

Abstract
Aberrant sugar metabolism is linked to an increased risk of pancreatic cancer. Previously, we found that high glucose induces genome instability and de novo oncogenic KRAS mutation preferentially in pancreatic cells through dysregulation of O-GlcNAcylation. Increasing O-GlcNAcylation by extrinsically supplying N-acetyl-D-glucosamine (GlcNAc) causes genome instability in all kinds of cell types regardless of pancreatic origin. Since many people consume excessive amount of sugar (glucose, fructose, and sucrose) in daily life, whether high sugar consumption directly causes genome instability in animals remains to be elucidated. In this communication, we show that excess sugar in the daily drink increases DNA damage and protein O-GlcNAcylation preferentially in pancreatic tissue but not in other kinds of tissue of mice. The effect of high sugar on the pancreatic tissue may be attributed to the intrinsic ratio of GFAT and PFK activity, a limiting factor that dictates UDP-GlcNAc levels. On the other hand, GlcNAc universally induces DNA damage in all six organs examined. Either inhibiting O-GlcNAcylation or supplementing dNTP pool diminishes the induced DNA damage in these organs, indicating that the mechanism of action is similar to that of high glucose treatment in pancreatic cells. Taken together, these results suggest the potential hazards of high sugar drinks and high glucosamine intake to genomic instability and possibly cancer initiation.
Keywords: Glucose, fructose, sucrose, UDP-GlcNAc, PFK, GFAT, GlcNAc, O-GlcNAcylation, DNA damage
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a devastating disease. Clinical observation reveals that several factors, including high blood glucose, obesity/overweight, high sugar/high-fat diet (Western diet), and smoking history, contribute to the increased risk of PDAC [1]. Of these, impaired glucose homeostasis is a common trait associated with PDAC. In epidemiological studies, higher post-load plasma glucose levels and higher dietary sugar are associated with a higher risk of pancreatic cancer mortality, especially in men [2,3]. Besides, pancreatic cancer patients with high blood glucose are associated with poor prognosis [4], and long-term hyperglycemia is associated with malignancy [5]. High glucose accelerates pancreatic tumor growth at the mechanistic level via the SREBP1-autophagy axis [6] and increases invasive activity via ERK and p38 induced by ROS [7]. Taken together, the discovery of the influence of aberrant sugar metabolism on the initiation and progression of cancer draws the attention of this aspect to cancer prevention and therapy.
Previously, we have established a mechanistic link between disturbed glucose metabolism and genomic instability that induces de novo oncogenic KRAS mutations and cell transformation preferably in pancreatic cells [8]. Under high glucose conditions, O-GlcNAcylation is significantly elevated in pancreatic cells due to low phosphofructokinase (PFK) activity. At least two key enzymes, PFK and RRM1, compromise their activities after O-GlcNAcylation, leading to dNTP pools deficiency, genomic DNA alterations with KRAS mutations, and cell transformation. This result highlights the importance of O-GlcNAcylation-mediated nucleotide perturbation in pancreatic tumorigenesis. However, it is unclear whether high sugar (glucose, fructose, or sucrose) consumption directly causes metabolic imbalance in animals, resulting in DNA damage in pancreatic tissue.
Like phosphorylation, O-GlcNAcylation modulates protein functions, including enzymatic activity, protein-protein interaction, subcellular localization, and transcription activity [9]. Emerging evidence reveals that the cellular O-GlcNAc level is linked to physiological and pathological conditions. Several studies show that O-GlcNAcylation is altered in multiple cancers, including breast, prostate, colon, leukemia, bladder, endometrial, pancreatic, lung, and thyroid, linking O-GlcNAcylation with cancer phenotype [10]. Indeed, our previous work showed that low PFK1 is a critical factor for the high-glucose-induced effects through dysregulation of O-GlcNAcylation in pancreatic cells [8]. However, since glucose is metabolized toward the substrate of O-GlcNAcylation through glycolysis and hexosamine biosynthesis pathways, other factors are likely to be involved in high glucose-induced effects.
Protein O-GlcNAcylation could be modulated by enzymes, including O-GlcNAc transferase (OGT) that adds O-GlcNAc to serine and threonine residues of protein and O-GlcNAcase (OGA) that removes the modification from them, and the substrate, UDP-GlcNAc, that provides fuels for OGT [11]. Interestingly, elevating O-GlcNAcylation by adding N-acetyl-D-glucosamine (GlcNAc) into the culture medium increases DNA damage in many non-tumorigenic human cell lines and induces oncogenic KRAS G12D mutation in non-tumorigenic pancreatic cells [8]. This result suggests that increasing O-GlcNAcylation by GlcNAc may cause universal effects on genomic instability and promote tumorigenesis. However, at present, GlcNAc or Glucosamine sulfate is considered as dietary supplement and consumed daily by the elderly. Therefore, the safety of these compounds in vivo needs to be further examined.
In this communication, we have assessed whether sugar consumption impacts genome stability directly through O-GlcNAcylation-mediated nucleotide perturbation in mice. We show that excess sugar in daily drinks preferentially increases DNA damage and protein O-GlcNAcylation in pancreatic tissue but not in other tissues examined. The high sugar-induced effects in the pancreatic tissue may be attributed to the intrinsic ratio of GFAT and PFK activity, a limiting factor that dictates UDP-GlcNAc levels. In contrast to sugar, high dose of GlcNAc universally induces DNA damage in all six organs examined. The DNA damage caused by GlcNAc in vivo could be blocked by either inhibiting O-GlcNAcylation or supplementing with dNTPs, suggesting that GlcNAc elicits DNA damage through a mechanism similar to that of glucose in the pancreas. In summary, these results suggest the potential hazards of high sugar drinks and high glucosamine intake to genomic instability and possibly cancer initiation.
Materials and methods
Animal protocol and treatment
All animal experiments were approved by the Institutional Animal Care and Utilization Committee of Academia Sinica, Taipei, Taiwan (IACUC#20-03-1451). All male C57BL/6JNarl mice at 7 weeks were purchased from National Laboratory Animal Center, Taiwan, Taipei. Mice were maintained in a specific pathogen-free animal facility at 20±2°C with a 12/12 hr light-dark cycle. They had free access to water and a standard laboratory diet (LabDiet 5010) for a week before being subjected to the following experiments. A blood glucose meter (Roche ACCU-CHEK AVIA) was used to monitor the level of blood glucose.
To evaluate the effect of DNA damage from sugar, we randomly allocated male mice with similar body weight and blood glucose levels to four groups: (1) vehicle control with distilled water; (2) high glucose; (3) high fructose; (4) high sucrose. All sugars were dissolved in distilled H2O and administered to mice three times daily by oral gavage. The dosing volume is 10 µl/g. The total amount of sugar for a mouse is 15000 mg/kg (mpk) per day. After 7 days, mice were sacrificed and dissected for analysis.
To analyze the long-term sugar effects, we randomly allocated male mice with similar body weight and blood glucose levels to two groups: (1) the chow diet (LabDiet 5010); (2) the high sugar diet (Labdiet, 57XL, Supplementary Table 1), which consists of 40% sugar in calorie intake. After 20 weeks, mice were sacrificed and dissected for analysis.
To perform isotope-labeled glucose tracing assay, we treated male mice with 5000 mpk U13C glucose (Sigma, 389374) using oral gavage. At indicated time points (15, 30, 45, 60, 120, 240 min) after administration, mice were sacrificed. The sera and six organs were collected for analysis.
To evaluate the effects of DNA damage from glucose and N-acetyl-D-glucosamine (GlcNAc), we randomly allocated male mice with similar body weight and blood glucose levels to three groups: (1) vehicle control with distilled water; (2) high glucose; (3) high GlcNAc. Glucose and GlcNAc were dissolved in distilled H2O and administered to mice three times daily by oral gavage. The total amount of glucose or GlcNAc for a mouse is 15,000 mpk or 7500 mpk, respectively, per day. After 7 days, mice were sacrificed and dissected for analysis.
To perform long-term GlcNAc administration, we randomly allocated male mice with similar body weight and blood glucose levels to three groups: (1) vehicle control with distilled water; (2) 1X GlcNAc (212 mpk); (3) 10X GlcNAc (2120 mpk). Mice were administered daily (P.O. and Q.D.) for 60 days. After treatment for 60 days, mice were sacrificed and dissected for analysis.
To test the effect of OSMI-1, we randomly allocated male mice with similar body weight and blood glucose levels to three groups: (1) vehicle control with distilled water and I.P. solution; (2) GlcNAc along with GlcNAc 7500 mpk P.O. and I.P. solution; (3) GlcNAc plus OSMI-1 (1 mpk I.P.). I.P. solution contains 10% DMSO, 40% PEG300, 5% tween 80, and 45% saline for dissolving OSMI-1. The injection volume is 5 µl/g. After 7 days, mice were sacrificed and dissected for analysis.
To test the effect of supplementing deoxynucleotides, we randomly allocated male mice with similar body weight and blood glucose levels to three groups: (1) vehicle control with distilled water; (2) GlcNAc (7500 mpk P.O.); (3) GlcNAc plus deoxynucleotide (240 mpk of each P.O.). Deoxynucleotide was dissolved in GlcNAc solution. After 7 days, mice were sacrificed and dissected for six organs collection.
