

Summary
Due to the low stoichiometry and highly transient nature of protein phosphorylation it is challenging to capture the dynamics and complexity of phosphorylation events on a systems level. Here, we present an optimized protocol to measure virus-induced phosphorylation events with high sensitivity using label free quantification-based phosphoproteomics. Specifically, we describe filter assisted protein digestion (FASP), enrichment of phosphopeptides, mass spectrometry, and subsequent bioinformatic analysis.
For complete details on the use and execution of this protocol, please refer to Hunziker et al. (2022).
Subject areas: Cell Biology, Microbiology, Signal Transduction, Protein Biochemistry, Proteomics, Mass Spectrometry, Systems biology
Graphical abstract
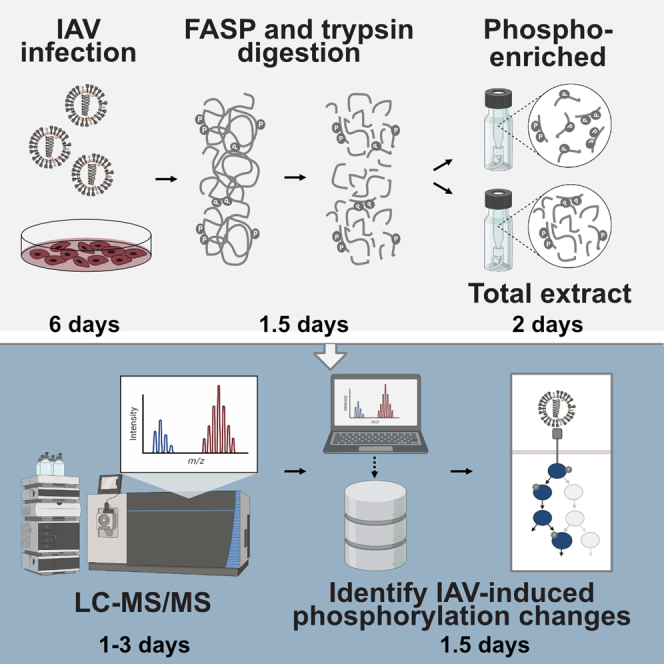
Highlights
-
•
Protocol to reveal virus-induced changes to the cellular phosphoproteome
-
•
Optimized automated phosphopeptide enrichment increases sensitivity
-
•
Protocol uses label-free quantification, allowing for large numbers of samples
Due to the low stoichiometry and highly transient nature of protein phosphorylation it is challenging to capture the dynamics and complexity of phosphorylation events on a systems level. Here, we present an optimized protocol to measure virus-induced phosphorylation events with high sensitivity using label free quantification-based phosphoproteomics. Specifically, we describe filter assisted protein digestion (FASP), enrichment of phosphopeptides, mass spectrometry, and subsequent bioinformatic analysis.
Before you begin
Mass spectrometry has become the method of choice to systematically analyze highly dynamic phosphorylation changes in an unbiased manner with high sensitivity and resolution (Aebersold and Mann, 2003). Initially, a method based on metabolic labeling of cells with stable isotopes (SILAC) was mostly used (Ong et al., 2002). However, SILAC is limited to three conditions per analysis. Its quantification is fully scan (MS1) based and although it is the most accurate method for quantitative phosphoproteomics, its quantification method leads to higher MS1 spectral complexity and therefore to over-sampling of peptide isotope–variants decreasing the total number of peptides identified (Li et al., 2012; Mertins et al., 2012). Therefore, other quantification techniques such as label free quantification (LFQ) (Huttlin et al., 2010; Lundby et al., 2012, 2013) and chemical labels, for example iTRAQ and TMT (Mertins et al., 2016) have gained popularity. LFQ based methods were shown to be equally accurate compared with isobaric tandem mass tags but to have a higher dynamic detection range (Trinh et al., 2013). Further, LFQ bypasses the need for metabolic or chemical labeling and is therefore the most cost effective (Cox et al., 2014; Neilson et al., 2011). It relies on reproducible chromatographic separations and different computational methods to compare peptide abundances in different conditions.
The method described here (Figure 1) was initially designed to measure influenza A virus (IAV) induced phosphorylation changes. However, it is not limited to IAV but can also be used with other stimuli, such as other viruses, bacteria, hormones, drugs and more. Since our protocol does not need metabolic labeling and works with a low amount of input material (300 μg), cell lines can be substituted by primary tissue and clinical samples. By substituting the databases, this method can be applied to any species and pathogen with an annotated genome. In this protocol, we give an automated alternative for phosphopeptide enrichment adapted from Tape et al. (2014), which allows invariable sample processing of a large number of replicates which further reduces variability (Cox et al., 2014). Standardized sample preparation also clears the way for inter-laboratory integration of data sets and enables large scale studies. We recommend to do at least five independent replicates per condition.
Figure 1.

Graphical procedure overview
(A) Graphical overview of the different steps of the protocol.
Generation of virus stocks
Timing: 14 days
-
1.
Because of low stoichiometry of phosphorylation, high titer infections in a small volume are recommended to maximize infection efficiency. Virus stocks should have high titers and be free of cellular cytokines or other contaminants since these can cause signaling events independent of viral infection. To obtain this, IAV can be grown in 11 day old embryonated chicken eggs. A detailed protocol is described by Brauer and Chen (2015).
CAUTION Always handle viruses under biological safety cabinets with the required biosafety measures in place.
Cultivation of cell line
-
2.
Thaw human adenocarcinoma A549 cells according to ATCC instructions: https://www.atcc.org/products/ccl-185#detailed-product-information
-
3.
