addition to the in vitro experiments described earlier, we also conducted a set of experiments to examine whether injection of the Piezo1 opener Yoda1 induces PH. Figure 8A depicts the experimental protocol in which we used 8-wk-old mice for the in vivo experiments. Piezo1 channels are expressed in both vascular SMC and endothelial cells (ECs), and stretch-mediated Ca2+ influx through Piezo1 in ECs has been shown to activate endothelial nitric oxide synthase (eNOS), increase nitric oxide (NO) synthesis and release, and results in vasodilation (26, 53, 65, 66). To differentiate the role of Yoda1-mediated activation of Piezo1 channels in SMCs and ECs, we used the eNOS inhibitor l-NAME in the experiment to minimize the contribution of eNOS, endothelium-derived NO, or EC-NO-mediated pulmonary vasodilation to the Yoda1 on pulmonary hemodynamics and vascular remodeling (Fig. 8, A and B). In mice intraperitoneally injected with Yoda1 alone (1 mg/kg, everyday) for 6 wk, we observed no significant changes in RVSP, mPAP, RV- ± dP/dt, and heart rate, as well as systolic systemic arterial pressure (sSAP) and mean SAP (Fig. 8, C, D, and F). Yoda1 alone also had no effect on Fulton Index, a ratio for RV hypertrophy (Fig. 8E). Intake of l-NAME (0.1% in drinking water, everyday) for 6 wk had little effect on RVSP, mPAP, RV- ± dP/dt, and heart rate, but significantly increased sSAP and mSAP (Fig. 8, C, D, and F). Combined administration of Yoda1 (ip) and l-NAME (in drinking water) for 6 wk, however, significantly increased RVSP, mPAP, RV- + (dP/dt)max, and Fulton Index (Fig. 8, C–F). Intraperitoneal injection of Yoda1 with intake of l-NAME in drinking water did not further increase sSAP and mSAP in these mice (Fig. 8F). The changes in pulmonary hemodynamics (e.g., RVSP and mPAP) and Fulton Index in mice treated with Yoda1, l-NAME, and Yoda1 + l-NAME were closely associated with the significant decreases in the total length of branches and the numbers of branches and branch junctions as indicated by angiogram (Fig. 8, G and H). These data indicate that activation of Piezo1 with Yoda1 is sufficient to induce PH in the presence of an eNOS inhibitor, l-NAME. These data also imply that Ca2+ influx through Piezo1 in pulmonary arterial ECs may activate eNOS and increase NO synthesis and release, which results in endothelium-dependent pulmonary vasodilation. Indeed, our preliminary data show that l-NAME significantly enhanced Yoda1-mediated pulmonary vasoconstriction in isolated perfused/ventilated mouse lungs (data not shown).
Figure 8.
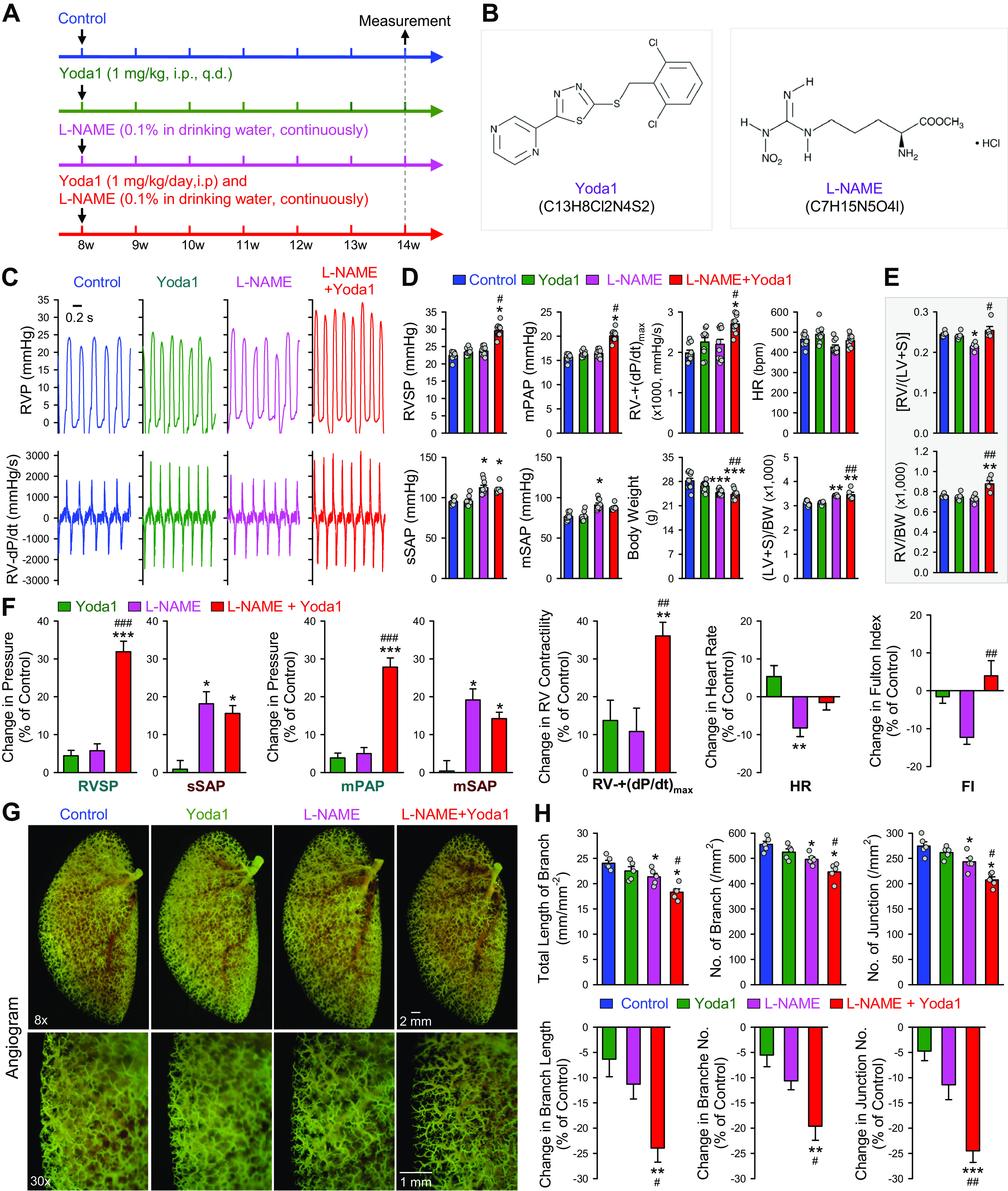
Yoda1-mediated activation of Piezo1 channels induces experimental pulmonary hypertension in mice when endothelial nitric oxide synthase (eNOS) is inhibited by nitro-l-arginine methyl ester (l-NAME). A: experimental protocol for the experiment using Yoda1 and l-NAME. Eight-week-old C57Bl/6 mice were divided into four groups: Control (vehicle), Yoda1 (1 mg/kg ip, everyday), l-NAME (0.1% in drinking water, everyday), and Yoda1 (1 mg/kg ip, everyday) and l-NAME (0.1% in drinking water, everyday). Pulmonary hemodynamics are measured after 6 wk of treatment with vehicle and Yoda1 or l-NAME. B: chemical structures of Yoda1 and l-NAME. C: representative records of right ventricular pressure (RVP) (top) and right ventricle (RV) contractility (RV- ± dP/dt) (bottom) in Control mice and mice receiving Yoda1, l-NAME, and Yoda1 + l-NAME. D: summarized data (means ± SE, n = 9 or 10/group) showing right ventricular systolic pressure (RVSP), estimated mean pulmonary arterial pressure (mPAP), RV- + (dP/dt)max, and heart rate (HR), as well as systolic systemic arterial pressure (sSAP), mean systemic arterial pressure (mSAP), body weight (BW), and the ratio of the weight of left ventricle (LV) and septum (S) to BW [(LV + S)/BW] in Control mice and mice receiving Yoda1, l-NAME, and Yoda1 + l-NAME. *P < 0.05 vs. Control; #P < 0.05 vs. l-NAME along (one-way ANOVA/Holm–Sidak). E: summarized data (means ± SE, n = 5 per group) showing the Fulton Index (FI), the ratio of the weight of right ventricle (RV) to the weight of left ventricle (LV) and septum (S) [RV/(LV + S)] (top) and the ratio of the weight of RV to body weight (RV/BW) (bottom) in Control mice and mice receiving Yoda1, l-NAME, and Yoda1 + l-NAME. F: summarized data (means ± SE) showing the Yoda1-mediated changes in RVSP, sSAP, mPAP, mSAP, RV- + (dP/dt)max, HR, and Fulton Index (FI) in mice receiving Yoda1, l-NAME, and Yoda1 + l-NAME. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Yoda1; ##P < 0.01, ###P < 0.001 vs. l-NAME alone (one-way ANOVA/Holm–Sidak). G: representative angiogram images of the left lung at ×8 and ×30 magnification from Control mice and mice receiving Yoda1, l-NAME, and Yoda1 + l-NAME. Images (×30) are enlarged images from the selected areas of the ×8 images. H: summarized data (means ± SE, n = 5 in each group) showing the total length of lung vascular branches (left), the number of lung vascular branches (middle), and the number of vascular branch junctions (right) per square millimeter of the selected lung area from Control mice and mice receiving Yoda1, l-NAME, and Yoda1 + l-NAME. *P < 0.05 vs. Control; #P < 0.05 vs. l-NAME alone (one-way ANOVA/Holm–Sidak). The Yoda1-mediated changes (means ± SE) in the total length of branches, the number of branches, and the number of junctions in Control mice and mice receiving Yoda1, l-NAME, and Yoda1 + l-NAME are shown in the bottom. **P < 0.01, ***P < 0.001 vs. Control; #P < 0.05, ##P < 0.01 vs. l-NAME (one-way ANOVA/Holm–Sidak).
It is unknown whether Yoda1, l-NAME, or the combination treatment of Yoda1 and l-NAME (Yoda1 + l-NAME) have cardiotoxic effect. Based on the data shown in Fig. 8D, we calculated the RV contractile index, the ratio of RV-(+dP/dt)max to RVSP, and found that the RV index was not significantly different (P = 0.233, one-way ANOVA) among the control group (88.7 ± 2.9 s−1), Yoda1 group (96.3 ± 12.5 s−1), l-NAME group (92.3 ± 11.7 s−1), and Yoda1 + l-NAME group (91.5 ± 7.5 s−1). These data imply that RV contractility was not significantly affected by the combination treatment with Yoda1 and l-NAME. The increased RVSP and mPAP in Yoda1 + l-NAME group (compared with control group) were likely due to pulmonary vasoconstriction (preliminary data not shown) and increased pulmonary vascular remodeling (Fig. 8, G and H).