Organs, including pancreas, brain, liver, lung, kidney, and colon, were collected from the experimental mice for immunohistochemistry, protein, or metabolites analysis. For protein or metabolite extraction, tissues were stored in cryogenic tubes (Biologix, 88-0150) under liquid nitrogen.
Histology and sample preparation
Body weight and food consumption were recorded every week in each animal experiment. At the indicated day, mice were euthanized with isoflurane, and the heart blood samples were collected to analyze complete cell count and serum chemistry. Next, the abdominopelvic cavity and thoracic cavity were opened, and most organs, including thymus, lung, heart, intestine, colon, liver, spleen, kidney, and reproductive system, were observed for gross abnormalities. The pancreas was dissected together with the duodenum and spleen to preserve orientation. Other organs, including the brain, liver, lung, kidney, and colon, were collected for analysis. All organs were formalin-fixed and paraffin-embedded. The processed pancreas was embedded in a mold with an orientation of the duodenum in the left and spleen in the right. After trimming tissue blocks, we cut them into 4 μm paraffin sections of the pancreas where most of the essential features of the pancreas, especially the main duct, could be observed. All other tissues were carefully oriented, embedded in paraffin blocks, and then cut into 4 μm paraffin sections.
Immunohistochemistry (IHC)
IHC was performed on BOND-MAX fully automated IHC staining system (Leica). Slices were baked, deparaffinized, and stained in this instrument. All slides were antigen-retrieved at PH 9, 95°C, 30 min. Dilutions and incubations of antibodies were 1:100, 60 min at RT for anti-γH2AX antibody (Cell Signaling #9718) and 1:1000, 30 min at RT for anti-O-GlcNAcylation antibody (CTD 110.6) (Cell Signaling #9875), followed by 10 min incubation with HRP-conjugated secondary antibody (Bond refine systems, Leica). Later, slides were stained with a DAB detection kit (Bond refine systems, Leica) for 5 min and counterstained with hematoxylin (Bond refine systems, Leica) for 7 min. All slides were mounted with coverslips and scanned at 40× magnification by Aperio GT450 (Leica). For γH2AX quantification, 30 ROIs (1 mm2) were randomly selected and quantified with positive cells detection algorithm from Qupath. In this algorithm, the threshold is determined by the Nucleus: DAB OD mean measurements, and a threshold of 0.1 was performed to count γH2AX positive cells. Statistical data are presented as positive cell percentage and relative folds, which all values are normalized to the average of the vehicle group. For O-GlcNAcylation quantification, 30 ROIs (1 mm2) were randomly selected and analyzed with a positive pixel counts v9 algorithm from Imagescope. H-score was assigned by [1× (% of weak positive pixel counts) + 2× (% of positive pixel counts) + 3× (% of strong positive pixel counts)]. Data are presented as a relative fold in which all values are normalized to the average of the vehicle control group.
PFK activity assay
PFK activity assay was measured following the manufacturer’s instruction (MAK093, Sigma Aldrich). In brief, six tissues, including pancreas, brain, liver, lung, kidney, and colon, were collected from 10-week-old male C57BL/6JNarl mice, and each tissue was homogenized in 700 μl of ice-cold PFK assay buffer by MagNA Lyser Instrument (Roche) and centrifuged at 6,500× g for 5 min at 4°C. After being centrifuged at 12000× g for 10 min at 4°C, 450 μl of supernatant was collected and measured with BCA protein assay (Thermo #23225). 0.1 μg of protein was used for each activity assay, and the assay was performed according to the manufacturer’s instructions. PFK activity was reported as nmol/min/μg at 37°C.
Antibodies
All antibodies used in this study were described in Supplementary Table 2.
GFAT activity assay
GFAT activity assay was modified from the previous method [12]. In brief, six tissues, including pancreas, brain, liver, lung, kidney, and colon, were collected from 10-week-old male C57BL/6JNarl mice. Each tissue was homogenized in 700 μl ice-cold PFK assay buffer with MagNA Lyser Instrument (Roche) and centrifuged at 6,500× g for 5 min at 4°C. After being clarified by centrifugation at 12000× g for 10 min at 4°C, the 450 μl supernatant of protein was collected and measured with BCA protein assay (Thermo 23225). Sample with 240 μg of protein was mixed with fructose-6-phosphate (F6P) and glutamine in 100 μl reaction solution (F6P, 10 mM; glutamine, 10 mM: Tris, 50 mM; EDTA, 5 mM; GSH, 5 mM; glucose-6-phosphate Na2, 5 mM; and KCl, 50 mM; pH 7.8). After 0, 30, 60 min at 37°C, each reaction was halted at 100°C for 3 min. Then, the metabolite was extracted by adding 400 μl of ice-cold methanol. After 30-sec vortex and 10 min centrifugation at 12000× g at 4°C, 450 μl of supernatant was collected and concentrated by Speedvac (Thermo). The amount of glutamine was measured by LC/MS analysis. GFAT activity was determined as nmol/min/g at 37°C.
Metabolite extraction from tissue
50 mg of tissue was homogenized with 700 ul distilled water and centrifuged at 12,000× g for 10 min at 4°C. The supernatant was collected, and the protein concentration was measured with a BCA protein assay. Then, 400 μl of methanol was added to the supernatant of 100 μg protein in 100 μl of water, and the mixture was vortexed 30 sec and centrifuged at 13000 rpm for 10 min at 4°C. 450 μl of supernatant was collected and concentrated by Speedvac (Thermo) for further analysis.
Metabolite extraction from cell
2~3×106 cells were collected in pellets, added with 300 ul distilled water, and sonicated with the amplitude of 80% on ice for 30 sec (Hielscher Ultrasound technology). After 10 min centrifugation at 12,000× g at 4°C, the supernatant was collected, and the protein concentration was measured by BCA protein assay. Then, 400 μl of methanol was added to the supernatant containing 100 μg of protein in 100 μl of water, and the mixture was vortexed for 30 sec and centrifuged at 13000 rpm for 10 min at 4°C. About 450 μl of supernatant was collected and concentrated by Speedvac (Thermo) for further analysis.
Liquid chromatography-mass spectrometry (LC-MS) analysis for the targeted metabolites
The vacuum-concentrated samples were dissolved in 100 ul of 0.1% formic acid and centrifuged at 14800 rpm for 5 min at 4°C. LC-MS experiments were done by an LTQ FT Ultra (Linear quadrupole ion trap Fourier transform ion cyclotron resonance) mass spectrometer (Thermo Electron, San Jose, CA) equipped with a standard ESI source, an Agilent 1100 Series binary high-performance liquid chromatography pump (Agilent Technologies, Palo Alto, CA), and a Famos autosampler (LC Packings, San Francisco, CA). 5 μl of the sample was injected at 50 μl/min flow rate on BioResolve RP mAb (1 mm I.D. ×150 mm, 2.7 µm). Chromatographic separation was done by 0.1% formic acid in water as mobile phase A and 0.1% formic acid in 80% acetonitrile as mobile phase B at 100 ul/min flow rate. The employed gradient was 2% buffer B at 2 min to 30% buffer B at 20 min. MS condition: mass range m/z 100-700, resolution 50,000 at m/z 400. Electrospray voltage was maintained at 4.0 kV, and the capillary temperature was set at 275°C.
Liquid chromatography-mass spectrometry (LC-MS) analysis for U13C glucose tracing
To perform a U13C-labeled glucose tracing experiment, we used Agilent 1290 Infinity II ultra-performance liquid chromatography (UPLC) system (Agilent Technologies, Palo Alto, CA, USA) coupled online to the Dual AJS electrospray ionization (ESI) source of an Agilent 6545XT quadrupole time-of-flight (Q-TOF) mass spectrometer (Agilent technologies, Palo Alto, CA, USA). The sample was separated using the ACQUITY UPLC BEH amide column (1.7 μm, 2.1×100 mm, Waters Corp., Milford, MA, USA). The column temperature was 40°C. The mobile phases were composed of double-distilled water (eluent A) and 10% double-distilled water in acetonitrile (eluent B), both of which were eluted with 15 mM ammonium acetate and 0.3% NH3∙H2O. The flow rate was 300 uL/min, and the injecting volume of the sample was 2 uL. The instrument was operated in positive and negative full-scan mode, collected in a range of 60-1500 m/z. The MS operating conditions were optimized as follows: Vcap voltage 3.5 kV, nozzle voltage 1 kV for negative mode or 0 V for positive mode, nebulizer 45 psi, gas temperature, 200°C sheath gas temperature 300°C, sheath gas flow (nitrogen) 8 L/min, and drying gas (nitrogen) 10 L/min. The chromatogram acquisition, mass spectral peaks detection, and waveform processing were performed using Agilent qualitative analysis 10.0 (Agilent, USA). Isotopologue extraction was performed, and natural isotope abundance was corrected using Agilent profinder 10.0 (Agilent, USA). The mass tolerance was 10 ppm and retention time tolerance was 0.5 min. Data represent 13C labeled pool fraction of the metabolite.
