

Abstract
A novel series of N-benzimidazol-2-yl-α-aryl nitrones 3a–j is synthesized via simple one-pot condensation/oxidation of 2-aminobenzimidazole, an aromatic aldehyde and m-chloro perbenzoic acid (m-CPBA) as an effective oxidant using Mn(NO3)2·6H2O as an efficient catalyst at room temperature. All synthesized N-benzimidazolyl nitrones were identified using FTIR, NMR and mass spectroscopy. Also, stability energy theory calculations were performed and 1H NMR computational spectra were generated for the isomeric structures of 3a; the results show that the stability order is oxaziridine (4) followed by the nitrones 3aE and 3aZ. Also, comparing the computational spectroscopy results with the experimental data shows great accordance with nitrone 3aE. Among the remarkable points of this protocol, stable N-heterocyclic nitrones were prepared for the first time from raw materials under mild oxidative conditions. Therefore, they can easily be applied as high-potential intermediates for synthesizing valuable heterocycles in mild conditions. Due to benefits such as the use of inexpensive and available catalysts, short reaction times, high yields, facile workup to obtain pure product, and facile separation of the side product (m-chlorobenzoic acid), this simple protocol complies greatly with the principles of green chemistry.
A novel series of N-benzimidazol-2-yl-α-aryl nitrones 3a–j is synthesized via one-pot condensation/oxidation of 2-aminobenzimidazole, an aromatic aldehyde and m-CPBA as an effective oxidant using Mn(NO3)2·6H2O as an efficient catalyst.
Introduction
The name “nitrone” is a brevity which was proposed by Pfeiffer in 1916 for a functional group in organic compounds composed of an N-oxide of an imine.1 The term is a contraction of the terms “nitrogen ketone”.2,3 Nitrones have a 1,3-dipole structure; they are used in 1,3-dipolar cycloadditions4 (especially for synthesis of oxaziridine, isoxazolines, oxadiazolidines, etc.5) and formal cycloadditions in the synthesis of many natural products.6 Increased reactivity of nitrones with metal derivatives has been discovered; therefore, one of the most important applications of nitrones is that some of them can be used as ligands in inorganic chemistry.7 Nitrones are also applied as spin traps in biological studies8 as well as therapies in age-related diseases.9 Many nitrone derivatives have pharmacological activity and form necessary parts of the molecular structures of main drugs.10–12 Also, they are forecasted to be effective inhibitors of acidic and microbial corrosion.13 Nitrones are found in many natural alkaloids, such as Huperzine J and K obtained from Huperzia serrata,14 Notoamide U,15 and Plakinamine isolated from the genus Corticium16 (Fig. 1).
Fig. 1. Natural structures with nitrone frameworks.

Resonance structures (1–3) can be considered for nitrone (Fig. 2). All the azometine N-oxide groups are dipolar, and typical nitrone reactions are dependent on this dipolar state. Due to the importance of nitrones, various methods for their synthesis have been reported. Generally, these are divided into four main methods, including (1) alkylation of oximes with various nitrogen reagents without using oxidant,2,17 (2) the condensation reaction of a carbonyl compound and a N-alkyl or aryl mono-substituted hydroxylamine,18 (3) oxidation of secondary amines or N,N-substituted hydroxylamines using oxidizing agents,19–22 and (4) oxidation of imines by oxidizing reagents such as m-CPBA, hydrogen peroxide (H2O2), and alkyl hydroperoxides23,24 (Fig. 2).
Fig. 2. Four main pathways of synthesis of nitrones and some of their synthetic applications.

Recently, a few methods have been developed for the preparation of nitrones through one-pot condensation/oxidation of primary amines and aldehydes in the presence of methyl trioxorhenium (MTO),25 Nafion-immobilized MoOCl4,26 silica-immobilized oxo-rhenium,27 or graphite oxide (and oxone as the oxidant) as the catalyst.28
There are also reports that isomerization of oxaziridines in the presence of Lewis acids propels the formation of nitrones.29 Although each of these synthetic strategies has valuable properties, in some cases, these methods present major drawbacks such as low selectivity, procedures that require harsh conditions, tedious workup and purification, inaccessibility of precursors (the hydroxylamines), formation of oxidation by-products, use of hazardous solvents, and expensive catalysts and oxidants. However, the contribution of new, more efficient procedures in this field can still be interesting and beneficial.
According to the above, this paper reports a facile and clean synthetic method for the preparation of novel nitrones containing the N-benzimidazole moiety under mild conditions for the first time. These N-heterocyclic nitrones, 3a–j, are obtained through one-pot reactions of 2-aminobenzimidazole, various aromatic aldehydes and m-CPBA as an oxidant reagent by applying Mn(NO3)2·6H2O as an effective catalyst at room temperature, with good to excellent yields and simple workup compared to previous reports.
Results and discussion
Firstly, to evaluate and optimize the synthesis conditions of target compounds, the reaction of 2-aminobenzimidazole with 4-nitro benzaldehyde and m-CPBA in ethanol in the presence of 10 mol% Cu(NO3)2 catalyst at room temperature was performed as a model reaction (Scheme 1).
Scheme 1. The model reaction for optimizing the reaction conditions.