DISCUSSION
In this study, we found that Piezo1, a mechanosensitive cation channel, was upregulated during the PASMC contractile-to-proliferative phenotypic transition. Compared with freshly isolated PA (containing mainly contractile PASMC), the upregulation of Piezo1 in primary cultured PASMCs in media containing 10% FBS and growth factors (containing mainly proliferative PASMCs) was also associated with increases in protein expression levels of CaSR (a GPCR that is activated by extracellular Ca2+), Notch2, Notch3, F-actin (a crucial cytoskeleton protein for cell mobility) (55), calpain-1 and calpain-2 (Ca2+-dependent cysteine proteases), and YAP (a transcription regulator that stimulates cell proliferation). In proliferative PASMCs, serum starvation not only downregulated Piezo1 but also decreased YAP level. siRNA-mediated downregulation of Piezo1 in proliferating PASMCs significantly inhibited PASMC proliferation by decreasing the ratios of pAKT/AKT and pmTOR/mTOR and by attenuating YAP nuclear translocation.
Furthermore, we also observed upregulation of Piezo1 in freshly isolated PA from rats with experimental PH compared with control rats. The upregulated Piezo1 in freshly isolated PA from animals with MCT-PH was positively associated with upregulation of YAP and increases in pAKT/AKT and pmTOR/mTOR ratios. These observations link the upregulated expression of Piezo1, along with upregulated CaSR (67, 68) and Notch (69–72), to PASMC contractile-to-proliferative phenotypic transition and PASMC proliferation via multiple intracellular signaling cascades including AKT/mTOR (73–78) and YAP (58–60, 79, 80). The close association between the “remodeling” of membrane receptors and ion channels and the phenotypic transition of PASMC is potentially an important contributor to the development and progression of pulmonary vascular remodeling in animals with experimental PH and, potentially, patients with PAH.
SMC Contractile-to-Proliferative Phenotypic Transition and SMC Proliferation Contribute to Pulmonary Vascular Remodeling
As indicated by the equation: mPAP = PVR × CO – PAWP (where mPAP is mean PAP, PVR is pulmonary vascular resistance, CO is cardiac output, and PAWP is pulmonary arterial wedge pressure) (81, 82), mean PAP is directly proportional to PVR in patients with precapillary PH (e.g., PAH, PH due to lung diseases or hypoxemia and chronic thromboembolic PH). According to Poiseuille equation, PVR is inversely proportional to the 4th power of the intraluminal radius (r) of pulmonary arteries: PVR = 8Lη/πr4, where L is the length of vessels, η is a viscosity constant, and π is a constant (= 3.14). The equation indicates that PVR is exquisitely sensitive to small changes in the pulmonary artery (PA) caliber radius. Pulmonary vasoconstriction (due to PASMC contraction), concentric pulmonary vascular remodeling (due partially to PASMC proliferation), and occlusive vascular lesions directly reduce the PA caliber radius and are thus the major contributor to the elevated PVR (and mPAP) in patients with PAH and other precapillary PH. Histological and pathological studies also show the presence of SMCs in the occlusive intimal lesions in the proximal and distal vessels (9, 10, 12). These data indicate that PASMC, regardless of its phenotype (i.e., contractile, or synthetic/proliferative), is involved in the development and progression of pulmonary vascular lesions (pathophysiological and pathological lesions) in patients (and animals) with severe PH. It is assumed that sustained pulmonary vasoconstriction (due to PASMC contraction) is the major cause for increasing PVR at the early stage of severe PH (e.g., idiopathic PAH) or in patients with mild PH (e.g., PH due to lung diseases or hypoxemia). Concentric pulmonary vascular remodeling and obliterative vascular lesions become predominant pathological changes in the vasculature to further increase PVR at the late stage of PAH or in patients with severe PH. The pathogenic contribution of PASMCs to pulmonary vasculopathy also relies on the coordination and interaction with other cells (e.g., ECs, myofibroblasts, inflammatory cells, and progenitor cells) via paracrine and juxtacrine mechanisms (16, 17, 83). One of the therapeutic effects of NO and cGMP enhancers (e.g., riociguat, selexipag, and sildenafil) is reported to be due to the inhibition of PASMC growth and proliferation and/or inhibition of SMC contractile-to-proliferative phenotypical transition (84), in addition to NO-mediated pulmonary vasodilation (85, 86).
PASMC in the pulmonary vasculature of normal subjects (and animals) is a highly differentiated cell with a major function for contraction. PASMC contraction and relaxation enable the precise regulation of pulmonary vascular tone, PVR, and PAP. The highly differentiated or contractile phenotype of PASMCs in the normal adult PA hardly proliferates or exhibits very low growth rate. Like other types of vascular SMCs, however, PASMCs retain a high level of phenotypic plasticity that enables rapid response to microenvironmental cues, such as rapid repair or remodeling in response to vascular injury and alveolar hypoxia (87–89). The synthetic or proliferative phenotype of PASMCs is believed to play an important role in the development and progression of concentric vascular remodeling, arteriole and precapillary muscularization, and obliterative vascular lesions in patients with idiopathic PAH, PH due to respiratory diseases or hypoxemia, and chronic thromboembolic PH (9, 10, 12, 13, 22, 90). Vascular SMCs undergo a phenotypic transition or switch during the development and progression of vascular diseases (19–21, 91–94), although the cellular and molecular mechanisms involved in vascular SMC phenotypic switch are well studied (21, 95–98). Various signaling pathways and transcription factors are revealed to be directly involved in the SMC phenotypic transition. Nevertheless, the therapeutic or beneficial effects of pulmonary arterial denervation (PADN) on pulmonary hemodynamics and functional tests in patients with PAH and other forms of precapillary PH imply that sustained pulmonary vasoconstriction due to PASMC contraction occurs in the late stage of the disease progression as well (99–103). The PASMC contractile-to-proliferative transition may occur in certain segments of the lung vasculature and/or certain subtypes of PASMCs in proximal and distal PA during the development and progression of PAH/PH.
Role of Upregulation of Mechanosensitive Channel Piezo1 in SMC Proliferation
In the current study, we observed a significant upregulation of Piezo1, a mechanosensitive cation channel (104–106), during the phenotypic transition. The upregulated Piezo1 was associated with increased protein expression levels of F-actin and BUBR1. It remains unknown whether the upregulation of Piezo1 is required for the phenotypic transition or the consequence of the phenotypic transition. The data from the study suggest that proliferative PASMC is more sensitive to mechanical stimulation, due to the high expression of Piezo1. In other words, PASMC proliferation can be further enhanced by mechanical stimulations, such as flow shear stress and physical stretch. These data also imply that, during mitosis as well as nuclear and cell division, the resultant cytoskeleton structural changes require or stimulate Ca2+ influx through mechanosensitive channels (e.g., Piezo1) to assure the normal progress of mitosis and cell division. It has been demonstrated that the G2 to M phase transition and the whole mitosis process are sensitive to Ca2+/CaM (107).
The cytoskeleton, a dynamic filament in the cytoplasm, plays an important role in regulating cell shape and morphology, cell movement (e.g., migration), cell contraction (such as in cardiac, skeletal, and smooth muscle cells), cytokinesis, and the organization of cytoplasmic organelles. The cytoskeleton includes mainly microfilaments (actin-like proteins), microtubules (tubulin-like proteins), and the intermediate filaments in eukaryotes. Cell proliferation is a process that increases the number of cells as a result of cell division, whereas cell growth is a process that enlarges the volume or size of cells in the absence of cell division and without the increase of cell numbers. Cell proliferation and growth are both involved in the development and progression of pulmonary vascular remodeling including concentric vascular wall thickening, muscularization of precapillary arterioles and capillary, and occlusive intimal lesions. During the cell division (and cell enlargement), a cell undergoes significant structural and dynamic changes in the cytoskeleton (e.g., upregulation of F-actin shown in this study). The dynamic and kinetic movement and (re)organization of the cytoskeleton during cell proliferation result in significant membrane stretch (29) that can open mechanosensitive cation channels, such as the upregulated Piezo1, which induces Ca2+ influx and increases [Ca2+]cyt. On the other hand, the proliferating cells require Ca2+ and Ca2+/CaM to fulfill and enable the dynamic and kinetic process occurring in the cell division or the cell cycle (mitosis and/or meiosis) (108–110). Furthermore, the cytoskeleton movement and reorganization during cell division and enlargement also require Ca2+ to assure and allow these necessary structural changes (111, 112). For example, cytoskeletal/cytoskeleton disorganization leads to more membrane stretch, which would increase Ca2+ influx through Piezo1 channels to fulfil Ca2+-dependent contraction of the contractile membrane enabling nuclear and cell division.
Furthermore, the cell-cell and cell-matrix adhesion is an important process in the tissue remodeling and the force developed along the adhesome is another mechanical stimulation to the plasma membrane of cells involved in the tissue network (29). The upregulated BUBR1, a cell cycle-related protein, is another evidence that the contractile-to-proliferative transition of PASMCs and/or enhanced PASMC proliferation are directly attributable to progression of the cell cycle and cell division. Budding uninhibited by benzimidazole 1 (BUB1) and BUBR1 (or BUB1B) are paralogs that are central components of the mitotic checkpoint for spindle assembly, an essential self-monitoring system of the cell cycle and cell division (56, 113). BUBR1 insufficiency has been demonstrated to inhibit neointimal hyperplasia via inhibition of SMC proliferation (114).
It is highly possible that the upregulated mechanosensitive cation channel Piezo1 in PASMCs is required for facilitating and promoting the dynamic and kinetic process in cell proliferation and enlargement, which stimulates or enhances PASMC proliferation and growth and, ultimately, contributes to the development and progression of pulmonary vascular remodeling (6–8, 115, 116). Indeed, blocking of Piezo1 with a potent blocker, GsMTx4, and downregulation of Piezo1 with siRNA both significantly inhibited PASMC proliferation in the presence of serum and growth factors. Activation of Piezo1 channels with an allosteric agonist or a small molecule opener, Yoda1 (36), enhanced PASMC proliferation and PASMC contractile-to-proliferative phenotypic transition (117). Although we focused on the role of Piezo1 channel in PASMCs and its potential contribution to the development and progression of pulmonary vascular remodeling, other types of cells (e.g., macrophages) are implicated in pulmonary vascular remodeling in PAH and experimental PH (118, 119). Piezo1 in macrophages is involved in a host response during infection (120) and a mechanical sensing of stiffness (121). These data imply that upregulated Piezo1 may occur in multiple cell types contributing to the development and progression of pulmonary vascular remodeling in PAH and experimental PH.