Cell culture
All cells were maintained in a 5% CO2, 37°C incubator (PHCBI). The non-transformed human pancreatic ductal epithelial cells (HPDE, a gift from Dr. Kelvin K. Tsai, National Health Research Institute, Taiwan) were grown in keratinocyte serum-free medium with 0.2 ng/ml EGF and 30 μg/ml BPE (Invitrogen). The non-transformed human pancreatic acinar-to-ductal epithelial-like cells (HPNE purchased from ATCC) were grown in a medium containing one volume of M3TM base medium (INCELL) and three volumes of glucose-free DMEM with 5% FBS, 1 g/L glucose, and 10 ng/ml EGF. The non-transformed human mammary epithelial cells MCF10A were purchased from ATCC and grown in a medium containing one volume of glucose-free DMEM and one volume of F12 medium with 5% horse serum, 10 μg/ml Insulin, 20 ng/ml EGF, 0.5 μg/ml hydrocortisone, and 100 ng/ml cholera toxin. The non-transformed lung epithelial cells NL20 were a gift from Dr. Michael Hsiao (Genomics Research Center, Taiwan) and were grown in a medium containing one volume of glucose-free DMEM and one volume of F12 medium with 4% FBS, 5 μg/ml insulin, 10 ng/ml EGF, 1 μg/ml transferrin, and 500 ng/ml hydrocortisone. The non-transformed mouse hepatocyte AML12 cells were purchased from ATCC (CRL-2254) and grown in a medium containing one volume of glucose-free DMEM and one volume of F12 with 10% FBS, 40 ng/ml dexamethasone, and 1X Insulin-Transferrin-Selenium (ITS-G, Gibco). The non-transformed mouse astrocyte C8-D1A cells were purchased from ATCC (CRL-2541) and grown in DMEM with 1 g/L glucose and 10% FBS. All cell lines were regularly tested for mycoplasma contamination.
Immunofluorescent staining
Cells were fixed with 100% methanol at -20°C for 20 min, blocked with MAXblockTM solution (Active Motif) at 37°C for 1 hr, and stained with anti-γH2AX antibody (1:1000, Merck Millipore #05-636) or anti-RL2 antibody (1:1000, Abcam ab2739) at 4°C overnight. After extensive wash, these cells were stained with Donkey anti-Mouse IgG Alexa Fluor 488 antibody (1:100, Invitrogen #A-21202) and Hoechst 33342 (10 µg/ml, Sigma #B2261) at room temperature for 1 hr. Cells were counted from ROIs randomly chosen in 20× magnification, and positive cells were defined as nuclei with at least 5 large foci. The intensity of O-GlcNAcylation (RL2) was determined by the average fluorescent intensity of at least 2000 cells. Images were captured by Image Xpress Micro XLS HCS System (Molecular Devices). Foci and fluorescent intensity were analyzed by Custom Module Editor (CME). Excel calculated the counted results.
Soft agar colony formation assay
500 cells were seeded in a layer of 0.35% agar/complete growth medium over a layer of 0.5% agar/complete growth medium in a 96-well plate. Cell medium containing indicated concentration of GlcNAc, OSMI-1 (Abcam, ab235455), and deoxynucleotides (Sigma) was replenished every 2 days. After 14 days, cells were stained with Hoechst 33342 to measure colony formation efficiency. Images were captured in Z stack mode (10 pieces with 150 μm interval) of 2× magnification and overlapped into one piece for each well. The size of each colony was then calculated by Custom Module Editor (CME). Colonies larger than 50 µm were counted and analyzed using Image Xpress Micro XLS HCS System (Molecular Devices).
RRM1 immunoprecipitation
Cells or tissues were sonicated or homogenized in RNR lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP40, 50 mM NaF, 1 mM Na2Vo3, 1 mM DTT, 0.1% SDS, 2 mM PMSF, and protease inhibitor). After being clarified by centrifugation at 12,000 g for 15 min at 4°C, the supernatant was collected and pre-cleaned with protein G agarose beads (Thermo Fisher) at 4°C for 10 min, followed by incubation of anti-RRM1 antibody (500 µg total protein using 1 ug antibody sc-11733 (Santa Cruz)) or anti-goat IgG antibody at 4°C overnight. After incubation of Protein G beads at 4°C for 1 hr, samples were washed three times with RNR lysis buffer, and proteins were eluted with Laemmli sample buffer for immunoblotting analysis.
Immunoblotting analysis
Cells or tissues were sonicated or homogenized in RIPA buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, and 1% sodium deoxycholate). After being clarified by centrifugation at 12,000 g for 15 min at 4°C, the supernatant was collected for BCA protein assay. Samples were mixed with Laemmli sample buffer. For gel electrophoresis, 15 μg protein was loaded in 4~12% of gradient gel, separated, and transferred to PVDF membrane (Millipore) by Biorad transblot system in transfer buffer (Tris-base: 50 mM, Glycine: 40 mM, SDS: 0.04%, Methanol 20%). After being blocked with 5% skimmed milk at RT for 1 hr, the membrane was incubated with primary antibodies at 4°C overnight and treated with horseradish peroxidase-conjugated goat anti-rabbit IgG and goat anti-mouse IgG antibodies at room temperature for 1 hr. Chemiluminescent detection of the horseradish peroxidase reaction was performed according to the manufacturer’s instruction (WBLUF0500, Merck Millipore) and filmed by ChemiDoc™ MP Imaging System (Biorad).
Statistical analyses
Immunohistochemistry, immunofluorescent staining, metabolites analysis, enzymatic activity, and soft agar colony formation studies were analyzed with a two-tailed unpaired student’s t-test. *, **, and *** indicate P<0.05, P<0.01, and P<0.001, respectively. N number used in the indicated section is shown in the figure legend.
Results
Sugar consumption increases DNA damage and O-GlcNAcylation in the pancreas but not in other tissues of mice
To investigate the effects of free sugar, we searched the intake of sugar-sweetened beverages in Taiwan [13] and the United States [14]. We concluded that the daily sugar intake of sugar-sweetened beverages for a young-aged, 60 kg man is 70~75 g/day. Converting human dose to mice, we set up an experiment in which mice were treated with glucose 15000 mpk, fructose 15000 mpk, and sucrose 15000 mpk by oral gavage T.I.D for 7 days (Supplementary Figure 1A). As γH2AX immunostaining is considered as an appropriate method for measuring DNA damage [15], six tissues, including pancreas, liver, lung, kidney, colon, and brain from each mouse, were stained with antibodies against γH2AX. The positive γH2AX staining images in the low power field of γH2AX staining in the pancreas were presented as representative images (Figure 1A). The percentage of cells with positive γH2AX staining (Figure 1B, 1D, 1F, 1H, 1J and 1L) and the relative fold of positive γH2AX staining (Figure 1C, 1E, 1G, 1I, 1K and 1M) of all six tissues were shown in Figure 1. These results suggest that the pancreas, but not the other five tissues, had 4 times more DNA damage than vehicle alone when mice were treated with glucose or fructose. In contrast, those treated with sucrose had a similar trend but did not reach statistical significance.
Figure 1.
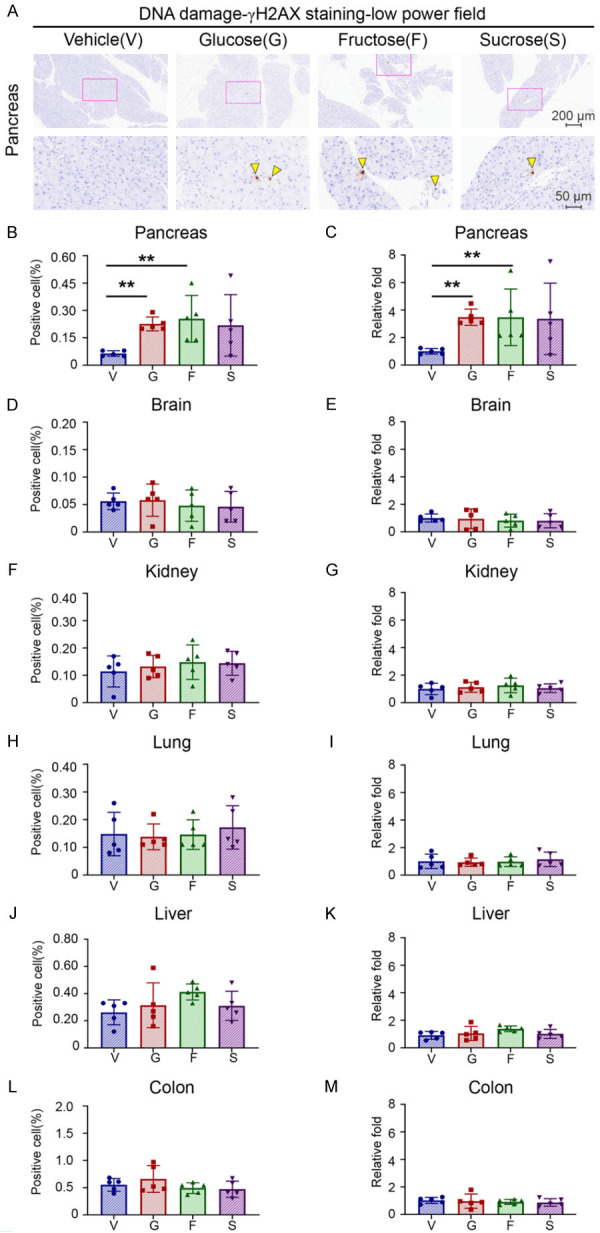
High sugar consumption preferentially induces DNA damage in the pancreatic tissue but not in other tissues. Five male mice of each group were treated with vehicle (water), glucose (G, 15000 mpk (mg/kg)), fructose (F, 15000 mpk), or sucrose (S, 15000 mpk) for 7 days. All sugars were dissolved in water and administered to mice three times daily by oral gavage at 10 μl/g. (A) Representative images of γH2AX staining by IHC in the pancreas. Upper panel: 10× magnification. Scale bar, 200 μm. Lower panel: 40× magnification. Scale bar, 50 μm. Yellow arrowhead: γH2AX positive nucleus (B-M). Percentage of positive cells (B, D, F, H, J and L) and relative folds (C, E, G, I, K and M) of γH2AX IHC staining in the pancreas (B and C), brain (D and E), kidney (F and G), lung (H and I), liver (J and K) and colon (L and M). Relative folds are calculated from the percentage of γH2AX positive cells in different groups and normalized to that of the vehicle control. Each dot represents the datum of one mouse. Values show mean ± SEM. **P<0.01 (two-tailed Student’s t-test).
