

Abstract
Tricarboxylic acid (TCA) cycle, also called Krebs cycle or citric acid cycle, is an amphoteric pathway, contributing to catabolic degradation and anaplerotic reactions to supply precursors for macromolecule biosynthesis. Oxoglutarate dehydrogenase complex (OGDHc, also called α-ketoglutarate dehydrogenase) a highly regulated enzyme in TCA cycle, converts α-ketoglutarate (αKG) to succinyl-Coenzyme A in accompany with NADH generation for ATP generation through oxidative phosphorylation. The step collaborates with glutaminolysis at an intersectional point to govern αKG levels for energy production, nucleotide and amino acid syntheses, and the resources for macromolecule synthesis in cancer cells with rapid proliferation. Despite being a flavoenzyme susceptible to electron leakage contributing to mitochondrial reactive oxygen species (ROS) production, OGDHc is highly sensitive to peroxides such as HNE (4-hydroxy-2-nonenal) and moreover, its activity mediates the activation of several antioxidant pathways. The characteristics endow OGDHc as a critical redox sensor in mitochondria. Accumulating evidences suggest that dysregulation of OGDHc impairs cellular redox homeostasis and disturbs substrate fluxes, leading to a buildup of oncometabolites along the pathogenesis and development of cancers. In this review, we describe molecular interactions, regulation of OGDHc expression and activity and its relationships with diseases, specifically focusing on cancers. In the end, we discuss the potential of OGDHs as a therapeutic target for cancer treatment.
Keywords: α-ketoglutarate dehydrogenase complex, 2-oxoglutarate dehydrogenase, tricarboxylic acid cycle, cancer metabolism, reactive oxygen species
Introduction
OGDH complex (OGDHc) is a rate-limiting enzyme in the tricarboxylic acid (TCA) cycle, catalyzing α-ketoglutarate (αKG) to yield succinyl-Coenzyme A (succinyl-CoA) and NADH. OGDHc is located in the matrix of mitochondria where it is binds to Complex I of electron transport chain (ETC) at the matrix side of the inner membrane [1,2]. The product, succinyl-CoA, can be used as the substrate for succinyl-CoA synthetase (SCS) within TCA cycle, but it also serves as the major precursor for heme biosynthesis [3]. Interestingly, the downstream enzyme succinate dehydrogenase (SDH, also called succinate-coenzyme Q reductase) in TCA cycle, is also an integral component of Complex II in ETC (Figure 1). These characteristics manifest the critical role of OGDHc in regulating mitochondrial reactive oxygen species (ROS) production and cellular redox homeostasis [4].
Figure 1.
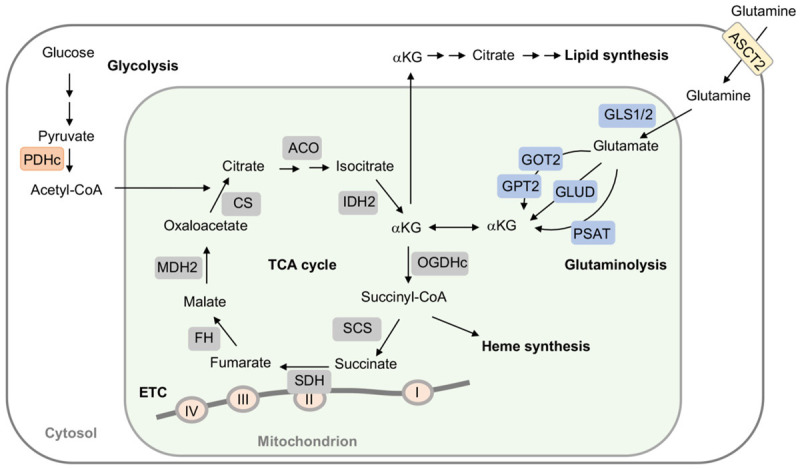
The role of OGDH and αKG in bioenergy synthesis. αKG is generated from glucose-derived citrate, namely glycolysis and glutaminolysis. Glutaminolysis initiates from deamination of glutamine via glutaminases (GLS1/GLS2) to produce glutamate and ammonia. Glutamate is then converted to αKG by glutamate dehydrogenase (GLUD) or through transamination by glutamate-oxaloacetate transaminase (GOT) and glutamate-pyruvate transaminase 2 (GPT2), as well as, phosphoserine transaminase (PSAT). αKG is oxidatively decarboxylated by 2-oxoglutarate dehydrogenase complex (OGDHc) to succinyl-CoA and CO2, followed by succinyl-CoA synthetase (SCS) converting to succinate. Succinyl-CoA is also forwarded to heme synthesis. Succinate is then oxidized to fumarate by succinate dehydrogenase (SDH). SDH is a part of both tricarboxylic acid (TCA) cycle and electron transport chain (ETC), where SDH is a component of Complex II. However, when pyruvate dehydrogenase complex (PDHc) activity is impeded, αKG is converted to citrate for lipid generation. ACO, aconitase; CS, citrate synthase; FH, fumarate hydratase; IDH2, isocitrate dehydrogenase 2; MDH2, malate dehydrogenase 2.
In TCA cycle, αKG, also referred to 2-oxoglutaric acid, is generated from isocitrate by isocitrate dehydrogenases (IDHs) via oxidative decarboxylation, while αKG also can be produced anaplerotically from glutamate through the glutaminolysis pathway to provide substrates for energy production and macromolecule syntheses [5,6]. Normally, αKG is oxidatively decarboxylated to succinyl-CoA and CO2 via OGDHc. Due to the need for fast growth and proliferation, however, glutamine is highly demanded and glutaminolysis process is greatly enhanced to support αKG supply for energy need and macromolecule biosynthesis in cancer cells [7]. Under hypoxia, a decrease of glucose-derived citrate and inhibition of OGDH activity cause αKG buildup, which in turn reverses IDH reaction and results in citrate accumulation and following lipid synthesis [8]. Thus, cancer cells greatly rely on reductive glutamine metabolism for lipid synthesis [9]. In addition, dysregulation of OGDH amplifies reductive carboxylation under hypoxia and defective IDH activity leading to accumulation of αKG and oncometabolites, such as 2-hydroxyglutarates (2-HG), fumarate, malate, enhanced hypoxia inducible factor-1α (HIF-1α) stabilization [10-12], and impaired DNA methylation to remodel intracellular environments optimal for cancer growth [11,13-15]. These results signify hypoxia to drive reductive glutamine metabolism [8]. Accordingly, OGDHc operates at the intersection connecting bioenergy synthesis and anaplerotic pathways in metabolism. Several human diseases such as cancers and neurodegeneration have been linked to dysregulation of OGDHc activity [16,17].
Although the precise mechanisms of OGDHc-regulated redox homeostasis and glutamine metabolism in cancer pathogenesis and progression remains fully understood, emerging data suggest a crucial role of OGDHc in cancer development. Herein, this review highlights the functional features of OGDHc in cancer development in respect to relevant metabolic pathways to delineate its potentials as a target in cancer therapy.
OGDH complex
The OGDH complex (OGDHc) is composed of OGDH (E1, also called α-ketoglutarate dehydrogenase; KGDH), dihydrolipoamide S-succinyltransferase (DLST, E2), and dihydrolipoamide dehydrogenase (DLDH, E3) (Figure 2A). E1 and E2 are unique for OGDHc, but E3 is also a part of pyruvate dehydrogenase complex (PDHc), branched-chain ketoacid dehydrogenase complex (BDKDHc), α-ketoadipate dehydrogenase (KADH), and glycine cleavage enzyme [18]. OGDH-derived succinyl-CoA is converted into succinate followed by oxidation to generate fumarate via SCS and SDH. SDH is also functions as a component of Complex II of ETC (Figure 1). OGDH is encoded by OGDH gene. In human, there are three major splice variants of OGDH gene, variant 1 (OGDH1, 115 kDa, here is referred to OGDH), variant 2 (OGDH2, 48 kDa) and variant 3 (OGDH3, 115 kDa) (Figure 2B). OGDH1 and OGDH3 share 99% identity except at amino acid fragment 153-169, while OGDH2 shares with OGDH1 identical amino acid 1-403 [8]. OGDH2 is also able to associate with E2 DLST and E3 DLDH and serves as an OGDH-similar function in TCA cycle [8]. The function of OGDH3 remains further investigations. Circular RNA circ-OGDH, a reverse splicing of OGDH gene, also has been identified [19,20]. The amino acid residue 320 of OGDH gene is required for OGDH activity [21]. The c.959A>G variant in OGDH gene leads to an amino acid change (p.Asn320Ser), causing a severe loss of OGDH activity. Patients with the mutation exhibited systemic delay in development, elevated serum lactate levels, ataxia, and seizure [21]. The deficiency of E3 subunit DLDH is rare and has been reported as an autosomal recessive genetic disorder [22].
Figure 2.
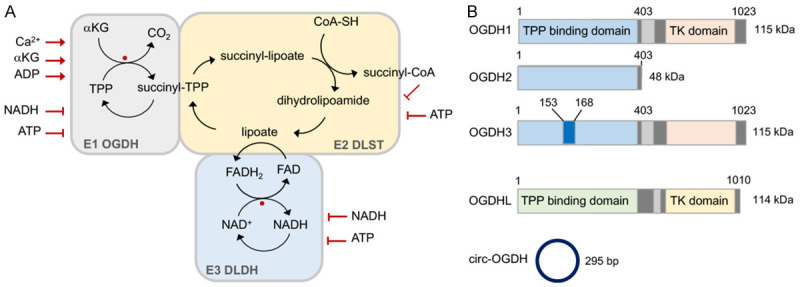
Regulation of OGDH complex. A. OGDH complex comprises three components, 2-oxoglutarate dehydrogenase (E1 OGDH), dihydrolipoamide succinyltransferase (E2 DLST), and dihydrolipoamide dehydrogenase (E3 DLDH), carrying out the coupled reaction to decarboxylate α-ketoglutarate to succinyl-CoA. E1 OGDH subunit is α-ketoglutarate decarboxylase that uses thiamine pyrophosphate (TPP) as a cofactor to decarboxylate α-ketoglutarate (αKG) for CO2 production and succinylation of TPP. The succinyl-moiety is then transferred to lipoamide in the E2 DLST subunit where yields succinyl-CoA and dihydrolipoamide. The dihydrolipoamide is re-oxidized by E3 DLDH subunit. Here, free electrons are transferred from FAD to reduce NAD+ to generate NADH. OGDH complex contains two susceptible sites for ROS generation (marked by red spots). All of the 3 subunits can be S-glutathionylation. Activity of OGDH complex is regulated by TPP, lipoic acid, CoA, FAD, NAD+, succinyl-CoA, and ROS. The susceptible target by ROS is the prosthetic lipoic acid of the E2 DLST subunit. B. The isoforms of OGDH, OGDH-like (OGDHL), and circ-OGDH. There are three major variants of OGDH in human, OGDH1, OGDH2 and OGDH3. All three OGDH isoforms contain TPP binding domain. OGDH2 lacks of transketolase (TK) domain. Circ-OGDH is the product of reverse splice of OGDH gene, 295 bp.
