

Abstract
Nuclear DNA viruses simultaneously access cellular factors that aid their life cycle while evading inhibitory factors by localizing to distinct nuclear sites. Adeno-associated viruses (AAVs), which are Dependoviruses in the family Parvovirinae, are non-enveloped icosahedral viruses, which have been developed as recombinant AAV vectors to express transgenes. AAV2 expression and replication occur in nuclear viral replication centers (VRCs), which relies on cellular replication machinery as well as coinfection by helper viruses such as adenoviruses or herpesviruses, or exogenous DNA damage to host cells. AAV2 infection induces a complex cellular DNA damage response (DDR), in response to either viral DNA or viral proteins expressed in the host nucleus during infection, where VRCs co-localized with DDR proteins. We have previously developed a modified iteration of a viral chromosome conformation capture (V3C-seq) assay to show that the autonomous parvovirus minute virus of mice localizes to cellular sites of DNA damage to establish and amplify its replication. Similar V3C-seq assays to map AAV2 show that the AAV2 genome co-localized with cellular sites of DNA damage under both non-replicating and replicating conditions. The AAV2 non-structural protein Rep 68/78, also localized to cellular DDR sites during both non-replicating and replicating infections, and also when ectopically expressed. Ectopically expressed Rep could be efficiently re-localized to DDR sites induced by micro-irradiation. Recombinant AAV2 gene therapy vector genomes derived from AAV2 localized to sites of cellular DNA damage to a lesser degree, suggesting that the inverted terminal repeat origins of replication were insufficient for targeting.
Introduction
Adeno-associated viruses (AAVs) are non-pathogenic human parvoviruses that have been effectively developed as gene therapy vectors for the treatment of numerous human diseases (1). AAV2 is a small non-enveloped icosahedral virus in the genus Dependoparvovirus that packages a linear genome of single-stranded DNA (2). The AAV2 genome is approximately 4.7 kilobases long with two large open reading frames, Rep and Cap, encoding the regulatory Rep and structural Capsid proteins respectively, flanked by inverted terminal repeats (ITRs), which serve as replication origins. The virus encodes four non-structural Rep proteins (Rep 78, Rep 68, Rep 52 and Rep 40), which are essential for numerous facets of the virus life cycle, as well as the assembly activating protein (AAP), membrane associated accessory protein (MAAP) and Protein X proteins (3–8). The large isoforms, Rep 78 and Rep 68, have origin binding, helicase and site-specific endonuclease activities and are essential for viral DNA replication, regulation of gene expression and site-specific integration (9–12). Rep 40 and Rep 52 play roles in loading the single-stranded DNA genomes into the capsid (13).
Due to their limited coding capacity, parvoviruses depend heavily on cellular functions for replication. Unlike most parvoviruses that replicate autonomously in their respective host, AAV2 requires the coinfection of a helper virus, such as adenovirus (Ad) or herpes virus (HSV), or exogenous DNA damage, in order to propagate (14,15). Interestingly, it has recently been suggested that AAV2 genome replication proceeds differently in the presence of HSV compared with Ad (16). In the absence of helper functions, viral replication and gene expression are limited. Under these conditions, AAV2 establishes a latent infection, persisting in cells either as episomes or as integrated genomes in rare cases (17–19). Integration of the AAV2 genome preferentially occurs at a specific locus on human chromosome 19 (19q13.42) known as AAVS1 (20,21). Site-specific integration is mediated by the large regulatory proteins Rep 68/78 that bind to Rep-binding elements (RBEs) present on the viral ITRs and within AAVS1 (22–24). More recently, an unbiased genome-wide analysis of AAV2 integration site revealed that integration is not restricted to AAVS1; rather Rep 68/78 targets AAV2 to integrate at numerous loci in the human genome in the vicinity of consensus Rep-binding sites in a cell type-specific manner (17,18,25,26). These findings suggested that AAV2 Rep 68/78, by virtue of their DNA binding and nicking activities, likely play a role in AAV2 genome localization and integration.
Upon helper virus coinfection, AAV2 can be rescued from latency and undergoes productive infection characterized by genome replication, expression of viral genes and progeny virus production (27). AAV2 replicates by a strand displacement mechanism, primed from the viral ITRs, likely utilizing the cellular DNA polymerase δ (28). The single-stranded DNA genome is converted into duplex replicative intermediate, which also serves as the primary template for viral transcription (29,30). Upon expression, Rep 68/78 binds site-specifically and introduces a single-strand nick at the terminal resolution site (TRS) within the ITR sequence to resolve the duplex structure and facilitate ongoing genome replication (31).
Many nuclear DNA viruses replicate in distinct subnuclear compartments, termed viral replication centers (VRCs), which accumulate essential viral and cellular factors creating an optimal environment for viral gene expression and genome replication (32,33). Similarly, during productive infection, AAV2 is recruited into helper virus VRCs taking advantage of helper as well as cellular functions to promote its own replication (34–36). AAV2 infection induces a DNA-PK-dependent DNA damage response (DDR) in cells that coincide with the nuclear VRCs (37). It has been reported that the viral genome itself can provoke a cellular DNA damage response characteristic of an aberrant replication fork, where the MRE11-RAD50-NBS1 (MRN) complex can bind the AAV2 ITRs (38). In addition, ectopic expression of the large Rep proteins, Rep 78 and Rep 68 in the absence of viral infection can trigger a DDR signal, perhaps due to their nicking capabilities and by inducing cell cycle arrest (37,39). However, DDR signals are more robust when the virus is replicating, suggesting that a combination of ITRs, replication intermediates, Rep and helper functions activate the full DNA damage response pathway (37).
Although prior studies have extensively mapped the cellular integration sites of the AAV2 genome, the specific location following infection, of unintegrated nuclear AAV2 viral genomes and Rep proteins relative to the host genome remains unknown. In order to map the localization sites of non-integrative DNA viruses, we have previously developed a generally adaptable high-throughput viral chromosome conformation capture assay (V3C-seq) for use in trans that allows genome-wide identification of the direct interactions of a lytic virus genome with distinct regions of the cellular chromosome. This assay allowed us to demonstrate that the autonomous parvovirus minute virus of mice (MVM) interacts with sites of cellular DNA damage to establish and amplify its lytic infection (40). Such sites contain DNA replication and gene expression factors (41), which could help support viral infection (4). A similar approach was recently used to examine the interaction of the pseudorabies virus (PRV) genome with the cellular genome during its infection (42). Additionally, we have shown that the major replication protein of MVM, NS1, aids in the localization of the MVM genome to sites of DNA damage (43). Here, using V3C-seq adapted to AAV2, together with chromatin immunoprecipitation followed by sequencing (ChIP-seq) for Rep 68/78 and the DNA damage marker gamma-H2AX (γH2AX), we have mapped the localization of AAV2 genomes and the large Rep proteins to distinct cellular sites, many of which coincide with Rep-occupied sites and cellular sites of DNA damage. These regions are, in general, distinct from that of recombinant AAV2 vector localization.
Results
The AAV2 genome and Rep localize to cellular sites of DNA damage during non-productive AAV2 infection
Previous work has associated AAV2 infection, recombinant AAV2 transduction and AAV2 Rep 68/78 with both cellular DNA damage and aspects of the cellular DDR (37–39,44–48). Other parvoviruses have shown similar traits (49), and we have recently developed a modified chromosome conformation capture assay (V3C-seq) showing that the autonomous parvovirus MVM localizes to sites of cellular DNA damage, via its large non-structural protein NS1, to establish and amplify its infection (40,43). To begin to investigate specific interactions between the AAV and cellular genomes during infection, we initially chose to determine sites to which the AAV2 genome localized within the infected cell nucleus in a non-productive AAV2 system, performing AAV2 V3C-seq with our viewpoint at the AAV2 Hind III site downstream of the P40 promoter (approximately in the middle of the AAV2 genome), following infection of non-synchronous U2OS human osteosarcoma cells in the absence of helper functions. All sites on the cellular genome to which AAV2 directly associated were compared with cellular sites occupied by γH2AX as a marker for sites of cellular DNA damage, determined by specific ChIP-seq. Analysis of two independent biological replicates indicated that approximately 45% of the cellular sites at which the AAV2 genome associated 24 h post infection (hpi), as identified by V3C-seq, overlapped with cellular sites exhibiting DNA damage (Fig. 1A; compare columns 1–3; quantification of genome-wide heatmaps in Supplementary Material, Fig. S1A). A recombinant AAV2 vector expressing a GFP transgene (rAAV2) showed significantly less frequent localization to γH2AX sites above background levels following transduction of U2OS cells (20% of the cellular γH2AX sites overlapped with rAAV2 V3C-seq; Fig. 1A; column 2; quantification of genome-wide heatmaps in Supplementary Material, Fig. S1A). These genome-wide studies indicated that following wild-type virus infection under non-productive conditions, the AAV2 genome preferentially localized to sites of DNA damage, whereas transduced recombinant AAV2 genomes did so at a significantly lesser frequency. These results suggested further, that the viral hairpins were not sufficient to target the AAV2 genome to cellular sites of damage. Importantly, we were unable to highly synchronize U2OS cells without causing additional DNA damage. Because we were restricted to analysis in non-synchronous populations rather than focusing on cells primarily in S-phase, our results likely underestimated the localization of the AAV2 genome to sites of DNA damage.
Figure 1.
