

Abstract
Objective:
Cherubism is a genetic disorder characterised by bilateral jawbone deformation. The associated jawbone lesions regress after puberty, whereas severe cases require surgical treatment. Although several drugs have been tested, fundamental treatment strategies for cherubism have not been established. The effectiveness of imatinib has recently been reported; however, its pharmaceutical mechanism remains unclear. In this study, we tested the effects of imatinib using a cherubism mouse model.
Methods:
We used Sh3bp2 P416R cherubism mutant mice, which exhibit systemic organ inflammation and osteopenia. The effects of imatinib were determined using primary bone marrow-derived macrophages. Imatinib was administered intraperitoneally to the mice, and serum tumour necrosis factor-α (TNFα), organ inflammation, and bone properties were examined.
Results:
The cherubism mutant macrophages produced higher levels of TNFα in response to lipopolysaccharide compared to wild-type macrophages, and imatinib did not significantly suppress TNFα production. Although imatinib suppressed osteoclast formation in vitro, administering it in vivo did not suppress organ inflammation and osteopenia.
Conclusion:
The in vivo administration of imatinib had a minimal therapeutic impact in cherubism mutant mice. To establish better pharmaceutical interventions, it is necessary to integrate new findings from murine models with clinical data from patients with a definitive diagnosis of cherubism.
Keywords: cherubism, imatinib, inflammation, osteopenia, murine model, SH3BP2
1. Introduction
Cherubism (OMIM#118400) is a genetic disorder characterised by bilateral multilocular cystic bone destruction in the mandibula and maxilla (Reichenberger, Levine, Olsen, Papadaki, & Lietman, 2012). The cystic bone lesions are filled with proliferative fibro-osseous tissue containing a large number of tartrate-resistant acid phosphatase (TRAP)-positive multinucleated giant cells, which indicates the presence of osteoclasts (Reichenberger et al., 2012). Cherubism-associated jaw lesions develop approximately between 2–5 years of age and progress with age, resulting in swelling and disfiguration in the lower portion of the face (Chrcanovic, Guimarães, Gomes, & Gomez, 2021; Kueper, Tsimbal, Olsen, Kaban, & Liao, 2021; Papadaki et al., 2012). In severe cases, the lesions expand into the orbital walls or upper airway, resulting in upward gazing, optic neuropathy, or respiratory obstruction (Battaglia, Merati, & Magit, 2000; Chrcanovic et al., 2021; Kueper et al., 2021). In most cases, these lesions spontaneously regress after puberty (Papadaki et al., 2012). Dislocation and eruption failure of teeth are irreversible, whereas eroded jawbone deformities can be restored spontaneously (Gupta et al., 2019; Papadaki et al., 2012). Since spontaneous regression is expected, “wait and see” is a basic treatment strategy for mild cherubism (Papadaki et al., 2012). However, therapeutic interventions such as surgical curettage and pharmacological treatment are required for aggressive cases. Although several drugs have been administered to patients with cherubism, fundamental treatment strategies have not yet been established (Chrcanovic et al., 2021; Kueper et al., 2021).
Cherubism is caused by mutations in the SH3BP2 gene, which encodes an adaptor protein termed as SH3 domain-binding protein 2 (SH3BP2) (Ueki et al., 2001). SH3BP2 is primarily expressed in immune cells, such as macrophages (Ueki et al., 2007), dendritic cells (Kawahara et al., 2021), B cells (de la Fuente, Kumar, Lu, & Geha, 2006), and T cells (Dimitriou et al., 2018) and regulates immune cell functions by interacting with various intracellular signalling proteins, including SYK (Hatani & Sada, 2008), PLCγ (Mukai, Ishida, et al., 2014), VAV (Foucault et al., 2005), and SRC (Levaot, Simoncic, et al., 2011). The pathogenesis of cherubism has been revealed by intensive analyses of a murine cherubism model with an Sh3bp2 P416R mutation (Ueki et al., 2007). The studies have revealed that heterozygous mutations in the RSPPDG hexapeptide sequence in exon 9 of SH3BP2 are responsible for cherubism (Levaot, Voytyuk, et al., 2011). The mutated sequences of SH3BP2 protein disrupt the binding of SH3BP2 with tankyrase, a poly(ADP-ribose) polymerase, resulting in the avoidance of tankyrase-mediated proteasomal degradation of SH3BP2 (Levaot, Voytyuk, et al., 2011). Consequently, aberrant accumulation of mutant SH3BP2 protein promotes macrophage and osteoclast activation in a gain-of-function manner via augmented SH3BP2-mediated intracellular signalling pathways (Mukai, Gallant, et al., 2014; Mukai, Ishida, et al., 2014; Ueki et al., 2007; Yoshitaka, Mukai, et al., 2014). The homozygous P416R cherubism mutation knock-in (KI) mouse model spontaneously develops macrophage-mediated systemic inflammation starting at approximately one week of age in a tumour necrosis factor-α (TNFα)-dependent manner (Yoshitaka, Ishida, et al., 2014), as well as severe osteopenia of systemic bones, including the spine, extremities, and jawbones (Ueki et al., 2007). Due to the nature of the inflammatory and osteopenic phenotypes developed in Sh3bp2KI/KI mice in the absence of T and B cells (Ueki et al., 2007), cherubism is considered to be an autoinflammatory bone disease, which comprises a new branch of autoinflammatory diseases (Bader-Meunier, Van Nieuwenhove, Breton, & Wouters, 2018; Morbach, Hedrich, Beer, & Girschick, 2013; Stern & Ferguson, 2013). In humans with cherubism, systemic inflammation is not observed, but local TNFα expression has been confirmed in jaw lesions (Mukai, Ishida, et al., 2014).
We explored potential drug candidates using a murine cherubism model. We have previously shown the effectiveness of tacrolimus, a calcineurin inhibitor (Mukai, Ishida, et al., 2014); etanercept, a TNF inhibitor (Yoshitaka, Ishida, et al., 2014); and SYK inhibitors (Yoshimoto et al., 2018) in vivo and in vitro using the murine cherubism model. For instance, we have shown that tacrolimus suppresses increased osteoclast formation by decreasing NFATc1 activation (Mukai, Ishida, et al., 2014), which was supported by clinical findings that the administration of tacrolimus ameliorated growing jaw lesions in a patient with aggressive cherubism (Kadlub et al., 2015). We have also shown that etanercept is effective when administered before the onset of cherubism lesions but not after the cherubism mutant mice develop a full-blown inflammation (Yoshitaka, Ishida, et al., 2014). In agreement with these findings, the clinical application of adalimumab, another TNF inhibitor, exhibited minimal therapeutic effects in two cases of active cherubism (Hero et al., 2013). These examples illustrate the importance of integrating findings from basic research and clinical trials to confirm the efficacy of drug candidates for cherubism.
Imatinib is a tyrosine kinase inhibitor that is used to target BCR-Abl tyrosine kinase in paediatric patients with leukaemia, especially Philadelphia chromosome-positive chronic myelogenous leukaemia or acute lymphoblastic lymphoma (Druker et al., 2006; Kantarjian et al., 2002). Imatinib has been recently reported to be effective against cherubism in two case reports (Eiden, Lausch, & Meckel, 2017; Ricalde, Ahson, & Schaefer, 2019); the authors reported that the administration of imatinib prevented the progression of cherubism-related symptoms and improved facial morphology. Although imatinib has been suggested to be clinically effective, its mechanisms have not been analysed at the cellular and molecular levels. Therefore, we investigated the effects of imatinib using a murine cherubism model.
2. Materials and Methods
2.1. Mice
Sh3bp2 P416R mutation KI mice were generated by introducing a Pro-to-Arg mutation into exon 9 of the murine Sh3bp2 gene (Ueki et al., 2007). The Sh3bp2 P416R mutation in mice is equivalent to a common SH3BP2 P418R mutation in human patients with cherubism (Ueki et al., 2001). All wild-type (Sh3bp2+/+), heterozygous (Sh3bp2KI/+), and homozygous (Sh3bp2KI/KI) mice on a C57BL/6J background were maintained in the animal facility of Kawasaki Medical School (Kurashiki, Okayama, Japan). All mice were housed in groups (2–5 mice/cage) and maintained at 22 °C under a 12 h/12 h light/dark cycle with free access to water and standard laboratory food (MF diet, Oriental Yeast Co., Tokyo, Japan). All animal experiments were approved by the Safety Committee for Recombinant DNA Experiments (No. 18–23, 1st Jan. 2019) and the Institutional Animal Care and Use Committee of Kawasaki Medical School (No. 20–072, 1st Jun. 2020). All experimental procedures were conducted in accordance with the institutional and National Institutes of Health guidelines for humane animal use.