Human adenocarcinoma A549 cells are cultured in Dulbecco’s modified Eagle medium (DMEM, Life technologies, #41966-029) supplemented with 1% Penicillin/streptomycin (Life technologies, #15140-122) and 10% fetal bovine serum (Life technologies, #10270106) and should be incubated at 37°C 5% CO2. Cells should be passaged at least two times before starting the protocol.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Virus strains | ||
| Egg grown influenza A virus A/WSN/1933 (H1N1) | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s modified Eagle medium (DMEM) | Life technologies | Cat#41966-029 |
| Penicillin/streptomycin | Life technologies | Cat#15140-122 |
| Fetal bovine serum | Life technologies | Cat#10270106 |
| cOmplete Mini EDTA-free protease inhibitor cocktail | Sigma-Aldrich | Cat#11836170001 |
| PhosSTOP | Sigma-Aldrich | Cat#4906845001 |
| Trypsin Gold, Mass Spectrometry Grade | Promega | Cat#VA9000 |
| MagReSyn Ti-IMAC | ReSyn Biosciences | Cat#MR-TIM005 |
| Synthetic peptides with indexed retention times (iRT) kit | BIOGNOSYS | Cat#Ki-3002-1 |
| Urea (UA) | Sigma-Aldrich | Cat#U5128 |
| Iodoacetamide (IAA) | Sigma-Aldrich | Cat#I6125 |
| Triethylammoniumbicarbonat (TEAB) buffer pH 8.5, 1 M | Sigma-Aldrich | Cat#T7408 |
| Trifluoroacetic acid (TFA) | Sigma-Aldrich | Cat#302031 |
| Formic acid (FA) – for LC-MS | Sigma-Aldrich | Cat#00940 |
| Sodium dodecyl sulfate (SDS) | Sigma-Aldrich | Cat#436143 |
| 1,4-Dithiothreitol (DTT) | Sigma-Aldrich | Cat#10708984001 |
| Glycolic acid (GA) | Sigma-Aldrich | Cat#124737 |
| Acetonitrile (ACN) – for LC-MS | Sigma-Aldrich | Cat#1.00029 |
| Tris/HCL | Sigma-Aldrich | Cat#PHG0002 |
| Ammonia water (NH4OH 28% aqueous solution) | Sigma-Aldrich | Cat#221228 |
| EtOH 100% | Sigma-Aldrich | Cat#51976 |
| NaCl | Sigma-Aldrich | Cat#S9888 |
| Critical commercial assays | ||
| Qubit Protein Assay Kit | Thermo Fisher Scientific | Cat#Q33211 |
| Experimental models: Cell lines | ||
| Human lung adenocarcinoma A549 cells | ATCC | ATCC CCL-185 |
| Software and algorithms | ||
| MaxQuant software v1.6.23 or higher | Cox et al. (2011) and Cox and Mann (2008) | www.maxquant.org |
| Uniprot database A/WSN/1933 | UniProt | https://www.uniprot.org/uniprot/?query=organism:382835&sort=score; Uniprot: 382835 |
| Uniprot data base human | UniProt | https://www.uniprot.org/proteomes/UP000005640; Proteome ID: UP000005640 |
| Uniprot data base chicken | UniProt | https://www.uniprot.org/proteomes/UP000000539; Proteome ID: UP000000539 |
| Other | ||
| Microcon-30 kDa Centrifugal Filter Units with Ultracel-30 membranes | Merck Millipore | Cat#MRCF0R030 |
| T-150 cell culturing flask | TPP | Cat#90151 |
| 1.5 mL Eppendorf tubes safe-lock | Eppendorf | Cat#0030121023 |
| p100 cell culture dish | TPP | Cat#93100 |
| Cell scraper | TPP | Cat#99003 |
| Invitrogen™ Countess™ 3 Automated Cell Counter | Fisher Scientific | Cat#16812556 |
| Invitrogen™ Countess™ Cell Counting Chamber Slides | Fisher Scientific | Cat#10399053 |
| Qubit Flex Fluorometer | Thermo Fisher Scientific | Cat#Q33327 |
| Micro centrifuge (1.5–2 mL tubes) | Various suppliers | N/A |
| 500 μL thin-walled PCR tubes | Thermo Fisher Scientific | CAT#AM12225 |
| Parafilm | Sigma-Aldrich | CAT#P7793-1EA |
| Incubator for cells | Various suppliers | N/A |
| Biological safety cabinet for cell culture and virus infection | Various suppliers | N/A |
| Chemical safety cabinet | Various suppliers | N/A |
| Eppendorf ThermoMixer® F1.5 or equivalent | Eppendorf | Cat#538400001 |
| Glass box for the wet chamber | Fisher Scientific | Cat#11766958 |
| SpeedVac concentrator | Thermo Fisher Scientific | Cat#SPD111V-115 |
| Ultrasonic bath | Various suppliers | N/A |
| Branson Ultrasonics Sonifier® SFX250 or equivalent dry sonicator | Fisher Scientific | Cat#15569624 |
| Eppendorf ThermoMixer® F1.5 or equivalent | Eppendorf | Cat#5384000012 |
| Computer station with minimum requirements of used MaxQuant version (http://www.coxdocs.org/doku.php?id=maxquant:common:download_and_installation) | Various suppliers | N/A |
| Optional: KingFisherFlex system | Thermo Fisher Scientific | Cat#A31508 |
| Optional: V-bottom micro tubes with screw cap | Merck | Cat#Z637432-1000EA |
| Optional: Magnetic separator | Merck | Cat#M1167-1EA |
| Optional: KingFisher Flex Microtiter deepwell 96 plate | Thermo Fisher Scientific | Cat#95040460 |
| C18 Stage Tips: P200 pipette tips with Empore Disk C18 | Merck; 3M | Cat#CLS4154-4X960EA; Cat#2215 |
| Optinal: Xcalibur Software to run the mass spectrometer | Thermo Scientific | N/A |
| Optional: Easy-nLC 1000 HPLC system | Thermo Scientific | N/A |
| Optional: Nano trap column (Thermo Scientific), 75 μm × 2 cm, packed with Acclaim PepMap100 C18 material, 3 μm, 100 A | Thermo Scientific | N/A |
| Optional: Analytical Easy-Spray column, 75 μm × 50 cm, packed with reverse-phase C18 material (Acclaim PepMap RSLC, 2 μm, 100 A) | Thermo Scientific | N/A |
| Optional: Thermo Scientific™ Orbitrap Fusion™ Tribrid™ mass spectrometer or any other mass spectrometer with high precision MS1 spectrum recording (e.g., Orbitrap) | Thermo Scientific, San Jose, Ca | N/A |
CRITICAL: All chemicals should be of highest purity.
Materials and equipment
-
•
Stop and go extraction (Stage) tip assembly
C18 Stage Tips: P200 pipette tips (Merck, #CLS4154-4X960EA) with Empore Disk C18 (3M, #2215) cores assembled according to Rappsilber et al. (2003).
-
•
Equipment setup mass spectrometer
Thermo Scientific™ Orbitrap Fusion™ Tribrid™ mass spectrometer (Thermo Scientific, San Jose, Ca) connected to an Easy-nLC 1000 HPLC system (Thermo Scientific). Peptides should be stored in a cooled (4°C) autosampler before being loaded on to a nano trap column (Thermo Scientific), 75 μm × 2 cm, packed with Acclaim PepMap100 C18 material, 3 μm, 100 A. Separation is done with an analytical Easy-Spray column (Thermo Scientific), 75 μm × 50 cm, packed with reverse-phase C18 material (Acclaim PepMap RSLC, 2 μm, 100 A).