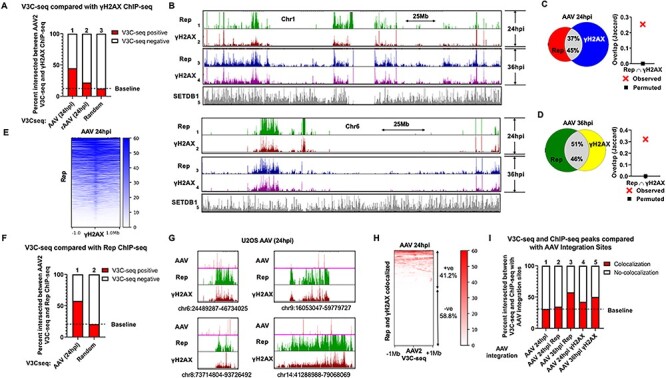
The AAV2 genome and Rep localize to cellular sites of DNA damage during non-productive AAV2 infection. (A) Genome-wide occupancy of γH2AX ChIP-seq peaks during the indicated conditions in U2OS cells were compared with the corresponding AAV2 genome localization (identified by V3C-seq with the viewpoint on the AAV2 or rAAV2 genomes). The presence of γH2AX peaks within 2 Mb windows of V3C-seq peaks were scored as ‘V3C-seq positive.’ The heatmaps underlying this data are presented in Supplementary Material, Figure S1A. (B) Rep and γH2AX ChIP-seq experiments in U2OS cells infected with AAV2 for 24 (tracks 1–2) or 36 h (tracks 3–4). Representative histograms for Rep and γH2AX binding to human chromosome 1 (top half) and chromosome 6 (bottom half) are shown. Genome-wide association of the chromatin modifier SETDB1 previously identified in U2OS cells was monitored as negative control (55). (C, D) Venn diagrams representing whole-genome comparison of the overlap of Rep and γH2AX ChIP-seq peaks in U2OS cells infected with AAV2 for 24 h (C, left panel) and 36 h (D, left panel). The statistical significance of the overlap is shown via Jaccard analysis (see Materials and Methods and (43,76)). (E) Heatmap of Rep association with 2 Mb windows of γH2AX peaks. Data are presented for Rep localization relative to γH2AX in AAV2 infected U2OS cells at 24 hpi. (F) V3C-seq during AAV2 infection of U2OS cells at 24 hpi was compared with the corresponding sites of Rep that were present in the sample. Findings were compared with a randomly permuted ‘baseline’ as described above. The heatmaps underlying this data are presented in Supplementary Material, Figure S1B. (G) The association of the AAV2 genome with sites on cellular DNA was mapped by V3C-seq assays (top panels) and was compared with Rep and γH2AX binding, both of which were monitored by respective ChIP-seq assays at 24 hpi in unsynchronized U2OS human osteosarcoma cells. Representative examples of the V3C-seq peaks and ChIP-seq peaks occupying distinct regions on human chromosomes 6, 8, 9 and 14 are shown. The data represent called peaks that are shared between two independent replicates of AAV2 infections of U2OS cells. The y-axis scale for V3C-seq is from 3 to 100 reads, and y-axis scale for the ChIP-seq is 0–400 reads. (H) Heatmap of AAV2 association relative to cellular sites containing intersected Rep and γH2AX peaks within a 2 Mb window. Regions that contained red signals in the heatmap were designated as V3C-seq positive (+ve) and lacking red signals were designated as V3C-seq negative (−ve). (I) AAV2 V3C-seq and ChIP-seq results at the indicated time points post infection with AAV2 in U2OS cells were intersected with 50 kilobase windows on either side of previously identified AAV2 integration sites using DeepTools software on the Galaxy project platform (25,26). Intersection of V3C-seq and ChIP-seq peaks with these integration sites were scored positive for ‘colocalization,’ whereas lack of interaction was scored as ‘no-colocalization.’ The heatmaps underlying this data are presented in Supplementary Material, Figure S1C. All datasets are intersection of two independent replicates of U2OS cell infections. A full explanation of how the intersection between the V3C-seq and ChIP-seq data was determined, as well as the analysis of their significance, for Figures 1–3, is presented in Materials and Methods.
Previous results have shown that the MVM NS1 protein localizes to sites of cellular DNA damage as shown by association with cellular sites occupied by γH2AX (43,50,51). A portion of that interaction was due to NS1 bound to the MVM genome as it associated with these sites—essentially another marker for genome localization; however, NS1 expressed by itself from stable cell lines also localized to damage sites (43). A similar strong correlation between sites occupied by the very low levels of Rep expressed during non-replicative infection of U2OS cells, as determined by specific ChIP assay, and sites occupied by γH2AX, could be seen at both 24 and 36 hpi (Fig. 1B shows the individual sites for representative chromosomes 1 and 6; compare tracks 1 and 2; 3 and 4; Supplementary Material, Fig. S2 shows localization sites throughout the human genome). Although the anti-Rep antibody used for these and subsequent studies interact with both large and small Rep proteins, we assume that these ChIP assays do not assess interactions of the small Rep proteins that lack DNA-binding capabilities (34,52–54). A genome-wide analysis of two independent biological replicates showed that approximately 45% of Rep-occupied sites overlapped with γH2AX-occupied sites at both 24 and 36 h post AAV2 infection of U2OS cells (Fig. 1C and D, respectively; Jaccard analysis is shown to validate the statistical significance of the overlap), similar to the overlap between V3C-seq results (Fig. 1A; column 1) and γH2AX. By 36 hpi, a greater percentage of γH2AX sites were occupied by Rep (37–51%, compare Fig. 1C–D; left panels). Genome-wide analysis of called peaks indicated that approximately 90% of the Rep peaks were centered in the 1 Megabase (Mb) vicinity of γH2AX peaks at 24 hpi (Fig. 1E). The negative control background for comparison was published ChIP-seq data for the epigenetic modifier SETDB1, a known genome-binding protein (55) (Fig. 1B and Supplementary Material, Fig. S2; track 5).
As both the AAV2 genome and Rep localized to specific cellular sites of DNA damage, it was of interest to determine the overlap between the two target sites. Comparison at the genomic level of two independent biological replicates of the AAV2 V3C-seq analysis with the Rep ChIP-seq analysis showed that approximately 55% of the cellular sites at which the AAV2 genome associated overlapped with sites of Rep binding (Fig. 1F; column 1; quantification of genome-wide heatmaps in Supplementary Material, Fig. S1B). A finer magnification (Fig. 1G) of representative interaction peaks at distinct cellular sites on human chromosomes 6, 8, 9 and 14 showed the AAV2 genome (represented as red peaks on the top tracks in Fig. 1G) localized to cellular regions that were bound by Rep (Fig. 1G; green middle tracks) and γH2AX (Fig. 1G; bottom dark red tracks). Under our experimental conditions, Rep ChIP-seq would be expected to detect both genome-associated, as well as non-genome-associated Rep. This will be addressed further, below. Comparison of Rep and γH2AX ChIP-seq with AAV2 V3C-seq revealed that 41% of cellular sites at which both Rep and γH2AX colocalized also had AAV2 genomes associated within a 2 Mb window of the overlapping ChIP-seq peaks (Fig. 1H).
Interestingly, we found that there was a significant correlation over background between cellular sites bound by both Rep and γH2AX (as determined by ChIP-seq), and AAV2 integration sites that have previously been identified in HeLa cells and human fibroblasts ((25,26)) (Fig. 1I; lanes 3–5; quantification of genome-wide heatmaps in Supplementary Material, Fig. S1C). This may be reflective of the observation that the percentage of γH2AX sites that were found to overlap with Rep-occupied sites increased between 24 and 36 hpi (compare 37% in Fig. 1C with 51% in 1D respectively; described above). However, surprisingly, there seemed to be no specific correlation between viral genome-associated sites and integration sites in U2OS cells (Fig. 1I; lane 1).
The AAV2 genome and Rep localize to cellular sites of DNA damage during productive AAV2 infection
To examine AAV2 localization under conditions where the virus was replicating, we chose to examine AAV2 infection of 293T cells transfected with the pHelper plasmid expressing the Adenovirus type 2 (Ad) E2A, E4 and VA-RNA (2), since following Ad viral coinfection, AAV2 localization is determined by formation of Ad replication centers (36). As shown in Figure 2A, AAV2 infection in 293T cells that express pHelper led to robust AAV2 genome replication and Rep expression at both 16 and 24 hpi.
Figure 2.
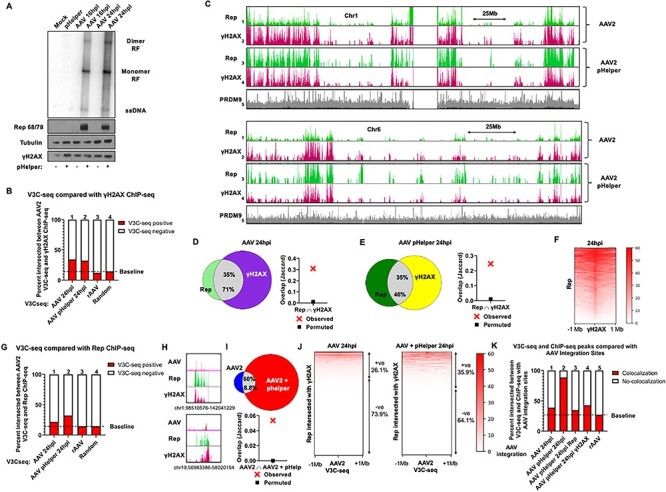
The AAV2 genome and Rep localize to cellular sites of DNA damage during productive AAV2 infection. (A) AAV2 replication and Rep expression during infection in the presence or absence of helper functions in human embryonic kidney 293T cells. Cells were mock transfected or transfected with pHelper plasmid for 24 h and infected with AAV2 at a MOI of 5000 viral genomes per cell. Cells were harvested at the indicated timepoint and processed for Southern blot (top panel) and western blot analysis (bottom panel) as described in Materials and Methods. The Southern blot was hybridized with radiolabeled AAV2 probe. The replicative intermediate forms monomer RF and dimer RF are indicated on the right. For western blot analysis, whole-cell lysates were assayed using antibodies directed against the indicated proteins. (B) Genome-wide occupancy of γH2AX ChIP-seq peaks during the indicated conditions in 293T cells were compared with the corresponding AAV2 genome localization (identified by V3C-seq with the viewpoint on the AAV2 genome) as described for Fig. 1A. The heatmaps underlying this data are presented in Supplementary Material, Fig. S3A. (C) Rep and γH2AX ChIP-seq assays were performed in 293T cells infected with AAV2 for 24 h in the presence (tracks 3–4) or absence of pHelper (tracks 1–2). Representative histograms for Rep and γH2AX binding to human chromosome 1 (top half) and chromosome 6 (bottom half) are shown. Genome-wide association of the chromatin modifier PRDM9 previously identified in 293T cells was monitored as negative control (56). (D, E) Venn diagrams representing whole-genome comparison of the overlap of Rep and γH2AX ChIP-seq peaks in 293T cells infected with AAV2 for 24 h in the absence (D, left panel) or presence of pHelper plasmid (E, left panel). The statistical significance of the overlap is shown via Jaccard analysis in the respective right panels. (F) Heatmap of the location of Rep ChIP-seq peaks relative to γH2AX within 2 Mb windows. Data are presented for Rep localization relative to γH2AX in AAV2 infected 293T cells at 24 hpi. (G) Summary of genome-wide V3C-seq of AAV2 during infection of 293T cells in the presence and in the absence of pHelper plasmid were compared with the corresponding sites of Rep that were present in each sample (as described in Fig. 2B). Samples were compared with 293T cells transduced with a control recombinant AAV2 vector expressing a GFP transgene. The heatmaps underlying this data are presented in Supplementary Material, Fig. S3B. (H) Representative examples of the V3C-seq peaks and ChIP-seq peaks occupying the indicated regions on human chromosomes 1 and 19. The y-axis scale for V3C-seq is from 3 to 100 reads, and y-axis scale for the ChIP-seq is 0–400 reads. (I) Intersection of AAV2 localization sites at 24 hpi in the absence (left) and presence (right) of pHelper expression in 293T cells. The statistical significance of the intersection was computed using Jaccard analysis and shown in the bottom half. (J) Heatmap of AAV2 association relative to cellular sites that contain both Rep and γH2AX peaks at 24 hpi in the absence (left heatmap) and presence of pHelper (right heatmap) within a 2 Mb window containing the peak. Data represent the magnitude of V3C-seq signals on either side of the common ChIP-seq peaks. Regions that contained red signals in the heatmap were designated as V3C-seq positive (+ve) and lacking red signals were designated as V3C-seq negative (−ve). (K) AAV2 V3C-seq and ChIP-seq results at the indicated conditions were intersected with 50 kilobase windows on either side of previously identified AAV2 integration sites (25,26). Intersection of V3C-seq and ChIP-seq peaks with these integration sites was scored positive for ‘colocalization,’ whereas lack of interaction was scored as ‘no-colocalization.’ All datasets are intersection of two independent replicates of 293T cell infections. The heatmaps underlying this data are presented in Supplementary Material, Fig. S3C.