Receptor and Ion Channel Remodeling during Contractile-to-Proliferative Transition
In the current study, we observed significant increases in protein expression levels of Piezo1, along with CaSR and Notch during the phenotypic transition. The molecular mechanisms or determinants for the upregulated Piezo1, CaSR, and Notch in proliferating PASMCs remain unclear; it is possible that the genes/transcription factors responsible for the SMC phenotypic switch or regulation of the contractile and proliferative marker genes are also responsible for the regulation of these channels and receptors (122, 123). One of the innovative concepts of this study is to suggest that the PASMC contractile-to-proliferative phenotypic transition requires a “reset” or remodeling of different membrane channels and receptors to ensure sufficient, for example, Ca2+, for cell proliferation or cell division. As discussed earlier, transition of quiescent cells into the cell cycle (G0 to G1), transitions of cells in G1 phase to S phase and cells from S phase to mitosis, and progression of mitosis are dependent on Ca2+/CaM (107, 110). Furthermore, the physical force required for nuclear and cell division requires sufficient Ca2+ to bring to fruition. The molecular mechanisms for the phenotypic transition-associated reset or adaptation of Piezo1 and other ion channels (e.g., TRPC6, CALHM1/2, and Orai/STIM2) (23, 24, 109, 124) and membrane receptors (e.g., CaSR, Notch2, Notch3) in SMC or PASMCs remain unclear. In the promoter of Piezo1 gene, there are many binding sites for different transcription factors. The transcription factors that can directly bind to the Piezo1 gene promoter include c-Ets-1/2 and YY-1, which are associated with SRF and implicated in the SMC contractile-to-proliferative phenotypic transition (1, 3, 21, 95, 116, 125). It is currently unknown whether the genes and proteins or transcription factors responsible for triggering or initiating the SMC phenotypic transition, concurrently and directly, upregulate Piezo1 to fulfill the contractile-to-proliferative phenotypic transition, the functional switch or adaptation of certain membrane channels (e.g., from voltage-dependent Ca2+ channels to voltage-independent cation channels) (126), and the upregulation of membrane receptors associated with cell proliferation. In addition, the Piezo1 promoter also contains binding sites for transcription factors, such as Oct-4 (127), p53 (128, 129), NF-κB (49, 130), RBP-Jκ (69–71), AP1/c-Jun (131–133), NF-AT (90, 134), STAT (135, 136), and CREB (137, 138), which are all implicated in regulating PASMC proliferation and the development of PAH or experimental PH.
The PASMC contractile-to-proliferative phenotypic switch seems to be associated with a transcriptional and functional “remodeling” or adaptation of various ion channels. The conversion from a dependence of Ca2+ influx on voltage-dependent Ca2+ channels in contractile PASMCs to a dependence on voltage-independent cation channels in proliferative PASMCs makes the proliferative cells more sensitive to receptor-operated and/or mechanosensitive Ca2+ influx in response to mitogenic factors and cytokinetic events. The voltage-insensitive, second messenger-sensitive, mechanosensitive, and nonselective cation channels would also be more effective to assure sufficient intracellular Ca2+ (or activation of Ca2+/CaM signaling cascades) required for propelling cells to go through the cell cycle (107, 108, 110). Indeed, the early nuclear separation at checkpoint requires Ca2+, provided by Ca2+ influx through mechanosensitive cation channels, to assure the physical division of mother cell to daughter cells. Primary-cultured proliferative PASMCs are more depolarized than freshly dissociated contractile PASMCs, whereas voltage-dependent Ca2+ channels are largely inactivated at the resting membrane potential in proliferative PASMCs. It thus makes sense for proliferating PASMCs to express more voltage-independent, nonselective cation channels regulated by intracellular second messengers (e.g., DAG, IP3, and cytosolic H+) and mechanical stimulation (e.g., membrane stretch due to cytoskeleton reorganization, flow shear stress) to assure Ca2+ influx for cell mitosis and nuclear/cell division. Indeed, blockade of the mechanosensitive cation channels (e.g., Piezo1) in proliferative PASMCs significantly attenuated PASMC proliferation and reduced the number of cells in mitosis (49). We and others have demonstrated that TRPC6, a mechanosensitive and receptor-operated cation channel, is also involved in regulating SMC phenotypical transition (23, 124, 139). Taken together with the observations in this study, mechanosensitive channels (e.g., Piezo1 and TRPC6) are required for PASMC proliferation, whereas voltage-dependent Ca2+ channel (VDCC) (e.g., L-type and T-type VDCC) are more involved in EC-coupling and PASMC contraction. It has been demonstrated that store-operated Ca2+ entry (SOCE) mediated by STIM1/Orai1 is inhibited during the mitosis of the cell cycle (140–143), whereas enhanced SOCE has also been implicated in cell proliferation. Given the fact that Ca2+ or Ca2+/CaM is required for the interphase transition from G2 to M phase and the whole mitosis (107, 108), it is possible that upregulated Piezo1 (this study) and TRPC6 (49, 130, 144), both are mechanosensitive Ca2+-permeable cation channels, are the major Ca2+ channels for the required cytosolic and nuclear Ca2+ in mitosis to propel the cell to go through the cell cycle and cell division. However, early studies also indicate that Ca2+ influx through L- and T-type VDCC is required for propelling systemic vascular SMCs go through different phases in the cell cycle (145, 146).
Mechanosensitive Yap/TAZ Signaling in PASMC Proliferation
The YES-associated protein (YAP)/transcriptional co-activator with PDZ-binding motif (TAZ) is regulated by mechanical cues such as shear stress, stretch and strain, cytoskeleton disorganization, and cell-cell adhesion (147) through mechanosensitive channels in the plasma membrane and nuclear envelope (148). Upregulation of YAP or increase of YAP/TAZ nuclear translocation (to stimulate gene expression and cell proliferation) is considered to be a downstream effector cascade of mechanosensitive channels and receptors in the plasma membrane (148). YAP/TAZ signaling has been reported to be an important downstream signaling pathway of the mechanosensitive cation channel Piezo1 (149), although the YAP/TAZ signaling is reported to be an upstream trigger for upregulating Piezo1 in cancer cells (80). The YAP/TAZ-associated transcriptional upregulation of Piezo1, through the Hippo signaling pathway, has been implicated in cancer cell proliferation (80). In oral squamous cell carcinoma (OSCC) cells, Hasegawa et al. (80) showed compelling evidence that Piezo1 was a transcriptional target of YAP signaling and elevated Piezo1 was required for the Ca2+-dependent OSCC cell proliferation. In our study, both YAP and phosphorylated YAP were significantly increased in proliferative PASMCs compared with contractile PASMCs. The increased pYAP was probably due to a compensatory mechanism for increased YAP and YAP-induced cell proliferation. It is possible that Piezo1-mediated Ca2+ influx (or increase in [Ca2+]cyt) may also enhance YAP translocation into nucleus stimulating cell proliferation and activate HIPPO signaling to phosphorylate YAP and sequester YAP in the cytoplasm during PASMC contractile-to-proliferative phenotypic transition and PASMC proliferation. Furthermore, our data in freshly isolated PA from rats with experimental PH (MCT-PH) showed that YAP protein level was significantly increased, along with pYAP. These data further indicate that upregulation of YAP is not only an important mechanism for PASMC contractile-to-proliferative phenotypic transition and PASMC proliferation but also a potential pathogenic mechanism for excessive PASMC proliferation and pulmonary vascular remodeling in, at least, experimental PH.
In cancer cells, mechanical factors such as nuclear softening and increased ECM stiffness can cause nuclear flattening and YAP1 translocation into the nucleus. Therefore, overexpression of YAP1 promotes cell proliferation in response to force or mechanical stimulation. Chan and coworkers have demonstrated that increased ECM stiffness promotes PASMC proliferation through HIPPO-YAP/TAZ signaling. Then, the YAP/TAZ-mediated transcriptional upregulation of Piezo1 would enhance mechanosensitive Ca2+ influx and Ca2+]cyt increases, which subsequently activate AKT/mTOR and ERK/MAPK signaling to promote PASMC proliferation. The AKT-mTOR-associated activation of YAP/TAZ can form a positive feedback circle/loop to further enhance or assure the proliferative response of PASMCs to mechanical stimulation. This mechanosensitive “network” may play an important role in the development and progression of pulmonary vascular remodeling in PAH and PH (58–60, 147).
Upregulation of Piezo1 in PASMCs Contributes to the Development of Experimental PH
Using freshly isolated PA, we found that Piezo1 was significantly upregulated in MCT-PH rats compared with control rats. The upregulated Piezo1 may represent solely an increased expression of Piezo1 in contractile PASMCs (or during PASMC contractile-to-proliferative phenotypic transition) and/or simply more proliferative PASMCs in PA in MCT-PH rats than in control rats. The upregulated Piezo1 in PA from MCT-PH rats was associated with a significant increase in protein levels of calpains (calpain-1 and calpain-2), Ca2+-sensitive proteins, and caveolin-1 (Cav1), an anchor protein required to form caveolae. In addition, the upregulated Piezo1 in PA from MCT-PH rats was associated with increases in pAKT, AKT, pmTOR, and mTOR, as well as the ratio of pAKT/AKT and pmTOR/mTOR. Furthermore, we also found that Cav1 protein expression level was significantly higher in PA (mainly containing PASMCs) isolated from MCT-PH rats than in PA isolated from control rats. One of the Cav1 functions is to form caveolae in the plasma membrane (150). The upregulated Cav1 in PA from MCT-PH rats is consistent with our previous observation that Cav1 protein level and caveolae number are increased in PASMCs from patients with idiopathic PAH compared with cells from healthy control subjects (151). These data direct us to speculate that the upregulated Cav1 may increase the formation of caveolae that result in clustering of upregulated receptors, ion channels, and chaperone proteins. The caveolae with functionally clustered receptors, channels, and regulatory proteins may subsequently enhance receptor- and stretch-mediated Ca2+ signaling by increasing Piezo1 interaction with proteins sensing mechanical stimulation (151–153).
Calpains are Ca2+-activated intracellular cysteine proteases or proteolytic enzymes. Once activated through Ca2+ binding, calpains can activate cell migration through their interaction with Talin and FAK (154, 155), cell invasion through MMP release and activation (156), and microvesicle formation and release through interaction with cortactin (157). Hypoxia- and VEGF-mediated activation of endothelial calpain-2 is implicated in angiogenesis via calpain-2-dependent activation of PI3K/AKT/eNOS pathway (158). Calpains are also demonstrated to activate pathological hypertrophy through the increase of nuclear translocation of NF-κB and NFAT and subsequent transcription of hypertrophic genes. ROS-mediated activation of calpains contribute to hypoxia-induced pulmonary vascular remodeling (159). Overexpression of calpain-2 significantly increases phosphorylated AKT in PASMCs, whereas inhibition of calpain-2 MDL28170 or siRNA attenuates PDGF-mediated PASMC proliferation via inhibition of AKT phosphorylation (160). The upregulated calpain-1/2 in freshly isolated PA from MCT-PH rats indicate that the calpain-associated cell migration and proliferation are important downstream effects resulting from the enhanced Ca2+ signaling in PASMCs during the development and progression of pulmonary vascular remodeling.