Within the OGDHc, thiamine pyrophosphate (TPP) serves as a cofactor to reduce NAD+ to NADH and catalyzes the reaction between αKG and CoA-SH to generate succinyl-CoA and CO2 along TCA cycle flux. Therefore, the complex activity is regulated by TPP, lipoic acid, CoA, FAD, NAD+, succinyl-CoA, and ROS [17]. The reaction initiates from E1 subunit utilizing the cofactor TPP to decarboxylate αKG to generate a succinyl-TPP intermediate, which in turn proceeds with reductive succinylation on the lipoyl group of the E2 subunit DLST. Then, the E2 subunit serves as a dihydrolipoamide succinyltransferase to transfer the succinyl group to CoA to produce succinyl-CoA and dihydrolipoamide. E3 DLDH subunit thereafter uses FAD to oxidize dihydrolipoamide to produce a disulfide bond for further catalyzing events. During this process, NAD+ is used to oxidize FADH2 back to FAD for NADH generation (Figure 2A). Similar to OGDHc, the catalytic mechanisms of PDHc and BDKDHc also utilize E1 and E2 subunits to specifically decarboxylate their respective substrates while the E3 subunit shares the same catalyzing reaction [18,23]. The lipoylation of OGDH thus controls the partition of carbon flux into TCA cycle [24]. Besides, calcium also acts as a key regulator of OGDH activity, which lowers the apparent Km of OGDH enzyme [25,26]. However, OGDH activity is inhibited under high calcium concentrations [27]. Similarly, the impact by high levels of mitochondrial calcium also occurs in PDH and IDH [25]. Several splice variants of OGDH with a Ca2+-insensitive characteristic have been identified [28], but their functional alterations and following impacts remain further investigations. Moreover, the sensibility of OGDH to Ca2+ relies on the concentration of NADH and ATP/ADP ratio, in a dose dependent manner to attenuate the stimulatory effect by Ca2+ [29].
Interestingly, translocation of TCA intermediates and enzymes, and operation of a TCA cycle subnetwork have been observed within the nucleus [30-32], but the function of OGDH in the nucleus require more intensive studies. Current studies suggest that nuclear OGDH can bind to the promoter regions, where it facilitates succinyl-CoA binding to lysine acetyltransferase 2 (KAT2A) and meanwhile, KAT2A exerts a succinyltransferase activity to succinylate histone H3 leading to epigenetic regulation in tumor cell proliferation [32,33]. In addition, OGDH also regulates the succinylation of histone H4 [34]. Whether DLST and/or DLDH co-exist with OGDH and form a functional complex for histone succinylation and following epigenetic regulation remain further studies.
The role of OGDH in redox homeostasis
OGDHc as a ROS generator
For a long time, the complexes of the respiratory chain are regarded as the major site of ROS production in the mitochondria. However, recently studies have demonstrated that OGDHc is an important source of mitochondrial ROS generation [4,35,36]. E3 DLDH subunit is the main site to generate ROS, whereas E1 OGDH subunit generates fewer ROS [4,37-39]. When NAD+/NADH ratio is low, FADH2 within the E3 DLDH subunit can be oxidized by O2, generating a semiquinone (FADH•) and superoxide (O2 •-) [37-39]. Besides, OGDHc can generate superoxide and hydrogen peroxide (H2O2) in response to high levels of αKG and free CoA [36-38] (Figure 3A).
Figure 3.
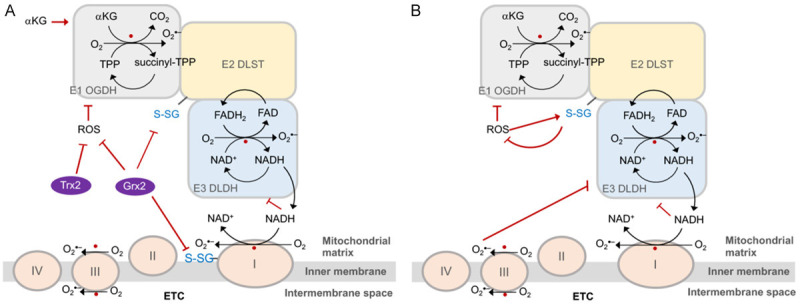
The role of OGDH complex in redox homeostasis. A. OGDH complex (OGDHc) is a potential ROS generator. E1 OGDH and E3 DLDH are two generation sites for ROS production (marked by red spots) within the OGDHc. E3 DLDH-derived NADH promotes ROS generation by Complex I located in electron transport chain (ETC). The glutathionylation (-S-SG) of OGDHc and Complex I enhances ROS generation, which can be suppressed by glutathione reductase 2 (Grx2) and thioredoxin 2 (Trx). OGDHc-generated ROS is also increased by α-ketoglutarate (αKG). B. OGDHc contributes to antioxidant defense. ROS generated from E3 DLDH can inhibit E1 OGDH and itself activities. ROS-enhanced glutathionylation of E2 DLSH protects peroxidative effects by ROS. The capability of ROS generation of E3 DLDH are regulated in response to cellular oxidative status, and thus, E3 DLDH-generated ROS is inhibited when a higher level of Complex III-derived ROS occurs.
The lipoyl moiety of E2 DLST enzyme can be glutathionylated, which inhibits the enzyme activity and protects lipoic acid from modification by the electrophilic lipid peroxidation [4,40,41]. Earlier studies showed that the glutathionylation of OGDHc and Complex I were down-regulated by glutathione reductase 2 (Grx2) [4,44,45]. In vivo studies further showed that Grx2 deficiency increased the glutathionylation of OGDHc and PDHc in accompany with decreased ROS generation from the enzymes [42]. Although, the glutathionylation of E2 DLST subunit can protect E1 subunit from irreversible oxidation, glutathione administration was shown to amplify O2 •-/H2O2 formation from E3 DLDH enzyme and decrease NADH production [4]. In addition, thioredoxin 2 was shown to protect OGDHc from self-inactivation in a low NAD+ status [43]. Moreover, early studies suggested that OGDHc interacts with Complex I of ETC, allowing a direct transfer of NADH to NADH-oxidation site of Complex I [2,46,47] (Figure 3A). Provocation of ROS due to oxidative metabolism of αKG accumulation was also confirmed in fumarate hydratase-deficient cancer cells [48]. The results suggest that OGDHc potentially contributes to mitochondrial electron leakage and following ROS production along its catalytic reactions and the potential greatly relies on the cellular redox status.
OGDHc contributes to antioxidant defense
As early in 1990s, a decline of OGDH activity has been identified in some neurodegenerative diseases such as Alzheimer’s and Huntington diseases [16,49,50]. The deficiency of OGDH function in neurodegenerative diseases is highly associated with oxidative stress, indicating OGDH as a critical regulator in cellular redox homeostasis. In early studies, exposure of cardiac mitochondria to HNE (4-hydroxy-2-nonenal), a fragmentation product of lipid peroxidation, was shown to impair OGDH and PDH activity and suppress the NADH-dependent state III respiration, while other mitochondrial dehydrogenases and complexes of ETC were not affected [51]. The target by HNE attack was attributed to the prosthetic lipoic acid, which covalently binds to the E2 subunit within OGDHc and PDHc [52]. Apparently, OGDH is the most vulnerable target in response to oxidative stress within mitochondria [53]. Furthermore, H2O2 was shown to directly interact with the sulfhydryl groups of OGDH, modulating its glutathionylation status and enzymatic activity [40,54]. During this modification process, lipoic acid requires cofactor covalently to attach to the enzyme and the glutathionylation of OGDH turn out to protect the prosthetic lipoic acid from peroxidation modification [4,40]. The results manifest a regulatory loop of OGDHc in antioxidant defense involving the modification of the prosthetic lipoic acid in response to cellular redox status. In spite, the E3 DLDH subunit of OGDH is a flavoenzyme responsible for the transfer of reducing equivalents from lipoic acid to NAD+ and contributes to most of ROS and H2O2 generation of OGDHc [36-38]. DLDH is vulnerable to peroxides; for example, inactivated by Complex III-derived H2O2 [55] (Figure 3B). Therefore, DLDH can induce or attenuate ROS production, depending on oxidative stress conditions. Aconitase is the most ROS-sensitive enzyme in TCA cycle [56], and its inhibition greatly interrupts the flux from pyruvate to αKG, thereby modulating OGDHc activity that limits NADH production and consequently ROS generation.
Several signaling pathways have been reported to participate in OGDH mediated redox homeostasis including mammalian target of rapamycin complex 1 (mTOR) and sirtuins (Sirt) [57]. In the crosstalk between mTOR and OGDH, a low dose of ROS exposure activates mTOR signaling, whereas high concentrations or long-term ROS treatment decreases mTOR activity [58]. The depletion of thiamine, a cofactor of OGDHc and PDHc, caused metabolic dysregulation and growth arrest in breast cancer and leukemia cells, which could be completely reversed by rapamycin treatment [59]. The results suggest that mTOR is involved in OGDH-related metabolic networks. A decrease of cellular αKG levels was shown to enhance the sensitivity of cancer cells to p53 inhibitor nutlin-3a-induced apoptosis, while the add-back of αKG analog dimethyl-αKG and genetic knockdown of OGDH rescued the cell viability [60]. Nardilysin, a metalloendopeptidase, is required for OGDH activity due to its facilitation on the folding of OGDH protein. A loss of nardilysin or OGDH resulted in αKG accumulation and consequently activated mTOR signaling and suppressed the autophagic process, while rapamycin treatment and a blockade of autophagy significantly ameliorated the neurodegeneration by nardilysin mutation [61]. The results highlight that mTOR serves as a downstream molecule of OGDH and its activity contributes to autophagic pathway. Genetic knockdown of OGDH-like (OGDHL), a variant of OGDH, has been shown to activate mTOR signaling pathway and consequently induce lipogenesis [62]. Sirt-5, was shown to repress cell growth and migration in gastric cancer by inhibiting OGDH activity due to its physical interaction with OGDH to impede the succinylation of OGDH [57].
The role of OGDHc and αKG in glutamine metabolism
Glucose and glutamine are the major nutrients for proliferation of mammalian cells, providing carbon and nitrogen for anabolism and macromolecule biosynthesis [64]. Cancer cells have a high demand of glutamine to meet the need for rapid growth and proliferation. Some cancers cells even require oncogene-dependent supplementations of glutamine in the culture systems [64]. In addition to glucose, glutamine also substantially contributes to citrate supply for fatty acid and glycerolipid synthesis in cancer cells [7,65]. αKG from glutamine metabolism can be integrated into TCA cycle flux either for oxidative decarboxylation forward to ATP generation or through reductive TCA cycle for de novo lipogenesis or metabolite production [5,66]. Accumulation of cellular αKG generally reflects an enhanced OGDHc activity and/or glutamine metabolism, namely glutaminolysis [5,6].
Glutaminolysis is a catabolic process of glutamine to generate TCA cycle metabolites, initiating from deamination of glutamine via glutaminases (GLS1/GLS2), which produces glutamate and ammonia. Glutamate is then converted to αKG via two different pathways. The first pathway is accomplished by glutamate dehydrogenase (GLUD) and the second pathway is mediated through several transaminases, including glutamate-oxaloacetate transaminase (GOT), glutamate-pyruvate transaminase (GPT), and phosphoserine transaminase (PSAT). The reaction by GLUD generates ammonium and NADH or NADPH, whereas the pathway by transaminases promotes the yield of some nonessential amino acids including asparate, alanine, and phosphoserine. Among these reactions, αKG serves as a resource for the anaplerotic reactions in TCA cycle [5,6] (Figure 1). Thus glutamine metabolism provides the substrates for ATP generation through the respiration chain and for the synthesis of nucleotides, amino acids, lipids, macromolecules. In contrast to normal cells, cancer cells display elevated ROS production and moderate ROS levels tend to promote cancer cell proliferation, while excessive ROS might cause oxidative damages [67]. Glutaminolysis also functions in regulating redox homeostasis via the production of glutathione and NADPH to scavenge ROS. Along the TCA flux, αKG can be metabolized to fumarate, which directly interacts with glutathione peroxidase 1 (GPx1) to activate its activity, but also can trigger Nrf2 antioxidant signaling for ROS scavenging [68,69].