-
•
Cell cultivation medium
| Reagent | Final concentration | Amount (555 mL) |
|---|---|---|
| Dulbecco’s modified Eagle medium | N/A | 500 mL |
| Penicillin /streptomycin | 1% (v/v) | 5 mL |
| Fetal bovine serum | 10% (v/v) | 50 mL |
Medium can be stored at 4°C for several weeks.
-
•
SDS lysis buffer
| Reagent | Final concentration | Amount (10 mL) |
|---|---|---|
| SDS | 4% (w/v) | 400 mg |
| Tris/HCL pH 8.2 (1 M) | 100 mM | 1 mL |
| cOmplete Mini EDTA free protease inhibitor cocktail | 1 tablet | |
| PhosSTOP | 1 tablet | |
| DTT | 0.1 M | 155 mg |
| ddH2O | N/A | 9 mL |
Prepare freshly before each use and keep at 4°C. Should be used within 1 day.
-
•
Urea buffer
| Reagent | Final concentration | Amount (10 mL) |
|---|---|---|
| Urea | 8 M | 4.8 g |
| Tris/HCL (pH 8.2) | 100 mM | 10 mL |
Prepare freshly before each use and keep at 4°C. Use within one day. ∼700 μL urea buffer are needed for each sample.
-
•
Alkylation buffer
| Reagent | Final concentration | Amount (1 mL) |
|---|---|---|
| Iodoacetamide (IAA) (0.5 M) | 50 mM | 100 μL |
| Urea buffer | 900 μL |
Prepare freshly before each use and keep at 4°C. Use within one day. ∼100 μL IAA buffer are needed for each sample.
-
•
TEAB buffer
| Reagent | Final concentration | Amount (1 mL) |
|---|---|---|
| TEAB (1 M, pH 8.5) | 50 mM | 50 μL |
| ddH2O | 950 μL |
Prepare freshly before each use. ∼500 μL TEAB buffer are needed for each sample.
-
•
Loading buffer
| Reagent | Final concentration | Amount (50 mL) |
|---|---|---|
| Glycolic acid (GA) | 1 M | 3.8 g |
| Acetonitrile (ACN) | 80% (v/v) | 40 mL |
| Trifluoroacetic acid (TFA) | 5% (v/v) aqueous solution | 2.5 mL |
| ddH2O | 7.5 mL |
Prepare freshly before each use.
-
•
Wash buffer 1
| Reagent | Final concentration | Amount (20 mL) |
|---|---|---|
| ACN | 80% (v/v) | 16 mL |
| TFA | 1% (v/v) aqueous solution | 200 μL |
| ddH2O | 3.8 mL |
Prepare freshly before each use.
-
•
Wash buffer 2
| Reagent | Final concentration | Amount (20 mL) |
|---|---|---|
| ACN | 10% (v/v) | 2 mL |
| TFA | 0.2% (v/v) aqueous solution | 40 μL |
| ddH2O | 17.96 mL |
Prepare freshly before each use.
-
•
Elution buffer
| Reagent | Final concentration | Amount (10 mL) |
|---|---|---|
| NH4OH 28% aqueous solution | 1% (v/v) | 360 μL |
| ddH2O | 9.64 mL |
Prepare freshly before each use.
-
•
Buffer 1 Stop and go extraction (Stage) tip clean up
| Reagent | Final concentration | Amount (20 mL) |
|---|---|---|
| ACN | 60% (v/v) | 12 mL |
| TFA | 0.1% (v/v) aqueous solution | 20 μL |
| ddH2O | 7.98 mL |
Buffer can be stored at 15°C–25°C for several weeks.
-
•
Buffer 2 Stop and go extraction (Stage) tip clean up
| Reagent | Final concentration | Amount (20 mL) |
|---|---|---|
| ACN | 3% (v/v) | 600 μL |
| TFA | 0.1% (v/v) aqueous solution | 20 μL |
| ddH2O | 19.38 mL |
Buffer can be stored at 15°C–25°C for several weeks.
-
•
MS buffer Stop and go extraction (Stage) tip clean up
| Reagent | Final concentration | Amount (20 mL) |
|---|---|---|
| ACN | 3% (v/v) | 600 μL |
| Formic acid (FA) | 0.1% (v/v) aqueous solution | 20 μL |
| ddH2O | 19.38 mL |
Prepare freshly before each use.
-
•
Buffers Mass Spectrometry measurement
| Reagent | Final concentration | Amount (20 mL) |
|---|---|---|
| Mass spectrometer buffer A | ||
| FA | 0.1% (v/v) | 20 μL |
| ddH2O | 19.98 mL | |
| Mass spectrometer buffer B | ||
| FA | 0.1% (v/v) | 20 μL |
| ACN | 19.98 mL | |
Buffers can be stored at 15°C–25°C for several days.
Note: Not needed if mass spectrometry measurement is outsourced.
Alternatives: Any other mass spectrometer with high precision MS1 spectrum recording can be used for measurement (e.g. Orbitrap). MS data analysis can also be done with other software packages allowing label free quantification. In this protocol, we describe the data analysis with MaxQuant, which is an open source program but subjected to license terms.
Step-by-step method details
Sample generation: virus infection and cell lysis
-
1.
Remove medium from cells and wash them once with phosphate salt buffered solution (PBS) (Life technologies, #14190-094).
-
2.
Add 1 mL of 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA) (Life technologies, #25200-056) and incubate them at 37°C 5% CO2 until cells are detached.
-
3.
Add 9 mL of pre-warmed cultivation medium and mix well.
-
4.
Count cells using a cell counter or Neubauer chamber (Merck, #BR717805-1EA):
Mix 10 μL of cell solution and 10 μL 0.4% (w/v) Trypan blue (Thermo Fisher Scientific, #15250-061) and inject into a Neubauer cell counting chamber or an equivalent slide used for a cell counter.
-
5.
Seed 2 × 106 A549 human lung epithelial cells (ATCC, #CCL-185) into a p100 dish (TPP, #93100) in 12 mL cell cultivation medium per dish.
-
6.
Incubate cells at 37°C 5% CO2 for 24 h.
-
7.
Wash the cells with PBS and incubate them in 12 mL DMEM (Life technologies, #41966-029) without supplements 16 h prior to infection.
-
8.
Prepare SDS lysis buffer freshly (see materials and equipment).
-
9.
Add 3 mL virus inoculum per plate (containing 25 PFU/cell) or up to 3 mL of allantoic liquid from mock-infected eggs as a control in a tube and pre-warm inoculum to 37°C.
-
10.
Suck off serum-free medium from the cells and add the inoculum directly to the cells and leave them at 37°C for 10 min.
