

Abstract
The COVID-19 pandemic generates a global threat to public health and continuously emerging SARS-CoV-2 variants bring a great challenge to the development of both vaccines and antiviral agents. In this study, we identified UA-18 and its optimized analog UA-30 via the hit-to-lead strategy as novel SARS-CoV-2 fusion inhibitors. The lead compound UA-30 showed potent antiviral activity against infectious SARS-CoV-2 (wuhan-HU-1 variant) in Vero-E6 cells and was also effective against infection of diverse pseudotyped SARS-CoV-2 variants with mutations in the S protein including the Omicron and Delta variants. More importantly, UA-30 might target the cavity between S1 and S2 subunits to stabilize the prefusion state of the SARS-CoV-2 S protein, thus leading to interfering with virus-cell membrane fusion. This study offers a set of novel SARS-CoV-2 fusion inhibitors against SARS-CoV-2 and its variants based on the 3-O-β-chacotriosyl UA skeleton.
Keywords: SARS-CoV-2, Membrane fusion, Pentacyclic triterpenoid saponins, SARs
Graphical abstract

1. Introduction
The worldwide outbreak of the life-threatening disease “coronavirus disease 2019” (COVID-19) is caused by infection with a new coronavirus SARS-CoV-2 that was first reported in Wuhan, China, in late 2019. SARS-CoV-2 is capable of inducing fever, coughing and difficulty breathing as does the well-known SARS-CoV [1,2], which probably becomes more serious in some cases [3]. Over the past one year, SARS-CoV-2 has continued to spread globally and led to 5,232,562 confirmed deaths among surpassing 263 million cases as of December 6, 2021 [4]. Therefore, it needs to develop vaccines and to identify effective antiviral agents able to prevent or treat COVID-19 complications. Vaccination is regarded as the major prophylactic and therapeutic treatment of SARS-CoV-2 infections [5]. However, constant updated knowledge of the antigenic properties of the new circulating strains being essential for timely production of vaccines is usually unavailable that weakens their potency against SARS-CoV-2 and its variants, despite of several vaccination programs with the FDA-approved Merck vaccine and others [6,7]. Notably, it was reported that antibodies to the SARS-CoV-2 virus faded quickly in asymptomatic people [8]. Therefore, there is the urgency for developing effective broad-spectrum small-molecule therapeutic agents to prevent or eradicate these lethal infections, which could be used alone or in combination with vaccines in current and future infections.
Currently, the RdRp inhibitor remdesivir is the only direct-acting antiviral drug approved by the United States Food and Drug Administration (FDA) for the treatment of COVID-19 [9]. It is worth noting that this molecule has demonstrated some efficacy during early or moderate COVID-19 infection, but more data are clinically needed to confirm its efficacy [10,11]. Apart from remdesivir, various small-molecule inhibitors that can target multiple mechanisms of the SARS-CoV-2 replication cycle have been developed [[12], [13], [14], [15], [16], [17], [18], [19], [20]]. For example, a set of ketone-based covalent inhibitors such as Paxlovid (PF-07321332) [17], and its diverse analogs [14,[18], [19], [20]] targeting 3CLpro of SARS-CoV-2 have been found to display both excellent inhibitory activity in the enzymatic assay and potent anti-SARS-CoV-2 infection activity in vitro/vivo. More recently, a set of potent PLpro inhibitors [13] such as XR8-23 and XR8-24 have been identified by a structure-based drug design campaign, which displayed efficacy in blocking infection of human cells by SARS-CoV-2 PLpro. Kim and co-workers designed novel D-peptide inhibitors of SARS-CoV-2 that are suggested to block the RBD-ACE2 interaction to exhibit strong inhibition toward SARS-CoV-2 as well as two variants B.1.1.7 and B.1.351 in vitro [21]. Two quinoline derivatives chloroquine and hydroxychloroquine have shown good inhibition against SARS-CoV-2 in Vero cells but no efficacy in patients with high levels of SARS-CoV-2 virus in the blood [22]. Interestingly, several natural small molecules are also being explored for the inhibition toward SARS-CoV-2 in vitro, as illustrated by gallinamide A [23] and andrographolide [24], while further tests are needed to assess their potency in vivo and druggability. Encouragingly, a few compounds have advanced to clinical trials, which show good therapeutic effect. For example, Molnupiravir was approved for medical use in the United Kingdom in November 2021 and Pfizer PF-07321332 has entered in phase 2/3 clinical trials, of which the effective rate is 89% [25,26].
As the first step of SARS-CoV-2 viral life cycle, viral entry into target cells is mainly mediated by the surface structural spike glycoprotein (S) that can be divided into two domains S1 subunit and S2 subunit. Specifically, SARS-CoV-2 S1 is responsible for angiotensin-converting enzyme II (ACE2) recognition that has been identified as host cell receptor, and S2 is capable of mediating membrane fusion [27,28]. Studies have shown that binding of the S1 subunit receptor-binding domain (RBD) to the ACE2 receptor can trigger multiple and sequential conformational changes in the S2 subunit, thereby bringing viral and target cell membranes together for fusion [27]. Therefore, the development of SARS-CoV-2 entry inhibitors by targeting S1 or S2 subunit is an attractive strategy to inhibit viral entry and infection. It is worth noting that SARS-CoV-2 S protein exhibited much more efficiency in mediating viral surface-fusion and entry into host cells than that of SARS-CoV [28]. Most importantly, the sequence of S2 subunit is more conserved in relation to that of S1 subunit [29,30]. Collectively, these data highlight S2 subunit as a better target for developing broad-spectrum and potent SARS-CoV-2 entry inhibitors.
Substantial efforts have been conducted to result in several potential SARS-CoV-2 fusion inhibitors including EK1-peptides [31] and several small molecules such as hernandezine, salvianolic acid, clobenztropine (Fig. 1 A) [[32], [33], [34], [35]]. Specifically, the lipopeptide EK1 and its derivative EK1C4 were found to exhibit highly potent inhibition against SARS-CoV-2 and its variants in vitro and in vivo [31]. Crystal structure revealed that EK1-peptides could directly target the HR1 domain in the SARS-CoV-2 S protein, which blocked the formation of six-helix bundle (6-HB) core of S, thus leading to inhibition of virus-cell fusion [31]. Huang and co-workers discovered a small set of bis-benzylisoquinoline alkaloids as small-molecule pan-coronavirus entry inhibitors through compound screening, as exampled by hernandezine [34]. These alkaloids effectively protect different cell lines (293T-ACE2, A549, and Calu-3) from infection by SARS-CoV-2 and other pan-coronavirus in vitro, which can block host calcium channels to inhibit Ca2+-mediated viral membrane and endosomal membrane fusion, thus suppressing virus entry [34]. Moreover, our group has previously reported that the natural polyphenol compound salvianolic acid C (Sal-C) from Danshen, is able to potently suppress SARS-CoV-2 infection in vitro by blocking formation of the 6-HB core [35]. Consequently, the development of small molecular blockers to directly disrupt the virus-cell fusion may be a feasible approach to inhibit the SARS-CoV-2 infection.
Fig. 1A.

Representative small-molecule SARS-CoV-2 fusion inhibitors.
Recently, certain small-molecule inhibitors structurally related to pentacyclic triterpenoid (PT) [[36], [37], [38]] have exhibited potential inhibition toward SARS-CoV and SARS-CoV-2, as illustrated by glycyrrhizic acid (GL) [37] and bardoxolone methyl [38], suggesting that PTs and their derivatives are worth exploring to identify potent antivirals against coronaviruses. Of note, ursolic acid (UA), a typical ursane type PT, and its derivatives have been shown to effectively block multiple enveloped viral entry such as human immunodeficiency virus (HIV), influenza virus (IFV) in test cells in the low micromolar range, apparently related with the fusion of the incoming virus to the host cell membrane [39,40]. These findings support the development and pharmacological validation of UA derivative as a novel kind of potential coronaviruses entry inhibitors.
In light of their chemical structural similarity coupled with anti-IFV activity as does GL, we inferred that a 3-O-β-chacotriosyl UA saponin library generated from semisynthesis by our group [39] are worth investigating to evaluate their potent inhibition against coronaviruses. To serve this purpose, herein, we report a focused screening of these saponins to identify potent SARS-CoV-2 entry inhibitors. From this effort, several 3-O-β-chacotriosyl UA saponins including two top hits UA-18 and UA-19 (Fig. 1 B) were obtained and characterized as novel SARS-CoV-2 entry inhibitors. Based on these results, optimization of the top hits UA-18 and UA-19 through chemically modifying the 28-position of UA with various aromatic heterocycles while maintaining the 3-O-β-chacotriosyl residue gave rise to the lead compound UA-30 (Fig. 1B). Furthermore, the cell-cell fusion assay, docking analysis coupled with multiple site-specific mutation studies on SARS-CoV-2-S suggests a novel binding mechanism that UA-30 might bind to S in the prefusion state to prevent S2-mediated viral/cell membrane fusion. These data have provided validation for the continued development of the 3-O-β-chacotriosyl UA saponin-based SARS-CoV-2 fusion inhibitors.
Fig. 1B.

Representative structures of UA saponins and workflow of this study.
2. Results and discussion
2.1. Screening UA saponins against pseudovirus model of SARS-CoV-2
To identify potential SARS-CoV-2 entry inhibitors, our efforts began with screening a carefully curated library of UA saponins on the basis of a luciferase-expressing pseudovirus encoding SARS-CoV-2 S protein (pSARS-CoV-2) [35], which was helpful in allowing for direct comparison of S function with a common lentiviral core and reporter. To screen for candidate compounds, individual saponins (at 40 or 10 μM, respectively) were mixed with SARS-CoV-2 S pseudotyped HIV and the mixture incubated with 293T-ACE2 cells (HEK293T cells overexpressing human angiotensin-converting enzyme 2). Salvianolic acid C [35], a small-molecule SARS-CoV-2 entry inhibitor previously shown to bind S directly, was used as the positive control in this assay. The biological data were summarized in Supplementary Tables S1 and S2. Of 28 saponins assayed, two top hits UA-18 and UA-19 showed good potency (inhibition rate>80% at 40 μM), which were retested to obtain accurate EC50 values. As illustrated in Fig. 2 A and Fig. S1, UA-18 and UA-19 exhibited the good inhibition against pSARS-CoV-2 with EC50 values of 10.69 μM and 12.71 μM, respectively, while both of them showed marginal inhibition toward VSV-G pseudovirus and cytotoxicity against 293T-ACE2 cells within the effective concentration range (Fig. 2B), suggesting that the observed inhibition against SARS-CoV-2 was highly specific. Then using infectious SARS-CoV-2 (wuhan-HU-1 variant) in Vero-E6 cells, we evaluated the potency of UA-18 in a BSL-3 facility to further confirm their antiviral activity. Interestingly, the hit UA-18 displayed a 5-fold improvement of inhibition against infectious SARS-CoV-2 (EC50 = 2.38 μM) relative to the potency observed in pseudoviruses (Fig. 2C), probably attributed to the use of different host cells in each assay. These results supported the validity of the S/HIV-based anti-SARS-CoV-2 assay used in this study. As shown in Fig. 2D, UA-18 exhibited a well-defined dose-dependent response against the S protein based on a surface plasmon resonance (SPR) assay. The high affinity between S and the hit UA-18 (K D = 13.9 μM) reveals that these 3-O-β-chacotriosyl UA saponins are able to directly bind to the S protein as predicted.
Fig. 2.

(A) Chemical structures of UA-18. (B) Dose-response curve for UA-18 in the antiviral assay against pSARS-CoV-2 and VSV-G infection on 293T-ACE2 cells. (C) Validation on the antiviral activity of UA-18 against authentic SARS-CoV-2 in Vero-E6 cells. (D) SPR analysis of the interaction between UA-18 with SARS-CoV-2 S-trimer.
2.2. Preliminary structure-activity relationship
Despite of the limited chemical diversity focused on common chemical features or pharmacophores with UA-18, a meaningful structure-activity relationship (SAR) could be drawn from these observations around the trisaccharide chain and aglycone, which furnished a preliminary understanding of receptor tolerances at three distinct sites for inhibitory activity (Fig. 3 ). Structurally, these saponins bear a hydrophilic branched trisaccharide α-l-rhamnopyranosyl-(1 → 2)-[α-l-rhamnopyranosyl-(1 → 4)]-β-d-glucopyranosyl residue, named known as a chacotriose, incorporated to the hydrophobic aglycone UA via β-glycosidic linkage, followed by different side chains at 17-COOH of UA. As can be seen from Supplementary Table S1, the β-chacotriosyl moiety of UA-18 was essential for potency since simplifying it into α-l-rhamnopyranosyl-(1 → 2)-β-d-glucopyranosyl or α-l-rhamnopyranosyl-(1 → 4)-β-d-glucopyranosyl residue, unfortunately, led to a significant loss of inhibition. This result illustrated the crucial nature of the β-chacotriosyl moiety for the anti-SARS-CoV-2 activity. However, on the other hand, neither chacotriose nor UA showed inhibition toward SARS-CoV-2 as single compounds at 40 μM (data not shown). This demonstrated that these saponins acted as an integral structure, of which the sugar chain or the aglycone alone was not enough to generate anti-SARS-CoV-2 activity.
Fig. 3.

The preliminary SARs and structural optimization of 3-O-β-chacotriosyl UA amide saponins as SARS-CoV-2 entry inhibitors.
After identifying a favorable β-chacotriosyl moiety pharmacophore on site A, we then analyzed the effect of subtle modifications of aglycone scaffold on the potency, wherein substantial differences in the potency of these saponins against SARS-CoV-2 were observed upon (1) varying the length and chemical composition of side chains at the 28-position of UA, (2) replacing the carboxyl oxygen atom with nitrogen in the linker group, and (3) substituting the C-3 configuration of UA. In analyzing the potency of ester analogs of UA (Supplementary Table S1), in which the structural variations were made in the 28-position of UA, it was observed that, in general, substitutions on site C were beneficial to both antiviral activity and selectivity index (SI). Notably, it seemed that the inhibitory activity of this series was sensitive to the length, size, and shape of side chains in the C-28 position of UA. First, the length of the linear alkyl groups had an important impact on SI since the cytotoxicity decreased with the increase in length of the substitutions, although with slight improvement in potency with a preferred rank order of potency n-pentyl > n-propyl > ethyl ≈ methyl. Interestingly, the C-28 position was well tolerated with a cycloalkyl group (e.g., cyclopentyl UA-6), presumably via increasing the hydrophobic interaction when binding to the active pocket of the S protein. Second, the aralkyl group was more potent than the alkyl group, as exemplified by the benzyl analog UA-7 that was the most potent of all of the ester analogs assessed in this study. Overall, the SARs suggested that these molecules with a lipophilic alkyl substitution at the 28-position of UA tend to have better potency, of which the lipophilic side-chains at region B might occupy a hydrophobic channel of S.