After several tests (Table 1), the best result was obtained when the reaction occurred in the presence of 10 mol% Mn(NO3)2·6H2O in ethanol. In this case, the reaction yield and time were 95% and 25 minutes, respectively (Table 1, entry 5).
Optimizing the reaction conditions for the synthesis of compound 3g.
| Entry | Catalyst | Catalyst loading (mol%) | Solvent | Temperature (°C) | Time (min) | Yielda (%) |
|---|---|---|---|---|---|---|
| 1 | Cu(NO3)2·6H2O | 10 | EtOH | rt | 30 | 65 |
| 2 | Co(NO3)2·6H2O | 10 | EtOH | rt | 30 | 70 |
| 3 | Zn(NO3)2·6H2O | 10 | EtOH | rt | 60 | 60 |
| 4 | Ni(NO3)2·6H2O | 10 | EtOH | rt | 30 | 87 |
| 5 | Mn(NO 3 ) 2 ·6H 2 O | 10 | EtOH | rt | 25 | 95 |
| 6 | Mn(NO3)2·6H2O | 5 | EtOH | rt | 40 | 70 |
| 7 | Mn(NO3)2·6H2O | 15 | EtOH | rt | 25 | 80 |
| 8 | Mn(NO3)2·6H2O | 10 | CH3CN | rt | 40 | 65 |
| 9 | Mn(NO3)2·6H2O | 10 | MeOH | rt | 35 | 78 |
| 10 | Mn(NO3)2·6H2O | 10 | CH2Cl2 | rt | 45 | 70 |
| 8 | MnCl2·6H2O | 10 | EtOH | rt | 30 | 85 |
| 9 | CoCl2·6H2O | 10 | EtOH | rt | 50 | 75 |
| 10 | ZnCl2·H2O | 10 | EtOH | rt | 80 | 50 |
| 11 | CuCl2·H2O | 10 | EtOH | rt | 35 | 72 |
| 12 | NiCl2·H2O | 10 | EtOH | rt | 35 | 70 |
| 13 | Mn(NO3)2·6H2O | 10 | H2O | rt | 60 | 0 |
| 14 | Mn(NO3)2·6H2O | 10 | H2O | 90 | 60 | 0 |
| 15 | Mn(NO3)2·6H2O | 10 | — | rt | 50 | 20 |
| 16 | Mn(NO3)2·6H2O | 10 | — | 80 | 50 | 25 |
| 17 | Mn(NO3)2·6H2O | 5 | — | 80 | 50 | 25 |
| 18 | CANb | 10 | EtOH | rt | 50 | 0 |
| 19 | Ca(IO3)2 | 10 | EtOH | rt | 50 | 0 |
| 20 | — | — | EtOH | rt | 50 | 0 |
Isolated yield.
Cerium(iv) ammonium nitrate.
These results created an incentive to develop an efficient and simple method to produce nitrones 3a–jvia the reaction of 2-aminobenzimidazole with various aromatic aldehydes containing electron withdrawing groups or electron donating groups and a per-acid (m-CPBA) under the same conditions (Scheme 2).
Scheme 2. The synthetic pathway for the preparation of nitrones 3a–j.

The results are summarized in Table 2, which shows that the yields of the one-pot condensation/oxidation method used for the production of nitrones from the raw materials are high to excellent. Moreover, the workup is simple, the amount of catalyst used is low and the reaction time is short in comparison to previous methods.
Synthesis of the nitrone derivatives 3a–ja.
| Entry | Compound | Time (min) | Mp (oC) | Yieldb (%) |
|---|---|---|---|---|
| 1 |

|
30 | 180–182 | 89 |
| 2 |

|
30 | 228 | 85 |
| 3 |

|
40 | 212–214 | 86 |
| 4 |

|
25 | 241–242 | 91 |
| 5 |

|
25 | 260–262 | 85 |
| 6 |

|
40 | 201–204 | 85 |
| 7 |

|
25 | 210–212 | 95 |
| 8 |

|
35 | 187–188 | 93 |
| 9 |

|
25 | 188–190 | 95 |
| 10 |

|
35 | 189–190 | 91 |
Reaction conditions: 0.1 mmol 2-aminobenzimidazole (1) with 0.1 mmol of the aromatic aldehyde in the presence of 10 mol% Mn(NO3)2·6H2O in ethanol (5 mL), addition of m-CPBA (0.12 mmol) and room temperature.
Isolated yield.
According to our previous knowledge, here, we suggest a probable mechanism for the synthesis of the nitrone in Scheme 3. Initially, an empty orbital of Mn2+ cation as a Lewis acid activates the carbonyl group of the aromatic aldehyde for nucleophilic amine (1) attack, followed by catalytic oxidation and the loss of a water molecule to form the intermediate I.34 The corresponding Schiff base, under catalytic activation, undergoes nucleophile attack by the third molecule, m-CPBA, instantly leading to the formation of intermediate II. Immediately, upon the third catalytic activation of the intermediate II following intermolecular nucleophile attack, cyclization of N-benzimidazolyl oxaziridines can be accomplished.29 Finally, in the presence of Mn2+ cation as a Lewis acid, the corresponding nitrone is formed through rearrangement.28
Scheme 3. Proposed mechanism for the synthesis of compounds 3a–j.