Note: Infection time can be varied dependent on your experimental design.
-
11.
After the desired infection time, directly lyse the cells in 400 μL of SDS-lysis buffer per p100 dish (TPP, #93100) and use a cell scraper (TPP, #99003) to remove lysed cells from the plate. Transfer lysate in a fresh 1.5 mL Eppendorf tube (Eppendorf, #0030121023).
-
12.
Put Eppendorf-tubes on ice.
-
13.
Incubate samples at 95°C for 5 min while shaking at 300 rpm in the ThermoMixer (Eppendorf, #538400001) and directly put them on ice again.
-
14.
Briefly sonicate the lysates to reduce viscosity using a dry-sonicator (5 times 15 s pulse with 75% intensity) (Branson Ultrasonics Sonifier® SFX250, FisherScientific, #15569624) and put samples on ice again.Problem 1
-
15.
Centrifuge at 14,000 × g and 4°C for 5 min to clarify the lysate and transfer lysates into a fresh 1.5 mL Eppendorf tube.
Pause point: Store the cell extracts at −80°C until you finished all replicates.
Filter aided sample preparation (FASP) and protein digestion
-
16.Quantify protein concentration using Qubit Flex Fluorometer (ThermoFisher Scientific, #Q33327) according to manufacturer’s protocol: (https://www.thermofisher.com/order/catalog/product/Q33211#/Q33211).
-
a.Add the Qubit buffer volume needed in an Eppendorf tube: 199 μL Qubit buffer per sample and 190 μL for each standard (3 in total).
-
b.Then add the Qubit reagent 1:200 (v/v) to the buffer and vortex mixture.
-
c.Aliquot 199 μL for samples and 190 μL for standards in Qubit tubes and add 1 μL sample or 10 μL of standard solutions to 500 μL thin-walled PCR tubes.
-
d.Vortex mixtures and incubate at 15°C–25°C for 15 min.
-
e.Measure according to Qubit instructions starting with the standard solution to calibrate the instrument.
-
a.
Note: Alternatively, the Invitrogen EZQ Protein Quantitation Kit (#R33200) or any other fluorescent dye based protein quantification method can be used. Biuret methods cannot be used because the lysis buffer contains DTT.
-
17.
Use 300 μg protein for each sample to start.
-
18.Prepare all solutions needed:
-
a.UA: 8 M urea in 100 mM Tris/HCl pH 8.2
-
b.IAA solution: 100 μL 0.05 M iodoacetamide in 900 μL UA
-
c.0.5 M NaCl in ddH2O
-
d.TEAB: 0.05 M Triethylammoniumbicarbonat in ddH2O
-
a.
-
19.
Label all tubes, lids and filters with the number from the randomized list of the corresponding sample.
-
20.Mix up to 50 μL of sample (containing 300 μg of protein) with 200 μL UA buffer and load onto the filter unit.CRITICAL: Samples should be at 15°C–25°C before you start centrifuging.
-
a.Centrifuge at 14,000 × g for 20 min at 15°C–25°C.
-
b.Discard flow-trough. Problems 2 and 3
-
a.
-
21.
Repeat step 20 until everything is loaded on the filter (in case several steps are needed to load the total amount of protein).
-
22.
Discard flow-trough.
-
23.Add 200 μL of UA to the filter unit.
-
a.Centrifuge at 14,000 × g for 20 min at 15°C–25°C.
-
b.Discard flow-trough.
-
a.
-
24.Add 100 μL IAA solution (0.05 M) to the filter unit.
-
a.Mix at 600 rpm in the ThermoMixer for 1 min (lid open).
-
b.Centrifuge at 14,000 × g for 20 min at 15°C–25°C.
-
c.Discard flow-trough.
-
a.
-
25.Add 100 μL of UA to the filter unit.
-
a.Centrifuge at 14,000 × g for 20 min at 15°C–25°C.
-
b.Discard flow-trough.
-
a.
-
26.
Repeat step 25 twice.
-
27.Add 100 μL of 0.5 M NaCl to the filter unit.
-
a.Centrifuge at 14,000 × g for 20 min at 15°C–25°C.
-
b.Discard flow-trough.
-
a.
-
28.
Repeat step 27 once.
-
29.Add 100 μL of TEAB to the filter unit.
-
a.Centrifuge at 14,000 × g for 20 min at 15°C–25°C.
-
b.Discard flow-trough.
-
a.
-
30.
Repeat step 29 twice.
-
31.
Transfer the filter units to new collection tubes.
-
32.
Reconstitute 20 μg lyophilized trypsin in 500 μL TEAB.
-
33.Add 120 μL of TEAB with 2% trypsin (5 μg trypsin per sample) to the filter unit.
-
a.Mix at 600 rpm in the thermo-mixer for 1 min.
-
b.Seal all tube-lids with parafilm.
-
a.
-
34.
Incubate the units for 16 h at 15°C–25°C in a wet-cell.
Elution and peptide drying
-
35.
Centrifuge the filter units at 14,000 × g for 20 min at 15°C–25°C.
-
36.
Re-elute the column by adding 80 μL of TEAB.
-
37.
Centrifuge the filter units at 14,000 × g for 20 min at 15°C–25°C.
-
38.
Transfer 10 μL of each sample into a new 1.5 mL Eppendorf tube for total extract samples and freeze them at -20°C.
-
39.
Completely dry the rest of the digested peptides in a SpeedVac condensator at 35°C. This can take 2–3 h.
Equilibration of MagReSyn® Ti-IMAC microspheres
-
40.Prepare all solutions freshly before starting the enrichment:
-
a.70% EtOH in ddH2O
-
b.Loading buffer: 1M glycolic acid in 80% ACN, 5% TFA
-
c.Wash buffer 1: aqueous solution of 80% ACN and 1% TFA
-
d.Wash buffer 2: aqueous solution of 10% ACN and 0.2% TFA
-
e.Elution buffer: 1% NH4OH in ddH2O
-
a.
-
41.
Resolve MagReSyn® Ti-IMAC thoroughly by vortex mixing or inversion to ensure a homogenous suspension.
-
42.Transfer 20 μL microspheres per sample to a 2 mL micro tube with a V bottom shape and screw cap.
-
a.Place the tube on a magnetic separator and allow microspheres to clear.
-
b.Remove the shipping solution (around 50% of the volume) by aspiration with a pipette and discard.
-
a.
-
43.Wash the microspheres in 200 μL of 70% EtOH with gentle agitation at 900 rpm in the ThermoMixer for 5 min.
-
a.Place the tube on the magnetic separator and allow the microspheres to clear.