2.2. Reagent
Imatinib was obtained from ChemScene LLC (Monmouth Junction, NJ, USA). Recombinant murine macrophage colony-stimulating factor (M-CSF) and receptor activator nuclear factor-κB ligand (RANKL) proteins were purchased from Peprotech (Rocky Hill, NJ, USA). Lipopolysaccharide (LPS) and FK506 were purchased from Sigma-Aldrich (St. Louis, MO, USA), and mouse anti-SH3BP2 antibody (H00006452-M01) was obtained from Abnova (Taipei City, Taiwan). Mouse anti-NFATc1 antibody (sc-7294) was obtained from Santa Cruz Biotechnology (Dallas, TX, USA), rabbit anti-actin antibody (A2066) was obtained from Sigma-Aldrich, and horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G (IgG) (#7074) and HRP-conjugated anti-mouse IgG (#7076) antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA).
2.3. Bone marrow-derived macrophage culture
Bone marrow cell isolation and culture were performed as described previously (Nagasu et al., 2019). Briefly, bone marrow cells were isolated from the long bones of 10-week-old female mice and cultured in Petri dishes for 2 h at 37 °C under 5% CO2. To minimise the contamination of stromal cells, only non-adherent bone marrow cells were collected. The non-adherent bone marrow cells were re-seeded on culture plates at a density of 5.0 × 105 cells/mL and incubated for 2 days in α-minimum essential medium containing 10% heat-inactivated foetal bovine serum and M-CSF (25 ng/mL). After the 2-day preculture, bone marrow-derived macrophages (BMMs) were stimulated with LPS for 24 h in the presence or absence of imatinib. Culture supernatants were collected and subjected to an enzyme-linked immunosorbent assay (ELISA) for TNFα and interleukin (IL)-6. For the culture supernatant collection for IL-1β measurement, BMMs were stimulated with LPS for 22 h and subsequently treated with ATP (5 mM) for 2 h to induce IL-1β secretion via inflammasome activation.
2.4. Osteoclast culture
Non-adherent bone marrow cells were seeded on 48-well plates at a density of 1.0 × 105 cells/mL and incubated for 2 days in α-minimum essential medium/10% foetal bovine serum containing M-CSF (25 ng/mL). After the 2-day preculture, BMMs were stimulated with RANKL at the indicated concentrations for 3 days. The formation of TRAP-positive multinucleated cells (TRAP+ MNCs) was visualised by TRAP staining (Sigma-Aldrich). TRAP+ MNCs with three or more nuclei were counted as osteoclasts. Biochemical assays for TRAP activity in the culture supernatant were performed as described previously (Mukai, Ishida, et al., 2014).
2.5. ELISA for TNFα, IL-6, and IL-1β
TNFα, IL-6, and IL-1β concentrations in culture supernatants and TNFα concentrations in serum samples were measured by sandwich ELISA kits (R&D Systems, Minneapolis, MN, USA) (Mukai et al., 2015). Each well of a 96-well plate was coated with goat anti-mouse cytokine-specific antibody as a capture reagent and incubated at 25 °C overnight, after which the wells were washed and blocked with 1% bovine serum albumin (BSA) in phosphate‐buffered saline (PBS) for 1 h at 25 °C. Samples and standards were added to the wells and incubated for 2 h at 25 °C. After washing, the wells were incubated with a biotinylated goat anti-mouse cytokine-specific antibody for 2 h at 25 °C, and then incubated with streptavidin-HRP substrate solution. The optical density of each well was measured at 450 nm using a microplate reader (Varioskan Flash, Thermo Fisher Scientific, Waltham, MA, USA), and the TNFα, IL-6, and IL-1β concentrations in each sample were calculated based on standard curves.
2.6. Immunofluorescent staining
Immunofluorescence staining was performed as described previously (Fujita et al., 2018). Non-adherent bone marrow cells were plated at a density of 1.0 × 105/mL onto 8-well chamber slides (BD Falcon, Franklin Lakes, NJ, USA) and stimulated with RANKL (50 ng/mL) for 3 days in the presence or absence of imatinib. The cells were fixed in 2% paraformaldehyde (PFA)/PBS, permeabilised using 0.2% Triton X-100, blocked in 2% normal goat serum/2.5% BSA/PBS, and incubated with an anti-NFATc1 antibody at 4 °C overnight. NFATc1 was detected by Alexa Fluor-555-conjugated goat anti-mouse IgG antibody (Thermo Fisher Scientific), and actin and nuclei were co-stained with Alexa Fluor-488-conjugated phalloidin (Thermo Fisher Scientific) and 4’, 6-diamidino-2-phenylindole (Santa Cruz Biotechnology), respectively. Fluorescent images of the cells were acquired using a BZX-700 microscope (Keyence, Osaka, Japan).
2.7. Western blotting
For immunoblot analysis, BMMs were washed with ice-cold PBS and lysed with RIPA lysis buffer (Sigma-Aldrich) containing protease and phosphatase inhibitor cocktails (Sigma-Aldrich) (Akagi et al., 2020). Protein concentrations were determined using a BCA Protein Assay Kit (Thermo Fisher Scientific). Protein samples were resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. After blocking with 5% skim milk in Tris-buffered saline with Tween 20, the membranes were incubated with primary antibodies, followed by incubation with appropriate HRP-conjugated species-specific secondary antibodies. Bands were detected using SuperSignal West chemiluminescent substrate (Thermo Fisher Scientific) and visualised using ImageQuant LAS-4000 (GE Healthcare, Little Chalfont, UK). Actin was used as a loading control to normalise the amount of protein.
2.8. In vivo imatinib administration
Ten-week-old Sh3bp2+/+ and Sh3bp2KI/KI female mice were injected intraperitoneally with 100 μg/g of imatinib five times a week for 4 weeks (n = 4–5/group). The dose of imatinib was the same or higher than that used in murine cancer models with significant efficacies (Hwang et al., 2003; Kurebayashi et al., 2006). After the 4-week treatment, the liver, lungs, hind limbs, and serum samples were obtained from the mice following euthanasia under sevoflurane-induced anaesthesia. We used these samples for histology, micro-computed tomography (micro-CT), and ELISA.
2.9. Histological analysis
The liver and lungs were fixed in 4% PFA/PBS for 2 days and then embedded in paraffin. Tissue sections (6 μm) were stained with haematoxylin and eosin (Kawahara et al., 2021). To evaluate the severities of inflammation in liver and lung tissues, the proportions of inflammatory infiltrates were calculated as described previously (Yoshitaka, Mukai, et al., 2014). Tissue images were taken with a 10× objective lens. Areas of inflammatory infiltrates were measured in pixels using ImageJ software (NIH, Bethesda, MD, USA) and divided by the total tissue areas. Three images were taken for each mouse, and the values were averaged.
2.10. Micro-CT analyses
Bone samples were fixed in 4% PFA/PBS for 2 days, and PFA-fixed hind limbs were immersed in 70% ethanol until micro-CT analyses. Three-dimensional cortical and trabecular microarchitectures of the right femurs were evaluated using a micro-CT system (SkyScan 1174, Aartselaar, Belgium) with a 1.0 mm-thick aluminium filter as described previously (Tosa et al., 2019). The voxel resolution of the femoral cortical and trabecular bone was 16.7 μm and 6.5 μm, respectively. The cortical and trabecular bone properties were analysed using SkyScan software (NRecon, CTAn, CTvol, and CTvox). The region of the cortical bone analysed in the femurs comprised 1 mm of the femoral midshaft and that of the trabecular bone comprised 1 mm of spongiosa (starting 0.8 mm from the growth plate peak). All micro-CT parameters were described according to the international guideline (Bouxsein et al., 2010).
2.11. Statistical analysis
All values are presented as mean ± standard deviation. Statistical analysis was performed by a one-way analysis of variance test (Tukey post-hoc test) to compare three or more groups using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). Statistical significance was set at p < 0.05.
3. Results
3.1. Imatinib does not significantly suppress inflammatory cytokines production in LPS-stimulated bone marrow-derived macrophages
In this study, we used Sh3bp2 P416R cherubism mutant mice as a murine model of cherubism. The Sh3bp2 P416R mutation contributes to gain-of-function by increasing SH3BP2 protein stability (Levaot, Voytyuk, et al., 2011; Ueki et al., 2007). In the Sh3bp2 mutant mice, SH3BP2 protein accumulates in the cytoplasm of their cells. As shown in Figure 1A, SH3BP2 protein was highly expressed in Sh3bp2KI/+ and Sh3bp2KI/KI mutant cells in an allele-dose-dependent manner.
Figure 1. Imatinib did not significantly suppress inflammatory cytokines production in LPS-stimulated bone marrow-derived macrophages (BMMs).