The critical role of OGDHc in cancer growth
OGDH in cancer growth
Due to the specific metabolic plasticity, cancer cells exhibit a great reliance on OGDH activity [70]. In gastric cancer patients, positive correlations between OGDH activity and several clinicopathological parameters were observed and the underlying mechanisms were attributed to enhanced mitochondrial function and Wnt/β-catenin signaling [63]. Succinyl phosphonate, an OGDH inhibitor, was shown to significantly affect cell cycle and suppress cell migration and cell viability in AGS and BGC823 gastric cancer cells [57]. Moreover, some cancer cells even displayed a high sensitivity to OGDH depletion [71]. Dysregulation of OGDH activity has been reported to contribute to fumarate accumulation in fumarate dehydratase-deficient cancer cells, which turns out to stabilize HIF-1α from degradation and enhance ROS generation [48]. Accordingly, OGDH-dependent cancer cells tend to exhibit lower asparate and malate-asparate shuttle activity [72]. Mutations in the catalytic subunit of phosphatidylinositol 3-kinase (PI3KCA) have been reported in a variety of cancer types including breast, colorectal endometrial, and lung cancer and the PI3KCA mutant cancer cells showed a great reliance on OGDH activity for their proliferation and growth [71,73]. Downregulation of OGDH by siRNA and αKG supplementation depleted cellular asparate levels, and desensitized malate-asparate shuttle, which in turn impeded cytoplasmic NAD+ generation leading to disrupted NAD+/NADH homeostasis [71] (Figure 4).
Figure 4.
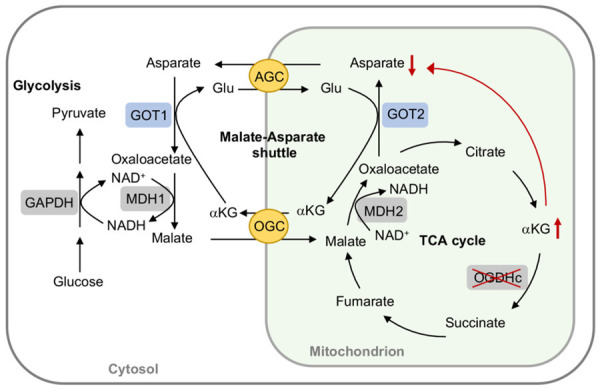
OGDH regulates malate-asparate shuttle activity. In OGDH-dependent or mutated catalytic subunit of phosphatidylinositol 3-kinase cancer cells, a lower OGDH activity leads to α-ketoglutarate (αKG) accumulation, which in turns to ameliorate the asparate and malate-asparate shuttle activity due to the depletion of asparate levels and disruption of NAD+/NADH homeostasis. AGC, asparate/glutamate carrier; GAPDH, glyceraldehyde-3-P-dehydrogenase; Glu, glutamate; GOT1/2, glutamate oxaloacetate transaminase 1/2 (asparate transaminase); MDH1/2, malate dehydrogenase 1/2; OGC, oxoglutarate/malate carrier.
CircRNA circ-OGDH (hsa_circ_0003340), located at chr7: 44684925-44687358, encoding a length of 295 bp, is produced by reverse splicing of OGDH gene. Circ-OGDH has been reported to act as an oncogene in esophageal squamous cell carcinoma [19]. Circ-OGDH affects cancer cell proliferation, migration, and invasion, by operating at glutamine metabolism for αKG and ATP production and by downregulating miR-615-5p to increase PDX1 (pancreatic and duodenal homeobox) expression [20]. In addition, circ-OGDH was shown to decoy miR-564 to promote cell invasion and proliferation by upregulating TPX2 (targeting protein for Xenopus kinesin like protein 2) expression [19]. Circ-OGDH displays a similar function to OGDH by operating glutamine metabolism and fuel utilization to promote cancer cell growth. In addition, circ-OGDH serves an up-regulator of oncogenes via modulating of specific miRNA levels.
OGDHL in cancer growth
OGDHL encoded by OGDHL gene is a variant of OGDH, sharing the similar nucleotide sequences with OGDH. Expression of OGDHL is tissue-specific mainly in the brain and liver [74,75]. Emerging data indicated that OGDHL is a tumor suppressor. Downregulation of OGDHL has been shown to contribute to the onset and progression of several cancers including colorectal, liver, pancreatic cancer, hepatocellular carcinoma, and cervical cancer cells [62,76-80]. The lower OGDHL expression in the cancer cells was attributed to aberrant promoter hypermethylation and DNA copy deletion of OGDHL [62,81], as well as, due to alternative splicing and downregulation by certain microRNA such as upregulated expression of miR-193a-59 in pancreatic cancer [79,80]. miR-193a-5p functions to promote pancreatic cancer cell migration and invasion by targeting serine/arginine-rich splicing factor 6 (SRSF6) for OGDHL alternative splicing and downregulation [80]. Furthermore, OGDHL is also a target gene of miR-214 and negatively regulated by miR-214 [79]. In liver cancer cells, the deficiency of OGDHL upregulates αKG/citrate ratio by inhibiting of OGDHL complex activity, which in turn induces reductive TCA cycle flux and drives reductive carboxylation of glutamine-derived αKG to support lipogenesis and cellular antioxidant system, and thereby influences the chemo-sensitization effect by sorafenib [62]. Furthermore, genetic silencing of OGDHL activated mTOR signaling pathway in an αKG-dependent manner, which turned out to upregulate the expression of several key enzymes involved in lipogenesis in accompany with an increase of NADPH and glutathione levels to assist the cellular antioxidant system [62]. On the other hand, enforced expression of OGDHL increased ROS production and resulted in cell apoptosis through caspase 3-mediated suppression of AKT signaling and inhibition of NF-κB activity [76]. In pancreatic cancer cells, enhanced expression of OGDHL inhibited TWIST1 expression through ubiquitin-mediated proteolysis of HIF-1α and by regulating AKT signaling, which thereafter downregulated miR-214 expression and mitigated cell migration and invasion [79]. Three deleterious variants (p.Val827Met, p.Pro839Leu, p.Phe836Ser) of OGDHL were predicated to associate with breast cancer risk [82]. However, in papillary thyroid cancer, the low-OGDHL patients had a better survival rate [83]. The discrepancy was attributed to different immune-related biological processes in regulating OGDHL expression, suggesting a crosstalk between OGDHL and microenvironments along tumor growth [83].
The aberrant of low-OGDHL expression was also observed in humans and in mouse models of Alzheimer’s disease. A homozygous deleterious variant of OGDHL (c.C2333T; p.Ser778Leu) has been identified in patients with neurodegenerative phenotypes [61]. An increase of OGDHL expression activated Wnt/β-catenin signaling pathway and ameliorated cognitive impairment and relevant pathology in mice [84]. Wnt/β-catenin signaling pathways have been shown with protective effects on neurodegenerative diseases by maintaining the mitochondrial homeostasis [85].
DLST in cancer growth
An aberrant upregulation of DLST has been observed in several types of cancers including leukemia, triple-negative cancer cells, and neuroblastoma [86-88], which was linked to poor overall and recurrence-free survival in human triple-negative cancer patients [88]. Depletion of DLST activity suppressed cancer cell growth by provoking ROS generation, lowering NADPH production, impairing oxidative phosphorylation reaction, as well as, apoptosis induction [86-88]. Several germline variants in DLST were also observed in patients with multiple pheochromocytomas and paragangliomas and the oncogenic mechanisms were shown operating at altered TCA cycle to support cancer development [89]. Similar alterations of TCA cycle intermediated by DLST to mediate MYC-driven leukemogenesis were also observed in human T-cell acute lymphoblastic leukemia [86]. In addition, DLST can activate the pseudohypoxia pathway by interacting with mutated-EPAS1 (endothelial PAS domain-containing protein 1) [87] and promoting epigenetic modification such as DNA methylation [90].
DLDH in cancer growth
In head and neck cancer, glutaminolysis enhances cystine deprivation-induced ferroptosis via increased OGDH activity. The genetic silencing of DLDH attenuated glutaminolysis-mediated ferroptosis, in accordance with relieved lipid peroxidation, ROS, and mitochondrial iron levels [91], implicating the potential role of OGDH in ferroptosis regulation. The inactivation of DLDH by oxidative stress was restored by cysteine and glutathione replenishment [55]. The results suggested that in the genetic OGDH deficiency patients, DLDH subunit activity is defective [92].
Based on these studies, an elevation of OGDH-formed complex activity and increased circ-OGDH levels play a synergistic role in oncogenesis, while OGDHL likely functions as a tumor suppressor. However, details of the mechanisms between OGDH and OGDHL in regulation of tumor metabolism require more studies.
αKG in cancer growth
In a very early study, more glutamine consumption than other amino acids by cancer cells has been reported [93]. Glutamine is the most abundant amino acid in the circulation and glutaminolysis is executed in cancer cells more than glutamine catabolites for the production of macromolecules [68,94]. The levels of αKG reflect the glutaminolysis activity and availability of glutamine. αKG has been shown to ameliorate autophagy by promoting mTOR signaling [61,95] and thereby, when autophagy is inhibited, αKG accumulation from enhanced glutamine metabolism promotes proliferation of cancer cells [96] while insufficient αKG levels may suppress mitochondrial respiration and ATP generation [95]. αKG from glutamine metabolism allows OGDH to sustain NADH-associated respiration [97]. Calcium also functions in the enhancement of activities of respiratory chain components to promote oxidative phosphorylation (OXPHOS) for ATP production [98,99]. Supplementation of αKG could rescue mitosis and viability of cancer cells due to the inhibition of Ca2+ transfer into mitochondria [100]. These results suggest that cancer cells greatly rely on functional TCA cycle for proliferation, in which αKG assists cancer cells to maintain a basal OXPHOS activity and adapt to the metabolic deficiencies by oxidative stress.
αKG and oncometabolites in cancer growth
Oncometabolites refer to the aberrantly accumulated metabolites, which possess pro-oncogenic capabilities to promote tumorigenesis via epigenetic dysregulation, hypoxia status, and switch the phenotype of cancers [15,101]. Oncometabolites are maintained in a limited quantity in normal tissues but persist at a higher level in cancer cells [15]. Somatic mutations of metabolic enzymes in TCA cycle including malate hydrogenase (MDH), fumarate hydratase (FH), and IDH1/IDH2 contribute to the accumulation of fumarate, succinate, L-2-HG and D-2-HG, which are generally recognized as oncometabolites [102]. Both L-2-HG and D-2-HG are derived from αKG and thus share the similar chemical structures and behaviors [102]. In normal cells, IDH converts isocitrate to αKG via a reversible oxidative decarboxylation reaction. D-2-HG is mainly generated from αKG by hydroxyacid-oxoacid transhydrogenase. However, in some cancer cells, mutations of IDH1 or IDH2 lead to the overproduction of D-2-HG [11]. Under hypoxia L-2-HG is produced by MDH or lactate dehydrogenase A (LDHA) through the reductive reaction of αKG [10,12]. Due to the structural similarity, the two molecules behave as competitive inhibitors of αKG-dependent dioxygenases but still retain the enzyme activities, which in turn exert effects on a wide range of biological functions, leading to epigenetic dysregulation and pseudohypoxia status, and pro-oncogenic capability [13-15] (Figure 5). Besides, D-2-HG has been shown with inhibitory effects on OGDH in rat heart [103].