In this project, we also synthesized nitrones 3a–j in accordance with general method 4 (Fig. 2) in two steps, but with lower yields (42% to 70%), longer reaction times (1.5 to 6 hours) and more complex and tight separation of the pure product (Scheme 4). The reason for the rapid formation and stability of these nitrones (compared to the formation of the oxaziridine ring or amide) may be the presence of intermolecular hydrogen bonds between the hydrogen of the imidazole30 and the nitrone oxygen31–33 (Scheme 4). On the other hand, the intense resonance between nitrone, aryl and the benzimidazole ring accelerates this process.
Scheme 4. Synthesis of compounds 3a–j in two steps without catalyst and intermolecular hydrogen bonding (III).

All the synthesized N-benzimidazolyl nitrones are new, and their structures were confirmed using FT-IR and NMR spectroscopy and mass spectral data. In the 1H-NMR spectra, the shift of the CH N (Schiff base) proton signals from 9.38 to 9.78 ppm34 to 7.82 to 8.80 ppm confirmed the formation of the nitrone structure. In general, in the IR spectrum for a nitrone system, it is expected that there will be five characteristic vibrational frequencies, including C N, N–O, and C–N stretching and C–H in-plane and out-of-plane bending frequencies.31 A red shift at the C NO-vibrational frequency in the N-benzimidazolyl nitrones (1535 to 1581 cm−1) relative to the corresponding Schiff bases (1601 to 1621 cm−1) as well as the appearance of a relatively strong band for N–O stretching in nitrones (1012 to 1103 cm−1) confirms the formation of these products. Other signals were revealed in the expected regions which are consistent with their structures. In the mass spectra of compounds 3a–j, molecular ion peaks (5% to 100%) and all expected fragments consistent with the structures of the annular products were seen.
To show the efficiency of this method, it was compared with reported results in the literature for the direct synthesis of nitrones. The obtained results are listed in Table 3. The results clearly show that this catalytic procedure is superior to other methods in terms of reaction time and yield, economical convenience, etc. Therefore, it can be considered as one of the best choices for facile and direct synthesis of N-heterocyclic nitrones under mild conditions.
Comparison of the catalytic procedure with other methods for the direct synthesis of N-aryl or alkyl nitrones.





Theoretical studies
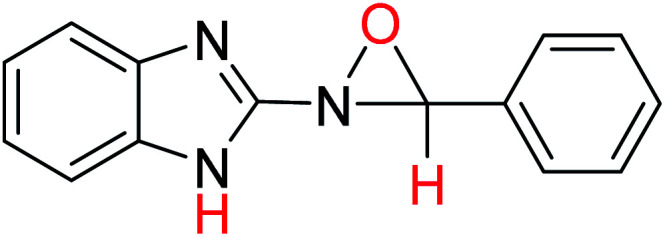
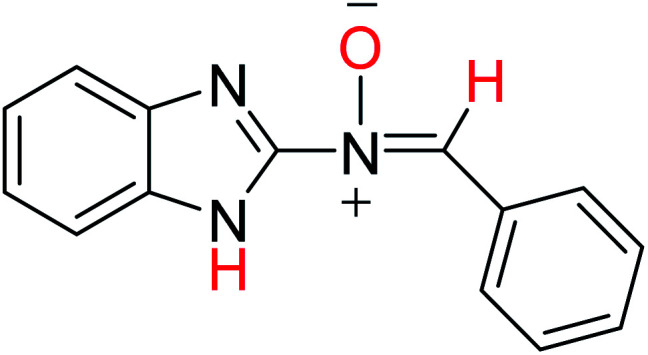
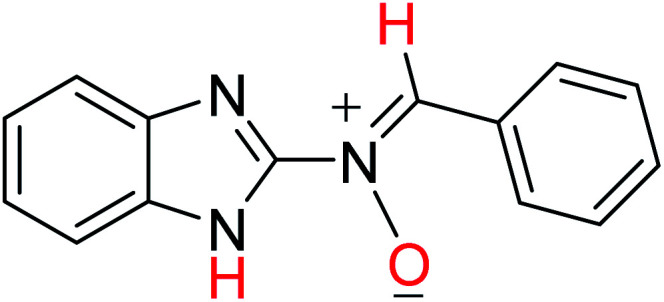
In order to investigate the different probable molecular structures of the nitrones and oxaziridine, a theoretical approach was applied.31,35 The optimized structures and relative energy comparison of these molecules are shown in Fig. 3. The figure indicates that the relative energies of the isomers are in the order oxaziridine (4) > 3aE > 3aZ. The stability of 3aE can be attributed to its full conjugation and aromatic rings.
Fig. 3. The optimized structures and relative energy comparison of the nitrone (3aZ, E) and oxaziridine (4) molecules.