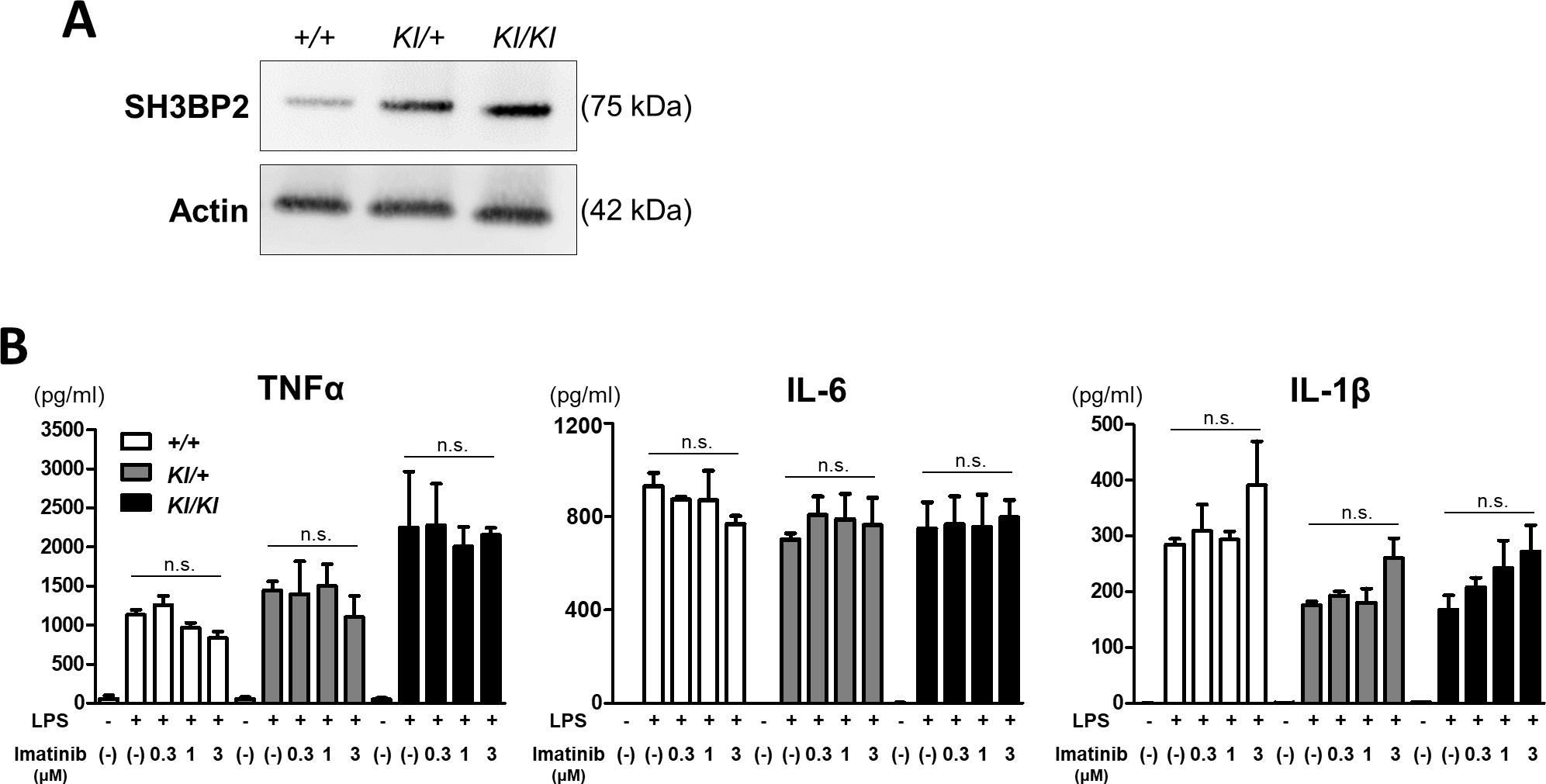
Bone marrow cells were isolated from Sh3bp2+/+, Sh3bp2KI/+, and Sh3bp2KI/KI mice. Non-adherent bone marrow cells were seeded at a density of 5.0 × 104/mL. After a 2-day preculture with M-CSF (25 ng/mL), BMMs were used for the following experiments. (A) Immunoblot analysis for SH3BP2. Protein samples were collected from the BMMs of Sh3bp2+/+, Sh3bp2KI/+, and Sh3bp2KI/KI mice. SH3BP2 was detected by western blotting. Actin was used as a loading control. (B) Inflammatory cytokines concentrations in the culture supernatant. BMMs were stimulated with LPS (100 ng/mL) in the presence or absence of imatinib for 24 h. TNFα and IL-6 concentrations in the culture medium were determined by ELISA. For IL-1β measurement, BMMs were stimulated with LPS (100 ng/mL) for 22 h, followed by treatment with ATP (5 mM) for 2 h. Values are presented as the mean ± SD. Note: n.s. = not significant. SH3BP2, SH3 domain-binding protein 2; +/+, wild-type; KI, knock-in; BMMs, bone marrow-derived macrophages; TNFα, tumour necrosis factor-α; M-CSF, macrophage colony-stimulating factor; LPS, lipopolysaccharide; IL, interleukin; ATP, adenosine triphosphate; ELISA, enzyme-linked immunosorbent assay.
Since the Sh3bp2 cherubism mutation enhances TNFα production in BMMs in response to Toll-like receptor ligands (Yoshitaka, Mukai, et al., 2014), we analysed the effect of imatinib on TNFα production in response to LPS. We found that the Sh3bp2 mutation enhanced TNFα production from LPS-stimulated macrophages in accordance with the findings from our previous study, and that the administration of imatinib did not significantly suppress TNFα production in all genotypes (Figure 1B). We also examined the concentrations of IL-6 and IL-1β in the culture supernatant. The increased IL-6 and IL-1β levels in response to LPS were not suppressed by imatinib treatment in all genotypes (Figure 1B). These findings indicate that imatinib does not have suppressive effects on inflammatory cytokines production regardless of the Sh3bp2 mutation.
3.2. Imatinib suppresses RANKL-induced osteoclast formation
Increased osteoclastogenesis is one of the characteristic features of cherubism (Mukai, Ishida, et al., 2014; Ueki et al., 2007). We have previously reported that the Sh3bp2 cherubism mutation enhances osteoclast formation in response to RANKL via increased NFATc1 activity (Ueki et al., 2007). We performed an osteoclast formation assay to investigate whether imatinib suppressed osteoclast formation. Primary murine BMMs were treated with RANKL, and osteoclast formation was determined by TRAP staining. We found that the Sh3bp2 mutation significantly enhanced RANKL-induced osteoclast formation, represented by increased numbers of TRAP+ MNCs and elevated TRAP activity in the culture supernatant (Figure 2), corroborating previous findings (Mukai, Ishida, et al., 2014; Ueki et al., 2007). The administration of imatinib significantly suppressed osteoclast formation in a dose-dependent manner. The inhibitory effect of imatinib was observed in all genotypes.
Figure 2. Imatinib suppresses RANKL-induced osteoclast formation.