At 24 hpi, first in the absence of pHelper, a significant number of AAV2-associated genomic sites (identified by V3C-seq and shared between two independent biological replicates) overlapped with cellular sites exhibiting DNA damage (identified by γH2AX ChIP-seq; Fig. 2B; column 1). This association with γH2AX-binding sites was maintained, but not increased, in the presence of pHelper (Fig. 2B; column 2; quantification of genome-wide heatmaps in Supplementary Material, Fig. S3A), suggesting, perhaps, saturation of target sites. As with the U2OS experiments described above, these analyses could only be performed under non-synchronous conditions and so may underestimate these associations. rAAV2 vectors did not show significant localization to γH2AX sites above background levels following transduction of 293T cells (Fig. 2B; lanes 3 and 4, respectively; quantification of genome-wide heatmaps in Supplementary Material, Fig. S3A), suggesting, as also mentioned above, that the viral hairpins were not sufficient to target the AAV2 genome to cellular sites of damage. Thus, in 293T cells, in both the absence and presence of helper functions, similar to U2OS cells, genome-wide analysis indicated that following infection, AAV2 genomes, but not transduced rAAV2 genomes, preferentially localized to cellular sites of DNA damage.
As shown in the representative genome browser tracks for AAV2 infected 293T cells, a strong correlation could be seen between Rep and γH2AX ChIP-seq profiles at 24 hpi both in the absence and presence of pHelper (Fig. 2C shows representative chromosomes 1 and 6; Supplementary Material, Fig. S4 shows all human chromosomes). As with U2OS cells described above, although only low levels of Rep below detection by western analysis were generated in the absence of pHelper, ChIP of Rep under these conditions could be readily analyzed. The background for comparison is published ChIP-seq analysis with the known genome binding histone methyltransferase PRDM9 (56). Genome-wide comparison of all Rep and γH2AX occupied sites revealed that in the absence of pHelper, under conditions in which AAV2 does not replicate, the percentage of sites shared between Rep and γH2AX was approximately 71% (Fig. 2D; left panel). Under replicating conditions in the presence of pHelper, approximately 46% of Rep-bound regions intersected with γH2AX at 24 hpi, which corresponded with 35% of all γH2AX sites at 24 hpi (Fig. 2E; left panel). This suggested that under replicating conditions as more Rep was produced, a smaller percentage of Rep was found associated with limiting γH2AX sites. Alternatively, following replication a greater percentage of Rep may have been directly associated with the increased number of viral genomes. Jaccard analysis revealed that these intersections were statistically significant (Fig. 2D and E; right panels). Genome-wide analysis of called peaks in AAV2 infected cells expressing pHelper indicated that approximately 90% of the Rep peaks were centered in the 1 Mb vicinity of γH2AX peaks at 24 hpi (Fig. 2F).
Comparison of the intersection of two biological replicates of AAV2 V3C-seq with the Rep ChIP-seq analysis following infection of 293T cells in the absence of pHelper as described above revealed minimal intersections between these regions at 24 hpi (Fig. 2G; column 1). However, in the presence of pHelper at 24 hpi, there was an increase in the percentage of Rep binding sites (shared between two independent biological replicates) associated with cellular sites of AAV2 interaction (Fig. 2G; column 2; quantification of genome-wide heatmaps in Supplementary Material, Fig. S3B). V3C-seq analysis of 293T cells transduced with rAAV2 showed minimal correlation with identified Rep-bound cellular regions, similar to previously seen following transduction of U2OS cells (Fig. 2G; column 3). A finer magnification (Fig. 2H) of representative interaction peaks on chromosomes 1 and 19 showed that the AAV2 genome (represented as red peaks on the top tracks in Fig. 2H) localized to cellular regions that were bound by Rep (Fig. 2H; green middle tracks) and γH2AX (Fig. 2H; bottom dark red tracks) at distinct cellular sites on human chromosomes 1 and 19. Genome-wide comparison of cellular sites occupied by AAV2 in the absence and presence of pHelper revealed that 60% of cellular sites occupied by AAV2 in the absence of pHelper at 24hpi was shared with replicating AAV2 in the presence of pHelper, suggesting that these cellular sites might be bona fide regions of AAV2 persistence. However, these common sites only make up approximately 9% of AAV2-associated sites during replication, suggesting that these might represent additional regions for amplification (Fig. 2I; top). Genome-wide comparison of overlapping Rep and γH2AX sites with V3C-seq analysis showed that approximately 26% of cellular sites occupied by both Rep and γH2AX in the absence of pHelper had AAV2 associated within a 2 Mb window of each peak (Fig. 2J; left panel), which increased to approximately 36% as AAV2 replicated in the presence of pHelper (Fig. 2J; right panel). As mentioned, the Rep ChIP-seq could potentially include both AAV2-genome associated Rep as the viral genome associated with the cellular genome, as well as non-AAV2-genome associated Rep expressed from virus.
We then compared V3C-seq and ChIP-seq data in 293T cells, in the absence and presence of pHelper, with AAV2 integration sites previously identified in HeLa cells and human fibroblasts (25,26). We observed a strong correlation between these integration sites and AAV2 genome localization sites in the presence of pHelper at 24 hpi (Fig. 2K; column 2; quantification of genome-wide heatmaps in Supplementary Material, Fig. S3C). This was substantially higher than that of AAV2 localization in the absence of pHelper (Fig. 2K; column 1), or following infection of U2OS cells as described above. Notably, Rep binding (Fig. 2K; column 3) and γH2AX binding (Fig. 2K; column 4) to these potential integration regions were lower, and comparable with that of non-replicating AAV2 localization (Fig. 2K; column 1), while slightly higher than that of rAAV2 localization (Fig. 2K; column 5).
Ectopically expressed Rep colocalizes with pre-existing and induced sites of cellular DNA damage
Because Rep 68/78 is associated with the AAV2 genome, and since the viral genome localized to cellular sites of DNA damage, we asked whether Rep 68/78 could also localize to cellular DDR sites when expressed independently. In order to investigate ectopically expressed Rep 68/78 localization to the host genome, we performed ChIP-seq for Rep and γH2AX in stable U2OS and 293T cells inducibly expressing Rep 68/78. These lines were generated using the pINDUCER20 retroviral-based transfer vector that features a very low uninduced expression level, and is strongly induced following treatment with doxycycline (57) (Fig. 3A). As shown in the representative ChIP-seq samples for human chromosomes 1 and 6 (and all other chromosomes on the human genome in Supplementary Material, Fig. S5), ectopically expressed Rep, in two biological repeat experiments in both 293T and U2OS cells, strongly co-localized with γH2AX (compare Fig. 3B tracks 2–3 and 6–7, respectively), similar to results seen following AAV2 infection of 293T cells in the presence of pHelper at 24 hpi (Fig. 3B; tracks 4 and 5—reproduced from Fig. 2C, for comparison). Interestingly, in uninfected cycling 293T cells, the endogenous cellular γH2AX sites identified by ChIP-seq revealed a similar pattern as that in induced and infected 293T cells (Fig. 3B; compare tracks 1 with tracks 3 and 5). These results suggested, as previously seen for MVM (40) that endogenous sites of damage can provide initial target sites for Rep and likely genome localization, prior to any damage caused by infection, and that ectopic Rep 68/78 was unlikely to induce extensive additional directed cellular DNA breaks at distinct cellular sites in these cells. Additionally, the profile of Rep and γH2AX are distinct between U2OS and 293T cells, suggesting that factors other than cognate Rep-binding sequences contribute to the localization of Rep to distinct sites on the cellular genome. The cell-type differences in Rep- and γH2AX-binding patterns are readily observable at the 5′ end and middle of chromosome 6 in U2OS cells as well as the 3′ end of chromosome 1 in 293T cells (Fig. 3B).
Figure 3.
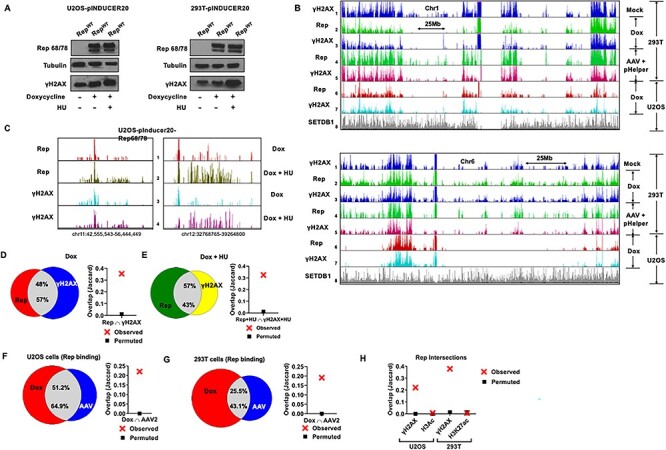
Ectopically expressed Rep colocalizes with pre-existing and induced sites of cellular DNA damage. (A) Stable doxycycline-inducible U2OS (left panel) and 293T (right panel) cell lines were induced for 24 h in the presence or absence of HU pulse for the last 12 h during induction and whole-cell lysates were subjected to western blot analysis using antibodies directed against the indicated proteins. (B) Representative Rep and γH2AX ChIP-seq tracks on human chromosome 1 (top panel) and chromosome 6 (bottom panel). ChIP-seq assays were carried out in 293T cells that were mock infected (track 1), transfected with pHelper and infected with AAV2 for 24 h (tracks 4–5) as well as in 293T and U2OS cells inducibly expressing Rep for 24 h (tracks 2–3 and 6–7, respectively). Genome-wide association of the chromatin modifier SETDB1 previously identified in U2OS cells was monitored as negative control (track 8). (C) Representative examples of Rep and γH2AX ChIP-seq peaks occupying distinct regions on human chromosomes 11 and 12 in U2OS cells inducibly expressing Rep for 24 h in the presence (tracks 2 and 4) or absence of HU pulse for the last 12 h during induction (tracks 1 and 3). (D, E) Venn diagrams representing whole-genome comparison of the overlap of Rep and γH2AX ChIP-seq peaks in stable Rep-expressing U2OS cells in the absence (D, left panel) or presence of HU (E, left panel). The statistical significance of the overlap is shown via Jaccard analysis (see Materials and Methods for description). (F, G) The overlap of Rep ChIP-seq peaks identified in the Rep-expressing stable U2OS (F, left panel) and 293T (G, left panel) cell lines was compared with Rep peaks identified at 24 hpi of AAV2 infection. The statistical significance of the respective overlap was evaluated by Jaccard analysis (F, G right panels). (H) The Rep ChIP-seq peaks identified in the stable doxycycline-inducible U2OS and 293T cells were compared with the corresponding γH2AX ChIP-seq peaks as well as published active chromatin marks acetylated histone H3 and Lysine 27-acetyled histone H3 respectively. The statistical significance of this overlap was determined by Jaccard analysis. The separation between red crosses and black squares reflect increased correlation of the intersections. All datasets are intersections of two independent biological replicates.