Also, 1H NMR data for the nitrone and oxaziridine molecules in the gas phase were calculated with the DFT method using the 6-311G (d) basis set. The results and experimental data are presented in Table 4. The table indicates that obtained experimental results for the N–H (11.07 ppm) bond and C–H (8.68 ppm) bond most resemble the 3aE form of nitrone in the theoretical approach.
The 1H NMR data (calculated and experimental) and total energies for the isomeric compounds 3a.
| No. | Compound | E a (E, kcal mol−1) | C–Hb (ppm) (experimental) | N–Hb (ppm) (experimental) |
|---|---|---|---|---|
| 4 |
|
−779.68669653 | 6.62 | 8.60 |
| (−489253.40207) | — | — | ||
| 3aE |
|
−779.69570020 | 8.52 | 10.62 |
| (−489259.05187) | (8.68) | (11.07) | ||
| 3aZ |
|
−779.71431871 | 9.55 | 10.13 |
| (−489270.73499) | (8.68) | (11.07) |
Total energies in hartree.
1H NMR data in the gas phase.
Conclusion
In this project, we succeeded in designing a new procedure for the mild and facile synthesis of novel N-benzimidazolyl-α-aryl nitrones for the first time. These compounds were obtained through one-pot reactions of 2-aminobenzimidazole, various aromatic aldehydes and m-CPBA as an effective oxidizing reagent in the presence of a catalytic amount of Mn(NO3)2·6H2O at room temperature in good to excellent yields. The method was demonstrated to be simple both in conducting the reaction and in isolating the pure products; thus, it is a useful procedure for the synthesis of the target compounds. Also, theoretical studies showed that the 3aE form of nitrone is close to the neutral structure of oxaziridine in terms of energy stability and is very similar to the experimental spectral data.
Experimental
All the consumed chemicals were purchased from Merck or Fluka. Melting points were determined using an electro-thermal digital apparatus and are uncorrected. A Galaxy Series FT-IR 5000 spectrometer (using KBr discs) was used to record the IR spectra. 1H and 13C NMR spectra were acquired in DMSO-d6 on a Bruker (300 and 500 MHz) spectrometer. Chemical shifts (δ) were presented in ppm using tetramethyl silane (TMS) as an internal standard. The mass spectra were recorded on an Agilent spectrometer, model 5975C VL MSD, with a Triple-Axis Detector at 70 eV. The reactions were monitored by thin layer chromatography (TLC) using silica gel on F254 aluminum sheets (Merck).
Computational methods
Thermodynamic data and investigation of the IR spectra of the nitrone and oxaziridine molecules in the gas phase were calculated with the DFT method using the B3LYP/6-311G (d) basis set.35,36 The study of the NMR spectra was carried out using the NMR = giao b3lyp/6-311+g(2d,p) level of theory. Theoretical calculations were performed using the Gaussian 09 package and the Gauss-View molecular visualization program.37
General preparation of nitrones 3a–j
To a solution of 2-aminobenzimidazole (1 mmol) and the corresponding aromatic aldehyde (1 mmol) in ethanol (5 mL), Mn(NO3)2·6H2O (10 mol%, 0.027 g) was added. The reaction mixture was stirred magnetically at room temperature. Then, m-CPBA (1.2 mmol) was added to the mixture. The reaction progress was monitored by TLC (petroleum ether : ethyl acetate 2 : 1). After completion of the reaction, aqueous NaHCO3 (10%, 15 mL) was added to the mixture, and the resulting precipitate was filtrated to afford pure nitrones 3a–j. For further purity, these products can be crystallized from a mixture of ether: n-hexane (1 : 1). Also, increasing the amount of HCl (10%) in the filtrate causes the m-chlorobenzoic acid to precipitate; then, the extraneous acid product is separated by filtration.