-
b.Remove the ethanol solution by aspiration with a pipette and discard.
-
a.
-
44.
Repeat the washing step 43.
-
45.Wash the microspheres in 100 μL of ddH2O (Millipore) with gentle agitation at 900 rpm in the ThermoMixer for 10 min.
-
a.Place the tube on the magnetic separator and allow the microspheres to clear.
-
b.Remove the buffer by aspiration with a pipette and discard.
-
a.
-
46.Equilibrate the microspheres in 50 μL loading buffer for 1 min.
-
a.Place the tube on the magnetic separator and allow the microspheres to clear.
-
b.Remove the loading buffer by aspiration with a pipette.
-
a.
-
47.
Repeat step 46 twice for a total number of three equilibrations. After the last one, dissolve microspheres in a total volume equaling the sample number (20 μL for each sample).
-
48.
Transfer 20 μL MagReSyn® Ti-IMAC to a 2 mL microcentrifuge for each enrichment.
Phosphopeptide enrichment using magnetic separator
-
49.
Prepare 10% FA solution in a chemical hood. To do so, calculate the amount you need and dilute formic acid 1:10 in ddH2O.
-
50.
Resuspend dried digested peptides in 100 μL loading buffer.
-
51.
Sonicate for 5 min in the water bath and shake 5 min in the ThermoMixer at 1000 rpm.
-
52.
Centrifuge briefly at 14,000 × g and add to the equilibrated MagReSyn® Ti-IMAC microspheres.
-
53.Incubate for 20 min at 15°C–25°C with continuous mixing at 900 rpm in the ThermoMixer to ensure adequate sample and microsphere interaction.
-
a.Place the tube on the magnetic separator and allow the microspheres to clear.
-
b.Remove and discard the coupling supernatant by aspiration with a pipette.
-
a.
-
54.Remove unbound sample by washing with 100 μL loading buffer for 1 min with gentle agitation at 900 rpm in the ThermoMixer.
-
a.Place the tube on a magnetic separator and allow microspheres to clear.
-
b.Remove the supernatant and discard.
-
a.
-
55.Remove non-specifically bound peptides by adding the microspheres to 100 μL wash buffer 1 for 2 min with gentle agitation at 900 rpm in the ThermoMixer.
-
a.Place the tube on a magnetic separator and allow microspheres to clear.
-
b.Remove the supernatant and discard.
-
a.
-
56.
Repeat step 55 once.
-
57.Wash once with 100 μL wash buffer 2 for 2 min with gentle agitation at 900 rpm in the ThermoMixer.
-
a.Place the tube on a magnetic separator and allow microspheres to clear.
-
b.Remove the supernatant and discard.
-
a.
Note: Washing with wash buffer 2 leads to better removal of hydrophilic peptides.
-
58.
Elute the bound phosphopeptides from the microspheres by adding 80 μL elution buffer for 15 min at 900 rpm in the ThermoMixer.
-
59.
Repeat step 58 twice for a final elution volume of 240 μL.
-
60.
Place the tube on the magnetic separator and allow the microspheres to clear.
-
61.
Remove the eluate containing the phosphopeptides and transfer to a new tube.
-
62.
Add 60 μL of 10% FA to the 240 μL eluate to acidify the solution.
-
63.
Dry peptides from phosphoenriched and total samples using SpeedVac condensator at 35°C (1–2 h).
-
64.
Reconstitute enriched peptides in 150 μL 3% ACN; 0.1% TFA.
-
65.
Sonicate peptides for 5 min in the water bath and shake them 5 min in the ThermoMixer at 1000 rpm.
-
66.
Centrifuge briefly at 140,000 × g.
Phosphoenrichment using the KingFisher Flex system
-
67.
After equilibration, transfer 20 μL MagReSyn® Ti-IMAC to a well in a KingFisher Flex Microtiter deepwell 96 plate and add 480 μL 100% ACN per well.
-
68.
Prepare all solutions according to step 40.
-
69.
Reconstitute dried peptides in 200 μL loading buffer.
-
70.
Sonicate in the water bath for 5 min.
-
71.
Mix solution well at 1000 rpm in the ThermoMixer for 1 min.
-
72.
Transfer each sample to a separate well of a KingFisher Flex Microtiter deepwell 96 plate.
-
73.
Prepare all KingFisher Flex Microtiter deepwell 96 plate with the amount and solution per well indicated in Table 1. Insert plates according to number into the KingFisher Flex instrument.
-
74.
Use the KingFisher Flex instrument with the following steps and timing (in total 42 min, Figure 2A). After elution, release beads into plate 2 (10 s release time) and tips in plate 1.
-
75.
Transfer eluted peptides from each well of plate 8 to a separate 1.5 mL Eppendorf tube.
-
76.
Add 50 μL of 10% FA to each sample.
-
77.
Dry peptides from phosphoenriched and total samples using SpeedVac condensator at 35°C (1–2 h).
-
78.
Reconstitute enriched peptides in 150 μL 3% ACN; 0.1% TFA.
-
79.
Sonicate peptides for 5 min in the water bath and shake them 5 min in the ThermoMixer at 1000 rpm.
-
80.
Centrifuge briefly at 14,000 × g.
Table 1.
Plates with their contents used for the automated phosphopeptide enrichment procedure
| KingFisher Flex Microtiter deepwell 96 plate order | ||
|---|---|---|
| Plate number and name | μL/well | Buffer |
| #1 Tip plate | ||
| #2 Equilibrated Ti-IMAC microspheres | 500 | In 100% ACN |
| #3 Microsphere wash | 500 | Loading buffer |
| #4 Sample | 200 | In loading buffer |
| #5 Wash 1 | 500 | Loading buffer |
| #6 Wash 2 | 500 | Wash buffer 1 |
| #7 Wash 3 | 500 | Wash buffer 2 |
| #8 Elution | 200 | Elution buffer |
Figure 2.

Graphical overview of the automated phosphopeptide enrichment procedure
(A) Graphical overview of the automated phosphopeptide enrichment.
Stop and go extraction (Stage) tip clean up
-
81.
Put Stage tips into a 2 mL Eppendorf tube with adaptors to hold Stage tips in place. Label tubes and Stage tips with sample number. Prepare two sets of fresh 1.5 mL Eppendorf tubes for phosphoenriched and total samples. Label the lids and the tubes.
-
82.Wet Stage tip with 150 μL 100% ACN.
-
a.Centrifuge at 2,000 × g for 1 min.
-
b.Discard flow-trough.
-
a.
-
83.Equilibrate Stage tip with 150 μL 60% ACN; 0.1% TFA.