It was noted that glucose-induced DNA damage in the pancreas is caused by increased O-GlcNAcylation [8]. We then immunostained these tissues with an anti-O-GlcNAcylation antibody (CTD 110.6). Similarly, we found that pancreas had 1.3 times more O-GlcNAcylation than vehicle alone when mice were treated with glucose or fructose. In contrast, those treated with sucrose had a similar trend but did not reach statistical significance (Figure 2A, 2B). These results suggest that mice consuming high free sugar have more DNA damage and higher O-GlcNAcylation preferentially in their pancreas.
Figure 2.
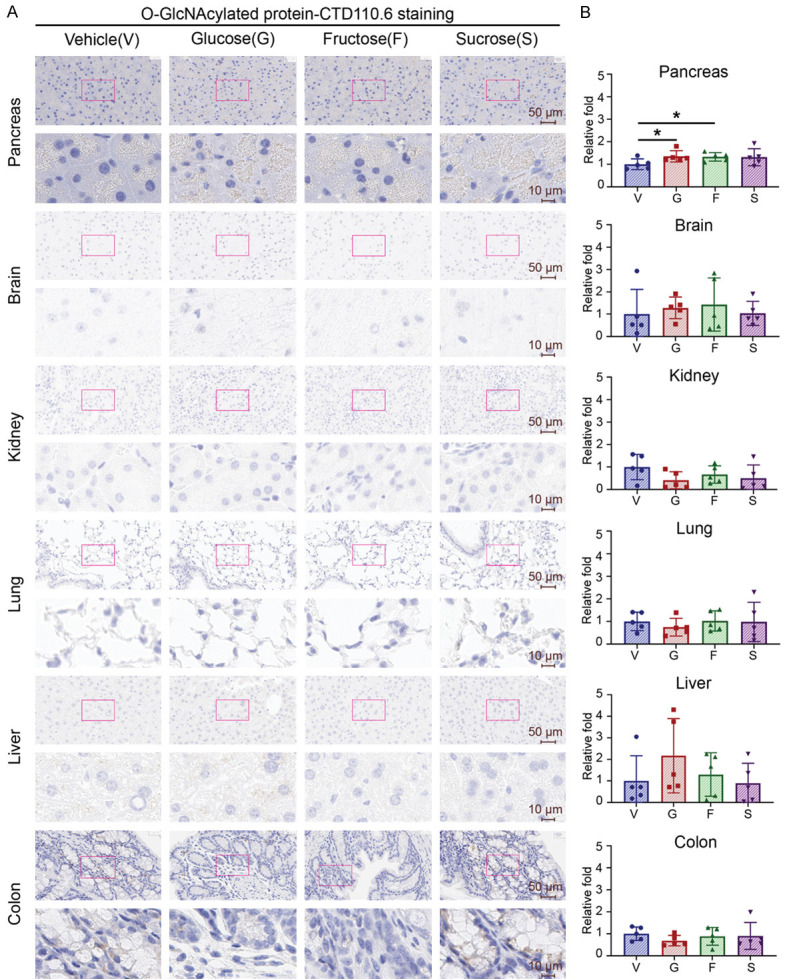
High sugar consumption preferentially increases protein O-GlcNAcylation in pancreatic tissue but not in other tissues. Five male mice of each group were treated with glucose (G, 15000 mpk), fructose (F, 15000 mpk), sucrose (S, 15000 mpk), or vehicle control (water) for 7 days. All sugars were dissolved in water and administered to mice three times daily by oral gavage at 10 μl/g. (A) Representative images and (B) quantifications of O-GlcNAcylation (CTD 110.6) staining by IHC in mouse pancreas, brain, kidney, lung, liver, and colon. (A) Upper panel: 40× magnification. Scale bar, 50 μm. Lower panel: 200× magnification. Scale bar, 10 μm. (B) Quantitative bar graphs show the relative folds of the H-score of O-GlcNAcylation. Each dot represents the datum of one mouse. Data are normalized to that from mice treated with vehicle control. Values show mean ± SEM. *P<0.05 (two-tailed Student’s t-test).
To explore the effects of long-term sugar uptake, we introduced a high sugar diet, of which sugar contained 40% calories (Supplementary Table 1), to male mice for 20 weeks (Figure 3A). Similar to the short-term results above, we found that a high sugar diet did not affect mice’s body weight and blood glucose levels (Figure 3B, 3C) but significantly increased O-GlcNAcylation, DNA damage, and cell proliferation in pancreas (Figure 3D-I). This result further suggests that long-term sugar consumption causes genome instability and may trigger cancer initiation in pancreas.
Figure 3.
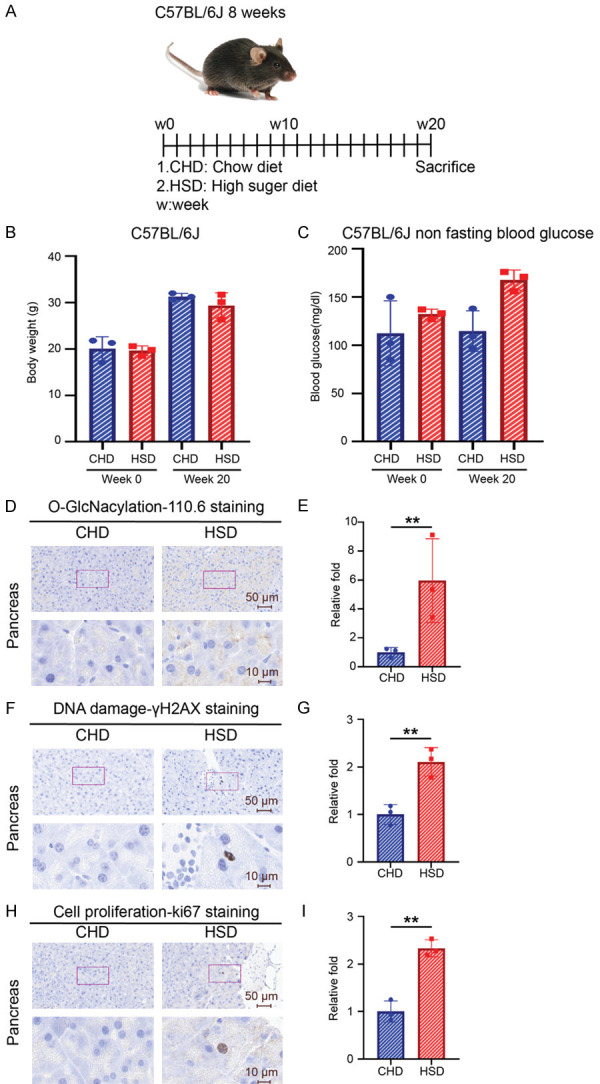
Long-term treatment of the high sugar diet increases DNA damage and protein O-GlcNAcylation in mouse pancreas. (A) Schematic model of mice high sugar diet experiment. Three male mice of each group were treated with a chow diet and a high sugar diet for 20 weeks. (B) Body weight and (C) non-fasting blood glucose levels at week 0 and week 20 of the treated mice. (D) Representative images and (E) quantification of O-GlcNAcylation (CTD 110.6) staining from each mouse pancreas. Data are normalized to that from mice treated with chow diet. (F) Representative images and (G) quantification of γH2AX staining of mouse pancreas from each group. Quantitative bar graphs showed the relative folds of the percentage of γH2AX positive cells. Data are normalized to that from mice treated with chow diet. (H) Representative images and (I) quantification of ki67 staining of mice pancreas. Upper panel: 40× magnification. Scale bar, 50 μm. Lower panel: 200× magnification. Scale bar, 10 μm. Quantitative bar graphs of the relative folds of the percentage of γH2AX positive cells. Data are normalized to that from mice treated with the chow diet. Each dot represents the datum of one mouse. Values show mean ± SEM. **P<0.005 (two-tailed Student’s t-test).