When additional DNA damage was introduced into the inducible U2OS cell lines with hydroxyurea (HU), we observed an increase in γH2AX adjacent to pre-existing γH2AX regions, as well as increased Rep binding to these regions (a detailed fine analysis of representative regions of chromosomes 11 and 12 are shown; Fig. 3C; compare tracks 3–4 and 1–2, respectively). When analyzed on a genome-wide basis, as new targets were induced by HU, the percentage of the limiting amount of Rep overlapping with γH2AX was reduced from 57% (Fig. 3D; left panel) to 43% (Fig. 3E; left panel). In the same experiments, the pre-existing 48% of γH2AX peaks that overlapped with Rep-binding sites (Fig. 3D; left panel) increased to 57% in the presence of HU (Fig. 3E; left panel). These results likely reflect that HU treatment both increases the number of sites of cellular DNA damage, as well as the abundance of ChIP-targeted γH2AX within, or adjacent, to existing damaged sites (58,59). These co-localizations were statistically significant, as determined by Jaccard analysis (Fig. 3D and E; right panels).
Comparison of the overlap between ectopically expressed Rep and γH2AX binding with the cellular genome, to the overlap seen during infection, allowed us to estimate the contribution of genome-associated Rep to this association during infection. As can be seen in Figure 3F, during AAV2 infection of U2OS cells, approximately 65% of Rep interactions at γH2AX sites during infection were at sites with which ectopically expressed Rep interacted with γH2AX in U2OS cells (intersection of two independent biological replicates with the respective statistical significance shown by Jaccard analysis on the right). For 293T cells, the congruency was approximately 43% (Fig. 3G). This suggested that during AAV2 infection, as much as 35% and 57% of Rep interactions with γH2AX, respectively, may have been dependent on its association with the viral genome, although there may be sites bound under both conditions.
Prior studies have suggested that the cellular RBEs are enriched in active chromatin sites that drive Rep-dependent AAV2 integration (25,26). To investigate this further, we computed the overlap of ectopically expressed Rep ChIP-seq peaks in U2OS and 293T cells with published active chromatin marks acetylated histone H3 (H3Ac) (60), and Lysine 27-acetyled histone H3 (H3K27ac) (61), respectively. As shown in Figure 3H, the Jaccard analysis of these comparisons revealed that Rep intersects more significantly with γH2AX than it does with H3Ac sites in U2OS cells (Fig. 3H; compare columns 1–2), or H3K27ac sites in 293T cells (Fig. 3H; compare columns 3–4).
Ectopically expressed Rep re-localizes to microirradiation-induced sites of cellular DNA damage
To confirm the localization of Rep 68/78 to sites of cellular DNA damage, we next assessed localization of ectopically expressed Rep 68/78 (RepWT) by immunofluorescence within seconds following the induction of DNA damage by laser microirradiation (40,43,62,63). As shown in Figure 4A, focused DNA damage generated by laser microirradiation 20 h post-induction of Rep expression led to the rapid re-localization of Rep from distinct nuclear foci to DNA breaks at induced stripes as defined by localization of γH2AX (Fig. 4A; compare panels 1–2). This confirmed that Rep could localize to cellular sites of DNA damage independent of infection. Treatment of cells with the pan ATM/ATR inhibitor caffeine (Fisher Biosciences), or the ATM inhibitor KU55933 (Selleck Chemicals) prior to laser microirradiation inhibited the re-localization of Rep to laser stripes (Fig. 4A; compare panel 2 with panels 3 and 4, respectively). The unrelated transcription factor NR5A2 (64), which does not have strong binding sites on the AAV2 genome or interact with the cellular DDR pathways, did not re-localize to laser microirradiated sites, and instead remained distinct from the induced γH2AX staining pattern (Fig. 4A; panel 5). Additionally, a previously characterized DNA binding-deficient mutant RepR107A/K136A (65), when expressed ectopically in parallel experiments (total expression levels of mutant R107A/K136A, and wild-type Rep shown in Fig. 4B), did not localize to DNA damage sites (Fig. 4C; compare panels 1–2). Taken together, these findings confirmed that AAV2 Rep 68/78 localized to cellular sites of DNA damage, and suggested that localization required its DNA-binding capacity, and may depend on local ATM-dependent signals in the vicinity of cellular DDR sites.
Figure 4.
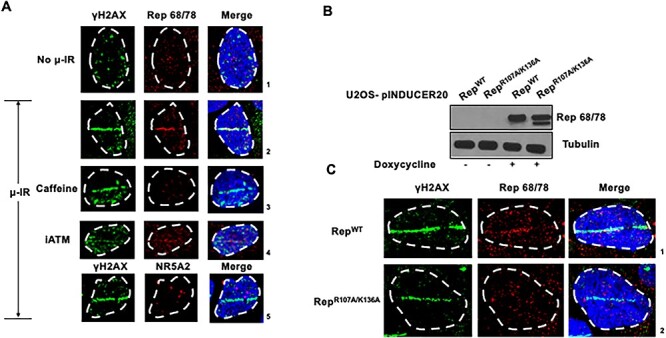
Ectopically expressed Rep re-localizes to microirradiation-induced sites of cellular DNA damage. (A) Laser micro-irradiation followed by immunofluorescence analysis of U2OS human osteosarcoma cells stably expressing Rep 68/78. Cells were induced with doxycycline for 20 h and either mock treated, treated with caffeine (2.5 mm) or treated with the ATM inhibitor (7.5 μm) for the duration of induction before focused DDR induction by laser micro-irradiation. The induced DDR site was monitored by γH2AX staining (green) and the expressed viral protein was visualized by Rep staining (red). The host transcription factor NR5A2, which do not have strong binding sites on the AAV2 genome, was monitored as a control (red). The cell nuclei were visualized by DAPI staining and the nuclear periphery was demarcated by dashed white line. Data are representative of two independent experiments, each imaging at least five fields of view containing 4–5 nuclei. (B) Stable doxycycline-inducible U2OS cell lines expressing wild-type Rep 68/78 (RepWT) or the DNA binding-deficient mutant RepR107A/K136A were induced for 24 h and whole-cell lysates were subjected to western blot analysis using antibodies directed against the indicated proteins. (C) U2OS cells that stably express RepWT or the DNA binding-deficient mutant RepR107A/K136A were induced with doxycycline for 20 h before focused DDR induction by laser micro-irradiation. Samples were then co-stained for γH2AX (green) and Rep (red). The cell nuclei were visualized by DAPI staining and the nuclear periphery was demarcated by dashed white line. Data are representative of two independent experiments, each imaging at least five fields of view containing 4–5 nuclei per field.
Discussion
Replication of AAV2 has been associated with the cellular DNA damage response. The viral genome has been shown to induce an ATR-dependent DDR on its own (38), and expression of Rep 68/78 themselves can trigger DDR signaling (37,39). During co-infection with Ad, in addition to AAV2 replication-induced DNA-PK-dependent effects on the cell, AAV2 replicates in the presence of a competing ATM-dependent DDR induced by Ad (37,66). In the absence of helper virus, under conditions of limited replication, a small fraction of AAV2 genomes are found integrated in the cellular chromosome (67), and these sites have been extensively mapped. However, how the majority of AAV2 genomes, both under replicative, and non-replicative conditions, directly interact with the cellular genome following infection is not known.
To begin to examine this question, we used a novel chromosome conformation capture assay adapted for use in-trans for lytic viruses (V3C-seq). Using this assay, we have previously shown that the autonomous parvovirus MVM localizes to cellular sites of DNA damage, many of which are subsets of early replicating fragile sites (ERFs), in order to establish and amplify its replication (40). Further we found that the MVM non-structural protein NS1 facilitates this localization following binding to the viral genome (43). Sites of DNA damage are typically replete with DNA replication and gene expression factors (41), and so may provide areas ready to immediately support virus replication. A similar approach was recently used to investigate the interaction of PRV with the cellular genome during its infection (42). In that study, the investigators found that the PRV genome was delivered to cellular sites of open chromatin and an active transcription zone by the host binding protein RUNX1, thus facilitating efficient access of PRV to host RNA polymerase II and aiding in its transcription (36).
In the present study, we found that following AAV2 infection of U2OS cells in the absence of helper functions, where the vast majority of the incoming genomes remained unintegrated, approximately 45% of the viral genomes localized to specific cellular regions exhibiting DNA damage as indicated by the presence of γH2AX. Because we were restricted to analysis in non-synchronous populations rather than focusing on cells primarily in S-phase, this number is likely an underestimate. In parallel experiments, transduced rAAV2 genomes associated substantially less frequently with cellular sites of DNA damage, implying that the viral hairpins were insufficient to target genomes to sites of DNA damage. During infection of U2OS cells, there was also a strong correlation between cellular sites of DNA damage and those sites occupied by Rep 68/78—approximately 90% of Rep ChIP-seq peaks were centered within a 1 Mb vicinity of γH2AX peaks at 24 hpi. Additionally, our results showed that approximately 55% of the cellular sites at which the AAV2 genome associated overlapped with sites exhibiting Rep binding.