-
a.Centrifuge at 2,000 × g for 1 min.
-
b.Discard flow-trough.
-
a.
-
84.Equilibrate Stage tip with 150 μL 3% ACN; 0.1% TFA.
-
a.Centrifuge at 2,000 × g for 1 min.
-
b.Discard flow-trough.
-
a.
-
85.
Repeat step 84 once.
-
86.
Put filters in fresh 1.5 mL Eppendorf tubes.
-
87.
Load sample on Stage tip.
-
88.
Centrifuge at 2,000 × g for 1.5 min.
-
89.
Put filters back in 2 mL Eppendorf tubes.
-
90.Wash with 150 μL 3% ACN; 0.1% TFA.
-
a.Centrifuge at 2,000 × g for 1 min.
-
b.Discard flow-trough.
-
a.
-
91.
Repeat step 90 twice for a total of three wash steps.
-
92.
Put filters in fresh 1.5 mL Eppendorf tubes.
-
93.
Elute sample with 150 μL 60% ACN; 0.1% TFA.
-
94.
Centrifuge at 2,000 × g for 1.5 min.
-
95.
Collect flow-through in a new 1.5 mL Eppendorf tube.
-
96.
Completely dry desalted peptides at 35°C using a SpeedVac condensator (2–6 h).
-
97.
Reconstitute phosphoenriched samples in 10 μL and total samples in 20 μL of 3% ACN; 0.1% FA.
-
98.
Sonicate for 5 min in the water bath and shake 5 min in the ThermoMixer at 1000 rpm.
-
99.
Centrifuge briefly at 14,000 × g.
Analysis of peptides using mass spectrometry
If mass spectrometry measurements are outsourced to a specialized facility, skip this paragraph and directly continue with LFQ-based quantification of phosphorylation changes.
-
100.
Transfer 6 μL of sample in a mass spectrometry tube.
-
101.Spike samples with iRTs (BIOGNOSYS) with a dilution of 1:60 (v/v) directly before the measurement:
-
a.Vortex iRT buffer for 1 min
-
b.Add 50 μL of buffer to the iRT peptides
-
c.Sonicate in water bath for 1 min
-
d.Vortex and add to samples
-
a.
-
102.Select the following parameters to measure on a Thermo Scientific™ Orbitrap Fusion™ Tribrid™ mass spectrometer coupled to an Easy-nLC 1000 HPLC system (Thermo Scientific):
-
a.Load 5 μL of peptides on the column from a cooled (4°C) autosampler.
-
b.Buffer A: 0.1% FA in H2O
-
c.Buffer B: 0.1% FA in ACN
-
a.
-
103.For the measurement of phospho-enriched samples, apply the LC gradient described in Table 2 and select the following parameters:
-
a.Acquire MS spectra in the Orbitrap mass analyzer with a mass range of 350–1800 (m/z), a resolution of 120,000 and a maximum injection time of 50 ms.
-
b.Acquire higher energy collisional dissociation (HCD) spectra in the ion trap mass analyzer using a normalized collision energy of 32% with a precursor ion isolation width of 2.0 (m/z) and a maximum injection time of 150 ms.
-
c.Set AGC target value = 2e4.
-
d.Enable charge state screening.
-
e.Include charge states 2–7.
-
f.Set the signal intensities threshold = 5e3.
-
g.Exclude precursor masses already selected for MS/MS acquisition for further selection during 30 s.
-
a.
-
104.For total protein samples, apply the LC gradient in Table 3 and select the following parameters:
-
a.Acquire MS spectra in the Orbitrap mass analyzer with a mass range of 300–2000 (m/z), with a resolution of 120,000, and a maximum injection time of 50 ms.
-
b.Acquire HCD spectra in the ion trap using a normalized collision energy of 30% with an isolation window of 1.6 (m/z) and a maximum injection time of 80 ms.
-
c.Set AGC target value = 8e3.
-
d.Set the intensity threshold = 5e3.
-
e.Set dynamic exclusion duration = 25 s. Problem 8
-
a.
Table 2.
Gradient used during mass spectrometry analysis of phospho-enriched samples
| LC gradient for phospho-enriched samples | |||
|---|---|---|---|
| Time | Duration | % of buffer B | Flow (nL/min) |
| 0 min | 0 min | 2 | 300 |
| 2 min | 2 min | 2 | 300 |
| 122 min | 120 min | 25 | 300 |
| 132 min | 10 min | 35 | 300 |
| 142 min | 10 min | 95 | 300 |
Table 3.
Gradient during mass spectrometry analysis of total protein extracts
| LC gradient for total protein samples | |||
|---|---|---|---|
| Time | Duration | % of buffer B | Flow (nL/min) |
| 0 min | 0 min | 2 | 300 |
| 120 min | 120 min | 25 | 300 |
| 130 min | 10 min | 35 | 300 |
| 140 min | 10 min | 95 | 300 |
LFQ-based quantification of phosphorylation changes
The data analysis can be performed with every software that supports label free based quantification of post-translational protein modifications such as Progenesis QI (Waters, https://www.waters.com/waters/de_CH/ProgenesisQI/nav.htm?locale=de_CH&cid=134790652). Here, we describe in detail the analysis workflow using the open source software MaxQuant. MaxQuant adds known contaminants to the fast file and generates decoy hits for the false discovery rate (FDR) estimation. Your data base is therefore not allowed to have any decoy entries. MaxQuant is coupled to the search engine Andromeda provided together with MaxQuant.
-
105.
Install MaxQuant on your computer.
-
106.
Open MaxQuant folder and open Andromeda executable file (.exe).
-
107.
Make sure that the database containing all annotated proteins of the organism you used is available by default in Andromeda. Problem 9
-
108.
Load either folder containing raw files of phosphoenriched or total samples into the software.
-
109.
Set experiment choosing “no fractions”.
Note: Only choose fractionation if your samples were obtained from a gel-based pre-fractionation.
-
110.
Group specific parameters: Choose cysteine carbamidomethylation as fixed modification.
Note: Fixed modifications occur on every specified amino acid and are often artificially introduced by sample preparation. Cysteine carbamidomethylation is introduced to prevent disulfide bridges.
-
111.
As variable modification, select protein N-terminal acetylation, methionine oxidation, N-terminal glutamate to pyroglutamate conversion and phosphorylation.
Note: Variable modifications increase the in silico peptide database. Only choose the ones necessary to keep computational time as short as possible.
-
112.
Choose modification “phopsho STY” for phosphoenriched but not for total samples.
-
113.
Choose label-free quantification.
-
114.
Select digestion enzyme Trypsin/P.
-
115.
Allow up to two missed cleavages.