We next explored the link moiety on site B by replacement of the ester linker group with its bioisosteric surrogate amide bond as seen in Table S2. It is noteworthy that such a small change remarkably enhanced the anti-SARS-CoV-2 activity since most of amide analogs had slightly and even significantly increased EC50 values and lower cytotoxicity in relation to corresponding ester analogs. This reveals that the potency toward SARS-CoV-2 is relatively tolerant to modifications in the link moiety. Specifically, replacement of the ester link in UA-2 by the amide moiety, furnishing NHMe and NMe2, improved the potency, as did the similar substitution in UA-13. However, compounds UA-14 and UA-15 with basic alkyl chains at the terminal end of N-ethyl group on site B had the poor activity, suggesting that this moiety was intolerant to alkyl chains containing more nitrogens. Interestingly, the installation of pyrrolidine (UA-16) or phenyl ring (UA-17) substituents at the C-28 position of UA via the amide linker resulted in substantial improvement in potencies compared with alkyl amide analogs, which further validated the SAR trend observed in the ester series. Then we further explored the consequences of incorporating diversified substituents into the phenyl ring of UA-17 to assess its functions in terms of electronic and positional contributions to anti-SARS-CoV-2 activity. As shown in Table S2, introduction of the methoxy group into the phenyl ring of UA-17 generated regioisomers UA-18, UA-20, and UA-22, all of which exhibited higher or comparable activity relative to UA-17, revealing the importance of this structural modification for anti-SARS-CoV-2 activity. It should be noted that the ortho-methoxy analogue UA-18 was much stronger in potency than the meta- and para-substituted analogs UA-20 and UA-22. However, the introduction of halogen atoms into the benzene ring of UA-17 resulted in significant drops in potency, with the exception of the ortho-Cl-substituted derivative UA-19. Among them, the analogue UA-19 displayed markedly increased inhibitory activity relative to UA-17, although with slightly less intensity than the ortho-CH3O substituted analogue UA-18. Thus, these data demonstrated that the substituent position in the phenyl ring played a crucial role in potency, with the replacement in the position ortho most favorable.
After identifying a favorable hydrophobic aromatic side chain incorporated into UA via an amide linker on site B, we turned our attention back to the aglycone on site C, wherein we reasoned that the C-3 configuration of UA might have a crucial effect on the potency. The results in Table S2 showed that the 3β-isomer UA-7 or UA-17 was substantially superior to the 3α-isomer UA-27 or UA-28, respectively. In this case, the lack of tolerance of the C-3 configuration of UA was presumably owing to the necessity of a preferred conformation at this position with S.
2.3. Optimization and synthesis of hit compounds
On the basis of the discovery of the top hits UA-18 and UA-19, our efforts were focused on the improvement of their binding affinity with S and selectivity index. In view of the importance of 3-O-β-chacotriosyl moiety attached to 3β-OH of UA, the C-28 carboxylic acid of UA was chosen as the only position for further modifications. Thus, we continued with a more focused SAR exploration on site B by varying the length of amide linker, the shape, steric and electronic properties of side chains at the 28-position of UA to generate saponins UA-29 -- UA-43 (Table 1 ), with the object of identifying a side chain that is able to lower the cytotoxicity while maintaining the potency.
Table 1.
Antiviral activities of the title saponins UA-29 – UA-43 screened against pSARS-CoV-2-S at 10 and 40 μM, respectively.
| No. |
Structure |
Inhibition rate (%)a |
|
|---|---|---|---|
| 40 μM 10 μM | |||
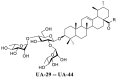 | |||
| UA-29 | 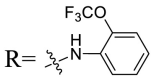 |
toxicb | toxicb |
| UA-30 | 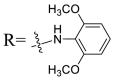 |
96.84 ± 5.40 | 46.81 ± 3.55 |
| UA-31 | 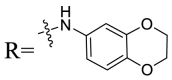 |
30.45 ± 2.13 | −2.49 ± 0.24 |
| UA-32 | 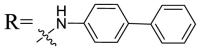 |
27.44 ± 4.11 | 16.94 ± 2.35 |
| UA-33 | 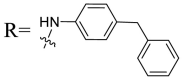 |
19.25 ± 1.03 | 10.32 ± 2.10 |
| UA-34 | 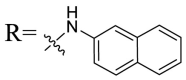 |
14.51 ± 1.06 | −7.27 ± 1.03 |
| UA-35 | 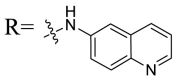 |
33.08 ± 1.02 | 27.35 ± 2.51 |
| UA-36 | 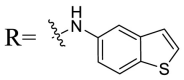 |
−21.66 ± 1.20 | −31.75 ± 4.32 |
| UA-37 | 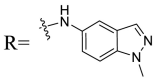 |
51.87 ± 2.23 | 41.08 ± 1.89 |
| UA-38 | 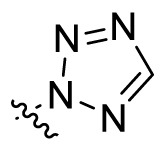 |
toxicb | toxicb |
| UA-39 | 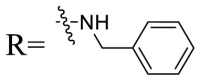 |
92.49 ± 3.78 | 59.23 ± 4.21 |
| UA-40 | 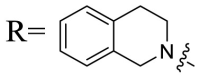 |
58.74 ± 5.13 | 32.52 ± 2.59 |
| UA-41 | 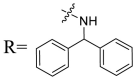 |
1.58 ± 0.34 | 0.47 ± 0.08 |
| UA-42 | 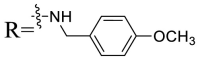 |
85.69 ± 1.33 | 36.26 ± 2.45 |
| UA-43 | 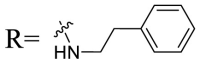 |
49.47 ± 3.08 | 28.16 ± 2.22 |
| Sal-C | / | 98.30 ± 2.62 | 80.25 ± 2.56 |
Data are expressed as the mean ± SD of three experiments.
Title saponins showed significant cytotoxicity at corresponding concentrations, and the inhibition rate could not be calculated.
Our general method for the synthesis of title saponins UA-29 -- UA-43 was outlined in Scheme 1 . The 4-methoxybenzyl ursolate 1 was obtained through the nucleophilic substitution reaction in the presence of potassium carbonate using the commercially available UA and 4-methoxybenzyl chloride as starting materials. The aglycone 1 went through a glycosylation reaction with the known donor 2, 3, 4, 6-tetra-O-benzoyl-d-glucopyranosyl trichloroacetimidate 2 [39] to furnish 3, where trimethylsilyl trifluoromethanesulfonate (TMSOTf) acted as an ideal catalyst and completed the reaction in good yield. Then the basic hydrolysis of the benzoyl ester in 3 produced β-d-glucopyranosyl-para-methoxybenzyl ursolate 4, which was subjected to 1-(benzoyloxy)-benzotriazole (1-BBTZ) to selectively protect the 3, 6-OHs of the β-glucopyranosyl residue, giving rise to the intermediate 5. The subsequent conjugation of the free 2, 4-OHs of the protected glucopyranoside in 5 with the known 2, 3, 4-tri-O-acetyl-l-rhamnopyranosyl trichloroacetimidate 6 [39] via a TMSOTf-catalyzed glycosylation reaction was carried out, followed by removing all the acyl groups under basic conditions to give the trisaccharide 7. As before, selective cleavage of 28-O-PMB group by palladium catalyst with hydrogen gas, followed by protection of all the hydroxyl groups using acetic anhydride in the presence of a catalytic amount of 4-dimethylaminopyridine (DMAP) to get the important intermediate 8. Treatment of 8 with oxalyl chloride led to 28-acyl chloride, followed by amide coupling with various amines and then hydrolysis of all the ethyl esters using MeONa in methanol to yield the final products UA-29 – UA-43, respectively.
Scheme 1.

Reagents and conditions: (a) PMBCl, K2CO3, DMF; (b) TMSOTf, CH2Cl2; (c) MeONa, CH3OH; (d)1-BBTZ, Et3N, CH2Cl2; (e) (i) TMSOTf, CH2Cl2; (ii) MeONa, MeOH; (f) (i) 10% Pd/C, H2, MeOH-THF; (ii) Ac2O, DMAP, pyridine; (g) (i) (COCl)2, CH2Cl2; (ii) various aromatic or aromatic alkyl amines, Et3N, CH2Cl2; (iii) MeONa, MeOH.
2.4. Structure-activity relationship development
Based on the preliminary SARs, further optimization was conducted on the C-17-COOH group of UA to investigate diverse substituted phenyl and other aromatic heterocycles linked by amide bond, and title saponins UA-29 – UA-43 were designed and synthesized. Based on the antiviral effect of compound UA-18 (EC50 = 10.69 μM), the inhibition rates of title saponins against pSARS-CoV-2 at 40 μM (high concentration) and 10 μM (EC50) concentrations were evaluated, respectively. Given trifluoromethoxy as the bioisostere of methoxy might form additional potential interaction with the S protein, we first incorporated the trifluoromethoxy group at the ortho-position of phenyl ring to generate UA-29, however, resulting in higher cytotoxicity compared with UA-18. Next, the impact of 2, 6-disubstitution was determined with compound UA-30. Encouragingly, despite of a slight improvement in potency relative to UA-18, the increased antiviral activity did not accompany cytotoxicity, as evidenced from CC50 of more than 100 μM against 293T-ACE2 cells with UA-30. In contrast, replacement of the phenyl ring of UA-17 with a 2, 3-dihydrobenzo[b] [1,4]dioxine residue (UA-31) was detrimental to antiviral activity.
Then we focused on the replacement of the relatively small Ph group with more sterically hindered biphenyl moieties to exploit more potential hydrophobic interactions. Our rationale was that the added phenyl linker could potentially extend the phenyl moiety deeper into the hydrophobic cavity in the S protein, probably leading to enhanced ligand affinity. Unfortunately, both of these linear diphenyl derivatives UA-32 and UA-33 suffered from remarkable loss of potency, suggesting that there is a limited steric tolerance in this side-chain position. These observations prompted us to probe the potential of more compact polycyclic aromatic and other heterocycles. With these results in hand, our SAR exploration continued with the replacement of the phenyl ring at the 28-position of UA with 6 + 6-fused aromatics or 6 + 5- fused heteroaryl residues. Although no marked improvement was observed with this substitute for the phenyl ring, we were pleased to notice that some structural modifications were tolerated. For example, replacement of the phenyl ring of UA-17 by naphthalene (UA-34) led to the dramatic drop in potency, while the corresponding quinolone-substituted analog UA-35 with similar steric hindrance of the substituent was equipotent to the Ph analog UA-17. A similar increase of inhibitory potency was also observed with other 6 + 5- fused heteroaryl derivatives containing nitrogen group. Specifically, 1-methyl-1H-indazole analogue (UA-37) was more potent than UA-17 in cellular assays while a complete loss in potency was observed for UA-36. These results reveal that larger substituents at the 28-position of UA sterically clashed with S, directing us toward smaller moieties. However, attempts to lower steric hindrance by substituting the phenyl side chain with a small tetrazole group (UA-38) induced strong toxicity toward 293T-ACE2 cells.
To acquire the optimal length of the alkyl spacer between amide group and the phenyl ring, we continued with a more focused SAR exploration, by targeting different aralkyl amines at the 28-position of UA. Notably, a dramatic enhancement of potencies was achieved through the insertion of a single methylene unit into the aromatic amine (benzyl spacer analogue UA-39) relative to UA-17. Subsequently, we found that conformational restriction of the benzyl group with 1, 2, 3, 4-tetrahydroisoquinoline restrained the flexibility of the NH-alkyl side chain, giving rise to UA-40, which showed a reduction in the anti-SARS-CoV-2 activity compared with UA-39. Furthermore, the attachment of a phenyl ring at the methylene position of the benzyl residue of compound UA-41 resulted in poor inhibition toward SARS-CoV-2, likely driven by the addition of steric clashes with an essentially hydrophobic binding pocket. These results highlight the superiority of the benzyl moiety with significantly increased rotatable bonds due to the flexible methylene unit, which may allow potential hydrophobic interactions with key residues in the S protein. Interestingly, the insertion of a methoxy group at the para benzyl position was tolerated, with compound UA-42 being equipotent to the parent UA-39. In addition, the ethyl spacer analogue UA-43 displayed significant decrease in potency at 40 μM compared with UA-39 (49% vs 92%), demonstrating that the aromatic substituent should be better linked via a methylene unit rather than a zero- or two-methylene unit. We inferred that the pendant phenyl substituent linked by a methylene unit favored the preferential conformation of the ligand bound to the active site in S.
2.5. Potent inhibitory activity of UA-30 against infectious SARS-CoV-2 and its variants
Of 15 title saponins assayed, only three compounds UA-30, UA-39 and UA-42 exhibited inhibition superior to 80% at 40 μM against pseudotyped SARS-CoV-2, which gave EC50 values of 9.84 μM, 12.00 μM, 11.81 μM, respectively. Notably, all these title saponins with relatively high antiviral activity showed low cytotoxicity (CC50 > 100 μM), and high specificity (SI > 10, VSV-G EC50 > 100 μM) as illustrated in Fig. 4 A and Fig. S1. Taking into account the selectivity, UA-30 stood out among all UA saponins evaluated, displaying both stronger inhibitory activity toward pSARS-CoV-2 and higher SI when compared to the hit UA-18, which was chosen as the lead compound.
Fig. 4.

(A) Dose-response curve for UA-30 in the antiviral assay against pSARS-CoV-2 and VSV-G infection in 293T-ACE2 cells. (B) Validation on the antiviral activity of UA-30 against authentic SARS-CoV-2 in Vero-E6 cells. (C) Antiviral activity of UA-30 against SARS-CoV-2 infection was detected by indirect immunofluorescence assay with chloroquine (CQ) as a positive control, scale bar = 200 mm. (D–E) Anti-SARS-CoV-2 efficacy of UA-30 on PsV variants in 293T-ACE2 cells, including E484K, D614G, N501Y (D) and Alpha, Beta, Gamma, Delta, Omicron (E).
As shown in Fig. 4B, the lead UA-30 showed slightly increased potency against wild-type SARS-CoV-2 (wuhan-HU-1 variant) infection of Vero-E6 cells (EC50 = 2.05 μM) than UA-18, consistent with that observed in pseudoviruses testing. Meanwhile, UA-30 displayed no cytotoxicity against Vero-E6 cells (CC50 > 100 μM) and a favorable SI value (SI > 48.78), signifying that UA-30 has a potent anti-SARS-CoV-2 activity with a high safety margin in cell culture models. Evidence has shown that SARS-CoV-2 S protein is an abundantly expressed protein during infection, which is also a multifunctional RNA-binding protein for viral RNA transcription and replication [24,41]. Consistently, fluorescence microscopy results exhibited that the expressions of SARS-CoV-2 N protein declined in a dose-dependent manner following the treatment with UA-30 (Fig. 4C). Overall, these data pointed out the good potency of UA-30 in the inhibition of SARS-CoV-2 infectivity in Vero-E6 cells and encouraged us to probe whether it also had antiviral effects against SARS-CoV-2 variants.