Summary
Piezo1, a mechanosensitive cation channel, is upregulated in proliferative PASMC compared with contractile PASMC. The upregulation of Piezo1 during PASMC contractile-to-proliferative phenotypic transition suggests that proliferative PASMC is more sensitive to mechanical stimulation. Physical stretch (e.g., membrane stretch) and flow shear stress can be important pathogenic stimuli for PASMC proliferation (161). In addition to Piezo1, other receptors (e.g., CaSR, Notch2, and Notch3) and channels (e.g., TRPC6, Orai2/STIM2, CALHM1, and CALHM2) (23, 24), along with AKT/mTOR signaling and caveolae-forming caveolin (Cav1) are upregulated during PASMC contractile-to-proliferative phenotypic transition and/or PASMC proliferation (compared with contractile PASMCs). During the contractile-to-proliferative phenotypic transition and during PASMC proliferation, reorganization of cytoskeletal network or cytoskeleton (e.g., F-actin) would increase intracellular and intercellular stretch or increase membrane stretch (54, 55, 162), and then activate the mechanosensitive complex, for example, Piezo1/TRPC6/Notch (all are reported to be mechanosensitive) (104, 163–167), in the plasma membrane of PASMCs, and contribute to propelling cells to go through the cell cycle mitosis and nuclear/cell division, and ultimately enhance PASMC proliferation. The “remodeling,” adaptation or upregulation of specific ion channels and membrane receptors in the plasma membrane of PASMCs associated to the contractile-to-proliferative phenotypic transition also takes place in pulmonary arterial remodeling from animals with experimental PH that are characterized by significantly increased PASMC proliferation. Development of specific and selective blockers for the mechanosensitive cation channel Piezo1 in proliferative PASMCs is potentially a novel therapeutic strategy for developing new drugs for treatment of precapillary PH.
GRANTS
This work was supported in part by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (R35 HL135807 and R01HL146764).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.C., A.M., and J.X.-J.Y. conceived and designed research; J.C., M.R., J.M., J.L., P.P.J., M.Z., T.Z., A.B., Z.W., S.P., R.P., M.M., and C.P. performed experiments; J.C., M.R., P.P.J., M.Z., T.Z., A.B., Z.W., S.P., R.P., M.M., C.P., Z.S., J.Y.-J.S., P.A.T., A.M., J.W., and J.X.-J.Y. analyzed data; J.C., M.R., J.M., J.L., P.P.J., M.Z., T.Z., A.B., Z.W., S.P., R.P., M.M., C.P., Z.S., J.Y.-J.S., P.A.T., A.M., J.W., and J.X.-J.Y. interpreted results of experiments; J.C., J.M., J.L., P.P.J., T.Z., Z.W., M.M., Z.S., A.M., J.W., and J.X.-J.Y. prepared figures; J.X.-J.Y. drafted manuscript; J.C., M.R., J.M., J.L., P.P.J., M.Z., T.Z., A.B., Z.W., S.P., R.P., M.M., C.P., Z.S., J.Y.-J.S., P.A.T., A.M., J.W., and J.X.-J.Y. edited and revised manuscript; J.C., M.R., J.M., J.L., P.P.J., M.Z., T.Z., A.B., Z.W. S.P., R.P., M.M., C.P., Z.S., J.Y.J.S., P.A.T., A.M., J.W., and J.X.-J.Y. approved final version of manuscript.
ACKNOWLEDGMENTS
Present addresses: M. Rodriguez, The University of Arizona, Tucson, AZ; P. P. Jain, Ottawa Hospital Research Institute, Ottawa, ON, Canada.
REFERENCES
- 1.Alencar GF, Owsiany KM, Karnewar S, Sukhavasi K, Mocci G, Nguyen AT, Williams CM, Shamsuzzaman S, Mokry M, Henderson CA, Haskins R, Baylis RA, Finn AV, McNamara CA, Zunder ER, Venkata V, Pasterkamp G, Bjorkegren J, Bekiranov S, Owens GK. Stem cell pluripotency genes Klf4 and Oct4 regulate complex SMC phenotypic changes critical in late-stage atherosclerotic lesion pathogenesis. Circulation 142: 2045–2059, 2020. doi: 10.1161/CIRCULATIONAHA.120.046672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin KA, Merenick BL, Ding M, Fetalvero KM, Rzucidlo EM, Kozul CD, Brown DJ, Chiu HY, Shyu M, Drapeau BL, Wagner RJ, Powell RJ. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem 282: 36112–36120, 2007. doi: 10.1074/jbc.M703914200. [DOI] [PubMed] [Google Scholar]
- 3.Nagao M, Lyu Q, Zhao Q, Wirka RC, Bagga J, Nguyen T, Cheng P, Kim JB, Pjanic M, Miano JM, Quertermous T. Coronary disease-associated gene TCF21 inhibits smooth muscle cell differentiation by blocking the myocardin-serum response factor pathway. Circ Res 126: 517–529, 2020. doi: 10.1161/CIRCRESAHA.119.315968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84: 767–801, 2004. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 5.Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg 45, Suppl A: A25–A32, 2007. doi: 10.1016/j.jvs.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Hassoun PM, Mouthon L, Barbera JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, Newman JH, Rabinovitch M, Schermuly R, Stenmark KR, Voelkel NF, Yuan JX, Humbert M. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol 54: S10–19, 2009. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 53: 1801887, 2019. doi: 10.1183/13993003.01887-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 54: S20–S31, 2009. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, Reid LM, Tuder RM. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol 43: 25S–32S, 2004. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 10.Pietra GG, Edwards WD, Kay JM, Rich S, Kernis J, Schloo B, Ayres SM, Bergofsky EH, Brundage BH, Detre KM. Histopathology of primary pulmonary hypertension. A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung, and Blood Institute, Primary Pulmonary Hypertension Registry. Circulation 80: 1198–1206, 1989. doi: 10.1161/01.cir.80.5.1198. [DOI] [PubMed] [Google Scholar]
- 11.Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet 361: 1533–1544, 2003. doi: 10.1016/S0140-6736(03)13167-4. [DOI] [PubMed] [Google Scholar]
- 12.Yi ES, Kim H, Ahn H, Strother J, Morris T, Masliah E, Hansen LA, Park K, Friedman PJ. Distribution of obstructive intimal lesions and their cellular phenotypes in chronic pulmonary hypertension. A morphometric and immunohistochemical study. Am J Respir Crit Care Med 162: 1577–1586, 2000. doi: 10.1164/ajrccm.162.4.9912131. [DOI] [PubMed] [Google Scholar]
- 13.Yi ES, Lee H, Yin S, Piguet P, Sarosi I, Kaufmann S, Tarpley J, Wang NS, Ulich TR. Platelet-derived growth factor causes pulmonary cell proliferation and collagen deposition in vivo. Am J Pathol 149: 539–548, 1996. [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu X, Aldred MA, McLean DL, Park H, Comhair SA, Greif DM, Erzurum SC, Chun HJ. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med 19: 74–82, 2013. doi: 10.1038/nm.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ntokou A, Dave JM, Kauffman AC, Sauler M, Ryu C, Hwa J, Herzog EL, Singh I, Saltzman WM, Greif DM. Macrophage-derived PDGF-B induces muscularization in murine and human pulmonary hypertension. JCI Insight 6: e139067, 2021. doi: 10.1172/jci.insight.139067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheikh AQ, Misra A, Rosas IO, Adams RH, Greif DM. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med 7: 308ra159, 2015. doi: 10.1126/scitranslmed.aaa9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheikh AQ, Saddouk FZ, Ntokou A, Mazurek R, Greif DM. Cell autonomous and non-cell autonomous regulation of SMC progenitors in pulmonary hypertension. Cell Rep 23: 1152–1165, 2018. doi: 10.1016/j.celrep.2018.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stevens T, Phan S, Frid MG, Alvarez D, Herzog E, Stenmark KR. Lung vascular cell heterogeneity: endothelium, smooth muscle, and fibroblasts. Proc Am Thorac Soc 5: 783–791, 2008. doi: 10.1513/pats.200803-027HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res 118: 692–702, 2016. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res 95: 156–164, 2012. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miano JM, Fisher EA, Majesky MW. Fate and state of vascular smooth muscle cells in atherosclerosis. Circulation 143: 2110–2116, 2021. doi: 10.1161/CIRCULATIONAHA.120.049922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musri MM, Coll-Bonfill N, Maron BA, Peinado VI, Wang RS, Altirriba J, Blanco I, Oldham WM, Tura-Ceide O, Garcia-Lucio J, de la Cruz-Thea B, Meister G, Loscalzo J, Barbera JA. MicroRNA dysregulation in pulmonary arteries from chronic obstructive pulmonary disease. Relationships with vascular remodeling. Am J Respir Cell Mol Biol 59: 490–499, 2018. doi: 10.1165/rcmb.2017-0040OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez RA, Wan J, Song S, Smith KA, Gu Y, Tauseef M, Tang H, Makino A, Mehta D, Yuan JX. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am J Physiol Cell Physiol 308: C581–C593, 2015. doi: 10.1152/ajpcell.00202.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez M, Chen J, Jain PP, Babicheva A, Xiong M, Li J, Lai N, Zhao T, Hernandez M, Balistrieri A, Parmisano S, Simonson T, Breen E, Valdez-Jasso D, Thistlethwaite PA, Shyy JY, Wang J, Garcia JGN, Makino A, Yuan JX. Upregulation of calcium homeostasis modulators in contractile-to-proliferative phenotypical transition of pulmonary arterial smooth muscle cells. Front Physiol 12: 714785, 2021. doi: 10.3389/fphys.2021.714785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kefauver JM, Ward AB, Patapoutian A. Discoveries in structure and physiology of mechanically activated ion channels. Nature 587: 567–576, 2020. doi: 10.1038/s41586-020-2933-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lhomme A, Gilbert G, Pele T, Deweirdt J, Henrion D, Baudrimont I, Campagnac M, Marthan R, Guibert C, Ducret T, Savineau JP, Quignard JF. Stretch-activated Piezo1 channel in endothelial cells relaxes mouse intrapulmonary arteries. Am J Respir Cell Mol Biol 60: 650–658, 2019. doi: 10.1165/rcmb.2018-0197OC. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, Sedo A, Hyman AJ, McKeown L, Young RS, Yuldasheva NY, Majeed Y, Wilson LA, Rode B, Bailey MA, Kim HR, Fu Z, Carter DA, Bilton J, Imrie H, Ajuh P, Dear TN, Cubbon RM, Kearney MT, Prasad RK, Evans PC, Ainscough JF, Beech DJ. Piezo1 integration of vascular architecture with physiological force. Nature 515: 279–282, 2014. doi: 10.1038/nature13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nonomura K, Lukacs V, Sweet DT, Goddard LM, Kanie A, Whitwam T, Ranade SS, Fujimori T, Kahn ML, Patapoutian A. Mechanically activated ion channel PIEZO1 is required for lymphatic valve formation. Proc Natl Acad Sci USA 115: 12817–12822, 2018. doi: 10.1073/pnas.1817070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asaro RJ, Lin K, Zhu Q. Mechanosensitivity occurs along the adhesome's force train and affects traction stress. Biophys J 117: 1599–1614, 2019. doi: 10.1016/j.bpj.2019.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jain PP, Zhao T, Xiong M, Song S, Lai N, Zheng Q, Chen J, Carr SG, Babicheva A, Izadi A, Rodriguez M, Rahimi S, Balistrieri F, Rahimi S, Simonson T, Valdez-Jasso D, Thistlethwaite PA, Shyy JY, Wang J, Makino A, Yuan JX. Halofuginone, a promising drug for treatment of pulmonary hypertension. Br J Pharmacol 178: 3373–3394, 2021. doi: 10.1111/bph.15442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang H, Babicheva A, McDermott KM, Gu Y, Ayon RJ, Song S, Wang Z, Gupta A, Zhou T, Sun X, Dash S, Wang Z, Balistrieri A, Zheng Q, Cordery AG, Desai AA, Rischard F, Khalpey Z, Wang J, Black SM, Garcia JGN, Makino A, Yuan JX. Endothelial HIF-2a contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol 314: L256–L275, 2018. doi: 10.1152/ajplung.00096.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfeiffer S, Leopold E, Schmidt K, Brunner F, Mayer B. Inhibition of nitric oxide synthesis by NG-nitro-l-arginine methyl ester (l-NAME): requirement for bioactivation to the free acid, NG-nitro-l-arginine. Br J Pharmacol 118: 1433–1440, 1996. doi: 10.1111/j.1476-5381.1996.tb15557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol 101: 746–752, 1990. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steudel W, Ichinose F, Huang PL, Hurford WE, Jones RC, Bevan JA, Fishman MC, Zapol WM. Pulmonary vasoconstriction and hypertension in mice with targeted disruption of the endothelial nitric oxide synthase (NOS 3) gene. Circ Res 81: 34–41, 1997. doi: 10.1161/01.RES.81.1.34. [DOI] [PubMed] [Google Scholar]
- 35.Wang D, Hsu K, Hwang CP, Chen HI. Measurement of nitric oxide release in the isolated perfused rat lung. Biochem Biophys Res Commun 208: 1016–1020, 1995. doi: 10.1006/bbrc.1995.1435. [DOI] [PubMed] [Google Scholar]
- 36.Botello-Smith WM, Jiang W, Zhang H, Ozkan AD, Lin YC, Pham CN, Lacroix JJ, Luo Y. A mechanism for the activation of the mechanosensitive Piezo1 channel by the small molecule Yoda1. Nat Commun 10: 4503, 2019. doi: 10.1038/s41467-019-12501-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans EL, Cuthbertson K, Endesh N, Rode B, Blythe NM, Hyman AJ, Hall SJ, Gaunt HJ, Ludlow MJ, Foster R, Beech DJ. Yoda1 analogue (Dooku1) which antagonizes Yoda1-evoked activation of Piezo1 and aortic relaxation. Br J Pharmacol 175: 1744–1759, 2018. doi: 10.1111/bph.14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Syeda R, Xu J, Dubin AE, Coste B, Mathur J, Huynh T, Matzen J, Lao J, Tully DC, Engels IH, Petrassi HM, Schumacher AM, Montal M, Bandell M, Patapoutian A. Chemical activation of the mechanotransduction channel Piezo1. eLife 4: e07369, 2015. doi: 10.7554/eLife.07369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chemla D, Castelain V, Humbert M, Hebert JL, Simonneau G, Lecarpentier Y, Herve P. New formula for predicting mean pulmonary artery pressure using systolic pulmonary artery pressure. Chest 126: 1313–1317, 2004. doi: 10.1378/chest.126.4.1313. [DOI] [PubMed] [Google Scholar]
- 40.Parasuraman S, Walker S, Loudon BL, Gollop ND, Wilson AM, Lowery C, Frenneaux MP. Assessment of pulmonary artery pressure by echocardiography—a comprehensive review. Int J Cardiol Heart Vasc 12: 45–51, 2016. doi: 10.1016/j.ijcha.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiong M, Jain PP, Chen J, Babicheva A, Zhao T, Alotaibi M, Kim NH, Lai N, Izadi A, Rodriguez M, Li J, Balistrieri A, Balistrieri F, Parmisano S, Sun X, Voldez-Jasso D, Shyy JY, Thistlethwaite PA, Wang J, Makino A, Yuan JX. Mouse model of experimental pulmonary hypertension: Lung angiogram and right heart catheterization. Pulm Circ 11: 20458940211041512, 2021. doi: 10.1177/20458940211041512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anwar T, Sinnett-Smith J, Jin YP, Reed EF, Rozengurt E. Ligation of HLA class I molecules induces YAP activation through Src in human endothelial cells. J Immunol 205: 1953–1961, 2020. doi: 10.4049/jimmunol.2000535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das A, Fischer RS, Pan D, Waterman CM. YAP nuclear localization in the absence of cell-cell contact is mediated by a filamentous actin-dependent, myosin II- and phospho-YAP-independent pathway during extracellular matrix mechanosensing. J Biol Chem 291: 6096–6110, 2016. doi: 10.1074/jbc.M115.708313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, Kosmalska AJ, Oria R, Kechagia JZ, Rico-Lastres P, Le Roux AL, Shanahan CM, Trepat X, Navajas D, Garcia-Manyes S, Roca-Cusachs P. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 171: 1397–1410.e4, 2017. doi: 10.1016/j.cell.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 45.Franklin JM, Ghosh RP, Shi Q, Reddick MP, Liphardt JT. Concerted localization-resets precede YAP-dependent transcription. Nat Commun 11: 4581, 2020. doi: 10.1038/s41467-020-18368-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li B, He J, Lv H, Liu Y, Lv X, Zhang C, Zhu Y, Ai D. c-Abl regulates YAPY357 phosphorylation to activate endothelial atherogenic responses to disturbed flow. J Clin Invest 129: 1167–1179, 2019. doi: 10.1172/JCI122440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J, Sinnett-Smith J, Stevens JV, Young SH, Rozengurt E. Biphasic regulation of Yes-associated protein (YAP) cellular localization, phosphorylation, and activity by G protein-coupled receptor agonists in intestinal epithelial cells: a novel role for protein kinase D (PKD). J Biol Chem 291: 17988–18005, 2016. doi: 10.1074/jbc.M115.711275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol 280: H746–H755, 2001. doi: 10.1152/ajpheart.2001.280.2.H746. [DOI] [PubMed] [Google Scholar]
- 49.Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol 284: C316–C330, 2003. doi: 10.1152/ajpcell.00125.2002. [DOI] [PubMed] [Google Scholar]
- 50.Bae C, Sachs F, Gottlieb PA. The mechanosensitive ion channel Piezo1 is inhibited by the peptide GsMTx4. Biochemistry 50: 6295–6300, 2011. doi: 10.1021/bi200770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Copp SW, Kim JS, Ruiz-Velasco V, Kaufman MP. The mechano-gated channel inhibitor GsMTx4 reduces the exercise pressor reflex in decerebrate rats. J Physiol 594: 641–655, 2016. doi: 10.1113/JP271714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gnanasambandam R, Ghatak C, Yasmann A, Nishizawa K, Sachs F, Ladokhin AS, Sukharev SI, Suchyna TM. GsMTx4: mechanism of inhibiting mechanosensitive ion channels. Biophys J 112: 31–45, 2017. doi: 10.1016/j.bpj.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhong M, Komarova Y, Rehman J, Malik AB. Mechanosensing Piezo channels in tissue homeostasis including their role in lungs. Pulm Circ 8: 2045894018767393, 2018. doi: 10.1177/2045894018767393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McNeill MC, Wray J, Sala-Newby GB, Hindmarch CCT, Smith SA, Ebrahimighaei R, Newby AC, Bond M. Nuclear actin regulates cell proliferation and migration via inhibition of SRF and TEAD. Biochim Biophys Acta Mol Cell Res 1867: 118691, 2020. doi: 10.1016/j.bbamcr.2020.118691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao JY, Hurst RE, Bales WD, Jones PL, Bass RA, Archer LT, Bell PB, Hemstreet GP III.. Cellular F-actin levels as a marker for cellular transformation: relationship to cell division and differentiation. Cancer Res 50: 2215–2220, 1990. [PubMed] [Google Scholar]
- 56.Bolanos-Garcia VM, Blundell TL. BUB1 and BUBR1: multifaceted kinases of the cell cycle. Trends Biochem Sci 36: 141–150, 2011. doi: 10.1016/j.tibs.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sato K, Kawashima S. Calpain function in the modulation of signal transduction molecules. Biol Chem 382: 743–751, 2001. doi: 10.1515/BC.2001.090. [DOI] [PubMed] [Google Scholar]
- 58.Bertero T, Cottrill KA, Annis S, Bhat B, Gochuico BR, Osorio JC, Rosas I, Haley KJ, Corey KE, Chung RT, Nelson Chau B, Chan SY. A YAP/TAZ-miR-130/301 molecular circuit exerts systems-level control of fibrosis in a network of human diseases and physiologic conditions. Sci Rep 5: 18277, 2015. doi: 10.1038/srep18277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, Hale A, Bhat B, Kaimal V, Zhang YY, Graham BB, Kumar R, Saggar R, Saggar R, Wallace WD, Ross DJ, Black SM, Fratz S, Fineman JR, Vargas SO, Haley KJ, Waxman AB, Chau BN, Fredenburgh LE, Chan SY. Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep 13: 1016–1032, 2015. doi: 10.1016/j.celrep.2015.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, Zhao J, Tai Y, Tang Y, Zhang YY, Rehman S, Sugahara M, Qi Z, Gorcsan J 3rd, Vargas SO, Saggar R, Saggar R, Wallace WD, Ross DJ, Haley KJ, Waxman AB, Parikh VN, De Marco T, Hsue PY, Morris A, Simon MA, Norris KA, Gaggioli C, Loscalzo J, Fessel J, Chan SY. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest 126: 3313–3335, 2016. doi: 10.1172/JCI86387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dasgupta I, McCollum D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J Biol Chem 294: 17693–17706, 2019. doi: 10.1074/jbc.REV119.007963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Yan X, Feng W, Wang Q, Shi W, Chai L, Zhang Q, Chen Y, Liu J, Qu Z, Xie X, Li M. S1P induces proliferation of pulmonary artery smooth muscle cells by promoting YAP-induced Notch3 expression and activation. J Biol Chem 296: 100599, 2021. doi: 10.1016/j.jbc.2021.100599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iglesias-Gonzalez J, Sanchez-Iglesias S, Beiras-Iglesias A, Soto-Otero R, Mendez-Alvarez E. A simple method for isolating rat brain mitochondria with high metabolic activity: effects of EDTA and EGTA. J Neurosci Methods 213: 39–42, 2013. doi: 10.1016/j.jneumeth.