Spectroscopic data for nitrones 3a–j
N-(1H-Benzo[d]imidazol-2-yl)-α-phenyl nitrone (3a)
Light yellow solid. IR (KBr, νmax): 3269 (N–H), 3095 (C–H), 1683, 1608 (C N), 1535 (–C N–O), 1419, 1222 (C C), 1382 (H–C N), 1263 (C–N), 1029 (N–O), 898, 759 (C–H), 619 cm−1; 1H NMR (300 MHz, DMSO-d6): δH 11.07 (br, 1H, NH, the NH protons disappeared upon D2O addition), 8.68 (s br, 1H, H–C N+–O−), 7.98 (s br, 2H, H–Ar), 7.49–7.08 (t br, 6H, H–Ar) ppm, 13C NMR (DMSO-d6, 75 MHz): δC 170.1, 153.6, 139.2, 133.2, 132.4, 130.8, 130.2, 129.3, 128.1, 122.2, 111.5 ppm; MS (m/z, %): 237.2 (M+, 5), 156.0 (75), 133.1 (100), 111.1 (58), 85.2 (40), 71.2 (46), 57.2 (62), 43.2 (43).
N-(1H-Benzo[d]imidazol-2-yl)-α-(2-chlorophenyl) nitrone (3b)
Gray solid. IR (KBr, νmax): 3377 (N–H), 1666, 1631 (C N), 1579 (–C N–O), 1519, 1435, 1298, 1273 (C C), 1456 (H–C N), 1176 (C–N), 1143, 1020 (N–O), 866 (C–Cl), 744 (C–H) cm−1; 1H NMR (DMSO-d6, 300 MHz): δH 12.27 (br, 1H, NH), 7.68 (d, J = 5.91 Hz, 1H, H–C N+–O−), 7.55–7.45 (m, 6H, H–Ar), 7.12 (s br, 2H, H–Ar) ppm, 13C NMR (DMSO-d6, 125 MHz): δC 167.2, 147.3, 135.8, 135.2, 131.4, 130.2, 129.7, 129.3, 127.0, 121.4, 119.9, 113.8 ppm, MS (m/z, %): 271.1 (M+, 46), 236.1 (52), 208.1 (48), 139.1 (100), 111.1 (71), 75.1 (34).
N-(1H-Benzo[d]imidazol-2-yl)-α-(3-chlorophenyl) nitrone (3c)
Gray solid. IR (KBr, νmax): 3307 (N–H), 1624, 1604 (C N), 1575 (–C N–O), 1596, 1344 (C C), 1473 (H–C N), 1263 (C–N), 1024 (N–O), 918 (C–Cl), 758 (C–H), 740 cm−1; 1H NMR (DMSO-d6, 300 MHz): δH 12.22 (br, 1H, NH), 7.68 (d, J = 5.49 Hz, 1H, H–C N+–O−), 7.55–7.48 (t br, 6H, H–Ar), 7.14 (s, 2H, H–Ar) ppm, 13C NMR (DMSO-d6, 75 MHz): δC 167.2, 147.2, 135.6, 135.0, 131.2, 129.9, 129.5, 129.0, 126.9, 124.0, 121.2, 113.6 ppm, MS (m/z, %): 271.1 (M+, 21), 243.1 (18), 160.1 (28), 139.1 (100), 111.1 (71), 75.1 (27).
N-(1H-Benzo[d]imidazol-2-yl)-α-(4-chlorophenyl) nitrone (3d)
Gray solid. IR (KBr, νmax): 3348 (N–H), 1653, 1641 (C N), 1571 (–C N–O), 1591, 1523, 1435 (C C), 1462 (H–C N), 1271 (C–N), 1093 (N–O), 844 (C–Cl), 750 (C–H) cm−1; 1H NMR (DMSO-d6, 500 MHz): δH 12.33 (br, 1H, NH, the NH protons disappeared upon D2O addition), 8.15 (s br, 1H, H–C N+–O−), 7.89 (s br, 2H, H–Ar), 7.65–7.53 (t br, 4H, H–Ar), 7.15 (s, 2H, H–Ar) ppm; 13C NMR (DMSO-d6, 125 MHz): δC 168.8, 150.1, 136.8, 136.4, 134.3, 132.8, 131.0, 130.2, 128.2, 121.8, 119.6, 112.7 ppm, MS (m/z, %): 271.1 (M+, 48), 236.2 (51), 208.2 (41), 139.2 (100), 111.2 (45), 75.2 (17).
N-(1H-Benzo[d]imidazol-2-yl)-α-(4-bromophenyl) nitrone (3e)
Gray solid. IR (KBr, νmax): 3311 (N–H), 1660, 1624 (C N), 1550 (–C N–O), 1571, 1394 (C C), 1340 (H–C N), 1273 (C–N), 1012 (N–O), 808 (C–Br), 769 (C–H), 736 cm−1; 1H NMR (DMSO-d6, 500 MHz): δH 12.36 (br, 1H, NH), 8.07 (s br, 2H, H–C N+–O− and H–Ar), 7.70 (br, 2H, H–Ar), 7.44 (br, 2H, H–Ar), 7.14 (br, 3H, H–Ar) ppm, 13C NMR (DMSO-d6, 125 MHz): δC 169.1, 150.2, 134.8, 132.7, 131.1, 130.4, 129.3, 128.3, 127.7, 125.5, 121.8, 119.0, 112.7 ppm, MS (m/z, %): 315.2 (M+, 57), 287.1 (53), 185.1 (100), 155.1 (50), 132 (14), 105.2 (28), 76.2 (28).