Recently, the emerged SARS-CoV-2 variants that bear vital mutations in their S proteins such as E484K, D614G or N501Y showed increased adaptability and transmissibility [30]. With the aim of investigating whether UA-30 can prevent infection mediated via S protein carrying these mutations, we evaluated potency of UA-30 by utilizing pseudoviruses with E484K, D614G or N501Y single mutation in their S proteins, respectively. We found that UA-30 effectively inhibited these pseudoviruses, respectively, similar to that against pSARS-CoV-2 (seen in Fig. 4D). It is worth noting that UA-30 also demonstrated appreciable inhibition of viral entry for the newly emerged variants with combinational mutations in S protein including pseudotyped Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.10), Delta (B.1.617.2) and Omicron with EC50 values ranging from 0.04 to 11.57 μM (Fig. 4E), respectively. It is worth noting that UA-30 exhibited a slight improvement in potency toward the pseudotyped Omicron relative to the pSARS-CoV-2 assays. Intriguingly, when compared to the pSARS-CoV-2 assays, UA-30 was 246-fold more potent against the pseudotyped P.10 and also displayed significantly increased inhibition toward pseudotyped B.1.617.2, while it showed comparable inhibition against pseudotyped B.1.1.7 and B.1.351. The overall high inhibitory rate of UA-30 implies that there is a general similarity in terms of the structure of the ligand binding pocket in different S proteins, and it is possible that the specific binding sites of UA-30 may be shared between SARS-CoV-2 and its variants. Collectively, these results indicated that UA-30 had broad-spectrum potency against SARS-CoV-2 and all variants tested that targeted the S-mediated entry process.
2.6. Target identification
Having confirmed these structure requirements of these UA saponins for maintaining high antiviral activity, we then designed experiments to identify the potential target. Consistent with the result of UA-18, a high affinity (K D = 0.78 μM) was also observed between the lead UA-30 and the SARS-CoV-2 S protein based on a SPR assay (Fig. 5 A), suggesting that the antiviral effect of UA-30 against SARS-CoV-2 infection derives from inhibition of viral entry into host cells by targeting the S protein. Structurally, the S protein comprises two functional subunits S1 and S2 that is responsible for ACE2 recognition (S1 subunit) and membranes fusion (S2 subunit), respectively. Considering this, we then conducted a Co-Immunoprecipitation (Co-IP) assay to explore whether UA-30 had any impact on the interaction of S1 subunit with its ACE2 receptor, the crucial step for attachment of SARS-CoV-2 to host cells for initiation of virus infection. As depicted in Fig. 5B, UA-30 was unable to block the receptor-binding domain of S1 subunit from interacting with the human ACE2 receptor. Consistent with the Co-IP results, UA-30 exhibited greatly decreased binding affinity with the S1 subunit (at millimolar level, data not shown) in relation to that with S protein according to the SPR analysis. These results suggest that UA-30 does not target the viral attachment step, and thus the S1 subunit can be ruled out as the potential target.
Fig. 5.

(A) SPR analysis of the interaction between UA-30 with SARS-CoV-2 S-trimer. (B) The binding of SARS-CoV-2 S protein and ACE2 (Anti-Flag) in the presence or absence of UA-30 (40 μM) were detected by Co-IP assays. IgG was included as a negative control. (C) The dose-dependent inhibitory effect of UA-30 on SARS-CoV-2 S mediated cell-cell fusion.
Motivated by these intriguing results, we turned our attention to probing whether UA-30 could inhibit viral infection by blocking the membrane fusion. This hypothesis was confirmed by measuring its effect on the fusion of SARS-CoV-2 S pseudoviruses as judged by a cell-cell fusion assay. As shown in Fig. 5C, UA-30 was able to potently suppress the fusion syncytium formation in Vero-E6 cells in a marked dose-dependent manner at 24 h, signifying that the antiviral property of UA-30 may correlate with its potency to block membrane fusion mediated by the SARS-CoV-2 S2 subunit. Taken together, UA-30 seemed to prevent fusion between the viral and endosome membranes by targeting the S2 subunit, thereby rendering the virus incapable of entering host cells.
2.7. UA-30 could target the prefusion state of spike protein
After receptor engagement by RBD, SARS-CoV-2 S2 will change its conformation from the pre-fusion state to a post-fusion trimer-of-hairpins conformation, leading to viral membrane fusion [30,31]. If UA-30 inhibits viral entry by maintaining S2 subunit in the pre-fusion or causing premature conversion of S2 to the post-fusion conformation, we would expect it to abolish viral fusion. Recently, our group and others have identified that the 6-HB formed by HR1 and HR2 domains of the S2 subunit plays a crucial role in the membrane fusion and viral infection mediated by SARS-CoV-2 S protein [31,35]. However, we found that UA-30 showed negligible impact on blocking viral 6-HB formation by using circular-dichroism (CD) spectroscopy analysis (Supplementary Fig. S2), which was contrasted with the action of Sal-C, a representative SARS-CoV-2 fusion inhibitor. Based on these results, we inferred that UA-30 appeared to interfere with the early stages (pre-fusion) but not with the later stages of viral membrane fusion.
To gather further insight into the binding mode of UA-30, we then performed docking studies of UA-30 using the X-ray crystal structures of SARS-CoV-2 S (PDB code: 6VXX) to identify the most probable binding site that is physically blocked in the SARS-CoV-2-S prefusion state. As illustrated in Fig. 6 A, we found that UA-30 was predicted to fit well into a cavity between the attachment (S1) and fusion (S2) subunits at the entrance to a large tunnel that links with equivalent tunnels from the other monomers of the trimer at the three-fold axis. Here, the chacotriosyl moiety of UA-30 formed multiple strong hydrogen bonds with the backbone of residues Thr302, Lys304, Thr315, Asn764, Arg765, Gln957 and Lys964 to result in improved interaction with S2, thus stabilizing the pre-fusion state of S2 to prevent conformational rearrangements in the S stem. At the other side of the pocket, the aromatic ring of UA-30 formed hydrophobic contacts with Val772, validating that incorporation of an aromatic ring favored the binding with S. These results suggest that UA-30 has a novel binding mechanism that can directly bind to S in the pre-fusion state and block S2-mediated fusion.
Fig. 6.

(A) Molecular docking of UA-30 to S protein (PDB: 6VXX). S1 subunit, S2 subunit, UA-30, and key residues are shown as green ribbon, blue ribbon, white sticks, and yellow stick, respectively. Light green dashes in the interaction plot indicating hydrogen bonds. (B) Antiviral efficacy of UA-30 against SARS-CoV-2 PsV mutants caused by site-directed mutation, including N764A/R765A, Q957A/K964A, and N764A/R765A/Q957A/K964A.
To provide support for the conclusions of the docking studies, we next used the amino acid mutagenesis of pSARS-CoV-2 S to investigate the molecular basis of entry inhibition by UA-30. Thus, base substitution was introduced into pSARS-CoV-2 S by site-mutation technique. As can be seen from Fig. 6B, the Q957A/K964 A S mutations resulted in a significant loss of potency toward UA-30 compared to WT S at different concentrations from 40 μM to 5 μM, respectively. There was a similar trend observed in the N764A/R765 A S mutations, although with slightly less intensity than Q957A/K964 A S mutations. With our expectation, we found that N764A/R765A/Q957A/K964 A S mutations caused more remarkable change in the response to inhibition of viral entry elicited by UA-30. This loss of potency reveals the interaction of UA-30 with the cavity between S1 and S2 subunits, leading to the inhibition of SARS-CoV-2 viral fusion. Thus, these data were in line with the molecular docking studies and provided further support for the proposed notion that our saponins could inhibit viral entry by targeting the SARS-CoV-2-S in a novel fashion.
3. Conclusions
In conclusion, inhibition of virus-cell membrane fusion is an appealing therapeutic target to block SARS-CoV-2 entry into host cells for antiviral drug discovery. This study presented here discovered a new SARS-CoV-2 entry inhibitor hit UA-18, via a screen of 3-O-β-chacotriosyl UA saponins against pseudotyped SARS-CoV-2. On the basis of UA-18, subsequent chemical optimization led to the development of the novel and potent lead compound UA-30, which had an excellent potency (EC50 = 2.05 μM) and a favorable SI value (SI > 49) when tested with infectious SARS-CoV-2, and displayed a broad-spectrum entry inhibition against recently emerged SARS-CoV-2 variants such as Delta and Omicron. Utilizing SPR, the Co-IP assay, cell-cell fusion assay, as well as dock in combination with mutagenesis studies, we confirmed that the lead UA-30 could occupy the cavity between S1 and S2 subunits in the SARS-CoV-2 S protein to interfere with virus-cell fusion, resulting in broad and effective antiviral activity in vitro. These results support further clinical development of UA-30-based fusion inhibitors against SARS-CoV-2 and its variants.
4. Experimental
4.1. Chemistry
Solvents were purified in a conventional manner. Thin layer chromatography (TLC) was performed on precoated E. Merck silica gel 60 F254 plates. Flash column chromatography was performed on silica gel (200–300 mesh, Qingdao, China). 1H NMR and 13C NMR spectra were taken on a JEOL JNM-ECP 600 spectrometer with tetramethylsilane as an internal standard, and chemical shifts are recorded in ppm values. Mass spectra were recorded on a Q-TOF Global mass spectrometer.
4.1.1. para-Methoxybenzyl-3β-hydroxy-urs-12-en-28-oic acid (1)
To a solution of ursolic acid (5.00 g, 10.96 mmol) in 100 mL dry DMF at r.t. was added K2CO3 (4.54 g, 32.88 mmol). After the mixture was stirred for 2 h, 4-methoxybenzyl chloride (2.28 mL, 16.44 mmol) was added and the reaction mixture was stirred for an additional 10 h. The solvents were concentrated in vacuo, diluted with CH2Cl2 (300 mL), washed with water (3 × 100 mL) and brine (2 × 100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, concentrated under diminished pressure and recrystallized (EtOH) to afford 1 (5.96 g, 94%) as a white solid. 1H NMR (600 MHz, CDCl3): δ 7.28 (d, 2H, J = 8.4 Hz, Ar–H), 6.89 (d, 2H, J = 8.4 Hz, Ar–H), 5.23 (t, 1H, J = 3.3 Hz, H-12), 5.06 (d, 1H, J = 12.1 Hz, Ar–CH2–1), 4.92 (d, 1H, J = 12.1 Hz, Ar–CH2–2), 3.82 (s, 3H, OCH3), 3.22 (dd, 1H, J = 11.2, 4.7 Hz, H-3), 2.26 (d, 1H, J = 11.3 Hz, H-18), 1.99 (td, 1H, J = 17.8, 4.3 Hz), 1.08, 1.00, 0.91, 0.79, 0.64 (each s, each 3H, CH3), 0.94 (d, 3H, J = 6.2 Hz, CH3), 0.86 (d, 3H, J = 6.6 Hz, CH3); 13C NMR (151 MHz, CDCl3): δ 177.3, 159.4, 138.0, 129.9 (two), 128.5, 125.6, 113.8 (two), 78.9, 65.8, 55.3, 55.2, 52.9, 48.1, 47.6, 42.0, 39.5, 39.1, 38.8, 38.6, 36.9, 36.6, 33.0, 30.7, 28.1, 27.9, 27.2, 24.2, 23.5, 23.3, 21.2, 18.3, 17.0, 15.6, 15.4; ESIMS (m/z): 599.5 [M+Na]+(calcd 599.4).
4.1.2. para-Methoxybenzyl-3β-O-(2, 3, 4, 6-tetra-O-benzoyl-β-d-glucopyranosyl)-urs-12-en-28-oic acid (3)
To a solution of 1 (1.00 g, 1.73 mmol), 2, 3, 4, 6-tetra-O-benzoyl-d-glucopyranosyl trichloroacetimidate (2) (1.93 g, 2.56 mmol) and 4 Å molecular sieves in dry CH2Cl2 was added TMSOTf (58 μL, 0.26 mmol) at −5 °C under argon. The reaction mixture was kept at −5 °C for 1 h and warmed to room temperature for 1 h. After completion of the reaction, the reaction was quenched by Et3N. The solid was removed, and the filtrate was concentrated in vacuo and purified by column chromatography (petroleum ether-EtOAc, 6:1) to yield the target compound 3 (1.76 g, 88%) as a white solid. 1H NMR (600 MHz, CDCl3): δ 8.02–6.85 (m, 24H, Ar–H), 5.90 (t, 1H, J = 9.7 Hz, H-3′), 5.60–5.55 (m, 2H, H-4′, H-2′), 5.25 (t, 1H, J = 3.5 Hz, H-12), 5.03 (d, 1H, J = 12.1 Hz, Ar–CH2–1), 4.88 (d, 1H, J = 12.1 Hz, Ar–CH2–2), 4.84 (d, 1H, J = 7.9 Hz, H-1′), 4.58–4.52 (m, 2H, H-6′), 4.15–4.13 (m, 1H, H-5′), 3.78 (s, 3H, OCH3), 3.06 (dd, 1H, J = 11.8, 4.6 Hz, H-3), 2.24 (d, 1H, J = 12.2 Hz, H-18), 1.02, 0.81, 0.68, 0.60, 0.56 (each s, each 3H, each CH3), 0.94 (d, 3H, J = 6.2 Hz, CH3), 0.88 (d, 3H, J = 6.6 Hz, CH3); 13C NMR (151 MHz, CDCl3): δ 177.3, 166.0, 165.8, 165.3, 165.0, 159.4, 138.0, 133.5, 133.2, 133.1, 129.9 (two), 129.8, 129.7 (two), 128.4 (two), 128.3 (two), 128.2 (two), 125.6, 113.7, 103.3 (C-1′), 90.5, 73.0, 72.1, 72.0, 70.3, 65.7, 63.4, 55.4, 55.2, 52.8, 48.0, 47.5, 42.0, 39.4, 39.4, 39.1, 38.7, 38.5, 36.6, 36.5, 32.9, 30.6, 27.8, 27.6, 25.8, 24.2, 23.5, 23.2, 21.2, 18.0, 16.9, 16.2, 15.3; ESIMS:calcd for [M+H]+ m/z 1155.5; found, 1155.6.