Figure 5.
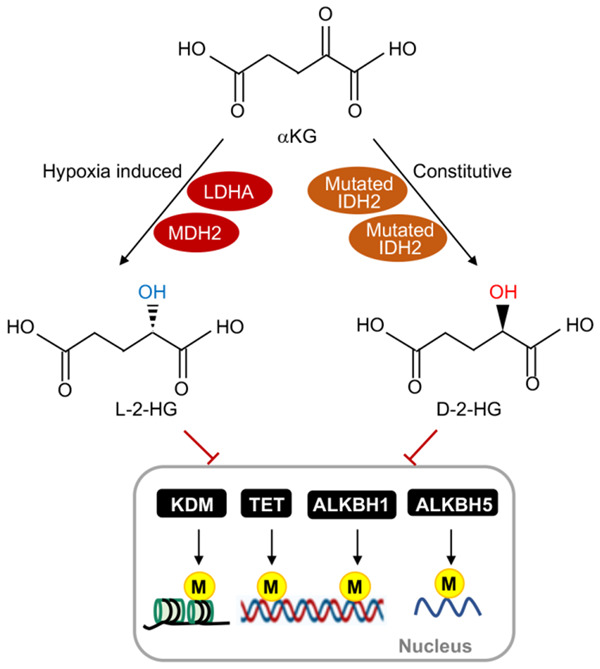
The role of αKG and oncometabolites in epigenetic regulation. In cancer cells, αKG can be converted to L-2-hydroxyglutarate (L-2-HG) through lactate dehydrogenase A (LDHA) and malate dehydrogenase 2 (MDH2) under hypoxia. Mutated IDH1/2 (isocitrate dehydrogenase1 and 2) can constitutively convert αKG to D-2-hydroxyglutarate (D-2-HG). Both L-2-HG and D-2-HG inhibit αKG-dependent dioxygenase activities, including histone lysine demethylases (KDMs), ten-eleven translocation proteins (TETs), and AlkB homologues (ALKBHs), which are involved in the transcriptional suppression of pro-oncogenes by maintaining the hypermethylation status of DNA and histones through epigenetic regulation.
Several mutations in IDH were identified and became a significant feature in acute myeloid leukemia (AML) and gliomas [104,105] and thus, inhibitors of mutated IDHs were developed for targeting therapy. Inhibitors of mutated IDH1 and IDH2, ivosidenib (AG-120) and enasidenib (AG-221), respectively, have been approved by FDA for IDH-mutant relapsed or refractory AML. Ivosidenib is also approved for cholangiocarcinoma and newly-diagnosed AML with a susceptible IDH1 mutation.
The family members of αKG-dependent dioxygenases include ten-eleven translocation proteins (TETs), histone lysine demethylases (KDMs), and AlkB homologues (ALKBHs). Under the inhibition of PDH activity, cellular HIF-1α is stabilized at a high level even when oxygen is abundant, termed pseudohypoxia, which can promote angiogenesis to support cancer growth [106]. Inhibition of TETs and KDMs by oncometabolites results in aberrant DNA and histone hypermethylation, respectively, and moreover affects epigenetic regulation in cancer cells [107,108]. ALKBH serves as an N 6-mA demethylase [109,110]. In the clinical case of SDH-mutated tumors, DNA hypermethylation and related gene silencing were observed, which reflects a result of succinate-mediated inhibition on OGDH activity [111].
Furthermore, glutamine-derived αKG has been shown to suppress M1 macrophage activation through a PHD (prolyl hydroxylase domain)-dependent post-translational downregulation of IKKβ activity, and meanwhile the polarization toward M2 macrophages is activated via engagement of fatty acid oxidation and JMJD3 (Jumonji domain-containing protein D3)-dependent epigenetic reprogramming [111]. Thereby, in this issue, elevated intracellular levels of αKG can attenuate tumor progression, suggesting that manipulations to raise cellular αKG levels may serve as a feasible strategy.
The crosstalk between OGDH and HIF-1α
An increase of cellular αKG levels can revise oncometabolite-mediated HIF-1α stabilization, in which αKG neutralizes succinate, fumarate and hypoxia-mediated activation of HIF-1α, leading to enhanced glycolysis and cell death [112,113]. Under hypoxia and ETC defects, citrate is generated from glutamine-derived αKG through reductive carboxylation catalyzed by IDH1 and IDH2 in the cytosol and mitochondria, respectively [9,114,115]. In OGDH2, a splice variant of the E1 subunit of the OGDH complex, HIF-1α greatly enhanced reductive carboxylation of αKG into citrate and the enhancement was attributed to SIAH2 E3 ligase to target OGDH2 for proteasomal degradation, which switched cancer cells to require exogenous citrate or lipids for proliferation, and thereby impeded tumor growth [8]. Suppression of TCA cycle flux by HIF-1α also can be achieved by inactivating pyruvate dehydrogenase kinase 1 (PDK1) and decreased citrate levels [106,116]. Therefore, HIF-1α intents to rewrite the metabolism by reversing TCA cycle flux from oxidation to reductive carboxylation. Inhibition of OGDH complex activity can promote reductive metabolism and induce αKG accumulation leading to reductive anabolism and L-2-HG generation, which in turn inhibits PHD activity and thus increases HIF-1α stability [24]. However, the oxidative or reductive carboxylation of αKG is reciprocally correlated depending on the citrate/αKG ratio to decide glutamine utilization. In normal conditions, glucose-derived citrate is the main source for fatty acid synthesis, while citrate shortage due to hypoxia and defects in ETC respiration can strength the reductive carboxylation of glutamine-derived αKG [66]. Enhanced reductive glutamine metabolism was observed in the cell growth of anchorage-independent tumor spheroids, which depends on the activity of cytosolic IDH1 to convert cytosolic αKG into isocitrate. Either isocitrate or isocitrate-metabolized citrate can be transported into mitochondria where they undergo oxidative carboxylation to synthesize NADPH and αKG, by which mitochondrial ROS toxicity raised by the detachment from extracellular matrix is neutralized, allowing the maximized growth of tumor spheroids [117]. Accordingly, OGDH activity is linked to HIF-α stabilization in regulating tumor growth.
The roles of OGDHc, OGDHL, and circ-OGDH in cancer cell growth are summarized in Table 1.
Table 1.
The roles of OGDHc, OGDHL, and circ-OGDH in cancer cell growth
| OGDH | Cancer growth | Mechanisms | References |
|---|---|---|---|
| OGDH | Oncogenesis (gastric cancer) | Sirt-5 inhibits cell growth and migration by inhibiting OGDH activity and hindering the succinylation of OGDH. | [57] |
| OGDH | Oncogenesis (gastric cancer) | OGDH enhances mitochondrial function and Wnt/β-catenin signaling | [63] |
| OGDH | Oncogenesis (OGDH-dependent, PI3KCA cancer cells) | OGDH suppression downregulates malate-asparate shuttle activity in OGDH-dependent and PI3KCA cancer cells. | [71,72] |
| OGDH | Tumor suppressor (Hela cells) | Inhibition of OGDHc activity promotes L-2-HG, which inhibits PHDs and TETs activities. | [24] |
| OGDH2 | Tumor suppressor (head and neck cancer cells, renal clear cell cancer cells) | Under hypoxia, OGDH2 is downregulated by HIF-1α-SIAH2. Resistant to degradation of mutated OGDH to inhibit tumor growth. | [8] |
| OGDHL | Tumor suppressor (pancreatic cancer cells) | Increased OGDHL inhibits TWIST1 and miR-214 via downregulation of HIF-1α, thereby impedes metastasis. | [79] |
| OGDHL | Tumor suppressor (pancreatic cancer cells) | MiR-193a-5p activates migration and invasion by downregulating SRSF6-OGDHL. | [80] |
| OGDHL | Tumor suppressor (hepatocellular carcinoma) | OGDHL suppression activates mTOR signaling, promotes lipogenesis and antioxidant capability to impair the chemo-sensitization to sorafenib. | [62] |
| OGDHL | Tumor suppressor (cervical cancer cells) | Increased OGDHL enhances caspase-3 and inhibits NF-κB. | [76] |
| Circ-OGDH | Oncogenesis (esophageal cancer cells) | Knockdown of circ-OGDH induces apoptosis and inhibits invasion via miR-564 downregulation- increased TPX2. | [19] |
| Circ-OGDH | Oncogenesis (esophageal cancer cells) | Knockdown of circ-OGDH induces apoptosis and inhibits invasion via miR-615-5p downregulation- increased PDX1. | [20] |
| DLST | Oncogenesis (T-cell acute lymphoblastic leukemia) | DLST mediates MYC-driven leukemogenesis. | [86] |
| DLST | Oncogenesis (neuroblastoma, pheochromocytoma-paraganglioma) | DLST activates pseudohypoxia pathway by interacting with mutated-EPAS1 and promoting DNA methylation. | [87,90] |
| DLST | Oncogenesis (pheochromocytoma-paraganglioma) | DLST alters TCA cycle to support cancer growth. | [89] |
Specific targeting on OGDH for cancer therapy (Figure 6)
Figure 6.
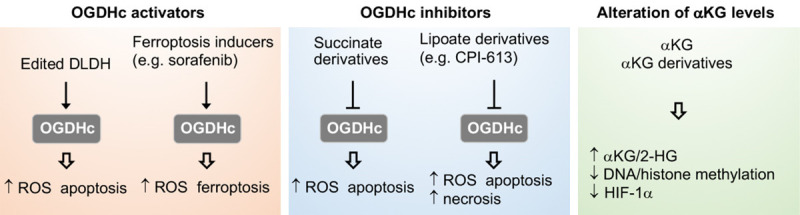
OGDHs serves as a target for cancer treatment. Engineered DLDH or ferroptosis inducers such as sorafenib acting on xCT can activate OGDH to promote ROS production and cancer cell death. Derivatives of succinate and lipoate induce cancer cell death via OGDH inhibition-mediated ROS provocation. Supplementation with exogenous αKG or αKG derivatives attenuates cancer cell growth by elevating αKG/2-hydroxyglutarate (2-HG) ratio followed by inhibiting DNA/histone methylation and/or HIF-1α stability.
DLDH as therapeutic target of cancer
DLDH is a component of several complex including OGDHc, PDHc, and BCKDHc [4,18]. With the oxidoreductive activity, DLDH is susceptible to peroxide and ROS attack but also capable of generating ROS, and thus serve as a potential anticancer target. Cancer cells often highly overexpress integrins, while DLDH can be bioengineered with Arg-Gly-Asp (RGD) motifs (DLDHRGD), allowing to interact with integrin-rich cancer cells being internalized through integrin-associated endocytosis into cells with intact enzyme activity and ROS-production capability. Treatment of DLDHRGD provoked ROS production, induced cell apoptosis, and suppressed tumor growth [118]. Due to the capability of moonlighting functions, DLDH is able to adhere to multiple materials and molecules such as metal-oxides and dsDNA [119]. Treatment combining DLDH and titanium dioxide (TiO2) was shown to elevate the selective destruction of tumor with a minimal harm to nearby normal cells [119,120]. Whether the interaction with DLDH affects the stability of DNA and genomic function remains further investigations. However, the moonlight activity of DLDH may act on DNA alkylation for cancer treatment [120]. Due to the vulnerability of DLDH, another anticancer approach utilizing UVA to suppress DLDH expression showed ROS provocation and decreased mitochondrial membrane potential, leading to autophagic cell death and mitigated tumor growth in melanoma cells [121]. In head and neck cancer, genetic silencing of DLDH blocked cystine deprivation-induced ferroptosis [91], suggesting that DLDH mediates the cystine-regulated ferroptosis. Enhancing DLDH activity thus may serve as a tool to potentiate the anticancer effects by ferroptosis-inducing agents.