N-(1H-Benzo[d]imidazol-2-yl)-α-(3-nitrophenyl) nitrone (3f)
Red solid. IR (KBr, νmax): 3317 (N–H), 1705, 1618 (C N), 1581 (–C N–O), 1531, 1348 (–NO2), 1597, 1469, 1419 (C C), 1400 (H–C N), 1263 (C–N), 1080 (N–O), 923, 752 (C–H), 731, 715 cm−1; 1H NMR (300 MHz, DMSO-d6): δH (the NH proton exchanges with deuterium in D2O, which is present in DMSO-d6), 8.96 (s, 1H, H–C N+–O−), 8.57–8.50 (m, 1H, H–Ar), 8.36 (d, J = 6.66 Hz, 1H, H–Ar), 7.91–7.74 (m, 2H, H–Ar), 7.34 (d, J = 7.50 Hz, 2H, H–Ar), 7.15 (br, 2H, H–Ar) ppm; 13C NMR (DMSO-d6, 75 MHz): δC 170.1, 137.6, 135.3, 135.0, 131.9, 131.4, 130.1, 128.9, 125.7, 124.4, 123.4, 122.4, 112.5 ppm, MS (m/z, %): 282.1 (M+, 50), 160.1 (50), 132.1 (100), 105.1 (50), 76.1 (37).
N-(1H-Benzo[d]imidazol-2-yl)-α-(4-nitrophenyl) nitrone (3g)
Red solid. IR (KBr, νmax): 3346 (N–H), 1670, 1651 (C N), 1575 (–C N–O), 1521, 1352 (–NO2), 1597, 1309 (C C), 1462 (H–C N), 1278 (C–N), 1103 (N–O), 850, 740 (C–H), 715 cm−1; 1H NMR (DMSO-d6, 300 MHz): δH 12.56 (br, 1H, NH), 8.34 (d, J = 6.99 Hz, 3H, H–C N+–O− and H–Ar), 8.13 (d, J = 8.07 Hz, 2H, H–Ar), 7.44–7.18 (d br, 4H, H–Ar) ppm; 13C NMR (DMSO-d6, 75 MHz): δC 170.6, 152.4, 151.0, 149.4, 143.3, 140.4, 131.0, 130.1, 124.6, 123.7, 122.8, 112.5 ppm; MS (m/z, %): 282 (M+, 100), 254 (57), 235 (12), 219 (12), 160 (99), 121, 104 (77).
N-(1H-Benzo[d]imidazol-2-yl)-α-(p-tolyl) nitrone (3h)
Gray solid. IR (KBr, νmax): 3396 (N–H), 2924 (C–H), 1658, 1633 (C N), 1573 (–C N–O), 1527, 1456, 1313, 1257 (C C), 1431 (H–C N), 1168 (C–N), 1020 (N–O), 842, 767, 750 (C–H), 611 cm−1; 1H NMR (300 MHz, DMSO-d6): δH (the NH proton exchanges with deuterium in D2O, which is present in DMSO-d6), 8.08 (d, J = 7.50 Hz, 2H, H–C N+–O− and H–Ar), 7.08–7.42 (m, 7H, H–Ar), 2.36 (s, 3H, CH3) ppm; 13C NMR (DMSO-d6, 75 MHz): δC 168.8, 151.1, 142.0, 135.7, 133.0, 129.2, 128.8, 121.3, 113.7 ppm, MS (m/z, %): 251.2 (M+, 96), 223.2 (56), 119.1 (100), 91.1 (87), 65.1 (25).
N-(1H-Benzo[d]imidazol-2-yl)-α-(4-methoxyphenyl) nitrone (3i)
Gray solid. IR (KBr, νmax): 3327 (N–H), 1660, 1624 (C N), 1556 (–C N–O), 1602, 1573, 1458, 1519 (C C), 1346 (H–C N), 1286 (C–O), 1273 (C–N), 1020 (N–O), 910, 767 (C–H), 731 cm−1; 1H NMR (DMSO-d6, 500 MHz): δH 12.18 (br, 1H, NH), 8.12 (d, J = 6.55 Hz, 1H, H–C N+–O−), 7.87 (t, J = 8.55 Hz, 1H, H–Ar), 7.66 (d, J = 7.75 Hz, 1H, H–Ar), 7.53–7.44 (q br, 2H, H–Ar), 7.12–6.90 (m, 4H, H–Ar), 3.83 (s, 3H, OMe) ppm; 13C NMR (DMSO-d6, 125 MHz): δC 166.9, 162.3, 148.5, 134.7, 131.6, 130.2, 125.8, 121.1, 114.4, 113.5, 55.3 ppm; MS (m/z, %): 267.1 (M+, 13), 263.1 (14), 220.1 (72), 133.1 (54), 105.1 (27), 77.1 (25), 44 (100).
N-(1H-Benzo[d]imidazol-2-yl)-α-(2-hydroxyphenyl) nitrone (3j)
Gray solid. IR (KBr, νmax): 3369 (N–H), 1683, 1635 (C N), 1560 (–C N–O) 1580, 1381 (C C), 1458 (H–C N), 1290 (C–O), 1273 (C–N), 1047 (N–O), 744 (C–H) cm−1; 1H NMR (DMSO-d6, 500 MHz): δH 11.95 (br, 1H, NH), 11.04 (br, 1H, OH), 8.11 (s, 1H, H–C N+–O−), 7.85 (s, 1H, H–Ar), 7.37–7.28 (d br, 2H, H–Ar), 6.97 (t br, 2H, H–Ar), 6.87–6.78 (m, 5H, H–Ar) ppm; 13C NMR (DMSO-d6, 125 MHz): δC 169.5, 154.0, 138.6, 133.68, 133.63, 133.2, 131.0, 130.3, 129.3, 128.1, 121.7, 111.6 ppm; EIMS (m/z, %): 253.1 (M+, 22), 225.1 (13), 160.0 (17), 121.1 (100), 93.1 (54), 65.1 (82).