4.1.3. para-Methoxybenzyl-3β-O-(d-glucopyranosyl)-urs-12-en-28-oic acid (4)
To compound 3 (1.50 g, 1.30 mmol) in 30 mL of anhydrous CH2Cl2 and MeOH (V:V = 1:1) at room temperature was added CH3ONa until pH = 10. After the reaction mixture was stirred at r.t. for 2 h, the solution was neutralized with Dowex 50 × 8 (H+) resin until pH = 7, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2–MeOH, 10:1) to provide 4 (0.62 g, 97%) as a white solid. 1H NMR (600 MHz, CDCl3): δ 7.27 (d, 2H, J = 8.4 Hz, Ar–H), 6.88 (d, 2H, J = 8.4 Hz, Ar–H), 5.23 (t, 1H, J = 3.0 Hz, H-12), 5.05 (d, 1H, J = 11.8 Hz, Ar–CH2–1), 4.93 (d, 1H, J = 11.9 Hz, Ar–CH2–2), 4.36 (d, 1H, J = 7.3 Hz, H-1′), 3.86 (dd, 1H, J = 12.0, 3.1 Hz, H-6′-1), 3.81 (s, 3H, OCH3), 3.80 (dd, 1H, J = 11.8, 4.5 Hz, H-6′-2), 3.64 (t, 1H, J = 9.2 Hz, H-4′), 3.56 (t, 1H, J = 8.6 Hz, H-3′), 3.45(t-like, 1H, J = 8.8, 8.2 Hz, H-2′), 3.30 (d, 1H, J = 8.6 Hz, H-3), 3.17–3.14 (m, 1H, H-5′), 2.24 (d, 1H, J = 11.0 Hz, H-18), 1.07, 1.01, 0.91, 0.83, 0.65 (each s, each 3H, CH3), 0.96 (d, 3H, J = 6.1 Hz, CH3), 0.86 (d, 3H, J = 6.2 Hz, CH3); 13C NMR (151 MHz, CDCl3): δ 177.3, 159.4, 138.1, 129.4 (two), 128.4, 125.4, 113.9 (two), 105.2 (C-1′), 90.0, 76.3, 75.3, 73.8, 69.4, 65.7 (two), 61.6, 55.6, 55.3, 52.9, 48.0, 47.6, 42.0, 39.5, 39.1, 38.8, 36.7 (two), 36.5, 33.1, 30.7, 28.1, 28.0, 26.2, 24.2, 23.6, 23.3, 21.2, 18.2, 17.0, 16.7, 15.5; ESIMS:calcd for [M+Na]+ m/z 738.6; found, 738.5.
4.1.4. para-Methoxybenzyl-3β-O-(3, 6-di-O-benzoyl-β-d-glucopyranosyl)-urs-12-en-28-oic acid (5)
To a solution of 4 (1.00 g, 1.35 mmol) and 1-(benzoyloxy)benzotriazole (1-BBTZ, 1.30 g, 5.40 mmol) in anhydrous CH2Cl2 (30 mL) was added triethylamine (1.13 mL, 18.0 mmol) drop-wise at 0 °C. Then the reaction was stirred at room temperature for 24 h. Upon completion, the solvent was removed in vacuo and purified by column chromatography (petroleum ether-EtOAc, 3:1) to give the white solid 5 (0.95 g, 74%). 1H NMR (600 MHz, CDCl3): δ 8.13–8.08 (m, 4H, Ar–H), 7.62–7.56 (m, 2H, Ar–H), 7.47 (t, 2H, J = 7.8, 1.2 Hz, Ar–H), 7.46 (t, 2H, J = 7.8, 1.2 Hz, Ar–H), 7.27 (d, 2H, J = 7.3 Hz, Ar–H), 5.25 (t, 1H, J = 3.2 Hz, H-12), 5.05 (d, 1H, J = 11.8 Hz, Ar–CH2–1), 4.93 (d, 1H, J = 11.9 Hz, Ar–CH2–2), 5.22 (t, 1H, J = 8.9 Hz, H-3′), 5.05 (d, 1H, J = 12.1 Hz, Ar–CH2–1), 4.93 (d, 1H, J = 12.0 Hz, Ar–CH2–2), 4.68–4.66 (m, 2H, H-6′), 4.50 (d, 1H, J = 7.8 Hz, H-1′), 3.82 (s, 3H, OCH3), 3.78–3.75 (m, 3H, H-4′, H-2′, H-5′), 3.15 (dd, 1H, J = 11.8, 4.6 Hz, H-3), 2.28 (d, 1H, J = 12.1 Hz, H-18), 2.01 (td, 1H, J = 17.7, 4.2 Hz), 1.07, 1.00, 0.87, 0.81, 0.63 (each s, each 3H, CH3), 0.97 (d, 3H, J = 6.2 Hz, CH3), 0.91 (d, 3H, J = 6.2 Hz, CH3); 13C NMR (151 MHz, CDCl3): δ 177.3, 167.9, 166.5, 159.4, 138.1, 133.5, 133.1, 130.0, 129.9, 129.8, 129.3, 128.5 (two), 128.4, 125.6, 113.8, 104.9 (C-1′), 90.1, 78.8, 74.1, 72.7, 70.2, 65.7, 63.8, 55.5, 55.3, 52.9, 48.1, 47.6, 42.0, 39.5, 39.1, 38.9, 38.8, 38.5, 36.6, 36.5, 33.0, 30.7, 28.2, 27.9, 25.9, 24.2, 23.5, 23.2, 21.2, 18.1, 17.0 (two), 16.6, 15.4; ESIMS:calcd for [M+K]+ m/z 985.6; found, 985.5.
4.1.5. para-Methoxybenzyl-3β-O-[2, 4-di-O-(α-l-rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oic acid (7)
To a mixture of 5 (560 mg, 0.59 mmol) and 4 Å molecular sieves in dried CH2Cl2 (10 mL) at −60 °C under argon was added TMSOTf (33 μL, 0.15 mmol), followed by a solution of the 2, 3, 4-tri-O-acetyl-l-rhamnopyranosyl trichloroacetimidate 6 (1.28 g, 2.96 mmol) in dry CH2Cl2 (5 mL). After stirring at - 60 °C for 3 h, the reaction mixture was warmed to room temperature and stirred overnight under argon. After the completion of the reaction, the reaction was quenched with Et3N. The solid was filtered, and the filtrate was concentrated under vacuum and purified by column chromatography (petroleum ether-EtOAc, 1:1) to yield the desired crude trisaccharide. Subsequently, this mixture was dissolved in dry CH2Cl2 and CH3OH (V:V = 1:1) and then NaOMe was added until pH = 10. After stirred at 45 °C for 12 h, the solution was neutralized with Dowex 50 × 8 (H+) resin until pH = 7, filtered, concentrated under vacuum and purified by column chromatography (CH2Cl2–CH3OH, 6:1) to afford a white solid 7 (462 mg, 76%). 1H NMR (600 Hz, CD3OD): δ 7.28 (d, 2H, J = 8.2 Hz, Ar–H), 6.90 (d, 2H, J = 8.4 Hz, Ar–H), 5.38 (s, 1H, Rha-H-1), 5.22 (t, 1H, J = 3.0 Hz, H-12), 5.01 (s, 1H, Ar–CH2–1), 4.87 (s, 1H, Ar–CH2–2), 4.86 (d, 1H, J = 1.1 Hz, Rha-H-1), 4.44 (d, 1H, J = 7.7 Hz, H-1′), 4.00–3.98 (m, 2H), 3.93–3.89 (m, 1H), 3.85 (dd, 1H, J = 3.1, 1.8 Hz, Rha-H-2), 3.81 (dd, 1H, J = 12.1, 1.7 Hz, H-6′-1), 3.80 (s, 3H, OCH3), 3.76 (dd, 1H, J = 9.4, 3.4 Hz, Rha-H-3), 3.67 (dd, 1H, J = 12.1, 4.1 Hz, H-6′-2), 3.64 (dd, 1H, J = 9.4, 3.3 Hz, Rha-H-3), 3.60 (t, 1H, J = 8.7 Hz), 3.55 (t, 1H, J = 9.3 Hz), 3.47 (t, 1H, J = 8.1 Hz), 3.44–3.38 (m, 2H), 3.34–3.32 (m, 1H), 3.19 (dd, 1H, J = 11.7, 4.1 Hz, H-3), 2.25 (d, 1H, J = 11.3 Hz, H-18), 1.28 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.23 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.13, 1.09, 0.93, 0.87, 0.86 (each s, each 3H, CH3), 0.92 (d, 3H, J = 6.0 Hz, CH3), 0.90 (d, 3H, J = 6.4 Hz, CH3); 13C NMR (151 Hz, CD3OD): δ 177.7, 159.8, 138.0, 130.0 (two), 128.3, 125.8, 113.3 (two), 104.0 (C-1′), 101.5 (Rha-C-1), 100.6 (Rha-C-1), 88.9, 79.1, 77.7, 76.8, 75.1, 72.6, 72.3, 71.0, 70.7, 70.6, 69.4, 68.6, 65.6, 60.6, 55.8, 54.3, 53.0, 41.7, 39.4, 39.0, 38.9, 38.8, 36.5, 36.4, 32.9, 30.3, 27.6, 27.1, 25.8, 23.9, 22.9, 22.6, 20.1, 17.8, 16.6, 16.5, 16.2, 15.8, 14.8; HRESIMS calcd for C56H87O17 1031.5872; found 1031.5865.
4.1.6. 3β-O-[2, 4-Di-O-(2, 3, 4-tri-O-acetyl-α-l-rhamnopyranosyl)-β-(3, 6-di-O-acetyl)-D-glucopyranosyl]-urs-12-en-28-oic acid (8)
To a solution of 7 (515 mg, 0.50 mmol) in anhydrous methanol (10 mL) was added 10% Pd/C (50 mg) under Ar at room temperature. The reaction was then stirred for 8 h at r.t. under hydrogen. After the reaction was completed, the mixture was filtered over a Celite pad and concentrated in vacuo. The residue was dissolved in anhydrous pyridine (10 mL) and were added acetic anhydride (0.56 mL, 6.0 mmol) and DMAP (12 mg, 0.10 mmol) at 0 °C. The mixture was warmed to 60 °C and stirred overnight under argon. Upon completion, the solvent was evaporated in vacuo, and the residue was re-dissolved in CH2Cl2.The organic phase was washed with 1 M HCl, saturated NaHCO3 solution and brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was subject to column chromatography on silica gel (CH2Cl2–MeOH, 5:1) to give 8 (588 mg, 94%) as a colorless solid. 1H NMR (600 Hz, CDCl3): δ 5.26–5.23 (m, 3H), 5.18 (dd, 1H, J = 10.1, 3.1 Hz, Rha-H-3), 5.11 (dd, 1H, J = 3.2, 17 Hz, Rha-H-2), 5.06–5.01 (m, 4H), 4.81 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.53 (d, 1H, J = 7.3 Hz, H-1′), 4.49 (d, 1H, J = 12.1 Hz, H-6′-1), 4.28–4.22 (m, 2H), 3.88–3.85 (m, 1H), 3.77 (t, 1H, J = 9.3 Hz), 3.68 (t, 1H, J = 8.3 Hz), 3.62–3.60 (m, 1H), 3.16 (dd, 1H, J = 11.5, 4.1 Hz, H-3), 2.55 (d, 1H, J = 12.4 Hz, H-18), 2.14, 2.13, 2.12, 2.11, 2.05, 2.02, 1.99, 1.98, (each s, each 3H, each CH3CO), 1.18 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.16 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.09, 1.03, 0.92, 0.81, 0.77 (each s, each 3H, each CH3), 0.95 (d, 3H, J = 6.0 Hz, CH3), 0.86 (d, 3H, J = 6.4 Hz, CH3); 13C NMR (151 Hz, CDCl3): δ 182.8, 170.5, 170.2, 170.1 (two), 170.0 (three), 169.6, 137.8, 125.8, 103.6 (C-1′), 99.4 (Rha-C-1), 97.0 (Rha-C-1), 90.0, 77.9, 75.7, 75.4, 72.1, 71.1, 70.6, 69.9, 69.7, 68.6, 68.5, 67.9, 66.7, 62.1, 55.9, 52.6, 47.9, 47.6, 42.0, 39.5, 39.0, 38.8, 36.7, 32.9, 30.6, 28.0, 27.8, 26.0, 24.1, 23.6, 23.3, 21.4, 21.2, 20.9 (two), 20.8 (two), 20.7 (two), 18.2, 17.2, 17.1, 17.0, 16.9, 16.2, 15.6; HRESIMS calcd for C64H94O24Na 1269.6033; found 1269.6042.
4.1.7. General procedure for the synthesis of all title saponins UA-29 -- UA-43
To a solution of 8 (1.0 mmol) in anhydrous CH2Cl2 (10 mL) was added oxalyl chloride (8.0 mmol), and the reaction mixture was allowed to stir at room temperature for 48 h under Ar. Then, the solvent was evaporated under the reduced pressure. Oxalyl chloride was removed by adding cyclohexane three times to yield the desired acid chloride, which was used directly in the next step. To the above acid chloride in 20 mL of anhydrous CH2Cl2 at 0 °C was added appropriate amine (1.5 mmol), followed by triethylamine (3.0 mmol). The reaction was stirred at room temperature under Ar for 5 h and then concentrated under reduced pressure. The obtained crude amide derivatives were re-dissolved in dry MeOH and CH2Cl2 (V:V = 1:1, 30 mL) and then NaOMe was added until pH = 10. After stirred at r.t. for 2 h, the solution was neutralized with Dowex 50 × 8 (H+) resin until pH = 7. The solid was removed, and the filtrate was concentrated under vacuum and purified by column chromatography (CH2Cl2–MeOH, 5:1) to yield UA-29 -- UA-43, respectively.
4.1.7.1. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-2-trifluoromethoxyaniline (UA-29)
Compound UA-29 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 7.76 (d, 1H, J = 8.1 Hz, Ar–H), 7.04 (dd, 2H, J = 14.1, 7.7 Hz, Ar–H), 6.93 (t, 1H, J = 7.8 Hz, Ar–H), 5.08 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.95 (t, 1H, J = 3.6 Hz, H-12), 4.62 (d, 1H, J = 1.1 Hz, Rha-H-1), 4.15 (d, 1H, J = 7.7 Hz, H-1′), 3.74–3.66 (m, 2H), 3.66–3.58 (m, 2H), 3.58–3.54 (m, 1H, Rha-H-2), 3.52 (d, 1H, J = 11.6 Hz, H-6′-1), 3.47 (dd, 1H, J = 9.5, 2.8 Hz, Rha-H-3), 3.41–3.32 (m, 2H), 3.29–3.22 (m, 1H), 3.20–3.06 (m, 3H), 1.94 (d, 1H, J = 11.2 Hz, H-18), 0.98 (d, 3H, J = 6.2 Hz, Rha-H-6), 0.93 (d, 3H, J = 5.6 Hz, Rha-H-6), 0.83 (s, 3H, CH3), 0.77 (s, 6H, 2 × CH3), 0.68 (s, 6H, 2 × CH3), 0.57 (d, 3H, J = 7.8 Hz, CH3), 0.48 (s, 3H, CH3); 13C NMR (151 Hz, CD3OD): δ 179.5, 178.4, 139.6, 139.2, 131.4, 128.2, 126.8, 125.2, 121.2, 105.2 (C-1′), 102.8 (Rha-C-1), 101.7 (Rha-C-1), 90.1, 80.0, 78.9, 77.9, 76.2, 73.6, 73.4, 72.2, 71.8, 71.7, 70.5, 69.7, 61.7, 57.0, 54.6, 54.1, 51.9, 50.0, 47.6, 43.1, 42.9, 40.6, 40.5, 40.1, 40.0, 37.5 (two), 28.9, 28.3, 27.0, 24.1, 23.9 (two), 21.3, 19.0, 17.8, 17.7, 17.5, 17.4, 17.3, 17.0 (two), 15.9; HRESIMS calcd for C55H83O16NF3 1070.5612; found 1070.5608.