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 64.Dela Paz NG, Frangos JA. Yoda1-induced phosphorylation of Akt and ERK1/2 does not require Piezo1 activation. Biochem Biophys Res Commun 497: 220–225, 2018. doi: 10.1016/j.bbrc.2018.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang S, Chennupati R, Kaur H, Iring A, Wettschureck N, Offermanns S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J Clin Invest 126: 4527–4536, 2016. doi: 10.1172/JCI87343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.John L, Ko NL, Gokin A, Gokina N, Mandala M, Osol G. The Piezo1 cation channel mediates uterine artery shear stress mechanotransduction and vasodilation during rat pregnancy. Am J Physiol Heart Circ Physiol 315: H1019–H1026, 2018. doi: 10.1152/ajpheart.00103.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang H, Yamamura A, Yamamura H, Song S, Fraidenburg DR, Chen J, Gu Y, Pohl NM, Zhou T, Jimenez-Perez L, Ayon RJ, Desai AA, Goltzman D, Rischard F, Khalpey Z, Black SM, Garcia JG, Makino A, Yuan JX. Pathogenic role of calcium-sensing receptors in the development and progression of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 310: L846–L859, 2016. doi: 10.1152/ajplung.00050.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yamamura A, Nayeem MJ, Al Mamun A, Takahashi R, Hayashi H, Sato M. Platelet-derived growth factor up-regulates Ca2+-sensing receptors in idiopathic pulmonary arterial hypertension. FASEB J 33: 7363–7374, 2019. doi: 10.1096/fj.201802620R. [DOI] [PubMed] [Google Scholar]
- 69.Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, Macias J, Yuan JX, Jamieson SW, Thistlethwaite PA. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med 15: 1289–1297, 2009. doi: 10.1038/nm.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith KA, Voiriot G, Tang H, Fraidenburg DR, Song S, Yamamura H, Yamamura A, Guo Q, Wan J, Pohl NM, Tauseef M, Bodmer R, Ocorr K, Thistlethwaite PA, Haddad GG, Powell FL, Makino A, Mehta D, Yuan JX. Notch activation of Ca2+ signaling in the development of hypoxic pulmonary vasoconstriction and pulmonary hypertension. Am J Respir Cell Mol Biol 53: 355–367, 2015. doi: 10.1165/rcmb.2014-0235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Song S, Babicheva A, Zhao T, Ayon RJ, Rodriguez M, Rahimi S, Balistrieri F, Harrington A, Shyy JY, Thistlethwaite PA, Makino A, Yuan JX. Notch enhances Ca2+ entry by activating calcium-sensing receptors and inhibiting voltage-gated K+ channels. Am J Physiol Cell Physiol 318: C954–C968, 2020. doi: 10.1152/ajpcell.00487.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamamura H, Yamamura A, Ko EA, Pohl NM, Smith KA, Zeifman A, Powell FL, Thistlethwaite PA, Yuan JX. Activation of Notch signaling by short-term treatment with Jagged-1 enhances store-operated Ca2+ entry in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 306: C871–C878, 2014. doi: 10.1152/ajpcell.00221.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Babicheva A, Makino A, Yuan JX. mTOR signaling in pulmonary vascular disease: Pathogenic role and therapeutic target. Int J Mol Sci 22: 2144, 2021. doi: 10.3390/ijms22042144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP, Tuder RM, Kawut SM, Goncharova EA. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 129: 864–874, 2014. doi: 10.1161/CIRCULATIONAHA.113.004581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goncharova EA, Chan SY, Ventetuolo CE, Weissmann N, Schermuly RT, Mullin CJ, Gladwin MT. Update in pulmonary vascular diseases and right ventricular dysfunction 2019. Am J Respir Crit Care Med 202: 22–28, 2020. doi: 10.1164/rccm.202003-0576UP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Goncharova EA, Simon MA, Yuan JX. mTORC1 in pulmonary arterial hypertension. At the crossroads between vasoconstriction and vascular remodeling? Am J Respir Crit Care Med 201: 1177–1179, 2020. doi: 10.1164/rccm.202001-0087ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR, Offermanns S, Ye RD, Bonini MG, Minshall RD, Garcia JG, Machado RF, Makino A, Yuan JX. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L208–L220, 2015. doi: 10.1152/ajplung.00242.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tang H, Wu K, Wang J, Vinjamuri S, Gu Y, Song S, Wang Z, Zhang Q, Balistrieri A, Ayon RJ, Rischard F, Vanderpool R, Chen J, Zhou G, Desai AA, Black SM, Garcia JGN, Yuan JX, Makino A. Pathogenic role of mTORC1 and mTORC2 in pulmonary hypertension. JACC Basic Transl Sci 3: 744–762, 2018. doi: 10.1016/j.jacbts.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183, 2011. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 80.Hasegawa K, Fujii S, Matsumoto S, Tajiri Y, Kikuchi A, Kiyoshima T. YAP signaling induces PIEZO1 to promote oral squamous cell carcinoma cell proliferation. J Pathol 253: 80–93, 2021. doi: 10.1002/path.5553. [DOI] [PubMed] [Google Scholar]
- 81.Naeije R. Pulmonary vascular function. In: Pulmonary Circulation Diseases and their Treatment, edited by Peacock AJ, Naeije R, Rubin LJ.. Boca Raton, FL: CRC Press, 2016, p. 11–24. [Google Scholar]
- 82.Naeije R, Westerhof N, Pulmonary vascular function. In: Textbook of Pulmonary Vascular Disease, edited by Yuan JX-J, Garcia JGN, Hales Ca Rich S, Archer SL, West JB. New York: Springer, 2011, p. 61–72. [Google Scholar]
- 83.Pu X, Du L, Hu Y, Fan Y, Xu Q. Stem/progenitor cells and pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 41: 167–178, 2021. doi: 10.1161/ATVBAHA.120.315052. [DOI] [PubMed] [Google Scholar]
- 84.Lehners M, Dobrowinski H, Feil S, Feil R. cGMP signaling and vascular smooth muscle cell plasticity. J Cardiovasc Dev Dis 5: 20, 2018. doi: 10.3390/jcdd5020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yuan XJ, Bright RT, Aldinger AM, Rubin LJ. Nitric oxide inhibits serotonin-induced calcium release in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 272: L44–L50, 1997. doi: 10.1152/ajplung.1997.272.1.L44. [DOI] [PubMed] [Google Scholar]
- 86.Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. NO hyperpolarizes pulmonary artery smooth muscle cells and decreases the intracellular Ca2+ concentration by activating voltage-gated K+ channels. Proc Natl Acad Sci USA 93: 10489–10494, 1996. doi: 10.1073/pnas.93.19.10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Biasin V, Crnkovic S, Sahu-Osen A, Birnhuber A, El Agha E, Sinn K, Klepetko W, Olschewski A, Bellusci S, Marsh LM, Kwapiszewska G. PDGFRa and aSMA mark two distinct mesenchymal cell populations involved in parenchymal and vascular remodeling in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 318: L684–L697, 2020. doi: 10.1152/ajplung.00128.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lagna G, Ku MM, Nguyen PH, Neuman NA, Davis BN, Hata A. Control of phenotypic plasticity of smooth muscle cells by bone morphogenetic protein signaling through the myocardin-related transcription factors. J Biol Chem 282: 37244–37255, 2007. doi: 10.1074/jbc.M708137200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu M, Gomez D. Smooth muscle cell phenotypic diversity. Arterioscler Thromb Vasc Biol 39: 1715–1723, 2019. doi: 10.1161/ATVBAHA.119.312131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu Y, Zhang J, Yi B, Chen M, Qi J, Yin Y, Lu X, Jasmin JF, Sun J. Nur77 suppresses pulmonary artery smooth muscle cell proliferation through inhibition of the STAT3/Pim-1/NFAT pathway. Am J Respir Cell Mol Biol 50: 379–388, 2014. doi: 10.1165/rcmb.2013-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ackers-Johnson M, Talasila A, Sage AP, Long X, Bot I, Morrell NW, Bennett MR, Miano JM, Sinha S. Myocardin regulates vascular smooth muscle cell inflammatory activation and disease. Arterioscler Thromb Vasc Biol 35: 817–828, 2015. doi: 10.1161/ATVBAHA.114.305218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Petsophonsakul P, Furmanik M, Forsythe R, Dweck M, Schurink GW, Natour E, Reutelingsperger C, Jacobs M, Mees B, Schurgers L. Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arterioscler Thromb Vasc Biol 39: 1351–1368, 2019. doi: 10.1161/ATVBAHA.119.312787. [DOI] [PubMed] [Google Scholar]
- 93.Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res 109: 880–893, 2011. doi: 10.1161/CIRCRESAHA.111.240150. [DOI] [PubMed] [Google Scholar]
- 94.Zeng Z, Xia L, Fan X, Ostriker AC, Yarovinsky T, Su M, Zhang Y, Peng X, Xie Y, Pi L, Gu X, Chung SK, Martin KA, Liu R, Hwa J, Tang WH. Platelet-derived miR-223 promotes a phenotypic switch in arterial injury repair. J Clin Invest 129: 1372–1386, 2019. doi: 10.1172/JCI124508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dandre F, Owens GK. Platelet-derived growth factor-BB and Ets-1 transcription factor negatively regulate transcription of multiple smooth muscle cell differentiation marker genes. Am J Physiol Heart Circ Physiol 286: H2042–H2051, 2004. doi: 10.1152/ajpheart.00625.2003. [DOI] [PubMed] [Google Scholar]
- 96.Murgai M, Ju W, Eason M, Kline J, Beury DW, Kaczanowska S, Miettinen MM, Kruhlak M, Lei H, Shern JF, Cherepanova OA, Owens GK, Kaplan RN. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat Med 23: 1176–1190, 2017. doi: 10.1038/nm.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Salmon M, Gomez D, Greene E, Shankman L, Owens GK. Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22a promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ Res 111: 685–696, 2012. doi: 10.1161/CIRCRESAHA.112.269811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yoshida T, Owens GK. Molecular determinants of vascular smooth muscle cell diversity. Circ Res 96: 280–291, 2005. doi: 10.1161/01.RES.0000155951.62152.2e. [DOI] [PubMed] [Google Scholar]