During AAV2 infection of 293T cells, although the viral genomes significantly localized to sites of DNA damage, there was little difference in the presence or absence of helper functions. This may have been because the endogenous levels of damage were high, supplying available targets under both conditions. This is consistent with observation that the same significant overlap of γH2AX- and Rep 68/78-occupied cellular sites was seen under both of these conditions and also as seen for U2OS infections. It is important to note that although AAV2 replicates well in the presence of expressed helper functions, interactions within the host nucleus would not necessarily be expected to be the same as in the presence of Ad. Ad’s dramatic effect on the host DDR and its synergy with AAV2 as it establishes replication centers adds a significant level of complexity likely to confound approaches such as ours when applied to such co-infections.
VRC’s accumulate essential viral and cellular factors for viral gene expression and genome replication (32,33). Similarly, during productive infection, AAV2-VRCs take advantage of helper as well as cellular functions to promote its own replication while also inducing a cellular DDR (34–36). Our findings show that AAV2–VRC’s associate with cellular sites of DNA damage, and binding regions of ectopic Rep 68/78. Since ectopic expression of the large Rep proteins, Rep 78 and Rep 68 in the absence of viral infection can trigger a DDR signal, perhaps due to their nicking capabilities, it remains possible that some of the cellular DNA damage at these sites are caused directly by Rep 68/78 (37,39).
Because during infection both the AAV2 genome and Rep 68/78 localized to sites of DNA damage, Rep 68/78 localization results would likely reflect both AAV2-genome-associated, and soluble Rep 68/78. Therefore, we examined localization of Rep 68/78 expressed ectopically in the absence of infection. There was a highly significant overlap of Rep 68/78 and γH2AX ChIP-seq binding sites in both U2OS and 293T cells inducibly expressing Rep 68/78, indicating that Rep 68/78 could localize specifically to sites of DNA damage in the absence of infection. Our comparison of these results with results of Rep interaction during infection (Fig. 3F and G) implied that as much as 35% and 57%, respectively, of Rep 68/78 occupancy during infection was the result of genome-associated Rep generated during infection, although there certainly could be an overlap between sites bound under both conditions.
Prior studies had indicated that a key feature of the host genome that drives Rep 68/78 localization is the existence of RBE (25,26,67), and our results, discussed further below, indicated that Rep localization to sites of DNA damage required its DNA binding capability. However, because the association profiles of the AAV2 genome and Rep 68/78 were different in U2OS compared with 293T cells, it is unlikely that RBEs are the sole factor driving Rep 68/78 localization.
Additionally, we found that during infection Rep 68/78 ChIP-seq peaks colocalized with pre-existing γH2AX peaks. This suggested that endogenous sites of cellular DNA damage could participate in the localization of Rep 68/78 to distinct nuclear sites. Since there was significant overlap between the localization of Rep, γH2AX and the viral genome, it may be that endogenous sites of DNA damage may be initial targets for AAV2 replication. If so, endogenous levels of DNA damage may play a role in permissivity to AAV2 infection, as it has been shown for MVM (40).
When additional DNA damage was introduced into both U2OS and 293T Rep-expressing lines with HU there was an increase in γH2AX binding to sites of DNA damage and an increase in Rep 68/78 occupancy. It is difficult to distinguish whether this implied continued delivery to expanding sites of damage, or perhaps alternatively, re-localization of Rep 68/78 to such sites. However, when U2OS cells that inducibly expressed Rep 68/78 were treated with laser-induced microirradiation, Rep 68/78 could clearly be re-localized from extant sites to sites of DNA damage. In these experiments, cells were fixed for processing within seconds of irradiation, making it unlikely that Rep 68/78 found at stripes came from new synthesis. Treatment with either caffeine or a specific ATM inhibitor inhibited the re-localization, suggesting that Rep 68/78 re-localization to sites of damage may require components of the ATM DDR signaling pathway. In parallel experiments, a previously characterized Rep 68/78 DNA binding-deficient mutant (RepR107A/K136A) (65), inducibly expressed in U2OS cells, did not re-localize to sites of DNA damage, suggesting this function of Rep 68/78 may be required for its localization to these sites. This is in contrast to MVM NS1. Similar experiments showed that a DNA-binding mutant of NS1 could re-localize to sites of DNA damage; however, its DNA-binding function was required to transport DNA molecules to such sites (43). Whether Rep 68/78 functions to target the AAV2 genome to sites of DNA damage is currently under investigation.
Recombinant AAV2 gene therapy vectors are flanked on either end by the AAV2 ITRs, which serve as origins of replication for AAV in the presence of Rep proteins. Prior studies have shown that cellular DDR factors such as the MRN complex associates with the rAAV2 genome in distinct nuclear foci (44,45), which are superficially similar to AAV2 VRCs, and MVM-autonomous parvovirus associated replication bodies. Our V3C-seq analyses suggest that rAAV2 genomes are associated with cellular sites distinct from those exhibiting cellular DDR—at least as revealed by binding of γH2AX, and from the predominant localization sites of AAV2. It remains to be investigated whether Rep, helper functions, or cellular factors might synergize with the ITRs to facilitate the transport of the rAAV2 genomes to new cellular sites.
Less than 1% of the incoming AAV2 genomes integrate into the host genome (67). Importantly, these integration sites can vary between cell types (25,26), suggesting that RBE-independent factors such as chromatin may play a role in driving AAV localization to potential integration sites. We have found that during non-productive infection in U2OS and 293T cells, AAV2 genomes localize with only a subset of the previously identified AAV2 integration sites (30% for U2OS and 38% for 293Ts). This suggested that AAV2 localization sites observed in our studies are primarily regions at which non-integrated AAV2 localized, rather than cis-interactions of AAV2 genomes leading to integration at regions that flank these sites (compare lanes 1 in Figs 1I and 2K). It is interesting in this regard, that we observed a greater correlation to previously identified integration sites under conditions in which the virus replicates, in 293T cells co-transfected with pHelper. However, these results do not necessarily extrapolate to higher-than-expected integration during active AAV2 replication in the presence of natural Ad coinfection, which likely results in a different association profile. In 293T cells in the presence of pHelper, perhaps the enriched RBEs in these cellular regions preferentially serve as substrates for Rep-binding and cleavage early in infection, which may generate a cellular DDR that attracts additional Rep-bound AAV2 genomes. This would be consistent with the requirement of DNA binding function of Rep 68/78 in DDR localization. Furthermore, we may not be able to detect Rep at all of these nuclear sites in our assays if they are very early events during AAV2 replication.
It is important to note that our findings do not provide insights about the structure of the AAV2 episomes, many of which can recombine using host DDR proteins to form extrachromosomal bodies that are proximal to these cellular sites (68). Strikingly, even in the absence of the AAV2 genome, the AAV2 integration sites correlate with sites of Rep 68/78 and γH2AX binding, suggesting that at some point, these regions may accrue Rep-induced DNA damage prior to AAV2 integration [as previously postulated by Huser et al. (25,26)].
Materials and Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Kinjal Majumder (kmajumder@wisc.edu).
Cell lines and drug treatments
Human U2OS and 293T/FT cells were propagated in 5% Serum Plus (Sigma Aldrich) containing DMEM media (Gibco) supplemented with Gentamicin at 37°C and 5% carbon dioxide. Cell lines were routinely authenticated for mycoplasma contamination, and background levels of DNA damage detected by γH2AX staining. Lentivirus constructs designed to inducibly express wild-type Rep 68/78 (RepWT) or the DNA binding-deficient mutant (RepR107A/K136A) were generated by co-transfecting equal concentrations of HIV Gag/Pol, vesicular stomatitis virus glycoprotein G (VSV-G) and relevant pINDUCER20 plasmids containing RepWT or RepR107A/K136A respectively into 293FT cells for 48 h (57). Stable doxycycline-inducible U2OS and 293T cell lines were selected with 300 and 500 μg/ml of Geneticin (Gibco), respectively. pINDUCER20 lentivirus transformed cell lines were induced with 500 ng/ml doxycycline hydrochloride (MP Biomedicals) for the indicated amount of time. For ChIP-seq experiments, U2OS and 293T cells containing an integrated RepWT-pINDUCER20 cassette were treated with 1 mm Hydroxyurea (Sigma Aldrich) at 12 h post induction for 12 h. When indicated, 293FT and 293T cells inducibly expressing Rep 68/78 were transfected with 1 μg pHelper plasmid 24 h prior to infection and induction respectively. For laser microirradiation experiments, stable RepWT-pINDUCER20 transformed cells were treated with caffeine (Fisher Biosciences) or with the ATM kinase inhibitor KU55933 (Selleck Chemicals) at a final concentration of 2.5 mm and 7.5 μm, respectively. Cells were pre-treated with the inhibitors 1 h before induction and for the duration of induction.
Virus and virus infections
Wild-type AAV2 (wtAAV2) virus was a gift from Jude Samulski (UNC-Chapel Hill). rAAV2 viral particles (pAAV2.CMV.PI.EGFP.WPRE.bGH) were purchased from Addgene (viral prep 105 530-AAV2) and used to transduce U2OS cells. wtAAV2 infections and rAAV2 transductions were carried out at a multiplicity of infection (MOI) of 5000 and cells were harvested at the indicated time points.
Production and purification of rAAV-GFP vector
rAAV2-GFP vectors used to transduce 293T cells were produced as described below. The 293T cells were cultured in three 10-floor cell factories in DMEM supplemented with penicillin/streptomycin and 10% fetal bovine serum (FBS; Thermo Fisher Scientific). When approximately 85% confluent, cells were transfected at equimolar ratios of the plasmids expressing Rep/Capsid proteins, pHelper and rAAV-GFP using 25-kDa polyethyleneimine (PEI; Polysciences) as the transfection reagent. After 48 h of post-transfection, cells were harvested and lysed by five rapid freeze–thaw cycles, treated with DNase and protease, and subjected to three rounds of CsCl density gradient ultracentrifugation. Vector titers were calculated after each round of centrifugation using qPCR as a previously described (44). CsCl was removed from final fractions by dialysis in PBS overnight. The vector was finally stored at 4°C.
Plasmids and transfections
The Rep 68/78 ORF was cloned into the mammalian expression vector pCDNA3.1 (Invitrogen) as previously described (69). This vector was used as a template to generate pINDUCER20 cassette containing the RepWT ORF. Although the Rep ORF also contains the p19 promoter, its expression was undetectable in this context. The R107A and K136A mutations were introduced to the RepWT-pINDUCER20 cassette by site directed mutagenesis to generate the RepR107A/K136A mutant. pINDUCER20 reagents were a gift from Guang Hu (NIH/NIEHS) (57). Transient transfections were performed using LipoD293 transfection reagent (SignaGen Laboratories) unless otherwise stated. pAAV.CMV.PI.EGFP.WPRE.bGH was a gift from James M. Wilson (Addgene plasmid # 105530; http://n2t.net/addgene:105530; RRID:Addgene_105 530).