4.1.7.2. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-2, 6-dimethoxyaniline (UA-30)
Compound UA-30 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 7.18 (t, 1H, J = 8.4 Hz, Ar–H), 6.63 (d, 2H, J = 8.5 Hz, Ar–H), 5.37 (d, 1H, J = 1.1 Hz, Rha-H-1), 5.35 (t, 1H, J = 3.2 Hz, H-12), 4.85 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.44 (d, 1H, J = 7.7 Hz, H-1′), 4.02–3.95 (m, 2H), 3.95–3.87 (m, 1H), 3.86–3.83 (m, 1H, Rha-H-2), 3.80 (d, 1H, J = 11.7 Hz, H-6′-1), 3.76 (s, 6H, 2 × OCH3), 3.66 (dd, 1H, J = 12.1, 3.2 Hz, H-6′-2), 3.63 (dd, 1H, J = 9.4, 3.0 Hz, Rha-H-3), 3.60–3.50 (m, 2H), 3.48–3.34 (m, 3H), 2.30 (d, 1H, J = 10.4 Hz, H-18), 1.27 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.2 Hz, Rha-H-6), 0.99 (d, 3H, J = 6.2 Hz, CH3), 0.93 (d, 3H, J = 6.2 Hz, CH3), 1.16, 1.06, 0.98, 0.92, 0.87 (each s, each 3H, each CH3); 13C NMR (151 Hz, CD3OD): δ 178.9, 157.1 (two), 139.4, 128.6, 126.9, 115.5, 105.2, 104.9 (C-1′), 102.8 (Rha-C-1), 101.7 (Rha-C-1), 90.1, 80.1, 78.9, 77.9, 76.2, 73.7, 73.4, 72.2, 71.9, 71.7, 70.5, 69.7, 61.7, 57.1, 56.0 (two), 54.6, 54.0, 49.7, 47.5 (two), 43.3, 40.7, 40.1, 40.0, 38.8, 37.6, 34.3, 32.0, 31.7, 30.5, 28.8, 28.3, 27.0, 25.1, 24.3, 23.7, 23.2, 21.5, 19.1, 17.8, 17.7, 17.5, 17.0, 16.1; HRESIMS calcd for C56H88O17N 1046.6046; found 1046.6042.
4.1.7.3. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-1, 4-benzodioxan-6-amine (UA-31)
Compound UA-31 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 7.05 (d, 1H, J = 2.2 Hz, Ar–H), 6.81 (dd, 1H, J = 8.7, 2.2 Hz, Ar–H), 6.73 (d, 1H, J = 8.7 Hz, Ar–H), 5.42 (t, 1H, J = 3.6 Hz, H-12), 5.36 (d, 1H, J = 1.6 Hz, Rha-H-1), 4.85 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.43 (d, 1H, J = 7.7 Hz, H-1′), 4.20 (s, 4H, OCH 2CH 2O), 4.01–3.96 (m, 2H), 3.96–3.87 (m, 2H), 3.87–3.83 (m, 1H, Rha-H-2), 3.80 (d, 1H, J = 11.4 Hz, H-6′-1), 3.75 (dd, J = 9.5, 3.1 Hz, 1H), 3.70–3.60 (m, 2H), 3.60–3.49 (m, 2H), 3.47–3.36 (m, 3H), 3.16 (dd, 1H, J = 11.5, 3.3 Hz, H-3), 2.28 (d, 1H, J = 10.6 Hz, H-18), 1.27 (d, 3H, J = 6.3 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.2 Hz, Rha-H-6), 0.98 (d, 3H, J = 5.9 Hz, CH3), 0.94 (d, 3H, J = 5.8 Hz, CH3), 1.14, 1.05, 0.92, 0.85, 0.75 (each s, each 3H, each CH3); 13C NMR (CD3OD): δ 178.1, 144.3, 141.7, 139.9, 132.8, 127.0, 117.5, 115.5, 111.7, 105.2 (C-1′), 102.7 (Rha-C-1), 101.7 (Rha-C-1), 90.1, 80.1, 79.0, 77.8, 76.2, 73.6, 73.4, 72.2, 71.9 (two), 71.7, 70.4, 69.7, 65.4, 65.2, 61.7, 57.0, 53.8, 43.1 (two), 40.6 (two), 39.9 (three), 38.0, 37.5 (two), 33.9, 31.7, 28.8, 28.3, 27.0, 25.0, 24.2, 23.9, 21.4, 19.0, 17.8, 17.7 17.6, 17.5, 17.0, 15.9; HRESIMS calcd for C56H86O17N 1044.5882; found 1044.5876.
4.1.7.4. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-4-biphenylamine (UA-32)
Compound UA-32 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 7.62–7.53 (m, 6H, Ar–H), 7.40 (t, 2H, J = 7.6 Hz, Ar–H), 7.29 (t, 1H, J = 7.4 Hz, Ar–H), 5.45 (t, 1H, J = 3.2 Hz, H-12), 5.36 (d, 1H, J = 1.1Hz, Rha-H-1), 4.86 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.42 (d, 1H, J = 7.7 Hz, H-1′), 4.00–3.96 (m, 2H), 3.96–3.88 (m, 1H), 3.86–3.83 (m, 1H, Rha-H-2), 3.80 (d, J = 11.2 Hz, H-6-1′), 3.75 (dd, 1H, J = 9.5, 3.3 Hz, Rha-H-3), 3.69–3.61 (m, 2H), 3.61–3.50 (m, 2H), 3.47–3.34 (m, 3H), 2.34 (d, 1H, J = 10.6 Hz, H-18), 1.27 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.20 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.00 (d, 3H, J = 6.1 Hz, CH3), 0.95 (d, 3H, J = 6.2 Hz, CH3), 1.15, 1.04, 0.90, 0.83, 0.75 (each s, each 3H, each CH3); 13C NMR (151 Hz, CD3OD): δ 178.4, 141.5, 139.9, 138.7, 138.0, 129.6 (two), 127.8 (two), 127.4 (three), 127.1, 122.4 (two), 105.2 (C-1′), 102.8 (Rha-C-1), 101.7 (Rha-C-1), 90.0, 80.1, 78.9, 77.9, 76.2, 73.6, 73.4, 72.2, 71.8, 71.7, 70.5, 69.7, 61.7, 57.0, 53.8, 47.6 (five), 43.1, 40.6, 39.9 (two), 38.0, 37.5, 33.8, 31.7, 28.8, 28.3, 27.0, 25.1, 24.2, 24.0, 21.4, 19.0, 17.8, 17.7, 17.6, 17.5, 17.0, 15.9; HRESIMS calcd for C60H886O15N 1062.6142; found 1062.6136.
4.1.7.5. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-4-benzylaniline (UA-33)
Compound UA-33 was obtained as a white solid. 1H NMR (CD3OD): δ 7.34 (d, 2H, J = 8.4 Hz, Ar–H), 7.28–7.21 (m, 2H, Ar–H), 7.17 (dd, 3H, J = 7.7, 2.3 Hz, Ar–H), 7.11 (d, 2H, J = 8.4 Hz, Ar–H), 5.43 (t, 1H, J = 3.6 Hz, H-12), 5.37 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.86 (s, 1H, Rha-H-1), 4.43 (d, 1H, J = 7.7 Hz, H-1′), 4.02–3.94 (m, 3H), 3.92 (s, 2H, Ar-CH 2-Ar), 3.91–3.89 (m, 1H), 3.84 (dd, 1H, J = 2.9, 1.7 Hz, Rha-H-2), 3.82–3.77 (m, 1H, H-6-1′), 3.74 (dd, 1H, J = 9.6, 3.2 Hz, Rha-H-3), 3.69–3.60 (m, 3H), 3.60–3.50 (m, 3H), 3.47–3.35 (m, 4H), 3.16–3.13 (m, 1H, H-3), 2.31 (d, 1H, J = 10.6 Hz, H-18), 1.27 (d, 3H, J = 6.3 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.2 Hz, Rha-H-6), 0.99 (d, J = 6.2 Hz, 3H), 0.94 (d, J = 6.5 Hz, 3H), 1.15 1.04, 0.91, 0.84, 0.73 (each s, each 3H, each CH3); 13C NMR (151 Hz, CD3OD): δ 179.5, 178.4, 142.4, 139.9, 138.7, 137.3, 129.8 (two), 129.6 (two), 129.2 (two), 127.1, 126.8, 122.4, 105.2 (C-1′), 102.8 (Rha-C-1), 101.8 (Rha-C-1), 90.1, 80.1, 78.9, 77.9, 76.2, 73.7, 73.4, 72.2, 71.9, 71.7, 70.5, 69.8, 61.7, 61.3, 57.0, 53.9, 51.9, 49.3, 47.6, 43.1, 42.0, 40.7, 40.0, 37.5, 33.9, 31.7, 28.8, 28.3, 27.0, 25.0, 24.2, 23.9, 21.3, 20.6, 19.0, 17.8, 17.7, 17.6, 17.5, 17.4, 17.0, 15.9, 14.2; HRESIMS calcd for C61H90O15N 1076.6302; found 1076.629.
4.1.7.6. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-2-naphthylamine (UA-34)
Compound UA-34 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 8.09 (s, 1H, Ar–H), 7.81–7.69 (m, 3H, Ar–H), 7.50 (dd, 1H, J = 8.8, 1.7 Hz, Ar–H), 7.46–7.32 (m, 2H, Ar–H), 5.45 (t, 1H, J = 3.6 Hz, H-12), 5.36 (d, 1H, J = 1.6 Hz, Rha-H-1), 4.85 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.41 (d, 1H, J = 7.7 Hz, H-1′), 4.00–3.96 (m, 1H), 3.96–3.87 (m, 1H), 3.87–3.83 (m, 1H, Rha-H-2), 3.80 (d, 1H, J = 11.1 Hz, H-6′-1), 3.75 (dd, 1H, J = 9.5, 3.2 Hz, Rha-H-3), 3.70–3.61 (m, 2H), 3.61–3.49 (m, 2H), 3.47–3.37 (m, 2H), 3.14 (dd, 1H, J = 11.5, 3.8 Hz, H-3), 2.35 (d, 1H, J = 10.5 Hz, H-18), 1.27 (d, 3H, J = 6.0 Hz, Rha-H-6), 1.20 (d, 3H, J = 6.2 Hz, Rha-H-6), 0.99 (d, 3H, J = 6.1 Hz, CH3), 0.95 (d, 3H, J = 6.1 Hz, CH3), 1.13, 1.03, 0.85, 0.81, 0.71 (each s, each 3H, each CH3); 13C NMR (151 Hz, CD3OD): δ 178.5, 139.9, 136.9, 134.9, 131.9, 129.0, 128.3 (two), 127.1, 125.7, 122.0, 118.7, 105.2 (C-1′), 102.8 (Rha-C-1), 101.7 (Rha-C-1), 90.1, 80.1, 79.0, 77.8, 76.2, 73.6, 73.4, 72.2, 71.8 (two), 71.7, 70.5, 69.7, 61.7, 57.0, 53.9, 49.4, 43.1 (two), 40.6 (two), 39.9 (two), 38.0, 37.5 (two), 33.8, 31.7, 28.8, 28.3, 26.9, 25.1, 24.2, 24.0, 21.4, 19.0, 17.8 (two), 17.7, 17.6, 17.5, 17.0, 15.9; HRESIMS calcd for C58H86O15N 1036.5997; found 1036.5983.
4.1.7.7. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-quinolin-6-amine (UA-35)
Compound UA-35 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 8.78–8.70 (m, 1H, Ar–H), 8.31–8.22 (m, 2H, Ar–H), 7.96 (d, 1H, J = 9.1 Hz, Ar–H), 7.81 (dd, 1H, J = 9.1, 2.2 Hz, Ar–H), 7.50 (dd, 1H, J = 8.4, 4.3 Hz, Ar–H), 5.47 (t, 1H, J = 3.6 Hz, H-12), 5.36 (d, 1H, J = 1.6 Hz, Rha-H-1), 4.85 (s, 1H, Rha-H-1), 4.42 (d, 1H, J = 7.7 Hz, H-1′), 4.01–3.95 (m, 2H), 3.95–3.87 (m, 1H), 3.87–3.83 (m, 1H, Rha-H-2), 3.80 (d, 1H, J = 12.2 Hz, H-6′-1), 3.74 (dd, 1H, J = 9.5, 3.3 Hz, Rha-H-3), 3.70–3.61 (m, 2H), 3.61–3.50 (m, 2H), 3.48–3.34 (m, 3H), 3.15 (dd, 1H, J = 11.8, 3.8 Hz, H-3), 2.40 (d, 1H, J = 11.1 Hz, H-18), 1.26 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.20 (d, 3H, J = 6.1 Hz, Rha-H-6), 1.15, 1.02, 1.00 (d, 3H, J = 6.3 Hz, CH3), 0.96 (d, 3H, J = 6.2 Hz, CH3), 0.85, 0.80, 0.70 (each s, each 3H, each CH3); 13C NMR (151 Hz, CD3OD): δ 178.7, 149.8, 145.7, 139.9, 138.1, 137.8, 130.0, 129.1, 127.1, 126.0, 122.6, 118.1, 105.2 (C-1′), 102.8 (Rha-C-1), 101.7 (Rha-C-1), 90.0, 80.2, 79.0, 77.9, 76.2, 73.7, 73.4, 72.2, 71.9 (two), 71.8, 70.5, 69.7, 61.7, 57.0, 53.8, 49.6, 47.6 (four), 43.1, 40.6 (two), 39.9, 38.0, 37.5, 33.8, 31.6, 28.8, 28.3, 26.9, 25.0, 24.2, 24.0, 21.3, 19.0, 17.8, 17.7, 17.5 (two), 17.0, 15.9; HRESIMS calcd for C57H85O15N2 1037.5938; found 1037.5932.