GFAT/PFK ratio determines glucose shunt toward UDP-GlcNAc and protein O-GlcNAcylation
It was noted that cellular O-GlcNAcylation homeostasis is achieved through coordinating the optimal level of UDP-GlcNAc, the end product of Hexosamine Biosynthesis Pathway (HBP), and the O-GlcNAc-cycling enzymes, OGT, and OGA [11]. This diverges from glycolysis and is controlled by the first and rate-limiting enzyme, glutamine: fructose-6-phosphate-amidotransferase (GFAT), which converts fructose-6-phosphate to glucosamine-6-phosphate [16]. Moreover, phosphofructokinases (PFKs) catalyze the first ‘committed’ step in glycolysis, converting fructose 6-phosphate and ATP to fructose 1,6 bisphosphate and ADP [17]. To explore potential mechanisms of how sugar mainly affects pancreas, we first analyzed the protein expression levels of GFAT1, GFAT2, PFKL, PFKM, PFKP, OGT, OGA among 6 tissues, including pancreas, brain, liver, lung, kidney, and colon, by immunoblotting analysis (Figure 4A). We found that pancreas had the highest GFAT1 expression level and the lowest PFKL, PFKM, and PFKP expression levels among these tissues examined, suggesting that the expression levels of GFAT and PFK are more relevant to the high sugar-induced effects in the pancreas than that of other enzymes. Accordingly, we measured both GFAT and PFK enzymatic activities and found that pancreas had the highest GFAT activity and the lowest PFK activity among these tissues (Figure 4B, 4C). Interestingly, the pancreas has the highest GFAT/PFK activity ratio if dividing GFAT activity value with PFK activity value (Figure 4D). This ratio may be likely to determine the level of glycolysis/HBP-related metabolite or protein O-GlcNAcylation.
Figure 4.

The PFK and GFAT activity ratio positively correlates with the cellular UDP-GlcNAc levels and protein O-GlcNAcylation in mouse tissues. Pancreas, brain, liver, lung, kidney, and colon were collected from 10-week-old male C57BL/6JNarl mice. A. Protein levels of GFAT1, GFAT2, PFKL, PFKM, PFKP, OGT, OGA, and p84 in various tissues were analyzed by immunoblotting analysis. Data are normalized to those from pancreatic tissues and the amount of internal control protein, p84. B. GFAT activity among pancreas, brain, liver, lung, kidney, and colon (n=3). C. PFK activity among pancreas, brain, liver, lung, kidney, and colon (n=3). D. GFAT/PFK activity ratio among pancreas, brain, liver, lung, kidney, and colon (n=3). Data are normalized to mice pancreas. E. UDP-GlcNAc levels among pancreas, brain, liver, lung, kidney, and colon (n=6). F. Protein O-GlcNAcylation was analyzed by immunoblotting analysis with an anti-RL2 antibody. Data are normalized to those from pancreatic tissues and the amount of internal control protein, p84. Each dot represents the datum of one mouse. Values show mean ± SEM.
To test this possibility, we analyzed the concentration of related metabolites by LC/MS analysis. It was noticed that pancreas had the highest UDP-GlcNAc level (Figure 4E), the end-product of the HBP pathway and the substrate of protein O-GlcNAcylation. However, there is no significant difference of F6P/G6P, glucosamine-6P, fructose-1,6-biphosphate, glutamine, and glutamate, the substrates or products of PFK and GFAT, among all tissues examined (Supplementary Figure 2A-E). Similarly, pancreas had the highest levels of O-GlcNAcylation among all tissues examined (Figure 4F). To further test whether pancreas metabolizes more glucose to UDP-GlcNAc, we performed a U13C glucose isotope tracing assay (Supplementary Figure 3A). In this experiment, blood glucose level peaked at 15 min and returned to normal at 120 min (Supplementary Figure 3B). All tissues examined had a similar amount of labeled glucose and lactate, indicating that all tissues had similar glucose uptake and glycolysis efficiency (Supplementary Figure 3C, 3D). Interestingly, more M + 6 UDP-GlcNAc was observed in pancreas than in other organs (Supplementary Figure 3E), indicating that pancreas metabolized more glucose to HBP and produced more UDP-GlcNAc than other organs. These results suggest that the high intrinsic ratio of GFAT and PFK activity in pancreas is critical in enhancing UDP-GlcNAc levels resulting in high sugar-induced effects.
N-acetyl-D-glucosamine (GlcNAc) universally triggers DNA damage
As noted above, the effect of glucose-induced DNA damage is mediated by the intrinsic ratio of GFAT and PFK activity through modulating UDP-GlcNAc levels. In contrast, the effect of GlcNAc may directly modulate UDP-GlcNAc levels as it bypasses the intrinsic ratio of GFAT and PFK activity [18]. To test this notion, we compared the effects of glucose (15000 mpk) and GlcNAc (7500 mpk) daily on mice through oral gavage for 7 days (Supplementary Figure 4A). With γH2AX IHC staining, both glucose and GlcNAc treatment increased DNA damage in pancreas, while GlcNAc also increased DNA damage in the other five organs, including brain, kidney, liver, lung, and colon (Figure 5A, 5B). These results suggest that the DNA-damaging effect of glucose is pancreas-specific, while the effect of GlcNAc is universal in all six tissues examined.
Figure 5.
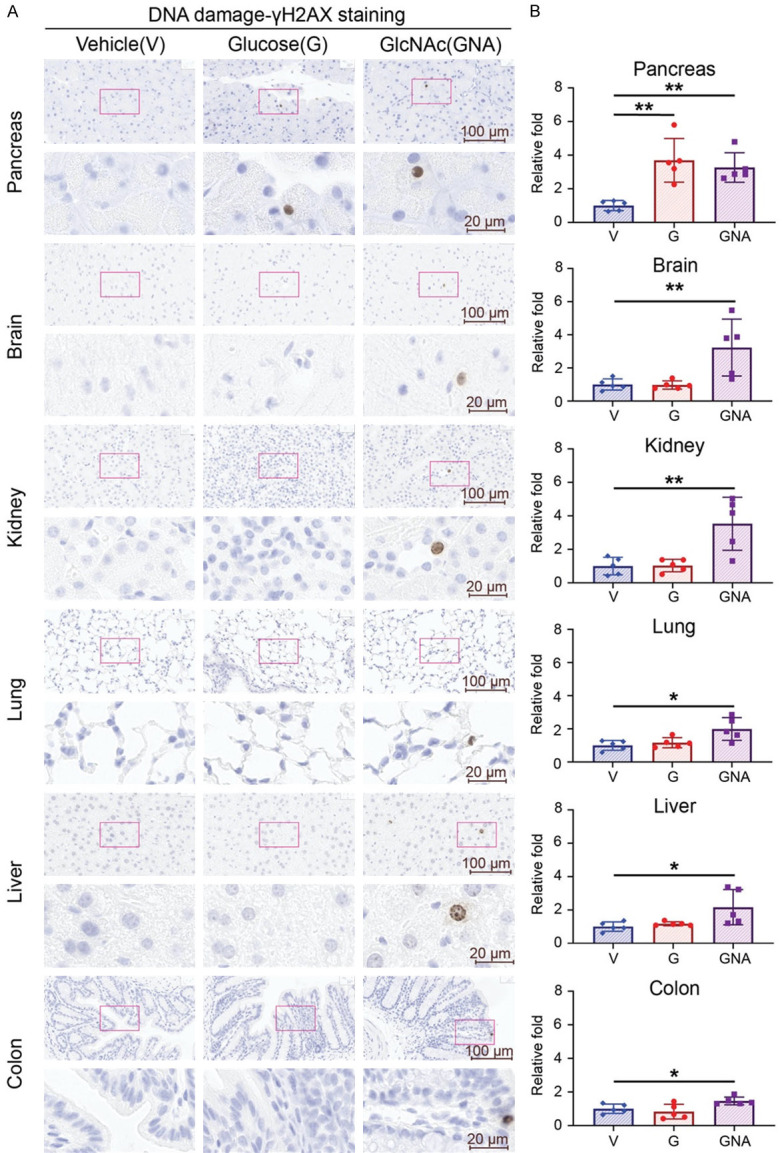
High dose of GlcNAc treatment induces DNA damage in tissues. Five male mice of each group were treated with glucose (G, 15000 mpk) or GlcNAc (GNA, 7500 mpk) or vehicle control (water) for 7 days. Glucose and N-Acetylglucosamine were dissolved in water and administered to mice three times daily by oral gavage at 10 μl/g. (A) Representative images and (B) quantifications of γ-H2AX staining by IHC of pancreas, brain, kidney, lung, liver, and colon from each group. (A) Upper panel: 40× magnification. Scale bar, 100 μm. Lower panel: 200× magnification. Scale bar, 20 μm. (B) Quantitative bar graphs of the relative folds of the percentage of γH2AX positive cells. Each dot represents the datum of one mouse. Data are normalized to that from mice treated with vehicle control. Values show mean ± SEM. *P<0.05; **P<0.01 (two-tailed Student’s t-test).
As GlcNAc is undergoing clinical trials for chondroprotective effect on healthy individuals [19], the potential side effect of long-term use remains unclear. Based on the above findings, we treated mice with equivalent human dose (212 mpk=1X) or 10 times dose (2120 mpk=10X) of GlcNAc daily for 60 days (Supplementary Figure 5A). IHC staining of γH2AX showed that 1X of GlcNAc treatment induced DNA damage among pancreas, lung, and colon. However, 10X treatment universally increased DNA damage among pancreas, brain, liver, lung, kidney, and colon (Figure 6A, 6B). These results show that the dosage of GlcNAc used in clinical trials may impact on genome instability in those tissues examined.
Figure 6.

Long-term GlcNAc treatment induces DNA damage in tissues. Five male mice of each group were treated with 1X GlcNAc (212 mpk=1X), or 10X GlcNAc (2120 mpk=10X) or vehicle control (water) daily for 60 days. GlcNAc was dissolved in water and administered to mice once daily by oral gavage at 10 μl/g. (A) Representative images and (B) quantifications of γ-H2AX staining by IHC of pancreas, brain, kidney, lung, liver, and colon from each group. (A) Upper panel: 40× magnification. Scale bar, 100 μm. Lower panel: 200× magnification. Scale bar, 20 μm. (B) Quantitative bar graphs of the relative folds of the percentage of γH2AX positive cells. Each dot represents the datum of one mouse. Data are normalized to that from mice treated with vehicle. Values show mean ± SEM. *P<0.05; **P<0.01 (two-tailed Student’s t-test).