Lipoate is an important cofactor for OGDHc activity by targeting to DLDH. CPI-613 (devimistat) is a lipoate analog capable to induce E3 DLDH activity for ROS generation, leading to E2 subunit inactivation, thus impeding OGDHc activity and TCA cycle flux [122]. CPI-613 has been shown to decrease cell viability and proliferation in various types of cancers including pancreatic, breast, colon, ovarian, lung cancer and clear cell sarcoma [123-127]. Although, CPI-613 primarily targets to DLDH, other anticancer mechanisms were also discovered. For example, CPI-613 enhances pancreatic apoptosis through AMPK-ACC signaling to rewires lipid metabolism [126]. In clear cell sarcoma, CPI-613 induces autophagosome formation followed by lysosome fusion, leading to cell necrosis [124]. CPI-613 is currently evaluated in clinical studies as a single agent or in combination with standard drug therapies for patients with pancreatic cancer (NCT03699319), T-cell non-Hodgkin lymphoma (NCT04217317), clear cell sarcoma (NCT04593758), Burkitt’s lymphoma (NCT03793140), and biliary trace cancer (NCT04203160).
Derivatives of succinate
Succinyl phosphonate and glutaryl phosphonate analogs of succinate have been used to blunt OGDH activity and tested for their anticancer capability in laboratory studies [57,70,107,128,129]. Another OGDH inhibitor, a derivative of succinate (S)-2-[(2,6-dichlorobenzoyl) amino] succinic acid (AA6), is also able to inhibit tumor growth by raising cellular αKG levels in an orthotopic mouse model of breast cancer 4T1 and human breast cancer cells [130].
Alterations of intracellular αKG levels
αKG is involved in a wide range of cellular processes in both physiological and pathological conditions. In cancer cells, αKG protects cells from oxidative insults, modulates protein and lipid synthesis, and participates in the β-oxidation of fatty acids. αKG can serve as a substrate of αKG-dependent dioxygenases to manipulate epigenetic modifications. Besides, αKG suppresses HIF-1α stability [24]. Due to the critical role of αKG in cancer cell metabolism and transcription programming, its metabolites as anticancer agents to modulate oncogenic process have been widely evaluated. An increase of αKG has been reported to reverse the inhibition of 2-HG on histone demethylase [15]. Accumulation of αKG by OGDH inhibition promoted TET1, TET3 protein expression, and enzyme activity in breast cancer, and impeded cell migration and epithelial-mesenchymal transition [130]. Furthermore, defective Complex I was shown to increase cellular αKG levels to destabilize HIF-1α under hypoxia, leading to suppress tumor growth in both in vitro and in vivo studies [131,132]. Under hypoxia conditions, treatment of exogenous αKG inhibited HIF-1α stability in a dose-dependent manner [133]. Exogenous αKG also suppressed tumor growth and ameliorated metastasis of breast cancer cells by switching metabolism from glycolytic and oxidative status [134]. Based on these findings, exogenous supplementation of αKG to raise its intracellular levels is proposed as a possible anticancer strategy. Due to low cell permeability, ester derivatives of αKG were developed to increase the permeability and availability within cells [112,113]. Treatment with these derivatives reversed pseudohypoxia induced by the loss of SDH or fumarate hydratase and prevented hypoxia-mediated invasion by reactivating PDH activity to impair HIF-1α stabilization [112,113]. Furthermore, supplementation of these derivatives in cancer cells significantly modulated HIF-1α-mediated effects such as angiogenesis and metabolic alterations, and more importantly induced cancer cell apoptosis [112,113,133,135,136]. Treatment with αKG was further shown to induce histone demethylation via JMJD-mediated senescence [137]. The supplementation of dimethyl-αKG could kill cancer cells when combined with anticancer agent or inhibitors of OXPHOS [138]. Collectively, αKG indeed exerts a positive anticancer effect on different cancer types. Since αKG is a native metabolic intermediate with a low toxicity to normal cells, αKG apparently serves as a potential candidate in pharmacological development for cancer treatment. However, more extensive investigations are required to evaluate the efficacy of αKG for clinical therapy. It has been demonstrated that αKG becomes a culprit to activate the αKG-dependent dioxygenase family enzymes and drive oncogenic transformation via hypoxia adapting and shaping of the epigenetic landscape [139].
Conclusions
In this review, we highlight OGDH as a key role to potentiate cancer cells against crucial alterations of the environments. With ROS production and antioxidative function, OGDH activity governs OXPHOS oscillations, manipulates the mitochondrial redox status through ROS and NADH generation, and guides the metabolite flux in TCA cycle towards energetic or anabolic process, as well as the corresponding signaling pathways. The consequent signaling pathways by altered OGDH activity may be amplified and extended outside of mitochondrion, which drives the systemic remodeling of cells. Next to glucose, glutamine works as a major energy source for cancer cells. Cancer cells rely on high levels of exogenous glutamine due to the catabolism into αKG as a fuel to replenish TCA cycle. αKG operates at the intersection linking TCA cycle and glutamine metabolism, coordinating the energy production, redox regulation, and macromolecule synthesis. αKG also functions as the substrate or co-factor to regulate biological processes. Dysfunctional αKG metabolism by hypoxia and mutated IDH enzymes promotes the generation of oncometabolites, which in turn can change epigenetic landscape and shape cell metabolism, allowing the adaption of cancer cells. In line with this, OGDH and αKG serve a hub in cancer growth and proliferation. Small molecules specifically targeting OGHD have been demonstrated with pronounced effects and efficacy against cancers in animal models and clinical trials. The key cancer metabolites have been proved as potential targets for anti-tumor therapies. However, since cancer cells may undergo a variety of regulatory loops to compensate the disturbed pathways and drought of metabolites, most single target therapies tend to fail. Therapies in combination with multiple targets, and more comprehensive studies are needed to improve the therapeutic effects.
Acknowledgements
This work was supported by the Ministry of Science and Technology, Taiwan (MOST 109-2320-B-039-016, MOST 110-2320-B-039-013-MY3) and China Medical University (CMU110-MF-03) to L.-C. Chang, MOST 110-2639-B-039-001-ASP to M.-C. Hung and by the grants to S.-E. Chen, including MOST 110-2313-B-005-050 from the Ministry of Science and Technology, Taiwan, the iEGG and Animal Biotechnology Center and the Innovation and Development Center of Sustainable Agriculture from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Disclosure of conflict of interest
None.
Abbreviations
- 2-HG
2-hydroxyglutarate
- ALKBH
AlkB homologue
- αKG
α-ketoglutarate
- BDKDHc
branched-chain ketoacid dehydrogenase complex
- CoA
Coenzyme A
- DLDH
dihydrolipoamide dehydrogenase
- D-2-HG
D-2-hydroxyglutarate
- DLST
dihydrolipoamine succinyltransferase
- ETC
electron transport chain
- FH
fumarate hydratase
- GLS
glutaminase
- GLUD
glutamate dehydrogenase
- GOT
glutamate-oxaloacetate transaminase
- GPT
glutamate-pyruvate transaminase
- Grx2
glutathione reductase 2
- GS
glutamine synthetase
- HIF-1α
hypoxia inducible factor-1α
- HNE
4-hydroxy-2-nonenal
- IDH
isocitrate dehydrogenase
- KAT2A
lysine acetyltransferase 2
- KDM
lysine demethylases
- KGDH
α-ketoglutarate dehydrogenase
- L-2-HG
L-2-hydroxyglutarate
- LDHA
lactate dehydrogenase A
- mTOR
mammalian target of rapamycin 1
- OGDHc
oxoglutarate dehydrogenase complex
- OGDHL
OGDH-like
- OXPHOS
oxidative phosphorylation
- PDHc
pyruvate dehydrogenase complex
- PHDs
prolyl hydroxylases
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- SCS
succinyl-CoA synthetase
- TCA
tricarboxylic acid
- TET
ten-eleven translocation hydroxylase
- TPP
thiamine pyrophosphate
References
- 1.Maas E, Bisswanger H. Localization of the alpha-oxoacid dehydrogenase multi-enzyme complexes within the mitochondrion. FEBS Lett. 1990;277:189–190. doi: 10.1016/0014-5793(90)80840-f. [DOI] [PubMed] [Google Scholar]
- 2.Sumegi B, Srere PA. Complex I binds several mitochondrial NAD-coupled dehydrogenases. J Biol Chem. 1984;259:15040–15045. [PubMed] [Google Scholar]
- 3.Burch JS, Marcero JR, Maschek JA, Cox JE, Jackson LK, Medlock AE, Phillips JD, Dailey HA Jr. Glutamine via α-ketoglutarate dehydrogenase provides succinyl-CoA for heme synthesis during erythropoiesis. Blood. 2018;132:987–998. doi: 10.1182/blood-2018-01-829036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mailloux RJ, Craig Ayre D, Christian SL. Induction of mitochondrial reactive oxygen species production by GSH mediated S-glutathionylation of 2-oxoglutarate dehydrogenase. Redox Biology. 2016;8:285–297. doi: 10.1016/j.redox.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L, Venneti S, Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng. 2017;19:163–194. doi: 10.1146/annurev-bioeng-071516-044546. [DOI] [PubMed] [Google Scholar]
- 6.Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Exp Mol Med. 2020;52:1496–1516. doi: 10.1038/s12276-020-00504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun RC, Denko NC. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014;19:285–292. doi: 10.1016/j.cmet.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rzem R, Vincent MF, Van Schaftingen E, Veiga-da-Cunha M. L-2-hydroxyglutaric aciduria, a defect of metabolite repair. J Inherit Metab Dis. 2007;30:681–689. doi: 10.1007/s10545-007-0487-0. [DOI] [PubMed] [Google Scholar]