4.1.7.8. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-1-benzothiophen-5-amine (UA-36)
Compound UA-36 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 8.03 (s, 1H, Ar–H), 7.80 (d, 1H, J = 8.6 Hz, Ar–H), 7.55 (d, 1H, J = 5.4 Hz, Ar–H), 7.37 (d, 1H, J = 8.7 Hz, Ar–H), 7.30 (d, 1H, J = 5.4 Hz, Ar–H), 5.45 (t-like, 1H, J = 3.6 Hz, H-12), 5.36 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.86 (d, 1H, J = 1.1 Hz, Rha-H-1), 4.42 (d, 1H, J = 7.7 Hz, H-1′), 4.01–3.96 (m, 2H), 3.96–3.87 (m, 1H), 3.87–3.83 (m, 1H, Rha-H-2), 3.80 (d, 1H, J = 11.6 Hz, H-6′-1), 3.75 (dd, 1H, J = 9.5, 2.7 Hz, Rha-H-3), 3.70–3.61 (m, 2H), 3.61–3.50 (m, 2H), 3.47–3.37 (m, 3H), 2.35 (d, 1H, J = 10.6 Hz, H-18), 1.27 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.20 (d, 3H, J = 6.1 Hz, Rha-H-6), 0.99 (d, 3H, J = 6.0 Hz, CH3), 0.95 (d, 3H, J = 6.1 Hz, CH3), 1.15, 1.04, 0.89, 0.82, 0.74 (each s, each 3H, each CH3); 13C NMR (151 Hz, CD3OD): δ 177.2, 140.0, 138.7, 135.6, 135.0, 127.1, 125.9, 123.5, 121.9, 118.8, 115.6, 104.1 (C-1′), 101.6 (Rha-C-1), 100.5 (Rha-C-1), 88.9, 78.9, 77.8, 76.7, 75.0, 72.5, 72.3, 71.0, 70.7 (two), 70.6, 69.3, 68.6, 60.5, 55.8, 52.7, 48.2, 46.4, 41.9, 39.5 (two), 38.8 (two), 36.9, 36.3, 32.7, 30.5, 27.7, 27.1, 25.8, 23.9, 23.1, 22.8, 20.2, 17.8, 16.6 (two), 16.5, 16.5, 16.4, 15.8, 14.8; HRESIMS calcd for C56H84O15NS 1042.5546; found 1042.5538.
4.1.7.9. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-1-methyl-5-indazolamine (UA-37)
Compound UA-37 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 7.93 (s, 1H, Ar–H), 7.90 (s, 1H, Ar–H), 7.49 (d, 1H, J = 9.1 Hz, Ar–H), 7.42 (d, 1H, J = 9.0 Hz, Ar–H), 5.43 (t-like, 1H, J = 3.6 Hz, H-12), 5.37 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.85 (d, 1H, J = 1.6 Hz, Rha-H-1), 4.42 (d, 1H, J = 7.7 Hz, H-1′), 4.04 (s, 3H, NCH3), 4.01–3.96 (m, 2H), 3.96–3.87 (m, 1H), 3.87–3.83 (m, 1H, Rha-H-2), 3.80 (d, 1H, J = 11.5 Hz, H-6′-1), 3.75 (dd, 1H, J = 9.5, 3.0 Hz, Rha-H-3), 3.70–3.61 (m, 2H), 3.61–3.50 (m, 2H), 3.47–3.35 (m, 3H), 2.36 (d, 1H, J = 10.8 Hz, H-18), 1.26 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.20 (d, 3H, J = 6.1 Hz, Rha-H-6), 0.99 (d, 3H, J = 6.0 Hz, CH3), 0.95 (d, 3H, J = 6.2 Hz, CH3), 1.14, 1.04, 0.88, 0.82, 0.75 (each s, each 3H, each CH3); 13C NMR (151 Hz, CD3OD): δ 178.5, 139.8, 138.7, 133.3, 132.8, 127.0, 124.7, 123.9, 113.9, 110.1, 105.2 (C-1′), 102.7 (Rha-C-1), 101.7 (Rha-C-1), 90.0, 80.0, 78.9, 77.8, 76.2, 73.6, 73.4, 72.2, 71.8, 71.7, 70.4, 69.7, 61.7, 56.9, 53.8, 49.2, 48.7, 47.6 (three), 43.0, 40.7, 40.0, 39.9, 39.9, 38.1, 37.5, 35.4, 33.9, 31.7, 28.8, 28.3, 26.9, 25.0, 24.2, 23.9, 21.4, 19.0, 17.8, 17.7, 17.5, 17.0, 15.9; HRESIMS calcd for C56H85O15N3 1040.6043; found 1040.6035.
4.1.7.10. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-tetrazole (UA-38)
Compound UA-38 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 9.03 (s, 1H, N = CH), 5.36 (d, 1H, J = 1.6 Hz, Rha-H-1), 5.23 (t-like, 1H, J = 3.7 Hz, H-12), 4.85 (s, 1H, Rha-H-1), 4.43 (d, 1H, J = 7.7 Hz, H-1′), 4.02–3.95 (m, 2H), 3.95–3.87 (m, 1H), 3.86–3.83 (m, 1H, Rha-H-2), 3.80 (d, 1H, J = 11.8 Hz, H-6′-1), 3.75 (dd, 1H, J = 9.5, 3.2 Hz, Rha-H-3), 3.70–3.63 (m, 2H), 3.58–3.51 (m, 2H), 3.48–3.36 (m, 3H), 3.17 (dd, 1H, J = 10.9, 3.2 Hz, H-3), 2.22 (d, 1H, J = 11.3 Hz, H-18), 1.27 (d, 3H, J = 6.1 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.1 Hz, Rha-H-6), 0.88 (d, 3H, J = 6.4 Hz, CH3), 0.96 (s, 6H, 2 × CH3), 1.11, 1.05, 0.86, 0.77 (each s, each 3H, each CH3); 13C NMR (151 Hz, CD3OD): δ 178.3, 143.8, 138.1, 125.6, 104.1 (C-1′), 101.6 (Rha-C-1), 100.6 (Rha-C-1), 88.9, 78.9, 77.8, 76.7, 75.0, 72.5, 72.2, 71.0, 70.7, 70.6, 69.3, 68.6, 60.5, 55.9, 53.4, 52.9, 50.7, 48.0, 41.7, 39.4, 38.9, 38.8, 36.4 (two), 32.8, 30.5, 30.2, 29.4, 27.7, 27.1, 25.8, 23.9, 22.9, 22.8, 20.2, 17.9, 16.6, 16.5, 16.3 (two), 15.8, 14.8; HRESIMS calcd for C49H80O15N5 978.5639; found 978.5633.
4.1.7.11. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-benzylamine (UA-39)
Compound UA-39 was prepared as a white; 1H NMR (600 Hz, CD3OD): δ 8.09 (t, 1H, J = 8.5 Hz, Ar–H), 7.95 (t, 1H, J = 8.6 Hz, Ar–H), 7.65–7.62 (m, 1H, Ar–H), 7.29–7.27 (m, 1H, Ar–H), 7.25–7.22 (m, 1H, Ar–H), 5.39 (d, 1H, J = 1.7 Hz, Rha-H-1), 5.30 (t, 1H, J = 3.6 Hz, H-12), 4.87 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.44 (d, 1H, J = 7.8 Hz, H-1′), 4.31 (t, 1H, J = 5.0 Hz), 4.02–3.98 (m, 2H), 3.96–3.92 (m, 1H), 3.86 (dd, 1H, J = 3.3, 1.1 Hz, Rha-H-2), 3.81 (dd, 1H, J = 12.2, 2.1 Hz, H-6-1′), 3.74 (dd, 1H, J = 9.7, 3.4 Hz, Rha-H-3), 3.68–3.64 (m, 4H), 3.60 (t, 1H, J = 8.7 Hz), 3.55 (t, 1H, J = 9.4 Hz), 3.47–3.41 (m, 2H), 3.39 (t, 1H, J = 8.8 Hz), 3.24–3.20 (m, 1H), 3.18 (dd, 1H, J = 11.8, 4.3 Hz, H-3), 2.17 (d, 1H, J = 9.7 Hz, H-18), 1.28 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.11, 1.05, 0.92, 0.86, 0.59 (each s, each 3H, CH3), 0.98 (d, 3H, J = 6.3 Hz, CH3), 0.91 (d, 3H, J = 6.4 Hz, CH3); 13C NMR (151 Hz, CD3OD): δ 178.6, 138.8, 138.5, 129.2, 128.0 (two), 127.5 (two), 126.7, 125.8, 104.0 (C-1′), 101.6 (Rha-C-1), 100.5 (Rha-C-1), 88.9, 79.0, 77.9, 76.7, 75.0, 72.5, 72.3, 71.0, 70.8 (two), 70.6, 69.3, 68.6, 60.6, 55.8, 53.0, 51.5, 48.5, 46.5, 42.9, 41.9, 39.5, 38.9, 38.8 (two), 37.3, 36.4, 32.8, 31.7, 30.5, 29.3, 27.5, 27.1, 25.8, 23.9, 23.0, 22.6, 22.3, 20.2, 17.8, 16.6, 16.5 (two), 16.3, 15.8, 14.7; HRESIMS calcd for C55H85O15NNa 1022.5817; found 1022.5822.
4.1.7.12. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-1, 2, 3, 4-tetrahydroisoquinolin (UA-40)
Compound UA-40 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 7.21–7.06 (m, 4H, Ar–H), 5.36 (d, 1H, J = 1.6 Hz, Rha-H-1), 5.26–5.06 (m, 2H), 4.86 (d, 1H, J = 1.7 Hz, Rha-H-1), 4.41 (d, 1H, J = 7.7 Hz, H-1′), 4.01–3.95 (m, 2H), 3.95–3.86 (m, 2H), 3.86–3.82 (m, 1H, Rha-H-2), 3.79 (d, 1H, J = 11.4 Hz, H-6′), 3.74 (dd, 1H, J = 9.5, 3.2 Hz, Rha-H-3), 3.70–3.59 (m, 3H), 3.59–3.50 (m, 2H), 3.48–3.33 (m, 3H), 3.14 (dd, 1H, J = 11.3, 3.6 Hz, H-3), 2.97–2.74 (m, 3H), 2.46 (d, 1H, J = 10.4 Hz, H-18), 1.26 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.1 Hz, Rha-H-6), 0.96 (d, 3H, J = 5.1 Hz, CH3), 1.10, 1.03, 0.98, 0.91, 0.84, 0.68 (each s, each 3H, each CH3); 13C NMR (151 Hz, CD3OD): δ 177.4, 139.7, 135.7, 134.5, 129.3, 127.6, 127.2, 127.1, 126.5, 105.2 (C-1′), 102.8 (Rha-C-1), 101.7 (Rha-C-1), 90.1, 80.0, 79.0, 77.9, 76.2, 73.6, 73.4, 72.2, 71.8, 71.7, 70.5, 69.7, 61.7, 61.3, 57.1, 54.6, 50.0, 45.0, 43.1, 40.4, 40.0 (two), 37.5, 35.0, 34.0, 32.8, 31.7, 31.3, 30.5, 30.2, 29.3, 29.1, 28.3, 26.9, 24.1, 23.5, 21.4, 20.6, 19.0, 17.8, 17.7, 17.2, 17.0, 16.0, 14.2; HRESIMS calcd for C57H88O15N 1026.6148; found 1026.6146.
4.1.7.13. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-α-phenylbenzylamine (UA-41)
Compound UA-41 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 7.54 (d, 1H, J = 7.8 Hz, NHCH), 7.34–7.22 (m, 10H, Ar–H), 6.11 (d, 1H, J = 7.5 Hz, NHCH), 5.38 (d, 1H, J = 1.7 Hz, Rha-H-1), 5.24 (t-like, J = 3.6 Hz, 1H, H-12), 4.87 (d, 1H, J = 1.2 Hz, Rha-H-1), 4.41 (d, 1H, J = 7.7 Hz, H-1′), 4.02–3.97 (m, 2H), 3.97–3.88 (m, 1H), 3.88–3.84 (m, 1H, Rha-H-2), 3.82–3.74 (m, 2H, H-6-1′, Rha-H-3), 3.70–3.62 (m, 2H), 3.62–3.50 (m, 2H), 3.47–3.34 (m, 4H), 3.14 (dd, 1H, J = 11.6, 3.9 Hz, H-3), 2.25 (d, 1H, J = 10.4 Hz, H-18), 1.27 (d, 3H, J = 6.3 Hz, Rha-H-6), 1.22 (d, 3H, J = 6.1 Hz, Rha-H-6), 0.98 (d, 3H, J = 6.3 Hz, CH3), 0.90 (d, 3H, J = 6.3 Hz, CH3), 1.08, 1.03, 0.99, 0.84, 0.79 (each s, each 3H, each CH3); 13C NMR (151 Hz, CD3OD): δ 179.1, 143.4, 142.5, 139.7, 129.5 (two), 129.1 (two), 128.9 (two), 128.4, 128.3, 127.8 (two), 127.2, 105.2 (C-1′), 102.6 (Rha-C-1), 101.5 (Rha-C-1), 90.0, 80.0, 78.9, 77.8, 76.1, 73.6, 73.4, 72.2, 71.9, 71.7, 70.4, 69.7, 61.6, 58.0, 56.9, 54.2, 48.7, 47.5 (three), 43.0, 40.7, 40.5, 39.9, 39.9, 38.2, 37.4, 34.0, 31.7, 28.7, 28.2, 26.9, 25.0, 23.9, 23.7, 21.3, 18.9, 17.9, 17.83 17.7, 17.4, 17.0, 15.9; HRESIMS calcd for C61H90O15N 1076.6302; found 1076.6300.
4.1.7.14. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-4-methoxybenzylamine (UA-42)
Compound UA-42 was prepared as a white solid; 1H NMR (600 Hz, CD3OD): δ 7.19 (d, 2H, J = 8.7 Hz, Ar–H), 6.83 (d, 2H, J = 9.1 Hz, Ar–H), 5.36 (d, 1H, J = 1.8 Hz, Rha-H-1), 5.26 (t, 1H, J = 3.7 Hz, H-12), 4.84 (d, 1H, J = 1.8 Hz, Rha-H-1), 4.42 (d, 1H, J = 7.8 Hz, H-1′), 4.26 (d, 1H, J = 14.4 Hz, NHCH2-1′), 4.18 (d, 1H, J = 14.4 Hz, NHCH2-2′), 4.01–3.93 (m, 2H), 3.94–3.86 (m, 1H), 3.83 (dd, 1H, J = 3.4, 1.8 Hz, Rha-H-2), 3.79 (dd, 1H, J = 12.0, 2.0 Hz, H-6-1′), 3.76 (s, 3H, OCH3), 3.74 (dd, 1H, J = 9.6, 3.3 Hz, Rha-H-3), 3.65 (dd, 1H, J = 11.9, 4.0 Hz, H-6-2′), 3.62 (dd, 1H, J = 9.4, 3.5 Hz, Rha-H-3), 3.58 (t, 1H, J = 8.7 Hz), 3.53 (t, 1H, J = 9.1 Hz), 3.48–3.35 (m, 3H), 3.33–3.27 (m, 1H), 3.15 (dd, 1H, J = 11.7, 4.4 Hz, H-3), 2.12 (d, 1H, J = 10.6 Hz, H-18), 1.26 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.09, 1.04, 0.90, 0.85, 0.53 (each s, each 3H, CH3), 0.96 (d, 3H, J = 6.2 Hz, CH3), 0.89 (d, 3H, J = 6.6 Hz, CH3); 13C NMR (151 Hz, CD3OD): δ 179.9, 160.4, 140.0, 132.2, 130.4, 130.0, 127.2, 119.4, 116.7, 114.8, 105.5 (C-1′), 103.1 (Rha-C-1), 102.0 (Rha-C-1), 90.3 (C-3), 80.5, 79.3, 78.2, 76.5, 74.0, 73.7, 72.5, 72.2, 72.1, 70.8 (two), 70.0, 62.0 (C-6′), 57.3, 55.7 (C-5), 54.5 (C-18), 49.9 (C-9), 43.9, 43.8, 43.3 (C-14), 40.9 (two), 40.3 (two), 40.2, 38.7, 37.8, 34.3, 32.0, 29.0, 28.5, 27.3, 25.3, 24.4, 24.0, 21.6, 19.3, 18.0, 17.9, 17.7, 17.3, 16.2; HRESIMS calcd for C56H87O16NNa 1052.5923; found 1052.5925.