GlcNAc treatment causes increased DNA damage and a cancer-like property through RRM1-O-GlcNAcylation-mediated dNTP pool imbalance
To further investigate the possible mechanism of action of GlcNAc in normal cells, we treated non-tumorigenic human cell lines, including HPDE, HPNE, MCF10A, NL20, AML12, and C8-D1A, with 2, or 10 mM GlcNAc for 3 days. The levels of UDP-GlcNAc were increased among examined cell lines at the indicated dose in the result of LC/MS analysis (Figure 7A). At the same time, protein O-GlcNAcylation levels were also increased in the results of immunoblotting analysis (Figure 7B) and immunofluorescent staining (Figure 7C). Accordingly, DNA damage was increased among these cell lines under the indicated dose of GlcNAc treatment (Figure 7D). To assess whether GlcNAc treatment enhances anchorage-independent growth, we performed a soft agar colony assay [20]. After 14 days of GlcNAc treatment, all six cell lines had higher colony formation activity in a dose-dependent manner (Figure 7E). Taken together, GlcNAc treatment universally increases O-GlcNAcylation, DNA damage, and anchorage-independent growth.
Figure 7.

GlcNAc treatment increases cellular UDP-GlcNAc levels, protein O-GlcNAcylation, DNA damage, and anchorage-independent growth in various non-tumorigenic cell lines. Non-tumorigenic cell lines, including human pancreatic cells (HPDE and HPNE), human mammary epithelial cells (MCF10A), human lung epithelial cells (NL20), mouse hepatocyte cells (AML12), and mouse astrocyte cells (C8D1A), were used in this study. GlcNAc (2 mM and 10 mM) was added to the culture medium and changed daily. A. The cellular levels of UDP-GlcNAc in various cell lines after treatment in the indicated concentration of GlcNAc for 3 days. The cellular levels of UDP-GlcNAc were determined by targeted LC/MS (n=3). B. Protein O-GlcNAcylation levels in various cell lines treated with the indicated concentration of GlcNAc for 3 days. Cells were analyzed by immunoblotting analysis with an anti-RL2 antibody. Data are normalized to those from vehicle treatment and the amount of internal control protein, p84. C. Quantitative results of the intranuclear intensity of protein O-GlcNAcylation. Various cell lines were treated with the indicated concentration of GlcNAc for 3 days and were subjected to immunofluorescent staining with an anti-RL2 antibody. 2,000 cells were counted in each experiment, and each assay was performed individually 5 times. D. Quantitative results of γH2AX positive cell percentage by immunofluorescent staining in various cell lines treated with the indicated concentration of GlcNAc for 3 days. 2,000 cells were counted in each experiment, and each assay was performed individually 5 times. E. Colony number of soft agar assay among HPDE, HPNE, MCF10A, NL20, AML12, and C8D1A after indicated concentration of GlcNAc treatment for 14 days. Each well contains 500 cells per well, n=5. Values show mean ± SEM. *P<0.05; **P<0.01; ***P<0.001 (two-tailed Student’s t-test).
If GlcNAc treatment induces DNA damage and cell transformation through RRM1-O-GlcNAcylation-mediated dNTP pool imbalance, the DNA damage should be abolished by inhibition of O-GlcNAcylation or dNTP pool restoration. To test this possibility, we immunoprecipitated RRM1 and performed immunoblotting analysis with anti-O-GlcNAcylation antibody (RL2) in four cell lines, including HPDE, HPNE, MCF10A, and NL20, treated with 10 mM of GlcNAc for 3 days. All these cell lines showed an increase of O-GlcNAcylated RRM1 (Figure 8A). To test whether either inhibition of O-GlcNAcylation or supplementation of dNTP pool reverses the increases in DNA damage and soft agar colony formation induced by GlcNAc, we supplemented GlcNAc-treated cell lines with a small molecule OGT inhibitor-OSMI-1 [21] or deoxynucleotides (dN) and measured their DNA damage and colony formation activity. As shown in Figure 8, both DNA damage and colony formation activities were suppressed after being treated with OSM-1 and dN (Figure 8B, 8C).
Figure 8.

GlcNAc induces DNA damage through RRM1 O-GlcNAcylation and dNTP pools imbalance. A. O-GlcNAcylated RRM1 determined by immunoprecipitation with anti-RRM1antibody and immunoblotting analysis with anti-RL2 antibody in various cells treated with 10 mM of GlcNAc for 3 days. Data are normalized to those from vehicle treatment and the amount of immunoprecipitated RRM1. B. Quantitative results of γH2AX positive cell percentage by immunofluorescence staining in various cell lines after 3-day culture with 10 mM of GlcNAc alone, GlcNAc with OGT inhibitor (OSMI-1, 5 μM), or GlcNAc with deoxynucleotide (dN, 5 μM of each). 2,000 cells were counted in each experiment, and each assay was performed individually 5 times. C. Quantitative results of soft agar colony formation in various cell lines after 14-day culture with 10 mM of GlcNAc alone, GlcNAc with OGT inhibitor (OSMI-1, 5 μM), or GlcNAc with deoxynucleotide (dN, 5 μM of each). Each replicate resembles 500 cells per well, n=5. D. Quantifications of γ-H2AX staining by IHC of pancreas, brain, kidney, lung, liver, and colon from GlcNAc and OSMI-1 experiment. Three male mice of each group were treated with GlcNAc (GNA, 7500 mpk) + I.P. solution, GlcNAc (GNA, 7500 mpk) combined with OSMI-1 (1 mpk I.P.), or vehicle (water) + I.P. solution as a control for 7 days as described in methods. Each dot represents the datum of one mouse. Values show mean ± SEM. E. Quantifications of γ-H2AX staining by IHC of the pancreas, brain, kidney, lung, liver, and colon from GlcNAc and deoxynucleotide experiment. Three male mice of each group were treated with GlcNAc (GNA, 7500 mpk), GlcNAc (GNA, 7500 mpk) combined with deoxynucleotide (240 mpk of each P.O.), or vehicle (water) for 7 days as described in methods. Each dot represents the datum of one mouse. Values show mean ± SEM. *P<0.05; **P<0.01; ***P<0.001 (two-tailed Student’s t-test).
To further confirm these findings, we performed similar experiments in GlcNAc-treated mice by giving them OSMI-1 (1 mpk I.P.) [22] (Supplementary Figure 6A), or dN (240 mpk, of each P.O.) [23] (Supplementary Figure 6C). While both administrations did not affect the body weight of these mice (Supplementary Figure 6B, 6D), the GlcNAc-induced DNA damage was diminished by either OSMI-1 or dN in all six tissues examined (Figure 8D, 8E). These results suggest that GlcNAc-induced DNA damage and cell transformation are caused by RRM1-O-GlcNAcylation-mediated dNTP pool imbalance, a mechanism similar to the previous one found in high glucose-treated pancreatic cells [8].
Discussion
The modern dietary pattern, characterized by high-fat dairy products, high-sugar drinks, and low intakes of vegetables, increases the incidence of type II diabetes, coronary heart disease, and cancer [24]. Here, we reported that high sugar consumption in mice induces DNA damage in pancreas but not in the other organs, including brain, liver, lung, kidney, and colon. Unlike sugar that induces pancreas-specific DNA damage, GlcNAc universally causes DNA damage in pancreas, brain, liver, lung, kidney, and colon. Owing to higher GFAT activity and lower PFK activity in pancreas, more glucose is metabolized to UDP-GlcNAc and RRM1-O-GlcNAcylation is increased in pancreas. The O-GlcNAcylated RRM1 triggers ribonucleotide reductase complex disruption, leading to the enzymatic activity reduction and dNTP pool imbalance. GlcNAc-induced DNA damage is also mediated by dNTP pool imbalance through a similar action mechanism of high sugar treatment in pancreas (Supplementary Figure 7).
Glucose is the primary source of energy for most tissues. Glucose consumption leads to an immediate flux in circulation after intestinal absorption. The glycemic surge stimulates insulin secretion of beta cells that regulates glucose absorption of peripheral tissues. In the United States, average Americans consume 126 g of sugar per day, which exceeds the emphasized 10% of energy intake in 2015-2020 Dietary Guidelines for Americans [25]. Those sugar sources mainly come from sweets and beverages which lack essential micronutrients and are processed with sucrose (table sugar) and high-fructose corn syrup. The excessive sugar intake is associated with the critical risk factor for obesity, insulin resistance, cardiovascular diseases, and cancer [26]. In type 2 diabetes, chronic hyperglycemia promotes pro-inflammatory cytokine secretion, NF-kappa B activation, Fas upregulation, DNA fragmentation, and impaired beta-cell function [27]. In addition, exposure to high glucose leads to mitochondria fragmentation and elevation of mitochondrial reactive oxygen species [28], and the advanced glycation end products caused by excessive sugar intake induce oxidative stress and promote cancer growth and metastasis [29]. Interestingly, we found that high glucose or fructose treatment induces DNA damage, specifically in pancreas of mice, suggesting that high sugar intake is the culprit causing diseases.