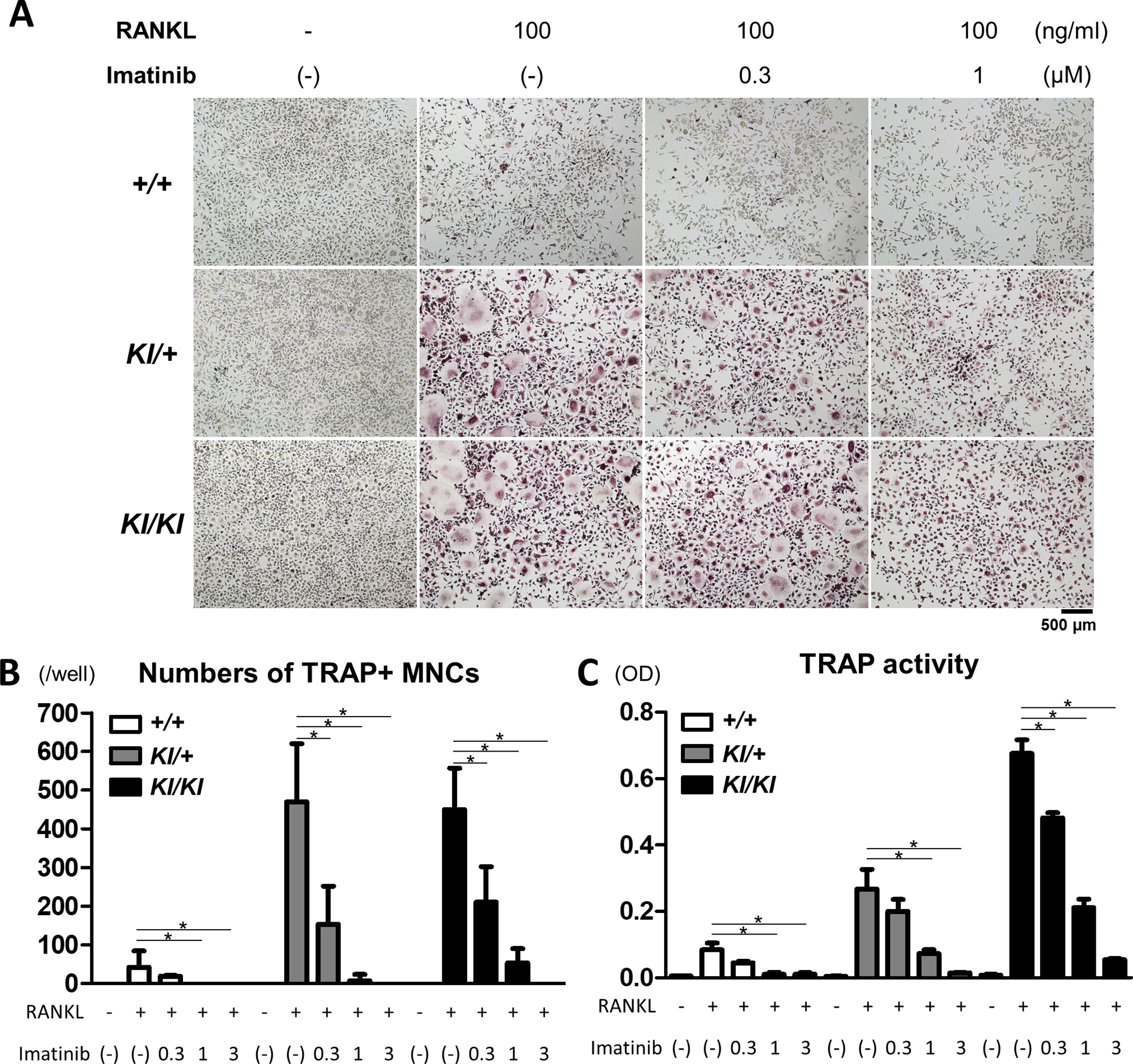
Bone marrow cells were isolated from Sh3bp2+/+, Sh3bp2KI/+, and Sh3bp2KI/KI mice. Non-adherent bone marrow cells were seeded at a density of 1.0 × 105/mL on 48-well plates. After a 2-day preculture with M-CSF (25 ng/mL), BMMs were stimulated with RANKL in the presence or absence of imatinib. (A) TRAP staining images of BMMs stimulated with RANKL in the presence of imatinib at indicated concentrations for 72 h. (B) Quantitation of TRAP-positive multinucleated cells (TRAP+ MNCs) per well. (C) TRAP activities in the culture supernatant. Values are presented as the mean ± SD. Note: * p < 0.05; n.s. = not significant. SH3BP2, SH3 domain-binding protein 2; +/+, wild-type; KI, knock-in; BMMs, bone marrow-derived macrophages; RANKL, receptor activator of nuclear factor-κB ligand; M-CSF, macrophage colony-stimulating factor; TRAP, Tartrate-resistant acid phosphatase; OD, optical density.
3.3. Imatinib decreases osteoclast formation via suppressed NFATc1 expression
Next, we examined osteoclast formation by fluorescent staining, in which osteoclasts were visualised as multinucleated giant cells with actin ring formation. We found that the administration of RANKL induced multinucleated osteoclast formation with actin rings and that osteoclast formation was significantly enhanced by the Sh3bp2 mutation (Figure 3A). The administration of imatinib suppressed RANKL-induced osteoclast formation in all the genotypes (Figure 3A).
Figure 3. Imatinib decreases osteoclast formation via suppressed NFATc1 expression.
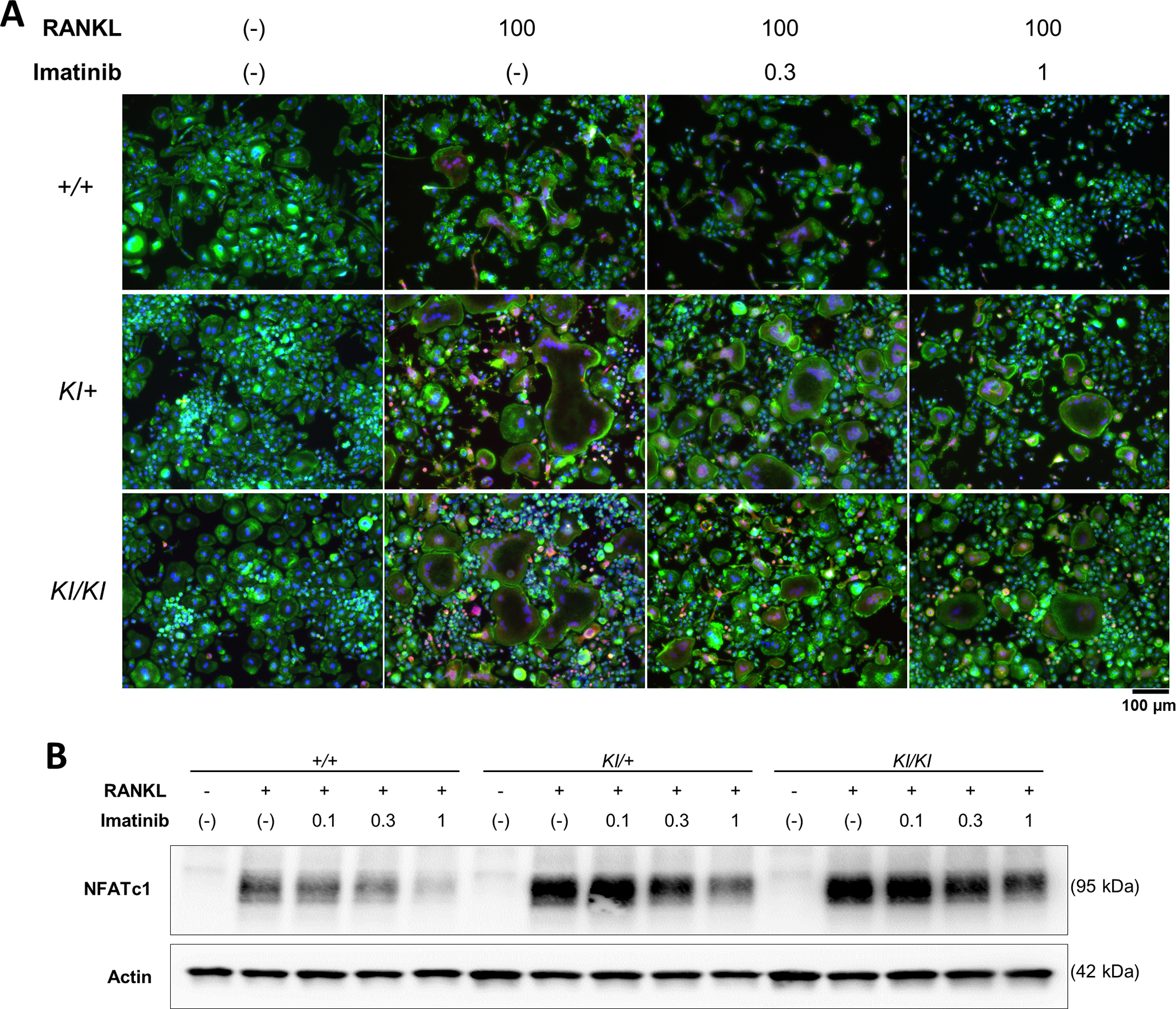
Bone marrow cells were isolated from Sh3bp2+/+, Sh3bp2KI/+, and Sh3bp2KI/KI mice. Non-adherent bone marrow cells were seeded at a density of 1.0 × 105/mL. After a 2-day preculture with M-CSF (25 ng/mL), BMMs were stimulated with RANKL (50 ng/mL) in the presence or absence of imatinib for 72 h. (A) Fluorescent staining of RANKL-stimulated BMMs. Cells were fixed with 2% PFA/PBS. Actin and nuclei were visualised with Alexa Fluor-488-conjugated phalloidin and 4’, 6-diamidino-2-phenylindole, respectively. NFATc1 was stained with an anti-NFATc1 antibody, followed by Alexa Fluor-555-conjugated anti-mouse antibody. Actin, nuclei, and NFATc1 were indicated in green, blue, and red, respectively. Original magnification, 20×. (B) Immunoblot analysis of NFATc1. Protein samples were collected at 48 h after the RANKL treatment. Actin was used as a loading control. SH3BP2, SH3 domain-binding protein 2; +/+, wild-type; KI, knock-in; BMMs, bone marrow-derived macrophages; RANKL, receptor activator of nuclear factor-κB ligand; PFA/PBS, paraformaldehyde/phosphate-buffered saline.