- 99.Kim CW, Aronow WS, Dutta T, Spevack DM, Frishman WH. Pulmonary artery denervation as an innovative treatment for pulmonary hypertension with and without heart failure. Cardiol Rev 29: 89–95, 2021. doi: 10.1097/CRD.0000000000000299. [DOI] [PubMed] [Google Scholar]
- 100.Le T, Makar C, Morway P, Hoftman N, Umar S. Pulmonary artery denervation: a novel treatment modality for pulmonary hypertension. J Thorac Dis 11: 1094–1096, 2019. doi: 10.21037/jtd.2019.02.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Romanov A, Cherniavskiy A, Novikova N, Edemskiy A, Ponomarev D, Shabanov V, Losik D, Elesin D, Stenin I, Mikheenko I, Zhizhov R, Kretov E, Pokushalov E, Po SS, Martynyuk TV, Steinberg JS. Pulmonary artery denervation for patients with residual pulmonary hypertension after pulmonary endarterectomy. J Am Coll Cardiol 76: 916–926, 2020. doi: 10.1016/j.jacc.2020.06.064. [DOI] [PubMed] [Google Scholar]
- 102.Rothman AMK, Vachiery JL, Howard LS, Mikhail GW, Lang IM, Jonas M, Kiely DG, Shav D, Shabtay O, Avriel A, Lewis GD, Rosenzweig EB, Kirtane AJ, Kim NH, Mahmud E, McLaughlain VV, Chetcuti S, Leon MB, Ben-Yehuda O, Rubin LJ. Intravascular ultrasound pulmonary artery denervation to treat pulmonary arterial hypertension (TROPHY1): multicenter, early feasibility study. JACC Cardiovasc Interv 13: 989–999, 2020. doi: 10.1016/j.jcin.2019.12.027. [DOI] [PubMed] [Google Scholar]
- 103.Zhang H, Zhang J, Chen M, Xie DJ, Kan J, Yu W, Li XB, Xu T, Gu Y, Dong J, Gu H, Han Y, Chen SL. Pulmonary artery denervation significantly increases 6-min walk distance for patients with combined pre- and post-capillary pulmonary hypertension associated with left heart failure: The PADN-5 study. JACC Cardiovasc Interv 12: 274–284, 2019. doi: 10.1016/j.jcin.2018.09.021. [DOI] [PubMed] [Google Scholar]
- 104.Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330: 55–60, 2010. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Murthy SE, Dubin AE, Patapoutian A. Piezos thrive under pressure: mechanically activated ion channels in health and disease. Nat Rev Mol Cell Biol 18: 771–783, 2017. doi: 10.1038/nrm.2017.92. [DOI] [PubMed] [Google Scholar]
- 106.Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, Xu J, Mathur J, Bandell M, Coste B, Li YS, Chien S, Patapoutian A. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci USA 111: 10347–10352, 2014. doi: 10.1073/pnas.1409233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Means AR. Calcium, calmodulin and cell cycle regulation. FEBS Lett 347: 1–4, 1994. doi: 10.1016/0014-5793(94)00492-7. [DOI] [PubMed] [Google Scholar]
- 108.Choi J, Husain M. Calmodulin-mediated cell cycle regulation: new mechanisms for old observations. Cell Cycle 5: 2183–2186, 2006. doi: 10.4161/cc.5.19.3265. [DOI] [PubMed] [Google Scholar]
- 109.Ichikawa J, Inoue R. TRPC6 regulates cell cycle progression by modulating membrane potential in bone marrow stromal cells. Br J Pharmacol 171: 5280–5294, 2014. doi: 10.1111/bph.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kahl CR, Means AR. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr Rev 24: 719–736, 2003. doi: 10.1210/er.2003-0008. [DOI] [PubMed] [Google Scholar]
- 111.Ellefsen KL, Holt JR, Chang AC, Nourse JL, Arulmoli J, Mekhdjian AH, Abuwarda H, Tombola F, Flanagan LA, Dunn AR, Parker I, Pathak MM. Myosin-II mediated traction forces evoke localized Piezo1-dependent Ca2+ flickers. Commun Biol 2: 298, 2019. doi: 10.1038/s42003-019-0514-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nourse JL, Pathak MM. How cells channel their stress: interplay between Piezo1 and the cytoskeleton. Semin Cell Dev Biol 71: 3–12, 2017. doi: 10.1016/j.semcdb.2017.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kapanidou M, Lee S, Bolanos-Garcia VM. BubR1 kinase: protection against aneuploidy and premature aging. Trends Mol Med 21: 364–372, 2015. doi: 10.1016/j.molmed.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 114.Kyuragi R, Matsumoto T, Harada Y, Saito S, Onimaru M, Nakatsu Y, Tsuzuki T, Nomura M, Yonemitsu Y, Maehara Y. BubR1 insufficiency inhibits neointimal hyperplasia through impaired vascular smooth muscle cell proliferation in mice. Arterioscler Thromb Vasc Biol 35: 341–347, 2015. [Erratum in Arterioscler Thromb Vasc Biol 35: e17, 2015]. doi: 10.1161/ATVBAHA.114.304737. [DOI] [PubMed] [Google Scholar]
- 115.Dorfmuller P, Gunther S, Ghigna MR, Thomas de Montpreville V, Boulate D, Paul JF, Jais X, Decante B, Simonneau G, Dartevelle P, Humbert M, Fadel E, Mercier O. Microvascular disease in chronic thromboembolic pulmonary hypertension: a role for pulmonary veins and systemic vasculature. Eur Respir J 44: 1275–1288, 2014. doi: 10.1183/09031936.00169113. [DOI] [PubMed] [Google Scholar]
- 116.Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol 8: 443–455, 2011. doi: 10.1038/nrcardio.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pathak MM, Nourse JL, Tran T, Hwe J, Arulmoli J, Le DT, Bernardis E, Flanagan LA, Tombola F. Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proc Natl Acad Sci USA 111: 16148–16153, 2014. doi: 10.1073/pnas.1409802111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li M, Riddle S, Kumar S, Poczobutt J, McKeon BA, Frid MG, Ostaff M, Reisz JA, Nemkov T, Fini MA, Laux A, Hu CJ, El Kasmi KC, D'Alessandro A, Brown RD, Zhang H, Stenmark KR. Microenvironmental regulation of macrophage transcriptomic and metabolomic profiles in pulmonary hypertension. Front Immunol 12: 640718, 2021. doi: 10.3389/fimmu.2021.640718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pugliese SC, Kumar S, Janssen WJ, Graham BB, Frid MG, Riddle SR, El Kasmi KC, Stenmark KR. A time- and compartment-specific activation of lung macrophages in hypoxic pulmonary hypertension. J Immunol 198: 4802–4812, 2017. doi: 10.4049/jimmunol.1601692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Geng J, Shi Y, Zhang J, Yang B, Wang P, Yuan W, Zhao H, Li J, Qin F, Hong L, Xie C, Deng X, Sun Y, Wu C, Chen L, Zhou D. TLR4 signalling via Piezo1 engages and enhances the macrophage mediated host response during bacterial infection. Nat Commun 12: 3519, 2021. doi: 10.1038/s41467-021-23683-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Atcha H, Jairaman A, Holt JR, Meli VS, Nagalla RR, Veerasubramanian PK, Brumm KT, Lim HE, Othy S, Cahalan MD, Pathak MM, Liu WF. Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat Commun 12: 3256, 2021. doi: 10.1038/s41467-021-23482-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lee MY, Park C, Ha SE, Park PJ, Berent RM, Jorgensen BG, Corrigan RD, Grainger N, Blair PJ, Slivano OJ, Miano JM, Ward SM, Smith TK, Sanders KM, Ro S. Serum response factor regulates smooth muscle contractility via myotonic dystrophy protein kinases and L-type calcium channels. PLoS One 12: e0171262, 2017. doi: 10.1371/journal.pone.0171262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Perisic Matic L, Rykaczewska U, Razuvaev A, Sabater-Lleal M, Lengquist M, Miller CL, Ericsson I, Rohl S, Kronqvist M, Aldi S, Magne J, Paloschi V, Vesterlund M, Li Y, Jin H, Diez MG, Roy J, Baldassarre D, Veglia F, Humphries SE, de Faire U, Tremoli E, Odeberg J, Vukojevic V, Lehtio J, Maegdefessel L, Ehrenborg E, Paulsson-Berne G, Hansson GK, Lindeman JH, Eriksson P, Quertermous T, Hamsten A, Hedin U. Phenotypic modulation of smooth muscle cells in atherosclerosis is associated with downregulation of LMOD1, SYNPO2, PDLIM7, PLN, and SYNM. Arterioscler Thromb Vasc Biol 36: 1947–1961, 2016. doi: 10.1161/ATVBAHA.116.307893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Numaga-Tomita T, Shimauchi T, Oda S, Tanaka T, Nishiyama K, Nishimura A, Birnbaumer L, Mori Y, Nishida M. TRPC6 regulates phenotypic switching of vascular smooth muscle cells through plasma membrane potential-dependent coupling with PTEN. FASEB J 33: 9785–9796, 2019. doi: 10.1096/fj.201802811R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Beck K, Wu BJ, Ni J, Santiago FS, Malabanan KP, Li C, Wang Y, Khachigian LM, Stocker R. Interplay between heme oxygenase-1 and the multifunctional transcription factor yin yang 1 in the inhibition of intimal hyperplasia. Circ Res 107: 1490–1497, 2010. doi: 10.1161/CIRCRESAHA.110.231985. [DOI] [PubMed] [Google Scholar]
- 126.Wamhoff BR, Bowles DK, McDonald OG, Sinha S, Somlyo AP, Somlyo AV, Owens GK. L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a Rho kinase/myocardin/SRF-dependent mechanism. Circ Res 95: 406–414, 2004. doi: 10.1161/01.RES.0000138582.36921.9e. [DOI] [PubMed] [Google Scholar]
- 127.Firth AL, Yao W, Remillard CV, Ogawa A, Yuan JX. Upregulation of Oct-4 isoforms in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 298: L548–L557, 2010. doi: 10.1152/ajplung.00314.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Si R, Zhang Q, Tsuji-Hosokawa A, Watanabe M, Willson C, Lai N, Wang J, Dai A, Scott BT, Dillmann WH, Yuan JX, Makino A. Overexpression of p53 due to excess protein O-GlcNAcylation is associated with coronary microvascular disease in type 2 diabetes. Cardiovasc Res 116: 1186–1198, 2020. doi: 10.1093/cvr/cvz216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Z, Yang K, Zheng Q, Zhang C, Tang H, Babicheva A, Jiang Q, Li M, Chen Y, Carr SG, Wu K, Zhang Q, Balistrieri A, Wang C, Song S, Ayon RJ, Desai AA, Black SM, Garcia JGN, Makino A, Yuan JX, Lu W, Wang J. Divergent changes of p53 in pulmonary arterial endothelial and smooth muscle cells involved in the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 316: L216–L228, 2019. doi: 10.1152/ajplung.00538.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yu Y, Keller SH, Remillard CV, Safrina O, Nicholson A, Zhang SL, Jiang W, Vangala N, Landsberg JW, Wang JY, Thistlethwaite PA, Channick RN, Robbins IM, Loyd JE, Ghofrani HA, Grimminger F, Schermuly RT, Cahalan MD, Rubin LJ, Yuan JX. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 119: 2313–2322, 2009. doi: 10.1161/CIRCULATIONAHA.108.782458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Birnhuber A, Biasin V, Schnoegl D, Marsh LM, Kwapiszewska G. Transcription factor Fra-2 and its emerging role in matrix deposition, proliferation and inflammation in chronic lung diseases. Cell Signal 64: 109408, 2019. doi: 10.1016/j.cellsig.2019.109408. [DOI] [PubMed] [Google Scholar]