Pancreas has lower PFK activity and higher GFAT activity, resulting in more UDP-GlcNAc metabolized from glucose, which increases O-GlcNAcylation and DNA damage. Apparently, the intrinsic ratio of GFAT and PFK activity is a critical factor dictating UDP-GlcNAc levels in pancreas. Consistently, either increasing PFK activity or decreasing GFAT1 expression suppresses high-glucose-induced effects in pancreatic cells [8]. It was noted that PFK serves as the gatekeeper of glycolysis, and three isoforms of PFK (PFKL, PFKM, and PFKP) modulate cell metabolic state and energy utilization [30]. Importantly, PFK activity is regulated by citrate acid, ATP, ADP, and AMP, suggesting its fine-tuning for cell energy utilization. The increased mechanical stimulus modulates PFK protein stability in normal lung cells by reducing its ubiquitination and glycolytic rate [31]. In lung cancer cells, PFK1 is O-GlcNAcylated, which decreases its enzymatic activity, redirects glucose flux through the pentose phosphate pathway, and promotes tumor growth [32]. In high-glucose-treated pancreatic cells, three PFK isoforms can also be O-GlcNAcylated and further reduce its activity, resulting in increased O-GlcNAcylation and DNA damage [8]. These data suggest that PFK activity may be regulated by protein expression levels and post-translational modification such as O-GlcNAcylation. However, the physiological role of low PFK activity in the pancreas remains to be explored.
On the other hand, GFAT, serving as the rate-limiting enzyme of HBP for UDP-GlcNAc synthesis, converts glutamine and fructose 6-phosphate to glucosamine-6-phosphate. There are two GFAT protein isoforms known as GFAT1 and GFAT2, encoded by separate genes, gfpt1 and gfpt2, respectively, and located on different chromosomes [33]. They share a 75% homology but have distinct tissue distributions. GFAT1 is highly expressed in the placenta, pancreas, and testis and GFAT2 is expressed throughout the central nervous system, particularly in the spinal cord [34]. In terms of gene regulation, GFAT is transcriptionally regulated by specificity protein 1 (Sp1) [35] and activating transcription factor 4 (ATF4) [36]. Furthermore, mammalian GFAT1 enzymatic activity is suppressed by UDP-GlcNAc [37]. In terms of tumorigenesis, the expression of GFAT1 is higher in human pancreatic cancer samples than adjacent normal tissue [38]. Targeting GFAT increases tumoral sensitivity to immune-checkpoint therapy in pancreatic cancer [39] and increases the cancer-killing activity of immune cells in lung cancer [40]. Since the GFAT inhibitors nowadays are glutamine analogs and have concerns of off-target effects on other glutamine utilizing enzymes, a specific inhibitor aimed at GFAT1 is promising for procrastinating tumor formation and treating cancer.
In recent decades, glucosamine has been used to improve the symptoms and delay the progression of osteoarthritis [41]. Besides, glucosamine is reported to have no adverse effect at very high doses (5000-15,000 mpk) among mice, dogs, rabbits, and horses [42]. In humans, glucosamine sulfate is as safe as a placebo in terms of adverse events and basic physiological characteristics in several randomized trials [43]. Similarly, GlcNAc has no adverse events reported [44]. Based on blood analytic results of our long-term GlcNAc experiment, the equivalent human dose of GlcNAc (212 mpk) did not affect blood cell count and biochemical composition of blood (data not shown).
Interestingly, pancreas has higher relative fold of DNA damage than other tissues under the long-term GlcNAc treatment. There are several possible explanations. Firstly, UDP-GlcNAc levels in the pancreas have already been higher than those in other tissues due to the higher intrinsic ratio of GFAT and PFK activity (Figure 4E). Additionally, the metabolic efficiency of UDP-GlcNAc may be different in various organs. After searching mRNA expression from the GTEx database [45], we found that enzymes that are involved in the GlcNAc salvage pathway (Supplementary Figure 8A) have different levels of RNA expression among the organs (Supplementary Figure 8B-E). The pancreas has a higher PGM3 expression and a lower GALE expression than the other organs, converting more GlcNAc to UDP-GlcNAc and metabolizing fewer UDP-GlcNAc to UDP-GalNAc. The accumulated UDP-GlcNAc can be easily higher in pancreas, leading to higher O-GlcNAcylation and more serious DNA damage in the long-term treatment of GlcNAc. Alternatively, pancreatic tissue may accumulate more DNA damage induced by GlcNAc treatment over time because of lower DNA repair efficiency [46].
On the contrary, epidemiological studies report that glucosamine plus chondroitin reduces colon cancer risk [47]. It is noted that such reduction in cancer risk was observed based on a shorter-duration use. As high O-GlcNAcylation is instrumental in pancreas, lung, and colon malignancy, long-term glucosamine consumption may increase O-GlcNAcylation, which draws the concern of cancer risk. Since pancreatic [48] and colon cancer [49] take more than 10 years to develop into recognizable malignancy, the long-term effect of glucosamine on cancer development needs further investigation.
Cells commonly face DNA damages generated by ROS, ionizing radiation, ultraviolet light, chemicals, hydrolysis, nucleotide imbalance, and replication error. Accordingly, they undergo DNA damage response (DDR) that determines its fate toward DNA repair or apoptosis [50]. However, if DDR is impaired, the genome will suffer mutations from damaged DNA, thereby providing a selective advantage for mutant cells to further develop into malignant cells [51]. As reported in our previous study, high glucose-induced DNA damage promotes oncogenic KRAS mutation and anchorage-independent growth through the increased O-GlcNAcylation in the pancreatic cells. In the communication, we also demonstrate that high glucosamine treatment induces DNA damage and anchorage-independent growth through the increased O-GlcNAcylation in various cells (Figure 8A, 8B). Furthermore, either OGT inhibition or deoxynucleotides supplementation rescues glucosamine-induced DNA damage in mice tissues (Figure 8C, 8D), suggesting that combining these approaches may minimize DNA damaging effects but sustain therapeutic benefits of glucosamine. Further development of useful OGT inhibitors or nucleotide precursors is of interest to pursue.
Acknowledgements
We thank Junglee Lin and Chialin Wu (Mass Core Facility, Genomics Research Center, Academia Sinica, Taiwan) for analyzing mouse tissue metabolites using LC-MS/MS and data processing. We thank Chihyu Lin and Gongmin Lin (Metabolomics Core Facility, Agricultural Biotechnology Research Center, Academia Sinica, Taiwan) for UPLC-MS/MS Label-U13C glucose analysis and data processing. We thank National Laboratory Animal Center (NARLabs, Taiwan) for animal supply. This research work was supported by funds from Academia Sinica and the Ministry of Science and Technology, Taiwan (AS-SUMMIT-109, MOST 110-2326-B-001-012, MOST 110-2311-B-001-016-MY3); and the higher education sprout project by the Ministry of Education, Taiwan via the “Drug Development Center of China Medical University” W-H.L.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.GBD 2017 Pancreatic Cancer Collaborators. The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2019;4:934–947. doi: 10.1016/S2468-1253(19)30347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gapstur SM, Gann PH, Lowe W, Liu K, Colangelo L, Dyer A. Abnormal glucose metabolism and pancreatic cancer mortality. JAMA. 2000;283:2552–2558. doi: 10.1001/jama.283.19.2552. [DOI] [PubMed] [Google Scholar]
- 3.Michaud DS, Liu S, Giovannucci E, Willett WC, Colditz GA, Fuchs CS. Dietary sugar, glycemic load, and pancreatic cancer risk in a prospective study. J Natl Cancer Inst. 2002;94:1293–1300. doi: 10.1093/jnci/94.17.1293. [DOI] [PubMed] [Google Scholar]
- 4.Li W, Liu H, Qian WK, Cheng L, Yan B, Han L, Xu QH, Ma QY, Ma JG. Hyperglycemia aggravates microenvironment hypoxia and promotes the metastatic ability of pancreatic cancer. Comput Struct Biotechnol J. 2018;16:479–487. doi: 10.1016/j.csbj.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Supabphol S, Seubwai W, Wongkham S, Saengboonmee C. High glucose: an emerging association between diabetes mellitus and cancer progression. J Mol Med (Berl) 2021;99:1175–1193. doi: 10.1007/s00109-021-02096-w. [DOI] [PubMed] [Google Scholar]