To examine the inhibitory mechanisms of imatinib on osteoclast formation, we examined NFATc1 expression, which is a master regulator of osteoclastogenesis (Takayanagi et al., 2002). We found that the administration of imatinib significantly suppressed NFATc1 expression in a dose-dependent manner (Figure 3B). The decreased expression and reduced nuclear localisation of NFATc1 were also observed after imatinib treatment in the immunofluorescent staining, as shown in Figure 3A. To confirm the importance of NFATc1 in osteoclastogenesis, we evaluated osteoclast formation in the presence of FK506, an NFATc1 inhibitor. We found that FK506 significantly suppressed osteoclast formation with diminished NFATc1 expression (Supplementary Figure 1), which is consistent with the results of our and other previous studies (Mukai, Ishida, et al., 2014; Takayanagi et al., 2002). These findings suggest that imatinib administration decreases osteoclast formation via reduced NFATc1 expression.
3.4. Imatinib does not ameliorate inflammatory phenotypes of the Sh3bp2KI/KI mice
Cherubism mutant homozygous mice exhibit severe osteopenia as well as systemic inflammation in organs such as the liver and lungs (Ueki et al., 2007). Inflammatory phenotypes were observed at 1 week of age at the earliest, and these became prominent at approximately 8 weeks of age, and progressed with age (Yoshitaka, Ishida, et al., 2014).
To investigate the therapeutic effect of imatinib on inflammation, we administered imatinib for 4 weeks to 10-week-old Sh3bp2KI/KI mice, at which time the mice already exhibited systemic organ inflammation. We found that treatment with imatinib did not rescue oedematous skin changes around the eyes, which was caused by skin inflammation (Figure 4A). Additionally, increased serum TNFα levels in Sh3bp2KI/KI mice were not lowered by imatinib treatment (Figure 4B). We also examined organ inflammation in the liver and lungs and found that the infiltration of inflammatory cells in Sh3bp2KI/KI mice was not rescued by imatinib (Figure 4C, 4D, 4E). These findings suggest that imatinib does not have a detectable anti-inflammatory potential in Sh3bp2KI/KI mice.
Figure 4. Imatinib does not ameliorate inflammatory phenotypes of the Sh3bp2KI/KI mice.
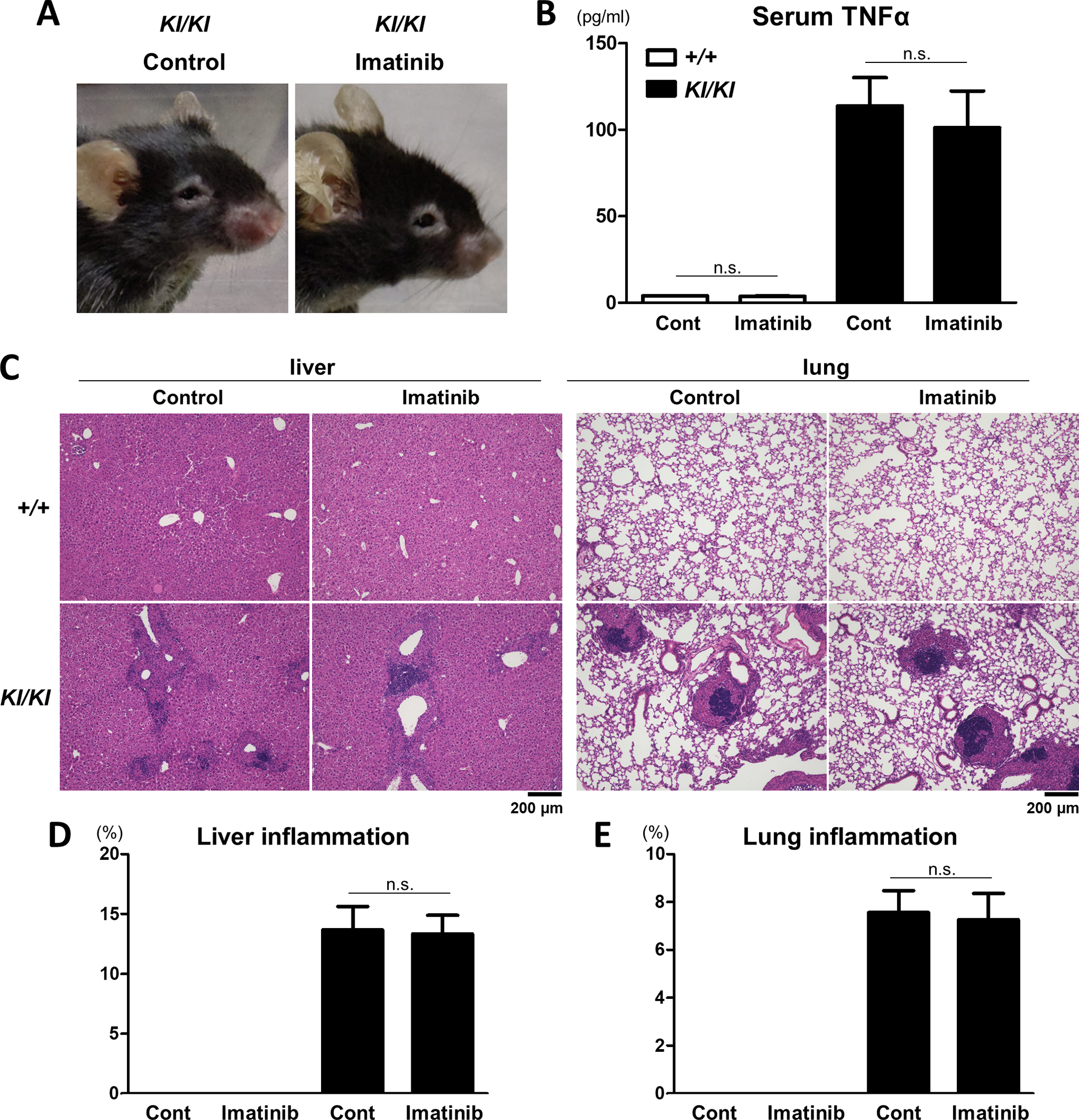
Imatinib was administered intraperitoneally to 10-week-old female Sh3bp2+/+ and Sh3bp2KI/KI mice daily 5 times per week for 4 weeks (n = 4–5/group). PBS was administered to the control mice. At the end of the experiments, serum, liver, and lung samples were collected. (A) Facial appearance of the Sh3bp2KI/KI mice at the age of 14 weeks. (B) Serum concentrations of TNFα. Serum levels of TNFα were determined by ELISA. (C) Representative images of haematoxylin and eosin-stained sections of the liver and lung. Original magnification, 10×. (D, E) Histomorphometric analyses of liver and lung inflammation. Proportions of inflammatory infiltrates per total tissue areas were quantified. SH3BP2, SH3 domain-binding protein 2; +/+, wild-type; KI, knock-in; ELISA, enzyme-linked immunosorbent assay; TNFα, tumour necrosis factor-α; PBS, phosphate-buffered saline.
3.5. Imatinib does not rescue the osteopenic phenotypes of the Sh3bp2KI/KI mice
Next, to examine the therapeutic effect of imatinib on osteopenia, we analysed the femoral bone properties using micro-CT. After a 4-week treatment with imatinib, the femurs of Sh3bp2+/+ and Sh3bp2KI/KI mice were collected, and the cortical and trabecular bones were analysed at the midshaft and distal regions of the femur, respectively. Micro-CT analyses showed that Sh3bp2KI/KI mice exhibited severe osteopenic phenotypes and that imatinib did not ameliorate bone loss (Figure 5). These findings indicate that imatinib does not have any protective effect in the osteopenic or inflammatory phenotypes of Sh3bp2KI/KI mice.
Figure 5. Imatinib does not rescue the osteopenic phenotypes of Sh3bp2KI/KI mice.