- 11.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, Rustenburg AS, Grinaway PB, Chodera JD, Cross JR, Thompson CB. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 2015;22:304–311. doi: 10.1016/j.cmet.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr Opin Struct Biol. 2010;20:659–672. doi: 10.1016/j.sbi.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 14.Rose NR, McDonough MA, King ON, Kawamura A, Schofield CJ. Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc Rev. 2011;40:4364–4397. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- 15.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H, Denton TT, Xu H, Calingasan N, Beal MF, Gibson GE. Reductions in the mitochondrial enzyme α-ketoglutarate dehydrogenase complex in neurodegenerative disease-beneficial or detrimental? J Neurochem. 2016;139:823–838. doi: 10.1111/jnc.13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vatrinet R, Leone G, De Luise M, Girolimetti G, Vidone M, Gasparre G, Porcelli AM. The α-ketoglutarate dehydrogenase complex in cancer metabolic plasticity. Cancer Metab. 2017;5:3. doi: 10.1186/s40170-017-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solmonson A, Deberardinis RJ. Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem. 2018;293:7522–7530. doi: 10.1074/jbc.TM117.000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hou Y, Liu H, Pan W. Knockdown of circ_0003340 induces cell apoptosis, inhibits invasion and proliferation through miR-564/TPX2 in esophageal cancer cells. Exp Cell Res. 2020;394:112142. doi: 10.1016/j.yexcr.2020.112142. [DOI] [PubMed] [Google Scholar]
- 20.Liang Z, Zhao B, Hou J, Zheng J, Xin G. CircRNA circ-OGDH (hsa_circ_0003340) acts as a ceRNA to regulate glutamine metabolism and esophageal squamous cell carcinoma progression by the miR-615-5p/PDX1 axis. Cancer Manag Res. 2021;13:3041–3053. doi: 10.2147/CMAR.S290088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yap ZY, Strucinska K, Matsuzaki S, Lee S, Si Y, Humphries K, Tarnopolsky MA, Yoon WH. A biallelic pathogenic variant in the OGDH gene results in a neurological disorder with features of a mitochondrial disease. J Inherit Metab Dis. 2021;44:388–400. doi: 10.1002/jimd.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quinonez SC, Thoene JG. In GeneReviews® (Internet) Seattle, WA, USA: University of Washington; 2014. Dihydrolipoamide dehydrogenase deficiency. Available online: https://www.ncbi.nlm.nih.gov/books/NBK220444/ (accessed on 3 April 2021) [Google Scholar]
- 23.Cronan JE. Assembly of lipoic acid on its cognate enzymes: an extraordinary and essential biosynthetic pathway. Microbiol Mol Bio Rev. 2016;80:429–450. doi: 10.1128/MMBR.00073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burr SP, Costa AS, Grice GL, Timms RT, Lobb IT, Freisinger P, Dodd RB, Dougan G, Lehner PJ, Frezza C, Nathan JA. Mitochondrial protein lipoylation and the 2-oxoglutarate dehydrogenase complex controls HIF1α stability in aerobic conditions. Cell Metab. 2016;24:740–752. doi: 10.1016/j.cmet.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J. 1979;180:533–544. doi: 10.1042/bj1800533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dneton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787:1309–1316. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Lai JC, Cooper AJ. Brain alpha-ketoglutarate dehydrogenase complex: kinetic properties, regional distribution, and effects of inhibitors. J Neurochem. 1986;147:1376–1386. doi: 10.1111/j.1471-4159.1986.tb00768.x. [DOI] [PubMed] [Google Scholar]
- 28.Denton RM, Pullen TJ, Armstrong CT, Heesom KJ, Rutter GA. Calcium-insensitive splice variants of mammalian E1 subunit of 2-oxoglutarate dehydrogenase complex with tissue-specific patterns of expression. Biochem J. 2016;473:1165–1178. doi: 10.1042/BCJ20160135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Armstrong CT, Anderson JL, Denton RM. Studies on the regulation of the human E1 subunit of the 2-oxoglutarate dehydrogenase complex, including the identification of a novel calcium-binding site. Biochem J. 2014;459:369–381. doi: 10.1042/BJ20131664. [DOI] [PubMed] [Google Scholar]
- 30.Nagaraj R, Sharpley MS, Chi F, Braas D, Zhou Y, Kim R, Clark AT, Banerjee U. Nuclear localization of mitochondrial TCA cycle enzymes as a critical step in mammalian zygotic genome activation. Cell. 2017;168:210–223. doi: 10.1016/j.cell.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kafkia E, Andres-Pons A, Ganter K, Seiler M, Jouhten P, Pereira F, Zaugg JB, Lancrin C, Beck M, Patil KR. Operation of a TCA cycle subnetwork in the mammalian nucleus. bioRxiv. 2020. https://doi.org/10.1101/2020.11.22.393413. [DOI] [PMC free article] [PubMed]
- 32.Liu X, Si W, He L, Yang J, Peng Y, Ren J, Liu X, Jin T, Yu H, Zhang Z, Cheng X, Zhang W, Xia L, Huang Y, Wang Y, Liu S, Shan L, Zhang Y, Yang X, Li H, Liang J, Sun L, Shang Y. The existence of a nonclassical TCA cycle in the nucleus that wires the metabolic-epigenetic circuitry. Signal Transduct Target Ther. 2021;6:375. doi: 10.1038/s41392-021-00774-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia Y, Tan L, Yang P, Lee JH, Li XJ, Hawke D, Zheng Y, Qian X, Lyu J, He J, Xing D, Tao YJ, Lu Z. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature. 2017;552:273–277. doi: 10.1038/nature25003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi S, Pfleger J, Jeon YH, Yang Z, He M, Shin H, Sayed D, Astrof S, Abdellatif M. Oxoglutarate dehydrogenase and acetyl-CoA acyltransferase 2 selectively associate with H2A. Z-occupied promoters and are required for histone modifications. Biochim Biophys Acta Gene Regul Mech. 2019;1862:194436. doi: 10.1016/j.bbagrm.2019.194436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andreyev AY, Kushnareva YE, Murphy AN, Starkov AA. Mitochondrial ROS metabolism: 10 Years later. Biochemistry (Mosc) 2015;80:517–531. doi: 10.1134/S0006297915050028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J Biol Chem. 2014;289:8312–8325. doi: 10.1074/jbc.M113.545301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ambrus A, Nemeria NS, Torocsik B, Tretter L, Nilsson M, Jordan F, Adam-Vizi V. Formation of reactive oxygen species by human and bacterial pyruvate and 2-oxoglutarate dehydrogenase multienzyme complexes reconstituted from recombinant components. Free Radic Biol Med. 2015;89:642–650. doi: 10.1016/j.freeradbiomed.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Applegate MA, Humphries KM, Szweda LI. Reversible inhibition of alpha-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry (Mosc) 2008;47:473–478. doi: 10.1021/bi7017464. [DOI] [PubMed] [Google Scholar]
- 41.McLain AL, Cormier PJ, Kinter M, Szweda LI. Glutathionylation of α-ketoglutarate dehydrogenase: the chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic Biol Med. 2013;61:161–169. doi: 10.1016/j.freeradbiomed.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chalker J, Gardiner D, Kuksal N, Mailloux R. Characterization of the impact of glutaredoxin-2 (GRX2) deficiency on superoxide/hydrogen peroxide release from cardiac and liver mitochondria. Redox Biol. 2018;15:216–227. doi: 10.1016/j.redox.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bunik VI. 2-oxo acid dehydrogenase complexes in redox regulation. Eur J Biochem. 2003;270:1036–1042. doi: 10.1046/j.1432-1033.2003.03470.x. [DOI] [PubMed] [Google Scholar]
- 44.Beer SM, Taylor ER, Brown SE, Dahm CC, Costa NJ, Runswick MJ, Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins. J Biol Chem. 2004;279:47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 45.Wu H, Xing K, Lou MF. Glutaredoxin 2 prevents H2O2-induced cell apoptosis by protecting complex I activity in the mitochondria. Biochim Biophys Acta. 2010;1797:1705–1715. doi: 10.1016/j.bbabio.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukushima T, Decker RV, Anderson WM, Spivey HO. Substrate channeling of NADH and binding of dehydrogenases to complex I. J Biol Chem. 1989;264:16483–16488. [PubMed] [Google Scholar]
- 47.Porpaczy Z, Sumegi B, Alkonyi I. Interaction between NAD-dependent isocitrate dehydrogenase, α-ketoglutarate dehydrogenase complex, and NADH: ubiquinone oxidoreductase. J Biol Chem. 1987;262:9509–9514. [PubMed] [Google Scholar]
- 48.Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, Licht JD, Deberardinis RJ, Chandel NS. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51:236–248. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, Perrino P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch Neurol. 1988;45:836–840. doi: 10.1001/archneur.1988.00520320022009. [DOI] [PubMed] [Google Scholar]
- 50.Butterworth RF, Besnard AM. Thiamine-dependent enzyme changes in temporal cortex of patients with Alzheimer’s disease. Metab Brain Dis. 1990;5:179–184. doi: 10.1007/BF00997071. [DOI] [PubMed] [Google Scholar]
- 51.Humphries KM, Yoo Y, Szweda LI. Inhibition of NADH-linked mitochondrial respiration by 4-hydroxy-2-nonenal. Biochemistry. 1998;37:552–557. doi: 10.1021/bi971958i. [DOI] [PubMed] [Google Scholar]
- 52.Humphries KM, Szweda LI. Selective inactivation of alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase: reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry. 1998;37:835–841. doi: 10.1021/bi981512h. [DOI] [PubMed] [Google Scholar]
- 53.Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360:2335–2345. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nulton-Persson AC, Starke DW, Mieyal JJ, Szweda LI. Reversible inactivation of alpha-ketoglutarate dehydrogenase in response to alterations in the mitochondrial glutathione status. Biochemistry (Mosc) 2003;42:4235–4242. doi: 10.1021/bi027370f. [DOI] [PubMed] [Google Scholar]
- 55.Yan LJ, Sumien N, Thangthaeng N, Forster MJ. Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic Res. 2013;47:123–133. doi: 10.3109/10715762.2012.752078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem. 2001;276:23357–23361. doi: 10.1074/jbc.M100320200. [DOI] [PubMed] [Google Scholar]
- 57.Lu X, Yang P, Zhao X, Jiang M, Hu S, Ouyang Y, Zeng L, Wu J. OGDH mediates the inhibition of SIRT5 on cell proliferation and migration of gastric cancer. Exp Cell Res. 2019;382:111483. doi: 10.1016/j.yexcr.2019.06.028. [DOI] [PubMed] [Google Scholar]
- 58.Li M, Zhao L, Liu J, Liu A, Jia C, Ma D, Jiang Y, Bai X. Multi-mechanisms are involved in reactive oxygen species regulation of mTORC1 signaling. Cell Signal. 2010;22:1469–1476. doi: 10.1016/j.cellsig.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 59.Liu S, Miriyala S, Keaton MA, Jordan CT, Wiedl C, Clair DK, Moscow JA. Metabolic effects of acute thiamine depletion are reversed by rapamycin in breast and leukemia cells. PLoS One. 2014;9:e85702. doi: 10.1371/journal.pone.0085702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duan L, Perez RE, Maki CG. Alpha ketoglutarate levels, regulated by p53 and OGDH, determine autophagy and cell fate/apoptosis in response to Nutlin-3a. Cacer Biol Ther. 2019;20:252–260. doi: 10.1080/15384047.2018.1523858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoon WH, Sandoval H, Nagarkar-Jaiswal S, Jaiswal M, Yamamoto S, Haelterman NA, Putluri N, Putluri V, Sreekumar A, Tos T, Aksoy A, Donti T, Graham BH, Ohno M, Nishi E, Hunter J, Muzny DM, Carmichael J, Shen J, Arboleda VA, Nelson SF, Wangler MF, Karaca E, Lupski JR, Bellen HJ. Loss of nardilysin, a mitochondrial co-chaperone for α-ketoglutarate dehydrogenase, promotes mTORC1 activation and neurodegeneration. Neuron. 2017;93:115–131. doi: 10.1016/j.neuron.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dai W, Xu L, Yu X, Zhang G, Guo H, Liu H, Song G, Weng S, Dong L, Zhu J, Liu T, Guo C, Shen X. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J Hepatol. 2020;72:909–923. doi: 10.1016/j.jhep.2019.12.015. [DOI] [PubMed] [Google Scholar]