- 6.Zhou CC, Qian WK, Li J, Ma JG, Chen X, Jiang ZD, Cheng L, Duan WX, Wang Z, Wu Z, Ma QY, Li XQ. High glucose microenvironment accelerates tumor growth via SREBP1-autophagy axis in pancreatic cancer. J Exp Clin Cancer Res. 2019;38:302. doi: 10.1186/s13046-019-1288-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li W, Ma ZH, Ma JG, Li XQ, Xu QH, Duan WX, Chen X, Lv YF, Zhou S, Wu EX, Ma QY, Huo XW. Hydrogen peroxide mediates hyperglycemia-induced invasive activity via ERK and p38 MAPK in human pancreatic cancer. Oncotarget. 2015;6:31119–31133. doi: 10.18632/oncotarget.5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu CM, Tien SC, Hsieh PK, Jeng YM, Chang MC, Chang YT, Chen YJ, Chen YJ, Lee E, Lee WH. High glucose triggers nucleotide imbalance through O-GlcNAcylation of key enzymes and induces KRAS mutation in pancreatic cells. Cell Metab. 2019;29:1334–1349. doi: 10.1016/j.cmet.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Chatham JC, Zhang J, Wende AR. Role of O-linked N-acetylglucosamine protein modification in cellular (Patho) physiology. Physiol Rev. 2021;101:427–493. doi: 10.1152/physrev.00043.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fardini Y, Dehennaut V, Lefebvre T, Issad T. O-GlcNAcylation: a new cancer hallmark? Front Endocrinol (Lausanne) 2013;4:99. doi: 10.3389/fendo.2013.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017;18:452–465. doi: 10.1038/nrm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye F, Maegawa H, Morino K, Kashiwagi A, Kikkawa R, Xie M, Shen Z. A simple and sensitive method for glutamine: fructose-6-phosphate amidotransferase assay. J Biochem Biophys Methods. 2004;59:201–208. doi: 10.1016/j.jbbm.2003.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Yeh CJ, Chang HY, Pan WH. Time trend of obesity, the metabolic syndrome and related dietary pattern in Taiwan: from NAHSIT 1993-1996 to NAHSIT 2005-2008. Asia Pac J Clin Nutr. 2011;20:292–300. [PubMed] [Google Scholar]
- 14.Stanhope KL. Sugar consumption, metabolic disease and obesity: the state of the controversy. Crit Rev Clin Lab Sci. 2016;53:52–67. doi: 10.3109/10408363.2015.1084990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mah LJ, El-Osta A, Karagiannis TC. Gamma H2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–686. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 16.Chiaradonna F, Ricciardiello F, Palorini R. The nutrient-sensing hexosamine biosynthetic pathway as the hub of cancer metabolic rewiring. Cells. 2018;7:53. doi: 10.3390/cells7060053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunaway GA. A review of animal phosphofructokinase isozymes with an emphasis on their physiological role. Mol Cell Biochem. 1983;52:75–91. doi: 10.1007/BF00230589. [DOI] [PubMed] [Google Scholar]
- 18.Akella NM, Ciraku L, Reginato MJ. Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019;17:52. doi: 10.1186/s12915-019-0671-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomonaga A, Watanabe K, Fukagawa M, Suzuki A, Kurokawa M, Nagaoka I. Evaluation of the effect of N-acetyl-glucosamine administration on biomarkers for cartilage metabolism in healthy individuals without symptoms of arthritis: a randomized double-blind placebo-controlled clinical study. Exp Ther Med. 2016;12:1481–1489. doi: 10.3892/etm.2016.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du F, Zhao XD, Fan DM. Soft agar colony formation assay as a hallmark of carcinogenesis. Bio Protoc. 2017;7:e2351. doi: 10.21769/BioProtoc.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ, Walker S. A small molecule that inhibits OGT activity in cells. ACS Chem Biol. 2015;10:1392–1397. doi: 10.1021/acschembio.5b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee KS, Lee J, Lee P, Jeon BC, Song MY, Kwak S, Lee J, Kim JS, Kim DJ, Kim JH, Tesh VL, Lee MS, Park SK. Inhibition of O-GlcNAcylation protects from Shiga toxin-mediated cell injury and lethality in host. EMBO Mol Med. 2022;14:e14678. doi: 10.15252/emmm.202114678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Gomez C, Levy RJ, Sanchez-Quintero MJ, Juanola-Falgarona M, Barca E, Garcia-Diaz B, Tadesse S, Garone C, Hirano M. Deoxycytidine and deoxythymidine treatment for thymidine kinase 2 deficiency. Ann Neurol. 2017;81:641–652. doi: 10.1002/ana.24922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tilman D, Clark M. Global diets link environmental sustainability and human health. Nature. 2014;515:518–522. doi: 10.1038/nature13959. [DOI] [PubMed] [Google Scholar]
- 25.DeSalvo KB. Public health 3.0: applying the 2015-2020 dietary guidelines for Americans. Public Health Rep. 2016;131:518–521. doi: 10.1177/0033354916662207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malik VS, Popkin BM, Bray GA, Despres JP, Hu FB. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation. 2010;121:1356–1364. doi: 10.1161/CIRCULATIONAHA.109.876185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110:851–860. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turner DP. Advanced glycation end-products: a biological consequence of lifestyle contributing to cancer disparity. Cancer Res. 2015;75:1925–1929. doi: 10.1158/0008-5472.CAN-15-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vora S. Isozymes of phosphofructokinase. Isozymes Curr Top Biol Med Res. 1982;6:119–167. [PubMed] [Google Scholar]
- 31.Park JS, Burckhardt CJ, Lazcano R, Solis LM, Isogai T, Li LQ, Chen CS, Gao BN, Minna JD, Bachoo R, DeBerardinis RJ, Danuser G. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature. 2020;578:621–626. doi: 10.1038/s41586-020-1998-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA 3rd, Peters EC, Driggers EM, Hsieh-Wilson LC. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKnight GL, Mudri SL, Mathewes SL, Traxinger RR, Marshall S, Sheppard PO, O’Hara PJ. Molecular cloning, cDNA sequence, and bacterial expression of human glutamine: fructose-6-phosphate amidotransferase. J Biol Chem. 1992;267:25208–25212. [PubMed] [Google Scholar]
- 34.Oki T, Yamazaki K, Kuromitsu J, Okada M, Tanaka I. cDNA cloning and mapping of a novel subtype of glutamine:fructose-6-phosphate amidotransferase (GFAT2) in human and mouse. Genomics. 1999;57:227–234. doi: 10.1006/geno.1999.5785. [DOI] [PubMed] [Google Scholar]
- 35.Sayeski PP, Wang D, Su K, Han IO, Kudlow JE. Cloning and partial characterization of the mouse glutamine:fructose-6-phosphate amidotransferase (GFAT) gene promoter. Nucleic Acids Res. 1997;25:1458–1466. doi: 10.1093/nar/25.7.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaveroux C, Sarcinelli C, Barbet V, Belfeki S, Barthelaix A, Ferraro-Peyret C, Lebecque S, Renno T, Bruhat A, Fafournoux P, Manie SN. Nutrient shortage triggers the hexosamine biosynthetic pathway via the GCN2-ATF4 signalling pathway. Sci Rep. 2016;6:27278. doi: 10.1038/srep27278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruegenberg S, Horn M, Pichlo C, Allmeroth K, Baumann U, Denzel MS. Loss of GFAT-1 feedback regulation activates the hexosamine pathway that modulates protein homeostasis. Nat Commun. 2020;11:687. doi: 10.1038/s41467-020-14524-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang C, Peng P, Li L, Shao M, Zhao J, Wang L, Duan F, Song S, Wu H, Zhang J, Zhao R, Jia D, Zhang M, Wu W, Li C, Rong Y, Zhang L, Ruan Y, Gu J. High expression of GFAT1 predicts poor prognosis in patients with pancreatic cancer. Sci Rep. 2016;6:39044. doi: 10.1038/srep39044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma NS, Gupta VK, Garrido VT, Hadad R, Durden BC, Kesh K, Giri B, Ferrantella A, Dudeja V, Saluja A, Banerjee S. Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. J Clin Invest. 2020;130:451–465. doi: 10.1172/JCI127515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen W, Saxton B, Tessema M, Belinsky SA. Inhibition of GFAT1 in lung cancer cells destabilizes PD-L1 protein. Carcinogenesis. 2021;42:1171–1178. doi: 10.1093/carcin/bgab063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silbert JE. Dietary glucosamine under question. Glycobiology. 2009;19:564–567. doi: 10.1093/glycob/cwp014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson JW, Nicolosi RJ, Borzelleca JF. Glucosamine effects in humans: a review of effects on glucose metabolism, side effects, safety considerations and efficacy. Food Chem Toxicol. 2005;43:187–201. doi: 10.1016/j.fct.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 43.Reginster JY, Neuprez A, Lecart MP, Sarlet N, Bruyere O. Role of glucosamine in the treatment for osteoarthritis. Rheumatol Int. 2012;32:2959–2967. doi: 10.1007/s00296-012-2416-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kubomura D, Ueno T, Yamada M, Tomonaga A, Nagaoka I. Effect of N-acetylglucosamine administration on cartilage metabolism and safety in healthy subjects without symptoms of arthritis: a case report. Exp Ther Med. 2017;13:1614–1621. doi: 10.3892/etm.2017.4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.GTEx Consortium. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yimit A, Adebali O, Sancar A, Jiang Y. Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nat Commun. 2019;10:309. doi: 10.1038/s41467-019-08290-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kantor ED, Lampe JW, Peters U, Shen DD, Vaughan TL, White E. Use of glucosamine and chondroitin supplements and risk of colorectal cancer. Cancer Causes Control. 2013;24:1137–1146. doi: 10.1007/s10552-013-0192-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iacobuzio-Donahue CA. Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut. 2012;61:1085–1094. doi: 10.1136/gut.2010.236026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonnington SN, Rutter MD. Surveillance of colonic polyps: are we getting it right? World J Gastroenterol. 2016;22:1925–1934. doi: 10.3748/wjg.v22.i6.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blanpain C, Mohrin M, Sotiropoulou PA, Passegue E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell. 2011;8:16–29. doi: 10.1016/j.stem.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 51.Rossi DJ, Jamieson CH, Weissman IL. Stems cells and the pathways to aging and cancer. Cell. 2008;132:681–696. doi: 10.1016/j.cell.2008.01.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.