-
116.
Select First search.
-
117.
Set fragment mass tolerance to 20 ppm.
-
118.
Set precursor ion mass tolerance to 8 ppm.
-
119.
Use sequences as global parameter: Minimum one unique peptide and a minimum of seven amino acids per identified peptide.
Note: If a peptide contains less than 7 or more than 9 amino acids it cannot be unique and is therefore not informative. Choose a value between 7 and 9.
-
120.
MS/MS and sequence: Choose your created database.
-
121.
Identifier: Up to first space - click identifier - identifierParseRule ([ˆ]∗).
-
122.
Identification and quantification: For identification use a protein FDR of 0.05.
-
123.
Miscellaneous: Choose Re-Quantify.
Note: Re-quantify allows peaks with no labeled pair to be re-searched based on their location or retention time.
-
124.
Go to advanced and choose “Match between runs”.
Note: Allows information on identification from one run to be used in another run searching for peptide peaks closely located in the same retention time range.
-
125.
Start analysis and check if steps are running fine in the performance menu.
-
126.
The result will be saved as text files in the same folder as your raw files in a folder called “combined”. proteinGroups.txt for total samples and Phospho(STY)sites.txt for phosphoenriched samples. To see first quality control data, open the PTXQC report. Problem 10
-
127.
Exported lists of differently phosphorylated factors (p value ≤ 0.05 and log2FC ≥ 1 or ≤ −1) and total identified factors as background list can be submitted to further analysis tools (Table 4).
Table 4.
Pathway analysis tools suitable for follow up analysis of the created phosphoproteomics data set
| Open source application | Source | References |
|---|---|---|
| WEB-based Gene Set Analysis Tool kit | www.WebGestalt.org | Wang et al. (2017) |
| DAVID Bioinformatics Resources | https://david.ncifcrf.gov | Huang da et al. (2009) |
| PhosphoPath (Cytoscape App) | http://apps.cytoscape.org/apps/phosphopath | Raaijmakers et al. (2015) |
| Panther | http://pantherdb.org/ | Mi et al. (2019) |
Expected outcomes
Using this protocol, phosphopeptide enrichment yielded 85% or 95% enrichment efficiency for manual respectively automated enrichment procedure (Figure 3A). We identified 7930 unique phosphopeptides belonging to 2991 phosphoproteins (Figure 3B). Phosphorylation of 9144 serine, 1335 threonine and 136 tyrosine were measured (Figure 3C) of which a substantial part was significantly changed between mock-infected and IAV infected conditions (p value ≤ 0.05, Log2FC ≥ 1 or ≤ -1) (Figure 3D). Comparing five different IAV subtypes allowed us to find phosphorylation sites equally or uniquely altered by different IAV strains (Figure 3E) indicating that the presented workflow allows a highly reproducible measurement of phosphorylation changes within minutes of infection. Obtained results can be further analyzed, for example with the Cytoscape plugin PhosphoPath (Raaijmakers et al., 2015), which analyses the involvement of the found phosphochanges in signaling pathways with respect to annotated phosphosites (Figure 3F).
Figure 3.

Anticipated results
(A) Phosphopeptide enrichment yield calculated using Scaffold PTM (version 4.0, Proteome Software, Portland USA). Data are means ± s.d. from n = 4 independent experiments.
(B) Depiction of phosphorylation acceptor site proportion.
(C) Depiction of total amount of identified phosphoproteins and unique phosphopeptides.
(D) Distribution of changes in cellular phosphorylation after 10 min of infection with A/WSN/1933 (25 PFU/cell) are shown. Log2 fold changes in phosphorylation of the identified phosphopeptides are presented in relation to their p value. Phosphopeptides with lower abundance in mock-infected versus infected samples are located in the upper left panel and significant ones are stained in red (p value ≤ 0.05, Log2FC ≤ −1). Hyper-phosphorylated peptides are located in the upper right panel, significant ones are highlighted in blue (p value ≤0.05, Log2FC ≥ 1). Horizontal dotted lines show Log2 fold change cut-off of ≥ 1 or ≤ −1.
(E) For the Venn-diagram, significantly changing phosphopeptides were used (p value ≤ 0.05, Log2FC ≥ 1 or ≤ −1). Number in brackets indicate total number of phosphopeptides fulfilling the mentioned criteria.
(F) Significantly changing phosphosites (p value ≤ 0.05, Log2FC ≥ 1 or ≤ −1) during A/WSN/1933 infection were mapped to pathways with the Cytoscape plugin PhosphoPath using all identified peptides as background list. Factors and their IAV-responsive phosphosites shown to be relevant for the PI3K signaling pathway (Wikipathways) are shown in blue and grey circles respectively (PhosphoSitePlus, BioGrid).
Limitations
During sample preparation for LFQ experiments, each sample has to be processed individually. Great care must be taken during sample preparation in order to not introduce variability. To overcome this caveat, automated versions of preparation steps should be considered. In this protocol, we describe an automated alternative for phosphopeptide enrichment adapted from Tape et al. (2014) which allows invariable sample processing of a large number of replicates to reduce variability. Standardized sample preparation also clears the way for inter-laboratory integration of data sets and enables large scale studies. We recommend to do at least five independent replicates per condition. One must consider that a LFQ based phosphoproteomics experiment can be more time consuming compared to label based methods since each sample has to be measured individually by mass spectrometry. LFQ quantification is based on comparing peptide abundances between separately measured conditions and missing values between runs can be an issue during data analysis. However, this can be minimized with a robust and reproducible workflow and adequate computational analysis tools presented in this protocol.
Troubleshooting
Problem 1
Samples are still viscous after sonication.
Potential solution
The DNA content is too high. Sonicate multiple times until you can properly pipette the solution.
Problem 2
Some liquid remains on the filter after 20 min of centrifugation.
Potential solution
Volumes added were most likely too high. When liquid is still going through, increase centrifugation duration for samples with increased backpressure but not for the others, filters should never dry out completely.
Problem 3
No liquid goes through the filter
Potential solution
It might be that the loaded protein amount is too high (max. 500 μg protein), sample is too viscous or sample is too cold. (1) Dilute your sample with urea buffer or/and sonicate again before loading onto the filter again. (2) Let samples warm up to 15°C–25°C before you start the FASP procedure.
Problem 4
Some samples do not fully dry in the SpeedVac condensator.
Potential solution
There is a vacuum leakage. (1) Check vacuum pump. (2) Empty condenser flask and replace it by an empty and dry one. (3) Check cover seal of the condenser flask or the machine itself.
Problem 5
No more liquid goes through the Stage tip.