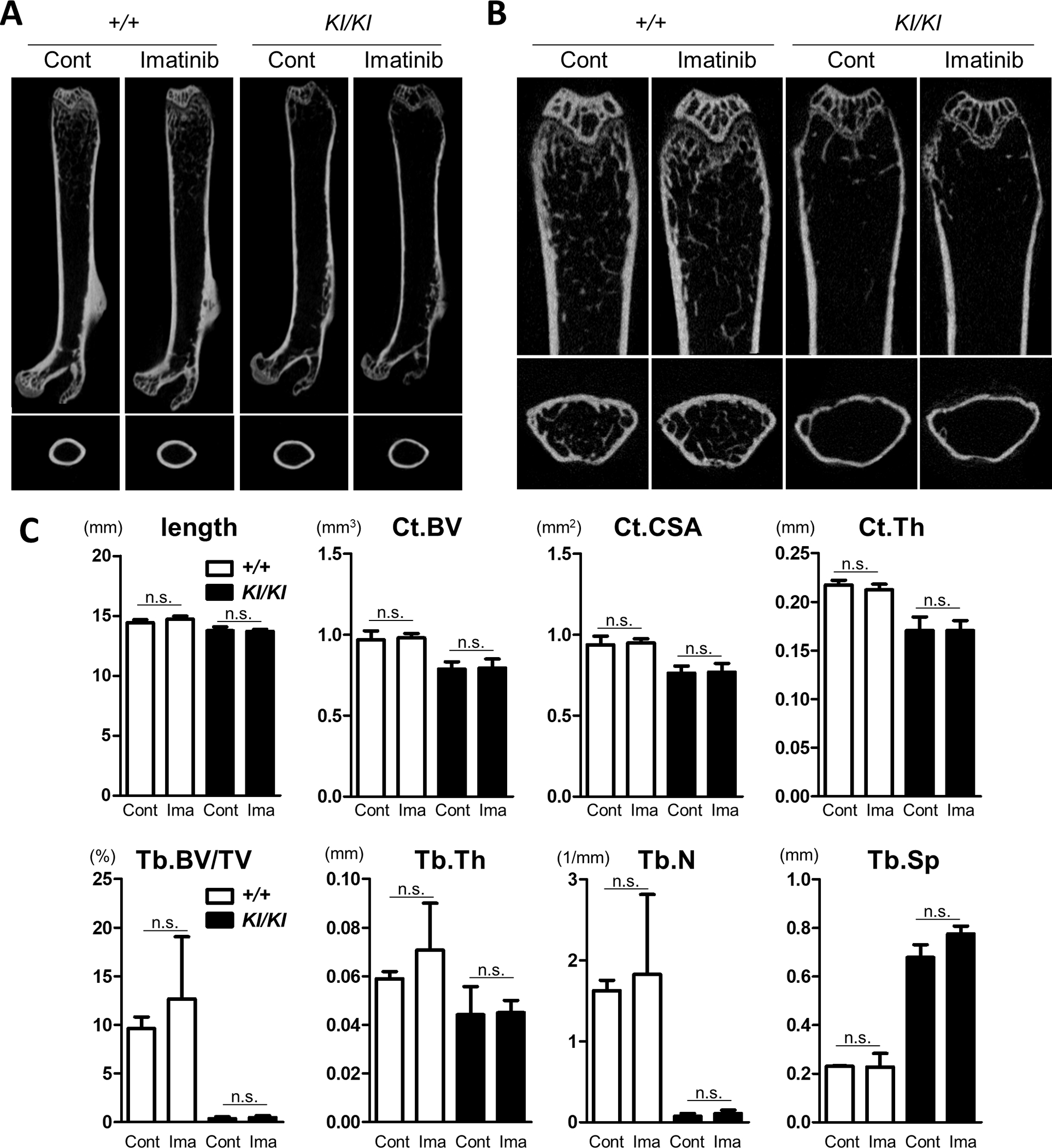
Imatinib was administered intraperitoneally to 10-week-old female Sh3bp2+/+ and Sh3bp2KI/KI mice daily 5 times per week for 4 weeks (n = 4–5/group). PBS was administered to the control mice. At the end of the experiments, the femurs were collected and subjected to micro-CT analysis. (A) Representative images of the whole femurs and cortical bones at the midshaft. (B) Representative images of the trabecular bone in the distal femurs. (C) Bone properties. Bone lengths were measured. Cortical bone volume (Ct.BV), cortical cross-sectional area (Ct.CSA), and cortical thickness (Ct.Th) of the midshaft of femurs were determined by micro-CT. Trabecular bone volume per total volume (Tb.BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular separation (Tb.Sp) were determined at the distal side of the femurs. Values are presented as the mean ± SD. Note: * p < 0.05; n.s. = not significant. SH3BP2, SH3 domain-binding protein 2; +/+, wild-type; KI, knock-in; Cont, control; Ima, Imatinib; PBS, phosphate-buffered saline; CT, computed tomography.
4. Discussion
The clinical effectiveness of imatinib has been reported in four patients with cherubism previously (Eiden et al., 2017; Ricalde et al., 2019). The current study was initially conducted to clarify the mechanisms by which imatinib exerts therapeutic effects on cherubism. In this study, we investigated the in vitro and in vivo effects of imatinib on inflammation and bone loss using Sh3bp2 cherubism mutant mice. We found that imatinib did not significantly suppress inflammatory phenotypes both in vitro and in vivo. Furthermore, we found that an in vivo administration of imatinib did not improve the osteopenic phenotypes of Sh3bp2KI/KI mice, although imatinib had a suppressive effect on the osteoclast formation in vitro. These findings indicate that the in vivo administration of imatinib has a minimal therapeutic impact on Sh3bp2 mutant cherubism mice. Possible reasons for the discrepancy between the therapeutic effects described in previous case reports and non-effectiveness in our murine study have been discussed in the following paragraphs.
The responsiveness to imatinib may differ between human patients and murine models, as there are several phenotypic differences between human patients and cherubism mice. In humans, bone lesions are restricted to the mandible and maxilla, while murine models develop generalised bone loss in the spine and extremities. Moreover, cherubism mice develop severe organ inflammation, while human patients do not develop any apparent systemic organ inflammation, except for an increased TNFα expression in the jaw lesions (Mukai, Ishida, et al., 2014). Furthermore, cherubism lesions in humans spontaneously regress after puberty, while murine inflammatory and osteopenic phenotypes progress with age (Papadaki et al., 2012; Ueki et al., 2007). Although the exact mechanisms underlying these differences have not been clarified, these phenotypic differences might have contributed to the difference in the therapeutic effect of imatinib in human patients and murine models.
Another possibility is that the spontaneous regression of cherubism lesions in humans might have incorrectly been assessed as a therapeutic effect of imatinib in previous case reports. Generally, most cherubism lesions regress after puberty (Papadaki et al., 2012). Since the timing of regression and disease severity can vary depending on the patient, judging whether a favourable clinical course is caused by the natural history of the disease or therapeutic effect of the drugs can be challenging. Furthermore, another concern is that patients in the previous case report might have been misdiagnosed with cherubism. Several other pathological entities mimic the clinical features of cherubism, including central giant cell granulomas, giant cell tumours of the jaws, fibrous dysplasia, and odontogenic myxoma (Reichenberger et al., 2012; Schreuder, van der Wal, de Lange, & van den Berg, 2021). Kleiber et al. reported the case of a patient with aggressive jaw swelling that mimicked cherubism (Kleiber, Skapek, Lingen, & Reid, 2014). The patient was finally diagnosed with myxoma, which was successfully treated with imatinib (Kleiber et al., 2014). This case illustrates that some entities that mimic cherubism can be treated with imatinib. In one of the two previous case reports showing the effectiveness of imatinib, one patient was confirmed to have a cherubism mutation in SH3BP2 (Eiden et al., 2017), while three patients in another study were diagnosed based on clinical features without genetic examinations (Ricalde et al., 2019). Although the authors of this study described that the histological findings of the patients were compatible with cherubism, actual histological images were not presented (Ricalde et al., 2019). Thus, due to the lack of sufficient clinical and genetic results, it is difficult to judge whether the patients who responded to imatinib had real cherubism or other conditions mimicking cherubism. This also illustrates the necessity of genetic testing to distinguish conditions similar to cherubism, which might respond differently to treatment. Notably, heterogeneity in the aetiologies of giant cell lesions of the jaw, which include cherubism, has been reported (Schreuder et al., 2021).
Differences in the route of administration of the drugs could also have caused a difference in the response to imatinib, as the drug was administered orally in humans and intraperitoneally in mice. However, it is less likely that intraperitoneal administration eliminated the efficacy of imatinib because this method has been widely applied to murine experiments and its effectiveness has been established (Hwang et al., 2003; Kurebayashi et al., 2006). Moreover, the dosage of imatinib used in our study was considered sufficient to exert its pharmacological activities. We used 100 μg/g body weight of imatinib; the same or smaller doses of imatinib have been used intraperitoneally for cancer models with significant efficacies (Hwang et al., 2003; Kurebayashi et al., 2006). In clinical practice, 400–600 mg of imatinib per body is prescribed orally, which is equivalent to approximately 5–10 μg/g body weight (Demetri et al., 2002; Druker et al., 2006; Kantarjian et al., 2002). Thus, it is unlikely that the drug delivery and dosage of imatinib used in this study were inappropriate to exert the therapeutic effects of imatinib in murine models.