4.1.7.15. N-{3β-O-[2, 4-Di-O-(α-l-Rhamnopyranosyl)-β-d-glucopyranosyl]-urs-12-en-28-oyl}-phenylethylamine (UA-43)
Compound UA-43 was obtained as a white solid. 1H NMR (600 Hz, CD3OD): δ 7.35–7.15 (m, 5H, Ar–H), 6.93 (t-like, 1H, NHCH2), 5.36 (d, 1H, J = 1.6 Hz, Rha-H-1), 5.09 (t, 1H, J = 3.6 Hz, H-12), 4.86 (s, 1H, Rha-H-1), 4.42 (d, 1H, J = 7.7 Hz, H-1′), 4.03–3.95 (m, 2H), 3.86–3.82 (m, 1H, Rha-H-2), 3.80 (d, 1H, J = 11.3 Hz, H-6′-1), 3.75 (dd, 1H, J = 9.5, 3.2 Hz, Rha-H-3), 3.70–3.59 (m, 2H), 3.59–3.49 (m, 3H), 3.48–3.37 (m, 3H), 3.15 (dd, 1H, J = 11.4, 3.8 Hz, H-3), 2.88–2.66 (m, 3H, NHCH 2CH 2), 1.27 (d, 3H, J = 6.2 Hz, Rha-H-6), 1.21 (d, 3H, J = 6.1 Hz, Rha-H-6), 0.89 (d, 3H, J = 7.9 Hz, CH3), 1.08, 1.04, 0.96, 0.92, 0.87, 0.68 (each s, each 3H, each CH3); 13C NMR (151 Hz, CD3OD): δ 180.0 (C-28), 140.3, 139.6 (C-13), 129.6 (two), 129.4 (two), 127.3 (C-12), 127.0, 105.2 (C-1′), 102.8 (Rha-C-1), 101.7 (Rha-C-1), 90.1, 80.1, 78.9, 77.9, 76.2, 73.6, 73.4, 72.2, 71.8 (two), 71.7, 70.5, 69.7, 61.7, 56.9, 54.6, 54.0, 43.0, 42.0, 40.6 (two), 40.0 (two), 39.9, 38.3, 37.5, 36.0, 33.8, 31.6, 30.5, 28.6, 28.3, 26.9, 25.1, 24.1, 23.8, 21.4, 19.0, 17.8, 17.7, 17.5 (two), 17.0, 16.0; HRESIMS calcd for C56H88O15N 1014.6138; found 1014.6135.
4.2. Biology assay
4.2.1. Cell lines and plasmids
HEK-293T, 293T-ACE2, and Vero-E6 cell lines (ATCC, USA) were cultured in Dulbecco's Modified Eagle Medium (DMEM, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Capricorn scientific, Germany) and 1% penicillin (100 units/ml)/streptomycin (100 μg/mL) (Gibco, USA). 293T cells stably expressing human-ACE2 (293T/ACE2) were constructed by our laboratory.
Plasmid pcDNA3.1-SARS-CoV-2 S and pAAV-IRES-GFP-SARS-CoV-2 S were presented by Professor Shibo Jiang kindly. Based on pcDNA3.1-SARS-CoV-2 S, the mutant plasmids (N764A, R765A, Q957A, K964A) were constructed by designing point mutant primer and PCR. PCR should be performed with high fidelity enzyme (Takara, Japan) and the fragments were recycled and relinked by homologous recombination. Primers include mutant points were used for PCR amplification with pcDNA3.1-SARS-CoV-2 S as a template, then homologous recombination was performed according to instructions from manufacturer (Vazyme, China). The relinked products were used to transform stabl3 and bacteria was seeded on the corresponding resistance plates. After incubation in 37 °C overnight, single colonies on plates were selected and sequenced. Mutation sites and corresponding primers were shown in followed table:
| 764/765-Foward | TTTGTACCCAGCTGGCTGCAGCCCTGACAGGCATCGCCGTGGA |
| 764/765-Reverse | CTGTCAGGGCTGCAGCCAGCTGGGTACAAAAGCTGCCGTACT |
| 957/964-Foward | GCGGCCCTGAATACCCTGGTGGCGCAGCTGAGCTCCAA |
| 957/964-Reverse | CGCCACCAGGGTATTCAGGGCCGCGGCGTTCTGATTCACCA |
| Circ-Foward | ACCTGGTTCCACGCCATCCACGTGA |
| Circ-Reverse | CCGCTCACGTGGATGGCGTGGAACC |
Plasmid pAAV-IRES-EGFP was purchased from Hedgehogbio Science and Technology Ltd. Expression plasmids for full-length vesicular stomatitis virus (VSV) glycoprotein (VSV-G) were obtained from Addgene (Cambridge, MA).
4.2.2. Seudotyped SARS-CoV-2 infection assay
Pseudovirus particles were produced as described [30]. Briefly, plasmids SARS-CoV-2 S, mutant E484K, D614G, N501Y, N764A/R765A, Q957A/K964A, and N764A/R765A/Q957A/K964A were transfected into 293T cells and supernatant containing PsV was collected after 48 h 293T-ACE2 cells were seeded in 96 wells plates. After 24 h, a mixture that consisted of 50 μL gradient concentrations of compounds and 50 μL PsV supernatant was added to each well and incubated with cells for 48 h. Cells were lysed and luciferase activities were quantified by Luciferase assay system (Promega, USA).
4.2.3. Cytotoxicity assay
The cytotoxicity effect of the compounds on 293T-ACE2 or Vero E6 cells were measured by methyl thiazolyl tetrazolium (MTT) assay. In brief, monolayers cells in 96-well plates were rinsed by PBS and incubated with indicated compounds for 48 h subsequently. Then, cells were treated with 0.5 mg/mL MTT for 4 h at 37 °C and formazan crystals in viable cells were dissolved in 150 μL DMSO. The absorbance of solubilized formazan was measured by Synergy multimode reader (BioTek, American) at 570 nm.
4.2.4. Authentic SARS-CoV-2 inhibition assay
Authentic SARS-CoV-2 inhibition assay was performed by Wuhan institute of virology, Chinese academy of sciences, as described previously [35]. Briefly, SARS-CoV-2 (Wuhan-HU-1) was incubated with different concentrations of UA-18 or UA-30 for 1 h before placing the mixture in vero-E6 cells for another 1 h incubation. After that, fresh medium with variant concentrations of compounds was added and viral total RNA was extracted by Viral RNA/DNA Extraction Kit (Takara, Japan) after 24 h, and the S gene copies were quantified on ABI 7500 (Takara TB Green® Premix Ex Taq™ II, Japan) by using the primers reported before [30,35].
4.2.5. Indirect immunofluorescence assay
After the authentic SARS-CoV-2 inhibition assay, we fixed the cells with 4% paraformaldehyde (Bio-Rad) and permeabilized by using Triton X-100 (Sigma, USA) [35]. Cells were stained with polyclonal rabbit anti-NP antibody and a secondary peroxidase-labeled goat anti-rabbit IgG H&L (Alexa Fluor® 488) (Abcam). DAPI was used to color nucleus and images were acquired by Axio Observer microscope (Zeiss, Germany).
4.2.6. Co-immunoprecipitation and western blotting
Plasmids pcDNA3.1-ACE2-Flag and pcDNA3.1-SARS-S were co-transfected into 293T cells. Total protein was extracted after 48 h and then incubated with protein A Sepharose in conjunction with anti-flag antibody or mouse IgG. The samples were incubated overnight and separated by 10% PAGE subsequently, followed by transfer to nitrocellulose membranes (Roche, Germany). ACE2 and SARS-S were detected by anti-Flag (Sigma, USA) and anti-SARS-CoV-S (Sinol biological Inc., China) with mouse anti-goat-horseradish peroxidase (HRP) (Fude biological Technology Co., LTD., China) as the secondary antibody, respectively [42].
4.2.7. Cell-cell fusion assays
HEK-293T cells were transfected with pAAV-IRES-GFP-SARS-CoV-2 S or vehicle plasmid pAAV-IRES-EGFP to construct effector cells by using Polyjet (SignaGen, USA). Targeted cells (Vero-E6) were seeded in 96 well plates 6 h prior to cell-cell fusion. Effector cells treated with or without gradient compounds for 30 min and were overlaid on targeted cells subsequently. After 24 h, three random fields were imaged by inverted fluorescence microscope (Zeiss, Germany) [35].
4.2.8. Surface plasmon resonance (SPR) measurement
Compound UA-18 or UA-30 was fixed on the chip by photo-crosslinking, then recombinant SARS-CoV-2 S-trimer protein (DRA 47, Novoprotein Inc. Shanghai) at indicated concentrations was injected sequentially into the chamber in buffer PBST (0.1% Tween 20, pH 7.4). The interaction of S-trimer with UA-18 or UA-30 fixed was detected by PlexArrayTM HT SPRi (Seattle, US). The reaction temperature was controlled at 4 °C, binding time was 600 s, disassociation time was 360 s, flow rate was 0.5 μL/s. The chip was regenerated with Glycine Hydrochloride (pH 2.0). The data of interaction signals was retrieved and analyzed with PlexeraDE software [42,43].
4.2.9. Circular dichroism (CD) spectroscopy
CD spectra were recorded on a Chirascan plus ACD (Applied Photophysics Ltd, England) [35]. HR1P and HR2P were dissolved in buffer (0.1 M KCl, 0.05 M KH2PO4, pH 7.2) at a final concentration of 10 μM. Briefly, HR1P was incubated with PBS or UA-30 (20 μM) at 25 °C for 30 min, followed by addition of HR2P (10 μM). After further incubation at 25 °C for 30 min, the CD wave scans were measured from 190 to 260 nm at 4 °C with the bandwidth of 2 nm and the step size of 1 nm.
4.2.10. Molecular docking
A molecular docking study was performed using Discovery Studio 3.0 [44,45]. The 3D crystal structure of SARS-CoV-2 spike glycoprotein was downloaded from RCSB Protein Date Bank (www.rcsb.org) using PDB ID of 6VXX, water and glycosyl molecules removed by manual. The protein and the ligand were prepared by minimization with CHARMM force field. Then the binding site of the protein was defined and prepared for docking by using Define Site (From Receptor Cavities) protocol. Molecular docking results were carried out using CDOCKER protocol without constraint and ranked by -CDOCKER_ENERGY.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (82073722 to G.S. and 82130101 to S.L.), Guangdong Basic and Applied Basic Research Foundation (2022A1515010016) and the special Project for prevention and control of SARS-CoV-2 from Educational Commission of Guangdong Province of China (2020KZDZX1039, 2020KZDZX1038) to G.S.; the Major scientific and technological projects of Guangdong Province (2019B020202002) and Chinese Academy of Traditional Chinese Medicine (ZZ13-035-02, 2019XZZX-LG04) to S. L.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2022.114426.