- 63.Lu X, Wu N, Yang W, Sun J, Yan K, Wu J. OGDH promotes the progression of gastric cancer by regulating mitochondrial bioenergetics and Wnt/β-catenin signal pathway. Onco Targets Ther. 2019;12:7489–7500. doi: 10.2147/OTT.S208848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportiunities. J Clin Invest. 2013;123:3678–3684. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 66.Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, Vokes NI, Guarente L, Vander Heiden MG, Stephanopoulos G. Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat Commun. 2013;4:2236. doi: 10.1038/ncomms3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21:363–383. doi: 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- 68.Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z, Boggon TJ, Jin P, Yi H, Wright ER, Duong D, Seyfried NT, Egnatchik R, DeBerardinis RJ, Magliocca KR, He C, Arellano ML, Khoury HJ, Shin DM, Khuri FR, Kang S. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell. 2015;27:257–270. doi: 10.1016/j.ccell.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jin L, Alesi GN, Kang S. Glutaminolysis as a target for cancer therapy. Oncogene. 2016;35:3619–3625. doi: 10.1038/onc.2015.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bunik VI, Mkrtchyan G, Grabarska A, Oppermann H, Daloso D, Araujo WL, Juszczak M, Rzeski W, Bettendorff L, Fernie AR, Meixensberger J, Stepulak A, Gaunitz F. Inhibition of mitochondrial 2-oxoglutarate dehydrogenase impairs viability of cancer cells in a cell-specific metabolism-dependent manner. Oncotarget. 2016;7:26400–26421. doi: 10.18632/oncotarget.8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ilic N, Birsoy K, Aguirre AJ, Kory N, Pacold ME, Singh S, Moody SE, DeAngelo JD, Spardy NA, Freinkman E, Weir BA, Tsherniak A, Cowley GS, Root DE, Asara JM, Vazquez F, Widlund HR, Sabatini DM, Hahn WC. PIK3CA mutant tumors depend on oxoglutarate dehydrogenase. Proc Natl Acad Sci USA. 2017;114:E3434–E3443. doi: 10.1073/pnas.1617922114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Allen EL, Ulanet DB, Pirman D, Mahoney CE, Coco J, Si Y, Chen Y, Huang L, Ren J, Choe S, Clasquin MF, Artin E, Fan ZP, Cianchetta G, Murtie J, Dorsch M, Jin S, Smolen GA. Differential aspartate usage identifies a subset of cancer cells particularly dependent on OGDH. Cell Rep. 2016;17:876–890. doi: 10.1016/j.celrep.2016.09.052. [DOI] [PubMed] [Google Scholar]
- 73.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 74.Bunik VI, Degtyarev D. Structure-function relationships in the 2-oxo acid dehydrogenase family: Substrate-specific signatures and functional predictions for the 2-oxoglutarate dehydrogenase-like proteins. Proteins Struct Funct Bioinform. 2008;71:874–890. doi: 10.1002/prot.21766. [DOI] [PubMed] [Google Scholar]
- 75.Bunik V, Kaehne T, Degtyarev D, Shcherbakova T, Reiser G. Novel isoenzyme of 2-oxoglutarate dehydrogenase is identified in brain, but not in heart. FEBS J. 2008;275:4990–5006. doi: 10.1111/j.1742-4658.2008.06632.x. [DOI] [PubMed] [Google Scholar]
- 76.Sen T, Sen N, Noordhuis MG, Ravi R, Wu TC, Ha PK, Sidransky D, Hoque MO. OGDHL is a modifier of AKT-dependent signaling and NF-κB function. PLoS One. 2012;7:e48770. doi: 10.1371/journal.pone.0048770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fedorova MS, Kudryavtseva AV, Lakunina VA, Snezhkina AV, Volchenko NN, Slavnova EN, Danilova TV, Sadritdinova AF, Melnikova NV, Belova AA, Klimina KM, Sidorov DV, Alekseev BY, Kaprin AD, Dmitriev AA, Krasnov GS. Downregulation of OGDHL expression is associated with promoter hypermethylation in colorectal cancer. Mol Biol (Mosk) 2015;49:678–688. doi: 10.7868/S0026898415040047. [DOI] [PubMed] [Google Scholar]
- 78.Jiao Y, Li Y, Fu Z, Hou L, Chen Q, Cai Y, Jiang P, He M, Yang Z. OGDHL expression as a prognostic biomarker for liver cancer patients. Dis Markers. 2019;2019:9037131. doi: 10.1155/2019/9037131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Y, Meng F, Wang J, Liu M, Yang G, Song R, Zheng T, Liang Y, Zhang S, Yin D, Wang J, Yang H, Pan S, Sun B, Han J, Sun J, Lan Y, Wang Y, Liu X, Zhu M, Cui Y, Zhang B, Wu D, Liang S, Liu Y, Song X, Lu Z, Yang J, Li M, Liu L. A novel oxoglutarate dehydrogenase-like mediated miR-214/TWIST1 negative feedback loop inhibits pancreatic cancer growth and metastasis. Clin Cancer Res. 2019;25:5407–5421. doi: 10.1158/1078-0432.CCR-18-4113. [DOI] [PubMed] [Google Scholar]
- 80.Li M, Wu P, Yang Z, Deng S, Ni L, Zhang Y, Jin L, Pan Y. miR-193a-5p promotes pancreatic cancer cell metastasis through SRSF6-mediated alternative splicing of OGDHL and ECM1. Am J Cancer Res. 2020;10:38–59. [PMC free article] [PubMed] [Google Scholar]
- 81.Khalaj-Kondori M, Hosseinnejad M, Hosseinzadeh A, Behroz Sharif S, Hashemzadeh S. Aberrant hypermethylation of OGDHL gene promoter in sporadic colorectal cancer. Curr Probl Cancer. 2020;44:100471. doi: 10.1016/j.currproblcancer.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 82.Guo X, Long J, Chen Z, Shu XO, Xiang YB, Wen W, Zeng C, Gao YT, Cai Q, Zheng W. Discovery of rare coding variants in OGDHL and BRCA2 in relation to breast cancer risk in Chinese women. Int J Cancer. 2020;146:2175–2181. doi: 10.1002/ijc.32825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mao M, Huang RZ, Zheng J, Liang HQ, Huang WH, Liu J, Li JH. OGDHL closely associates with tumor microenvironment and can serve as a prognostic biomarker for papillary thyroid cancer. Cancer Med. 2021;10:728–736. doi: 10.1002/cam4.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yao L, Xu X, Xu Y, Li C, Xie F, Guo M, Liu Z, Liu X. OGDHL ameliorates cognitive impairment and Alzheimer’s disease-like pathology via activating Wnt/β-catenin signaling in Alzheimer’s disease mice. Behav Brain Res. 2022;418:113673. doi: 10.1016/j.bbr.2021.113673. [DOI] [PubMed] [Google Scholar]
- 85.Arrázola MS, Silva-Alvarez C, Inestrosa NC. How the Wnt signaling pathway protects from neurodegeneration: the mitochondrial scenario. Front Cell Neurosci. 2015;9:166. doi: 10.3389/fncel.2015.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anderson NM, Li D, Peng HL, Laroche FJ, Mansour MR, Gjini E, Aioub M, Helman DJ, Roderick JE, Cheng T, Harrold I, Samaha Y, Meng L, Amsterdam A, Neuberg DS, Denton TT, Sanda T, Kelliher MA, Singh A, Look AT, Feng H. The TCA cycle transferase DLST is important for MYC-mediated leukemogenesis. Leukemia. 2016;30:1365–1374. doi: 10.1038/leu.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anderson NM, Qin X, Finan JM, Lam A, Athoe J, Missiaen R, Skuli N, Kennedy A, Saini AS, Tao T, Zhu S, Nissim I, Look AT, Qing G, Simon MC, Feng H. Metabolic enzyme DLST promotes tumor aggression and reveals a vulnerability to OXPHOS inhibition in high-risk neuroblastoma. Cancer Res. 2021;81:4417–4430. doi: 10.1158/0008-5472.CAN-20-2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shen N, Korm S, Karantanos T, Li D, Zhang X, Ritou E, Xu H, Lam A, English J, Zong WX, Liu CT, Shirihai O, Feng H. DLST-dependence dictates metabolic heterogeneity in TCA-cycle usage among triple-negative breast cancer. Commun Biol. 2021;4:1289. doi: 10.1038/s42003-021-02805-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Remacha L, Pirman D, Mahoney CE, Coloma J, Calsina B, Curras-Freixes M, Leton R, Torres-Pérez R, Richter S, Pita G, Herraez B, Cianchetta G, Honrado E, Maestre L, Urioste M, Aller J, García-Uriarte O, Galvez MÁ, Luque RM, Lahera M, Moreno-Rengel C, Eisenhofer G, Montero-Conde C, Rodriguez-Antona C, Llorca O, Smolen GA, Robledo M, Cascon A. Recurrent germline DLST mutations in individuals with multiple pheochromocytomas and paragangliomas. Am J Hum Genet. 2019;104:651–664. doi: 10.1016/j.ajhg.2019.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Buffet A, Zhang J, Rebel H, Corssmit EPM, Jansen JC, Hensen EF, Bovee JVMG, Morini A, Gimenez-Roqueplo AP, Hes FJ, Devilee P, Favier J, Bayley JP. Germline DLST variants promote epigenetic modifications in pheochromocytoma-paraganglioma. J Clin Endocrinol Metab. 2021;106:459–471. doi: 10.1210/clinem/dgaa819. [DOI] [PubMed] [Google Scholar]
- 91.Shin D, Lee J, You JH, Kim D, Roh JL. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. 2020;30:101418. doi: 10.1016/j.redox.2019.101418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Odievre MH, Chretien D, Munnich A, Robinson BH, Dumoulin R, Masmoudi S, Kadhom N, Rotig A, Rustin P, Bonnefont JP. A novel mutation in the dihydrolipoamide dehydrogenase E3 subunit gene (DLD) resulting in an atypical form of alpha-ketoglutarate dehydrogenase deficiency. Hum Mutat. 2005;25:323–324. doi: 10.1002/humu.9319. [DOI] [PubMed] [Google Scholar]
- 93.Eagle H. The minimum vitamin requirements of the L and HeLa cells in tissue culture, the production of specific vitamin deficiencies, and their cure. J Exp Med. 1955;102:595–600. doi: 10.1084/jem.102.5.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, Stevens M, Fischer R, Carmeliet P, Maxwell PH, Pugh CW, Frizzell N, Soga T, Kessler BM, El-Bahrawy M, Ratcliffe PJ, Pollard PJ. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, Hall MN. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012;47:349–358. doi: 10.1016/j.molcel.2012.05.043. [DOI] [PubMed] [Google Scholar]
- 96.Duran RV, Hall MN. Glutaminolysis feeds mTORC1. Cell Cycle. 2012;11:4107–4108. doi: 10.4161/cc.22632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Coloff JL, Murphy JP, Braun CR, Harris IS, Shelton LM, Kami K, Gygi SP, Selfors LM, Brugge JS. Differential glutamate metabolism in proliferating and quiescent mammary epithelial cells. Cell Metab. 2016;23:867–980. doi: 10.1016/j.cmet.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 98.Murphy AN, Kelleher JK, Fiskum G. Submicromolar Ca2+ regulates phosphorylating respiration by normal rat liver and AS-30D hepatoma mitochondria by different mechanisms. J Biol Chem. 1990;265:10527–10534. [PubMed] [Google Scholar]