- 132.Yu Y, Platoshyn O, Zhang J, Krick S, Zhao Y, Rubin LJ, Rothman A, Yuan JX. c-Jun decreases voltage-gated K+ channel activity in pulmonary artery smooth muscle cells. Circulation 104: 1557–1563, 2001. doi: 10.1161/hc3801.095662. [DOI] [PubMed] [Google Scholar]
- 133.Zabini D, Crnkovic S, Xu H, Tscherner M, Ghanim B, Klepetko W, Olschewski A, Kwapiszewska G, Marsh LM. High-mobility group box-1 induces vascular remodelling processes via c-Jun activation. J Cell Mol Med 19: 1151–1161, 2015. doi: 10.1111/jcmm.12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.He RL, Wu ZJ, Liu XR, Gui LX, Wang RX, Lin MJ. Calcineurin/NFAT signaling modulates pulmonary artery smooth muscle cell proliferation, migration and apoptosis in monocrotaline-induced pulmonary arterial hypertension rats. Cell Physiol Biochem 49: 172–189, 2018. doi: 10.1159/000492852. [DOI] [PubMed] [Google Scholar]
- 135.Roger I, Milara J, Montero P, Cortijo J. The role of JAK/STAT molecular pathway in vascular remodeling associated with pulmonary hypertension. Int J Mol Sci 22: 4980, 2021. doi: 10.3390/ijms22094980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yerabolu D, Weiss A, Kojonazarov B, Boehm M, Schlueter BC, Ruppert C, Gunther A, Jonigk D, Grimminger F, Ghofrani HA, Seeger W, Weissmann N, Schermuly RT. Targeting Jak-Stat signaling in experimental pulmonary hypertension. Am J Respir Cell Mol Biol 64: 100–114, 2021. doi: 10.1165/rcmb.2019-0431OC. [DOI] [PubMed] [Google Scholar]
- 137.Garat CV, Majka SM, Sullivan TM, Crossno JT Jr, Reusch JEB, Klemm DJ. CREB depletion in smooth muscle cells promotes medial thickening, adventitial fibrosis and elicits pulmonary hypertension. Pulm Circ 10: 2045894019898374, 2020. doi: 10.1177/2045894019898374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Song S, Carr SG, McDermott KM, Rodriguez M, Babicheva A, Balistrieri A, Ayon RJ, Wang J, Makino A, Yuan JX. STIM2 (stromal interaction molecule 2)-mediated increase in resting cytosolic free Ca2+ concentration stimulates PASMC proliferation in pulmonary arterial hypertension. Hypertension 71: 518–529, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fernandez RA, Sundivakkam P, Smith KA, Zeifman AS, Drennan AR, Yuan JX. Pathogenic role of store-operated and receptor-operated Ca2+ channels in pulmonary arterial hypertension. J Signal Transduct 2012: 951497, 2012. doi: 10.1155/2012/951497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kito H, Yamamura H, Suzuki Y, Yamamura H, Ohya S, Asai K, Imaizumi Y. Regulation of store-operated Ca2+ entry activity by cell cycle dependent up-regulation of Orai2 in brain capillary endothelial cells. Biochem Biophys Res Commun 459: 457–462, 2015. doi: 10.1016/j.bbrc.2015.02.127. [DOI] [PubMed] [Google Scholar]
- 141.Smyth JT, Petranka JG, Boyles RR, DeHaven WI, Fukushima M, Johnson KL, Williams JG, Putney JW Jr.. Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat Cell Biol 11: 1465–1472, 2009. [Erratum in Nat Cell Biol 12:199, 2010]. doi: 10.1038/ncb1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tani D, Monteilh-Zoller MK, Fleig A, Penner R. Cell cycle-dependent regulation of store-operated ICRAC and Mg2+-nucleotide-regulated MagNuM (TRPM7) currents. Cell Calcium 41: 249–260, 2007. doi: 10.1016/j.ceca.2006.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yu F, Hubrack SZ, Chakraborty S, Sun L, Alcantara-Adap E, Kulkarni R, Billing AM, Graumann J, Taylor CW, Machaca K. Remodeling of ER-plasma membrane contact sites but not STIM1 phosphorylation inhibits Ca(2+) influx in mitosis. Proc Natl Acad Sci USA 116: 10392–10401, 2019. doi: 10.1073/pnas.1821399116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 101: 13861–13866, 2004. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cribbs LL. T-type Ca2+ channels in vascular smooth muscle: multiple functions. Cell Calcium 40: 221–230, 2006. doi: 10.1016/j.ceca.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 146.Kuga T, Kobayashi S, Hirakawa Y, Kanaide H, Takeshita A. Cell cycle-dependent expression of L- and T-type Ca2+ currents in rat aortic smooth muscle cells in primary culture. Circ Res 79: 14–19, 1996. doi: 10.1161/01.res.79.1.14. [DOI] [PubMed] [Google Scholar]
- 147.Dieffenbach PB, Haeger CM, Coronata AMF, Choi KM, Varelas X, Tschumperlin DJ, Fredenburgh LE. Arterial stiffness induces remodeling phenotypes in pulmonary artery smooth muscle cells via YAP/TAZ-mediated repression of cyclooxygenase-2. Am J Physiol Lung Cell Mol Physiol 313: L628–L647, 2017. doi: 10.1152/ajplung.00173.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mosaddad SA, Salari Y, Amookhteh S, Soufdoost RS, Seifalian A, Bonakdar S, Safaeinejad F, Moghaddam MM, Tebyanian H. Response to mechanical cues by interplay of YAP/TAZ transcription factors and key mechanical checkpoints of the cell: a comprehensive review. Cell Physiol Biochem 55: 33–60, 2021. doi: 10.33594/000000325. [DOI] [PubMed] [Google Scholar]
- 149.Duchemin AL, Vignes H, Vermot J. Mechanically activated piezo channels modulate outflow tract valve development through the Yap1 and Klf2-Notch signaling axis. Elife 8: e44706, 2019. doi: 10.7554/eLife.44706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Parton RG, Hanzal-Bayer M, Hancock JF. Biogenesis of caveolae: a structural model for caveolin-induced domain formation. J Cell Sci 119: 787–796, 2006. doi: 10.1242/jcs.02853. [DOI] [PubMed] [Google Scholar]
- 151.Patel HH, Zhang S, Murray F, Suda RY, Head BP, Yokoyama U, Swaney JS, Niesman IR, Schermuly RT, Pullamsetti SS, Thistlethwaite PA, Miyanohara A, Farquhar MG, Yuan JX, Insel PA. Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. FASEB J 21: 2970–2979, 2007. doi: 10.1096/fj.07-8424com. [DOI] [PubMed] [Google Scholar]
- 152.Hardin CD, Vallejo J. Caveolins in vascular smooth muscle: form organizing function. Cardiovasc Res 69: 808–815, 2006. doi: 10.1016/j.cardiores.2005.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Isshiki M, Anderson RG. Function of caveolae in Ca2+ entry and Ca2+-dependent signal transduction. Traffic 4: 717–723, 2003. doi: 10.1034/j.1600-0854.2003.00130.x. [DOI] [PubMed] [Google Scholar]
- 154.Chan KT, Bennin DA, Huttenlocher A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J Biol Chem 285: 11418–11426, 2010. doi: 10.1074/jbc.M109.090746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kerstein PC, Patel KM, Gomez TM. Calpain-mediated proteolysis of Talin and FAK regulates adhesion dynamics necessary for axon guidance. J Neurosci 37: 1568–1580, 2017. doi: 10.1523/JNEUROSCI.2769-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hoskin V, Szeto A, Ghaffari A, Greer PA, Cote GP, Elliott BE. Ezrin regulates focal adhesion and invadopodia dynamics by altering calpain activity to promote breast cancer cell invasion. Mol Biol Cell 26: 3464–3479, 2015. doi: 10.1091/mbc.E14-12-1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Suzuki K, Sorimachi H, Yoshizawa T, Kinbara K, Ishiura S. Calpain: novel family members, activation, and physiologic function. Biol Chem Hoppe Seyler 376: 523–529, 1995. doi: 10.1515/bchm3.1995.376.9.523. [DOI] [PubMed] [Google Scholar]
- 158.Zhang Y, Liu NM, Wang Y, Youn JY, Cai H. Endothelial cell calpain as a critical modulator of angiogenesis. Biochim Biophys Acta Mol Basis Dis 1863: 1326–1335, 2017. doi: 10.1016/j.bbadis.2017.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhu J, Kovacs L, Han W, Liu G, Huo Y, Lucas R, Fulton D, Greer PA, Su Y. Reactive oxygen species-dependent calpain activation contributes to airway and pulmonary vascular remodeling in chronic obstructive pulmonary disease. Antioxid Redox Signal 31: 804–818, 2019. doi: 10.1089/ars.2018.7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Abeyrathna P, Kovacs L, Han W, Su Y. Calpain-2 activates Akt via TGF-b1-mTORC2 pathway in pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 311: C24–C34, 2016. doi: 10.1152/ajpcell.00295.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hyman AJ, Tumova S, Beech DJ. Piezo1 channels in vascular development and the sensing of shear stress. Curr Top Membr 79: 37–57, 2017. doi: 10.1016/bs.ctm.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 162.Ladoux B, Nicolas A. Physically based principles of cell adhesion mechanosensitivity in tissues. Rep Prog Phys 75: 116601, 2012. doi: 10.1088/0034-4885/75/11/116601. [DOI] [PubMed] [Google Scholar]
- 163.Gottlieb P, Folgering J, Maroto R, Raso A, Wood TG, Kurosky A, Bowman C, Bichet D, Patel A, Sachs F, Martinac B, Hamill OP, Honore E. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch 455: 1097–1103, 2008. doi: 10.1007/s00424-007-0359-3. [DOI] [PubMed] [Google Scholar]
- 164.Gudipaty SA, Lindblom J, Loftus PD, Redd MJ, Edes K, Davey CF, Krishnegowda V, Rosenblatt J. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature 543: 118–121, 2017. doi: 10.1038/nature21407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Paolini A, Fontana F, Pham VC, Rodel CJ, Abdelilah-Seyfried S. Mechanosensitive Notch-Dll4 and Klf2-Wnt9 signaling pathways intersect in guiding valvulogenesis in zebrafish. Cell Rep 37: 109782, 2021. doi: 10.1016/j.celrep.2021.109782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ridone P, Vassalli M, Martinac B. Piezo1 mechanosensitive channels: what are they and why are they important. Biophys Rev 11: 795–805, 2019. doi: 10.1007/s12551-019-00584-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stassen O, Ristori T, Sahlgren CM. Notch in mechanotransduction—from molecular mechanosensitivity to tissue mechanostasis. J Cell Sci 133: jcs250738, 2020. doi: 10.1242/jcs.250738. [DOI] [PMC free article] [PubMed] [Google Scholar]