Potential solution
It is likely that particles in the sample clog the Stage tip. Distribute your sample to multiple Stage tips with larger diameter.
Problem 6
Peptides are not binding C18 Stage tip and run through.
Potential solution
Concentration of organic solvent higher than 5% will abolish binding of most or all peptides. Check your buffers and repeat Stage tip clean up with lower concentration of organic solvent.
Problem 7
Stage tip dried out.
Potential solution
There were too many samples processed at the same time and/or sample processing was too slow. If a Stage tip dried out before loading the sample, equilibration procedure must be repeated. If it happens after sample loading, immediately add some liquid to the filter.
Problem 8
No phosphopeptides are detected.
Potential solution
It might be that there are not enough abundant phosphopeptides in the sample due to low MOI infection or because the processing or mass spectrometry measurement did not work. (1) Repeat digestion with a standard phospho-protein and measure without enrichment by mass spectrometry to ensure that your digestion protocol works. (2) Then, enrich sample for the standard phosphopeptide and measure to see if enrichment worked. (3) Repeat Stage tip clean up with enriched phosphopeptides of the standard phospho-protein to validate your protocol and measure. (4) Measure standard phospho-peptides to validate that the mass spectrometry measurement works. (5) If your protocols work, repeat your experiment using a higher MOI for infection.
Problem 9
Andromeda does not have a database of the used organism or digestion enzyme.
Potential solution
You can add proteome databases of any organism with an annotated genome. To do so, open the Andromeda.exe file in the MaxQuant folder and select sequences or enzymes and "+". You can rename your database and choose the organism or enzyme you want to add to the Andromeda default menu.
Problem 10
MaxQuant gives an error.
Potential solution
Go to www.maxquant.org for troubleshooting.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Silke Stertz Ph.D., stertz.silke@virology.uzh.ch
Materials availability
No new material was created in this study.
Acknowledgements
This study was supported by the Swiss National Science Foundation (grants 31003A_156805 and 31003A_176170 to S.S.) and by University of Zurich through the Candoc Forschungskredit (Project K-41902-0 to A.H.). We thank Peter Gehrig and Jonas Grossmann from the Functional Genomic Center Zurich for their input and support. Figures were created with BioRender.com.
Author contributions
A.H. designed and conducted the experiments, analyzed data and wrote the manuscript. S.S. acquired funding, planned and supervised the work of this study and co-wrote the paper.
Declaration of interests
The authors declare no competing interests.
Data and code availability
Data sets generated during this study will be available in Hunziker et al. (Cell Reports, revised manuscript under review) upon its publication.
References
- Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Brauer R., Chen P. Influenza virus propagation in embryonated chicken eggs. J. Vis. Exp. 2015 doi: 10.3791/52421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hunziker, A., Glas, I., Pohl, M.O., and Stertz, S. (2022). Phosphoproteomic profiling of influenza virus entry reveals infection-triggered filopodia induction counteracted by dynamic cortactin phosphorylation. Cell Reports 38, S2211-1247(22)00018-3. 10.1016/j.celrep.2022.110306. [DOI] [PubMed]
- Huttlin E.L., Jedrychowski M.P., Elias J.E., Goswami T., Rad R., Beausoleil S.A., Villen J., Haas W., Sowa M.E., Gygi S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Adams R.M., Chourey K., Hurst G.B., Hettich R.L., Pan C. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J. Proteome Res. 2012;11:1582–1590. doi: 10.1021/pr200748h. [DOI] [PubMed] [Google Scholar]
- Lundby A., Andersen M.N., Steffensen A.B., Horn H., Kelstrup C.D., Francavilla C., Jensen L.J., Schmitt N., Thomsen M.B., Olsen J.V. In vivo phosphoproteomics analysis reveals the cardiac targets of beta-adrenergic receptor signaling. Sci. Signal. 2013;6:rs11. doi: 10.1126/scisignal.2003506. [DOI] [PubMed] [Google Scholar]
- Lundby A., Secher A., Lage K., Nordsborg N.B., Dmytriyev A., Lundby C., Olsen J.V. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012;3:876. doi: 10.1038/ncomms1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P., Mani D.R., Ruggles K.V., Gillette M.A., Clauser K.R., Wang P., Wang X., Qiao J.W., Cao S., Petralia F., et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P., Udeshi N.D., Clauser K.R., Mani D.R., Patel J., Ong S.E., Jaffe J.D., Carr S.A. iTRAQ labeling is superior to mTRAQ for quantitative global proteomics and phosphoproteomics. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.M111.014423. M111 014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Muruganujan A., Huang X., Ebert D., Mills C., Guo X., Thomas P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0) Nat. Protoc. 2019;14:703–721. doi: 10.1038/s41596-019-0128-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhjavani M., Nikounezhad N., Ashtarinezhad A., Shirazi F.H. Human lung carcinoma reaction against metabolic serum deficiency stress. Iran J. Pharm. Res. 2016;15:817–823. [PMC free article] [PubMed] [Google Scholar]
- Neilson K.A., Ali N.A., Muralidharan S., Mirzaei M., Mariani M., Assadourian G., Lee A., van Sluyter S.C., Haynes P.A. Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics. 2011;11:535–553. doi: 10.1002/pmic.201000553. [DOI] [PubMed] [Google Scholar]
- Ong S.E., Blagoev B., Kratchmarova I., Kristensen D.B., Steen H., Pandey A., MANN M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Raaijmakers L.M., Giansanti P., Possik P.A., Mueller J., peeper D.S., Heck A.J., Altelaar A.F. PhosphoPath: visualization of phosphosite-centric dynamics in temporal molecular networks. J. Proteome Res. 2015;14:4332–4341. doi: 10.1021/acs.jproteome.5b00529. [DOI] [PubMed] [Google Scholar]
- Rappsilber J., Ishihama Y., Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Tape C.J., Worboys J.D., Sinclair J., Gourlay R., VOGT J., Mcmahon K.M., Trost M., Lauffenburger D.A., Lamont D.J., Jorgensen C. Reproducible automated phosphopeptide enrichment using magnetic TiO2 and Ti-IMAC. Anal. Chem. 2014;86:10296–10302. doi: 10.1021/ac5025842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh H.V., Grossmann J., Gehrig P., Roschitzki B., Schlapbach R., Greber U.F., Hemmi S. iTRAQ-based and label-free proteomics approaches for studies of human adenovirus infections. Int. J. Proteomics. 2013;2013:581862. doi: 10.1155/2013/581862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Vasaikar S., Shi Z., Greer M., Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45:W130–W137. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sets generated during this study will be available in Hunziker et al. (Cell Reports, revised manuscript under review) upon its publication.