Laser microirradiation for immunofluorescence
Laser microirradiation experiments were performed on 1 million U2OS cells containing an integrated RepWT- or RepR107A/K136A-pINDUCER20 cassette cultured on glass bottomed dishes (MatTek Corporation) induced with doxycycline for 20 h and either mock treated, treated with caffeine (2.5 mm) or treated with the ATM inhibitor KU55933 (7.5 μm) for the duration of induction. Cells were sensitized with 3 μl Hoechst dye (Thermo Fisher Scientific) 5 min prior to microirradiation. Samples were irradiated as previously described (43) using a Leica TCP SP8 confocal microscope using 40× oil objective and 3× digital zoom with a 405 nm laser using 25% power at 40 Hz frequency for 1 frame per field of view. Regions of interest (ROIs) were selected across the nucleus, such that they did not traverse the nuclear membrane and processed immediately for immunofluorescence as described below.
Immunofluorescence assays
Cells were pre-extracted with CSK buffer (10 mm PIPES pH 6.8, 100 mm sodium chloride, 300 mm sucrose, 1 mm EGTA, 1 mm magnesium chloride) and CSK with 0.5% Triton X-100 for 3 min each before being fixed with 4% paraformaldehyde for 10 min at room temperature. Samples were washed briefly in PBS and permeabilized with 0.5% Triton X-100 in PBS for 10 min at room temperature. The samples were then blocked with 3% BSA in PBS for 1 h, incubated with primary antibody diluted in 3% BSA solution for 1 h, briefly washed in PBS and incubated with secondary antibody (Alexa Fluor-conjugated, Life Technologies) for 1 h. Samples were washed in PBS and mounted on slides with DAPI Fluoromount (Southern Biotech).
Antibodies
Mouse anti-γH2AX (EMD Millipore; 05-636), rabbit anti-γH2AX (Abcam; ab11174), rabbit anti-NR5A2 (Abcam; ab189876), anti-mouse AF488 secondary (Life Technologies; A11029), anti-rabbit AF488 secondary (Life Technologies; A11034), anti-mouse AF568 secondary (Life Technologies; A11031), anti-rabbit AF568 secondary (Life Technologies; A11036). Mouse anti-Rep monoclonal antibody IF11 (27), which detects all four AAV Rep proteins, was a gift from Nick Muzyczka (University of Florida) and was purified for ChIP-experiments using a GE HiTrap Protein G column according to manufacturer’s instructions. ChIP experiments with the IF11 antibody were optimized by ChIP-qPCR at AAVS1 in AAV2 infected 293T cells transfected with pHelper for 24 hpi.
Chromatin immunoprecipitation followed by sequencing
Cells were crosslinked with 1% formaldehyde for 10 min on a rocker at room temperature before being quenched with glycine (0.125 M) for 5 min on ice. Cells were collected in cold PBS followed by lysis in ChIP lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris–HCl, pH 8, protease inhibitors). The whole-cell lysates were sonicated using a Diagenode Bioruptor for 90 cycles (U2OS) or 105 cycles (293T) with 30 s on and 30 s off. The samples were centrifuged at 4°C for 10 min before the lysate was added to suspensions of antibodies and Protein A Dynabeads (Thermo Scientific). The samples were incubated by rotation at 4°C overnight before being washed with low salt wash (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris–HCl pH 8, 150 mm NaCl), high salt wash (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris–HCl pH 8, 500 mm NaCl), lithium chloride wash (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mm EDTA, 10 mm Tris–HCl pH 8) and twice with TE buffer before being eluted in SDS elution buffer (1% SDS, 0.1 M Sodium Bicarbonate). Following elution, the chromatin-antibody complexes were reverse-crosslinked with 100 mm sodium chloride heated at 65°C and Proteinase K for 2 h. The eluted DNA was purified using a Qiagen PCR purification kit and samples were eluted in 50 μl of Buffer EB (Qiagen).
Sequencing libraries were generated from ChIP DNA using the NEBNext Ultra II Library Prep Kit for Illumina, and the sonication quality was verified using Agilent Bioanalyzer. For ChIP-seq assays, up to 12 samples were pooled and sequenced on an Illumina Next Seq 500 instrument using 75 base-pair Single End Sequencing.
ChIP-seq samples were aligned to the human genome (build hg38) using Bowtie2 (70). The resultant SAM files were converted to BAM files using Samtools (71). BED files were generated using BEDtools (72). Peaks were called with EPIC analysis software using the SICER algorithm (73–75) according to default parameters. Called peaks that were shared between two independent biological replicates were identified using BEDtools software (72). Statistical significance of overlapping ChIP-seq peaks were performed using the Jaccard function on BEDtools software as previously described (43,76). The Jaccard analysis represents the ratio of the intersection of two sets (in this case, EPIC-called ChIP-seq peaks) to that of the union of the same sets. The resultant overlap is represented as a decimal between 0 (reflecting no overlap) and 1 (complete overlap). ‘Random’ BED files were permuted using BEDtools, such that they contained comparable number of peaks of similar size as determined by Rep ChIP-seq assays, ensuring that the size of the sets being compared were similar, which were subsequently sorted before calculation by Jaccard analysis.
Viral chromosome conformation capture followed by sequencing (V3C-seq)
V3C-seq assays were performed on 107 cultured U2OS or 293T cells infected with wtAAV2 or transduced with rAAV2 as previously described (4,40,43). Briefly, at the indicated time points, cells were cross-linked in 2% formaldehyde for 10 min with rotation, before quenching them in 0.125 M glycine for 5 min. Cells were washed in PBS, pelleted and lysed in NP40 lysis buffer (0.1% NP40, NaCl, Tris–HCl) and the resulting nuclei were resuspended in NEB 2.1 restriction enzyme buffer. The nuclei were permeabilized in 0.3% SDS for 1 h at 37°C, followed by sequestration of SDS in 2% Triton X-100. The samples were then digested in 400 U of Hind III restriction enzyme overnight. Digestion was continued on the next day with additional 300 U of Hind III, before inactivating the enzyme with 1% SDS at 65°C. SDS was sequestered with 1% Triton X-100, and chromatin was resuspended in 1.15× T4 DNA ligase reaction buffer. Intramolecular ligation of samples was carried out at room temperature for 4 h in the presence of 50 U of T4 DNA ligase. Crosslinks were reversed with Proteinase K overnight at 65°C. The 3C DNA was purified by phenol:chloroform:isoamyl alcohol extraction, isopropanol precipitation and resuspended in 200 μl Buffer EB (Qiagen). The 3C DNA was secondary digested with 100 U Nla III overnight at 37°C, before heat inactivated and circularized with 100 U of T4 DNA Ligase at room temperature overnight in 15 ml ligation reactions. Samples were then purified by phenol:chloroform:isoamyl alcohol, precipitated in isopropanol and resuspended in 200 μl Buffer EB. V3C DNA was further purified using a PCR purification kit (Qiagen) and resuspended in 100 μl Buffer EB. Inverse PCR was performed on the circularized DNA using primers within the Hind III- Nla III fragments on the AAV2/rAAV2 genomes described in Supplementary Material, File S1. Inverse PCR products were diluted 1:100 in TE buffer and used as templates for nested PCR reactions with primers described in Supplementary Material, File S1. Nested PCR products were used to prepare sequencing libraries using the NEBNext Ultra II Library Prep kit for Illumina (NEB). Samples were sequenced on a Illumina Next Seq 500 instrument using 75 base-pair single end sequencing.
V3C-seq samples were trimmed and aligned to the human genome (build hg38) using Bowtie2 (70). Bioinformatic codes are provided in Supplementary Material, File S2.
ChIP-seq and V3C-seq intersection analysis
The intersection of V3C-seq (described below) with ChIP-seq was performed using the deeptools package on the Galaxy project server (77). V3C-seq and ChIP-seq peaks, which were common between multiple biological replicates, were first computed using BEDtools (72). Deeptools was used to generate heatmaps that computed the presence of ChIP-seq peaks within 2 Mb window of the V3C-seq peaks. The 2 Mb window (1 Mb upstream and 1 Mb downstream) was chosen to detect the multi-megabase spans of γH2AX that span either side of DNA breaks on the host genome (78). The presence of V3C-seq peaks within this window was indicated by positive heatmap signals within the 2 Mb window. In order to account for statistical significance of the overlap within these windows, the bigwig files were compared with a randomly computed library of ChIP-seq peaks that contained the same number of sites and spanned the same average length as that of the Rep and γH2AX ChIP-seq experiments (labeled as ‘random’). The bigwig files that correlated within these windows were used to designate the ‘baseline’ levels in the analysis, and correlations above this were designated as statistically significant. All ChIP-seq and V3C-seq analysis was performed in two independent biological replicates as prescribed by the ENCODE consortium (79) and the common peaks for each timepoint/treatment was first determined using BEDtools. Additional bioinformatic codes used for the analysis have been provided in Supplementary Material, File S2.
Jaccard analysis
The statistical significance of the overlap of ChIP-seq peaks were computed using Jaccard analysis on the BEDtools program (72). This utilized ChIP-seq peaks that were shared between two independent biological replicates of the assay. In all shown figures, the intersection of the shared ChIP-seq peaks were represented as a red cross and labeled as ‘observed.’ This intersection was compared with bioinformatically called ChIP-seq peaks that represented an equivalent number of randomly generated locations on the human genome of the same size (labeled as ‘Permuted’; black square). Jaccard values range from 0 to 1, with 0 representing no correlation and 1 representing complete correlation, as described previously (43,76). In our Jaccard analysis, the overlap of ‘observed’ and ‘permuted’ Jaccard values represent absence of statistical significance, and separation between them represent statistical significance.
Data availability
All high-throughput sequencing data have been uploaded to the Gene Expression Omnibus (GEO) repository and are available under the accession number GSE178316.
Immunoblot analysis
Cells were harvested at the indicated timepoints, lysed in 1×dye (25 mm Tris pH 7.5, 2% SDS, 2 mm EDTA, 6% glycerol, 20 mm DTT, bromophenol blue) and sheared using a 25 G × 5/8-inch, 1-ml needle-syringe (BD Biosciences). The whole-cell lysates were boiled for 10 min at a 100°C-heat block and equal volumes of samples were loaded per well for western blot analysis.