A possible link between imatinib and SH3BP2 was reported in a previous study on gastrointestinal stromal tumour (GIST) (Serrano-Candelas et al., 2018). Since imatinib inhibits c-Kit and platelet-derived growth factor receptor (PDGFR) tyrosine kinases, it has been applied to the treatment of GIST in clinical practice (Demetri et al., 2002). In a previous study, Serrano-Candelas et al. investigated the mechanisms by which imatinib suppresses the growth of GIST (Serrano-Candelas et al., 2018). They found that imatinib suppressed SH3BP2 expression in human GIST-derived cells, and that the knockdown of SH3BP2 decreased KIT and PDGFRA mRNA expression, resulting in decreased cell proliferation (Serrano-Candelas et al., 2018). In our study, we initially speculated that the effects of imatinib on cherubism could be due to a similar mechanism. However, we found that treatment with imatinib did not significantly decrease SH3BP2 protein expression in Sh3bp2+/+ and Sh3bp2 mutant mouse embryo fibroblasts (Supplementary Figure 2A). Additionally, imatinib did not affect the mRNA expression of Sh3bp2, Kit, and Pdgfra in Sh3bp2+/+ and Sh3bp2 mutant mouse embryo fibroblasts (Supplementary Figure 2B).
Our in vitro analyses revealed that imatinib decreased RANKL-induced osteoclast formation. In contrast, the in vivo administration of imatinib did not exhibit significant suppressive effects on the bone loss in cherubism mice. There are several possibilities to account for this discrepancy. First, imatinib might not be effectively distributed to osteoclasts. If this is true, cell-specific drug delivery might be required for imatinib to exert a bone-protective effect. Second, the effect of imatinib on cells other than osteoclasts might have eliminated its suppressive effect on osteoclast formation in vivo since bone mass is controlled by several mechanisms, such as osteoclast-mediated bone resorption, osteoblast-mediated bone formation, and stromal cell-mediated regulation of both osteoclasts and osteoblasts (Gao, Patil, & Jia, 2021).
Randomised control trials represent an optimal strategy to evaluate the efficacy of drug candidates in clinical settings. However, in the case of cherubism, performing randomised control trials is not practical since cherubism is a rare disease and patients with cherubism are scattered all over the world. In cases of rare diseases, murine models represent useful tools for validating drug candidates. Considering this, using in vitro and in vivo analyses of the cherubism murine model, entospletinib (SYK inhibitor) (Yoshimoto et al., 2018), tacrolimus (calcineurin inhibitor) (Mukai, Ishida, et al., 2014), and etanercept (TNF inhibitor) (Yoshitaka, Ishida, et al., 2014) have all been shown to have potential beneficial effects on cherubism. To date, several drugs have been tested in practice with variable responses, such as TNF inhibitors, tacrolimus, calcitonin, corticosteroids, interferon α, bisphosphonate, and denosumab (Chrcanovic et al., 2021; Kueper et al., 2021). Obtaining experimental data using murine models and accumulating clinical data, including non-effective cases, are necessary to develop better pharmaceutical treatment strategies.
In conclusion, our study did not show the beneficial therapeutic effects of imatinib in a murine cherubism model. To develop better pharmacological treatment strategies, it is necessary to obtain new knowledge from murine models and human cellular studies, as well as to accumulate clinical data from patients with a definite diagnosis.
Supplementary Material
Acknowledgements
We would like to thank K. Maitani, M. Yoshimoto, and A. Kusumoto (Department of Rheumatology, Kawasaki Medical School) for their technical assistance. We are also indebted to the staff at the Central Research Institute of Kawasaki Medical School. This work was supported in part by grants from Health Labour Sciences Research Grant (20FC1047 to T.M.), JSPS KAKENHI (18K08398 and 21K08484 to T.M., 20K08814 to Y.M., 20K17442 to S.H.A.), and the National Institute of Health (R01DE025870, R01DE025870-06S1, and R21DE030561 to Y.U.), as well as Research Project Grants from Kawasaki Medical School (R03-B046 and R02-B063 to T.M. and R02-B062 to Y.M.), Teraoka Foundation, and the Mishima-Kaiun foundation to T.A., GSK Japan Research Grant to S.H.A., and UCB Japan to T.M.
Footnotes
Conflict of interest
T.M., T.A., S.H.A., and Y.M. received scholarship donations from Chugai, AYUMI, Asahi Kasei, and Abbvie. Y.M. received speaker honoraria from Eli Lilly. The funders had no role in the design of the study, collection, analyses, interpretation of data, writing of the manuscript, or in the decision to publish the results.
Ethics approval statement
All animal experiments were approved by the Safety Committee for Recombinant DNA Experiments (No. 18–23) and the Institutional Animal Care and Use Committee of Kawasaki Medical School (No. 20–072). All experimental procedures were conducted in accordance with the institutional and National Institutes of Health guidelines for humane animal use.
References
- Akagi T, Mukai T, Mito T, Kawahara K, Tsuji S, Fujita S, . . . Morita Y (2020). Effect of Angiotensin II on Bone Erosion and Systemic Bone Loss in Mice with Tumor Necrosis Factor-Mediated Arthritis. Int J Mol Sci, 21(11). doi: 10.3390/ijms21114145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader-Meunier B, Van Nieuwenhove E, Breton S, & Wouters C (2018). Bone involvement in monogenic autoinflammatory syndromes. Rheumatology (Oxford), 57(4), 606–618. doi: 10.1093/rheumatology/kex306 [DOI] [PubMed] [Google Scholar]
- Battaglia A, Merati A, & Magit A (2000). Cherubism and upper airway obstruction. Otolaryngol Head Neck Surg, 122(4), 573–574. doi: 10.1067/mhn.2000.103534 [DOI] [PubMed] [Google Scholar]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, & Müller R (2010). Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res, 25(7), 1468–1486. doi: 10.1002/jbmr.141 [DOI] [PubMed] [Google Scholar]
- Chrcanovic BR, Guimarães LM, Gomes CC, & Gomez RS (2021). Cherubism: a systematic literature review of clinical and molecular aspects. Int J Oral Maxillofac Surg, 50(1), 43–53. doi: 10.1016/j.ijom.2020.05.021 [DOI] [PubMed] [Google Scholar]
- de la Fuente MA, Kumar L, Lu B, & Geha RS (2006). 3BP2 deficiency impairs the response of B cells, but not T cells, to antigen receptor ligation. Mol Cell Biol, 26(14), 5214–5225. doi: 10.1128/mcb.00087-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, . . . Joensuu H (2002). Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. New England Journal of Medicine, 347(7), 472–480. doi: 10.1056/NEJMoa020461 [DOI] [PubMed] [Google Scholar]
- Dimitriou ID, Lee K, Akpan I, Lind EF, Barr VA, Ohashi PS, . . . Rottapel R (2018). Timed Regulation of 3BP2 Induction Is Critical for Sustaining CD8(+) T Cell Expansion and Differentiation. Cell Rep, 24(5), 1123–1135. doi: 10.1016/j.celrep.2018.06.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, . . . Investigators I (2006). Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. New England Journal of Medicine, 355(23), 2408–2417. doi: 10.1056/NEJMoa062867 [DOI] [PubMed] [Google Scholar]
- Eiden S, Lausch E, & Meckel S (2017). [Not Available]. Rofo, 189(7), 675–677. doi: 10.1055/s-0043-105074 [DOI] [PubMed] [Google Scholar]
- Foucault I, Le Bras S, Charvet C, Moon C, Altman A, & Deckert M (2005). The adaptor protein 3BP2 associates with VAV guanine nucleotide exchange factors to regulate NFAT activation by the B-cell antigen receptor. Blood, 105(3), 1106–1113. doi: 10.1182/blood-2003-08-2965 [DOI] [PubMed] [Google Scholar]