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The gratitude of the authors goes to the research community of the Chemistry Department of Payame Noor University, who provided financial and technical support for this project.
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08570j
References
- Padwa A. and Pearson W. H., in Synthetic application of 1,3-dipolar cycloaddition chemistry toward heterocycles and natural products, John Wiley and Sons, Hoboken/NJ, vol. 59, 2003, ISBN: 978-0-471-28061-3 [Google Scholar]
- Smith L. Chem. Rev. 1938;23:193–285. doi: 10.1021/cr60075a001. [DOI] [Google Scholar]
- Hamer J. M. Macaluso A. Chem. Rev. 1964;64:473–495. doi: 10.1021/cr60230a006. [DOI] [Google Scholar]
- Zhu T. Xiang J. Liu Z. Dang Q. Bai X. Synlett. 2015;26:238–242. doi: 10.1055/s-0034-1380120. [DOI] [Google Scholar]
- (a) Yang J. Synlett. 2012;23:2293–2297. doi: 10.1055/s-0032-1317096. [DOI] [Google Scholar]; (b) Murahashi S.-I. Imada Y. Chem. Rev. 2019;119:4684–4716. doi: 10.1021/acs.chemrev.8b00476. [DOI] [PubMed] [Google Scholar]
- Goti A. Fedi V. Nannelli L. Sarlo F. D. Brandi A. Synlett. 1997;5:577–579. doi: 10.1055/s-1997-3222. [DOI] [Google Scholar]
- (a) Masson G. Py S. Vallée Y. Angew. Chem. 2002;41:1772–1775. doi: 10.1002/1521-3773(20020517)41:10<1772::AID-ANIE1772>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]; (b) Young I. S. Kerr M. A. A. Angew. Chem. 2003;26:3023–3026. doi: 10.1002/anie.200351573. [DOI] [PubMed] [Google Scholar]; (c) Cardona F. Goti A. Angew. Chem. 2005;44:7832–7835. doi: 10.1002/anie.200502640. [DOI] [PubMed] [Google Scholar]
- (a) Zhang H. Joseph J. Vasquez-Vivar J. Karoui H. Nsanzumuhire C. Martásek P. Tordo P. Kalyanaraman B. FEBS Lett. 2000;473:58–62. doi: 10.1016/S0014-5793(00)01498-8. [DOI] [PubMed] [Google Scholar]; (b) Durand G. Polidori A. Salles J. P. Pucci B. Bioorg. Med. Chem. Lett. 2003;13:859–862. doi: 10.1016/S0960-894X(02)01079-X. [DOI] [PubMed] [Google Scholar]; (c) Morandat S. Durand G. Polidori A. Desigaux L. Bortolato M. Roux B. Pucci B. Langmuir. 2003;19:9699–9705. doi: 10.1021/la034470t. [DOI] [Google Scholar]
- Floyd R. A. Aging Cell. 2006;5:51–57. doi: 10.1111/j.1474-9726.2006.00189.x. [DOI] [PubMed] [Google Scholar]
- Floyd R. A. Kopke R. D. Choi C. H. Foster S. B. Doblas S. Towner R. A. Free Radical Biol. Med. 2008;45:1361–1374. doi: 10.1016/j.freeradbiomed.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D. P. Zhang K. Zhang Z. J. Sun Y. W. Guo B. J. Wang Y. Q. Hoi P. M. Han Y. F. Lee S. M. Neurochem. Int. 2014;78:76–85. doi: 10.1016/j.neuint.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Floyd R. A. Neto H. C. C. F. Zimmerman G. A. Hensley K. Towner R. A. Free Radical Biol. Med. 2013;62:145–156. doi: 10.1016/j.freeradbiomed.2013.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. Zhao K. Chen G. J. Chem. 2015;2015:1–6. [Google Scholar]
- Gao W. Li Y. Jiang S. Zhu D. Planta Med. 2000;66:664–667. doi: 10.1055/s-2000-8630. [DOI] [PubMed] [Google Scholar]