Southern blot analysis
Cells were harvested at the indicated timepoints, pelleted and resuspended in Southern lysis buffer (2% SDS, 150 mm NaCl, 10 mm Tris pH 8.0, 1 mm EDTA). Cells were proteinase K treated for 2 h at 37°C, and sheared using 25 G × 5/8-inch, 1-ml needle-syringe (BD Biosciences). Total DNA content in the samples was quantified using Nanodrop, equal amount of DNA loaded per well and electrophoresed on a 1% agarose gel for 16 h at 35 V. Samples were transferred to a nitrocellulose membrane and hybridized with randomly primed radiolabeled AAV2 probe.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank members of the Pintel Lab for valuable discussion and Lisa Burger for expert technical assistance. Laser micro-irradiation and subsequent confocal images were acquired at the University of Missouri Molecular Cytology Core facility. High-throughput sequencing services were performed at the University of Missouri DNA Core Facility.
Conflict of Interest statement. The authors declare that they have no conflict of interest.
Contributor Information
Maria Boftsi, Pathobiology Area Graduate Program, University of Missouri-Columbia, Columbia, MO 65211, USA; Christopher S. Bond Life Sciences Center, University of Missouri-Columbia, Columbia, MO 65211, USA.
Fawn B Whittle, Christopher S. Bond Life Sciences Center, University of Missouri-Columbia, Columbia, MO 65211, USA.
Juexin Wang, Christopher S. Bond Life Sciences Center, University of Missouri-Columbia, Columbia, MO 65211, USA; Department of Electrical Engineering and Computer Science, University of Missouri-Columbia, Columbia, MO 65211, USA.
Phoenix Shepherd, Institute for Molecular Virology, University of Wisconsin-Madison, Madison, WI 53706, USA.
Lisa R Burger, Christopher S. Bond Life Sciences Center, University of Missouri-Columbia, Columbia, MO 65211, USA.
Kevin A Kaifer, Christopher S. Bond Life Sciences Center, University of Missouri-Columbia, Columbia, MO 65211, USA; Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri-Columbia, Columbia, MO 65211, USA.
Christian L Lorson, Christopher S. Bond Life Sciences Center, University of Missouri-Columbia, Columbia, MO 65211, USA; Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri-Columbia, Columbia, MO 65211, USA.
Trupti Joshi, Christopher S. Bond Life Sciences Center, University of Missouri-Columbia, Columbia, MO 65211, USA; Department of Electrical Engineering and Computer Science, University of Missouri-Columbia, Columbia, MO 65211, USA; MU Informatics Institute, University of Missouri-Columbia, Columbia, MO 65211, USA; Department of Health Management and Informatics, School of Medicine, University of Missouri-Columbia, Columbia, MO 65211, USA.
David J Pintel, Christopher S. Bond Life Sciences Center, University of Missouri-Columbia, Columbia, MO 65211, USA; Molecular Microbiology and Immunology, School of Medicine, University of Missouri-Columbia, Columbia, MO 65211, USA.
Kinjal Majumder, Institute for Molecular Virology, University of Wisconsin-Madison, Madison, WI 53706, USA; McArdle Laboratory for Cancer Research, University of Wisconsin School of Medicine and Public Health, Madison, WI, 53706, USA; University of Wisconsin-Carbone Cancer Center, Madison, WI, USA.
Funding
Computational analysis was performed on the high-performance computing infrastructure provided by Research Computing Support Services and in part by the National Science Foundation under grant number CNS-1429294 at the University of Missouri, Columbia MO. Work in the Pintel Laboratory was supported by National Institute of Health grants AI046458 and AI116595 to D.J.P. K.M. was supported during this work by a Ruth L. Kirschstein Postdoctoral Individual National Research Service Award (AI131468), National Institute of Health K99/R00 Pathway to Independence Award (AI148511), Office of Vice Chancellor for Research and Graduate Education and the Human Cancer Virology Program at the University of Wisconsin- Carbone Cancer Center.
References
- 1. Wang, D., Tai, P.W.L. and Gao, G. (2019) Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov., 18, 358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berns, K.I. and Giraud, C. (1996) Biology of adeno-associated virus. Curr. Top. Microbiol. Immunol., 218, 1–23. [DOI] [PubMed] [Google Scholar]
- 3. Srivastava, A., Lusby, E.W. and Berns, K.I. (1983) Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol., 45, 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Majumder, K., Boftsi, M. and Pintel, D.J. (2019) Viral chromosome conformation capture (V3C) assays for identifying trans-interaction sites between lytic viruses and the cellular genome. Bio. Protoc., 9, e3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Earley, L.F., Powers, J.M., Adachi, K., Baumgart, J.T., Meyer, N.L., Xie, Q., Chapman, M.S. and Nakai, H. (2017) Adeno-associated virus (AAV) assembly-activating protein is not an essential requirement for capsid assembly of AAV serotypes 4, 5, and 11. J. Virol., 91, e01980–e02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao, M., You, H. and Hermonat, P.L. (2014) The X gene of adeno-associated virus 2 (AAV2) is involved in viral DNA replication. PLoS One, 9, e104596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hermonat, P.L., Santin, A.D., De Greve, J., De Rijcke, M., Bishop, B.M., Han, L., Mane, M. and Kokorina, N. (1999) Chromosomal latency and expression at map unit 96 of a wild-type plus adeno-associated virus (AAV)/Neo vector and identification of p81, a new AAV transcriptional promoter. J. Hum. Virol., 2, 359–368. [PubMed] [Google Scholar]
- 8. Ogden, P.J., Kelsic, E.D., Sinai, S. and Church, G.M. (2019) Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science, 366, 1139–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chejanovsky, N. and Carter, B.J. (1989) Mutagenesis of an AUG codon in the adeno-associated virus rep gene: effects on viral DNA replication. Virology, 173, 120–128. [DOI] [PubMed] [Google Scholar]
- 10. Im, D.S. and Muzyczka, N. (1989) Factors that bind to adeno-associated virus terminal repeats. J. Virol., 63, 3095–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Im, D.S. and Muzyczka, N. (1992) Partial purification of adeno-associated virus Rep78, Rep52, and Rep40 and their biochemical characterization. J. Virol., 66, 1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Surosky, R.T., Urabe, M., Godwin, S.G., McQuiston, S.A., Kurtzman, G.J., Ozawa, K. and Natsoulis, G. (1997) Adeno-associated virus Rep proteins target DNA sequences to a unique locus in the human genome. J. Virol., 71, 7951–7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. King, J.A., Dubielzig, R., Grimm, D. and Kleinschmidt, J.A. (2001) DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. EMBO J., 20, 3282–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cotmore, S.F. and Tattersall, P. (1987) The autonomously replicating parvoviruses of vertebrates. Adv. Virus Res., 33, 91–174. [DOI] [PubMed] [Google Scholar]
- 15. Geoffroy, M.C. and Salvetti, A. (2005) Helper functions required for wild type and recombinant adeno-associated virus growth. Curr. Gene Ther., 5, 265–271. [DOI] [PubMed] [Google Scholar]
- 16. Meier, A.F., Tobler, K., Leisi, R., Lkharrazi, A., Ros, C. and Fraefel, C. (2021) Herpes simplex virus co-infection facilitates rolling circle replication of the adeno-associated virus genome. PLoS Pathog., 17, e1009638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Janovitz, T., Klein, I.A., Oliveira, T., Mukherjee, P., Nussenzweig, M.C., Sadelain, M. and Falck-Pedersen, E. (2013) High-throughput sequencing reveals principles of adeno-associated virus serotype 2 integration. J. Virol., 87, 8559–8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petri, K., Gabriel, R., Agundez, L., Fronza, R., Afzal, S., Kaeppel, C., Linden, R.M., Henckaerts, E. and Schmidt, M. (2015) Presence of a trs-like motif promotes rep-mediated wild-type adeno-associated virus type 2 integration. J. Virol., 89, 7428–7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schnepp, B.C., Jensen, R.L., Chen, C.L., Johnson, P.R. and Clark, K.R. (2005) Characterization of adeno-associated virus genomes isolated from human tissues. J. Virol., 79, 14793–14803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kotin, R.M., Siniscalco, M., Samulski, R.J., Zhu, X.D., Hunter, L., Laughlin, C.A., McLaughlin, S., Muzyczka, N., Rocchi, M. and Berns, K.I. (1990) Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA., 87, 2211–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Samulski, R.J., Zhu, X., Xiao, X., Brook, J.D., Housman, D.E., Epstein, N. and Hunter, L.A. (1991) Targeted integration of adeno-associated virus (AAV) into human chromosome 19. EMBO J., 10, 3941–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feng, D., Chen, J., Yue, Y., Zhu, H., Xue, J. and Jia, W.W. (2006) A 16bp rep binding element is sufficient for mediating rep-dependent integration into AAVS1. J. Mol. Biol., 358, 38–45. [DOI] [PubMed] [Google Scholar]
- 23. Linden, R.M., Winocour, E. and Berns, K.I. (1996) The recombination signals for adeno-associated virus site-specific integration. Proc. Natl. Acad. Sci. USA., 93, 7966–7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weitzman, M.D., Kyöstiö, S.R., Kotin, R.M. and Owens, R.A. (1994) Adeno-associated virus (AAV) rep proteins mediate complex formation between AAV DNA and its integration site in human DNA. Proc. Natl. Acad. Sci. USA., 91, 5808–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hüser, D., Gogol-Döring, A., Lutter, T., Weger, S., Winter, K., Hammer, E.M., Cathomen, T., Reinert, K. and Heilbronn, R. (2010) Integration preferences of wildtype AAV-2 for consensusrep-binding sites at numerous loci in the human genome. PLoS Pathog., 6, e1000985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hüser, D., Gogol-Döring, A., Chen, W. and Heilbronn, R. (2014) Adeno-associated virus type 2 wild-type and vector-mediated genomic integration profiles of human diploid fibroblasts analyzed by third-generation PacBio DNA sequencing. J. Virol., 88, 11253–11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Henckaerts, E. and Linden, R.M. (2010) Adeno-associated virus: a key to the human genome? Future Virol., 5, 555–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nash, K., Chen, W. and Muzyczka, N. (2008) Complete in vitro reconstitution of adeno-associated virus DNA replication requires the minichromosome maintenance complex proteins. J. Virol., 82, 1458–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cotmore, S.F. and Tattersall, P. (2014) Parvoviruses: small does not mean simple. Annu. Rev. Virol., 1, 517–537. [DOI] [PubMed] [Google Scholar]
- 30. Ni, T.H., McDonald, W.F., Zolotukhin, I., Melendy, T., Waga, S., Stillman, B. and Muzyczka, N. (1998) Cellular proteins required for adeno-associated virus DNA replication in the absence of adenovirus coinfection. J. Virol., 72, 2777–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Im, D.S. and Muzyczka, N. (1990) The AAV origin binding protein Rep68 is an ATP-dependent site-specific endonuclease with DNA helicase activity. Cell, 61, 447–457. [DOI] [PubMed] [Google Scholar]