- Fujita S, Mukai T, Mito T, Kodama S, Nagasu A, Kittaka M, . . . Morita Y (2018). Pharmacological inhibition of tankyrase induces bone loss in mice by increasing osteoclastogenesis. Bone, 106, 156–166. doi: 10.1016/j.bone.2017.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Patil S, & Jia J (2021). The Development of Molecular Biology of Osteoporosis. Int J Mol Sci, 22(15). doi: 10.3390/ijms22158182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Singh K, Garg A, Bhandari PS, Sah SK, Reichenberger E, . . . Trehanpati N (2019). Clinicoradiologic follow up of cherubism with aggressive characteristics: a series of 3 cases. Oral Surg Oral Med Oral Pathol Oral Radiol, 128(5), e191–e201. doi: 10.1016/j.oooo.2019.01.082 [DOI] [PubMed] [Google Scholar]
- Hatani T, & Sada K (2008). Adaptor protein 3BP2 and cherubism. Curr Med Chem, 15(6), 549–554. [DOI] [PubMed] [Google Scholar]
- Hero M, Suomalainen A, Hagström J, Stoor P, Kontio R, Alapulli H, . . . Mäkitie O (2013). Anti-tumor necrosis factor treatment in cherubism--clinical, radiological and histological findings in two children. Bone, 52(1), 347–353. doi: 10.1016/j.bone.2012.10.003 [DOI] [PubMed] [Google Scholar]
- Hwang RF, Yokoi K, Bucana CD, Tsan R, Killion JJ, Evans DB, & Fidler IJ (2003). Inhibition of Platelet-Derived Growth Factor Receptor Phosphorylation by STI571 (Gleevec) Reduces Growth and Metastasis of Human Pancreatic Carcinoma in an Orthotopic Nude Mouse Model. Clinical Cancer Research, 9(17), 6534–6544. [PubMed] [Google Scholar]
- Kadlub N, Vazquez MP, Galmiche L, L’Herminé AC, Dainese L, Ulinski T, . . . Coudert AE (2015). The calcineurin inhibitor tacrolimus as a new therapy in severe cherubism. J Bone Miner Res, 30(5), 878–885. doi: 10.1002/jbmr.2431 [DOI] [PubMed] [Google Scholar]
- Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, . . . Int STICMLSG (2002). Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. New England Journal of Medicine, 346(9), 645–652. doi: 10.1056/NEJMoa011573 [DOI] [PubMed] [Google Scholar]
- Kawahara K, Mukai T, Iseki M, Nagasu A, Nagasu H, Akagi T, . . . Morita Y (2021). SH3BP2 Deficiency Ameliorates Murine Systemic Lupus Erythematosus. Int J Mol Sci, 22(8). doi: 10.3390/ijms22084169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiber GM, Skapek SX, Lingen M, & Reid RR (2014). Odontogenic myxoma of the face: mimicry of cherubism. J Oral Maxillofac Surg, 72(11), 2186–2191. doi: 10.1016/j.joms.2014.05.027 [DOI] [PubMed] [Google Scholar]
- Kueper J, Tsimbal C, Olsen BR, Kaban L, & Liao EC (2021). SH3BP2-related fibro-osseous disorders of the maxilla and mandible: A systematic review. Int J Oral Maxillofac Surg. doi: 10.1016/j.ijom.2021.04.001 [DOI] [PubMed] [Google Scholar]
- Kurebayashi J, Okubo S, Yamamoto Y, Ikeda M, Tanaka K, Otsuki T, & Sonoo H (2006). Additive antitumor effects of gefitinib and imatinib on anaplastic thyroid cancer cells. Cancer chemotherapy and pharmacology, 58(4), 460–470. [DOI] [PubMed] [Google Scholar]
- Levaot N, Simoncic PD, Dimitriou ID, Scotter A, La Rose J, Ng AH, . . . Rottapel R (2011). 3BP2-deficient mice are osteoporotic with impaired osteoblast and osteoclast functions. J Clin Invest, 121(8), 3244–3257. doi: 10.1172/jci45843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levaot N, Voytyuk O, Dimitriou I, Sircoulomb F, Chandrakumar A, Deckert M, . . . Rottapel R (2011). Loss of Tankyrase-mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell, 147(6), 1324–1339. doi: 10.1016/j.cell.2011.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morbach H, Hedrich CM, Beer M, & Girschick HJ (2013). Autoinflammatory bone disorders. Clin Immunol, 147(3), 185–196. doi: 10.1016/j.clim.2012.12.012 [DOI] [PubMed] [Google Scholar]
- Mukai T, Gallant R, Ishida S, Kittaka M, Yoshitaka T, Fox DA, . . . Ueki Y (2015). Loss of SH3 domain-binding protein 2 function suppresses bone destruction in tumor necrosis factor-driven and collagen-induced arthritis in mice. Arthritis Rheumatol, 67(3), 656–667. doi: 10.1002/art.38975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T, Gallant R, Ishida S, Yoshitaka T, Kittaka M, Nishida K, . . . Ueki Y (2014). SH3BP2 gain-of-function mutation exacerbates inflammation and bone loss in a murine collagen-induced arthritis model. PLoS One, 9(8), e105518. doi: 10.1371/journal.pone.0105518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T, Ishida S, Ishikawa R, Yoshitaka T, Kittaka M, Gallant R, . . . Ueki Y (2014). SH3BP2 cherubism mutation potentiates TNF-alpha-induced osteoclastogenesis via NFATc1 and TNF-alpha-mediated inflammatory bone loss. J Bone Miner Res, 29(12), 2618–2635. doi: 10.1002/jbmr.2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasu A, Mukai T, Iseki M, Kawahara K, Tsuji S, Nagasu H, . . . Morita Y (2019). Sh3bp2 Gain-Of-Function Mutation Ameliorates Lupus Phenotypes in B6.MRL-Fas(lpr) Mice. Cells, 8(5). doi: 10.3390/cells8050402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadaki ME, Lietman SA, Levine MA, Olsen BR, Kaban LB, & Reichenberger EJ (2012). Cherubism: best clinical practice. Orphanet J Rare Dis, 7 Suppl 1(Suppl 1), S6. doi: 10.1186/1750-1172-7-s1-s6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenberger EJ, Levine MA, Olsen BR, Papadaki ME, & Lietman SA (2012). The role of SH3BP2 in the pathophysiology of cherubism. Orphanet J Rare Dis, 7 Suppl 1(Suppl 1), S5. doi: 10.1186/1750-1172-7-s1-s5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricalde P, Ahson I, & Schaefer ST (2019). A Paradigm Shift in the Management of Cherubism? A Preliminary Report Using Imatinib. J Oral Maxillofac Surg, 77(6), 1278.e1271–1278.e1277. doi: 10.1016/j.joms.2019.02.021 [DOI] [PubMed] [Google Scholar]
- Schreuder WH, van der Wal JE, de Lange J, & van den Berg H (2021). Multiple versus solitary giant cell lesions of the jaw: Similar or distinct entities? Bone, 149, 115935. doi: 10.1016/j.bone.2021.115935 [DOI] [PubMed] [Google Scholar]
- Serrano-Candelas E, Ainsua-Enrich E, Navinés-Ferrer A, Rodrigues P, García-Valverde A, Bazzocco S, . . . Martin M (2018). Silencing of adaptor protein SH3BP2 reduces KIT/PDGFRA receptors expression and impairs gastrointestinal stromal tumors growth. Mol Oncol, 12(8), 1383–1397. doi: 10.1002/1878-0261.12332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern SM, & Ferguson PJ (2013). Autoinflammatory bone diseases. Rheum Dis Clin North Am, 39(4), 735–749. doi: 10.1016/j.rdc.2013.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, . . . Taniguchi T (2002). Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell, 3(6), 889–901. doi: 10.1016/s1534-5807(02)00369-6 [DOI] [PubMed] [Google Scholar]
- Tosa I, Yamada D, Yasumatsu M, Hinoi E, Ono M, Oohashi T, . . . Takarada T (2019). Postnatal Runx2 deletion leads to low bone mass and adipocyte accumulation in mice bone tissues. Biochem Biophys Res Commun, 516(4), 1229–1233. doi: 10.1016/j.bbrc.2019.07.014 [DOI] [PubMed] [Google Scholar]
- Ueki Y, Lin CY, Senoo M, Ebihara T, Agata N, Onji M, . . . Olsen BR (2007). Increased myeloid cell responses to M-CSF and RANKL cause bone loss and inflammation in SH3BP2 “cherubism” mice. Cell, 128(1), 71–83. doi: 10.1016/j.cell.2006.10.047 [DOI] [PubMed] [Google Scholar]
- Ueki Y, Tiziani V, Santanna C, Fukai N, Maulik C, Garfinkle J, . . . Reichenberger E (2001). Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat Genet, 28(2), 125–126. doi: 10.1038/88832 [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Hayashi T, Kondo T, Kittaka M, Reichenberger EJ, & Ueki Y (2018). Second-Generation SYK Inhibitor Entospletinib Ameliorates Fully Established Inflammation and Bone Destruction in the Cherubism Mouse Model. J Bone Miner Res, 33(8), 1513–1519. doi: 10.1002/jbmr.3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitaka T, Ishida S, Mukai T, Kittaka M, Reichenberger EJ, & Ueki Y (2014). Etanercept administration to neonatal SH3BP2 knock-in cherubism mice prevents TNF-alpha-induced inflammation and bone loss. J Bone Miner Res, 29(5), 1170–1182. doi: 10.1002/jbmr.2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitaka T, Mukai T, Kittaka M, Alford LM, Masrani S, Ishida S, . . . Ueki Y (2014). Enhanced TLR-MYD88 signaling stimulates autoinflammation in SH3BP2 cherubism mice and defines the etiology of cherubism. Cell Rep, 8(6), 1752–1766. doi: 10.1016/j.celrep.2014.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.