Appendix A. Supplementary data
The following is the supplementary data to this article:
References
- 1.Bedford J., Enria D., Giesecke J., Heymann D.L., Ihekweazu C., Kobinger G., Lane H.C., Memish Z., Oh M.D., Sall A.A., Schuchat A., Ungchusak K., Wieler L.H. COVID-19: towards controlling of a pandemic. Lancet. 2020;395:1015–1018. doi: 10.1016/S0140-6736(20)30673-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Jiang R.D., Liu M.Qi., Chen Y., Shen X.R., Wang X., Zheng X.S., Zhao K., Chen Q.J., Deng F., Liu L.L., Yan B., Zhan F.X., Wang Y.Y., Xiao G.F., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin, Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollán M., Pérez-Gómez B., Pastor-Barriuso R., Oteo J., Hernán M.A., Pérez-Olmeda M., Sanmartín J.L., Fernández-García A., Cruz I., de Larrea N.F., Molina M., Rodríguez-Cabrera F., Martín M., Merino-Amador P., Paniagua J.L., Muñoz-Montalvo J.F., Blanco F., Yotti R. Prevalence of SARS-CoV-2 in Spain (ENE-COVID): a nationwide, population-based seroepidemiological study. Lancet. 2020;396:535–544. doi: 10.1016/S0140-6736(20)31483-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coronavirus W.H.O. (COVID-19) dashboard. 2021. https://covid19.who.int
- 5.Krammer F. SARS-CoV-2 vaccines in development. Nature. 2020;586:516–527. doi: 10.1038/s41586-020-2798-3. [DOI] [PubMed] [Google Scholar]
- 6.Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Marc G.P., Moreira E.D., Zerbini C., Bailey R., Swanson K.A., Roychoudhury S., Koury K., Li P., Kalina W.V., Cooper D., Hammitt L.L., Türeci Ö., Nell H., Schaefer A., Ünal S., Tresnan D.B., Mather S., Dormitzer P.R., Şahin U., Jansen K.U., Gruber W.C. Safety and efficacy of the BNT162b2 mRNA covid-19, N. Engl. J. Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poland G.A., Ovsyannikova I.G., Kennedy R.B. SARS-CoV-2 immunity: review and applications to phase 3 vaccine candidates. Lancet. 2020;396:1595–1606. doi: 10.1016/S0140-6736(20)32137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forni G., Mantovani A. On behalf of the Covid-19 commission of accademia nazionale dei Lincei, R, COVID-19 vaccines: where we stand and challenges ahead. Cell Death Differ. 2021;28:626–639. doi: 10.1038/s41418-020-00720-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pardo J., Shukla A.M., Chamarthi G., Gupte A. The journey of remdesivir: from Ebola to COVID-19. Drugs Context (US) 2020;9:1–9. doi: 10.7573/dic.2020-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y., Zhang D., Du G., Du R., Zhao J., Jin Y., Fu S.Z., Gao L., Cheng Z.S., Lu Q.F., Hu Y., Luo G.W., Wang K., Lu Y., Li H.D., Wang S.Z., Ruan S.N., Yang C.Q., Mei C.L., Wang Y., Ding D., Wu F., Tang X., Ye X.Z., Ye Y.C., Liu B., Yang J., Yin W., Wang A.L., Fan G.H., Zhou F., Liu Z.B., Gu X.Y., Xu J.Y., Shang L.H., Zhang Y., Cao L.J., Guo T.T., Wan Y., Qin H., Jiang Y.S., Jaki T., Hayden F.G., Horby P.W., Cao B., Wang C. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569–1578. doi: 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., Hohmann E., Chu H.Y., Luetkemeyer A., Kline S., de Castilla D.L., Finberg R.W., Dierberg K., Tapson Vi., Hsieh L., Patterson T.F., Paredes R., Sweeney D.A., Short W.R., Touloumi G., Lye D.C., Ohmagari N., Oh M.D., Ruiz-palacios G.M., Benfield T., Fätkenheuer G., Kortepeter M.G., Atmar R.L., Creech C.B., Lundgren J., Babiker A.G., Pett S., Neaton J.D., Burgess T.H., Bonnett T., Green M., Makowski M., Osinusi A., Nayak S., Lane H.C. ACTT-1 study group members. Remdesivir for the treatment of covid-19-final report. N. Engl. J. Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The RECOVERY Collaborative Group Dexamethasone in hospitalized patients with Covid-19. N. Engl. J. Med. 2021;384:693–704. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen Z.N., Ratia K., Cooper L., Kong D.Y., Lee H., Kwon Y.J., Li Y.F., Alqarni S., Huang F., Dubrovskyi O., Rong L.J., Thatcher G.R.J., Xiong R. Design of SARS-CoV-2 PLpro inhibitors for COVID-19 antiviral therapy leveraging binding cooperativity. J. Med. Chem. 2022;65:2940–2955. doi: 10.1021/acs.jmedchem.1c01307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffman R.L., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S., He M.Y., Hogan R.J., Kozminski K., Li L.L.Y., Lockner J.W., Lou J.H., Marra M.T., M L.J., Jr., Murray B.W., Nieman J.A., Noell S., Planken S.P., Rowe T., Ryan K., Smith G.J., 3rd, Solowiej J.E., Steppan C.M., Taggart B. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of covid-19. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iketani S., Forouhar F., Liu H., Hong S.J., Lin F.Y., Nair M.S., Zask A., Huang Y.X., Xing L., Stockwell B.R., Chavez A., Ho D.D. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat. Commun. 2021;12:2016. doi: 10.1038/s41467-021-22362-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chien M.C., Anderson T.K., Jockusch S., Tao C.J., Li X.X., Kumar S., Russo J.J., Kirchdoerfer R.N., Ju J.Y. Nucleotide analogues as inhibitors of SARS-CoV-2 polymerase, a key drug target for COVID-19. J. Proteome Res. 2020;19:4690–4697. doi: 10.1021/acs.jproteome.0c00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin W.C., Mao C.Y., Luan X.D., Shen D.D., Shen Q.Y., Su H.X., Wang X.X., Zhou F.L., Zhao W.F., Gao M.Q., Chang S.H., Xie Y.C., Tian G.H., Jiang H.W., Tao S.C., Shen J.S., Jiang Y., Jiang H.L., Xu Y.C., Zhang S.Y., Zhang Y., Xu H.E. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020;368:1499–1504. doi: 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiao J.X., Li Y.S., Zeng R., Liu F.L., Luo R.H., Huang C., Wang Y.F., Zhang J., Quan B.X., Shen C.J., Mao X., Liu X.L., Sun W.N., Yang W., Ni X.C., Wang K., Xu L., Duan Z.L., Zou Q.C., Zhang H.L., Qu Wa, Long Y.H.P., Li M.H., Yang R.C., Liu X.L., You J., Zhou Y.L., Yao R., Li W.P., Liu J.M., Chen P., Liu Y., Lin G.F., Yang X., Zou J., Li L.L., Hu Y.G., Lu G.W., Li W.M., Wei Y.Q., Zheng Y.T., Lei J., Yang S.Y. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin Z.M., Du X.Y., Xu Y.C., Deng Y.Q., Liu M.Q., Zhao Y., Zhang B., Li X.F., Zhang L.K., Peng C., Duan Y.K., Yu J., Wang L., Yang K.L., Liu F.J., Jiang R.D., Yang X.L., You T., Liu X.C., Yang X.N., Bai F., Liu H., Liu X., Guddat L.W., Xu W.Q., Xiao G.F., Qin C.F., Shi Z.L., Jiang H.L., Rao Z.H., Yang H.T. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 20.Dai W.H., Zhang B., Jiang X.M., Su H.X., Li J., Zhao Y., Xie X., Jin Z.M., Peng J.J., Liu F.J., Li C.P., Li Y., Bai F., Wang H.F., Cheng X., Cen X.B., Hu S.L., Yang X.N., Wang J., Liu X., Xiao G.F., Jiang H.L., Rao Z.H., Zhang L.K., Xu Y.C., Yang H.T., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valiente P.A., Wen H., Nim S., Lee J.A., Kim H.J., Kim J., Perez-Riba A., Paudel Y.P., Hwang I., Kim K.D., Kim S., Kim P.M. Computational design of potent D-peptide inhibitors of SARS-CoV-2. J. Med. Chem. 2021;64:14955–14967. doi: 10.1021/acs.jmedchem.1c00655. [DOI] [PubMed] [Google Scholar]
- 22.Hoffmann M., Mösbauer K., Hofmann-Winkler H., Kaul A., Kleine-Weber H., Krüger N., Gassen N.C., Müller M.A., Drosten C., Pöhlmann S. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature. 2020;585:588–590. doi: 10.1038/s41586-020-2575-3. [DOI] [PubMed] [Google Scholar]
- 23.Ashhurst A.S., Tang A.H., Fajtová P., Yoon M.C., Aggarwal A., Bedding M.J., Stoye A., Beretta L., Pwee D., Drelich A., Skinner D., Li L.F., Meek T.D., McKerrow J.H., Hook V., Tseng C.T., Larance M., Turville S., Gerwick W.H., O'Donoghue A.J., Payne R.J. Potent anti-SARS-CoV-2 activity by the natural product gallinamide A and analogues via inhibition of cathepsin L. J. Med. Chem. 2022;65:2956–2970. doi: 10.1021/acs.jmedchem.1c01494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sa-Ngiamsuntorn K., Suksatu A., Pewkliang Y., Thongsri P., Kanjanasirirat P., Manopwisedjaroen S., Charoensutthivarakul S., Wongtrakoongate P., Pitiporn S., Chaopreecha J., Kongsomros S., Jearawuttanakul K., Wannalo W., Khemawoot P., Chutipongtanate S., Borwornpinyo S., Thitithanyanont A., Hongeng S. Anti-SARS-CoV-2 activity of andrographis paniculata extract and its major component andrographolide in human lung epithelial cells and cytotoxicity evaluation in major organ cell representatives. J. Nat. Prod. 2021;84:1261–1270. doi: 10.1021/acs.jnatprod.0c01324. [DOI] [PubMed] [Google Scholar]
- 25.Jayk Bernal A., Gomes da Silva M.M., Musungaie D.B., Kovalchuk E., Gonzalez A., Delos Reyes V., Martín-Quirós A., Caraco Y., Williams-Diaz A., Brown M.L., Du J.J., Pedley A., Assaid C., Strizki J., Grobler J.A., Shamsuddin H.H., Tipping R., Wan H., Paschke A., Butterton J.R., Johnson M.G., De Anda C., MOVe-OUT Study Group Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N. Engl. J. Med. 2022;386:509–520. doi: 10.1056/NEJMoa2116044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drożdżal S., Rosik J., Lechowicz K., Machaj F., Szostak B., Przybyciński J., Lorzadeh S., Kotfis K., Ghavami S., Łos M.J. An update on drugs with therapeutic potential for SARS-CoV-2 (COVID-19) treatment. Drug Resist. Update. 2021;59 doi: 10.1016/j.drup.2021.100794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia S., Liu M.Q., Wang C., Xu W., Lan Q.S., Feng S.L., Qi F.F., Bao L.L., Du L.Y., Liu S.W., Qin C., Sun F., Shi Z.L., Zhu Y., Jiang S.B., Lu L. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shang J., Wan Y.S., Luo C.M., Ye G., Geng Q.B., Auerbach A., Li F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2020;117:11727–11734. doi: 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xia S., Lan Q.S., Zhu Y., Wang C., Xu W., Li Y.T., Wang L.J., Jiao F., Zhou J., Hua C., Wang Q., Cai X., Wu Y., Gao J., Liu H., Sun G., Münch J., Kirchhoff F., Yuan Z.H., Xie YoH., Sun F., Jiang S.B., Lu L. Structural and functional basis for pan-CoV fusion inhibitors against SARS-CoV-2 and its variants with preclinical evaluation. Signal Transduct. Targeted Ther. 2021;6:288. doi: 10.1038/s41392-021-00712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu S., Zheng X., Zhou B., Li J., Chen M., Deng R., Wong G., Lavillette D., Meng G. SARS-CoV-2 spike engagement of ACE2 primes S2' site cleavage and fusion initiation. Proc. Natl. Acad. Sci. U. S. A. 2022;119 doi: 10.1073/pnas.2111199119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xia S., Liu M.Q., Wang C., Xu W., Lan Q.S., Feng S.L., Qi F.F., Bao L.L., Du L.Y., Liu S.W., Sun C.Qin F., Shi Z.L., Zhu Y., Jiang S.B., Lu L. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu X., Chen C.Z., Xu M., Hu Z.Y., Guo H., Itkin Z., Shinn P., Ivin P., Leek M., Liang T.J., Shen M., Zheng W., Hall M.D. Discovery of small molecule entry inhibitors targeting the fusion peptide of SARS-CoV-2 spike protein. ACS Med. Chem. Lett. 2021;12:1267–1274. doi: 10.1021/acsmedchemlett.1c00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin W.R., Cheng F.X. Repurposing of FDA-approved toremifene to treat COVID-19 by blocking the spike glycoprotein and NSP14 of SARS-CoV-2. J. Proteome Res. 2020;19:4670–4677. doi: 10.1021/acs.jproteome.0c00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He C.L., Huang L.Y., Wang K., Gu C.J., Hu J., Zhang G.J., Xu W., Xie Y.H., Tang N., Huang A.L. Identification of bis-benzylisoquinoline alkaloids as SARSCoV-2 entry inhibitors from a library of natural products. Transduct. Target. Ther. 2021;1:131. doi: 10.1038/s41392-021-00531-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang C., Pan X.Y., Xu X.F., Cheng C., Huang Y., Li L., Jiang S.B., Xu W., Xiao G.F., Liu S.W. Salvianolic acid C potently inhibits SARS-CoV-2 infection by blocking the formation of six-helix bundle core of spike protein. Signal Transduct. Targeted Ther. 2020;5:220. doi: 10.1038/s41392-020-00325-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevaert A., Krasniqi B., Loy B.V., Nguyen T., Thomas J., Vandeput J., Jochmans D., Thiel V., Dijkman R., Dehaen W., Voet A., Naesens L. Betulonic acid derivatives interfering with human coronavirus 229E replication via the nsp15 endoribonuclease. J. Med. Chem. 2021;64:632–5644. doi: 10.1021/acs.jmedchem.0c02124. [DOI] [PubMed] [Google Scholar]
- 37.Yu S.P., Zhu Y.Y., Xu J.R., Yao G.T., Zhang P., Wang M.G., Zhao Y.F., Lin G.Q., Chen H.Z., Chen L.L., Zhang J.G. Glycyrrhizic acid exerts inhibitory activity against the spike protein of SARS-CoV-2. Phytomedicine. 2021;85 doi: 10.1016/j.phymed.2020.153364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Q., Ye F., Liang H., Liu H.B., Li C.M., Lu R.J., Huang B.Y., Zhao L., Tan W.J., Lai L.H. Bardoxolone and bardoxolone methyl, two Nrf2 activators in clinical trials, inhibit SARS-CoV-2 replication and its 3C-like protease. Signal Transduct. Targeted Ther. 2021;6:212. doi: 10.1038/s41392-021-00628-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song G.P., Shen X.T., Li S.M., Li Y.B., Liu Y.P., Zheng Y.S., Lin R.H., Fan J.H., Ye H.M., Liu S.W. Structure-activity relationships of 3-O-β-chacotriosyl ursolic acid derivatives as novel H5N1 entry inhibitors. Eur. J. Med. Chem. 2015;93:431–442. doi: 10.1016/j.ejmech.2015.02.029. [DOI] [PubMed] [Google Scholar]
- 40.Xiao S.L., Tian Z.Y., Wang Y.F., Si L.L., Zhang L.H., Zhou D.M. Recent progress in the antiviral activity and mechanism study of pentacyclic triterpenoids and their derivatives. Med. Res. Rev. 2018;38:951–976. doi: 10.1002/med.21484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y.T., Yang C., Wu Y., Lv J.J., Feng X., Tian X.F., Zhou Z.Z., Pan X.Y., Liu S.W., Tian L.W. Axial chiral binaphthoquinone and perylenequinones from the stromata of hypocrella bambusae are SARS-CoV-2 entry inhibitors. J. Nat. Prod. 2021;84:436–443. doi: 10.1021/acs.jnatprod.0c01136. [DOI] [PubMed] [Google Scholar]
- 42.Li H., Cheng C., Li S.M., Wu Y., Liu Z.H., Liu M.J., Chen J.X., Zhong Q.Y., Zhang X.S., Liu S.W., Song G.P. Discovery and structural optimization of 3-O-β-chacotriosyl oleananetype triterpenoids as potent entry inhibitors of SARS-CoV-2 virus infections. Eur. J. Med. Chem. 2021;215 doi: 10.1016/j.ejmech.2021.113242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Y.X. Liao, Y.L. Ye, S.M. Li, Y.L. Zhuang, L.Y. Chen, J.X. Chen, Z.N. Cui, L.J. Huo, S.W. Liu, G.P. Song, Synthesis and SARs of dopamine derivatives as potential inhibitors of influenza virus PAN endonuclease, Eur. J. Med. Chem. 189 (202) 112048. [DOI] [PubMed]
- 44.Song G.P., Shen X.T., Li S.M., Li Y.B., Si H.Z., Fan J.H., Li J.H., Gao E.Q., Liu S.W. Structure-activity relationships of 3-O-β-chacotriosyl oleanane-type triterpenoids as potential H5N1 entry inhibitors. Eur. J. Med. Chem. 2016;119:109–121. doi: 10.1016/j.ejmech.2016.04.061. [DOI] [PubMed] [Google Scholar]
- 45.Liu Z.H., Gu S.Y., Zhu X., Liu M.J., Cao Z.Q., Qiu P.S., Li S.M., Liu S.W., Song G.P. Discovery and optimization of new 6, 7-dihydroxy-1, 2, 3, 4-tetrahydroisoquinoline derivatives as potent influenza virus PAN inhibitors. Eur. J. Med. Chem. 2022;227 doi: 10.1016/j.ejmech.2021.113929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.