- 32. Charman, M. and Weitzman, M.D. (2020) Replication compartments of DNA viruses in the nucleus: location, location, location. Viruses, 12, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmid, M., Speiseder, T., Dobner, T. and Gonzalez, R.A. (2014) DNA virus replication compartments. J. Virol., 88, 1404–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hunter, L.A. and Samulski, R.J. (1992) Colocalization of adeno-associated virus Rep and capsid proteins in the nuclei of infected cells. J. Virol., 66, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meier, A.F., Fraefel, C. and Seyffert, M. (2020) The interplay between adeno-associated virus and its helper viruses. Viruses, 12, 662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weitzman, M.D., Fisher, K.J. and Wilson, J.M. (1996) Recruitment of wild-type and recombinant adeno-associated virus into adenovirus replication centers. J. Virol., 70, 1845–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schwartz, R.A., Carson, C.T., Schuberth, C. and Weitzman, M.D. (2009) Adeno-associated virus replication induces a DNA damage response coordinated by DNA-dependent protein kinase. J. Virol., 83, 6269–6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jurvansuu, J., Raj, K., Stasiak, A. and Beard, P. (2005) Viral transport of DNA damage that mimics a stalled replication fork. J. Virol., 79, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berthet, C., Raj, K., Saudan, P. and Beard, P. (2005) How adeno-associated virus Rep78 protein arrests cells completely in S phase. Proc. Natl. Acad. Sci. USA., 102, 13634–13639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Majumder, K., Wang, J., Boftsi, M., Fuller, M.S., Rede, J.E., Joshi, T. and Pintel, D.J. (2018) Parvovirus minute virus of mice interacts with sites of cellular DNA damage to establish and amplify its lytic infection. Elife, 7, e37750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hashiguchi, K., Matsumoto, Y. and Yasui, A. (2007) Recruitment of DNA repair synthesis machinery to sites of DNA damage/repair in living human cells. Nucleic Acids Res., 35, 2913–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xiao, K., Xiong, D., Chen, G., Yu, J., Li, Y., Chen, K., Zhang, L., Xu, Y., Xu, Q., Huang, X. et al. (2021) RUNX1-mediated alphaherpesvirus-host trans-species chromatin interaction promotes viral transcription. Sci. Adv., 7, eabf8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Majumder, K., Boftsi, M., Whittle, F.B., Wang, J., Fuller, M.S., Joshi, T. and Pintel, D.J. (2020) The NS1 protein of the parvovirus MVM Aids in the localization of the viral genome to cellular sites of DNA damage. PLoS Pathog., 16, e1009002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cervelli, T., Palacios, J.A., Zentilin, L., Mano, M., Schwartz, R.A., Weitzman, M.D. and Giacca, M. (2008) Processing of recombinant AAV genomes occurs in specific nuclear structures that overlap with foci of DNA-damage-response proteins. J. Cell Sci., 121, 349–357. [DOI] [PubMed] [Google Scholar]
- 45. Lentz, T.B. and Samulski, R.J. (2015) Insight into the mechanism of inhibition of adeno-associated virus by the Mre11/Rad50/Nbs1 complex. J. Virol., 89, 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raj, K., Ogston, P. and Beard, P. (2001) Virus-mediated killing of cells that lack p53 activity. Nature, 412, 914–917. [DOI] [PubMed] [Google Scholar]
- 47. Vogel, R., Seyffert, M., Strasser, R., de Oliveira, A.P., Dresch, C., Glauser, D.L., Jolinon, N., Salvetti, A., Weitzman, M.D., Ackermann, M. and Fraefel, C. (2012) Adeno-associated virus type 2 modulates the host DNA damage response induced by herpes simplex virus 1 during coinfection. J. Virol., 86, 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zentilin, L., Marcello, A. and Giacca, M. (2001) Involvement of cellular double-stranded DNA break binding proteins in processing of the recombinant adeno-associated virus genome. J. Virol., 75, 12279–12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Luo, Y. and Qiu, J. (2013) Parvovirus infection-induced DNA damage response. Future Virol., 8, 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adeyemi, R.O., Landry, S., Davis, M.E., Weitzman, M.D. and Pintel, D.J. (2010) Parvovirus minute virus of mice induces a DNA damage response that facilitates viral replication. PLoS Pathog., 6, e1001141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ruiz, Z., Mihaylov, I.S., Cotmore, S.F. and Tattersall, P. (2011) Recruitment of DNA replication and damage response proteins to viral replication centers during infection with NS2 mutants of minute virus of mice (MVM). Virology, 410, 375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Seyffert, M., Glauser, D.L., Schraner, E.M., de Oliveira, A.P., Mansilla-Soto, J., Vogt, B., Büning, H., Linden, R.M., Ackermann, M. and Fraefel, C. (2017) Novel mutant AAV2 rep proteins support AAV2 replication without blocking HSV-1 Helpervirus replication. PLoS One, 12, e0170908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smith, R.H. and Kotin, R.M. (2000) An adeno-associated virus (AAV) initiator protein, Rep78, catalyzes the cleavage and ligation of single-stranded AAV ori DNA. J. Virol., 74, 3122–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chiorini, J.A., Wiener, S.M., Owens, R.A., Kyöstió, S.R., Kotin, R.M. and Safer, B. (1994) Sequence requirements for stable binding and function of Rep68 on the adeno-associated virus type 2 inverted terminal repeats. J. Virol., 68, 7448–7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Iyengar, S., Ivanov, A.V., Jin, V.X., Rauscher, F.J. and Farnham, P.J. (2011) Functional analysis of KAP1 genomic recruitment. Mol. Cell. Biol., 31, 1833–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Imbeault, M., Helleboid, P.Y. and Trono, D. (2017) KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature, 543, 550–554. [DOI] [PubMed] [Google Scholar]
- 57. Meerbrey, K.L., Hu, G., Kessler, J.D., Roarty, K., Li, M.Z., Fang, J.E., Herschkowitz, J.I., Burrows, A.E., Ciccia, A., Sun, T. et al. (2011) The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. USA., 108, 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barlow, J.H., Faryabi, R.B., Callén, E., Wong, N., Malhowski, A., Chen, H.T., Gutierrez-Cruz, G., Sun, H.W., McKinnon, P., Wright, G. et al. (2013) Identification of early replicating fragile sites that contribute to genome instability. Cell, 152, 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lyu, X., Chastain, M. and Chai, W. (2019) Genome-wide mapping and profiling of γH2AX binding hotspots in response to different replication stress inducers. BMC Genomics, 20, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Walz, S., Lorenzin, F., Morton, J., Wiese, K.E., von Eyss, B., Herold, S., Rycak, L., Dumay-Odelot, H., Karim, S., Bartkuhn, M. et al. (2014) Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature, 511, 483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schuijers, J., Manteiga, J.C., Weintraub, A.S., Day, D.S., Zamudio, A.V., Hnisz, D., Lee, T.I. and Young, R.A. (2018) Transcriptional dysregulation of MYC reveals common enhancer-docking mechanism. Cell Rep., 23, 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kong, X., Ball, A.R. and Yokomori, K. (2017) The use of laser microirradiation to investigate the roles of Cohesins in DNA repair. Methods Mol. Biol., 1515, 227–242. [DOI] [PubMed] [Google Scholar]
- 63. Mistrik, M., Vesela, E., Furst, T., Hanzlikova, H., Frydrych, I., Gursky, J., Majera, D. and Bartek, J. (2016) Cells and stripes: a novel quantitative photo-manipulation technique. Sci. Rep., 6, 19567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cobo, I., Martinelli, P., Flández, M., Bakiri, L., Zhang, M., Carrillo-de-Santa-Pau, E., Jia, J., Sánchez-Arévalo Lobo, V.J., Megías, D., Felipe, I. et al. (2018) Transcriptional regulation by NR5A2 links differentiation and inflammation in the pancreas. Nature, 554, 533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Alex, M., Weger, S., Mietzsch, M., Slanina, H., Cathomen, T. and Heilbronn, R. (2012) DNA-binding activity of adeno-associated virus rep is required for inverted terminal repeat-dependent complex formation with herpes simplex virus ICP8. J. Virol., 86, 2859–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shah, G.A. and O'Shea, C.C. (2015) Viral and cellular genomes activate distinct DNA damage responses. Cell, 162, 987–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hüser, D., Weger, S. and Heilbronn, R. (2002) Kinetics and frequency of adeno-associated virus site-specific integration into human chromosome 19 monitored by quantitative real-time PCR. J. Virol., 76, 7554–7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Choi, H.S., Kim, J.W., Cha, Y.N. and Kim, C. (2006) A quantitative nitroblue tetrazolium assay for determining intracellular superoxide anion production in phagocytic cells. J. Immunoassay Immunochem., 27, 31–44. [DOI] [PubMed] [Google Scholar]
- 69. Miller, C.L. and Pintel, D.J. (2001) The NS2 protein generated by the parvovirus minute virus of mice is degraded by the proteasome in a manner independent of ubiquitin chain elongation or activation. Virology, 285, 346–355. [DOI] [PubMed] [Google Scholar]
- 70. Langmead, B. and Salzberg, S.L. (2012) Fast gapped-read alignment with bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Subgroup, G. P. D. P (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Quinlan, A.R. and Hall, I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stovner, E.B. and Sætrom, P. (2019) epic2 efficiently finds diffuse domains in ChIP-seq data. Bioinformatics, 35, 4392–4393. [DOI] [PubMed] [Google Scholar]
- 74. Xu, S., Grullon, S., Ge, K. and Peng, W. (2014) Spatial clustering for identification of ChIP-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. Methods Mol. Biol., 1150, 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zang, C., Schones, D.E., Zeng, C., Cui, K., Zhao, K. and Peng, W. (2009) A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics, 25, 1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kim, J., Sturgill, D., Sebastian, R., Khurana, S., Tran, A.D., Edwards, G.B., Kruswick, A., Burkett, S., Hosogane, E.K., Hannon, W.W. et al. (2018) Replication stress shapes a protective chromatin environment across fragile genomic regions. Mol. Cell, 69, 36–47.e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ramírez, F., Ryan, D.P., Grüning, B., Bhardwaj, V., Kilpert, F., Richter, A.S., Heyne, S., Dündar, F. and Manke, T. (2016) deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res., 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rogakou, E.P., Pilch, D.R., Orr, A.H., Ivanova, V.S. and Bonner, W.M. (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem., 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- 79. Landt, S.G., Marinov, G.K., Kundaje, A., Kheradpour, P., Pauli, F., Batzoglou, S., Bernstein, B.E., Bickel, P., Brown, J.B., Cayting, P. et al. (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res., 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All high-throughput sequencing data have been uploaded to the Gene Expression Omnibus (GEO) repository and are available under the accession number GSE178316.