- (a) Kagiyama I. Kato H. Nehira T. Frisvad J. C. Sherman D. H. Williams R. M. Tsukamoto S. Angew. Chem. 2016;55:1128–1132. doi: 10.1002/anie.201509462. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Choo C. Y. Hirasawa Y. Karimata C. Koyama K. Sekiguchi M. Kobayashid J. Morita H. Bioorg. Med. Chem. 2007;15:1703–1707. doi: 10.1016/j.bmc.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Lee H. S. Seo Y. Rho J. R. Shin J. Paul V. J. J. Nat. Prod. 2001;64:1474–1476. doi: 10.1021/np0101649. [DOI] [PubMed] [Google Scholar]
- Smith P. A. S. Robertson J. E. J. Am. Chem. Soc. 1962;84:1197–1204. doi: 10.1021/ja00866a027. [DOI] [Google Scholar]
- Sandler S. R. and Karo W., Organic Functional Group Preparations, E-Book, 1989, vol. 3, pp. 351–376, ISBN: 9780080925585 [Google Scholar]
- Zajac J. W. W. Walters T. R. Darcy M. G. J. Org. Chem. 1988;53:5856–5860. doi: 10.1021/jo00260a012. [DOI] [Google Scholar]
- Mitsui H. Zenki S. Shiota T. Murahashi S. I. J. Chem. Soc., Chem. Commun. 1984;13:874–875. doi: 10.1039/C39840000874. [DOI] [Google Scholar]
- Murahashi S. Mitsui H. Shiota T. Tsuda T. J. Org. Chem. 1990;55:1736–1744. doi: 10.1021/jo00293a013. [DOI] [Google Scholar]
- Murahashi S. I. Shiota T. Imada Y. Org. Synth. 1991;70:265. [Google Scholar]
- Yamazaki S. Bull. Chem. Soc. Jpn. 1997;70:877–883. doi: 10.1246/bcsj.70.877. [DOI] [Google Scholar]
- (a) Gella C. Ferrer E. Alibes R. Busqu E. F. De March P. Figueredo M. Font J. J. Org. Chem. 2009;74:6365–6367. doi: 10.1021/jo901108u. [DOI] [PubMed] [Google Scholar]; (b) Saladino R. Neri V. Cardona F. Goti A. Adv. Synth. Catal. 2004;346:639–647. doi: 10.1002/adsc.200303223. [DOI] [Google Scholar]; (c) Soldaini G. Cardona F. Goti A. Org. Lett. 2007;9:473–676. doi: 10.1021/ol062862w. [DOI] [PubMed] [Google Scholar]
- Cardona F. Bonanni M. Soldaini G. Goti A. ChemSusChem. 2008;1:327–332. doi: 10.1002/cssc.200700156. [DOI] [PubMed] [Google Scholar]
- Singh B. Jain S. L. Khatri P. K. Sain B. Green Chem. 2009;11:1941–1944. doi: 10.1039/B914402A. [DOI] [Google Scholar]
- Singh B. Jain S. L. Rana B. S. Khatri P. K. Sinha A. K. Sain B. ChemCatChem. 2010;2:1260–1264. doi: 10.1002/cctc.201000121. [DOI] [Google Scholar]
- Mirza-Aghayan M. Molaee Tavana M. Boukherroub R. Tetrahedron Lett. 2014;55:5471–5474. doi: 10.1016/j.tetlet.2014.08.049. [DOI] [Google Scholar]
- Williamson K. S. Michaelis D. J. Yoon T. P. Chem. Rev. 2014;114:8016–8036. doi: 10.1021/cr400611n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nájera C. Yus M. Tetrahedron Lett. 2015;56:2623–2633. doi: 10.1016/j.tetlet.2015.03.099. [DOI] [Google Scholar]
- Shindo H. Umezawa B. Chem. Pharm. Bull. 1962;10:492–503. doi: 10.1248/cpb.10.492. [DOI] [PubMed] [Google Scholar]
- Tanekazu K. Masumi Y. Yutaka M. Bull. Chem. Soc. Jpn. 1960;33:109–114. doi: 10.1246/bcsj.33.109. [DOI] [Google Scholar]
- Petkova E. G. Domasevitch K. Gorichkob M. V. Zuba V. Y. Lampekaa R. D. Z. Naturforsch., B: Chem. Sci. 2014;56:1264–1270. [Google Scholar]
- Mobinikhaledi A. Forughifar N. Kalhor M. Turk. J. Chem. 2010;34:367–373. [Google Scholar]
- Lee C. Yang W. Parr R. G. Phys. Rev. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- Francl M. M. Pietro W. J. Hehre W. J. Binkley J. S. Gordon M. S. DeFrees D. J. Pople J. A. J. Chem. Phys. 1982;77:3654–3665. doi: 10.1063/1.444267. [DOI] [Google Scholar]
- Frisch A., Gaussian 09 Reference, Gaussian, Inc., Wallingford, CT, 2009 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.