- 99.Territo PR, Mootha VK, French SA, Balaban RS. Ca2+ activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol Cell Physiol. 2000;278:C423–C435. doi: 10.1152/ajpcell.2000.278.2.C423. [DOI] [PubMed] [Google Scholar]
- 100.Kingsbury JM, Shamaprasad N, Billmyre RB, Heitman J, Cardenas ME. Cancer-associated isocitrate dehydrogenase mutations induce mitochondrial DNA instability. Hum Mol Genet. 2016;25:3524–3538. doi: 10.1093/hmg/ddw195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yong C, Stewart GD, Frezza C. Oncometabolites in renal cancer. Nat Rev Nephrol. 2020;16:156–172. doi: 10.1038/s41581-019-0210-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pollard PJ, Brière JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 103.Karlstaedt A, Zhang X, Vitrac H, Harmancey R, Vasquez H, Wang JH, Goodell MA, Taegtmeyer H. Oncometabolite D-2-hydroxyglutarate impairs α-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc Natl Acad Sci U S A. 2016;113:10436–10441. doi: 10.1073/pnas.1601650113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27:599–608. doi: 10.1093/annonc/mdw013. [DOI] [PubMed] [Google Scholar]
- 105.Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21:373–380. [PubMed] [Google Scholar]
- 106.Kim J, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 107.Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De Reyniès A, Gimenez-Roqueplo AP, Favier J. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 108.Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. 2017;49:e324. doi: 10.1038/emm.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, Liu T. The critical role of RNA m(6)A methylation in cancer. Cancer Res. 2019;79:1285–1292. doi: 10.1158/0008-5472.CAN-18-2965. [DOI] [PubMed] [Google Scholar]
- 110.Zhang M, Yang S, Nelakanti R, Zhao W, Liu G, Li Z, Liu X, Wu T, Xiao A, Li H. Mammalian ALKBH1 serves as an N 6-mA demethylase of unpairing DNA. Cell Res. 2020;30:197–210. doi: 10.1038/s41422-019-0237-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M, Muret C, Debackere K, Mazzone M, Huang HD, Fendt SM, Ivanisevic J, Ho PC. α-Ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18:985–994. doi: 10.1038/ni.3796. [DOI] [PubMed] [Google Scholar]
- 112.MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG, Gottlieb E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol. 2007;27:3282–3289. doi: 10.1128/MCB.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tennant DA, Frezza C, MacKenzie ED, Nguyen QD, Zheng L, Selak MA, Roberts DL, Dive C, Watson DG, Aboagye EO, Gottlieb E. Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene. 2009;28:4009–4021. doi: 10.1038/onc.2009.250. [DOI] [PubMed] [Google Scholar]
- 114.Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM, Chandel NS, DeBerardinis RJ. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014;7:1679–1690. doi: 10.1016/j.celrep.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, Arreola A, Rathmell WK, Olumi A, Lopez-Larrubia P, Stephanopoulos G, Iliopoulos O. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17:372–385. doi: 10.1016/j.cmet.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, Schmidt S, Metallo CM, Dranka BP, Schwartz B, DeBerardinis RJ. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532:255–258. doi: 10.1038/nature17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dayan A, Fleminger G, Ashur-Fabian O. Targeting the Achilles’ heel of cancer cells via integrin-mediated delivery of ROS-generating dihydrolipoamide dehydrogenase. Oncogene. 2019;38:5050–5061. doi: 10.1038/s41388-019-0775-9. [DOI] [PubMed] [Google Scholar]
- 119.Fleminger G, Dayan A. The moonlighting activities of dihydrolipoamide dehydrogenase: Biotechnological and biomedical applications. J Mol Recognit. 2021;34:e2923. doi: 10.1002/jmr.2924. [DOI] [PubMed] [Google Scholar]
- 120.Dayan A, Yeheskel A, Lamed R, Fleminger G, Ashur-Fabian O. Dihydrolipoamide dehydrogenase moonlighting activity as a DNA chelating agent. Proteins. 2020 doi: 10.1002/prot.25991. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 121.Yumnam S, Kang MC, Oh SH, Kwon HC, Kim JC, Jung ES, Lee CH, Lee AY, Hwang JI, Kim SY. Downregulation of dihydrolipoyl dehydrogenase by UVA suppresses melanoma progression via triggering oxidative stress and altering energy metabolism. Free Radic Biol Med. 2021;162:77–87. doi: 10.1016/j.freeradbiomed.2020.11.037. [DOI] [PubMed] [Google Scholar]
- 122.Dorsam B, Fahrer J. The disulfide compound α-lipoic acid and its derivatives: a novel class of anticancer agents targeting mitochondria. Cancer Lett. 2016;371:12–19. doi: 10.1016/j.canlet.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 123.Wenzel U, Nickel A, Daniel H. α-Lipoic acid induces apoptosis in human colon cancer cells by increasing mitochondrial respiration with a concomitant O2-*-generation. Apoptosis. 2005;10:359–368. doi: 10.1007/s10495-005-0810-x. [DOI] [PubMed] [Google Scholar]
- 124.Egawa Y, Saigo C, Kito Y, Moriki T, Takeuchi T. Therapeutic potential of CPI-613 for targeting tumorous mitochondrial energy metabolism and inhibiting autophagy in clear cell sarcoma. PLoS One. 2018;13:e0198940. doi: 10.1371/journal.pone.0198940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang L, Wen Y, Lv G, Lin Y, Tang J, Lu J, Zhang M, Liu W, Sun X. α-Lipoic acid inhibits human lung cancer cell proliferation through Grb2-mediated EGFR downregulation. Biochem Biophys Res Commun. 2017;494:325–331. doi: 10.1016/j.bbrc.2017.10.030. [DOI] [PubMed] [Google Scholar]
- 126.Gao L, Xu Z, Huang Z, Tang Y, Yang D, Huang J, He L, Liu M, Chen Z, Teng Y. CPI-613 rewires lipid metabolism to enhance pancreatic cancer apoptosis via the AMPK-ACC signaling. J Exp Clin Cancer Res. 2020;39:73. doi: 10.1186/s13046-020-01579-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lang L, Wang F, Ding Z, Zhao X, Loveless R, Xie J, Shay C, Qiu P, Ke Y, Saba NF, Teng Y. Blockade of glutamine-dependent cell survival augments antitumor efficacy of CPI-613 in head and neck cancer. J Exp Clin Cancer Res. 2021;40:393. doi: 10.1186/s13046-021-02207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Biryukov AI, Bunik VI, Zhukov YN, Khurs EN, Khomutov RM. Succinyl phosphonate inhibits alpha-ketoglutarate oxidative decarboxylation, catalyzed by alpha-ketoglutarate dehydrogenase complexes from E. coli and pigeon breast muscle. FEBS Lett. 1996;382:167–170. doi: 10.1016/0014-5793(96)00166-4. [DOI] [PubMed] [Google Scholar]
- 129.Artiukhov AV, Kazantsev AV, Lukashev NV, Bellinzoni M, Bunik VI. Selective inhibition of 2-oxoglutarate and 2-oxoadipate dehydrogenases by the phosphonate analogs of their 2-oxo acid substrates. Front Chem. 2021;8:596187. doi: 10.3389/fchem.2020.596187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Atlante S, Visintin A, Marini E, Savoia M, Dianzani C, Giorgis M, Sürün D, Maione F, Schnütgen F, Farsetti A, Zeiher AM, Bertinaria M, Giraudo E, Spallotta F, Cencioni C, Gaetano C. α-ketoglutarate dehydrogenase inhibition counteracts breast cancer-associated lung metastasis. Cell Death Dis. 2018;9:756. doi: 10.1038/s41419-018-0802-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Calabrese C, Iommarini L, Kurelac I, Calvaruso MA, Capristo M, Lollini PL, Nanni P, Bergamini C, Nicoletti G, Giovanni CD, Ghelli A, Giorgio V, Caratozzolo MF, Marzano F, Manzari C, Betts CM, Carelli V, Ceccarelli C, Attimonelli M, Romeo G, Fato R, Rugolo M, Tullo A, Gasparre G, Porcelli AM. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab. 2013;1:11. doi: 10.1186/2049-3002-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kurelac I, Iommarini L, Vatrinet R, Amato LB, De Luise M, Leone G, Girolimetti G, Umesh Ganesh N, Bridgeman VL, Ombrato L, Columbaro M, Ragazzi M, Gibellini L, Sollazzo M, Feichtinger RG, Vidali S, Baldassarre M, Foriel S, Vidone M, Cossarizza A, Grifoni D, Kofler B, Malanchi I, Porcelli AM, Gasparre G. Inducing cancer indolence by targeting mitochondrial Complex I is potentiated by blocking macrophage-mediated adaptive responses. Nat Commun. 2019;10:903. doi: 10.1038/s41467-019-08839-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Matsumoto K, Imagawa S, Obara N, Suzuki N, Takahashi S, Nagasawa T, Yamamoto M. 2-Oxoglutarate downregulates expression of vascular endothelial growth factor and erythropoietin through decreasing hypoxia-inducible factor-1alpha and inhibits angiogenesis. J Cell Physiol. 2006;209:333–340. doi: 10.1002/jcp.20733. [DOI] [PubMed] [Google Scholar]
- 134.Tseng CW, Kuo WH, Chan SH, Chan HL, Chang KJ, Wang LH. Transketolase regulates the metabolic switch to control breast cancer cell metastasis via the α-ketoglutarate signaling pathway. Cancer Res. 2018;78:2799–2812. doi: 10.1158/0008-5472.CAN-17-2906. [DOI] [PubMed] [Google Scholar]
- 135.Koivunen P, Fell SM, Lu W, Rabinowitz JD, Kung AL, Schlisio S. The 2-oxoglutarate analog 3-oxoglutarate decreases normoxic hypoxia-inducible factor-1α in cancer cells, induces cell death, and reduces tumor xenograft growth. Hypoxia. 2016;4:15–27. doi: 10.2147/HP.S96366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tennant DA, Gottlieb E. HIF prolyl hydroxylase-3 mediates alpha-ketoglutarate-induced apoptosis and tumor suppression. J Mol Med. 2010;88:839–849. doi: 10.1007/s00109-010-0627-0. [DOI] [PubMed] [Google Scholar]
- 137.Efimova EV, Takahashi S, Shamsi NA, Wu D, Labay E, Ulanovskaya OA, Weichselbaum RR, Kozmin SA, Kron SJ. Linking cancer metabolism to DNA repair and accelerated senescence. Mol Cancer Res. 2016;14:173–184. doi: 10.1158/1541-7786.MCR-15-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sica V, Bravo-San Pedro JM, Izzo V, Pol J, Pierredon S, Enot D, Durand S, Bossut N, Chery A, Souquere S, Pierron G, Vartholomaiou E, Zamzami N, Soussi T, Sauvat A, Mondragón L, Kepp O, Galluzzi L, Martinou JC, Hess-Stumpp H, Ziegelbauer K, Kroemer G, Maiuri MC. Lethal poisoning of cancer cells by respiratory chain Inhibition plus dimethyl α-ketoglutarate. Cell Rep. 2019;27:820–834. doi: 10.1016/j.celrep.2019.03.058. [DOI] [PubMed] [Google Scholar]
- 139.Abla H, Sollazzo M, Gasparre G, Iommarini L, Porcelli AM. The multifaceted contribution of α-ketoglutarate to tumor progression: an opportunity to exploit? Semin Cell Dev Biol. 2020;98:26–33. doi: 10.1016/j.semcdb.2019.05.031. [DOI] [PubMed] [Google Scholar]